

Abstract
Ketamine is the first exemplar of a rapid-acting antidepressant with efficacy for treatment-resistant symptoms of mood disorders. Its discovery emerged from a reconceptualization of the biology of depression. Neurobiological insights into ketamine efficacy shed new light on the mechanisms underlying antidepressant efficacy.
The rapid, profound, and sustainable antidepressant effects of ketamine seem poised to transform the treatment of depression, while mechanisms through which it may work are overturning the received wisdom regarding the underlying neurobiology.
The problem: an overly narrow focus on monoamine signaling
Depression is among the most disabling medical conditions. Yet, in America, 12.5% of individuals over the age of 12 recently filled an antidepressant prescription (Pratt et al., 2017). Thus, shortcomings in the effectiveness of antidepressant treatments probably contribute to the enormous public health burden of depression. Despite progress, too few patients respond to antidepressants, improvement is too slow among eventual responders, and too many patients relapse after having achieved response. Further, subgroups of depressed patients, particularly those with bipolar disorder, respond poorly to traditional antidepressants and they are treated predominately with alternative treatments (Duman et al., 2016; Krystal et al., 2013).
The premature conclusion that all antidepressants worked by ameliorating deficits in monoamine signaling contributed to the failure to identify fundamentally new treatment mechanisms since the discovery of antidepressants in the late 1950s. Limitations of this “monoamine hypothesis of depression” emerged in the 1990s. While depleting the body of monoamines transiently reversed the therapeutic effects of antidepressants, monoamine depletion did not reliably induce depression in healthy people nor did it consistently worsen depression in unmedicated depressed patients. Further, studies focused increasingly on the postsynaptic response to antidepressants, involving signaling mechanisms that were downstream from monoamine receptors and not specific to monomine signal transduction. These mechanisms included alterations in neurotrophin signaling, transcriptional alterations, and epigenetic changes. Thus, there had long been clues that critical elements of the neurobiology of depression and its treatment were extrinsic to monoamine neurons (Duman et al., 2016).
The opportunity: a broader circuitry perspective
We wondered, how could we target the non-monoaminergic mechanisms more directly? In a simple perspective shift, illustrated in figure 1A, we and others reasoned that the pathology of depression might “reside” in the intrinsic circuitry of the cortex and limbic system (Duman et al., 2016; Krystal et al., 2013), where neurons predominately released glutamate and GABA rather than monoamines. We (JK, DC) had studied glutamate pathophysiology related to schizophrenia and alcohol use disorders by evaluating the response to the N-methyl-D-aspartate (NMDA) glutamate receptor antagonist, ketamine. In light of evidence of cortico-limbic pathology in depression, we (JK, DC) decided to explore glutamate synaptic alterations using the response to ketamine as a probe. We were aware of preclinical studies suggesting that NMDA receptor antagonists had antidepressant effects, particularly those of Dr. Phil Skolnick and his colleagues. However, we did not expect much benefit from single doses of ketamine in depressed patients. Our study was not motivated by clinical reports of antidepressant effects of ketamine in the anesthesia context. The dose and manner of ketamine infusion in our depression study (0.5 mg/kg ketamine infused intravenously over 40 minutes) was derived from our psychosis studies. In those studies, we selected a dose and rate of infusion that produced transient schizophrenia-like symptoms and cognitive impairments without producing delirium or an anesthetized state.
Figure 1.
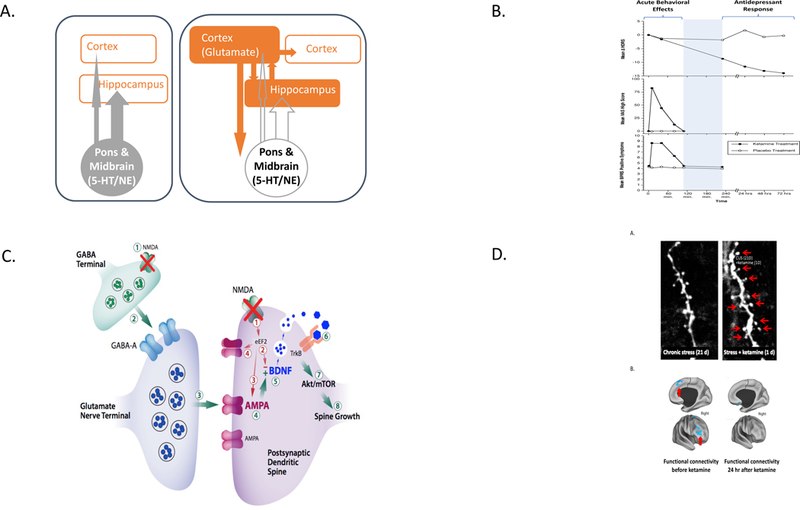
The Path to the Identification of the Antidepressant Effects of Ketamine and Its Therapeutic Mechanisms of Action (A) This figure illustrates the perspective shift that set the stage for testing ketamine effects in depressed patients. The left side of the figure presents the historically dominant theory, i.e., that deficits in serotonin (5-HT) and norepinephrine (NE) signaling contribute to the biology of depression and that pharmacotherapy aims to reverse these deficits. 5-HT and NE neurons are based in the midbrain and pons, respectively, and modulate the activity of higher brain centers. The right side of the figure highlights the shift in perspective to cortical and limbic mechanisms. The neurons in the higher brain centers release glutamate and GABA predominately. If one viewed depression as a disorder of cortico-limbic function, then glutamatergic and GABAergic signaling would be implicated. This perspective shift led us to test the effects of the NMDA glutamate receptor antagonist as a probe of alterations in glutamate signaling associated with depression. (B) This figure highlights the temporal dissociation of the acute behavioral effects of ketamine, which are observed in healthy humans and patient populations, and the rapidly emerging antidepressant effects of ketamine, which occur only in individuals with psychiatric symptoms (n = 7, modified from Berman et al., 2000). It presents mean changes from baseline in the 25-item Hamilton Depression Rating Scale scores (HDRS), the mean Visual Analog Scale “high” scores (VAS-high, 0–100 mm), and mean positive symptom scores of the Brief Psychiatric Rating Scale (BPRS-positive) after ketamine (0.5 mg/kg over 40 min) and saline infusions. The emergence of antidepressant effects after the abatement of the transient pharmacologic effects of ketamine is consistent with the hypothesis that these antidepressant effects reflect a reaction to ketamine exposure rather than a property of ketamine intoxication. (C) This cartoon illustrates emerging mechanistic hypotheses related to the antidepressant effects of ketamine. Some effects may emerge directly as a downstream consequence of NMDA glutamate receptor antagonism. These effects are illustrated by blockade of postsynaptic, presumably GluN2B-containing NMDA receptors. When overstimulated, these receptors activate eukaryotic elongation factor-2 (eEF2) and depress BDNF levels. Blockade of these NMDA receptors raises BDNF levels and shuttles AMPA glutamate receptors to the synapse, enhancing synaptic efficacy. Ketamine also may generate its antidepressant effects indirectly by blocking NMDA receptors on GABA interneurons. In this way, ketamine reduces inhibition of glutamate release and, in turn, results in enhanced stimulation of AMPA glutamate receptors. AMPA receptor activation activates a signaling cascade that raises BDNF levels. Local release of BDNF is thought to stimulate TrkB receptors, engaging relevant signaling cascades and resulting in the activation of the molecular target of rapamycin complex 1 (mTORC1). This step, in turn, activates local protein synthesis necessary for increasing dendritic spine formation and restoring synaptic connectivity. In this figure, AMPA, BDNF, and mTORC1 are highlighted because blockade of these steps in the pathway prevents the emergence of the antidepressant effects of ketamine (Duman et al., 2016, Krystal et al., 2013). Please note the convergence of the direct and indirect effects of ketamine on some common mechanisms may constitute elements of a common pathway for antidepressant efficacy, i.e., enhancement of synaptic efficacy and connectivity in key circuits involved in the regulation of mood. (D) These figures present data suggesting that the ability of ketamine to increase cortical structural connectivity in animals and restore cortical functional connectivity in depressed patients is related to its clinical efficacy. (Da) This figure illustrates the depletion of dendritic spines on prefrontal cortical pyramidal (glutamate-releasing) neurons in animals exposed to repeated stresses (Li et al., 2010). (Db) This figure presents functional MRI data collected in symptomatic depression patients prior to (left figure) and 24 h after (right figure) ketamine. Areas in blue show reduced functional connectivity (degree of correlation of activity with other brain regions). Following ketamine administration, the reductions in functional connectivity (areas in blue) are ameliorated, associated with alleviation of depression symptoms (Abdallah et al., 2017)
To the amazement of our patients and ourselves, we found that ketamine produced rapid, profound, and surprisingly durable antidepressant effects that were temporally dissociated from the brief acute behavioral effects of the drug, i.e., the initial euphoria produced by ketamine was not a part of its antidepressant effect ((Berman et al., 2000), figure 1B). These antidepressant effects had not been observed in healthy subjects. Our initial findings were widely replicated (Krystal et al., 2013; Zarate et al., 2006). These replications found that a single dose of ketamine produced antidepressant effects that began within hours, peaked within 24–72 hours, and then dissipated typically within 2 weeks if ketamine was not repeated. Also, the subsequent studies showed that ketamine was effective in antidepressant non-responders, including patients with bipolar disorder. Further, the antidepressant effects of ketamine were meaningful clinically, with one third of patients with treatment-resistant symptoms achieving remission and approximately 50%−75% of patients demonstrating clinical response from a single dose, with higher rates of response and remission with repeated administrations (Wilkinson et al., 2017). Lastly, ketamine reduced all symptoms of depression, notably suicidal ideation (Krystal et al., 2013).
Many questions remain regarding the optimal prescription of ketamine. For example, there is great interest in finding a sub-dissociative therapeutic dose. This aim is challenging because the dose-response relationship for subanesthetic ketamine is very steep, with 0.2 mg/kg being subtherapeutic and 0.5 mg/kg being dissociative and effective (Fava et al., 2017; Su et al., 2017), although one study reported positive effects with 0.1 mg/kg (Fava et al., 2017). This effort is further complicated by variability in plasma levels at each dose and differential sensitivity to ketamine across patients. Some patients show improvement with minimal dissociation. In other patients, increasing dissociative symptoms by administering a higher ketamine dose (1.0 mg/kg; ~70 mg) does not enhance the rapid therapeutic response (Fava et al., 2017). Thus, dissociative symptoms may not mediate clinical benefits, but they may signal adequate target engagement by ketamine. The optimal frequency of ketamine administration is also evolving. Treatment is initiated typically twice per week. The frequency of infusions is tapered gradually, with as many as 40% of patients maintained with monthly or less frequent infusions in our clinic. However, there is relatively little comparative effectiveness data to guide the tapering of administration frequency. In practice, the timing of infusions is adapted to the needs of particular patients.
Mechanism of action: ketamine as “the tip of the iceberg”
The identification of the antidepressant effects of ketamine stimulated basic and translational neuroscience research. Paradoxically, ketamine increases neuroplasticity despite blocking NMDA receptors, a critical mediator of plasticity (Duman et al., 2016; Krystal et al., 2013). By blocking GluN2B-containing NMDA receptors, ketamine may prevent the phosphorylation of eEF2, raise BDNF levels, and promote the shuttling of AMPA receptors to the synapse, enhancing synaptic connectivity and plasticity (figure 1C). To date, there is at least one study suggesting that a GluN2B-prefering NMDA antagonist showed evidence of antidepressant efficacy at a dose that also produced dissociative symptoms (Krystal et al., 2013). It remains unclear whether NMDA receptor antagonist properties, including subtype selectivity, competitive vs uncompetitive antagonism, or high vs low trapping within the cation channel will be strategies to meaningfully improve efficacy or safety.
The ability of ketamine to disinhibit glutamate release also may contribute to its antidepressant efficacy (figure 1C) (Duman et al., 2016; Krystal et al., 2013). In humans, ketamine stimulates the cortical rate of conversion of 13C-glutamate to 13-C glutamine, a measure stoichiometrically related to glutamate release (Abdallah et al., 2018). In animal studies, by transiently enhancing glutamate release and stimulating AMPA glutamate receptors, ketamine promotes BDNF release, enhances TrkB receptor stimulation, activates mTORC1, and stimulates local protein synthesis. Most remarkably, this cascade of processes results in the rapid proliferation of dendritic spines that share a temporal profile with antidepressant effects. As illustrated in figure 1D, these effects in animals are paralleled by restoration of functional connectivity in an fMRI study of depressed patients (Abdallah et al., 2018).
In the time window where ketamine enhances synaptic connectivity, it also enhances synaptic plasticity. In animals, fear extinction is enhanced 24 hours after ketamine administration (Duman et al., 2016). In depressed patients, there is preliminary evidence that cognitive behavioral therapy may extend the duration of ketamine efficacy. Thus, there may be specific ways to combine ketamine, psychotherapies, and other interventions that might heighten their clinical impact.
We do not yet have closure on the mechanisms through which ketamine produces its antidepressant effects. For example, ketamine has anti-inflammatory effects, epigenetic effects, and it alters activity within circuits implicated in reward and motivation. Ketamine effects at sites other than NMDA receptors also may contribute to its efficacy. The antidepressant effects of both isomers of ketamine are being studied, as well as the ketamine metabolites, (S)-norketamine and (2R,6R)-hydroxynorketamine (HNK). If (S)-ketamine, (R)-ketamine, and HNK differ in their mechanisms of action, it is possible that they have complementary or even additive antidepressant effects (Duman et al., 2016).
Ketamine may serve as a prototype for an entirely new class of antidepressant medications. This view is based on the hypothesis that selectively targeting elements of ketamine’s effects could preserve efficacy while producing greater tolerability. For example, other putative antidepressants that increase glutamate release might include serotonergic hallucinogens (LSD, psilocybin), muscarinic receptor antagonists (scopolamine), mGluR2 antagonists and GABAA subtype selective negative allosteric modulators or inverse agonists. AMPAkines might have antidepressant effects via AMPA receptor facilitation. Similarly, one could imagine developing drugs to raise BDNF levels, enhance TrkB receptor signaling, or promote mTORC1 activation (Duman et al., 2016; Krystal et al., 2013).
Changing expectations
The surprisingly rapid and profound efficacy of ketamine revealed that our expectations for antidepressant treatment were constrained by limitations in our understanding of what might be possible. Further, the fascinating biology of the antidepressant effects of ketamine showed how little we understood the most intensively studied class of medications in psychiatry.
Where does ketamine belong in the treatment algorithm for depression?
In light of growing long-term safety and efficacy data, particularly for esketamine, ketamine can be viewed as a long-term treatment. It was reserved initially for patients who failed electroconvulsive therapy. However, increasingly, it is prescribed to patients who have failed two adequate antidepressant treatments. The distinctive rapidity of onset and efficacy of ketamine and esketamine for the treatment of suicidal ideation raise the possibility that they could be used in urgent care contexts, including Emergency Rooms or other medical contexts, to rapidly manage suicide risk and to mitigate or shorten psychiatric hospitalization.
What does long-term ketamine treatment look like?
Ketamine treatment must include protections against the abuse liability of ketamine and provide for the management of its acute dissociative effects. Both of these risks may be managed by limiting drug administration to clinical settings. However, alternative treatment approaches, such as home administration by a visiting nurse, might be explored to see whether they could increase access and reduce costs of treatment without substantially increasing risks.
Could ketamine change what it means to have depression?
Depression is highly stigmatized. When people disclose their depression, they may have difficulty obtaining jobs, getting promotions, and maintaining relationships. When disabled by depression, they may be unable to perform at work or to care for their families. They may lose hope of recovery and attempt suicide, particularly if treatments have been ineffective in producing remission or preventing relapse. Ketamine may become a transformative treatment. Transformative treatments have a powerful impact on stigma, as exemplified by the emergence of anti-retroviral treatments for AIDS. Thus, ketamine may not only be a source of hope for patients and their families, but also a powerful weapon in the fight against stigma and for parity in the support for depression prevention, treatment, and research.
Was the discovery of ketamine’s antidepressant serendipitous?
Of course. However, its discovery emerged from the testing of a novel mechanistic hypothesis related to the pathophysiology of depression. Without that hypothesis and a pharmacologic tool to test that hypothesis, the fortuitous clinical observation would not have been made. Once the clinical observation occurred, advances in basic neuroscience led to more specific mechanistic hypotheses. These hypotheses are now driving a new generation of human translational neuroscience studies. Thus, the “ketamine story” is a step toward an era when psychiatric neuroscience more routinely identifies novel treatments based on a progressively deeper understanding of the brain and the pathophysiology of psychiatric disorders. We must be cautious about what we claim to understand about the brain, the biology of depression, and treatment. Nonetheless, this increasingly mature scientific foundation for psychiatry bodes well for the future of the overall enterprise aimed at reducing the burden of mental illness.
Acknowledgements
Dr. Krystal is supported by the Department of Veterans Affairs (National Center for PTSD), the NIAAA Center for the Translational Neuroscience of Alcohol P50AA12870), and the the Yale Center for Clinical Investigation (UL1RR024139).
Dr. Krystal Financial Disclosures:
NON Federal Research Support AstraZeneca Pharmaceuticals provides the drug, Saracatinib, for research related to NIAAA grant “Center for Translational Neuroscience of Alcoholism [CTNA-4]. Dr. Sanacora: Dr. Sanacora has received consulting fees from Alkermes, Allergan, AstraZeneca, Avanier Pharmaceuticals, Axsome Therapeutics, Biohaven Pharmaceuticals, Boehringer Ingelheim, Bristol-Myers Squibb, Hoffmann–La Roche, Intra-Cellular Therapies, Janssen, Merck, Minerva Neurosciences, Naurex, Navitor Pharmaceuticals, Novartis, Noven Pharmaceuticals, Otsuka, Perception Neuroscience, Praxis Therapeutics, Sage Pharmaceuticals, Servier Pharmaceuticals, Taisho Pharmaceuticals, Teva, Valeant, and Vistagen Therapeutics. He has also received research contracts from AstraZeneca, Bristol-Myers Squibb, Eli Lilly, Johnson & Johnson, Hoffmann–La Roche, Merck, Naurex, and Servier Pharmaceuticals. No-cost medication was provided to Dr. Sanacora for an NIH-sponsored study by Sanofi-Aventis. In addition, he holds shares in Biohaven Pharmaceuticals Holding Company and is a co-inventor on the patent “Glutamate agents in the treatment of mental disorders” (patent 8778979) and is a co-inventor on. U.S. Provisional Patent Application No. 047162–7177P1 (00754) Combination Therapy for Treating or Preventing Depression or Other Mood Diseases filed on August 20, 2018 by Yale University Office of Cooperative Research OCR 7451 US01. Dr. Charney is supported by P01HL131478 (PI: Fayad) and grants from the Brain and Behavior Research Foundation to collaborating faculty. He is Dean of the Icahn School of Medicine at Mount Sinai (ISMMS) and co-inventor on patents related to ketamine outlined below. If ketamine gains regulatory approval both Dr. Charney and ISMMS stand to benefit financially. ISMMS also is seeking to license ketamine as a treatment for PTSD. Dr. Charney holds a share of the following patents: 9,592,207 - Intranasal Administration of Ketamine to Treat Depression (Issued March 14, 2017), 9,539,220 - Methods for Treating Suicidal Ideation (Issued January 10, 2017), 8,785,500 - Intranasal Administration of Ketamine to Treat Depression (Issued July 22, 2014), 10,123,737 - Systems and Methods for Treating a Psychiatric Disorder (Issued November 13, 2018), US CON Patent Appl No. 16/189,059 – and related foreign patent applications - Systems and Methods for Treating a Psychiatric Disorder, Provisional Patent Appl No. 62/649,469 – Brain Plasticity Following Cognitive-Emotional Training, US CON Patent Appl No. 14/974,576 and related foreign patent applications - Method for Treating Post Traumatic Stress Disorder (PTSD), US Serial No. 14/889,746 and a related foreign patent application - Treatment of Mood and Anxiety Disorders, US CON Patent Appl Nos. 15/379,013 and 15/417,689 - Intranasal Administration of Ketamine to Treat Depression. Dr. Duman is supported by NIMH grants MH045481 (R.S.D.), MH093897 (R.S.D.), MH077681, and the state of Connecticut. Dr. Duman has served as a consultant for Janssen, Taisho, Naurex, and Aptinyx, and has received research support from Lilly, Taisho, Allergan, Janssen, Naurex, Aptynix, Navitor, and Relmada.
Footnotes
Disclosures:
The Individual Consultant Agreements listed below are less than $10,000 per year: AstraZeneca Pharmaceuticals, Biogen, Idec, MA, Biomedisyn Corporation, Bionomics, Limited (Australia) Boehringer Ingelheim International, Concert Pharmaceuticals, Inc., Epiodyne, Inc., Heptares Therapeutics, Limited (UK), Janssen Research & Development, L.E.K. Consulting, Otsuka America Pharmaceutical, Inc., Perception Neuroscience Holdings, Inc., Spring Care, Inc., Sunovion Pharmaceuticals, Inc., Takeda Industries,Taisho Pharmaceutical Co., Ltd.
Scientific Advisory Board: Bioasis Technologies, Inc., Biohaven Pharmaceuticals, BioXcel Therapeutics, Inc. (Clinical Advisory Board), BlackThorn Therapeutics, Inc., Broad Institute of MIT and Harvard, Cadent Therapeutics, Lohocla Research Corporation, Stanley Center for Psychiatric Research at the Broad Institute
Stock: ArRETT Neuroscience, Inc., BlackThorn Therapeutics, Inc., Biohaven Pharmaceuticals Medical Sciences Spring Care, Inc., Sage Pharmaceuticals.
Stock Options: Biohaven Pharmaceuticals Medical Sciences, Storm Biosciences, Inc.
Income Greater than $10,000: Editorial Board - Editor - Biological Psychiatry.
Patents and Inventions 1) Seibyl JP, Krystal JH, Charney DS. Dopamine and noradrenergic reuptake inhibitors in treatment of schizophrenia. US Patent #5,447,948. September 5, 1995. 2) Vladimir, Coric, Krystal, John H, Sanacora, Gerard – Glutamate Modulating Agents in the Treatment of Mental Disorders US Patent No. 8,778,979 B2 Patent Issue Date: July 15, 2014. US Patent Application No. 15/695,164: Filing Date: 09/05/2017 3) Charney D, Krystal JH, Manji H, Matthew S, Zarate C., - Intranasal Administration of Ketamine to Treat Depression United States Application No. 14/197,767 filed on March 5, 2014; United States application or Patent Cooperation Treaty (PCT) International application No. 14/306,382 filed on June 17, 2014 4) Zarate, C, Charney, DS, Manji, HK, Mathew, Sanjay J, Krystal, JH, Department of Veterans Affairs “Methods for Treating Suicidal Ideation”, Patent Application No. 14/197.767 filed on March 5, 2014 by Yale University Office of Cooperative Research. 5) Arias A, Petrakis I, Krystal JH. – Composition and methods to treat addiction. Provisional Use Patent Application no.61/973/961. April 2, 2014. Filed by Yale University Office of Cooperative Research. 6) Chekroud, A., Gueorguieva, R., & Krystal, JH. “Treatment Selection for Major Depressive Disorder” [filing date 3rd June 2016, USPTO docket number Y0087.70116US00]. Provisional patent submission by Yale University. 7) Gihyun, Yoon, Petrakis I, Krystal JH – Compounds, Compositions and Methods for Treating or Preventing Depression and Other Diseases. U. S. Provisional Patent Application No. 62/444,552, filed on January10, 2017 by Yale University Office of Cooperative Research OCR 7088 US01. 8) Abdallah, C, Krystal, JH, Duman, R, Sanacora, G. Combination Therapy for Treating or Preventing Depression or Other Mood Diseases. U.S. Provisional Patent Application No. 047162–7177P1 (00754) filed on August 20, 2018 by Yale University Office of Cooperative Research OCR 7451 US01
Citations
- Abdallah CG, Averill LA, Collins KA, Geha P, Schwartz J, Averill C, DeWilde KE, Wong E, Anticevic A, Tang CY, et al. (2017). Ketamine Treatment and Global Brain Connectivity in Major Depression. Neuropsychopharmacology 42, 1210–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdallah CG, De Feyter HM, Averill LA, Jiang L, Averill CL, Chowdhury GMI, Purohit P, de Graaf RA, Esterlis I, Juchem C, et al. (2018). The effects of ketamine on prefrontal glutamate neurotransmission in healthy and depressed subjects. Neuropsychopharmacology 43, 2154–2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, and Krystal JH (2000). Antidepressant effects of ketamine in depressed patients. Biol Psychiatry 47, 351–354. [DOI] [PubMed] [Google Scholar]
- Duman RS, Aghajanian GK, Sanacora G, and Krystal JH (2016). Synaptic plasticity and depression: new insights from stress and rapid-acting antidepressants. Nat Med 22, 238–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava M, Freeman M, Flynn M, Judge H, Hoeppner B, Cusin C, Ionescu D, Mathew S, Chang L, Iosifescu D, et al. (2017). Double-blind, placebo-controlled trial of ketamine therapy in treatment resistant depression (TRD). In American Society of Clinical Psychopharmacology (Miami, Florida), p. 84 (#W28). [Google Scholar]
- Krystal JH, Sanacora G, and Duman RS (2013). Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry 73, 1133–1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pratt LA, Brody DJ, and Gu Q (2017). Antidepressant use among persons aged 12 and over: United States, 2011–2014 In NCHS Data Brief, no 283 (Hyattsville, MD: National Center for Health Statistics; ), pp. 1–8. [PubMed] [Google Scholar]
- Su TP, Chen MH, Li CT, Lin WC, Hong CJ, Gueorguieva R, Tu PC, Bai YM, Cheng CM, and Krystal JH (2017). Dose-Related Effects of Adjunctive Ketamine in Taiwanese Patients with Treatment-Resistant Depression. Neuropsychopharmacology 42, 2482–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson ST, Toprak M, Turner MS, Levine SP, Katz RB, and Sanacora G (2017). A Survey of the Clinical, Off-Label Use of Ketamine as a Treatment for Psychiatric Disorders. Am J Psychiatry 174, 695–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA Jr., Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, Charney DS, and Manji HK (2006). A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry 63, 856–864. [DOI] [PubMed] [Google Scholar]