

ABSTRACT
A number of environmental factors from nutrition to toxicants have been shown to promote the epigenetic transgenerational inheritance of disease and phenotypic variation. This requires alterations in the germline (sperm or egg) epigenome. Previously, the agricultural fungicide vinclozolin was found to promote the transgenerational inheritance of sperm differential DNA methylation regions (DMRs) termed epimutations that help mediate this epigenetic inheritance. The current study was designed to investigate the developmental origins of the transgenerational DMRs during gametogenesis. Male control and vinclozolin lineage F3 generation rats were used as a source of embryonic day 13 (E13) primordial germ cells, embryonic day 16 (E16) prospermatogonia, postnatal day 10 (P10) spermatogonia, adult pachytene spermatocytes, round spermatids, caput epididymal spermatozoa, and caudal sperm. The DMRs between the control versus vinclozolin lineage samples were determined for each developmental stage. The top 100 statistically significant DMRs for each stage were compared. The developmental origins of the caudal epididymal sperm DMRs were assessed. The chromosomal locations and genomic features of the different stage DMRs were investigated. In addition, the DMR associated genes were identified. Previous studies have demonstrated alterations in the DMRs of primordial germ cells (PGCs). Interestingly, the majority of the DMRs identified in the current study for the caudal sperm originated during the spermatogenic process in the testis. A cascade of epigenetic alterations initiated in the PGCs appears to be required to alter the epigenetic programming during spermatogenesis to modify the sperm epigenome involved in the transgenerational epigenetic inheritance phenomenon.
KEYWORDS: Epigenetic, transgenerational, inheritance, primordial germ cells, spermatogenesis, sperm, epimutation
Introduction
One of the first observations that environmental factors could promote the epigenetic transgenerational inheritance of disease involved the agricultural fungicide vinclozolin, and an analysis of testis spermatogenic cell viability [1]. Subsequently, a large number of environmental factors such as caloric restriction, high-fat diets, stress, and a variety of different toxicants have been shown to promote transgenerational epigenetic inheritance [2–4]. This non-genetic form of inheritance allows the environment to impact the health and evolution of species [3–5]. The epigenetic transgenerational inheritance of phenotypic variation requires epigenetic alterations of the germline (sperm or egg) to mediate this epigenetic inheritance phenomenon. This transgenerational germline transmission of epigenetic information involves a number of different types of epigenetic processes [6]. A germline with an altered epigenome has the capacity to alter the early embryo’s stem cell’s epigenome and transcriptome that can subsequently impact the epigenomes and transcriptomes of all derived somatic cells [2]. Therefore, an altered sperm epigenome has the capacity to transmit phenotypes transgenerationally.
Epigenetics is defined as ‘molecular factors and processes around the DNA that regulate genome activity independent of DNA sequence and that are mitotically stable’ [7]. Different epigenetic processes including DNA methylation [8–10], non-coding RNAs [11,12], and histone modifications and retention [10,13,14] have been shown to be involved in the transgenerational phenomenon. Recently, alterations in all three of these processes have been observed in the transgenerational transmission of epimutations via sperm [6,15]. The majority of observations have been provided using sperm due to the ability to isolate large numbers of purified sperm. In contrast, the inability to isolate large numbers of oocytes has limited molecular studies in the female germline. However, experiments have demonstrated that epigenetic inheritance can also be transmitted through the female germline [16,17]. The current study investigates the developmental origins of transgenerational sperm DNA methylation alterations.
The male embryonic germline is derived from its stem cell population, the primordial germ cells (PGCs), that develop in the epiblast and migrate to the genital ridge to colonize the indifferent gonad [18]. Upon gonadal sex determination, the PGCs develop into the male or female germline lineage based on the sex of the gonad [19]. The early developing testis germ cells develop into prospermatogonia during the male gonadal sex differentiation period by embryonic day 16 (E16) in the rat gonad [20,21]. Following birth, the prospermatogonia differentiate into spermatogonia by postnatal day 10 (P10). Following the onset of puberty, the initial wave of spermatogenesis in the testis begins and the spermatogonia differentiate into the spermatocyte stages of development, including the meiotic stage of pachytene spermatocytes. Following meiosis, the round spermatid stage is initiated which then leads to the final stages of spermiogenesis and spermiation when the spermatozoa are released into the lumen of the seminiferous tubule [22]. The spermatozoa then enter the epididymis and undergo further maturation beginning at the caput portion of the epididymis and during transit to the final caudal stage of the epididymis where the sperm develop the capacity for motility [23,24]. The mature caudal epididymal sperm are then stored in the vas deferens prior to ejaculation or degradation. The male germline developmental periods used for the current study were the E13 primordial germ cells (PGCs), E16 prospermatogonia, P10 spermatogonia, and adult pachytene spermatocytes, round spermatids, caput epididymal spermatozoa, and caudal epididymal sperm in order to investigate the developmental origins of the transgenerational sperm differential DNA methylation regions (DMRs).
Epigenetic programming occurs during these male germline developmental periods. The migrating PGCs undergo an erasure of DNA methylation that will then be restored and re-methylated during gonadal sex determination to create the male germline and prospermatogonia [25]. In addition, following fertilization there is a DNA methylation erasure to generate the stem cells in the early embryo, which then remethylate in a cell type-specific manner [25,26]. The DNA methylation erasure is thought to, in part, reset deleterious epigenetics in the germline [27]. However, imprinted gene DNA methylation sites and induced transgenerational epimutations appear to be protected from this DNA methylation erasure [28]. During spermatogenesis in the adult testis, there are additional epigenetic programming events [29,30]. Some of the first examples of this male germline epigenetic programming were observed with imprinted genes [31,32]. Some imprinted gene DMR develop early in the embryonic period, while others develop later during spermatogenesis. The current study was designed to further investigate [33] the developmental programming, or origins, of the environmentally induced transgenerational epigenetic alterations observed in sperm.
The developmental programming and origins of vinclozolin induced epigenetic transgenerational inheritance of sperm DMRs termed epimutations are investigated. The hypothesis tested is that a dynamic cascade of epigenetic changes occurs during development, such that the majority of sperm DMRs originate at later spermatogenic stages. A previous study has demonstrated epigenetic alterations in the PGCs, however, these PGC DMRs were distinct from the epimutations observed in sperm [34]. Observations also indicated distinct regulation of the different members of the DNA methyltransferase (DNMT) gene family [34]. Therefore, sperm epimutations appear to develop at later stages of epigenetic programming. A recent study using DDT (dichlorodiphenyltrichloroethane) induced epigenetic transgenerational inheritance of sperm epimutations identified the developmental origins of DMRs during different stages of germline development [33]. Therefore, the induction of a cascade of epigenetic programming throughout gametogenesis appears to be required to promote the sperm epigenome alterations. The current study with vinclozolin induced transgenerational inheritance demonstrates that sperm DMRs also originate during both spermatogenesis and earlier stages of germline development, but at distinct developmental stages. This is a genome-wide analysis of epigenetic programming during gametogenesis for transgenerational sperm epimutations.
Results
The experimental design involved the development of F3 generation control and vinclozolin lineage male rats in order to isolate various germ cell stages for epigenetic analysis. The F0 generation, gestating female rats at 90 days of age, were transiently exposed to vinclozolin or a vehicle control during embryonic days 8–14 when the PGCs were migrating to colonize the indifferent gonad and during the early stages of gonadal sex determination, as previously described [18,25,34]. The F1 generation offspring were obtained, and at 90 days of age were bred within the control and vinclozolin lineages, respectively, to generate the F2 generation (grand offspring), which were then bred at 90 days of age to obtain the F3 generation (great-grand offspring) for each control and vinclozolin lineage. Since epigenetics can be transmitted in a parent of origin allelic manner through the germline [31], an intercross within the lineage was used to optimize the epigenetic alterations and phenotypes developed in the transgenerational F3 generation. No cousin or sibling breeding was used to avoid any inbreeding artefacts. Only the F0 generation gestating females were transiently exposed to vinclozolin, which also directly exposed the F1 generation fetus and the germline within the F1 generation fetus that will generate the F2 generation. Therefore, the transgenerational F3 generation is the first generation not directly exposed [35], and was used to study the developmental origins of the transgenerational sperm epimutations. The F3 generation control and vinclozolin lineage male rats were aged to the embryonic day 13 (E13) stage for primordial germ cell isolation, to the embryonic day 16 (E16) stage for prospermatogonia cell isolation, to postnatal day 10 (P10) prepubertal stage for spermatogonial cell isolation, and to adult 120 days for pachytene spermatocyte isolation, round spermatid isolation, and caput epididymal spermatozoa and cauda sperm isolation. The isolation procedure involved a gravity sedimentation StaPut protocol described in the Methods [36,37], and direct isolation for the epididymal sperm collections [8,13]. Testis pools, each from different individual sets of animals from different litters, were obtained from 5 to 6 males per pool for the E13 or E16 stage, 6–7 males per pool for the P10 stage, 3 adult males per pool for the spermatocyte or spermatid stages, and 3 adult males per pool for the caput or cauda epididymal sperm stages. Therefore, 3–7 different males from different litters were used in each of the different pools analysed for both the control and vinclozolin lineages. Three different pools were used for all stages except the PGC stage that used two pools. The DNA was isolated from each pool for subsequent epigenetic analysis.
The analysis of DMRs was accomplished with a methylated DNA immunoprecipitation (MeDIP) procedure, followed by next-generation sequencing for an MeDIP-Seq protocol, as described in the Methods [8]. The control versus vinclozolin lineage sequence data was compared to identify the DMRs for each developmental stage of the male germline. The DMRs at various p-value thresholds are presented for each developmental stage in Figure 1. An EdgeR p-value of p < 1e−05 was selected for all stages, except the pachytene spermatocytes, for which a p < 1e−07 was used for further analysis of the DMRs. The altered p-value for the pachytene spermatocytes was selected to allow a more balanced comparative DMR analysis for the DMR characteristics, but was not altered for the subsequent developmental analysis. These p-value DMRs correlated with an FDR (false discovery rate) adjusted p-values of 0.1 or less for all developmental stages, except the pachytene spermatocytes, which had an FDR of <0.001. Not all DMRs met this adjusted p-value threshold with the percent of DMR meeting this FDR p-value 0.1 being 47% in the E16, 25% in the P10, 100% in pachytene, 67% in round spermatids, 62% in caput, and 100% in cauda sperm. The All Windows involves all DMRs with at least one 100 bp region (window) with statistical significance while the Multiple Windows represents ≥2 significant 100 bp windows, Figure 1. The All Window DMRs were used for all subsequent analyses. Most developmental stages showed approximately 100 DMRs except the cauda sperm, which showed 356 DMRs, and the pachytene spermatocytes, which showed greater than 10-fold more DMRs at p < 10−5 than the other stages, Supplemental Tables S1–S7. Due to this increase in the pachytene spermatocyte DMRs, the remaining DNA was used to repeat the MeDIP-Seq analysis and similar data was obtained, indicating that the increase was not due to a technical issue. As mentioned, for the pachytene spermatocytes, a p < 1e−07 threshold was selected to provide a more balanced comparison between the stages for subsequent data analysis. Other data analysis such as the cauda sperm DMR developmental origins did not use this adjusted p-value. Clearly the epigenetic alteration was higher at the pachytene spermatocyte stage, which may reflect a high variation in DNA methylation at this stage of development.
Figure 1.
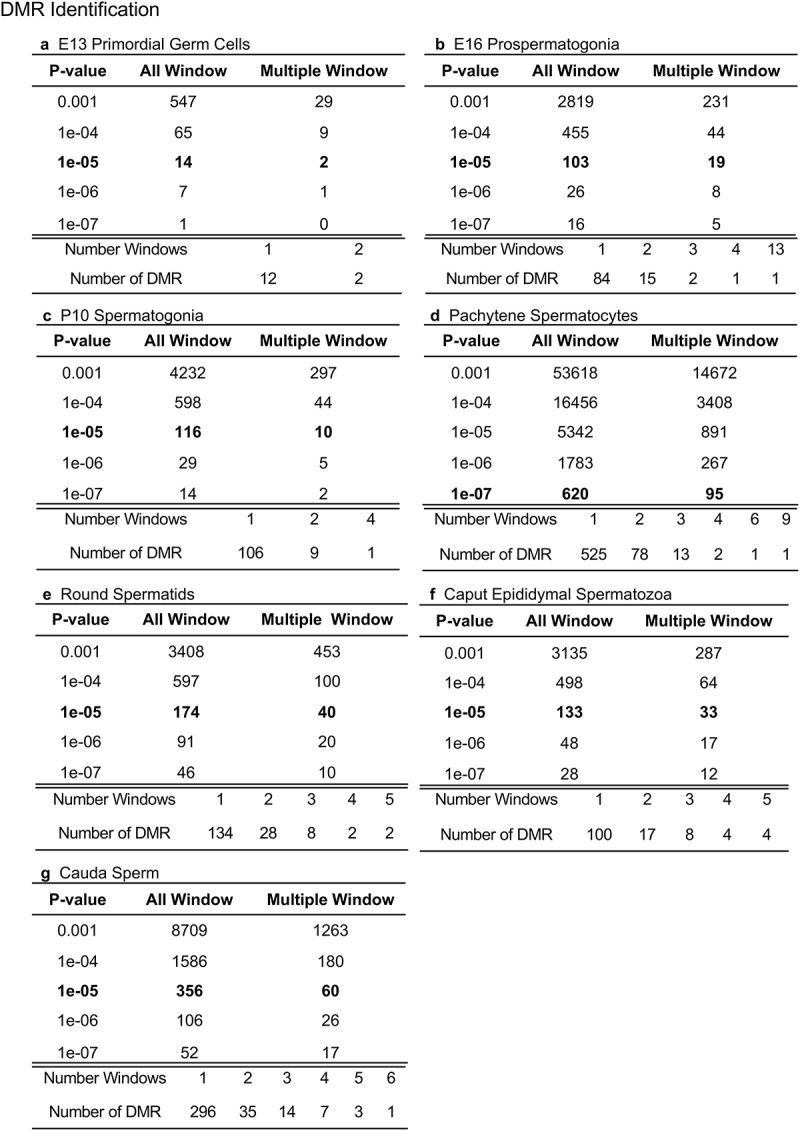
DMR analysis. The number of DMRs found using different p-value cut-off thresholds. The All Window column shows all DMRs. The Multiple Window column shows the number of DMRs containing ≥2 significant windows. The number of DMR with the number of significant windows at a p-value threshold bolded is presented. (a) E13 primordial germ cells. (b) E16 prospermatogonia. (c) P10 spermatogonia. (d) Pachytene spermatocytes. (e) Round spermatids. (f) Caput epididymal spermatozoa. (g) Cauda sperm.
The chromosomal locations of the DMRs at each developmental stage are presented in Figure 2. The red arrowheads indicate the location of a DMR with the black boxes indicating clusters of DMRs. Generally, all the chromosomes contained DMRs. The genomic features of the DMRs for each stage of development were investigated. The CpG density of the DMRs at all stages was 1–5 CpG per 100 bp with 1 CpG per 100 bp being predominant, Figure 3. This is characteristic of a low-density CpG desert [38], which has been previously correlated with transgenerational DMRs. The lengths of the DMRs at all the developmental stages were between 1 and 3 kb, with 1 kb length being predominant, Figure 4. Therefore, the DMRs were generally 1 kb in size with approximately 10 CpGs, as previously observed [38]. The overlap between the various developmental stage DMRs was examined and is presented in Figure 5. Interestingly, the DMRs were found to be primarily stage specific with only a few DMRs overlapping between the stages with a p < 1e−05. The caput and cauda epididymal sperm had the highest level of overlap with 45 DMRs, Figure 5(b) and Supplemental Table S8. This represented over 30% of the caput epididymal spermatozoa DMRs. Observations suggest a cascade of epigenetic programming occurs throughout male germline development, such that DMRs developed at all the different stages.
Figure 2.
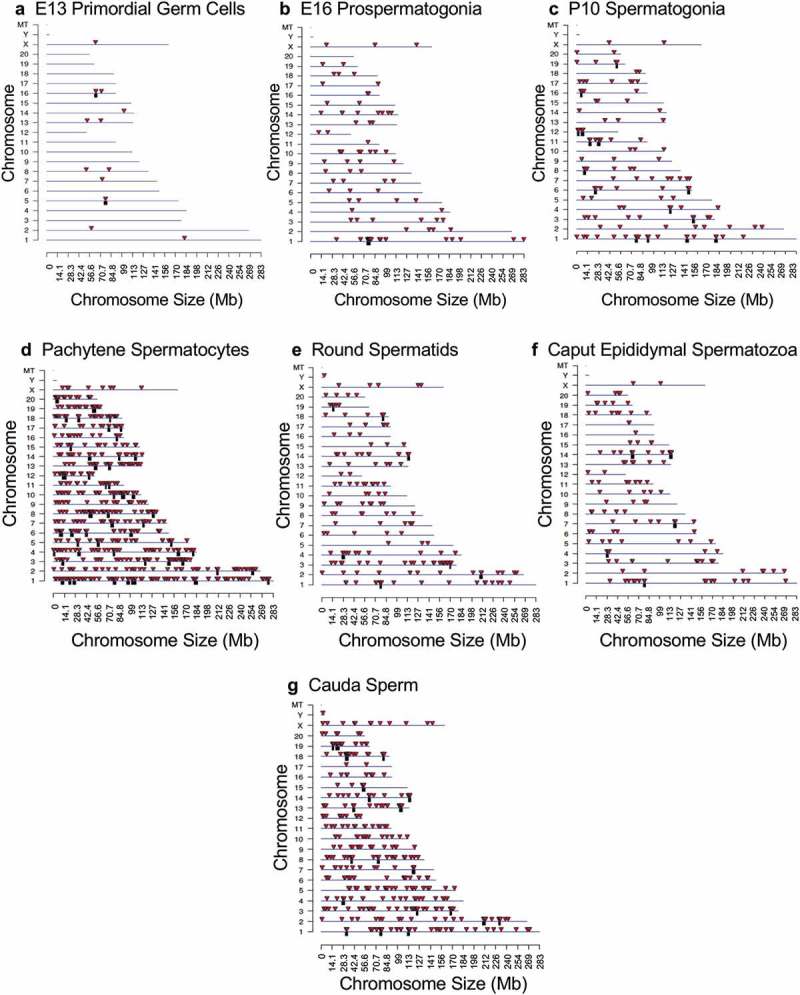
DMR chromosome location. The DMR locations on the individual chromosomes are presented with red arrowheads and DMR clusters indicated with black boxes. The chromosome number versus the chromosome size (megabase) is presented. (a) E13 primordial germ cells. (b) E16 prospermatogonia. (c) P10 spermatogonia. (d) Pachytene spermatocytes. (e) Round spermatids. (f) Caput epididymal spermatozoa. (g) Cauda sperm.
Figure 3.
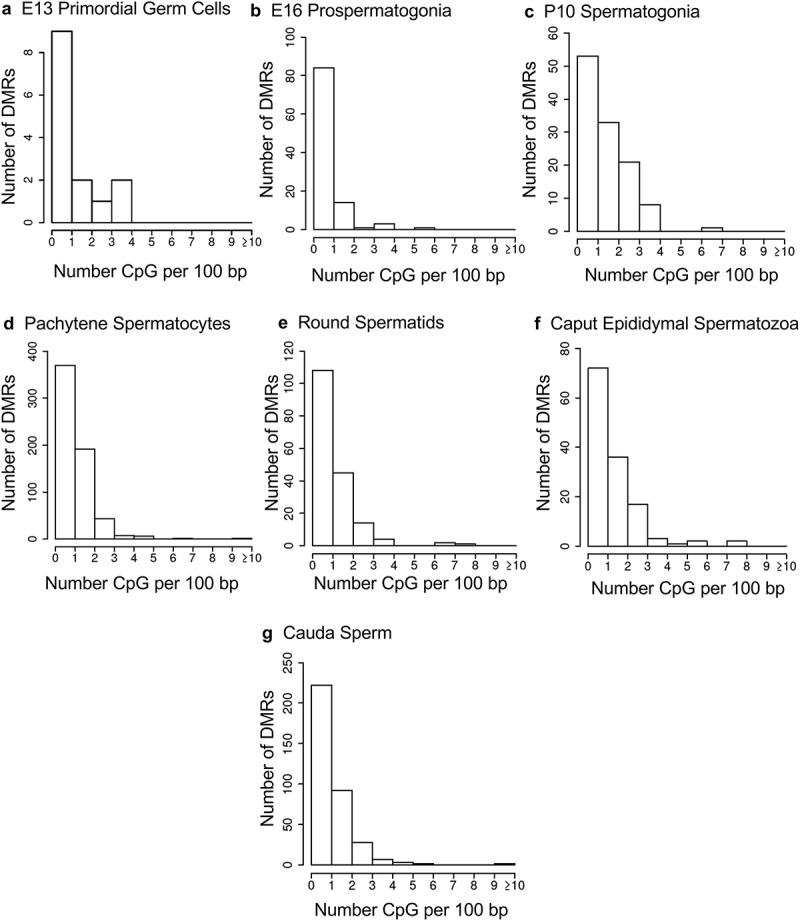
DMR CpG density. The number of DMRs at different CpG densities (CpG per 100 bp) are presented. (a) E13 primordial germ cells. (b) E16 prospermatogonia. (c) P10 spermatogonia. (d) Pachytene spermatocytes. (e) Round spermatids. (f) Caput epididymal spermatozoa. (g) Cauda sperm.
Figure 4.
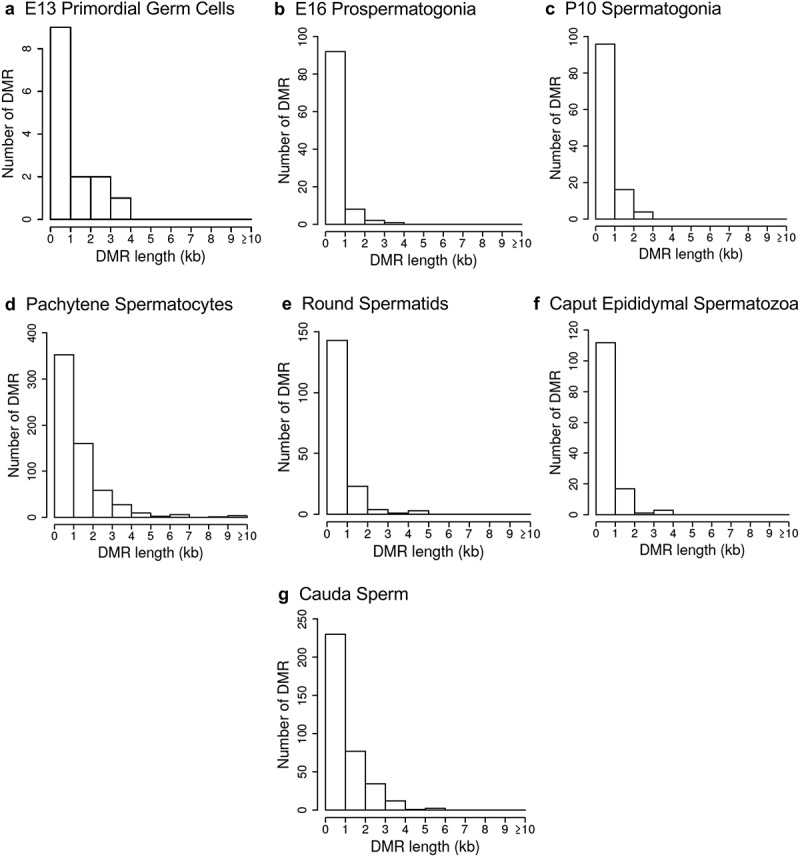
DMR length. The number of DMR at different DMR lengths in kilobases (kb). (a) E13 primordial germ cells. (b) E16 prospermatogonia. (c) P10 spermatogonia. (d) Pachytene spermatocytes. (e) Round spermatids. (f) Caput epididymal spermatozoa. (g) Cauda sperm.
Figure 5.
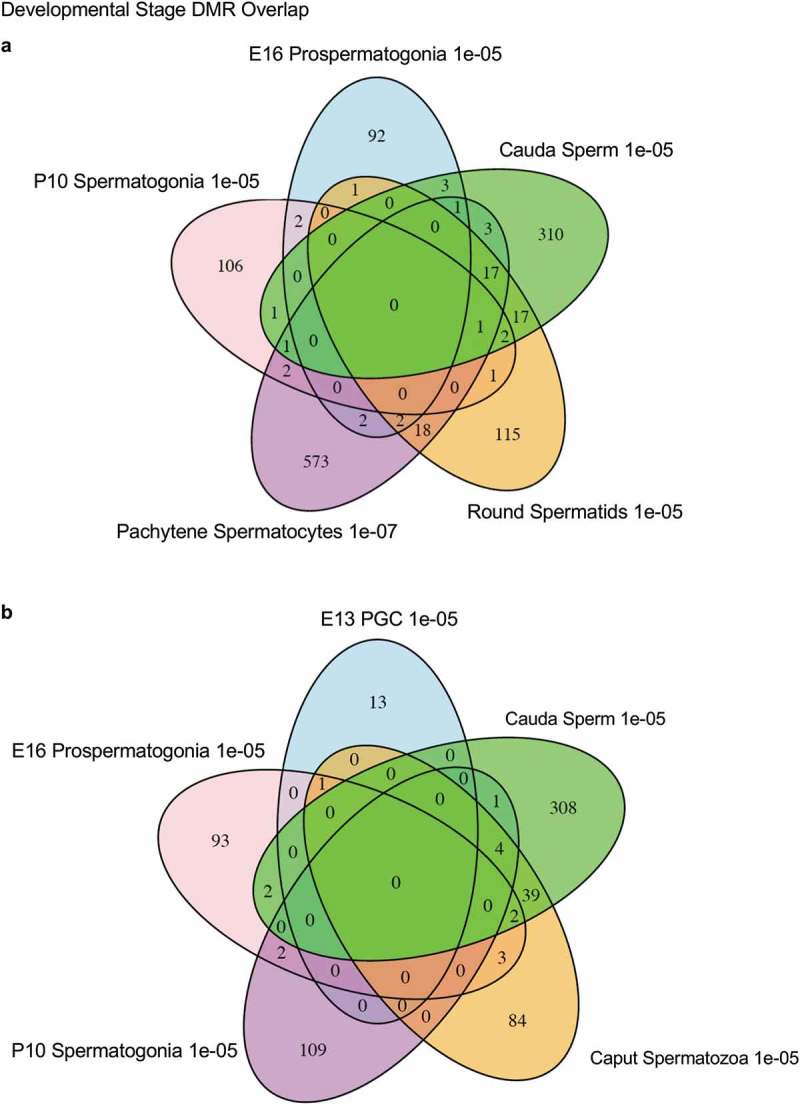
DMR developmental stage overlaps. The DMRs for the different developmental stages (a) are compared to identify overlapping DMRs in the Venn diagram. (b) The comparison of caput and cauda epididymal sperm DMRs in a Venn diagram with PGCs and spermatogonia.
Specific DNA methylation alterations of the DMRs for each stage of development were investigated to identify the patterns of change developmentally. The top 100 statistically significant DMRs at each stage of development were examined in regard to the percent mean (i.e., RPKM) read depth between the control versus vinclozolin lineage DMR data. The percent mean read depth values for each DMR were separated into control greater than vinclozolin data (representing a decrease in DNA methylation) and vinclozolin greater than control data (representing an increase in DNA methylation), which are presented as a percent scaled mean read depth, Figures 6 and 7. The early stage E13 primordial germ cells, E16 prospermatogonia, P10 spermatogonia and pachytene spermatogonia data are presented in Figure 6. The 100 most significant PGC DMRs included 60 DMRs with decreased DNA methylation (control > vinclozolin) and 40 DMRs with increased DNA methylation (vinclozolin > control), Figure 6(a,b). The E16 prospermatogonia DMRs included 64 DMRs with decreased DNA methylation (control > vinclozolin) and 36 DMRs with increased DNA methylation (vinclozolin > control), Figure 6(c,d). For both data sets the PGC or E16 DMR methylation alterations generally increased or decreased by the next stage, and then became mixed with no distinct patterns at later stages. Although a few DMRs of both populations remained elevated, the majority showed a reduced presence at later stages of development. The P10 spermatogonia DMRs included 44 DMRs with decreased DNA methylation (control > vinclozolin) and 56 with increased DNA methylation (vinclozolin > control) in regard to percent mean read depth, Figure 6(e,f). There was a dramatic alteration in DNA methylation between the prospermatogonia to spermatogonia stages, and between the spermatogonia stage and later stages of development. Therefore, the E16 prospermatogonia and PGCs or P10 spermatogonia stage DMRs were for the most part unique between those stages of development. The pachytene spermatocyte DMRs had predominantly an increase in DNA methylation (vinclozolin > control) with 89 DMRs compared to only 11 DMRs with a decrease in DNA methylation (control > vinclozolin), Figure 6(g,h). Therefore, DNA methylation was primarily increased in the pachytene spermatocyte DMRs. Interestingly, both pachytene spermatocyte data set DMRs were more consistent with fewer changes in DNA methylation than observed at earlier and later developmental stages. The round spermatid DMRs also showed increased DNA methylation in 68 DMRs (vinclozolin > control), and decreased DNA methylation in 32 DMRs (control > vinclozolin), Figure 7(a,b). Similar to the pachytene spermatocytes, the round spermatid DMRs were also more consistent between the developmental stages for both data sets, with the exception of the prospermatogonia stage. The caput epididymal spermatozoa DMRs included 65 DMRs with decreased DNA methylation (control > vinclozolin) and 35 DMRs with increased DNA methylation (vinclozolin > control), Figure 7(c,d). The caput spermatozoa DMRs were relatively consistent with DMRs in the spermatocytes, round spermatids and cauda sperm, but were distinct from those in the prospermatogonia and spermatogonia stages of development. The cauda epididymal sperm DMRs included 55 DMRs with decreased DNA methylation (control > vinclozolin) and 45 DMRs with increased DNA methylation (vinclozolin > control), Figure 7(e,f). As seen with the caput spermatozoa DMRs, the cauda epididymal sperm DMRs were also relatively consistent with DNA methylation characteristics for the pachytene spermatocytes, round spermatids, and caput spermatozoa DMRs. Although the caput spermatozoa and cauda sperm DMRs were generally more consistent between the developmental stages, the earlier stages were more dynamic and showed examples of DMRs that appear, are lost, and reappear later. Therefore using the top 100 significant DMRs the cauda sperm DMRs have more consistent DNA methylation patterns during spermatogenesis and epididymal sperm maturation, but are generally distinct from the PGC, spermatogonia and prospermatogonia stages.
Figure 6.
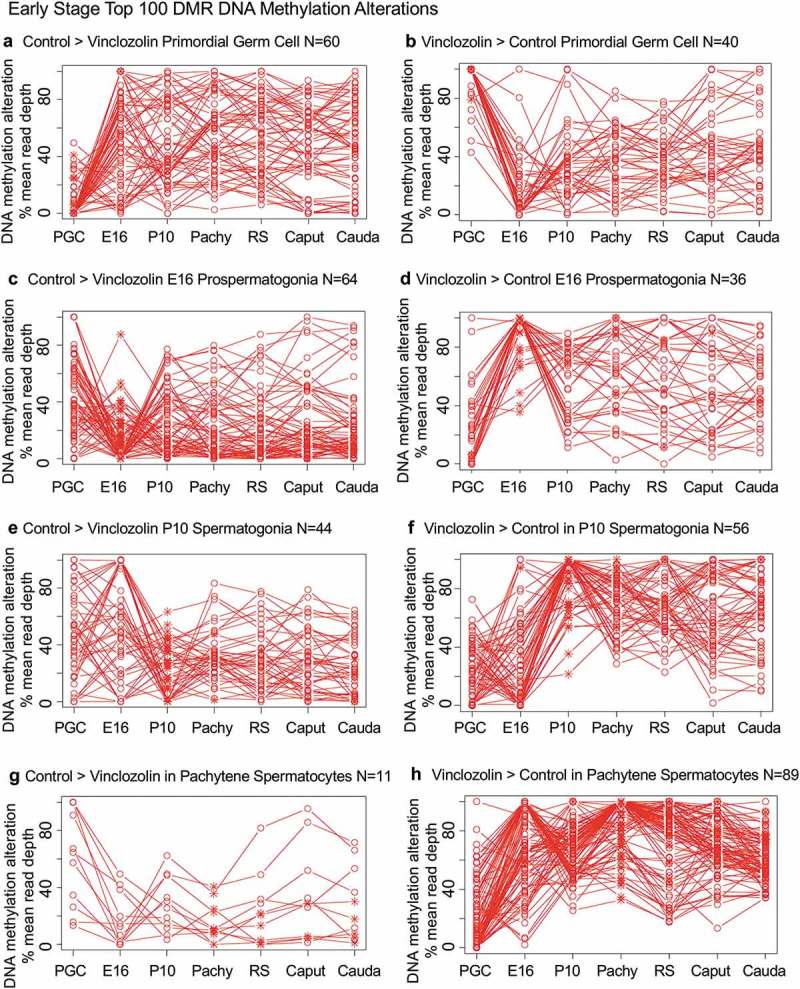
DMR developmental timelines. Top 100 most statistically significant DMR developmental alterations (E13 primordial germ cells, E16 prospermatogonia, P10 spermatogonia, pachytene spermatocyes). The DMRs are separated into two groups based on whether scaled RPKM read depth is elevated in the control or in the exposure lineage. The vinclozolin percent (%) scaled RPKM read depth developmental timelines are presented. (a,b) Top 100 DMRs in the primordial germ cell comparison. (c,d) Top 100 DMRs in the E16 prospermatogonia comparison. (e,f) Top 100 DMRs in the P10 spermatogonia comparison. (g,h) Top 100 DMRs in the pachytene spermatocytes comparison.
Figure 7.
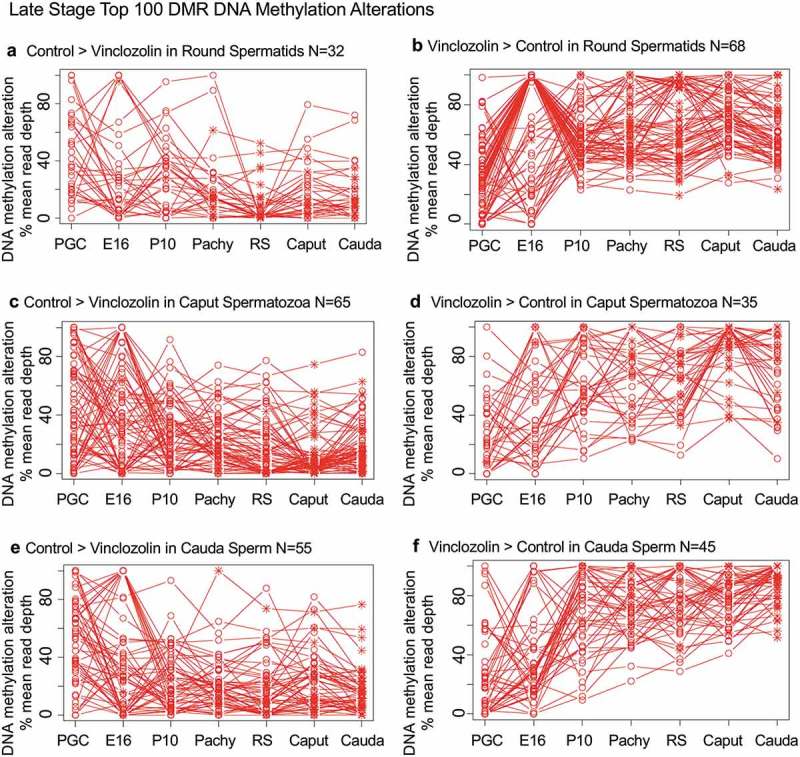
DMR developmental timelines. Top 100 most statistically significant DMR developmental alterations (round spermatids, caput, cauda). The DMRs are separated into two groups based on whether scaled RPKM read depth is elevated in the control or in the exposure. The vinclozolin percent (%) scaled RPKM read depth developmental timelines are presented. (a,b) Top 100 DMRs in the round spermatids comparison. (c,d) Top 100 DMRs in the caput comparison. (e,f) Top 100 DMRs in the cauda comparison.
The developmental origins of the cauda sperm DMRs were investigated by determining when these DNA methylation alterations developed within the germline stages examined. In the cauda sperm, there were 356 DMRs at p < 1e−05. For the analysis of the developmental origins of these 356 DMRs the statistical stringency was reduced to p < 0.05 in order to identify the first stage the DMR appeared. Figure 8(a) shows that 43 of the DMRs were detected in the PGCs, 84 of the DMRs detected in the cauda sperm originated at the E16 prospermatogonia stage, while 69 DMRs originated at the P10 spermatogonia stage, 86 DMRs originated at the spermatocyte stage, 17 DMRs originated at the round spermatid stage, 9 DMRs originated at the epididymal caput spermatozoa stage, and 48 DMRs originated at the cauda sperm stage, Supplemental Table S9. Figure 8(a) depicts the developmental origins and number of DMR at each stage of development using a less stringent (p < 0.05) criteria than the previous analyses. Although the majority of DMR originated and were maintained, a number appeared, were lost, and reappeared at later stages of development, Supplemental Table S10. The various groups were most diverse for the prospermatogonia, but the majority were simply maintained. The origins of the cauda sperm DMRs were dynamic during these developmental periods, Supplemental Table S10. Therefore, the majority of cauda sperm DMRs originated in the prospermatogonia, spermatogonia and spermatocyte stages, and fewer originated in the round spermatids and caput epididymal spermatozoa stages, Figure 8(a). This correlated with the dramatic shifts in DNA methylation that occur between the prospermatogonia stage, spermatogonia stage and spermatocyte stage, Figure 6, as well as the more consistent DNA methylation patterns that are maintained between the spermatocytes through cauda sperm stages, Figure 7. There were 48 DMRs that developed between the epididymal caput spermatozoa and cauda sperm stages. This indicates that a cascade (i.e., unique developmental origins) of epigenetic and genetic transcriptome changes occurs during these stages of male germline development to generate the transgenerational sperm epimutations, Figure 8(b).
Figure 8.
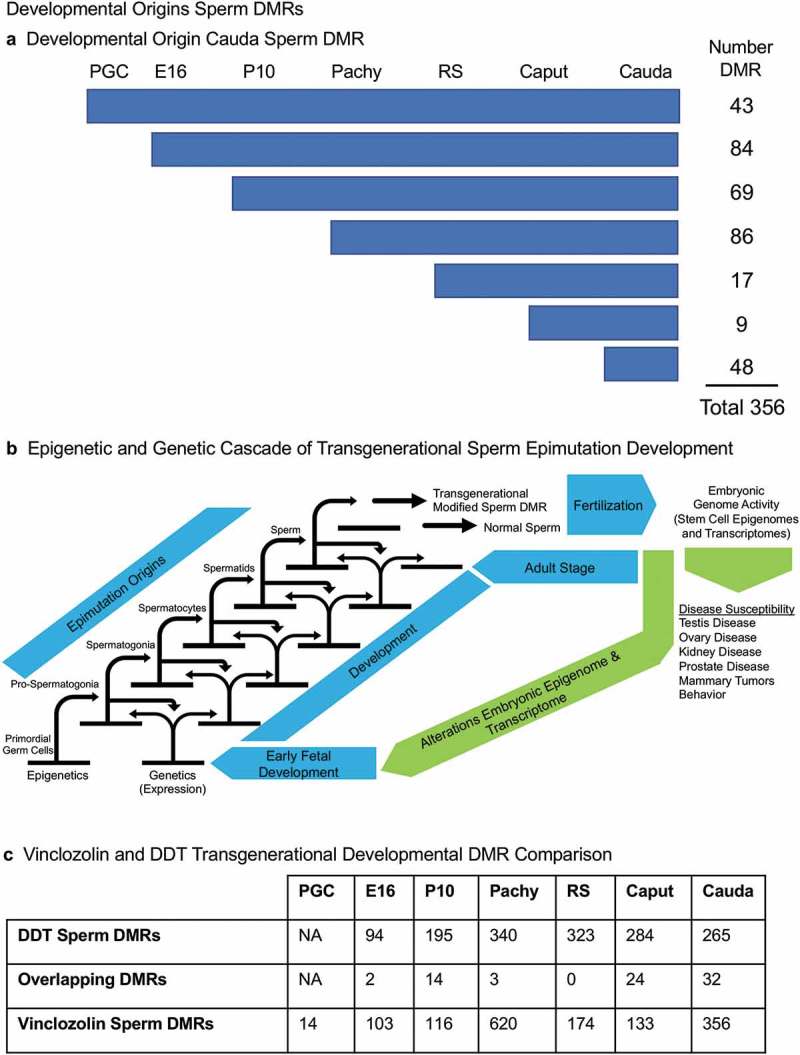
Developmental origins sperm DMR. (a) The origin of DMRs at specific developmental stages for (PGC) primordial germ cells, (E16) prospermatogonia, (P10) spermatogonia, (Pachy) pachytene spermatocytes, (RS) round spermatids, Caput epididymal spermatozoa, and Cauda sperm. The initial developmental stage the cauda sperm DMRs appeared was determined using a relaxed 0.05 edgeR p-value. The numbers of DMRs originating at the different stages are indicated and add to the total 356 DMRs in cauda sperm. (b) Epigenetic and genetic cascade of transgenerational sperm epimutation development scheme. The stages of development for epigenetic and genetic cascades are indicated with epimutation origins correlated to genetic normal or transgenerational modified sperm. Following fertilization, the hypothesis is that the transgenerational epimutations modify early embryonic transcriptomes and epigenomes to re-establish the cascade for the next generation. As the individual develops all somatic cells have altered epigenomes and transcriptomes to promote disease susceptibility later in life. (c) Vinclozolin and DDT transgenerational developmental DMR comparisons with the overlapping DMRs indicated. The DMRs at each stage of development are listed and overlapping numbers. The DMR p-value is p < 1e-05 for all except the vinclozolin pachytene at p < 1e-07.
Observations in a recent publication on the developmental origins of DDT transgenerational sperm epimutations [33] were compared to the current study of vinclozolin transgenerational sperm DMRs, Figure 8(c). The DDT analysis did not involve PGCs, but the comparison of the other developmental stage DMRs showed some overlapping DMRs. The majority of the stage-specific DMRs were unique for the exposure lineage and did not overlap, Figure 8(c).
The final aspect of the study involved the identification of DMR associated genes at each of the developmental stages examined. The DMRs that had a gene within 10 kb distance were identified and the associated genes and gene functional categories identified, Supplemental Tables S1–S7. The DMRs with a p < 1e−05 were used in this analysis for all the developmental stages, except the pachytene spermatocyte stage, that used p < 1e−07. The number of genes associated with specific gene functional categories at each stage is presented in Figure 9(a). The signalling, transcription, receptors, and metabolism were the major gene categories associated with the DMRs at the different developmental stages. Additional categories that were detected at all of the developmental stages include transport, cytoskeleton, development, and extracellular matrix, Figure 9(a). The DMR associated genes for each developmental stage were also used in a gene pathway analysis using the KEGG pathway association, as described in the Methods. The pathways with the highest number of correlated genes were identified, and the numbers of DMR associated genes in each of the major pathways impacted are presented in Figure 9(b). The most predominant pathways in two or more different developmental stages are presented. The primary E16 prospermatogonia associated pathways were metabolism, thermogenesis, oxidative phosphorylation and endocytosis, while in P10 spermatogonia metabolism, transcription and misregulation in cancer were observed. The pachytene spermatocyte and round spermatid DMR associated genes were in the papillomavirus infection, metabolism, pathways in cancer, and endocytosis pathways. The caput and cauda epididymal sperm DMR associated genes were in the metabolism, actin cytoskeleton, endocytosis, and papillomavirus infection pathways. Interestingly, the only DMR associated gene pathways that were present in nearly all the stages were metabolism and endocytosis. The round spermatids, caput epididymal spermatozoa, and cauda sperm DMR associated gene pathways were more consistent between each other than the other stages.
Figure 9.
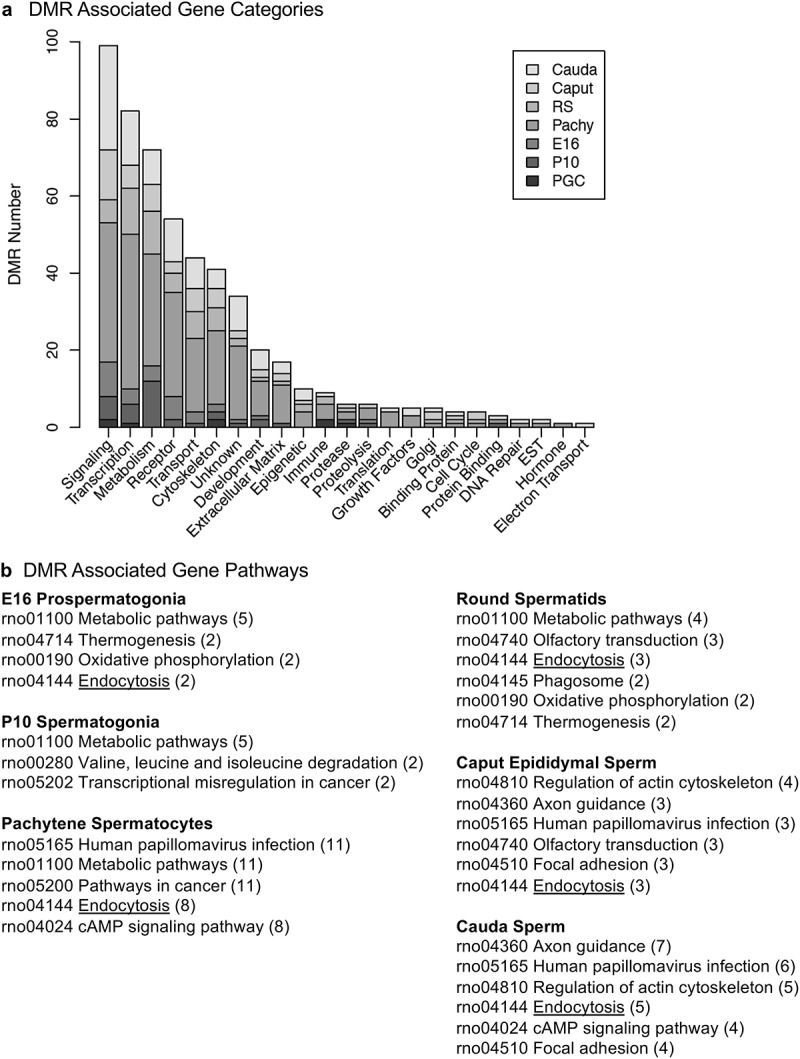
DMR associated gene categories and pathways. (a) The DMR associated gene functional categories are presented for each stage of development, indicated in the inset, with number of DMRs for each category. (b) The DMR associated gene pathways for each developmental stage with the pathway indicated and number of genes in brackets.
Discussion
The objective of the current study was to identify the developmental origins of the cauda sperm transgenerational DMRs that in part transmit the epigenetic transgenerational inheritance of disease and phenotypic variation. The F0 generation gestating female exposure to vinclozolin was during the time of primordial germ cell (PGC) migration and colonization of the indifferent fetal gonad, embryonic day 8–14 in the rat [18,25,34]. The subsequent F3 generation male germ cell developmental stages examined involved the E13 PGCs, E16 prospermatogonia, P10 spermatogonia, and adult pachytene spermatocytes, round spermatids, epididymal caput spermatozoa, and cauda sperm. The epididymal caput spermatozoa and cauda sperm were directly isolated as outlined in the Methods, then sonicated to destroy any contaminating somatic cells, leading to pure preparations of sperm cells. The other cell populations were isolated by gravity sedimentation on a StaPut apparatus, as previously described [37–39]. The E13 PGCs, E16 prospermatogonia, and P10 spermatogonia were found to have a greater than >85% purity. The isolated pachytene spermatocytes were greater than >95% spermatocytes, with >90% of these cells being pachytene spermatocytes and the rest other spermatocyte stages. This is due to the longer life span or developmental period of the pachytene spermatocytes compared to the other spermatocyte stages [40] and efficient separation of the cells on the StaPut gradient. The isolated round spermatids were >95% spermatids with >90% of these being round spermatids and <10% being elongating spermatids [41]. The purity of these developmental stage male germline cell populations was high, but needs to be considered in the data interpretations. The E13 PGCs and E16 prospermatogonia were isolated from the fetal gonad, the P10 were from the prepubertal age, and the pachytene spermatocytes, round spermatids, epididymal caput spermatozoa and cauda sperm were all isolated from adult, 120 days of age male rats. Previously, vinclozolin induced transgenerational inheritance of disease has been primarily shown to develop between 6 and 12 months of age [2]. Therefore, negligible disease is present at 120 days of age or earlier, such that no disease artefacts were anticipated or observed in the current study.
The transgenerational F3 generation control and vinclozolin lineage germline samples were compared at each of the developmental stages. The F1 and F2 generations also have somatic cell and germline epigenetic alterations [8], but these are primarily due to the F1 generation fetal direct exposure and F1 generation fetal germline direct exposure that will generate the F2 generation. Therefore, the F3 generation is the first transgenerational generation not having any direct exposures, so was the focus of the current study [35]. Future studies will need to examine the directly exposed F1 and F2 generations to compare with the F3 generation and assess the distinction between direct exposure and transgenerational epimutations. The different F3 generation developmental stages all showed differential DNA methylation regions (DMRs) that were found to be primarily distinct from each other, except for the caput spermatozoa and cauda sperm, that showed a subset of common DMRs, Figure 5. These different developmental stage germline populations were compared using the top 100 most statistically significant DMRs at each developmental stage separately. Analysis of these DNA methylation alterations for the different developmental stage DMRs demonstrated that E13 PGCs, E16 prospermatogonia, and P10 spermatogonia had distinct patterns of DNA methylation compared to the other stages, Figure 6. The top 100 DMRs for both these developmental stages differed dramatically from those at adjacent stages, and typically showed reduced statistical significance. In contrast, the pachytene spermatocytes, round spermatids, caput spermatozoa and caudal sperm were more consistent between these different stages in regards to DNA methylation alterations and statistical significance, Figures 6 and 7. Observations suggest the developing primordial germ cells, prospermatogonia, and spermatogonial stem cells have largely unique DNA methylation profiles associated with the cascade of epigenetic programming. As the spermatogonia initiate spermatogenesis, more consistent and persistent epigenetic programming and DNA methylation is observed among the spermatogenic cell populations, as well as in sperm undergoing epididymal maturation.
Previous studies have demonstrated significant epigenetic reprogramming in the primordial germ cells (PGCs) involving a DNA methylation erasure during migration and colonization of the fetal gonad [18]. This creates the stem cell population to facilitate the generation of the male or female germline associated with gonadal sex determination [18,25]. As the prospermatogonia develop and give rise to spermatogonia postnatally, there appears to be an epigenetic cascade (i.e., distinct developmental origins) of events that are stable and specific to obtain the spermatogonial stem cell population in the adult testis. Previous studies have demonstrated the ability of vinclozolin to promote epigenetic DNA methylation alterations in the PGCs and prospermatogonia [34]. These transgenerational PGC DMRs were found to be distinct from the caudal epididymal sperm DMRs [34]. PGC DMR gene associations also indicated epigenetic regulatory genes such as DNA methyltransferase (DNMTs) are also altered with DNMT3 being predominant at this stage of development [34]. The current study supports this observation and demonstrates that the origins of the sperm DMRs do not only originate in the fetal period, but appear to promote a dynamic cascade of epigenetic alterations in the adult that eventually impact the mature sperm epigenome, Figure 8(b). Therefore, the fetal exposure initiates a developmental cascade (i.e., distinct developmental origins) of aberrant epigenetic programming, and does not simply induce a specific number of DMRs that are maintained throughout development.
The origins of the DMRs in the cauda sperm were investigated, and demonstrated the majority arise throughout the development of prospermatogonia, spermatogonia and pachytene spermatocytes, Figure 8(a) and Supplemental Table S9 and S10. A smaller number of DMRs also originated in the PGCs, round spermatids and caput epididymal spermatozoa. The highest number arise in the prospermatogonia, which is also the stage of development the initial F0 generation gestating female and F1 generation developing fetus, was exposed. The DMRs in the cauda sperm originated in all the preceding developmental stages including epididymal maturation. This correlated with the DNA methylation alteration of the top 100 DMRs between the different developmental stages, shown in Figures 6 and 7, using a more stringent set of DMRs. Therefore, the primary origins of the transgenerational sperm epimutations are not induced DMRs in early PGC development or the embryonic stem cell population, but instead developed throughout subsequent gametogenesis and during epididymal sperm maturation. Although the induction of a cascade of epigenetic programming in the PCGs is essential, as previously suggested [7,34], the cascade of epigenetic alterations and programming that ultimately alter spermatogonia and spermatogenesis is a critical aspect of the origins of the transgenerational sperm DMRs, Figure 8(b).
A recent study from our laboratory reported similar observations following DDT (dichlorodiphenyltrichloroethane) ancestral exposures [33]. In contrast, approximately 25% of the DMR originated in the prospermatogonia, 25% in the spermatogonia, 18% in the pachytene spermatocytes, 5% in the round spermatids, 12% in the caput spermatozoa, and 14% in the cauda epididymal sperm [33], Figure 5(c). Minimal overlap between the vinclozolin and DDT developmental stage DMRs was observed, Figure 5(c). Therefore, the same trends for developmental origins of the transgenerational sperm epimutations were observed with some shifts in the percentages. The largest difference was in the current study with vinclozolin approximately 30% of the sperm DMR originated in prospermatogonia and 30% in the pachytene spermatocytes. This previous study supports the overall conclusions of the current study and shows differences in the developmental profiles and distinct sets of exposure-specific epimutations [33].
The DMR associated genes were identified for DMRs at p < 1e−05 for each stage of development, except pachytene spermatocytes at p < 1e−07. This more stringent p-value was used for some subsequent analysis to allow a more balanced DMR analysis and comparison, but other analysis such as the DMR origins did not alter the p-value. The analysis of DMR associated gene functional categories identified signalling, transcription, metabolism and receptor as the major categories for all stages of development. The pathway analysis also identified a number of stage-specific pathways with negligible overlap, except in metabolism and endocytosis. Those pathways similar between the stages generally involved different genes within the pathway, such that common pathways are impacted but distinct genes are involved. Future studies will need to correlate the DMRs identified with gene transcriptome data and altered non-coding RNA expression data to better understand how the DMRs identified may regulate genome activity.
An interesting observation was that the caput epididymal spermatozoa showed some DMRs that were unique, and others that were in common with the cauda sperm. In addition, the cauda epididymal sperm acquired DMRs that were not present at earlier stages of development. Therefore, some of the transgenerational sperm epimutations were acquired during cauda epididymal sperm maturation. Although the majority of the cauda epididymal sperm DMRs appear during prior developmental stages, 9 originated in the caput epididymal spermatozoa, Supplemental Table S8. Interestingly, 48 DMRs arose independently during epididymal cauda sperm maturation, Supplemental Table S9. Previous studies have suggested epididymal maturation (epididymosomes) may contribute to transmission of transgenerational sperm epimutations, but no direct data have been shown [42,43]. The current study supports a role for epididymal maturation altering sperm DMRs, however, the majority of DMRs originated during earlier developmental stages and spermatogenesis in the testis and not the epididymis, Figure 8(a). Further research is needed to elucidate the molecular mechanisms in the epididymis that alter the sperm epigenome.
Conclusion
Environmentally induced epigenetic transgenerational inheritance of disease and phenotypic variation requires the epigenetic alterations in the germline (sperm or egg) to mediate the phenomenon. These epigenetic alterations are termed epimutations. The current study investigated the developmental origins of the transgenerational sperm epimutations. The differential DNA methylation regions (DMRs) were found to originate in the fetal gonad PGCs, prospermatogonia, the prepubertal spermatogonia, the adult pachytene spermatocytes and round spermatids, and during epididymal maturation in the caput spermatozoa and cauda sperm. Therefore, vinclozolin induced transgenerational sperm epimutations originate throughout development, with the majority developing in prospermatogonia and during testicular spermatogenesis. Although previous studies have demonstrated epigenetic alterations in the primordial germ cells (PGCs), the DMRs identified in caudal sperm originated at later stages during gametogenesis. However, a cascade of epigenetic alterations initiated in the PGCs is required to alter the dynamic epigenetic programming during gametogenesis in order to modify the sperm epigenome. The developmental programming of the transgenerational epigenetic inheritance phenomenon occurs throughout the development of the germline.
Methods
Animal studies and breeding
Sprague Dawley®™SD®™ male and female rats were fed a standard diet with water ad lib and mated. On gestational day 8 through 14 [44], the pregnant F0 generation females were exposed to vinclozolin or dimethylsulfoxide (DMSO) vehicle control. The offspring were subsequently bred to the F2 and F3 generations, as described in the Supplemental Methods. All experimental protocols for the procedures with rats were pre-approved by the Washington State University Animal Care and Use Committee (protocol IACUC # 6252), and methods performed in accordance with the relevant guidelines and regulations.
Developing germ cell stage isolation
The F3 generation control and vinclozolin lineage individuals were obtained from embryonic day 13 (E13), embryonic day 16 (E16), postnatal day 10 (P10) or adult 120 days of age. The E13 primordial germ cells (PGCs), prospermatogonia E16 germ cells, spermatogonia P10 germ cells, adult pachytene spermatocytes, and adult round spermatids were isolated by using a mini StaPut gradient sedimentation method, as described in the Supplemental Methods. The adult epididymal caput spermatozoa and cauda sperm were isolated as previously described [33] and presented in the Supplemental Methods.
Epigenetic analysis, statistics and bioinformatics
DNA was isolated from E16 prospermatogonia, P10 spermatogonia, adult pachytene spermatocytes, adult round spermatids, caput epididymal spermatozoa, and cauda sperm as previously described [33]. Three different pools generated from different sets of F3 generation control and vinclozolin lineage males were analysed, except for the PGC stage which involved two pools for both the vinclozolin and control lineages. Methylated DNA immunoprecipitation (MeDIP) followed by next-generation sequencing (MeDIP-Seq) was performed. MeDIP-Seq, sequencing libraries, next-generation sequencing, and bioinformatics analysis were performed as described in the Supplemental Methods, as previously described [33]. All molecular data have been deposited into the public database at NCBI (GEO # GSE114032) and R code computational tools available at GitHub (https://github.com/skinnerlab/MeDIP-seq) and www.skinner.wsu.edu.
Funding Statement
This study was supported by John Templeton Foundation (50183 and 61174) grants to MKS and NIH (ES012974) grant to MKS. The funders had no role in the design of the study, collection, analysis, interpretation of data or in writing the manuscript;National Institutes of Health [ES012974].
Ethics approval
All experimental protocols for the procedures with rats were pre-approved by the Washington State University Animal Care and Use Committee (IACUC approval # 6252).
Consent for publication
Not applicable
Availability of data and material
All data are presented in the manuscript and attached Supplemental Material section. All raw sequencing data are publicly available at NCBI GEO site listed. (GEO # GSE117995).
Acknowledgments
We acknowledge Ms. Margaux McBirney, Ms. Deepika Kubsad, Mr. Ryan Thompson for technical assistance and Ms. Amanda Quilty for editorial assistance and Ms. Heather Johnson for assistance in preparation of the manuscript. We thank the Genomics Core laboratory at WSU Spokane.
Disclosure statement
No potential conflict of interest was reported by the authors.
Author contributions
MKS conceived the research, designed the study, wrote the manuscript and acquired funding. EN performed animal and cell isolation studies. ISR performed molecular studies. DB performed data analysis and computational studies. MBM performed molecular studies and data analysis. JRM performed cell isolation studies. All authors performed data analysis and edited the manuscript.
Supplementary material
Supplementary material data can be accessed here
References
- [1].Anway MD, Cupp AS, Uzumcu M, et al. Epigenetic transgenerational actions of endocrine disruptors and male fertility. Science. 2005;308(5727):1466–1469. PubMed PMID: 15933200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Skinner MK. Endocrine disruptor induction of epigenetic transgenerational inheritance of disease. Mol Cell Endocrinol. 2014;398(1–2):4–12. PubMed PMID: 25088466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Soubry A. Epigenetic inheritance and evolution: A paternal perspective on dietary influences. Prog Biophys Mol Biol. 2015;118(1–2):79–85. PubMed PMID: 25769497 [DOI] [PubMed] [Google Scholar]
- [4].Vaiserman AM, Koliada AK, Jirtle RL. Non-genomic transmission of longevity between generations: potential mechanisms and evidence across species. Epigenetics Chromatin. 2017;10(1):38 PubMed PMID: 28750655; PubMed Central PMCID: PMCPMC5531095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Skinner MK. Environmental Epigenetics and a unified theory of the molecular aspects of evolution: a neo-lamarckian concept that facilitates neo-darwinian evolution. Genome Biol Evol. 2015;7(5):1296–1302. PubMed PMID: 25917417; PubMed Central PMCID: PMC4453068 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Skinner MK, Ben Maamar M, Sadler-Riggleman I, et al. Alterations in sperm DNA methylation, non-coding RNA and histone retention associate with DDT-induced epigenetic transgenerational inheritance of disease. Epigenetics Chromatin. 2018;11(1):8, 1–24. PubMed PMID: 29482626; PubMed Central PMCID: PMCPMC5827984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Skinner MK. Environmental epigenetic transgenerational inheritance and somatic epigenetic mitotic stability. Epigenetics. 2011;6(7):838–842. PubMed PMID: 21637037; Eng. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Beck D, Sadler-Riggleman I, Skinner MK. Generational comparisons (F1 versus F3) of vinclozolin induced epigenetic transgenerational inheritance of sperm differential DNA methylation regions (epimutations) using MeDIP-Seq. Environ Epigenet. 2017;3(3):1–12, dvx016. PubMed PMID: 29147574 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Quadrana L, Colot V. Plant transgenerational epigenetics. Annu Rev Genet. 2016;50:467–491. PubMed PMID: 27732791 [DOI] [PubMed] [Google Scholar]
- [10].Sharma U, Rando OJ. Metabolic inputs into the epigenome. Cell Metab. 2017;25(3):544–558. PubMed PMID: 28273477 [DOI] [PubMed] [Google Scholar]
- [11].Gapp K, Jawaid A, Sarkies P, et al. Implication of sperm RNAs in transgenerational inheritance of the effects of early trauma in mice. Nat Neurosci. 2014;17(5):667–669. PubMed PMID: 24728267 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Yan W. Potential roles of noncoding RNAs in environmental epigenetic transgenerational inheritance. Mol Cell Endocrinol. 2014;398(1–2):24–30. PubMed PMID: 25224488; PubMed Central PMCID: PMC4262681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Ben Maamar M, Sadler-Riggleman I, Beck D, et al. Epigenetic transgenerational inheritance of altered sperm histone retention sites. Sci Rep. 2018;(8):5308, 1–10. doi: 10.1038/s41598-018-23612-y PubMed PMID: 29593303; PubMed Central PMCID: PMCPMC5871750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Furuhashi H, Kelly WG. The epigenetics of germ-line immortality: lessons from an elegant model system. Dev Growth Differ. 2010;52(6):527–532. PubMed PMID: 20646025; PubMed Central PMCID: PMCPMC4098867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Ben Maamar M, Sadler-Riggleman I, Beck D, et al. Alterations in sperm DNA methylation, non-coding RNA expression, and histone retention mediate vinclozolin-induced epigenetic transgenerational inheritance of disease. Environ Epigenet. 2018;4(2):1–19, dvy010. PubMed PMID: 29732173; PubMed Central PMCID: PMCPMC5920293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Skinner MK, Manikkam M, Tracey R, et al. Ancestral dichlorodiphenyltrichloroethane (DDT) exposure promotes epigenetic transgenerational inheritance of obesity. BMC Med. 2013;11:228, 1–16. PubMed PMID: 24228800 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Manikkam M, Haque MM, Guerrero-Bosagna C, et al. Pesticide methoxychlor promotes the epigenetic transgenerational inheritance of adult onset disease through the female germline. PloS One. 2014;9(7):1–19, e102091. PubMed PMID: 25057798 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Seisenberger S, Andrews S, Krueger F, et al. The dynamics of genome-wide DNA methylation reprogramming in mouse primordial germ cells. Mol Cell. 2012;48(6):849–862. PubMed PMID: 23219530; PubMed Central PMCID: PMC3533687 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Jost A, Vigier B, Prepin J, et al. Studies on sex differentiation in mammals. Recent Prog Horm Res. 1973;29:1–41. PubMed PMID: 4584366 [DOI] [PubMed] [Google Scholar]
- [20].McCarrey JR. Toward a more precise and informative nomenclature describing fetal and neonatal male germ cells in rodents. Biol Reprod. 2013;89(2):47 PubMed PMID: 23843236; PubMed Central PMCID: PMCPMC4076367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].McCarrey JR. The epigenome as a target for heritable environmental disruptions of cellular function. Mol Cell Endocrinol. 2012;354(1–2):9–15. PubMed PMID: 21970811 [DOI] [PubMed] [Google Scholar]
- [22].Dym M, Fawcett DW. Further observations on the numbers of spermatogonia, spermatocytes, and spermatids connected by intercellular bridges in the mammalian testis. Biol Reprod. 1971;4(2):195–215. PubMed PMID: 4107186. [DOI] [PubMed] [Google Scholar]
- [23].Orgebin-Crist MC, Danzo BJ, Cooper TG. Re-examination of the dependence of epididymal sperm viability on the epididymal environment. J Reprod Fertil Suppl. 1976. Sep(24 suppl):115–128. PubMed PMID: 1069848 [PubMed] [Google Scholar]
- [24].Cornwall GA. New insights into epididymal biology and function. Hum Reprod Update. 2009;15(2):213–227. PubMed PMID: 19136456; PubMed Central PMCID: PMC2639084 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tang WW, Kobayashi T, Irie N, et al. Specification and epigenetic programming of the human germ line. Nat Rev Genet. 2016;17(10):585–600. PubMed PMID: 27573372 [DOI] [PubMed] [Google Scholar]
- [26].Surani MA, Hajkova P. Epigenetic reprogramming of mouse germ cells toward totipotency. Cold Spring Harb Symp Quant Biol. 2010;75:211–218. PubMed PMID: 21139069 [DOI] [PubMed] [Google Scholar]
- [27].Iqbal K, Tran DA, Li AX, et al. Deleterious effects of endocrine disruptors are corrected in the mammalian germline by epigenome reprogramming. Genome Biol. 2015;16:59 PubMed PMID: 25853433; PubMed Central PMCID: PMC4376074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Plasschaert RN, Bartolomei MS. Genomic imprinting in development, growth, behavior and stem cells. Development. 2014;141(9):1805–1813. PubMed PMID: 24757003; PubMed Central PMCID: PMCPMC3994769 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Ku HY, Gangaraju VK, Qi H, et al. Tudor-SN interacts with piwi antagonistically in regulating spermatogenesis but synergistically in silencing transposons in drosophila. PLoS Genet. 2016;12(1):e1005813 PubMed PMID: 26808625; PubMed Central PMCID: PMCPMC4726654 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].La Salle S, Oakes CC, Neaga OR, et al. Loss of spermatogonia and wide-spread DNA methylation defects in newborn male mice deficient in DNMT3L. BMC Dev Biol. 2007;7:104 PubMed PMID: 17875220; PubMed Central PMCID: PMCPMC2212652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Mann JR. Imprinting in the germ line. Stem Cells. 2001;19(4):287–294. PubMed PMID: 11463948 [DOI] [PubMed] [Google Scholar]
- [32].Barlow DP, Bartolomei MS. Genomic imprinting in mammals. Cold Spring Harb Perspect Biol. 2014;6(2):1–21. PubMed PMID: 24492710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ben Maamar M, Nilsson E, Sadler-Riggleman I, et al. Developmental origins of transgenerational sperm DNA methylation epimutations following ancestral DDT exposure. Dev Biol. 2018;445(2):280–293. PubMed PMID: 30500333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Skinner M, Guerrero-Bosagna C, Haque MM, et al. Environmentally induced transgenerational epigenetic reprogramming of primordial germ cells and subsequent germline. PLoS One. 2013;8(7):1–15, e66318. PubMed PMID: 23869203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Skinner MK. What is an epigenetic transgenerational phenotype? F3 or F2. Reprod Toxicol. 2008;25(1):2–6. PubMed PMID: 17949945; PubMed Central PMCID: PMC2249610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Ariel M, McCarrey J, Cedar H. Methylation patterns of testis-specific genes. Proc Natl Acad Sci USA. 1991;88(6):2317–2321. PubMed PMID: 2006171; PubMed Central PMCID: PMC51222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Hermann BP, Mutoji KN, Velte EK, et al. Transcriptional and translational heterogeneity among neonatal mouse spermatogonia. Biol Reprod. 2015;92(2):54 PubMed PMID: 25568304; PubMed Central PMCID: PMCPMC4342790 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Skinner MK, Guerrero-Bosagna C. Role of CpG deserts in the epigenetic transgenerational inheritance of differential DNA methylation regions. BMC Genomics. 2014;15(1):692 PubMed PMID: 25142051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].McCarrey JR, Berg WM, Paragioudakis SJ, et al. Differential transcription of Pgk genes during spermatogenesis in the mouse. Dev Biol. 1992;154(1):160–168. PubMed PMID: 1426623 [DOI] [PubMed] [Google Scholar]
- [40].Ventela S, Ohta H, Parvinen M, et al. Development of the stages of the cycle in mouse seminiferous epithelium after transplantation of green fluorescent protein-labeled spermatogonial stem cells. Biol Reprod. 2002;66(5):1422–1429. PubMed PMID: 11967206 [DOI] [PubMed] [Google Scholar]
- [41].Suresh R, Aravindan GR, Moudgal NR. Quantitation of spermatogenesis by DNA flow cytometry: comparative study among six species of mammals. J Biosci. 1992;17(4):413–419. [Google Scholar]
- [42].Sharma U, Conine CC, Shea JM, et al. Biogenesis and function of tRNA fragments during sperm maturation and fertilization in mammals. Science. 2016;351(6271):391–396. PubMed PMID: 26721685; PubMed Central PMCID: PMCPMC4888079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Rompala GR, Mounier A, Wolfe CM, et al. Heavy chronic intermittent ethanol exposure alters small noncoding RNAs in mouse sperm and epididymosomes. Front Genet. 2018;9:32 PubMed PMID: 29472946; PubMed Central PMCID: PMCPMC5809758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Nilsson E, Sadler-Riggleman I, Skinner MK. Environmentally induced epigenetic transgenerational inheritance of disease. Environ Epigenet. 2018;4(2):1–13, dvy016. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are presented in the manuscript and attached Supplemental Material section. All raw sequencing data are publicly available at NCBI GEO site listed. (GEO # GSE117995).