

SUMMARY
Heterotypic interactions across diverse cell types can enable tumor progression and hold the potential to expand therapeutic interventions. Here, combined profiling and functional studies of glioma cells in glioblastoma multiforme (GBM) models establish that PTEN deficiency activates YAP1, which directly upregulates lysyl oxidase (LOX) expression. Mechanistically, secreted LOX functions as a potent macrophage chemoattractant via activation of the β1 integrin-PYK2 pathway in macrophages. These infiltrating macrophages secrete SPP1, which sustains glioma cell survival and stimulates angiogenesis. In PTEN-null GBM models, LOX inhibition markedly suppresses macrophage infiltration and tumor progression. Correspondingly, YAP1-LOX and β1 integrin-SPP1 signaling correlates positively with higher macrophage density and lower overall survival in GBM patients. This symbiotic glioma-macrophage interplay provides therapeutic targets specifically for PTEN-deficient GBM.
Keywords: Glioblastoma, Lysyl oxidase, Macrophages, PTEN, SPP1 and YAP1
IN BRIEF
Chen et al. find that PTEN deficiency in glioblastoma (GBM) increases macrophage infiltration via a YAP1-LOX-β1 integrin-PYK2 axis, the infiltrated macrophages in turn secrete SPP1 to support GBM survival. In PTEN-null GBM xenograft mouse models, inhibition of LOX reduces macrophage infiltration and tumor growth.
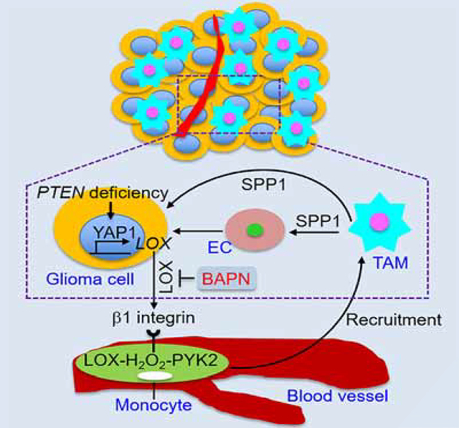
Graphical Abstract
INTRODUCTION
GBM is a lethal primary brain cancer in adults, with a median survival averaging 1 year following diagnosis (Lim et al., 2018; Maher et al., 2001; Zhu and Parada, 2002). Genomic profiling has defined GBM subgroups and identified alterations in core signaling pathways including the RTK/RAS/PI3K/PTEN, P53/ARF/MDM2 and RB/CDKN2A pathways (Brennan et al., 2013; Cancer Genome Atlas Research, 2008; Dunn et al., 2012; Zheng et al., 2008). The mesenchymal, classical and proneural subtypes are enriched for mutational alterations in PTEN, TP53, NF1 and RB1; in EGFR; and in PDGFRA and TP53, respectively (Verhaak et al., 2010). There is an urgent need to identify effective therapeutic strategies as current therapies have failed to improve survival meaningfully over the last 40 years (Dunn et al., 2012; Hambardzumyan and Bergers, 2015; Khosla, 2016; Roth and Weller, 2014).
The basis for therapeutic failure relates to significant inter- and intra-tumoral genetic instability and resultant heterogeneity, which generate diverse aberrant signaling pathways within and across tumors (Dunn et al., 2012; Patel et al., 2014). Heterogeneity also extends to the cellular level, with significant variability observed in the spectrum of stromal and immune cell types across different tumors (Quail and Joyce, 2013). The interplay between cancer cells and TME components is known to play pivotal roles in the maintenance of cancer hallmarks including angiogenesis and immune sequestration (Hambardzumyan and Bergers, 2015). The TME can be shaped by cancer cell-intrinsic signaling pathways and secreted factors (Parsa et al., 2007; Quail and Joyce, 2013; Wen et al., 2001; Zerrouqi et al., 2012). Infiltrating stromal and immune cells are prominent TME components that can affect tumor progression (Quail and Joyce, 2013). At the same time, the promise of immunotherapy has not yet been realized in GBM, prompting further study of the genetics and biology of infiltrating immune cells in specific GBM subsets and genotypes (Butowski et al., 2016).
Here, we explore whether and how specific genetic alterations in glioma cells might influence the immune composition of the TME and how such infiltrating immune cells might in turn function to inhibit or support the growth of glioma cells. Our goal was to identify therapeutically actionable targets functioning at the level of these TME heterotypic interactions between glioma cells and immune cells.
RESULTS
PTEN deletion/mutation facilitates macrophage infiltration in GBM
To identify specific genetic alterations and immune cell types that might influence GBM tumor biology, gene expression signature analyses were used to identify stromal and immune cell populations (Yoshihara et al., 2013) that might correlate with patient survival and track with specific genotypes. Using The Cancer Genome Atlas (TCGA) GBM datasets, we showed that high stromal and immune signatures were correlated with poor outcomes (Figure S1A), enriched in mesenchymal patients (Figure S1B), and correlated with genetic alterations of the PTEN-PI3K pathway, but not with other signature pathway alterations (Figures 1A,B and Table S1).
Figure 1. PTEN deletion/mutation facilitates macrophage infiltration in GBM.

(A, B) The stroma score (A) and immune score (B) of PTEN wild-type (WT) and deleted/mutated (Del/Mut) patients in TCGA GBM database (HG-UG133A, n=528; and Agilent4502A, n=489). The stroma and immune scores were determined based on expression data (Yoshihara et al., 2013).
(C) RNA microarray experiments and GSEA analysis in PTEN-WT and PTEN-KO SF763 cells.
(D) GSEA analysis for various types of immune cells in PTEN-WT and PTEN-deficient patients in TCGA GBM database (n=528).
(E) Quantification of relative migration of THP-1 macrophages following stimulation with conditioned media from PTEN-KO (#4 and #6) and PTEN-WT SF763 cells (#1 and #5). n=3 biological replicates.
(F) Quantification of relative migration of THP-1 macrophages following stimulation with conditioned media from PTEN-WT (#3) and PTEN-KO (#5) LN229 cells. n=3 biological replicates.
(G) Quantification of relative macrophage infiltration (CD206 and CD68 positive cells) in tumors established from U87 cells with or without PTEN overexpression (OE). n=3 biological replicates.
(H) Left panels, representative images showing the low, medium and high expression levels of phospho-AKT (P-AKT) and CD68 in human GBM TMA. Scale bar, 100 μm. Right panel, correlation analysis between phospho-AKT and CD68 expression in TMA (n=35). Pearson’s correlation test.
Data from multiple replicates are presented as mean. Error bars indicate mean ± SD. *p<0.05, ***p<0.001, Student’s t test. See also Figure S1; Tables S1 and S2.
To assess more directly the relevance of PTEN in modulating the TME, we conducted gene expression profiling and Gene Set Enrichment Analysis (GSEA) of PTEN null (CRISPRKO) versus parental PTEN-WT SF763 glioma cell lines. Specifically, in these isogenic lines, relative to WT controls, GSEA of PTEN-null cells exhibited a profile resembling the mesenchymal subtype with markedly increased expression of CTGF, FN1, VIM, CD44 and OSMR (Figures S1C,D), and showed prominent representations of immune response networks including TNFα/NF-κB signaling, inflammatory response and IL2/STAT5 signaling (Figure 1C).
To identify specific immune cells linked to PTEN deletions/mutations in GBM, we examined the TCGA GBM dataset for 17 types of immune cells using validated gene set signatures (Bindea et al., 2013; Engler et al., 2012). These analyses demonstrated that PTEN mutated/deleted GBMs correlated with significant enrichment of macrophages (total, M1 and M2) and, to a lesser extent, dendritic cells, while microglia and other immune cell types were not significantly changed (Figure 1D). Given the prominent representation of macrophages in PTEN-null GBM, we assessed two gene sets known to be responsible for macrophage chemoattraction by GSEA (Subramanian et al., 2005), showing enrichment of macrophage chemoattraction gene sets in PTEN-null GBM tumors and SF763 cells compared to PTEN-WT controls (Table S2). The gene sets for macrophages (total, M1 and M2) and for macrophage chemoattraction were not altered by other GBM signature alterations such as TP53 and EGFR (Table S2). Consistent with these in silico findings, transwell migration assays showed greatly enhanced macrophage migration with conditioned media (CM) from the PTEN-KO SF763 or PTEN-KO LN229 cells compared with CM from PTEN-WT cells (Figures 1E,F and S1E–H). Conversely, re-expression of PTEN in PTEN-null U87 tumor model significantly reduced macrophage infiltration (Figures 1G and S1I,J). Finally, activated AKT (phospho-AKT at Ser473) induced by PTEN deficiency showed a strong positive correlation with macrophage marker expression (CD68 and Mac-2) in human GBM tissue microarrays (TMAs) (Figures 1H and S1K). Together, these findings suggest that PTEN deficiency enhances recruitment of presumed tumor promoting macrophages into the GBM TME.
LOX, a potent macrophage chemoattractant, is secreted abundantly by PTEN-deficient glioma cells
To elucidate the factors governing macrophage recruitment in PTEN-deficient GBM, we examined putative macrophage recruiting factors exhibiting a •2.0-fold change in PTEN-KO SF763 cells using a secreted protein database (Chen et al., 2005) (Table S3). RT-qPCR and immunoblot analysis showed increased expression and presence in the CM of PTEN-KO SF763 cells for LOX, CXCL5, FABP5 and SERPINE2, with LOX showing a dramatic increase (Figures 2A–C). Enhanced production of LOX, but not CXCL5, FABP5 or SERPINE2, was confirmed in a second PTEN-WT GBM cell line, LN229, upon PTEN deletion (Figure S2A). Moreover, LOX expression levels were decreased upon re-expression of PTEN in two PTEN-deficient GBM cell lines, U87 and U251 (Figures 2D and S2B) and in the PTEN-deficient human PDX line GSC23 (Figure S2C). The relationship between PTEN and LOX was reinforced further by the positive correlation between phospho-AKT (Ser473) and LOX in a panel of human PDX lines, including of GSC17, TS603, GSC107, GSC7–10 and GSC23 (Figure 2E). In contrast to PTEN alterations, CRISPR-directed homozygous deletion of TP53 in SF763 cells or overexpression of EGFR mutant (EGFRvIII) in distinct cell lines (including LN229 cells, astrocytes and NSCs) did not increase LOX expression and/or secretion (Figures S2D,E). Finally, CRISPR-directed homozygous deletion of PTEN in the prostate cancer cell line DU145 and breast cancer cell line T-47D had minimal impact on LOX expression and secretion (Figure S2F), consistent with cell type specificity. Together, these findings demonstrate that LOX is preferentially and abundantly secreted by PTEN-deficient glioma cells.
Figure 2. LOX is preferentially secreted by PTEN-deficient cancer and promotes macrophage migration.

(A) RT-qPCR validation of upregulated genes encoding secreted proteins in PTEN-KO SF763 cells based on the microarray data from PTEN-WT and PTEN-KO SF763 cells. The values were expressed as the fold change between PTEN-KO and WT SF763 cells. The top 10 genes (with fold changes >8.5) were chosen for further validation.
(B) RT-qPCR validation of upregulated genes encoding secreted proteins in PTEN-KO SF763 cells based on the microarray data from PTEN-WT and PTEN-KO SF763 cells. Values are expressed as relative expression levels to housekeeping gene GAPDH.
(C) Immunoblots for LOX, CXCL5, SEMA3E, FABP5 and SERPINE2 in the cell lysates and conditioned media of PTEN-KO and WT SF763 cells.
(D) Immunoblots and RT-qPCR for the expression of LOX protein (left panel) and mRNA (right panel) in control and PTEN-overexpressing (OE) U87 cells.
(E) Immunoblots for LOX and phospho-Akt (Ser473) in cell lysates of GSC17, TS603, GSC107, GSC7–10 and GSC23.
(F) Quantification of relative migration of THP-1 macrophages following stimulation with recombinant LOX, MCP-1, CXCL5, FABP5 and SERPINE2 proteins (10 ng/ml). n=3–4 biological replicates.
(G) Left panel, representative immunofluorescence for F4/80 in growth factor-reduced Matrigel plugs supplemented with PBS or LOX (200 ng/ml) and subcutaneously injected into WT C57BL/6 mice. Scale bar, 100 μm. Right panel, quantification of F4/80+ macrophages in Matrigel plugs. n=3 biological replicates.
(H) Representative transwell analysis (left two panels) and quantification of relative migration (right panel) of THP-1 macrophages following stimulation with conditioned media from PTEN-KO SF763 cells pretreated with or without LOX inhibitor BAPN (200 μM). Scale bar, 100 μm. n=3 biological replicates.
(I) Quantification of relative migration of THP-1 macrophages following stimulation with conditioned media from PTEN-KO SF763 cells expressing LOX shRNA (shLOX) or control shRNA (shC). n=4 biological replicates.
Data from multiple replicates are presented as mean. Error bars indicate SD. *p<0.05, **p<0.01, **p<0.001, n.s., no significant difference, Student’s t test. See also Figures S2; Tables S3 and S4.
Next, to validate the capacity of LOX to function as a macrophage chemoattractant in vitro, we utilized the transwell assay and showed that recombinant LOX-supplemented media increased macrophage migration to a level comparable to MCP-1 (aka, CCL2), a known potent macrophage chemokine (Figures 2F). To confirm the chemoattractant ability of LOX in vivo, matrigel plugs with or without LOX supplementation were implanted subcutaneously, revealing that F4/80+ macrophage density was significantly increased in the LOX-supplemented matrigel plugs compared with controls (Figure 2G). The Gene Ontology Enrichment Analysis (GOEA) on the sub-ontology of Cell Compartment in TCGA GBM patients showed that LOX promotes extracellular matrix remodeling (Table S4), which is consistent with previous findings (Barker et al., 2012). The GOEA on the sub-ontologies of Biological Process and Molecular Function, as well as KEGG Enrichment Analysis in TCGA GBM patients, demonstrated that the migration of leukocytes and/or myeloid cells, as well as the chemokine/cytokine activity were the top LOX-regulated processes (Table S4). Thus, LOX functions as a potent chemokine for the recruitment of macrophages to the GBM TME.
Finally, to further confirm the role of LOX in the enhanced macrophage migration induced by PTEN-deficient glioma cells, we employed both genetic (LOX-specific shRNAs) and pharmacologic (LOX inhibitor β-aminopropionitrile [BAPN]) approaches. Notably, neither LOX shRNA nor BAPN affected the proliferation and apoptosis of either PTEN-WT or PTEN-KO glioma cells in vitro (Figures S2H–J). In contrast, CM from PTEN-KO SF763 cells pretreated with BAPN or expressing LOX shRNA induced significantly less macrophage migration than CM from untreated PTEN-KO SF763 cells (Figures 2H,I and S2K). Tumor-associated macrophages (TAMs) consist of M1 and M2 subtypes, and are usually biased to the M2 phenotype which is associated with GBM progression (Chen et al., 2017a; Hambardzumyan et al., 2016). We found that the PTEN-LOX pathway was not associated with the degree of M2 polarization in GBM (Figures S2L7–O), i.e., GBM TAMs were consistently of M2 phenotype regardless of PTEN status, and LOX did not show an effect on macrophage polarization (Figures S2P,Q). In summary, these results reinforce that increased LOX expression is associated with PTEN-deficient glioma and that LOX functions as a potent macrophage chemoattractant in this GBM subtype.
YAP1 is activated in PTEN-null glioma cells and regulates LOX expression
To explore PTEN regulation of LOX expression, GSEA was utilized to catalogue signaling pathways modulated by PTEN status. We found that SRC, AKT, NOTCH1 and YAP1 were the top pathways activated in PTEN-null GBM cells compared to PTEN-WT controls, which was confirmed by Western blot analysis and/or RT-qPCR analysis of signature genes in these pathways (Figures 3A,B and S3A–D). To investigate the functional relevance of these pathways, PTEN-KO SF763 cells were treated with various pathway inhibitors including SRC inhibitor KX2–391, AKT inhibitor MK2206, NOTCH1 inhibitor LY3039478 or YAP-TEAD interaction inhibitor verteporfin (Johnson and Halder, 2014). The SRC inhibitor blocked YAP1 and LOX up-regulation, but had no effect on AKT phosphorylation or NOTCH1 expression. Conversely, the AKT inhibitor repressed YAP1, LOX, and NOTCH1, and had no impact on SRC phosphorylation in the PTEN-KO cells (Figures 3B and S3E). However, the NOTCH1 inhibitor had no effect on YAP1 or LOX in PTEN-KO cells (Figure S3D), suggesting that the NOTCH1 signaling pathway is not involved in PTEN-dependent LOX regulation. Finally, YAP1-TEAD inhibitor verteporfin repressed LOX expression in PTEN-KO GBM cells (Figure 3C). As LOX is a secreted protein, the impact of these inhibitors on LOX expression in glioma cells mirrored the effect on LOX levels in CM (Figure 3D). Together, these in silico and pharmacological findings suggest that SRC/AKT-YAP1 signaling pathways are involved in PTEN-dependent regulation of LOX expression in glioma cells.
Figure 3. PTEN deficiency induced LOX production is regulated by SRC- and AKT-YAP1 pathway.

(A) GSEA for SRC, AKT and YAP1 signatures in PTEN-KO comparing to WT SF763 cells. NES, normalized enrichment score; FDR, false discovery rate.
(B) Immunoblots of LOX, YAP1, NOTCH1, phospho-AKT, AKT, phospho-SRC and SRC, in SF763 cells, and PTEN-KO SF763 cells treated with or without SRC inhibitor (SRCi) KX2–391 (50 nM) or AKT inhibitor (AKTi) MK2206 (2 μM).
(C) Immunoblots of LOX in SF763 cells and PTEN-KO SF763 cells treated with or without YAPTEAD interaction inhibitor (YAP1i) verteporfin (1 μM).
(D) Immunoblots of LOX in conditioned media from SF763 cells, and PTEN-KO SF763 cells treated with or without SRCi KX2–391 (50 nM), AKTi MK2206 (2 μM), YAPi verteporfin (1 μM) or NOTCH1 inhibitor (NOTCH1i) LY3039478 (1 μM).
(E) Quantification of YAP1 and H3K27ac ChIP-PCR in the LOX promoter of SF763 (Ctrl) and PTEN-KO SF763 cells, as well as U87 (Ctrl) and U87 PTEN-overexpression (PTEN-OE) cells. n=3 biological replicates.
(F) ChIP-seq analysis of H3K27ac-enriched profiles at LOX promotor in U87 and PTEN-KO SF763 cells.
(G) Immunoblots of LOX and YAP1 in PTEN-KO SF763 cells and U343 cells expressing shC or shYAP1.
(H) Quantification of the relative migration of THP-1 macrophages following stimulation with conditioned media from U343 cells expressing shC or shYAP1. n=3–4 biological replicates.
(I) Quantification of relative migration of primary mouse bone-marrow-derived macrophages (BMDMs) following stimulation with conditioned media from PTEN-KO SF763 cells pretreated with verteporfin (1 μM). n=4 biological replicates.
Data from multiple replicates are presented as mean. Error bars indicate SD. *p<0.05, **p<0.01, Student’s t test. See also Figure S3.
The above circumstantial evidence prompted us to obtain more direct evidence of a role for YAP1 in LOX regulation. YAP1 is a transcriptional coactivator, whose transcriptional activity is mediated by TEAD transcription factors; in addition, YAP1 largely exerts its transcriptional control via distal enhancers that are marked by H3K27 acetylation (H3K27ac) (Stein et al., 2015). Correspondingly, we performed ChIP-PCR analysis in PTEN-null U87 cells and PTEN-KO SF763 cells as well as their PTEN-WT controls and observed specific YAP1 and H3K27ac binding to the LOX promoter in these cells. Interestingly, the binding was enhanced in PTEN-deficient cells compared to PTEN-WT cells (Figure 3E). Moreover, ChIP-seq analysis of H3K27ac marks on PTEN-deficient cells exhibited specific peaks at the LOX promoter (Figure 3F). In addition, on the genetic level, shRNA-mediated YAP1 depletion in PTEN-mutated/deleted glioma cells reduced LOX expression (Figures 3G and S3F). With respect to macrophage recruitment, CM from PTEN-mutated/deleted glioma cells transfected with shRNAs against YAP1, or pretreated with verteporfin, showed decreased macrophage migration (Figures 3H,I and S3G,H). Together, these findings reinforce the role of SRC/AKT-YAP1 in regulation of LOX expression and subsequent increased macrophage recruitment specifically in PTEN-deficient GBM.
LOX activates β1 integrin-PYK2 signaling to recruit macrophages
To explore the molecular mechanisms underlying LOX-directed macrophage recruitment, we first documented LOX protein uptake by macrophages using Western blot, flow cytometry and immunofluorescence (Figures 4A,B and S4A,B). Although the caveolae/lipid raft and clathrin-dependent pathways play important roles in endocytosis of many cargoes (Mayor et al., 2014; Nabi and Le, 2003), inhibition of these pathways showed no impact on LOX internalization (Figures S4C,D). Since previous work has shown that LOX action can be mediated by β1 integrin (Gao et al., 2010; Levental et al., 2009; Matsuura et al., 2016), we hypothesized that LOX internalization into macrophages might be regulated by β1 integrin. Immunofluorescence and co-immunoprecipitation (co-IP) studies demonstrated that β1 integrin was highly expressed by macrophages (Figure 4C), and interacted with LOX in macrophages (Figure 4D). Moreover, shRNA-mediated depletion of β1 integrin abolished the internalization of LOX into macrophages (Figures 4E), demonstrating that LOX internalization into macrophages occurs in part via the β1 integrin receptor.
Figure 4. LOX promotes macrophage recruitment via β1 integrin-PYK2 pathway.

(A) Immunoblots of LOX in THP1 macrophages incubated with human recombinant LOX (100 ng/ml) for the indicated time. The cell surface binding of LOX was removed by washing with acid buffer.
(B) Flow cytometry for LOX in Raw264.7 macrophages treated with FITC- labelled LOX protein.
(C) Representative immunofluorescence for β1 integrin in Raw264.7 (green) and THP-1 (red) macrophages. Scale bar, 20 μm.
(D) Immunoprecipitation with a β1 integrin antibody (β1 Ab) and immunoblotting for the interaction between LOX and β1 integrin in Raw264.7 and THP-1 macrophages.
(E) Representative shRNA knockdown efficiency and immunoblots for LOX in shC and shItgb1 Raw264.7 macrophages treated with LOX protein.
(F) Immunoblots for phospho-PYK2 and PYK2 in Raw264.7 macrophages treated with LOX at the indicated concentrations and time points.
(G) Representative shRNA knockdown efficiency, as well as representative images and quantification of relative migration of Raw264.7 macrophages from a transwell analysis following stimulation with LOX in macrophages expressing shC or shPtk2b. Scale bar, 100 μm. n=3 biological replicates.
(H) Quantification of relative migration index of Raw264.7 macrophages treated with LOX in the absence or presence of increasing doses of PYK2/FAK inhibitor PF-00562271 (PF-271). n=3 biological replicates.
Data from multiple replicates are presented as mean. Error bars indicate SD. **p<0.01, ***p<0.001, Student’s t test. See also Figure S4.
Following internalization, LOX may trigger the activation of downstream signaling pathways that are responsible for LOX-induced macrophage migration. Thus, we performed an unbiased analysis using a human phospho-kinase antibody array and identified that PYK2 was one of the top three signals significantly activated upon treatment with recombinant LOX protein (Figures S4E,F), which has been shown as a key signaling regulator of macrophage migration (Okigaki et al., 2003). In our glioma system, Western blotting confirmed that phosphorylation of PYK2 (Y402), but not the structurally related tyrosine kinase FAK (Y397), was induced by LOX in a time- and dose-dependent manner (Figures 4F and S4G). It has been shown that the reactive oxygen species hydrogen peroxide (H2O2) is a direct byproduct of LOX activity, which is able to activate downstream signaling pathways (Cox et al., 2015; Pez et al., 2011) and is required for the chemotactic ability of LOX (Li et al., 2000). To investigate whether LOX-induced macrophage migration is regulated by H2O2, we performed macrophage transwell migration with recombinant LOX protein in the presence or absence of catalase, which can rapidly degrade H2O2. We found that LOX-induced macrophage migration was significantly inhibited by catalase (Figure S4H). Based on these findings and previous studies showing that H2O2 is able to activate PYK2 (Yamamoto et al., 2008), we hypothesized that LOX-induced macrophage migration in GBM might be regulated by the PYK2 signaling pathway. A macrophage transwell migration assay showed that LOX-induced macrophage migration was inhibited by shRNA knockdown of PYK2 (Figure 4G), and by PF-00562271 (a dual inhibitor for PYK2 and FAK) at 16 nM, a dose inhibiting both kinases, but not at 3 nM, which only inhibits FAK (Roberts et al., 2008) (Figures 4H and S4I). Together, these findings support a role for β1 integrin-PYK2 signaling in LOX-directed macrophage migration into the GBM TME.
Inhibition of LOX reduces TAM recruitment, inhibits tumor growth and extends survival in various GBM models
We recently generated a de novo GBM model using human neural stem/progenitor cells (hNSCs) engineered with a constitutively active myristoylated form of AKT (myr-AKT) and dominant-negative mutant of P53 (P53DN) (Hu et al., 2016). Using this model, we observed a prominent YAP1 signature and high LOX expression in myr-AKT/P53DN hNSCs relative to P53DN-hNSCs controls (Figures S5A–D). GBM tumors derived from the myr-AKT/P53DN hNSCs showed high expression of YAP1, β1 integrin, LOX and macrophage markers (Mac-2 and CD68) relative to tumors derived from glioma cells with lower levels of AKT (GSC TS543) (Hu et al., 2016) (Figure 5A). These correlative results prompted us to assess more rigorously the impact of LOX inhibition on macrophage recruitment and tumor growth in vivo.
Figure 5. Inhibition of LOX in cancer cells reduces TAM recruitment and inhibits GBM growth in vivo.

(A) IHC for YAP1, LOX, β1 integrin, Mac-2 and CD68 in GBM established from P53DN-AKT-hNSCs (high AKT) and GSC TS543 (low AKT) in SCID mice. Scale bar, 100 μm.
(B) Representative in vivo bioluminescence-based images of SCID mice at day 28 post-orthotopic injection of U87 (4×105 cells) transduced with shC or shLOX.
(C) Representative in vivo bioluminescence-based images of SCID mice at day 28 post-orthotopic injection of U87 cells (4×105 cells). The mice were treated with LOX inhibitor BAPN (2 g/L in drinking water) starting at 4 days post-orthotopic injection of U87 cells.
(D) Quantification of tumor volume based on bioluminescence (n=5/group).
(E, F) Immunofluorescence (left panel) and quantification (right panel) of macrophage marker CD68 in U87 tumors expressing shC or shLOX (E) or treated with LOX inhibitor BAPN (F). Tumors were harvested at day 28 post-orthotopic injection of cells. Scale bar, 50 μm. n=3 biological replicates.
(G) Survival curves of SCID mice implanted with U87 cells (4×105 cells) expressing shC or shLOX (n=10–12/group).
(H) Survival curves of SCID mice implanted with U87 cells (4×105 cells). Mice were treated with or without LOX inhibitor BAPN (2 g/L in drinking water) starting at 4 days post-orthotopic injection of U87 cells (n=10/group).
(I, J) Survival curves of SCID mice implanted with P53DN-AKT-hNSCs (2×105 cells) (I) or C57BL/6 mice implanted with QPP7 GSCs (2×104 cells) (J). Mice were treated with or without LOX inhibitor BAPN (2 g/L in drinking water) starting at 4 days post-orthotopic injection of P53DN-AKT-hNSCs or QPP7 GSCs (n=5–8/group).
Data from multiple replicates are presented as mean. Error bars indicate SD, *p<0.05, **p<0.01, ***p<0.001, Student’s t test. In (G) to (J), **p<0.01, ***<0.001, log-rank test. See also Figure S5.
Using shRNA-mediated depletion of LOX, small molecule LOX inhibitor BAPN, or neutralizing anti-LOX antibody, LOX depletion/inhibition significantly impaired tumor growth in both the PTEN-null U87 orthotopic model (Figures 5B–D and S5E) and the 005 GSC orthotopic model (Figure S5F) that is a GSC line isolated from GBMs with lentiviral transduction of brains with activated AKT and H-Ras in Trp53+/− mice (Marumoto et al., 2009; Saha et al., 2017). Accordingly, the macrophage infiltration was significantly impaired upon LOX inhibition in both models (Figures 5E,F and S5G,H). As a result, LOX inhibition using either shRNA or BAPN dramatically extended survival in several PTEN-deficient GBM mouse models, including human models in SCID mice and murine models in C57BL/6J mice. The human models included U87, PDX GSC23, and myr-AKT/P53DN hNSC models (Figures 5G–I and S5I). The immune intact murine models included the 005 GSC model and the QPP7 GSC model, a line derived from an engineered mouse GBM model with null alleles for Pten, Qki and Trp53 (Shingu et al., 2017) (Figures 5J and S5J). To confirm that macrophages were the critical target of LOX inhibition in impairing tumor growth, we compared the effect of LOX inhibitor BAPN and clodronate liposomes (which deplete macrophages in mice). Each agent equally extended survival, and their combined use did not exhibit additional antitumor effects (Figure S5J). In contrast, we found that BAPN did not impair the tumor growth of PTEN-intact GBM tumors (including the PTEN-WT GL261 model, PTEN-overexpressing U87 model and PTEN-WT Ink4a/Arf-null NSCs with EGFRvIII overexpression model) (Figures S5K–P). Together, these in vivo results validate the importance of TAMs in promoting glioma growth and support LOX as a therapeutic target specifically for PTEN-deficient/high AKT-activated GBM.
LOX inhibition promotes glioma cell apoptosis and impairs angiogenesis by decreasing TAM-derived SPP1
To understand how LOX-regulated TAMs contribute to GBM progression, tumor biological hallmarks were assessed in LOX-inhibited tumors. While LOX inhibition had no impact on glioma cell proliferation (Figures S6A–C), anti-LOX treatment was associated with increased glioma cell apoptosis and decreased tumor angiogenesis (Figure 6A), prompting speculation that these TAMs may secrete factors supporting glioma survival and angiogenesis. To identify such factors, we first analyzed RNA-seq data from mouse glioma-associated bone marrow-derived macrophages (BMDMs) and blood-derived monocytes (Bowman et al., 2016), which revealed 16 genes encoding secreted proteins (Chen et al., 2005) that were overexpressed in glioma-associated BMDMs compared to blood-derived monocytes (Figures S6D,E). Next, we performed unsupervised clustering with a well-established macrophage signature (Bindea et al., 2013) of 489 TCGA GBMs to generate three subtypes: Macrophage-high (n=201), Macrophage-medium (n=153) and Macrophage-low (n=135) (Figure S6F). In these subtypes, we identified 8 genes with expression correlating positively with TAM infiltration in GBM patients (Figure S6G). Among these 8 genes, Spp1 was the highest upregulated gene in IL4-polarized M2 BMDMs compared to PBS-treated controls (Figure 6B), and in human GBM TAMs compared to blood-derived monocytes isolated from GBM patients or from healthy controls (Gabrusiewicz et al., 2016) (Figure 6C). The relationship between SPP1 and TAMs was further reinforced by the positive correlation between SPP1 and TAM gene signatures (including total and M2 macrophage gene sets) in TCGA GBM patient datasets (Figures S6H,I), as well as by their co-localization in mouse 005 GSC tumors and human GBM tumors (Figure 6D). TAM infiltration has been shown to promote angiogenesis and correlate negatively with patient survival (Hambardzumyan et al., 2016; Zhu et al., 2017). Correspondingly, SPP1 expression was up-regulated in GBM patients (Figure S6J), negatively correlated with patient survival (Figure S6K), and positively correlated with angiogenesis (Figure S6L). Higher angiogenesis correlated with increased macrophages and lower overall survival in GBM patients (Figures S6M,N). More importantly, SPP1 expression was dramatically decreased in LOX-inhibited GBM tumors (Figure 6E), and positively correlated with LOX expression in GBM patients (Figure 6F). Furthermore, SPP1 inhibited LPS-induced glioma cell apoptosis (Figure 6G). Finally, co-implantation of glioma cells and M2-polarized macrophages harboring Spp1 shRNA significantly extended mouse survival, enhanced apoptosis and decreased angiogenesis when compared with M2 macrophages harboring shRNA control (Figures 6H–J). Together, these results demonstrate that LOX inhibition impairs GBM progression in part by decreasing TAM-derived SPP1.
Figure 6. LOX inhibition promotes glioma cell apoptosis and inhibits angiogenesis by impairing TAM-derived SPP1.

(A) Immunofluorescence and IHC analysis (left and middle panels) and quantification (right panel) of cleaved caspase-3 (CC3) and CD31 in 005 GSC mouse tumors treated with LOX antibodies and in U87 tumors treated with LOX inhibitor BAPN. Scale bars, 50 μm. n=3 biological replicates.
(B) RT-qPCR validation of 8 upregulated genes encoding secreted proteins in primary mouse BMDMs treated with or without IL4 (10 ng/ml) for 16 hr. Values are provided as relative expression levels to housekeeping gene Actb.
(C) Heat map representation of 8 upregulated genes in GBM patient blood monocytes, GBM TAMs and healthy blood monocytes. Red signal denotes higher expression and blue signal indicates lower expression.
(D) Immunofluorescence for CD206 (red) and SPP1 (green) in mouse 005 GSC tumors (left panel) and human GBM tumors (right panel). Scale bar, 50 μm.
(E) Immunofluorescence (left panels) and quantification (right panel) of SPP1 in 005 GSC mouse tumors treated with LOX antibodies and in U87 tumors treated with LOX inhibitor BAPN. Scale bars, 50 μm. n=3 biological replicates.
(F) The correlation of LOX and SPP1 in TCGA GBM patients. r and p values are shown. p<0.001, Pearson’s correlation test.
(G) Immunoblots for CC3 in QPP7 GSCs treated with LPS (10 μg/ml) in the presence or absence of SPP1 (300 ng/ml) for 14 hr.
(H) RT-qPCR to assess Spp1 shRNA knockdown efficiency in Raw264.7 macrophages. n=3 biological replicates.
(I) Survival curves of C57BL/6 mice implanted with GL261 cells (1×104 cells) and IL4-polarized Raw264.7 cells (1×104 cells) expressing shC or shSpp1 (n=5/group). *p<0.05, log-rank test.
(J) Immunofluorescence (left panels) and quantification (right panel) of CC3 and CD31 in tumors from mice implanted with GL261 cells and IL4-polarized Raw264.7 cells expressing shC or shSpp1. Scale bars, 50 μm. n=3 biological replicates.
Data from multiple replicates are presented as mean. Error bars indicate SD. **p<0.01, ***p<0.001, Student’s t test unless otherwise indicated. See also Figure S6.
An active YAP1-LOX-β1 axis tracks with macrophage prominence in human GBM
The clinical relevance of our experimental findings was supported by bioinformatics and TMA analyses showing that the expression of YAP1, LOX, β1 integrin (ITGB1) and Mac-2 (LGALS3) was enhanced in GBM tumors, which negatively correlated with overall survival of GBM patients (Figures S7A–F). Using the macrophage clustering shown in Figure S6F, we found that patients in the Macrophage-high group harbored more frequent PTEN mutation/deletion and exhibited poorer survival compared to patients in the Macrophage-low group (Figures 7A–C). GSEA demonstrated that two YAP1 gene signatures (including Cordenonsi_YAP_Conserved_Signature and YAP1_UP) were highly enriched in Macrophage-high samples compared with Macrophage-low samples (Figure 7D). Correspondingly, higher YAP1, LOX and ITGB1 expression was present in the Macrophage-high versus Macrophage-low patients (Figures 7E–G). Finally, we observed positive expression correlations among YAP1, LOX, β1 integrin (ITGB1) and Mac-2 (or a macrophage gene signature) in both GBM TMAs (protein) and TCGA GBM database (mRNA) (Figures 7H,I). Together, these correlative human GBM findings are consistent with the hypothesis that PTEN deficiency-induced activation of the YAP1-LOX-β1 integrin axis drives macrophage recruitment.
Figure 7. PTEN-YAP1-LOX-β1 integrin axis is activated and tracks with macrophage signature in human GBM.

(A) Distribution of PTEN mutated/deleted patients in Macrophage-high, -medium and -low groups of TCGA GBM patients.
(B) Percentage of PTEN mutated/deleted patients in Macrophage-high, -medium and -low groups of TCGA GBM patients. Chi-squared test.
(C) Analysis of correlation between macrophage levels and overall survival of IDH1-WT TCGA GBM patient. p<0.001, log-rank test.
(D) GSEA for two YAP1 signatures in Macrophage-high versus -low TCGA GBM samples.
(E-G) The expression of YAP1 (E), LOX (F), and ITGB1 (G) in Macrophage-high (n=201), medium (n=153) and -low (n=135) groups of TCGA GBM patients. Data are presented as mean ± SD, Student’s t test.
(H) IHC staining for YAP1, LOX, β1 integrin and Mac-2 in human GBM tissue microarray. Scale bar, 100 μm. The correlations among these four proteins was determined by Spearman correlation analysis (r and p values are shown). Pearson’s correlation test.
(I) The correlation analysis among YAP1, LOX, ITGB1 and average macrophage signature in TCGA GBM patients. r and p values are shown. Pearson’s correlation test.
See also Figure S7.
DISCUSSION
In this study, we explored the mechanisms underlying the recruitment of TAMs, the most abundant cell type accounting for up to 50% of live cells within a GBM tumor (Chen et al., 2017b), and assessed their role in gliomagenesis. We established that PTEN deficiency promotes YAP1-driven LOX expression in glioma cells and that LOX activates the β1 integrin-PYK2 pathway in macrophages to promote their infiltration into GBM TME. In addition to affecting the function of other immune cells (Hussain et al., 2006; Morford et al., 1999), TAMs are known to promote glioma cell survival and angiogenesis (Hambardzumyan et al., 2016; Kanamori et al., 2006; Samaras et al., 2007). We identified that these infiltrated TAMs promote tumor growth in part through SPP1 secretion which promotes glioma cell survival and angiogenesis. Consonant signaling patterns in human GBM supports the hypothesis that targeting these heterotypic interactions could provide clinical benefit specifically for PTEN-deficient GBM patients (Figure 8).
Figure 8. Working model.

Schematic representation of the role of the SRC/AKT-YAP1 pathway in PTEN deficiencyinduced LOX expression, and the role of β1 integrin-PYK2 pathway in LOX-induced macrophage migration. The infiltrated TAMs promote glioma cell survival and angiogenesis via secretion of SPP1. Inhibition of LOX is a promising therapeutic strategy for GBM.
Recent comprehensive genomic profiling and functional studies of GBM have yielded a paucity of druggable driver mutations, with such alterations occurring against the backdrop of extreme genetic and functional heterogeneity across the glioma cell compartment (Palumbo et al., 2015; Patel et al., 2014). Detailed explorations of the cellular and molecular composition of the GBM TME have catalogued the diverse signaling molecules and cell types including immunocytes and endothelial cells (Quail and Joyce, 2013) which are increasingly recognized as a potential source of therapeutic targets (Chen et al., 2015; Hambardzumyan et al., 2016; Quail and Joyce, 2013). Mounting evidence also points to an important role of glioma genetics in the regulation of the GBM TME, including PTEN deficiency-mediated upregulation of PD-L1 which promotes immune escape (Parsa et al., 2007), P14ARF-mediated upregulation of metalloproteinase-3 which inhibits angiogenesis (Zerrouqi et al., 2012), rearrangement and activation of EGFRviii which promotes angiogenesis via the NF-κB-interleukin-8 (IL-8) pathway (Bonavia et al., 2012). In our study, we established that PTEN-dependent LOX-driven regulation of macrophage infiltration supports GBM progression in animal models, and that this axis tracks with poor clinical outcomes. Our work reinforces the importance of the TME, particularly TAMs, in GBM biology, and identifies potential targets along with specific responder population for such intervention.
In exploring the link between PTEN and LOX, we observed that PTEN inactivation triggers activation of multiple downstream signaling pathways in glioma cells, most prominently AKT and SRC, consistent with previous work (Dey et al., 2008). Our study demonstrates that these two pathways play key roles in LOX upregulation in glioma cells. In contrast, while activating mutations in EGFR are common in GBM and can also activate AKT (Brennan et al., 2013; Cancer Genome Atlas Research, 2008), the failure of EGFR activation to increase LOX expression and macrophage infiltration may relate to the compensatory regulation of other pathways. YAP1 is a key regulator of LOX transcription in GBM, and the Hippo-YAP1 pathway is known to play an important role in cancer progression (Johnson and Halder, 2014; Kapoor et al., 2014). In the context of our study, it is interesting that previous work has shown that YAP1 downregulates PTEN via induction of miR-29 (Tumaneng et al., 2012). Together with our findings, this points to a reciprocal regulatory interaction between YAP1 and PTEN which may induce a potent feedback loop to promote GBM progression through LOX production. The importance of YAP1 in gliomagenesis is supported by recent studies demonstrating that YAP1 activation promotes the recruitment of macrophages and myeloid-derived suppressor cells via directly transcriptional regulation of chemokines (including CCL2, CSF1 and CXCL5) in liver and prostate cancer (Guo et al., 2017; Wang et al., 2016). These distinct YAP1-driven mechanisms of macrophage recruitment in different cancers underscores the need for precision medicine approaches in various cancer types and genotypes.
Our work showing that LOX inhibition impairs GBM progression by blocking macrophage infiltration yet does not directly affect glioma cells aligns with previous studies showing that LOX is essential for bone marrow cell recruitment and stimulating osteoclasts/osteoblasts to form a pre-metastatic niche with no direct impact on cancer cells (Cox et al., 2015; Erler et al., 2009; Erler et al., 2006; Rachman-Tzemah et al., 2017). Our work further reinforces the view that LOX can act as a potent chemokine produced by glioma cells to attract macrophages into GBM TME. LOX has been shown to trigger the activation of several downstream signaling pathways, such as β1 integrin and FAK, to promote cancer metastasis (Baker et al., 2011; Erler et al., 2006; Gao et al., 2010; Levental et al., 2009). While our work also shows that macrophage β1 integrin can mediate LOX uptake and drive the LOX responsiveness of macrophages, we showed that PYK2, but not FAK, is required for LOX-induced macrophage infiltration in GBM. This role of PYK2 is consistent with previous studies showing that PYK2 is crucial for macrophage migration in response to SDF1α (Okigaki et al., 2003). Lysosomal degradation may affect LOX-regulated downstream pathways, however LOX secretion from PTEN-deficient GBM cells and internalization into macrophages would be continuous processes that can activate the PYK2 signaling. Together, our study reveals that LOX specifically recruits macrophages in GBM via the β1 integrin-PYK2 pathway.
The identification of SPP1 as an abundant TAM-secreted factor in GBM, coupled with the anti-tumor impact of SPP1 inhibition, encourages the development of therapeutic agents targeting SPP1-expressing TAMs in GBM patients. Interestingly, a recent study showed that SPP1 is required for macrophage infiltration and M2 maintenance in GBM (Wei et al., 2018), which in turn can induce a positive feedback loop for the production and GBM-promoting effect of TAM-derived SPP1. However, our work establishes that this effect is context and cell type dependent, since previous studies demonstrated that endogenous SPP1 in non-transformed cells at tumor-free context can promote microglia M1 polarization (Ellert-Miklaszewska et al., 2016), and loss of stroma-derived SPP1 creates a GBM-promoting microenvironment, but decreases angiogenesis and does not affect BMDM infiltration (Szulzewsky et al., 2018). Given the known immune suppressive role of TAMs, it is tempting to speculate that a combination of targeting M2 macrophages and T cell checkpoint blockade could benefit GBM patients. However, while CSF1R inhibitors impede GBM progression and decrease M2 macrophages (Hambardzumyan et al., 2016; Pyonteck et al., 2013; Quail et al., 2016), the anti-tumor responses have been shown to be transient in GBM mouse models (Quail et al., 2016), and a phase II clinical trial with CSF1R inhibitor PLX3397 has shown minimal activity in recurrent GBM patients which could relate to the loss of TAMs (Butowski et al., 2016). Notably, two of 37 GBM patients with extended progression free survival (Butowski et al., 2016) are the mesenchymal subtype in which PTEN deficiency is common. Our findings that LOX inhibition increases survival in PTEN deficient GBM models, coupled with the fact that the LOX inhibitor is bloodbrain barrier penetrable, should motivate the testing of BAPN specifically in PTEN deficient GBM patients.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ronald A. DePinho (RDePinho@mdanderson.org).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture
The GBM cell lines SF763, U87, U251, U343 and LN229, as well as J714 macrophages and 293T cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM). DU145 cells were cultured in Eagle’s Minimum Essential Medium (EMEM). THP-1 and Raw264.7 macrophage cell lines and T-47D were cultured in RPMI 1640 medium (RPMI). All cell lines were cultured in indicated medium containing 10% FBS (Sigma) and 1:100 antibiotic-antimycotic (Gibco), and purchased from the American Type Culture Collection (ATCC). All human cell lines have been validated through fingerprinting by the MD Anderson Cell Line Core Facility. The mouse GBM tumor-derived glioma stem cell lines 005 GSC (Saha et al., 2017) and QPP7 GSC was provided by Dr. S.D. Rabkin (Massachusetts General Hospital, Harvard Medical School, Boston) and Dr. J. Hu (MD Anderson Cancer Center, Houston), respectively. P53DN-hNSCs and P53DN-AKT-hNSCs were generated by our laboratory as described recently (Hu et al., 2016). Ink4a/Arf−/− astrocytes and NSCs with or without EGFRviii overexpression were provided by Dr. R.M. Bachoo (UT Southwestern Medical Center, Dallas). Astrocytes were cultured in DMEM containing 10% FBS and 1:100 antibiotic-antimycotic. All the GSCs and NSCs were cultured in NSC proliferation media (Millipore Corporation, Billerica, MA) containing 20 ng/ml EGF and 20 ng/ml bFGF. All cells were confirmed to be mycoplasma-free, and maintained at 37 °C and 5% CO2. BMDMs from C57BL/6 mice were cultured as previously described (Chen et al., 2017a). Conditioned media were collected from treated or untreated cells as indicated after culturing for 24 hr in FBS-free culture medium.
Mice and intracranial xenograft tumor models
Female ICR SCID and C57BL/6 mice (3–4 weeks age) were purchased from Taconic Biosciences and The Jackson Laboratory, respectively. Mice were grouped by 5 animals in large plastic cages and were maintained under pathogen-free conditions. All animal experiments were performed with the approval of MD Anderson Cancer Center’s Institutional Animal Care and Use Committee (IACUC).
The intracranial xenograft tumor models in SCID mice or C57BL/6 mice were established as we described recently (Hu et al., 2016). The mice were bolted and intracranially implanted with cells as indicated at MD Anderson’s Brain Tumor Center Animal Core. Briefly, the mice were anesthetized by intraperitoneal injection of ketamine/xylazine (200 mg ketamine and 20 mg xylazine in 17 mL of saline) at a dosage of 0.15 mg/10 g body weight. The plastic screw was rotated into a small drill hole made 2.5 mm lateral and 1 mm anterior to the bregma and the central hole of the guide screw was closed by placing a cross-shaped stylet inside it. After one week recovery, mice were placed into group of four or five animals for cells implantation. The cells were injected in 5 μl culture medium. Mice with neurological deficits or moribund appearance were sacrificed. Following the transcardial perfusion with 4% paraformaldehyde (PFA), brains were removed and fixed in formalin, and were processed for paraffin embedded blocks.
Human Samples
Tissue microarrays (TMA) containing 32 GBM, 3 anaplastic astrocytoma samples and 5 normal brain tissues were purchased from US Biomax (Cat#GL806e). These materials were commercially available anonymized and de-identified. According to MD Anderson’s Institutional Review Board the conducted research meets the criteria for exemption #4 (45 CFR 46.101(b) Categories of Exempt Human Subjects Research) and does not constitute human research.
METHOD DETAILS
shRNA knockdown
shRNA hairpins targeting human LOX and YAP1 as well as mouse Ptk2b, Itgb1 and Spp1, were used in this study. We screened five to seven hairpins of each gene and chose the sequences that reduced protein levels by >70%. These selected hairpins were in the pLKO.1 vector. Recombinant lentiviral particles were produced by transient transfection of plasmids into 293T cells. In brief, 8 μg of the shRNA plasmid, 4 μg of the psPAX2 plasmid, and 2 μg of the pMD2.G plasmid were transfected using Lipofectamine 2000 into 293T cells plated in 100-mm dishes. Viral supernatant was collected 48 hr and 72 hr after transfection and filtered. Cells were infected twice in 48 hr with viral supernatant containing 10 μg/ml polybrene, and then selected using 2 μg/ml puromycin and tested the expression of LOX, YAP1, PYK2 and β1 integrin by immunoblot and SPP1 by RT-qPCR.
Knockout using CRISPR
sgRNA plasmids targeting human PTEN (sc-400103) and TP53 (sc-416469) were purchased from Santa Cruz Biotechnology. The plasmids with sgRNA were transiently transfected into cells (SF763, LN229, DU145 or T-47D cells) using Lipofectamine 2000. Cells were harvested 72 hr later, and ten GFP-positive cells were sorted into each well of a 96-well plate by flow cytometry, followed by immunoblotting for PTEN and P53 protein. PCR sequencing was also performed using genomic DNA extracted from PTEN- or TP53-deficient single clones to identify genetic alteration at PTEN or TP53 allele. Finally, we chose the single clone in which one or more premature stop codons were introduced in coding exons by sgRNA-induced mutations.
Immunoprecipitation and immunoblotting
For β1 integrin immunoprecipitation, cells were lysed in NP-40 buffer containing 50 mM Tris-HCl (pH 7.5), 150 mM NaCl, 0.3% Nonidet P-40, and protease inhibitor cocktail (Sigma-Aldrich). Cell lysates (500 μl) were incubated with anti-β1 integrin antibody (Abcam #ab183666) or control IgG and Protein A/G PLUS-Agarose (40 μl, Santa Cruz Biotechnology, sc-2003) overnight at 4 °C. The beads were washed three times with NP-40 buffer, followed by immunoblotting. Immunoblotting was performed following standard protocol. Antibodies were purchased from the indicated companies, including Cell Signaling (PTEN, #9188S; S473P-AKT, #3787S; AKT, #2910S; T416P-SRC, #6943S; SRC, #2109S; Notch1, #4380S; YAP1, #14074S; T402P-PYK2, #3291S; PYK2, #3090S; T397P-FAK, #3283S;FAK, #3285S; T1068P-EGFR, #3777S and EGFRviii, #64952S), Abcam (LOX, #ab174316; CXCL5, #ab9802; SEMA3E, #ab128619, SERPINE2, #ab134905 and β1 integrin, #ab179471), R&D system (FABP5, #AF3077), EMD Millipore (Vinculin, #05–386), Santa Cruz (P53, #sc-126) and Sigma (ȕ-actin, #A3854).
Immunohistochemistry and immunofluorescence
Immunohistochemistry was performed using standard protocol as we previously described (Chen et al., 2017a; Zhao et al., 2017). In brief, a pressure cooker (95 °C for 30 min followed by 120 °C for 10 s) was used for antigen retrieval using antigen unmasking solution (Vector Laboratories). Antibodies specific to Mac-2 (BioLegend, #125403), S473P-AKT, PTEN, YAP1, LOX, β1 integrin, CD68 (Abcam, #ab53444), cleaved caspase 3 (CST, #9661S), Ki67 (Thermo Scientific, #RM-9106-S1), CD31 (Abcam, #ab28364) were used in this study. The human and mouse tumor tissue sections were reviewed and scored as reported recently (Chen et al., 2017a). Slides were scanned using Pannoramic 250 Flash III (3DHISTECH Ltd) and images were captured through Pannoramic Viewer software (3DHISTECH Ltd). The studies related to human specimens were approved by the MD Anderson Institutional Review Board under protocol #PA14–0420.
Immunofluorescence was performed using the standard protocol in cells and tissues. Antibodies specific to F4/80 (Abcam, #ab6640), CD68, CD206 (R&D system, #AF2535), LOX, YAP1, β1 integrin, CD31, cleaved caspase 3 and SPP1 (Santa Cruz, #sc-21742) were used. Images were captured using a fluorescence microscope (Leica DMi8).
Migration assay
Macrophages (1 × 104 for Raw264.7 and 5 × 105 for THP-1 macrophages) were suspended in serum-free culture medium and seeded into 24-well Transwell inserts (5.0 μm). Medium with indicated factors or conditioned media was added to the remaining receiver wells. After 8 (Raw264.7 macrophages) or 24 h (THP-1 macrophages), the migrated macrophages were fixed and stained with crystal violet (0.05%, sigma), and then counted as cells per field of view under microscope or measured as OD590 nm (expressed as migration index) after crystal violet was dissolved.
Colony formation assay
Glioma cell proliferation in vitro was assayed through colony formation. 1 × 104 cells were seeded in each well of 6-well plates and cultured for 5–7 days. At the end point, cells were fixed and stained with 0.5% crystal violet in 25% methanol for 1 hr. These experiments were performed in triplicate.
Flow cytometry
LOX recombinant protein was labelled with FITC using a kit (BioVision, #K832–5) according to manufacturer’s protocol. Macrophages were treated with FITC-labeled LOX for 1 hr. After washing twice using cold PBS, cells were analyzed by flow cytometer (Becton Dickinson).
ChIP–seq and ChIP-PCR
ChIP was performed as we described recently (Zhao et al., 2017). Briefly, chromatin from PFA-fixed U87, U87-PTEN OE, SF763 or SF763-PTEN KO cells were cross-linked using 1% PFA (10 min) and then reactions were quenched using 0.125 M glycine (5 min) at room temperature. Cells were lysed with ChIP lysis buffer [10 mM Tris-HCl (pH 8.0), 140 mM NaCl, 1 mM EDTA (pH 8.0), 1% Triton X-100, 0.2% SDS and 0.1% deoxycholic acid] for 30 min on ice. Chromatin fragmentation was performed using a Diagenode BioruptorPico sonicator (30 s on and 30 s off, 45 cycles). Solubilized chromatin was then incubated with the appropriate mixture of antibody [YAP1 (Novus, #NB110–58358) or H3K27ac (Abcam, ab4729)] and Dynabeads (Life Technologies) overnight. Immune complexes were then washed with RIPA buffer (three times), once with RIPA-500 (RIPA with 500 mM NaCl), and once with LiCl wash buffer [10 mM Tris-HCl (pH 8.0), 1 mM EDTA (pH 8.0), 250 mM LiCl, 0.5% NP-40 and 0.5% deoxycholic acid]. Elution and reverse-crosslinking were performed in direct elution buffer [10 mM Tris-Cl (pH 8.0), 5 mM EDTA, 300 mM NaCl, 0.5% SDS] containing proteinase K (20 mg/ml) at 65 °C overnight. Eluted DNA was purified using AMPure beads (Beckman-Coulter), which then was used to generate libraries using NEBNext Ultra DNA Library kit (E7370), or to perform qPCR. Sequencing was performed using an Illumina HiSeq 2500 instrument to generate dataset.
mRNA expression analysis and microarray
RNA was isolated from cells using Trizol (Invitrogen) following the manufacturer’s instructions. RNA (200–500 ng) was first treated with RNase-free DNase I using the DNA-free kit (Ambion) to remove all genomic DNA, and then reverse-transcribed into cDNA using an ABI High Capacity cDNA RT Kit (Invitrogen). The cDNA was analyzed using real-time quantitative PCR (SYBR Green, Invitrogen) with an Applied Biosystems 7900 Sequence Detection System. Each reaction was performed in triplicate. The expression of each gene was normalized by that of mouse Actin or human GAPDH.
RNA was isolated from cells using TRIzol Reagent (Invitrogen; #15596–026) according to the manufacturer’s instructions. RNA was then further purified with RNeasy (QIAGEN) and then was reverse-transcribed into cDNA using the SuperScript III First-Strand Synthesis System (Invitrogen). RT-qPCR was performed to analyze the expression of targeted genes using the SYBR Green PCR Master Mix (Applied Biosystems). Primers are listed in Table S5. Microarray analysis was performed on RNA prepared from PTEN-WT and PTEN-KO SF763 cells (biological triplicates for control and PTEN-KO SF763 cells) at the MD Anderson Microarray Core facility using the GeneChip Human Clariom D array (Affymetrix) to generate dataset. Genes that were differentially expressed between control and PTEN-depleted SF763 cells were subjected to gene set enrichment analysis (GSEA). The raw data were processed and analyzed by GenePattern using Transcriptome Analysis Console.
Computational analysis of human GBM TCGA data
For analysis of human GBM data, we downloaded the gene expression and survival data of TCGA data from GlioVis: http://gliovis.bioinfo.cnio.es/ (Bowman et al., 2017) or cBioPortal: http://www.cbioportal.org (Cerami et al., 2012). The stromal and immune score of each patient was downloaded from ESTIMATE: http://bioinformatics.mdanderson.org/estimate/ (Yoshihara et al., 2013). We analyzed the correlation between patient stromal/immune score and survival. Based on availability of genetic backgrounds of each patient, we analyzed the potential role of each core signaling pathway and genetic event in modulation of stromal/immune score in the TME and gene expression in tumor. The expression of interesting genes in GBM with different subtypes or grades was analyzed using GlioVis. We clustered TCGA GBM patients into macrophage-high, macrophage -low and macrophage-medium subgroups based a list of 38 human macrophage signature genes (Bindea et al., 2013). Differentially expressed genes between macrophage-high and macrophage-low were analyzed by GSEA. The survival of patients, and the expression of YAP1, LOX and ITGB1 was analyzed in these three subgroups.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed with Student’s t-test and represented as mean ± SD. The analysis of GBM TCGA database and TAM IHC staining for the correlation among YAP1, LOX, β1 integrin and macrophage markers/signature, as well as the correlation between stromal/immune score and patient survival was performed using pearson’s correlation test (GraphPad Prism 7). The analysis of PTEN mutation/deletion in Macrophage-high and Macrophage-low groups of TCGA patients was performed using the chi-squared test. The analysis of the survival data from GBM TCGA database was performed using log-rank (MantelCox) test (GraphPad Prism 7). The p values were designated as: *, p<0.05; **, p<0.01 and ***, p<0.001; n.s. non-significant (p>0.05).
DATA AND SOFTWARE AVAILABILITY
RNAseq and microarray data have been submitted to the GEO repository; the accession numbers are GSE122283 and GSE122284, respectively.
Supplementary Material
HIGHLIGHTS.
PTEN-deficiency in GBM drives macrophage infiltration via upregulation of LOX
LOX is directly regulated by YAP1 in PTEN-deficient GBM
LOX recruits macrophages into GBM via the β1 integrin-PYK2 pathway
LOX inhibition impairs PTEN-deficient GBM progression by decreasing TAM-derived SPP1
SIGNIFICANCE.
The GBM tumor microenvironment (TME) contains a large number of infiltrated macrophages. Here, we show that PTEN deficiency in GBM is a powerful driver of macrophage infiltration via YAP1-regulated LOX expression. Secreted LOX recruits macrophages into the GBM TME via activation of the β1 integrin-PYK2 pathway. These infiltrating macrophages in turn sustain glioma cell survival and stimulate angiogenesis via secretion of SPP1. In PTEN-deficient GBM models, LOX inhibition markedly inhibits tumor progression and macrophage infiltration. Collectively, our work identifies key macrophage-cancer cell interdependencies and validates LOX inhibition as a promising therapeutic strategy specifically in PTEN-deficient GBM.
ACKNOWLEDGMENTS
We thank Peoples MD for providing shRNAs, Lang FF, Hendriks WJAJ, Rabkin SD, Hu J, Bachoo RM and Tasciotti E for providing cell lines; Hu B for providing P53DN-AKT-hNSC tumor sections; Kingsley CV and Tran V for assistance with imaging; and Jiang S for mouse husbandry. This work was supported in part by the Cancer Research Institute Irvington Postdoctoral Fellowship (P.C.), the Caroline Ross Endowed Fellowship (P.C.), The Harold C. and Mary L. Daily Endowment Fellowship (P.C.), the Burkhart III Distinguished University Chair in Cancer Research Endowment (R.A.D.), Clayton & Modesta William Cancer Research Fund (R.A.D.), NIH P01 CA117969 (R.A.D.), NIH R01 CA084628 (R.A.D.), Emerson Collective Award (Y.A.W.), NIH R01 CA231349 (Y.A.W.), NIH K99 CA226360 (D.Z.) and NIH R01 NS094615 (G.R.). The core facilities at MD Anderson are supported by P30CA16672.
Footnotes
DECLARATION OF INTERESTS
RAD is a co-founder, advisor and director of Tvardi Therapeutics.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Baker AM, Cox TR, Bird D, Lang G, Murray GI, Sun XF, Southall SM, Wilson JR, and Erler JT (2011). The role of lysyl oxidase in SRC-dependent proliferation and metastasis of colorectal cancer. Journal of the National Cancer Institute 103, 407–424. [DOI] [PubMed] [Google Scholar]
- Barker HE, Cox TR, and Erler JT (2012). The rationale for targeting the LOX family in cancer. Nature reviews Cancer 12, 540–552. [DOI] [PubMed] [Google Scholar]
- Bindea G, Mlecnik B, Tosolini M, Kirilovsky A, Waldner M, Obenauf AC, Angell H, Fredriksen T, Lafontaine L, Berger A, et al. (2013). Spatiotemporal dynamics of intratumoral immune cells reveal the immune landscape in human cancer. Immunity 39, 782–795. [DOI] [PubMed] [Google Scholar]
- Bonavia R, Inda MM, Vandenberg S, Cheng SY, Nagane M, Hadwiger R, Tan P, Sah DW, Cavenee WK, and Furnari FB (2012). EGFRvIII promotes glioma angiogenesis and growth through the NF-kappaB, interleukin-8 pathway. Oncogene 31, 4054–4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman RL, Klemm F, Akkari L, Pyonteck SM, Sevenich L, Quail DF, Dhara S, Simpson K, Gardner EE, Iacobuzio-Donahue CA, et al. (2016). Macrophage Ontogeny Underlies Differences in Tumor-Specific Education in Brain Malignancies. Cell reports 17, 2445–2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowman RL, Wang Q, Carro A, Verhaak RG, and Squatrito M (2017). GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro-oncology 19,139–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan CW, Verhaak RG, McKenna A, Campos B, Noushmehr H, Salama SR, Zheng S, Chakravarty D, Sanborn JZ, Berman SH, et al. (2013). The somatic genomic landscape of glioblastoma. Cell 155, 462–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, Cloughesy TF, Marimuthu A, Haidar S, Perry A, et al. (2016). Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neurooncology 18, 557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research, N. (2008). Comprehensive genomic characterization defines human glioblastoma genes and core pathways. Nature 455,1061–1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer discovery 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhuang X, Lin L, Yu P, Wang Y, Shi Y, Hu G, and Sun Y (2015). New horizons in tumor microenvironment biology: challenges and opportunities. BMC medicine 13, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P, Zuo H, Xiong H, Kolar MJ, Chu Q, Saghatelian A, Siegwart DJ, and Wan Y (2017a). Gprl32 sensing of lactate mediates tumor-macrophage interplay to promote breast cancer metastasis. Proceedings of the National Academy of Sciences of the United States of America 114, 580–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Zhang Y, Yin Y, Gao G, Li S, Jiang Y, Gu X, and Luo J (2005). SPD--a web-based secreted protein database. Nucleic acids research 33, D169–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Z, Feng X, Herting CJ, Garcia VA, Nie K, Pong WW, Rasmussen R, Dwivedi B, Seby S, Wolf SA, et al. (2017b). Cellular and Molecular Identity of Tumor-Associated Macrophages in Glioblastoma. Cancer research 77, 2266–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox TR, Rumney RM, Schoof EM, Perryman L, Hoye AM, Agrawal A, Bird D, Latif NA, Forrest H, Evans HR, et al. (2015). The hypoxic cancer secretome induces pre-metastatic bone lesions through lysyl oxidase. Nature 522,106–110. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Dey N, Crosswell HE, De P, Parsons R, Peng Q, Su JD, and Durden DL (2008). The protein phosphatase activity of PTEN regulates SRC family kinases and controls glioma migration. Cancer research 68, 1862–1871. [DOI] [PubMed] [Google Scholar]
- Dunn GP, Rinne ML, Wykosky J, Genovese G, Quayle SN, Dunn IF, Agarwalla PK., Chheda MG, Campos B, Wang A, et al. (2012). Emerging insights into the molecular and cellular basis of glioblastoma. Genes & development 26, 756–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellert-Miklaszewska A, Wisniewski P, Kijewska M, Gajdanowicz P, Pszczolkowska D, Przanowski R, Dabrowski M, Maleszewska M, and Kaminska B (2016). Tumour-processed osteopontin and lactadherin drive the protumorigenic reprogramming of microglia and glioma progression. Oncogene 35, 6366–6377. [DOI] [PubMed] [Google Scholar]
- Engler JR, Robinson AE, Smirnov I, Hodgson JG, Berger MS, Gupta N, James CD, Molinaro A, and Phillips JJ (2012). Increased microglia/macrophage gene expression in a subset of adult and pediatric astrocytomas. PloS one 7, e43339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erler JT, Bennewith KL, Cox TR, Lang G, Bird D, Koong A, Le QT, and Giaccia AJ (2009). Hypoxia-Induced Lysyl Oxidase Is a Critical Mediator of Bone Marrow Cell Recruitment to Form the Premetastatic Niche. Cancer cell 15, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erler JT, Bennewith KL, Nicolau M, Dornhofer N, Kong C, Le QT, Chi JTA, Jeffrey SS, and Giaccia AJ (2006). Lysyl oxidase is essential for hypoxia-induced metastasis. Nature 440,1222–1226. [DOI] [PubMed] [Google Scholar]
- Gabrusiewicz K, Rodriguez B, Wei J, Hashimoto Y, Healy LM, Maiti SN, Thomas G, Zhou S, Wang Q, Elakkad A, et al. (2016). Glioblastoma-infiltrated innate immune cells resemble M0 macrophage phenotype. JCI insight 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Y, Xiao Q, Ma H, Li L, Liu J, Feng Y, Fang Z, Wu J, Han X, Zhang J, et al. (2010). LKB1 inhibits lung cancer progression through lysyl oxidase and extracellular matrix remodeling. Proceedings of the National Academy of Sciences of the United States of America 107,18892–18897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Zhao Y, Yan H, Yang Y, Shen S, Dai X, Ji X, Ji F, Gong XG, Li L, et al. (2017). Single tumor-initiating cells evade immune clearance by recruiting type II macrophages. Genes & developmental, 247–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, and Bergers G (2015). Glioblastoma: Defining Tumor Niches. Trends in cancer 1, 252–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hambardzumyan D, Gutmann DH, and Kettenmann H (2016). The role of microglia and macrophages in glioma maintenance and progression. Nature neuroscience 19, 20–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu B, Wang Q, Wang YA, Hua S, Sauve CG, Ong D, Lan ZD, Chang Q, Ho YW, Monasterio MM, et al. (2016). Epigenetic Activation of WNT5A Drives Glioblastoma Stem Cell Differentiation and Invasive Growth. Cell 167,1281–1295 el218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hussain SF, Yang D, Suki D, Aldape K, Grimm E, and Heimberger AB (2006). The role of human glioma-infiltrating microglia/macrophages in mediating antitumor immune responses. Neuro-oncology 8, 261–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson R, and Haider G (2014). The two faces of Hippo: targeting the Hippo pathway for regenerative medicine and cancer treatment. Nature reviews Drug discovery 13, 63–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanamori M, Kawaguchi T, Berger MS, and Pieper RO (2006). Intracranial microenvironment reveals independent opposing functions of host alphaVbeta3 expression on glioma growth and angiogenesis. The Journal of biological chemistry 281, 37256–37264. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu CJ, Sadanandam A, Hu B, et al. (2014). Yapl activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khosla D (2016). Concurrent therapy to enhance radiotherapeutic outcomes in glioblastoma. Annals of translational medicine 4, 54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, Fong SF, Csiszar K, Giaccia A, Weninger W, et al. (2009). Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell 139, 891–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Liu G, Chou IN, and Kagan HM (2000). Hydrogen peroxide-mediated, lysyl oxidase-dependent chemotaxis of vascular smooth muscle cells. J Cell Biochem 78, 550–557. [PubMed] [Google Scholar]
- Lim M, Xia Y, Bettegowda C, and Weller M (2018). Current state of immunotherapy for glioblastoma. Nature reviews Clinical oncology. [DOI] [PubMed] [Google Scholar]
- Maher EA, Furnari FB, Bachoo RM, Rowitch DH, Louis DN, Cavenee WK, and DePinho RA (2001). Malignant glioma: genetics and biology of a grave matter. Genes & development 15, 1311–1333. [DOI] [PubMed] [Google Scholar]
- Mai WX, Gosa L, Daniels VW, Ta L, Tsang JE, Higgins B, Gilmore WB, Bayley NA, Harati MD, Lee JT, et al. (2017). Cytoplasmic p53 couples oncogene-driven glucose metabolism to apoptosis and is a therapeutic target in glioblastoma. Nature medicine 23,1342–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marumoto T, Tashiro A, Friedmann-Morvinski D, Scadeng M, Soda Y, Gage FH, and Verma IM (2009). Development of a novel mouse glioma model using lentiviral vectors. Nature medicine 15,110–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuura S, Mi R, Koupenova M, Eliades A, Patterson S, Toselli P, Thon J, Italiano JE Jr., Trackman PC, Papadantonakis N, et al. (2016). Lysyl oxidase is associated with increased thrombosis and platelet reactivity. Blood 127,1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayor S, Parton RG, and Donaldson JG (2014). Clathrin-independent pathways of endocytosis. Cold Spring Harbor perspectives in biology 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morford LA, Dix AR, Brooks WH, and Roszman TL (1999). Apoptotic elimination of peripheral T lymphocytes in patients with primary intracranial tumors. Journal of neurosurgery 91, 935–946. [DOI] [PubMed] [Google Scholar]
- Nabi IR, and Le PU (2003). Caveolae/raft-dependent endocytosis. The Journal of cell biology 161, 673–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okigaki M, Davis C, Falasca M, Harroch S, Felsenfeld DP, Sheetz MP, and Schlessinger J (2003). Pyk2 regulates multiple signaling events crucial for macrophage morphology and migration. Proceedings of the National Academy of Sciences of the United States of America 100, 10740–10745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palumbo A Jr., Da Costa Nde O, Bonamino MH, Pinto LF, and Nasciutti LE (2015). Genetic instability in the tumor microenvironment: a new look at an old neighbor. Molecular cancer 14, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parsa AT, Waldron JS, Panner A, Crane CA, Parney IF, Barry JJ, Cachola KE, Murray JC, Tihan T, Jensen MC, et al. (2007). Loss of tumor suppressor PTEN function increases B7-H1 expression and immunoresistance in glioma. Nature medicine 13, 84–88. [DOI] [PubMed] [Google Scholar]
- Patel AP, Tirosh I, Trombetta JJ, Shalek AK, Gillespie SM, Wakimoto H, Cahill DP, Nahed BV, Curry WT, Martuza RL, et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344,1396–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pez F, Dayan F, Durivault J, Kaniewski B, Aimond G, Le Provost GS, Deux B, Clezardin P, Sommer P, Pouyssegur J, et al. (2011). The HIF-l-inducible lysyl oxidase activates HIF-1 via the Akt pathway in a positive regulation loop and synergizes with HIF-1 in promoting tumor cell growth. Cancer research 71, 1647–1657. [DOI] [PubMed] [Google Scholar]
- Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, Olson OC, Quick ML, Huse JT, Teijeiro V, et al. (2013). CSF-1R inhibition alters macrophage polarization and blocks glioma progression. Nature medicine 19,1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, Bowman RL, Akkari L, Quick ML, Schuhmacher AJ, Huse JT, Holland EC, Sutton JC, and Joyce JA (2016). The tumor microenvironment underlies acquired resistance to CSF-1R inhibition in gliomas. Science 352, aad3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quail DF, and Joyce JA (2013). Microenvironmental regulation of tumor progression and metastasis. Nature medicine 19, 1423–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rachman-Tzemah C, Zaffryar-Eilot S, Grossman M, Ribero D, Timaner M, Maki JM, Myllyharju J, Bertolini F, Hershkovitz D, Sagi I, et al. (2017). Blocking Surgically Induced Lysyl Oxidase Activity Reduces the Risk of Lung Metastases. Cell reports 19, 774–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts WG, Ung E, Whalen P, Cooper B, Hulford C, Autry C, Richter D, Emerson E, Lin J, Kath J, et al. (2008). Antitumor activity and pharmacology of a selective focal adhesion kinase inhibitor, PF-562,271. Cancer research 68,1935–1944. [DOI] [PubMed] [Google Scholar]
- Roth P, and Weller M (2014). Challenges to targeting epidermal growth factor receptor in glioblastoma: escape mechanisms and combinatorial treatment strategies. Neuro-oncology 16 Suppl 8, viiil4–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saha D, Martuza RL, and Rabkin SD (2017). Macrophage Polarization Contributes to Glioblastoma Eradication by Combination Immunovirotherapy and Immune Checkpoint Blockade. Cancer cell 32, 253–+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samaras V, Piperi C, Korkolopoulou P, Zisakis A, Levidou G, Themistocleous MS, Boviatsis EI, Sakas DE, Lea RW, Kalofoutis A, et al. (2007). Application of the ELISPOT method for comparative analysis of interleukin (IL)-6 and IL-10 secretion in peripheral blood of patients with astroglial tumors. Molecular and cellular biochemistry 304, 343–351. [DOI] [PubMed] [Google Scholar]
- Shingu T, Ho AL, Yuan L, Zhou X, Dai CX, Zheng SY, Wang QH, Zhong Y, Chang Q, Horner JW, et al. (2017). Qki deficiency maintains sternness of glioma stem cells in suboptimal environment by downregulating endolysosomal degradation. Nat Genet 49, 75–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, Agarinis C, Schmelzle T, Bouwmeester T, Schubeler D, et al. (2015). YAP1 Exerts Its Transcriptional Control via TEAD-Mediated Activation of Enhancers. PLoS genetics 11, el005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proceedings of the National Academy of Sciences of the United States of America 102,15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szulzewsky F, Schwendinger N, Guneykaya D, Cimino PJ, Hambardzumyan D, Synowitz M, Holland EC, and Kettenmann H (2018). Loss of host-derived osteopontin creates a glioblastoma-promoting microenvironment. Neuro-oncology 20, 355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tumaneng K, Schlegelmilch K, Russell RC, Yimlamai D, Basnet H, Mahadevan N, Fitamant J, Bardeesy N, Camargo FD, and Guan KL (2012). YAP mediates crosstalk between the Hippo and PI(3)K-TOR pathways by suppressing PTEN via miR-29. Nature cell biology 14,1322–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, Miller CR, Ding L, Golub T, Mesirov JP, et al. (2010). Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer cell 17, 98–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang G, Lu X, Dey P, Deng P, Wu CC, Jiang S, Fang Z, Zhao K, Konaparthi R, Hua S, et al. (2016). Targeting YAP-Dependent MDSC Infiltration Impairs Tumor Progression. Cancer Discov 6, 80–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei J, Marisetty A, Schrand B, Gabrusiewicz K, Hashimoto Y, Ott M, Grami Z, Kong LY, Ling X, Caruso HG, et al. (2018). Osteopontin mediates glioblastoma-associated macrophage infiltration and is a therapeutic target. The Journal of clinical investigation. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen S, Stolarov J, Myers MP, Su JD, Wigler MH, Tonks NK, and Durden DL (2001). PTEN controls tumor-induced angiogenesis. Proceedings of the National Academy of Sciences of the United States of America 98,4622–4627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Shimizu S, Kiyonaka S, Takahashi N, Wajima T, Hara Y, Negoro T, Hiroi T, Kiuchi Y, Okada T, et al. (2008). TRPM2-mediated Ca2+influx induces chemokine production in monocytes that aggravates inflammatory neutrophil infiltration. Nature medicine 14, 738–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martinez E, Vegesna R, Kim H, Torres-Garcia W, Trevino V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nature communications 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zerrouqi A, Pyrzynska B, Febbraio M, Brat DJ, and Van Meir EG (2012). P14ARF inhibits human glioblastoma-induced angiogenesis by upregulating the expression of TIMP3. The Journal of clinical investigation 122,1283–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao D, Lu X, Wang G, Lan Z, Liao W, Li J, Liang X, Chen JR, Shah S, Shang X, et al. (2017). Synthetic essentiality of chromatin remodelling factor CHD1 in PTEN-deficient cancer. Nature 542,484–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Ying H, Yan H, Kimmelman AC, Hiller DJ, Chen AJ, Perry SR, Tonon G, Chu GC, Ding Z, et al. (2008). Pten and p53 converge on c-Myc to control differentiation, self-renewal, and transformation of normal and neoplastic stem cells in glioblastoma. Cold Spring Harbor symposia on quantitative biology 73, 427–437. [DOI] [PubMed] [Google Scholar]
- Zhu CB, Kros JM, Cheng C, and Mustafa D (2017). The contribution of tumor-associated macrophages in glioma neo-angiogenesis and implications for anti-angiogenic strategies. Neuro-oncology 19,1435–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, and Parada LF (2002). The molecular and genetic basis of neurological tumours. Nature reviews Cancer 2, 616–626. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNAseq and microarray data have been submitted to the GEO repository; the accession numbers are GSE122283 and GSE122284, respectively.