

ABSTRACT
Post-fertilization epigenome reprogramming erases epigenetic marks transmitted through gametes and establishes new marks during mid-blastula stages. The mouse embryo undergoes dynamic DNA methylation reprogramming after fertilization, while in zebrafish, the paternal DNA methylation pattern is maintained throughout the early embryogenesis and the maternal genome is reprogrammed in a pattern similar to that of sperm during the mid-blastula transition. Here, we show DNA methylation dynamics in medaka embryos, the biomedical model fish, during epigenetic reprogramming of embryonic genome. The sperm genome was hypermethylated and the oocyte genome hypomethylated prior to fertilization. After fertilization, the methylation marks of sperm genome were erased within the first cell cycle and embryonic genome remained hypomethylated from the zygote until 16-cell stage. The DNA methylation level gradually increased from 16-cell stage through the gastrula. The 5-hydroxymethylation (5hmC) levels showed an opposite pattern to DNA methylation (5-mC). The mRNA levels for DNA methyltransferase (DNMT) 1 remained high in oocytes and maintained the same level through late blastula stage and was reduced thereafter. DNMT3BB.1 mRNA levels increased prior to remethylation. The mRNA levels for ten-eleven translocation methylcytosine dioxygenases (TET2 & TET3) were detected in sperm and embryos at cleavage stages, whereas TET1 and TET3 mRNAs decreased during gastrulation. The pattern of genome methylation in medaka was identical to mammalian genome methylation but not to zebrafish. The present study suggests that a medaka embryo resets its DNA methylation pattern by active demethylation and by a gradual remethylation similar to mammals.
KEYWORDS: Epigenetic reprogramming, medaka embryo, DNA methylation
Introduction
DNA methylation is one of the major forms of epigenetic modifications which is associated with transcriptional regulation [1], genomic imprinting [2], suppression of repetitive elements [3,4], and DNA–protein interaction during the development of organisms. In vertebrates, DNA methylation occurs predominantly at the CpG sites and is catalysed by DNA methyltransferases (DNMT). In mammals, there are two types of DNA methyltransferases involved in DNA methylation regulation: DNMT1 and DNMT3 family [5]. The function of DNMT1 is to maintain the methylation of genomic DNA after DNA replication [6]. DNMT3A and DNMT3B are responsible for de novo methylation of genomic DNA [7]. DNMT3C is a duplication of DNMT3B and found in germ cells [8], while DNMT3L is an important cofactor of DNMT3A and DNMT3B [9–11]. Methylated DNA is demethylated in two ways – passive demethylation (also known as replication-based demethylation) and active demethylation. Passive demethylation occurs due to a dilution of methylation signal on DNA strands which do not receive methylation marks through DNMT-mediated recruitment process [12–15]. Active DNA demethylation is achieved through TET-mediated oxidation of 5-methylcytosine (5mC) to 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC), followed by replication-dependent dilution of oxidized 5mC or thymine DNA glycosylase (TDG)-mediated excision of 5-fC and 5-caC coupled with base excision repair [16]. The current known mechanism of active demethylation involves TET/TDG and GADD45A/B proteins [17–20].
The sperm and oocyte are highly specialized cells and possess a capacity to form an entire embryo upon fertilization. After fertilization, the epigenetic modifications are reprogrammed to totipotent status by blastula stage [21–23]. DNA methylation plays an important role in embryonic development and undergoes dramatic changes during embryogenesis. In mammals, global genomic DNA methylation levels of the zygote decrease after fertilization and reach the lowest level at blastula stage [24–27]. After fertilization, the paternal nucleus undergoes rapid demethylation during the first cell cycle and then continues demethylation during cleavage, whereas the maternal nucleus undergoes the process of gradual demethylation. Evidences suggest that both passive and positive demethylation are involved in this process [26,28]. When the mammalian embryos develop into the blastocyst stage, the parental nucleus gains the similar methylation pattern and reaches the lowest level during rest of the cleavage stages [13,24,26]. In zebrafish, the zygotic genome does not undergo demethylation process. Unlike in mice and humans, the overall methylation level of the paternal nuclear genome remains stable during early embryonic development, while the overall methylation level of the maternal nuclear genome increases beginning from the 32-cell stage. When the embryo is at initial blastocyst (1k-cell) stage, the methylation of the maternal genome reaches a pattern equivalent to the overall methylation of the paternal genome [29–31], suggesting that zebrafish reprogram the parental epigenetic modifications through the way different from mammals.
Medaka (Oryzias latipes) is an important biomedical research model organism [32–34]. Advantages include genetic sex determination in Hd-rR strain [35], external fertilization, daily spawning, availability of large numbers of eggs and sperm, short generation time (2–3 months per reproductive cycle) and easy maintenance, sequenced genome, and smaller genome size [36]. Additionally, molecular mechanisms underlying early embryogenesis, germ cell migration, and differentiation are believed to be complementary to mouse [37]. Despite being an excellent model organism, the medaka epigenetic programming has not been studied except for a study which reported that the genomic DNA is highly methylated at CCGG sites in embryos [38] and warrant investigation to generate a baseline epigenetic information on epigenetic reprogramming of embryo. In the present study, the global DNA methylation/demethylation changes and the expression of methylation/demethylation-related genes were measured to provide an overall profile of DNA methylation dynamics during the epigenetic programming of medaka embryo. We demonstrate that medaka zygote completes DNA demethylation of genome within the first cell cycle (1-cell stage). Embryos keep the genome hypomethylated for the first several cleavage stages and achieve hypermethylation from the stage-13 through the gastrula. The expression of DNMT genes and TET demethylase genes further confirm the genome methylation and demethylation data. Our results provide a DNA methylation reprogramming model during early embryonic stages in medaka, demonstrating that the medaka uses active demethylation process to erase the parental methylation, and overall epigenetic reprogramming is identical to that of mammal but not zebrafish.
Methods
Fish care and embryo collection
The medaka strain Hd-rR was used and maintained under standard conditions at the University of North Carolina Greensboro according to Institutional Animal Care and Use Committee (IACUC)-approved methods established for this species (Protocol #16-003). Briefly, medaka fishes were raised on a light–dark cycle of 14 h:10 h. During the first 10 d of post-hatching development, juveniles were supplied with ground food and thereafter with flake food twice and brine shrimp once a day. Tanks were siphon cleaned periodically, and water temperature was maintained at 25 ± 1°C. The eggs were collected immediately after fertilization, cleaned and kept in embryo-rearing medium at 25 ± 1°C in petri dishes. The samples were collected at various stages between fertilization and gastrulation. The embryos were staged by developmental time and morphology under brightfield microscope according to Iwamatsu (2004) [39].
Genomic DNA and total RNA extraction
Genomic DNA and total RNA from each embryo samples were extracted using ZR-Duet™ DNA/RNA MiniPrep Plus kit (Zymo Research, D7003) according to the manufacturer’s instruction. Genomic DNA and total RNA were quantified using Nanodrop 2000, and stored at −80°C until further analysis.
Whole genome bisulfite sequencing of methylated DNA and sequence data analysis
Whole genome bisulfite sequencing (WGBS) libraries were prepared with NEBNext® Ultra™ II FS DNA Library Prep Kit (NEB, E7805S) according to the manufacturer’s user manual. For each sample, 100 ng genomic DNA added with 0.5% unmethylated E. coli DNA as a control of bisulfite conversion efficiency, and was incubated 12 min with Ultra II FS Enzyme. DNA fragments were ligated with methylated adaptor. The adapter-ligated DNA fragments were purified with NEBNext Sample Purification Beads supplied with kit. Purified DNA fragments were bisulfite-treated with EZ DNA Methylation-Lightning Kit (Zymo Research, D5030) according to the manufacturer’s handbook. The bisulfite-treated DNA fragments were amplified with 13 cycles using Taq DNA polymerase (NEB). Finally, the MethylC-Seq libraries were used to perform 150-bp single-end sequencing with Illumina NextSeq 500 instrument at the DNA Core Facility of the University of Missouri, USA.
Bismark (Version: v0.19.0) was used to map bisulfite sequencing reads onto medaka genome (Ensembl release-89) with default parameters [40]. To guarantee to get the most accurate mapping result, we only retained the reads being mapped on unique position of the genome. Then, a custom Bash script was used to calculate the sequencing depth and the genome distribution of filtered mapped results. In following analysis, reads only mapped on chromosomes were considered. The utility ‘bismark_methylation_extractor’ wrapped in Bismark was used to extract methylation levels on each site. The mapped reads on both of strands were considered to calculate methylation level. CpG methylation calls were analysed using SeqMonk software (Version 1.43.0, https://www.bioinformatics.babraham.ac.uk/projects/seqmonk/). The genome was divided into consecutive 5-kb probes with a step size of 2.5 kb. Probes contain at least 5 CpG sites, and each CpG site was covered by at least 5 reads which were retained for further analysis. Methylation percentages were calculated using the bisulfite feature methylation pipeline in SeqMonk. For analysis of promoters, the Ensembl gene set annotations for medaka (Ensembl release 89) was used, and 2kb upstream of a transcription start site (TSS) was defined as a promoter. Only promoters containing at least five CpG sites, each of which covered by at least five reads, were considered for further analysis. To identify differentially methylated probes or promoters, we first calculated the difference of methylation levels of a prober or promoter in two samples and constructed the Chi-Squared Test with SeqMonk software. If the difference of methylation level of the prober or promoter was larger than 0.2 and the p-value was less than 0.001, the probes or promoter was identified as differentially methylated. The sperm WGBS data have been submitted to the following website: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE128797.
Global DNA methylation quantification assay
Global DNA methylation was measured using 5mC DNA ELISA Kit (Zymo Research, D5326) according to the manufacturer’s instruction manual. A 100 ng genomic DNA was used for each technical replicate, two technical replicates were used for each biological replicate, and at least three biological replicates were used for each stage. The DNA was denatured at 98°C for 5 min in a thermal cycler, then transferred immediately to ice for 10 min. The entire denatured DNAs were transferred to the wells of the plate and used to coat the plate wells with 5mC coating buffer by incubation at 37°C for 1 h. After washing three times with 200 μl of 5mC ELISA Buffer, the wells were incubated at 37°C for 30 min in 200 μl of 5mC ELISA Buffer. Antibody mix was prepared by diluting anti-5-methylcytosine (1:1000) and secondary antibody (1:1000) in 5mC ELISA Buffer. Total of 100 μl antibody mix was added to each well and incubated at 37ºC for 1 h. After washing three times with 200 μl of 5mC ELISA buffer, 100 μl of HRP Developer was added to each well, and then, incubated at room temperature for 30–60 min to develop colour. Absorbance at 405nm was measured using BioTek Synergy2 plate reader.
To quantify the percentage of 5mC, a standard curve was generated. The Negative Control (100 ng/μl) and Positive Control (100 ng/μl) were premixed to generate a set of standards with known 5mC percentage and assayed in parallel with the samples. A logarithmic second-order regression was generated to determine the relation of 5mC percentage and absorbance. Then the equation below was used to determine the 5mC percentage for DNA samples based on their absorbance.
Then the 5mC percentage was corrected with the medaka CpG density. According to the manufacturer, the E. coli CpG density/genome length is 0.075, and medaka CpG density/genome length is 0.019 (13,320,763/700,386,597, calculated according Ensembl genome release 89). The fold difference between E. coli and medaka CpG density is 3.94, and all genome DNA methylation ratios were corrected.
Global DNA 5-hydroxymethylcytosine quantification assay
Global DNA genomic 5-hydroxymethylcytosine was measured by using a Quest 5hmC™ DNA ELISA Kit (ZymoResearch, D5426) according to the manufacturer’s instruction manual. A total of 25 ng genome DNA was used for each technical replicate, two technical replicates were used for each biological replicate, and at least three biological replicates were used for each stage. Briefly, anti-5-Hydroxymethylcytosine Polyclonal Antibody was diluted in coating buffer (1 ng/μl) and incubated with wells at 37ºC for 1 h. Each well was washed with 200 μl of 1X ELISA Buffer three times and incubated with 1X ELISA buffer at 37ºC for 30 min. DNA samples and standards were denatured at 98°C for 5 min in a thermal cycler and then transferred immediately to ice for 10 min. The denatured DNAs were diluted into 1 ng/μl with 1X ELISA buffer, then transferred to the wells, and incubated at 37°C for 1 h. Anti-DNA HRP antibody was diluted in 1X ELISA buffer with the ratio 1:100 and incubated at 37ºC for 30 min after removing DNA solutions. Each well was washed with 200 μl of 1X ELISA buffer for three times. A 100 μl of HRP Developer was added to each well and allowed for colour development at room temperature for 60 min. BioTek Synergy2 plate reader was used to measure the absorbance at 405 nm.
To quantitate the 5hmC percentage in a DNA sample, a standard curve was generated using the provided controls. The control data as absorbance (Y-axis) vs. per cent 5hmC (X-axis) was plotted and the linear regression (equation below) was used to determine the ‘% 5hmC’ for the DNA samples (unknowns).
Real-time qRT-PCR
Real-time qRT-PCR analysis was performed for DNA methylation- and demethylation-related genes, 4 DNMTs and 3 TETs. Specific primer pairs used in the qRT-PCR were designed using Primer3web [41] and are shown in Supplemental Table 1. Primer sequence spanned at least one junction of exon and intron to avoid amplification of genomic DNA. β-actin was used as an endogenous reference gene to determine the relative expression. Target gene expression was analysed by 2−ΔΔCt method [42]. qRT-PCR was performed by using PowerUp SYBR™ Green Master Mix (Applied Biosystems, A25742) using a QuantStudio 3 Real-Time PCR System with the following temperature regimes: 2 min at 50°C and 10 min at 95°C for pre-incubation, 40 cycles at 95°C (15 s) and 60°C (1 min). Expression levels in all the stages were normalized to the expression levels in sperm.
Statistical analysis
ELISA and qRT-PCR results were analysed and plotted using R language, RStudio and Excel. Statistical differences between different stages were determined using one-way ANOVA followed by Tukey’s multiple comparisons test. For important sample pairs, we also used two-sample t-test to further justify on differences. Data are presented as mean ± standard error of the mean (SEM), and p < 0.05 was considered as significant. Correlation coefficient was calculated to measure the correlation of methylation and hydroxymethylation.
Results
DNA methylation (5-mC) dynamics during early embryonic development
The global genomic DNA methylation levels at various embryonic stages are shown in Figure 1(a). To confirm the 5-mC ELISA results, we sequenced sperm methylome by WGBS and used published WGBS data at blastula stage [43,44]. We found no significant differences on global methylation in two methods and confirming the validity of ELISA results (Supplemental Figure 1A). One-way ANOVA test showed significant difference in global methylation among stages examined (p < 0.005). As Tukey’s multiple comparisons test did not show significant difference between each sample pairs, we used two samples t-test to justify the differences between important stages. In gametes, paternal genome was highly hypermethylated (83.03%), while maternal genome was hypomethylated (26.59%). The methylation levels in the sperm were higher than that in the oocytes (two samples t-tests, p < 0.05). After fertilization, the global genome methylation level in zygotes (Stage 2) had no significant difference compared with oocyte (22.91% vs. 26.59%) but was significantly lower than sperm (22.91% vs. 83.03%, two samples t-tests, p < 0.05), indicating that there was an immediate demethylation process in paternal genome after fertilization within the first cell cycle. The genome was globally hypomethylated during the first four cleavages, and gradually increased from 16-cell to late gastrulation. The global methylation level in gastrulation stages was close to sperm, but higher than oocyte, zygote, 2-cell, and 4-cell embryos. From late gastrulation to early neurula, there was a significant decrease in global DNA methylation level (88.83% vs. 58.23%, two samples t-test, p < 0.05), indicating that tissue differentiation was associated with demethylation in medaka embryo.
Figure 1.
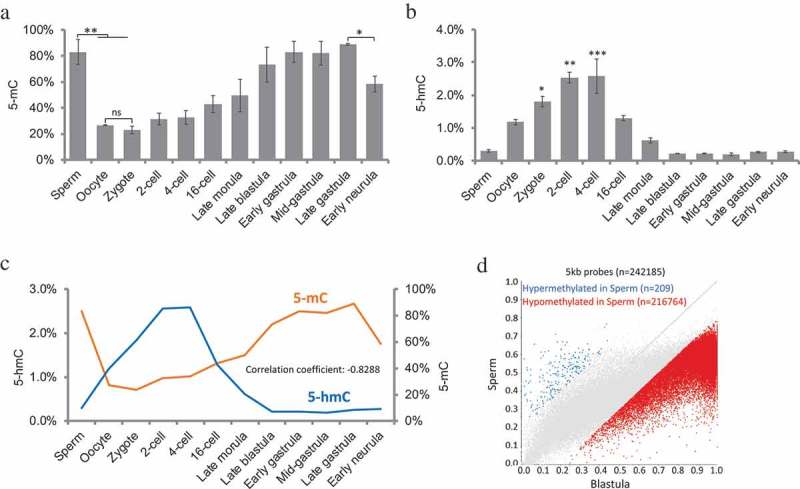
Genome methylation (5-mC) and hydroxymethylation (5-hmC) in medaka during embryogenesis. (a): Constitutive global DNA methylation; one-way ANOVA analysis showed p < 0.001; two-sample t-test showed significant differences between Sperm vs. Oocyte, Sperm vs. Zygote and Late gastrula vs. Early neurula; (b): constitutive global DNA hydroxymethylation levels; one-way ANOVA analysis showed p < 0.0001, Tukey’s multiple comparisons test showed significant differences between Zygote, 2-cell, 4-cell vs. Sperm, blastula stages, and gastrula stages; (c): correlation between 5-mC and 5-hmC levels during embryogenesis. (d): differentially methylated probes between Sperm and Blastula embryos. Data represent mean ± SEM in Figure 1(a) and (b). Asterisk indicates statistical significance (*p < 0.05; **p < 0.01).
DNA hydroxymethylation (5-hmC) dynamics during embryonic development
To determine if positive demethylation mediated by TET enzyme involved in embryonic DNA methylation reprogramming, the levels of DNA hydroxymethylation (5-hmC) were determined by 5-hmC ELISA. The global genomic 5-hmC level in different stages are shown in Figure 1(b). One-way ANOVA test showed that there was a significant difference in global hydroxymethylation among examined stages (p < 0.0001). The maximum level of hydroxymethylation was observed in zygote (Stage 2), 2-cell (Stage 3), and 4-cell (Stage 4) (Tukey’s multiple comparisons test), which were significantly higher than sperm, blastula, and gastrulation stages. The 5-hmC significantly accumulated during demethylation, when 5-mC levels were low. There was a negative correlation between 5-mC levels and 5-hmC levels (Figure 1(c) and Supplemental Figure 1B, correlation coefficient = −0.8288), suggesting DNA hydroxymethylation was involved in DNA demethylation during early embryos stages in medaka.
Methylome of paternal DNA was not inherited by blastula embryo
In zebrafish, the global methylation of the paternal nuclear genome remains stable during early embryonic development, while the methylation of maternal genome is reprogrammed to a pattern equivalent to the paternal genome [29–31], suggesting that methylome of sperm is inherited by blastula embryo in zebrafish. To address if the methylome of medaka sperm was inherited by blastula embryo, we sequenced sperm epigenome by WGBS and analysed the published blastula stage epigenome in Hd-rR strain of medaka [44] with the same pipeline. To obtain better genome coverage, we combined three sperm sample replicates together in this analysis. Using a 5kb probe strategy, we found that the majority of 5kb probes were hypomethylated in sperm compared with blastula embryo (Figure 1(d)). For functional genomic feature, we observed a similar pattern in promoter analysis in which the majority of promoters were hypomethylated in sperm compared to blastula embryo (Supplemental Figure 1C), demonstrating blastula embryos obtain methylome through dramatic reprogramming but not directly inherited from sperm.
Expression of DNA methyltransferase genes during early embryonic development
The expression of DNA methyltransferase genes in different stages was determined by real-time quantitative RT-PCR method (Figure 2). Results show a distinct expression pattern for four DNMT transcripts in medaka embryos. The expression of DNMT1 was detected in all stages examined with a maternal expression in unfertilized eggs (Figure 2(a)). The DNMT1 transcripts remained high from the cleavage to blastula stage, dropped at early gastrula stage (Stage 13), and remained at a low level through gastrulation and early neurula stages. DNMT3AA was maternally provided in oocyte and the mRNAs were detected in all the stages examined (Figure 2(b)). The expression of DNMT3BA was high in embryos at early gastrula stage (Stage 13), which coincides with the timing of zygotic genome activation (Figure 2(c)). DNMT3BB.1 expression was detected in unfertilized eggs and in all the stages examined with an increased expression from 16-cell (Stage 6) through early gastrula stage (Stage 13) (Figure 2(d)). The expression of DNMT3BB.1 showed a pattern similar to the trend observed prior to global DNA methylation until gastrulation stage (Figure 2(e)), indicating that DNMT3BB.1 might play a critical role in the genome de novo methylation during embryogenesis.
Figure 2.
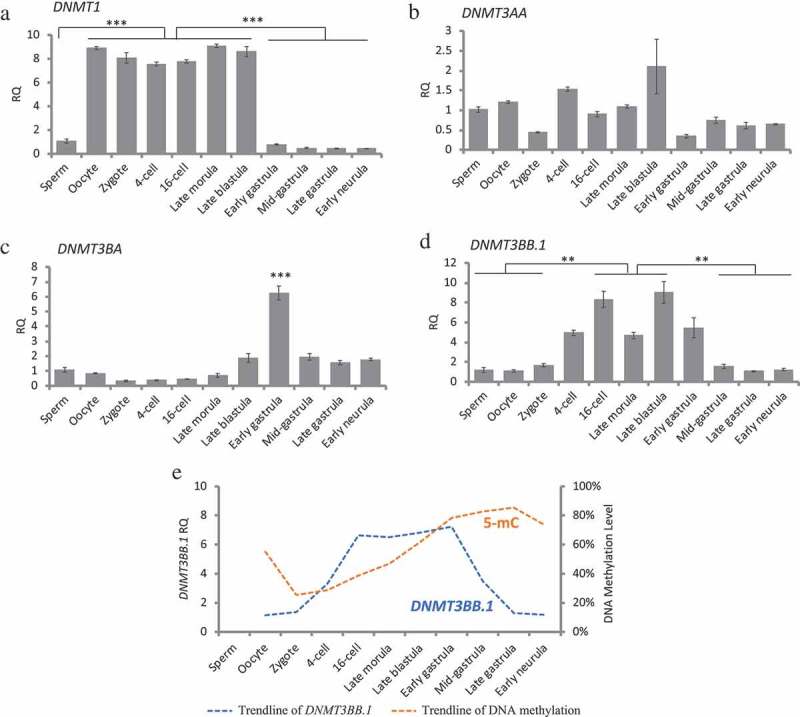
DNA methyltransferases expression during embryogenesis measured by real-time qRT-PCR. Gene expressions in all the embryonic stages were normalized against expression in sperm (set as 1), RQ: Relative quantification. (a): DNMT1, one-way ANOVA analysis showed p < 0.0001, Tukey’s multiple comparisons test showed significant differences between Oocyte to blastula stages vs. Sperm, gastrula stages and Early neurula. (b): DNMT3AA, one-way ANOVA analysis showed no significant differences among examined stages. (c): DNMT3BA, one-way ANOVA analysis showed p < 0.0001, Tukey’s multiple comparisons test showed significant differences between Early gastrula vs. all other examined stages. (d): DNMT3BB.1, one-way ANOVA analysis showed p < 0.0001, Tukey’s multiple comparisons test showed significant differences between 16-cell, Late blastula vs. Sperm, Oocyte, Zygote, Mid gastrula, Late gastrula, and Early neurula. (e): A relationship between DNMT3BB.1 expression and global 5-mC levels. Data represent mean ± SEM. Asterisk indicates statistical significance (**p < 0.01, ***p < 0.001).
Expression of TET methylcytosine dioxygenase genes during early embryonic development
The expression of TET1 was detected in all stages examined and was expressed with high abundance during the cleavage and blastula stage (Figure 3(a)). The expression of TET1 decreased at early gastrula stage (Stage 13) and remained at a low level through gastrulation and early neurula stages (Figure 3(a)). In gametes, transcripts of TET2 only expressed in sperm but not oocyte, showing that TET2 was paternally expressed (Figure 3(b)). After fertilization, the expression of TET2 initiated from early gastrula (Stage 13, Figure 3(b)), showing that it did not involve DNA methylation reprogramming during embryogenesis. The expression of TET3 was detected in gametes, cleavage, and blastula stages, and dramatically decreased from early gastrula (Figure 3(c)), indicating that TET3 might have a specific function in reprogramming during embryogenesis.
Figure 3.
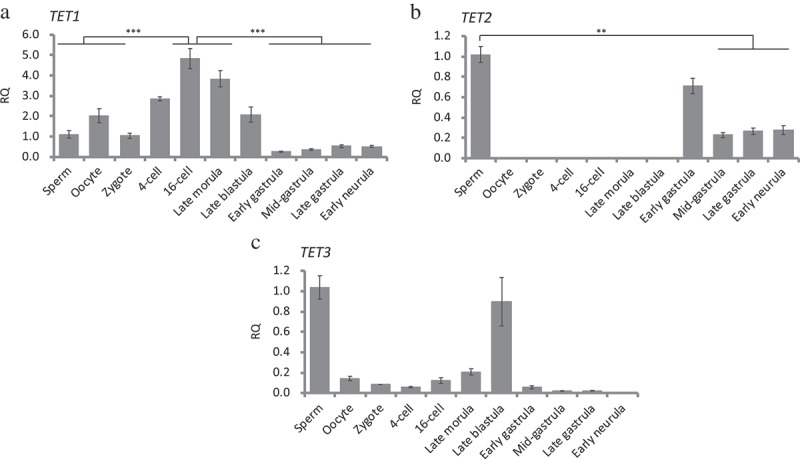
TET methylcytosine dioxygenases expression during early embryonic development measured by real-time qRT-PCR. Gene expressions in all the embryonic stages were normalized against expression in sperm (set as 1). RQ: Relative quantification. (a): TET1, one-way ANOVA analysis showed p < 0.0001, Tukey’s multiple comparisons test showed significant differences between 16-cell, Late morula vs. Sperm, Oocyte, Zygote, blastula stages, gastrula stages, and Early neurula. (b): TET2, one-way ANOVA analysis showed p < 0.0005, Tukey’s multiple comparisons test showed significant differences between Sperm vs. Mid gastrula, Late gastrula, and Early neurula. (c): TET3, one-way ANOVA analysis showed no significant differences among examined stages. TET1, TET2, and TET3 are paternally expressed and TET2 expression began after genome-zygotic transition. TET3 expression was absent after genome-zygotic transition. Data represent mean ± SEM. Asterisk indicates statistical significance (**p < 0.01, ***p < 0.001).
Discussion
After fertilization, the mammalian zygote undergoes several cleavages to develop into blastula, in which the paternal and maternal epigenetic modifications reach the identical patterns and the cells of embryo reach the pluripotent status after reprogramming. In medaka, sperm and oocyte fuse together to form a blastodisc within the first hour post fertilization, and then the embryo enters the cleavage stage. Blastulation completes between Stage 10 and Stage 11, and the gastrulation continues from Stage 11 to Stage 16 [39]. The present study mapped DNA methylation and demethylation in sperm, eggs, and embryos of medaka undergoing cleavage, morula, blastulation, and gastrulation including expression profile of the DNA methylation-related genes. This is the first time to comprehensively map these key processes during development in a vertebrate other than mice, zebrafish, and human.
In medaka, the sperm genome was highly methylated which is similar to that of human, mouse, and zebrafish, whereas the oocyte genome was hypomethylated. We found that the embryonic DNA methylation reprogramming in medaka completed in four steps after fertilization (Figure 4(a)). Step 1 (from gametes to zygotes): genome DNA methylation level decreased to 20%. This process is unique as it is different from mice and zebrafish [13,29]. All three model species (mice, medaka, and zebrafish) seem to undergo a different level of DNA methylation dynamics immediately after fertilization. In medaka, sperm genome was hypermethylated and the majority of genome features were hypermethylated (methylation level >0.5), except 5ʹ-UTR (Supplemental Figure 2A and 2B). In mice, DNA methylation marks that are inherited from parental gametes are gradually erased by both passive and active demethylation processes, and it takes several cell cycles to completely remove the parental methylation marks [13,26,45]. In zebrafish, no demethylation was found, and no TET genes were detected during this period [29,46]. The methylation level in zebrafish zygotes equals to the average of sperm and oocyte [29]. In medaka, DNA methylation in sperm and oocyte was erased within the first cell cycle, and TET1 and TET3 expression were detectable in the zygote, demonstrating that active demethylation plays an important role in this process. This erasure suggests the beginning of reprogramming towards pluripotency in medaka at 1-cell stage of embryo development, which is similar with mice and human. During this level of reprogramming, the genome still maintained 20% methylation. In mice, bisulfite sequencing of cells undergoing reprogramming, particularly during preimplantation development, revealed some erasure-resistant genomic regions. These protected genomic regions were intracisternal A particles (IAPs), long terminal repeats of endogenous retrovirus 1 (LTR-ERV1) elements, and a few single-copy sequences [47,48]. It is important to further clarify what genes are resistant to reprogramming at this stage in medaka.
Figure 4.
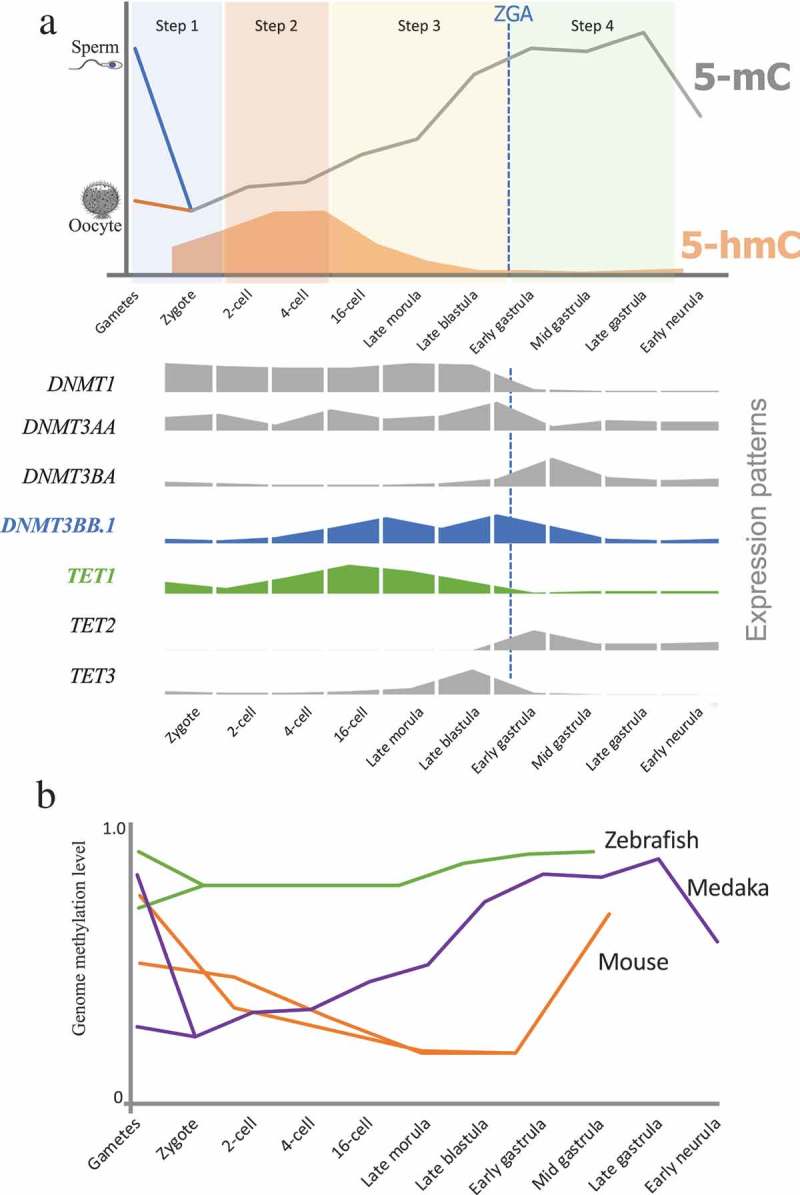
DNA methylation reprogramming model during embryogenesis in medaka. (a): Sperm was hypermethylated and oocyte was hypomethylated. Paternal genomic methylation was erased in zygote (Step 1). Embryos stayed in hypomethylation status during first several cell cycles (Step 2). Global DNA methylation levels increased from 16-cell stage to Late blastula stage (Step 3). Embryos maintained hypermethylation during gastrula stages (Step 4). Global DNA methylation level decreased from gastrula to neurula stage. Relative quantification expression of genes involved in DNA methylation and hydroxymethylation were showed on lower panel. (b): A comparative epigenetic programming during embryogenesis in three model species – zebrafish [29,30], medaka, and mice [23,26].
Step 2, 2-cell (Stage 3) to 8-cell (Stage 5): the genome maintains hypomethylation. No significant changes in global DNA methylation levels were observed within Stage 3, Stage 5, and zygote, indicating that embryos had already finished the demethylation process in Step 1, which was similar to human [24]. The DNMT1 was highly expressed during this period probably to maintain the DNA methylation status which was consistent with the fast DNA replication and short cell cycles. In mice, genome DNA demethylation continues during this period [13,49], whereas in zebrafish there are no significant changes in global DNA methylation during this period [29], and the genome remained in hypermethylated state as compared to mice, medaka, and human. Step 3, 16-cell (Stage 6) to early gastrula (Stage 13): embryo underwent a remethylation process and maintained a hypermethylated status, which was confirmed by WGBS results at blastula stage [43]. Medaka fish have three DNMT3 genes: DNMT3AA, DNMT3BA, and DNMT3BB.1 [50]. All of the DNMT3 genes were detected in all the stages during this period. Intriguingly, the expression of DNMT3BB.1 showed the expression pattern coinciding with the pattern of global 5mC levels until early gastrula stage indicating an important role of DNMT3BB.1 in de novo methylation of the medaka genome DNA. In medaka, the mid-blastula transition (MBT) occurs at late blastula (Stage 11) [44]. Due to the zygotic genome activation and depletion of maternal transcripts, several genes show dramatic changes in expression patterns from late blastula to early gastrula. For instance, DNMT1, DNMT3AA, TET1, and TET3 expression decreased from late blastula to early gastrula, while DNMT3BA expression increased from late blastula to early gastrula. Intriguingly, in our gene expression results, both DNMT3BB.1 and TET1 were significantly upregulated at 16-cell stage compared to zygote (Figures 2(d) and 3(a)). 16-cell stage was also the initial of remethylation (Figure 1(a)) and the erasure of 5-hmC (Figure 1(b)), indicating 16-cell being a specific stage during medaka embryogenesis. Previous studies showed, by the end of this period, the mouse genome also has a dramatic DNA remethylation, while in zebrafish the global genome methylation still maintains the highest level. These discrepancies are indicative of dissimilar epigenetic reprogramming between mice and zebrafish, whereas the pattern of medaka epigenome reprogramming is somewhat similar to that in mice. Step 4, early gastrula (Stage 13) to late gastrula (Stage 16): during this period, the genome still maintained hypermethylation from early gastrula to late gastrula but comparatively lower than earlier stages and reached the highest level at late gastrula. The expression of DNMT1 significantly decreased, indicating that the need for maintenance of fast cleavage was complete. Intriguingly, the expression of TET3 was terminated; while the expression of TET2 was initiated, showing that TET2 and TET3 may have complementary functions during embryogenesis in medaka. In both mice and zebrafish, genome DNA methylation maintains the highest level during this period [23,26,29,30]. All three animal models maintain genome hypermethylation during gastrula, indicating that embryonic epigenetic reprogramming and pluripotency are maintained for a defined period of embryogenesis in a species-specific manner.
Post-fertilization epigenome reprogramming modulates the outcome of transgenerational inheritance by erasing epigenetic marks transmitted through gametes and creating new marks [49]. Medaka has been demonstrated to be an excellent model for studying transgenerational inheritance of phenotypes [51,52]. Embryonic exposure to environmental estrogenic chemicals results in transgenerational inheritance of reproductive dysfunction in medaka [51]. The present study showed medaka genome undergoes reprogramming of DNA methylation marks during early embryogenesis. However, it is not clearly understood if environmental estrogenic chemicals inducing DNA methylation marks could escape the four stages of reprogramming described herein. In mammals, the basic principles of epigenetic reprogramming in embryos and germ cells have been known and studied for many years; major aspects, including the dynamics of these processes, remain enigmatic [45]. Understanding this process in another model organism opens the door for further investigation into the mechanisms underlying epigenetic reprogramming and transgenerational inheritance, especially when the model organism allows studies due to its external fertilization and embryo development ex ovo. Furthermore, similarities in these epigenetic processes between human, mice, and medaka further strengthen the possibility for medaka to serve as an ideal model for epigenetic and transgenerational inheritance research (Figure 4(b)).
In conclusion, this study provides dynamics of genome DNA methylation and demethylation during early embryogenesis in medaka together with the dynamics of expression pattern of genes involved in genome methylation and demethylation. Medaka embryos utilize active demethylation strategy to erase the paternal genome methylation pattern within the first cell cycle, and then the global DNA methylation levels gradually increase from 16-cell stage to gastrula stages. Unlike zebrafish, in which only maternal genome has a slight increase in DNA methylation during embryogenesis, medaka has a DNA methylation reprogramming process similar to mammals including human [24,25]. Although medaka undergoes genome demethylation immediately after fertilization and completes reprogramming quickly, the pattern of de novo methylation and completion of reprogramming event during gastrulation make medaka ideal model for studying embryo development.
Funding Statement
This work was supported by the National Institute of Environmental Health Sciences [R21ES027123].
Accession Number
Sequencing data reported in this paper have been deposited into the public database at NCBI Gene Expression Omnibus (GEO) under accession number GSE128797.
Acknowledgments
Authors have no financial conflict of interests. The study was supported by exploratory research grant (ES027123) from National Institute of Environmental Health Sciences, National Institute of Health, USA to RKB.
Author contributions
X.W. and R.K.B designed research; X.W. performed research; RKB contributed reagents/analytic tools; X.W. and R.K.B. analysed data, and X.W. and R.K.B. wrote the paper.
Disclosure statement
No potential conflict of interest was reported by the authors.
Supplementary material
Supplemental data for this article can be accessed here.
References
- [1].Kass SU, Pruss D, Wolffe AP.. How does DNA methylation repress transcription? Trends Genet. 1997;13(11):444–449. [DOI] [PubMed] [Google Scholar]
- [2].Li E, Beard C, Jaenisch R.. Role for DNA methylation in genomic imprinting. Nature. 1993;366(6453):362. [DOI] [PubMed] [Google Scholar]
- [3].Jähner D, Stuhlmann H, Stewart CL, et al. De novo methylation and expression of retroviral genomes during mouse embryogenesis. Nature. 1982;298(5875):623. [DOI] [PubMed] [Google Scholar]
- [4].Iida A, Shimada A, Shima A, et al. Targeted reduction of the DNA methylation level with 5-azacytidine promotes excision of the medaka fish Tol2 transposable element. Genet Res (Camb). 2006;87(3):187–193. [DOI] [PubMed] [Google Scholar]
- [5].Bestor TH. The DNA methyltransferases of mammals. Hum Mol Genet. 2000;9(16):2395–2402. [DOI] [PubMed] [Google Scholar]
- [6].Li E, Bestor TH, Jaenisch R. Targeted mutation of the DNA methyltransferase gene results in embryonic lethality. Cell. 1992;69(6):915–926. [DOI] [PubMed] [Google Scholar]
- [7].Okano M, Bell DW, Haber DA, et al. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell. 1999;99(3):247–257. [DOI] [PubMed] [Google Scholar]
- [8].Barau J, Teissandier A, Zamudio N, et al. The DNA methyltransferase DNMT3C protects male germ cells from transposon activity. Science. 2016;354(6314):909–912. [DOI] [PubMed] [Google Scholar]
- [9].Chédin F, Lieber MR, Hsieh C-L. The DNA methyltransferase-like protein DNMT3L stimulates de novo methylation by Dnmt3a. Proc Nat Acad Sci. 2002;99(26):16916–16921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Hata K, Okano M, Lei H, et al. Dnmt3L cooperates with the Dnmt3 family of de novo DNA methyltransferases to establish maternal imprints in mice. Development. 2002;129(8):1983–1993. [DOI] [PubMed] [Google Scholar]
- [11].Suetake I, Shinozaki F, Miyagawa J, et al. DNMT3L stimulates the DNA methylation activity of Dnmt3a and Dnmt3b through a direct interaction. J Biol Chem. 2004;279(26):27816–27823. [DOI] [PubMed] [Google Scholar]
- [12].Rougier N, Bourc’his D, Gomes DM, et al. Chromosome methylation patterns during mammalian preimplantation development. Genes Dev. 1998;12(14):2108–2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Guo F, Li X, Liang D, et al. Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell. 2014;15(4):447–458. [DOI] [PubMed] [Google Scholar]
- [14].Inoue A, Shen L, Dai Q, et al. Generation and replication-dependent dilution of 5fC and 5caC during mouse preimplantation development. Cell Res. 2011;21(12):1670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Inoue A, Zhang Y. Replication-dependent loss of 5-hydroxymethylcytosine in mouse preimplantation embryos. Science. 2011;334(6053):194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Wu X, Zhang Y. TET-mediated active DNA demethylation: mechanism, function and beyond. Nat Rev Genet. 2017;18(9):517–534. Epub 2017 May 31 PubMed PMID: 28555658. [DOI] [PubMed] [Google Scholar]
- [17].Kohli RM, Zhang Y. TET enzymes, TDG and the dynamics of DNA demethylation. Nature. 2013;502(7472):472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hackett JA, Sengupta R, Zylicz JJ, et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339(6118):448–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Li Z, Gu T-P, Weber AR, et al. Gadd45a promotes DNA demethylation through TDG. Nucleic Acids Res. 2015;43(8):3986–3997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Kienhöfer S, Musheev MU, Stapf U, et al. GADD45a physically and functionally interacts with TET1. Differentiation. 2015;90(1–3):59–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Labbé C, Robles V, Herraez MP. Epigenetics in fish gametes and early embryo. Aquaculture. 2017;472:93–106. [Google Scholar]
- [22].Seisenberger S, Peat JR, Hore TA, et al. Reprogramming DNA methylation in the mammalian life cycle: building and breaking epigenetic barriers. Philos Trans R Soc B. 2013;368(1609):20110330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Dean W, Santos F, Reik W. Epigenetic reprogramming in early mammalian development and following somatic nuclear transfer. Semin Cell Dev Biol. 2003;14(1):93–100. Epub 2003 Jan 14 PubMed PMID: 12524012. [DOI] [PubMed] [Google Scholar]
- [24].Guo H, Zhu P, Yan L, et al. The DNA methylation landscape of human early embryos. Nature. 2014;511(7511):606. [DOI] [PubMed] [Google Scholar]
- [25].Smith ZD, Chan MM, Humm KC, et al. DNA methylation dynamics of the human preimplantation embryo. Nature. 2014;511(7511):611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Wang L, Zhang J, Duan J, et al. Programming and inheritance of parental DNA methylomes in mammals. Cell. 2014;157(4):979–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Smith ZD, Chan MM, Mikkelsen TS, et al. A unique regulatory phase of DNA methylation in the early mammalian embryo. Nature. 2012;484(7394):339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Peat JR, Dean W, Clark SJ, et al. Genome-wide bisulfite sequencing in zygotes identifies demethylation targets and maps the contribution of TET3 oxidation. Cell Rep. 2014;9(6):1990–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Jiang L, Zhang J, Wang -J-J, et al. Sperm, but not oocyte, DNA methylome is inherited by zebrafish early embryos. Cell. 2013;153(4):773–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Potok ME, Nix DA, Parnell TJ, et al. Reprogramming the maternal zebrafish genome after fertilization to match the paternal methylation pattern. Cell. 2013;153(4):759–772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Fang X, Corrales J, Thornton C, et al. Global and gene specific DNA methylation changes during zebrafish development. Comp Biochem Physiol Part B Biochem Mol Biol. 2013;166(1):99–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Papoulias DM, Noltie DB, Tillitt DE. An in vivo model fish system to test chemical effects on sexual differentiation and development: exposure to ethinyl estradiol. Aquatic Toxicol. 2000;48(1):37–50. [Google Scholar]
- [33].Ishikawa Y. Medakafish as a model system for vertebrate developmental genetics. Bioessays. 2000;22(5):487–495. [DOI] [PubMed] [Google Scholar]
- [34].Wittbrodt J, Shima A, Schartl M. Medaka—a model organism from the far East. Nat Rev Genet. 2002;3(1):53. [DOI] [PubMed] [Google Scholar]
- [35].Matsuda M, Nagahama Y, Shinomiya A, et al. DMY is a Y-specific DM-domain gene required for male development in the medaka fish. Nature. 2002;417(6888):559. [DOI] [PubMed] [Google Scholar]
- [36].Kasahara M, Naruse K, Sasaki S, et al. The medaka draft genome and insights into vertebrate genome evolution. Nature. 2007;447(7145):714. [DOI] [PubMed] [Google Scholar]
- [37].Shinomiya A, Tanaka M, Kobayashi T, et al. The vasa‐like gene, olvas, identifies the migration path of primordial germ cells during embryonic body formation stage in the medaka, Oryzias latipes. Dev Growth Differ. 2000;42(4):317–326. [DOI] [PubMed] [Google Scholar]
- [38].Walter RB, Li H-Y, Intano GW, et al. Absence of global genomic cytosine methylation pattern erasure during medaka (Oryzias latipes) early embryo development. Comp Biochem Physiol Part B Biochem Mol Biol. 2002;133(4):597–607. [DOI] [PubMed] [Google Scholar]
- [39].Iwamatsu T. Stages of normal development in the medaka Oryzias latipes. Mech Dev. 2004;121(7–8):605–618. [DOI] [PubMed] [Google Scholar]
- [40].Krueger F, Andrews SR. Bismark: a flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics. 2011;27(11):1571–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Untergasser A, Cutcutache I, Koressaar T, et al. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012;40(15):e115–e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods. 2001;25(4):402–408. [DOI] [PubMed] [Google Scholar]
- [43].Qu W, Hashimoto S-I, Shimada A. Genome-wide genetic variations are highly correlated with proximal DNA methylation patterns. Genome Res. 2012;22:1419–1425. Epub 2012 June 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Aizawa K, Shimada A, Naruse K, et al. The medaka midblastula transition as revealed by the expression of the paternal genome. Gene Expression Patterns. 2003;3(1):43–47. [DOI] [PubMed] [Google Scholar]
- [45].Messerschmidt DM, Knowles BB, Solter D. DNA methylation dynamics during epigenetic reprogramming in the germline and preimplantation embryos. Genes Dev. 2014;28(8):812–828. Epub 2014 Apr 17 PubMed PMID: 24736841; PubMed Central PMCID: PMCPMC4003274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Almeida RD, Loose M, Sottile V, et al. 5-hydroxymethyl-cytosine enrichment of non-committed cells is not a universal feature of vertebrate development. Epigenetics. 2012;7(4):383–389. [DOI] [PubMed] [Google Scholar]
- [47].Lane N, Dean W, Erhardt S, et al. Resistance of IAPs to methylation reprogramming may provide a mechanism for epigenetic inheritance in the mouse. Genesis. 2003;35(2):88–93. PubMed PMID: 12533790. [DOI] [PubMed] [Google Scholar]
- [48].Hackett JA, Sengupta R, Zylicz JJ, et al. Germline DNA demethylation dynamics and imprint erasure through 5-hydroxymethylcytosine. Science. 2013;339(6118):448–452. PubMed PMID: 23223451; PubMed Central PMCID: PMC3847602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Guo F, Li X, Liang D, et al. Active and passive demethylation of male and female pronuclear DNA in the mammalian zygote. Cell Stem Cell. 2014;15(4):447–459. Epub 2014 Sep 16 PubMed PMID: 25220291. [DOI] [PubMed] [Google Scholar]
- [50].Dasmahapatra AK, Khan IA. DNA methyltransferase expressions in Japanese rice fish (Oryzias latipes) embryogenesis is developmentally regulated and modulated by ethanol and 5-azacytidine. Comp Biochem Physiol Part C Toxicol Pharmacol. 2015;176:1–9. [DOI] [PubMed] [Google Scholar]
- [51].Bhandari RK, Vom Saal FS, Tillitt DE. Transgenerational effects from early developmental exposures to bisphenol A or 17alpha-ethinylestradiol in medaka, Oryzias latipes. Sci Rep. 2015;5:9303 Epub 2015 Mar 21 PubMed PMID: 25790734; PubMed Central PMCID: PMCPMC4366817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bhandari RK. Medaka as a model for studying environmentally induced epigenetic transgenerational inheritance of phenotypes. Environ Epigenet. 2016;2(1):dvv010 Epub 2016 Jan 30 PubMed PMID: 29492282; PubMed Central PMCID: PMCPMC5804509. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.