

Summary
Kinase and phosphatase overexpression drives tumorigenesis and drug resistance. We previously developed a mass-cytometry-based single-cell proteomics approach that enables quantitative assessment of overexpression effects on cell signaling. Here, we applied this approach in a human kinome- and phosphatome-wide study to assess how 649 individually overexpressed proteins modulated cancer-related signaling in HEK293T cells in an abundance-dependent manner. Based on these data, we expanded the functional classification of human kinases and phosphatases and showed that the overexpression effects include non-catalytic roles. We detected 208 previously unreported signaling relationships. The signaling dynamics analysis indicated that the overexpression of ERK-specific phosphatases sustains proliferative signaling. This suggests a phosphatase-driven mechanism of cancer progression. Moreover, our analysis revealed a drug-resistant mechanism through which overexpression of tyrosine kinases, including SRC, FES, YES1, and BLK, induced MEK-independent ERK activation in melanoma A375 cells. These proteins could predict drug sensitivity to BRAF-MEK concurrent inhibition in cells carrying BRAF mutations.
Keywords: single-cell proteomics, mass cytometry, human kinome and phosphatome, functional classification, overexpression effects, signaling dynamics, signaling relationships, cancer signaling networks, drug resistance, BP-R2 analysis
Graphical Abstract
Highlights
-
•
Human kinome- and phosphatome-wide screen of overexpression-altered signaling network
-
•
Classification of the kinome and phosphatome based on abundance-dependent effects
-
•
Phosphatase overexpression sustains MAPK-ERK signaling by halting negative feedback
-
•
SRC, FES, YES1, and BLK lead to drug resistance in BRAF-MEK combined inhibition
Lun et al. perform mass-cytometry-based single-cell analysis to study how the expression levels of 649 kinases and phosphatases affect the intracellular signaling network. They expand the functional classification of the kinome and phosphatome based on abundance-dependent effects and identify novel signaling mechanisms underlying overexpression-driven diseases and drug resistance.
Introduction
Kinases and phosphatases control the reversible process of phosphorylation. Signaling networks involving these enzymes compute extracellular signals into transcriptional, functional, and phenotypical responses. Deregulation of signaling networks can lead to the initiation and progression of many types of human disease, including cancer (Fleuren et al., 2016, Julien et al., 2007). Signaling network structure has been studied by mapping physical interactions of kinases and phosphatases in steady and dynamic states using biochemical approaches and reporter assays (Barrios-Rodiles et al., 2005, Breitkreutz et al., 2010, Couzens et al., 2013, Horn et al., 2011). Using in vitro kinase assays and motif-based predictions, the specificity and targets of many kinases have been revealed (Linding et al., 2007, Mok et al., 2010, Yu et al., 2009). Kinase and phosphatase perturbations have been applied to systematically determine network responses in yeast and human cells (Bodenmiller et al., 2010, Ochoa et al., 2016, Sacco et al., 2012a).
Mutation-induced signaling network rewiring and modulation of signaling dynamics have also been characterized for many kinases (Creixell et al., 2015, Pawson and Warner, 2007), providing a basis for the identification of targeted therapies in cancer (Hennessy et al., 2005, Logue and Morrison, 2012). Independently of mutations, kinase overexpression drives tumorigenesis in multiple cancer types and is a critical factor in drug resistance (Eralp et al., 2008, Santarius et al., 2010, Shaffer et al., 2017). Recently, overexpression of phosphatases has been shown to mediate cancer progression and has been linked to the poor prognosis of patients (Julien et al., 2011, Liu et al., 2016, De Vriendt et al., 2013). Overexpression-induced signaling modulation remains largely uncharacterized because factors such as genetic instability induce highly heterogeneous quantities of deregulated signaling proteins in cancer cells (Abbas et al., 2013), making conventional cell population-based analysis inapplicable. Only recently have technologies emerged that account for such heterogeneity and that can comprehensively quantify signaling network behavior with single-cell resolution. This resolution is required to characterize abundance-related cellular signaling states (measured as phosphorylation levels of signaling proteins) and phenotypical alterations caused by a given kinase or phosphatase of interest (Bendall et al., 2011, Lun et al., 2017). Mass cytometry allows simultaneous quantification of >40 proteins or protein modifications at single-cell resolution, enabling the profiling of complex cellular behaviors in highly heterogeneous samples (Bendall et al., 2011, Bodenmiller et al., 2012, Chevrier et al., 2017, Levine et al., 2015). We have recently established and thoroughly validated an approach that couples transient protein overexpression with mass-cytometry-based, single-cell analysis and have revealed that protein overexpression induces complex signaling network modulations in an abundance-dependent manner (Lun et al., 2017).
Here, we applied this technique in a human kinome- and phosphatome-wide screen to determine kinase and phosphatase abundance-dependent effects on 30 phosphorylation sites known to be involved in the regulation of growth, proliferation, survival, and stress signaling pathways. Over 10 million individual cells were analyzed, covering 649 overexpression conditions with or without 10-min epidermal growth factor (EGF) stimulation. Assessing the effects of kinase and phosphatase on the signaling network, we expanded the functional classification of the kinome and phosphatome. Our analysis identified 1,323 pairs of overexpression-dependent signaling relationships, including 208 pairs that were previously unknown. By characterizing signaling dynamics in a follow-up EGF stimulation time course and a kinase-phosphatase combinatorial overexpression assay, we found a pro-cancer signaling response in which the overexpression of ERK-specific phosphatases sustained cell proliferative signals. Further analysis of our dataset revealed a drug-resistant mechanism through which the overexpression of tyrosine kinases, including SRC, FES, YES1, and BLK, induced MEK-independent ERK activation in melanoma A375 cells. The expression levels of these proteins could predict drug sensitivity to BRAF-MEK concurrent inhibition in patients with BRAF mutations and may be suggestive of alternative treatments.
Results
Abundance-Dependent Effects of Human Kinases and Phosphatases on Cell Signaling
Protein abundance variance on the single-cell level is often observed in tumors as heterogeneous genomic abnormalities accumulate (Du and Elemento, 2015). Inter-tumoral heterogeneity presumably results in highly variable signaling responses to stimuli or drug treatments. In addition, a high degree of intra-tumoral heterogeneity further challenges cancer therapeutic interventions (Patel et al., 2014, Roth et al., 2016). To understand the signaling network modulation in cells that overexpress a defined kinase or phosphatase at various levels, we applied our abundance-dependent signaling network assessment system (Lun et al., 2017) in a kinome- and phosphatome-wide screen.
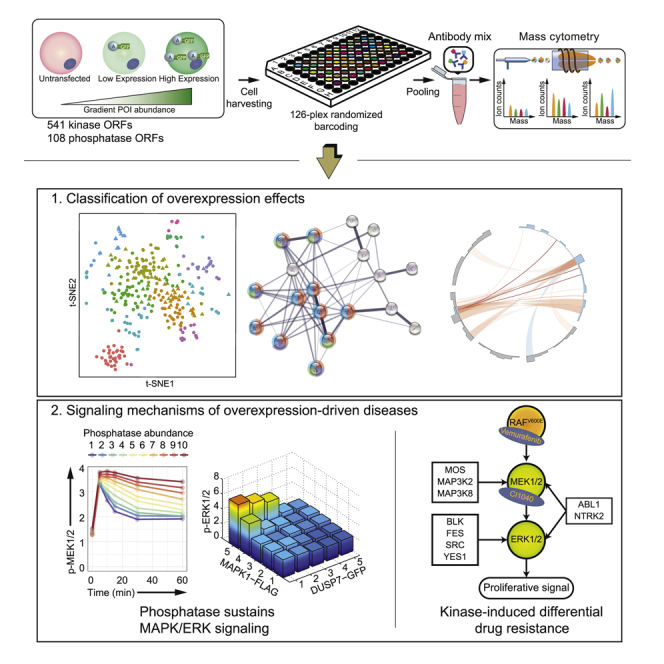
We cloned open reading frames (ORFs) from the human kinase library (Johannessen et al., 2010) and the human phosphatase library into a vector, enabling the expression of GFP-tagged proteins (Couzens et al., 2013). The generated 541 kinase and 108 phosphatase expression clones (Table S1) were individually transiently transfected into human embryonic kidney HEK293T cells. Unstimulated cells and cells stimulated for 10 min with EGF were harvested and processed with a 126-plex barcoding strategy (adapted from Bodenmiller et al., 2012, Zunder et al., 2015) for simultaneous antibody staining followed by multiplexed mass cytometry measurements (Figure 1A).
Figure 1.

Kinome- and Phosphatome-wide Screen for Effects of Protein Abundance on Signaling States and Dynamics
(A) The experimental workflow: ORFs of 541 human kinases and 108 human phosphatases were cloned into a vector encoding GFP-tagged fusion proteins upon transient transfection into HEK293T cells. Cells with or without 10-min EGF stimulation were harvested, barcoded, and stained with antibody mix before mass-cytometry-based single-cell analysis.
(B) Plot of counts versus BP-R2 values for control and experimental samples. Cutoff value was determined by analysis of the BP-R2 values in all of the control samples. Square-root transformation was applied on the y axis.
(C) Venn diagram showing the quantification of POIs with abundance-dependent influences on the AKT pathway (p-PDK1, p-GSK3β, β-catenin, p-mTOR, p-p70S6K, p-4EBP1, and p-S6), MAPK-ERK pathways (p-RAF, p-MEK1/2, p-ERK1/2, p-p90RSK, p-CREB, and p-SMAD2/3), stress response pathways (p-MKK3/6, p-MKK4/7, p-p38, p-JNK, p-MAPKAPK2, p-AMPKα, and p-p53), and PKC and STAT pathways (grouped for illustration purposes; p-SRC, p-FAK, p-BTK, p-PLCγ2, p-MARCKS, p-NFκB, p-STAT1, p-STAT3, and p-STAT5).
(D) Shape-based clustering on all strong signaling.
Transient transfection generates a single-cell gradient of the GFP-tagged protein of interest (POI) expression levels with up to 1,000-fold enhancement relative to the endogenous POI expression range (Lun et al., 2017). The abundance variation of each overexpressed kinase or phosphatase was quantified on the single-cell level by mass cytometry with detection by a metal-conjugated anti-GFP antibody. Simultaneously, we quantified 30 phosphorylation states of proteins involved in key cancer-related signaling pathways, including the AKT, protein kinase C (PKC), signal transducer and activator of transcription (STAT), mitogen-activated protein kinase (MAPK)/ERK, and stress pathways, and 5 non-signaling markers indicative of cell physiological states (Table S2). Over 10 million individual cells were analyzed in the 659 overexpression conditions with or without 10-min EGF stimulation, averaging ∼7,000 measured cells per sample. That the GFP-tagging system rarely influenced protein overexpression effects has been previously reported (Lun et al., 2017).
The dependence of phosphorylation levels on the abundance of GFP-tagged POI was quantified by the binned pseudo-R2 (BP-R2) method, a density-independent measure of signaling relationship strength (Figures S1A and S1B) (Lun et al., 2017). We confirmed that signaling relationships assessed with our approach were reproducible in five different cell lines from multiple tissues of origin (Figure S1C). We analyzed 108 control samples (FLAG-GFP overexpression or untransfected cells) and used the highest BP-R2 score (0.13) of all of the controls as the cutoff to consider a signaling relationship as “strong.” In total, our human kinome and phosphatome analysis detected 1,323 pairs of strong relationships between POIs and phosphorylation sites (Figure 1B). Among the 649 kinases and phosphatases, 327 (50.4%) had at least 1 strong signaling relationship to the cancer-related signaling network when overexpressed. Of these, 245 had narrow influences with the modulation of 1–5 signaling nodes, and 26 overexpressed proteins had broad effects on the network with >10 measured phosphorylation sites influenced (Figure S1D). We identified 49 proteins that affected all of the measured signaling pathways, including 11 receptor proteins (e.g., MET, fibroblast growth factor receptor 1 [FGFR1], and platelet-derived growth factor receptor A [PDGFRA]) and many MAPK cascade activators (e.g., MAP4K1, MAP4K2, and MAP4K5) (Figure 1C).
To characterize how POI abundance modulates intracellular signaling, we performed shape-based clustering (see STAR Methods) on all of the detected strong signaling relationships. We classified these relationship shapes into 10 shape clusters (Figure 1D). Shapes 1–5 involve overexpression-induced signal upregulation, with sensitivity to abundance changes increasing from shape 1 to shape 5. Shape 6 is non-monotonic signaling relationships, as phosphorylation levels initially increase and then decrease as a function of POI abundance. Shapes 7–10 reflect overexpression-induced signaling downregulation, with sensitivity to POI abundance changes increasing from shape 7 to shape 10 (Figure 1D). Overexpression of a particular POI can affect different phosphorylation sites with dissimilar shapes of relationship (Table S3). We also found that 250 pairs of signaling relationships had different shapes before and after EGF stimulation, indicating that POI levels determine the strength of the signaling response to EGF stimulation.
Altered responses to EGF stimulation are the potential results of network topology modulations (Koseska and Bastiaens, 2017, Santos et al., 2007) that are induced by POI abundance changes. To verify this, we focused on two pairs of EGF-induced shape-switching relationships, KSR2 to p-MAPKAPK2 (MAPKAPK2 signaling is essential for tumor cell survival; Morandell et al., 2013) and TEC to p-ERK1/2 (a pair of non-monotonic relationship). We performed additional perturbation experiments using MEK inhibitor CI1040 and characterized signaling network variations over a large dynamic range of POI concentrations. We found that at the medium expression levels of KSR2, MAPKAPK2 phosphorylation was MAPK-ERK cascade-dependent and that it could be highly induced by EGF stimulation; high KRS2 expression levels contributed to MAPK-ERK-independent MAPKAPK2 signaling that had a weak response to EGF stimulation and was insensitive to MEK inhibition (Figures S1E–S1G). Increased TEC abundance led to non-monotonic ERK1/2 phosphorylation that was partially diminished by MEK inhibition (Figure S1H), indicating the presence of both MEK-dependent and MEK-independent pathways for the TEC overexpression-induced ERK activation. The MEK-dependent signaling was reduced at high TEC expression levels, potentially due to a negative regulatory mechanism that is only activated in the presence of high concentrations of TEC (Figures S1H–S1J). Here, by characterizing signaling network variations over a large dynamic range of POI concentration, our analysis revealed complex modulations of signaling network topology in a protein abundance-dependent manner.
Functional Classification of Kinases and Phosphatases Based on Signaling Network Modulations
To understand the regulatory and functional similarity of overexpressed POIs, we indicated the sign for signaling relationships (according to their directionality) to the BP-R2 (Table S4; STAR Methods). Then, we applied the dimensional reduction algorithm t-distributed stochastic neighbor embedding (t-SNE) (van der Maaten and Hinton, 2008) to the matrix of all 60 measured signaling parameters (as signed-BP-R2 scores) over the 327 signaling network-influential kinases and phosphatases (Figure 2A). As expected, homologous groups of kinases and phosphatases showed nearly identical influences on signaling and overlapped with each other on the t-SNE plot (Figure 2A, green boxes). This demonstrates that our method sensitively, specifically, and reproducibly detected abundance-dependent signaling behaviors. All eight overexpressed SRC family members—SRC, YES1, BLK, LCK, LYN, HCK, FGR, and FRK—co-localized in the t-SNE analysis (Figure 2A, purple box), indicating that these kinases have similar abundance-dependent signaling effects, despite the previously revealed differential patterns of expression (Parsons and Parsons, 2004). Members of protein tyrosine phosphatase (PTPN1, PTPN2, and PTPN5) and dual-specificity phosphatase (DUSP3, DUSP4, DUSP6, DUSP7, DUSP10, and DUSP16) families were grouped together, suggesting similarities in regulating the measured cancer signaling network (Figure 2A, orange box).
Figure 2.

Kinase and Phosphatase Classification Based on Abundance-Dependent Effects on Cancer Signaling Network
(A) t-SNE analysis of overexpressed kinases and phosphatases performed on signed-BP-R2 of all measured phosphorylation sites with and without EGF stimulation, color coded by hierarchical clusters.
(B) The mean signed-BP-R2 values of all measured phosphorylation sites in each cluster of kinases or phosphatases shown in literature-guided canonical signaling network visualizations.
We then applied hierarchical clustering based on signed-BP-R2 scores of all of the measured phosphorylation sites (Figures S2A and S2B) to further analyze functional similarities among all of the kinases and phosphatases. This led to the identification of 10 major signaling response clusters (color coded on the t-SNE plot in Figure 2A). Correspondence analysis was performed between these identified clusters and classes of kinases and phosphatases previously established based on catalytic domain sequences (Johannessen et al., 2010, Sacco et al., 2012b) (Figure S2C). In certain cases, proteins with partial sequence identity had similar influences on signaling. For example, all of the kinases in cluster 1 are receptor or non-receptor tyrosine kinases (Figure S2C). These kinases are early responders to stimuli, as shown in the literature-based graph of canonical EGF receptor (EGFR) networks (Figure 2B). Clusters 5, 9, and 10 include non-receptor serine or threonine kinases and kinases classified in the group of “other” (i.e., kinases that do not fit into any of the major groups) (Figure S2C). Despite conserved catalytic domain sequences, kinases in clusters 5, 9, and 10 induced different cellular responses (Figure 2B). Cluster 7 proteins had negative relationships with the mediators of the MAPK-ERK pathway when cells were treated with EGF (Figure 2B). Cluster 7 mostly consists of protein tyrosine phosphatases, but also includes a few proteins from the classes of non-receptor serine or threonine kinase and lipid kinases (Figure S2C). Comparing our identified clusters to the phylogenetic tree of the human kinome (Eid et al., 2017, Manning et al., 2002), we observed the enrichment of cluster 1 in the tyrosine kinase group (Figure S2D, orange arrow). In addition, PKC family members are enriched in cluster 5 (Figure S2C, brown arrow), and MAP3Ks are enriched in cluster 9 (Figure S2D, blue arrow). In summary, the human kinome- and phosphatome-wide overexpression analysis identified 10 clusters of kinases and phosphatases, with distinct signaling patterns found for each cluster. These clusters partially matched the sequence-based classification and expanded the functional classification of the human kinases and phosphatases based on their abundance-dependent modulations to the cancer signaling network.
Functional Enrichment Analysis of Kinase and Phosphatase Clusters
Our analysis indicated that signaling proteins with different catalytic domain sequences may affect signaling networks similarly when overexpressed. To understand the functional relationship between proteins with similar overexpression effects, we performed a functional enrichment analysis using the STRING database (Szklarczyk et al., 2017) on the 10 identified clusters (Figures 2A and 3A). We found that 7 of the 10 clusters had significant functional enrichment (p < 0.01; statistical details in STAR Methods). Physical and functional interaction enrichments are shown as protein-protein association networks for cluster 7 in Figure 3B and for all other clusters in Figure S3A.
Figure 3.

Functional Enrichment Analysis of Kinases and/or Phosphatases in Each Cluster
(A) An unrooted tree shows the hierarchical clustering of the kinases and phosphatases based on their signed-BP-R2 scores. Terms of enriched functions (p < 0.05) from each cluster are annotated, with circle color indicating the p value and circle size showing the coverage of cluster components. The percentage of associated proteins is indicated by the size of the adjacent circle.
(B) Functional association analysis performed with the STRING database (Szklarczyk et al., 2017) for cluster 7. Confident edges are shown in the network. Functional enrichments are shown as color-coded pies, with the pie radius indicating the p value.
Cluster 7 is enriched for protein tyrosine phosphatases that negatively regulate MAPK pathways. Several MAPK regulating kinases are present in this cluster, including KSR1 and ARAF, which have overexpression effects similar to those of the phosphatases (Figure 3B). KSR1 and ARAF are core components of the KSR-RAF dimeric protein complex that transduces signal in the MAPK-ERK cascade (Lavoie and Therrien, 2015). Overexpressing one subunit of this protein complex may result in competitive inhibition, diminishing the downstream signal activities in a manner similar to that of phosphatase overexpression. These analyses demonstrated that proteins with different catalytic functions can mediate highly related signaling responses when overexpressed and that kinase overexpression does not affect signaling networks in the same manner as direct kinase activation.
To assess the relationship between overexpression effects and protein catalytic activities, we chose to overexpress five kinase-dead mutants: AKT3K177M, AXLK567R, MAPK3K71R, PRKCEK437W, and MAP2K1K97M. Unlike wild-type kinases, the overexpression of kinase-dead mutants AXLK567R and PRKCEK437W had almost no network effect (Figure S3B), indicating that the detected abundance-dependent network modulations of these kinases are related to their catalytic functions. In contrast, the main network effects of AKT3, MAPK3, and MAP2K1 were also observed when the kinase-dead mutants were overexpressed (Figure S3B). This suggests that overexpression-induced signaling network modulations for these kinases are non-catalytic. In addition, 26 kinases in our screen were previously predicted to be catalytically inactive (Manning et al., 2002). We found that 6 of these 26 proteins influenced the measured network, with a total of 17 pairs of strong signaling relationships detected (Table S5), also demonstrating that our analysis captured non-catalytic network modulations determined by kinase abundance. In summary, these data show that protein overexpression effects can be catalytic or non-catalytic and suggest that activity-based modeling of signaling network deregulation for drug target discovery alone is likely insufficient.
Signaling Relationships Detected in Kinome- and Phosphatome-wide Analysis
The functions of many kinases and phosphatases analyzed in our screen are unknown or only poorly characterized. We hypothesized that our global analysis could lead to the identification of signaling relationships. To assess this, we performed a systematic comparison between all identified overexpression-induced signaling relationships and records in OmniPath, an integrated database of literature-curated signaling interaction information (Türei et al., 2016). We first mapped all pairs of relationships to the OmniPath signaling network and then computed the signed, directed paths for each pair of relationship (Krumsiek et al., 2011, Perfetto et al., 2016). The distance between an overexpressed protein and a measured phosphorylation site is represented by the length of the path (Figure 4A). For example, a distance of 0 indicates the relationship between the overexpressed POI and its own phosphorylation levels. Of 14 pairs of signaling relationships with a known distance of 0, 12 had strong BP-R2 values with and without 10-min EGF stimulation (Figure 4A), revealing that the phosphorylation level of a particular kinase is often determined by its own abundance, even in the absence of additional perturbation.
Figure 4.

Prediction of Potential Signaling Connections by Comparison with Literature Evidence in the Signaling Interaction Database OmniPath
(A) Abundance-dependent relationship strength for each pair of overexpressed POIs and measured phosphorylation site, as quantified with signed-BP-R2, plotted on the length of shortest signed, directed path between the two extracted from the OmniPath database (Türei et al., 2016).
(B) Occurrences of strong signaling relationships (BP-R2 > 0.13), with path length from 0 to 5 or infinite path length (OmniPath) in each individual hierarchical cluster.
(C and D) For clusters 8 (C) and 5 (D), respectively, the shortest signed directed path length for each determined strong signaling relationship is shown in Circos plots (Krzywinski et al., 2009).
We detected 208 (16%) strong relationships (BP-R2 > 0.13) with infinite distance (Figure 4A; Table S6), which is indicative of connections not described previously. In total, 93 overexpressed POIs contributed to these signaling relationships, which were enriched (in absolute count) in clusters 2, 3, and 4 and to a lesser extent in cluster 6 (Figure 4B). We did not detect any relationships with infinite distance in clusters 9 or 10 (Figure 4B); POIs from these clusters participate in MAPK signal transduction (Figure 3A), which is well characterized. We also assessed the distribution of infinite paths for each kinase and phosphatase class and did not detect any enrichment (Figure S4A). There were 132 pairs of strong relationships between proteins with length of signed directed path >3 in OmniPath, suggesting potentially undiscovered direct or short-range connections (Figure 4A).
Many potential signaling relationships were related to disease and to poorly characterized kinases (Figures 4C, 4D, and S4B). For instance, high levels of RIOK2 (highlighted in Figure 4C) have been recently shown to correlate with the poor prognosis of patients with non-small-cell lung cancer, but the underlying signaling mechanisms are unclear (Liu et al., 2016). We discovered that RIOK2 overexpression affected several phosphorylation sites, most strongly Thr172 on adenosine 5′ monophosphate-activated protein kinase α (AMPKα), Ser257/Thr261 on MKK4/7, and Thr180/Tyr182 on p38 (Figure 4C), indicating the activation of the AMPK-p38 axis upon RIOK2 overexpression. The AMPK-p38 axis regulates cellular energy metabolism, contributing to cancer cell survival in nutrient-deficient conditions (Chaube et al., 2015, Zadra et al., 2015). In cancer proteome data from the Clinical Proteomic Tumor Analysis Consortium (Koboldt et al., 2012), we found that expression levels of RIOK2 were highly correlated with levels of AMPK subunits β and γ and the AMPK activator LKB1 (STK11), confirming RIOK2 as a co-regulatory kinase in the AMPK signaling pathway (Figures S4C and S4D). To further illustrate the clinical relevance of overexpression-induced AMPK activation, we coupled our data of kinase and phosphatase overexpression effects with the proteome data of breast cancer and ovarian cancer patients and their prognosis information (Mertins et al., 2016, Zhang et al., 2016) (Figures S4E and S4F). We found three kinases (CSNK1A1, NEK7, and TLK1) and a phosphatase (CDC25C) inducing AMPK activation when overexpressed and affecting patient outcomes; patients overexpressing any of these kinases had significantly worse prognoses in comparison to the patients underexpressing the same kinase (Figures S4G and S4H). Our data suggest that kinase overexpression-induced AMPK activation is related to the prognosis in cancer patients and that AMPK is a potential therapeutic target for patients overexpressing proteins, such as RIOK2, CSNK1A1, CDC25C, NEK7, and TLK1.
Overexpression of a poorly characterized kinase, MGC42105 (NIM1K), in cluster 5 (highlighted in Figure 4D) modulated the phosphorylation of Ser241 on PDK1, Thr389 on p70S6K, and Ser235/Ser236 on S6, suggesting a role in growth signaling. MGC42105 also induced abundance-dependent activation of stress pathways, as strong relationships to p-p53 (Ser15) and p-AMPKα (Thr172) were observed (Figure 4D). In summary, mapping our identified signaling relationships to the OmniPath database enabled the assignment of signaling functions to a number of kinases and phosphatases and shed light on potential signaling mechanisms associated with the poor prognosis of cancer patients.
In-Depth Analysis of Signaling Dynamics Reveals Overexpression-Dependent MAPK-ERK Activity
An understanding of signaling dynamics is essential for identifying diseased signaling circuits within a network and in the prediction of drug efficiency (Du and Elemento, 2015, Hughey et al., 2010). We have previously shown that altering the expression levels of central signaling proteins in the EGFR signaling network results in complex changes in network dynamics (Lun et al., 2017). Given the key role of signaling dynamics on cell proliferation, growth, and differentiation (Koseska and Bastiaens, 2017), we systematically evaluated kinases and phosphatases from the 10 identified clusters for overexpression-induced signaling dynamic modulations. We calculated the differences in signed-BP-R2 scores between the EGF-stimulated and -unstimulated conditions to identify cases in which overexpression modulated signaling dynamics (i.e., altered the strength or the shape of abundance-dependent signaling relationships between the unstimulated and the 10-min EGF-stimulated conditions). We found that POIs in clusters 1, 6, 7, 9, and 10 strongly modulated signaling network dynamics when overexpressed (Figure S5A). We then analyzed the overexpression effects of the top 39 dynamic-modulating POIs over a 1-h EGF stimulation time course. The dynamic responses of all of the measured phosphorylation sites are shown in Figures 5A and 5B. Example signaling relationship shapes at each time point during the time course and the POI abundance-dependent signaling trajectories over the time course are shown in Figures 5C–5G. Features of the signaling amplitudes (see STAR Methods) are shown in Figures S5F–S5H.
Figure 5.

Effects of EGF Stimulation on 39 Kinases and Phosphatases
(A) Heatmap of signed-BP-R2 scores for measured signaling relationships over a 1-h EGF stimulation time course. Six identified groups of kinases and phosphatases are labeled in color codes.
(B) For one representative POI from each group, signaling relationships to all measured phosphorylation sites, as quantified by signed-BP-R2, are shown in the literature-guided canonical signaling network map.
(C–G) Violin plots show cell distribution in each of 10 bins based on GFP-tagged POI expression levels (see STAR Methods) for EGFR-GFP to p-ERK1/2 (C), DUSP4-GFP to p-ERK1/2 (D), PTPN2-GFP to p-ERK1/2 (E), DUSP4-GFP to p-MEK1/2 (F), and PTPN2-GFP to p-MEK1/2 (G) over the 1-h EGF stimulation time course. Medians of all 10 bins are connected to indicate the shape of signaling relationships (black lines), with the relationship strength quantified by signed-BP-R2, as shown on top of each individual plot. In the plot on the far right, medians of each bin are connected over the time course to demonstrate the POI abundance-dependent signaling trajectories.
(H) Schematic illustration of how two sets of phosphatases induce different abundance-dependent influences on the signaling dynamics of the MAPK-ERK cascade.
Hierarchical clustering of the overexpression-induced EGFR signaling dynamics classified the 39 selected proteins into 6 groups (Figures 5A and S5B). Signaling network responses for one representative kinase or phosphatase from each of the six identified groups are illustrated in Figure 5B. We showed that the network responses to EGF stimulation were highly specific to cell lines (Figure S5C). In HEK293T cells, EGF stimulation strongly activated the MAPK-ERK signaling pathway and had weak effects on the AKT, PKC, STAT, or stress response pathways (Figure S5C). Similarly, we observed that EGF stimulation primarily influenced the POI abundance-dependent dynamics of the MAPK-ERK signaling cascade rather than the AKT, PKC, and STAT pathways (Figure 5A). As the MAPK-ERK proliferative pathway is known to be involved in tumor progression and drug response, we focused our subsequent analyses on this pathway in HEK293T cells.
Phosphorylation of Thr202/Tyr204 on ERK1/2 was elevated in cells with high levels of GFP-tagged EGFR in the absence of EGF stimulation (Figures 5B and 5C). These cells did not respond to EGF stimulation, indicating ligand-independent ERK activation (Figures 5C and S5G). Since the ligand-independent ERK activation is known as a cancer-driving mechanism (Guo et al., 2015), we next sought to systematically identify signaling proteins causing similar abundance-dependent ligand insensitivity. Applying shape-based clustering, we classified p-ERK1/2 signaling trajectories during the 1-h EGF stimulation time course, over the range of expression levels of each analyzed POI. We found that overexpression of kinases TYRO3, TEC, MST1R, MOS, MET, MAP3K8, FGF1R, and ABL1 also led to prolonged ERK1/2 activation (Figures S6A–S6D). These proteins have been previously shown to mediate oncogenic signaling (Duan et al., 2016, Johannessen et al., 2010, Paul and Mukhopadhyay, 2004, Salgia, 2017).
In the absence of EGF stimulation, overexpression of phosphatases DUSP4 and PTPN2 did not affect the levels of phosphorylation in the MAPK-ERK pathway (Ser221 on MEK1/2, Thr202/Tyr204 on ERK1/2, or Ser380 on p90RSK) (Figures 5B, 5D–5G, S5D, and S5E). This suggests either a mechanism that compensates for phosphatase overexpression to maintain basal MAPK-ERK signaling or that the overexpressed phosphatases are inactive without EGF stimulation. Upon EGF stimulation, signaling dynamics on phosphorylation sites of the MAPK-ERK pathway were modulated by DUSP4 and PTPN2 in an abundance-dependent manner as negative signaling relationships to p-ERK1/2 and p-p90RSK were detected (Figures 5B, 5D, 5E, S5D, and S5E). DUSP4 or PTPN2 overexpression also resulted in reduced p-ERK1/2 and p-p90RSK amplitudes (Figure S5H). Different p-MEK1/2 dynamics were observed in cells overexpressing these two phosphatases. In response to the EGF stimulation, cells with DUSP4 overexpression had more sustained MEK1/2 activation (Figure 5F) compared to cells with PTPN2 overexpression (Figure 5G). DUSP4 has been shown to specifically target ERK1 and ERK2 (Guan and Butch, 1995). Our data indicate that by diminishing ERK1/2 phosphorylation, the overexpressed DUSP4 attenuates the negative feedback from ERK1/2 to MEK1/2, resulting in constant activation of MEK1/2. Substrates of PTPN2 are primarily membrane kinases, including EGFR (Mattila et al., 2005). As expected, overexpression of PTPN2 downregulated the activation of all measured signaling proteins known to be downstream of EGFR, including MEK1/2 and ERK1/2 (Figure 5H).
To systematically classify all overexpressed phosphatases based on the signaling dynamics, we performed shape-based clustering on p-MEK1/2 trajectories during the 1-h time course after EGF addition, over the expression levels of each POI (Figures S6E–S6H). Abundance-dependent prolonged MEK1/2 phosphorylation was observed with other phosphatases in group E, including DUSP6, DUSP7, DUSP10, DUSP16, and PTPN7, indicating that Thr202 and Tyr204 on ERK1/2 are substrates of these enzymes (Figure 5H). DUSP10 and DUSP16 have been previously reported to be JNK- and p38-specific phosphatases (Finch et al., 2012, Masuda et al., 2003). Here, we show that DUSP10 and DUSP16, expressed at high abundance, also dephosphorylate p-ERK1/2 and attenuate the MAPK-ERK signaling, thereby likely decreasing the negative feedback from ERK1/2 to MEK1/2 and causing sustained MEK1/2 activation (Figures 5A and 5H).
Pairwise Overexpression Analysis Reveals that Phosphatases Sustain Kinase-Induced MAPK-ERK Signaling
Phosphatase overexpression is oncogenic in different tumor types, but the signaling mechanisms remain unclear (Julien et al., 2007, Julien et al., 2011). Recent work indicates that overexpressed phosphatases increase the malignancy of cancers that have a hyperactivated MAPK-ERK pathway (Julien et al., 2007, Low and Zhang, 2016, De Vriendt et al., 2013). Our data suggest a mechanism through which overexpression of ERK-specific phosphatases sustains MEK phosphorylation levels (Figures 5F and 5H). To assess whether an additional, secondary signaling input that increases MAPK pathway activity could lead to phosphatase-driven oncogenic-like signaling, we developed a combinatorial transfection assay in which overexpression of a kinase and a phosphatase were detected via an FLAG-tag and a GFP-tag, respectively, providing a two-dimensional analysis of abundance-dependent signaling modulations on the single-cell level (Figure 6A). Using this approach, we analyzed the MAP2K2, MAPK1, and RPS6KA1 (also known as MEK2, ERK2, and p90RSK1) kinases and the DUSP4, DUSP7, and PTPN2 phosphatases in 9 combinations of double overexpression over a 1-h EGF stimulation time course (Figure 6B).
Figure 6.

Effect of Pairwise Overexpression of a Kinase and a Phosphatase on Signaling
(A) Workflow of the pairwise overexpression. Two plasmids encoding an FLAG-tagged kinase and a GFP-tagged phosphatase were transfected into HEK293T cells successively. Cells were binned into 25 groups according to their FLAG and GFP abundances. The median level of each measured phosphorylation site was computed for each bin.
(B), Kinases MAP2K2, MAPK1, and RPS6KA1 and phosphatases DUSP4, DUSP7, and PTPN2 were selected for the pairwise overexpression, generating nine overexpression combinations in total.
(C) In cells with overexpression of MAP2K2 and DUSP4, median phosphorylation levels of p-MEK1/2 and p-ERK1/2 are plotted for all of the bins over the 1-h EGF stimulation time course.
(D) Signaling trajectories of p-MEK1/2 and p-ERK1/2 plotted as the medians of each individual bin over the 1-h EGF stimulation time course.
(E) Schematic illustrating the modulation of RAF-MEK-ERK cascade signaling states and dynamics upon pairwise overexpression.
(F and G) Analysis of p-ERK1/2 and p-MEK1/2 phosphorylation levels (F) and signaling trajectories (G) on MAPK1-FLAG and DUSP7-GFP abundances.
When overexpressed individually, we observed that DUSP4 overexpression sustained the phosphorylation of Ser221 on MEK1/2 over the 1-h EGF stimulation time course, likely due to the weakened ERK-to-MEK negative feedback (Figures 6C and 6D). MAP2K2-FLAG overexpression led to an increased MEK1/2 phosphorylation (Figure 6C). MAP2K2-FLAG and DUSP4-GFP co-overexpression further increased the hyperactivated states of MEK1/2 over the 1-h EGF stimulation time course compared to the activation induced by MAP2K2-FLAG overexpression alone (Figures 6C–6E). Moreover, in cells with simultaneously overexpressed MAP2K2-FLAG and DUSP4-GFP, the downstream ERK1/2 phosphorylation on Thr202 and Tyr204 were inhibited (Figures 6C–6E). Previously, highly activated MEK1/2 was observed to lead to ERK-independent oncogenic-like signaling (Burgermeister and Seger, 2008, Takahashi-Yanaga et al., 2004).
The overexpression of FLAG-tagged MAPK1 (ERK2) drastically augmented ERK1/2 phosphorylation during EGF stimulation (Figure 6F), increased p-ERK1/2 amplitudes, and delayed p-ERK1/2 peak times (Figure 6G) in agreement with a previous study of the effect of MAPK1 overexpression (Lun et al., 2017). The simultaneous overexpression of MAPK1-FLAG and DUSP7-GFP decreased p-ERK1/2 levels at all time points and reduced the signaling amplitudes. Furthermore, DUSP7 overexpression delayed p-ERK1/2 peak times upon EGF stimulation: in cells with the highest MAPK1 abundance and mid-level overexpression of DUSP7, ERK1/2 phosphorylation peaked at 30 min after the addition of EGF (Figures 6F and 6G, purple arrows), whereas in untransfected cells, p-ERK1/2 peaked at the 5-min time point (Figures 6F and 6G). As expected, DUSP7 overexpression also resulted in constant MEK1/2 phosphorylation (Figures 6F and 6G, green arrows). Compared to cells overexpressing only MAPK1 (ERK2), which induced strong but transient ERK activation, the additional low-to-mid levels of DUSP7 decreased the ERK1/2 phosphorylation amplitude and partially limited the negative feedback signal from ERK to MEK, inducing a sustained MEK activation and a prolonged ERK signal. Thus, our analysis indicates that overexpression of certain phosphatases, such as DUSP4 and DUSP7, led to sustained activation of ERK due to the reduced negative feedback strength. This mechanism may underlie the pro-cancer effects of phosphatase overexpression.
Analysis of the Abundance-Dependent Signaling Relationship Characterizes Drug-Resistance Mechanisms
As protein overexpression has been correlated with drug resistance of cancer cells (Johannessen et al., 2010, Johannessen et al., 2013, Shaffer et al., 2017), we next sought to determine whether our kinome- and phosphatome-wide signaling network profiles could identify kinases or phosphatases that, when overexpressed, induce drug resistance and could characterize underlying signaling mechanisms. In melanoma cells carrying the BRAFV600E mutation, the overexpression of certain kinases is associated with de novo or acquired resistance to MAPK-ERK inhibition; Johannessen et al., 2010, Johannessen et al., 2013) have identified resistance-driving candidate genes in viability assays using melanoma A375 cells. Seventeen of these candidates were analyzed in our screen, and the overexpression of 14 caused abundance-dependent signaling modulations to p-ERK1/2 (Thr202/Tyr204) in unstimulated cells (Figure S7A). The 10-min EGF stimulation reduced relationship strengths for each of these kinases (Figure S7B), indicating that these overexpression-related ERK activations were ligand-binding independent (described above with Figure 5C), which has been previously suggested to be a drug-resistance mechanism (Guo et al., 2015).
In our kinome- and phosphatome-wide study, we detected 54 POIs that activated ERK in the absence of EGF (Table S7). To determine whether the overexpression of these proteins is predictive of drug resistance in cells with BRAF mutations, we transfected A375 cells, a melanoma cell line with the BRAFV600E mutation, using vectors encoding proteins with the strongest abundance-dependent signaling relationship to p-ERK1/2; ABL1, BLK, FES, MAP3K2, MAP3K8, MOS, NTRK2, SRC, and YES1. MEK1DD, a constitutively active kinase, was used as a positive control (Johannessen et al., 2010). Cells were subsequently treated for 48 h with the BRAFV600E inhibitor vemurafenib or with DMSO (Figure 7A). The strong signaling relationships between these POIs and p-ERK1/2 were observed only in cells treated with vemurafenib, not in control cells treated with DMSO (Figures 7B, S7C, and S7D). This suggests that (1) the constitutive BRAF activation caused by the BRAFV600E mutation leads to strong MAPK-ERK activation that overrides the overexpression effects, and (2) POI abundance-dependent ERK signaling is independent of RAF activity. To determine whether overexpression of these candidate POIs reduced sensitivity to the BRAFV600E inhibitor vemurafenib, we assigned each single cell into one of four bins based on the abundance of GFP-tagged POI and calculated the percentage of cells in each bin relative to the total cell count (Figure 7B). As expected, the positive control cells that expressed MEK1DD-GFP had significant cell enrichment in the fourth bin (i.e., the bin with the highest expression level of the GFP-tagged POI), with vemurafenib treatment compared to the DMSO-treated control (Figure 7C). This indicates that MEK1DD overexpression contributes to cell survival. Similarly, all nine examined kinases showed an enrichment of cell abundance in the fourth bin; in six cases, this enrichment was statistically significant (Figure 7C). These results reveal that in melanoma A375 cells, the overexpression of kinases capable of ligand-independent ERK activation reduces cellular dependency on signaling inputs from BRAFV600E.
Figure 7.

Kinase Overexpression Induces Resistance to BRAF-MEK Concurrent Inhibition in Melanoma Cells by MEK-Independent ERK Reactivation
(A) Selected kinases were transfected into A375 cells and cultured in the inhibitor- or DMSO-containing media, and signaling states and cell viability were assessed.
(B) Single-cell data from each sample were divided into four bins, depending on the expression level of the GFP-tagged kinase. Signed-BP-R2 analysis was performed to quantify signaling relationships with and without vemurafenib.
(C) The proportion of cells in bin 4, cells with the highest levels of POI, for each individual overexpressed kinase in vemurafenib-treated cells and DMSO-treated cells. (∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001, n = 3).
(D and E) Overexpression-induced signaling relationships to p-ERK1/2 (D) and p-MEK1/2 (E) under the treatment conditions indicated by line colors.
(F) Illustrations of the diverse MAPK-ERK reactivation mechanisms induced by different assessed POIs and the targets of applied inhibitors, vemurafenib and CI1040.
(G) Dot plot shows signed-BP-R2 between p-MEK1/2 to p-ERK1/2 for all assessed kinases in HEK293T cells.
Three of the tested kinases, MAP3K8, MOS, and SRC, were previously suggested to mediate MAPK-ERK inhibition resistance in A375 cells (Johannessen et al., 2013). In analyzing the cell viability data in the same study, we noted that MAP3K8 and MOS only induced resistance to RAF inhibition, whereas SRC overexpression caused resistance to both RAF and MEK individual inhibitions and the concurrent RAF-MEK inhibition (Figure S7E) (Johannessen et al., 2013). We hypothesize that the difference in drug responses are due to the MEK dependency of the POI overexpression-induced ERK activation (i.e., an overexpressed POI directly activating ERK will not be influenced by upstream RAF and/or MEK inhibition). To test this, A375 cells were transfected individually with vectors encoding ABL1, BLK, FES, MAP3K2, MAP3K8, MOS, NTRK2, SRC, YES1, and MEK1DD and were treated with the BRAFV600E inhibitor vemurafenib, MEK inhibitor CI1040, or the combination of both inhibitors for 3 h. As expected, cells overexpressing MEK1DD were not sensitive to vemurafenib, whereas treatment with CI1040 or the combination treatment completely blocked ERK activation (Figures 7D and S7F). Similarly, cells that overexpressed MAP3K8, MAP3K2, or MOS were resistant to vemurafenib but sensitive to CI1040, suggesting that MEK activity is necessary for the MAPK-ERK reactivation induced by these three kinases (Figure 7D, purple boxes). Other POIs, including ABL1, BLK, FES, NTRK2, SRC, and YES1, showed abundance-dependent ERK activation, even with the combination treatment of vemurafenib and CI1040, indicating that these proteins activate ERK in an MEK-independent manner (Figures 7D and S7F).
Analysis of the POI abundance-dependent p-MEK1/2 levels confirmed that MAP3K8, MAP3K2, and MOS induced MEK1/2 activity (Figures 7E and S7F). Kinases BLK, FES, SRC, and YES1 did not cause abundance-dependent MEK1/2 phosphorylation in all conditions (Figure 7E), validating that the overexpression of these kinases did not activate MEK (Figure S7F). Abundances of both ABL1 and NTRK2 showed positive correlations with p-MEK1/2 levels only when cells were treated simultaneously with CI1040 and vemurafenib (Figure 7E). This suggests that ABL1 and NTRK2 activate both MEK and ERK. The addition of CI1040 blocked the MEK-ERK binding (Allen et al., 2003), leading to the reduced ERK activity and the ERK-MEK negative feedback, whereas the POI-induced signal inputs on MEK were constant, resulting in the increased MEK phosphorylation levels (Figure S7F). The diverse MAPK-ERK reactivation mechanisms induced by these kinases are illustrated in Figure 7F. We note that the data in our kinome and phosphatome screen with HEK293T cells were indicative of MEK dependency in MAPK-ERK reactivation, as the kinases MAP3K8, MOS, and MAP3K2 had high signaling relationship strength to p-MEK1/2 (Figure 7G). Our analysis predicted potential biomarkers of MAPK-ERK reactivation and identified a key mechanism for drug resistance in melanoma cells carrying the BRAFV600E mutation. We further classified MAPK-ERK reactivation mechanisms and revealed kinases that induce resistance to BRAF-MEK combined inhibition.
Discussion
The data described here are unique for the broad coverage of the human kinome and phosphatome, the multiplexed measurement of cellular phosphorylation states and dynamics at single-cell resolution, and the wide continuous abundance range (over three orders of magnitude) over which proteins of interest were studied. Our analyses enabled protein abundance-determined functional classification, signaling kinetics quantification, and the identification of potential biomarkers of drug resistance.
Protein abundance and mRNA expression levels of kinases and phosphatases have been quantified in normal and diseased tissues by multiple approaches (Petryszak et al., 2016, Uhlen et al., 2017, Wang et al., 2015). Our analysis, for the first time, characterized at kinome- and phosphatome-wide scope how these proteins differentially modulate signaling network behaviors when expressed over a concentration gradient. In the overexpression effect-based functional classification, we assigned kinases and phosphatases into 10 clusters that partly agreed with the kinase and phosphatase catalytic specificities, indicating the dissimilar network alterations between signaling protein overexpression and activation. Functions of these signaling proteins include non-catalytic roles such as allosteric regulation and scaffolding (Kung and Jura, 2016). We showed that our unique analysis is able to capture these non-catalytic effects (Figure S3B). This finding is also highly relevant in cancer therapeutics in that targeting the catalytic function of a kinase may not affect the deregulated signaling caused by abundance changes. Future work with a kinome- and phosphatome-scale catalytically inactive mutant library would allow global characterization of signaling protein non-catalytic effects.
Phosphatase overexpression can drive tumor progression, but the underlying signaling mechanisms have been unclear (Julien et al., 2011). Our data indicate that rather than directly activating a cancer-driving signaling pathway, the overexpression of ERK-specific phosphatases modulates signaling dynamics, resulting in a prolonged proliferative signal in cells. This supports the suggestion that phosphatases should be considered therapeutic targets for cancer treatment (Bollu et al., 2017, Julien et al., 2011, Low and Zhang, 2016).
The MAPK-ERK pathway reactivation induced by protein overexpression is one cause of drug resistance to BRAF inhibitors in melanoma (Johannessen et al., 2010). Therapies that concurrently inhibit BRAF and MEK have been developed to simultaneously target the active BRAF signal and the MAPK-ERK reactivation signal. However, drug resistance still occurs (Carlino et al., 2014, Eroglu and Ribas, 2016). In our kinome and phosphatome analysis, we discovered a group of tyrosine kinases, including SRC, FES, YES1, and BLK, that led to hyper-ERK activation independent of MEK activity, suggesting a mechanism underlying drug resistance to the combined BRAF and MEK inhibition in melanoma patients with BRAF mutations. The identified kinases could be used as biomarkers to predict the drug response to BRAF-MEK combined inhibition and to screen for patients to be treated with alternative therapies. Compared to previous population-based assays (Johannessen et al., 2010, Johannessen et al., 2013), our screening method is more sensitive and robust in identifying drug resistance-related protein overexpression, as it assesses signaling variances over a large POI range and can be applied to highly heterogeneous samples.
Our analysis has several limitations. First, the measured effects of overexpression may be indirect; for example, protein overexpression may lead to cellular stress that activates MAPK-p38 or MAPK-JNK cascades. However, even if indirect, these signaling responses may be typical of such overexpression in diseased conditions. Second, our mass-cytometry-based analysis used antibodies targeting 30 specific phosphorylation sites. This antibody panel does, however, cover the most critical and informative phosphorylation sites known to be involved in the cancer-related signaling network. Third, GFP-tag can disrupt the localization of a kinase or phosphatase. In a previous study, we cross-validated our results with multiple protein tagging systems and showed that perturbations on overexpression effects due to the tag were rare (Lun et al., 2017). Fourth, the catalytic functionality of many phosphatases requires the co-presence of a phosphatase catalytic subunit and a phosphatase regulatory subunit (Chen et al., 2017). Individually overexpressing one of these subunits may not result in phosphatase activation; rather, it may affect the kinetics of related dephosphorylating reaction in cells, and this is what we characterized in the present study.
In summary, we demonstrated, in a human kinome- and phosphatome-scale analysis, how the overexpression of individual signaling proteins modulates signaling networks in an abundance-dependent manner and how the provided datasets can reveal biological insights underlying diseased conditions. Our data established that protein expression levels can result in different signaling states in a population of cells treated identically. Our analysis expands the functional classification of the human kinases and phosphatases and suggests 208 signaling relationships that can be interrogated to improve our understanding of signaling causality and network structure. Our data are also suitable for the inference of signaling pathway kinetics using mathematical models and for the development of network reconstruction methods.
STAR★Methods
Key Resources Table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| p-CREB/ATF1 (Ser133 of CREB/Ser63 of ATF1), Clone J151-21 | BD Biosciences | Cat# 558436; RRID: AB_647204 |
| p-STAT5 (Tyr694), Clone 47/Stat5 | BD Biosciences | Cat# 612567; RRID: AB_399858 |
| p-SRC (Tyr418), Clone SC1T2M3 | eBioscience | Cat# 14-9034-82; RRID: AB_2572916 |
| p-FAK (Tyr397), Polyclonal | Cell Signaling Technology | Cat# 3283; RRID: AB_2173659 |
| p-MEK1/2 (Ser221), Clone 166F8 | Cell Signaling Technology | Cat# 2338; RRID: AB_490903 |
| p-MAPKAPK2 (Thr334), Clone 27B7 | Cell Signaling Technology | Cat# 3007; RRID: AB_490936 |
| p-p70S6K (Thr389), Clone 1A5 | Cell Signaling Technology | Cat# 9206; RRID: AB_2285392 |
| p-MKK4 (Ser257/Thr261), Clone C36C11 | Cell Signaling Technology | Cat# 4514; RRID: AB_2140946 |
| p-STAT1 (Ser727), Polyclonal | Cell Signaling Technology | Cat# 9177; RRID: AB_2197983 |
| p-p53 (Ser15), Clone 16G8 | Cell Signaling Technology | Cat# 4030; RRID: AB_10694347 |
| p-NFκB (Ser529), Clone K10-895.12.50 | BD Biosciences | Cat# 558393; RRID: AB_647284 |
| p-p38 (Thr180/Tyr182), Clone 36/p38 | BD Biosciences | Cat# 612289; RRID: AB_399606 |
| p-AMPKα (Thr172), Clone 40H9 | Cell Signaling Technology | Cat# 5256; RRID: AB_10705605 |
| p-AKT (Ser473), Clone D9E | Cell Signaling Technology | Cat# 5012; RRID: AB_2224726 |
| p-ERK1/2 (Thr202/Tyr204), Clone 20A | BD Biosciences | Cat# 612359; RRID: AB_399648 |
| p-MARCKS (Ser167/170), Clone D13E4 | Cell Signaling Technology | Cat# 8722; RRID: AB_10999091 |
| cyclin B1, Clone GNS-11 | BD Biosciences | Cat# 554178; RRID: AB_395289 |
| p-GSK3β (Ser9), Clone D85E12 | Cell Signaling Technology | Cat# 5558; RRID: AB_10013750 |
| GAPDH, Clone 6C5 | Thermo Fisher Scientific | Cat# AM4300; RRID: AB_2536381 |
| p-MKK3/6 (Ser189 of MKK3/Ser207 of MKK6), Clone D8E9 | Cell Signaling Technology | Cat# 12280 |
| p-PDK1 (Ser241), Clone J66-653.44.22 | BD Biosciences | Cat# 558395; RRID: AB_647291 |
| p-BTK/ITK (Tyr551 of BTK/Tyr551 of ITK), Clone 24a/BTK | BD Biosciences | Cat# 558034; RRID: AB_2067823 |
| p-p90RSK (Ser380), Clone D5D8 | Cell Signaling Technology | Cat# 12032 |
| p-SMAD2/3 (Ser465/467 of SMAD2/Ser423/425 of SMAD3), Clone D27F4 | Cell Signaling Technology | Cat# 8828; RRID: AB_2631089 |
| β-catenin (Non-phospho Ser33/37/Thr41), Clone D13A1 | Cell Signaling Technology | Cat# 8814; RRID: AB_11127203 |
| p-STAT3 (Tyr705), Clone 4/P-STAT3 | BD Biosciences | Cat# 612356; RRID: AB_399645 |
| p-JNK (Thr183/Tyr185), Clone G9 | Cell Signaling Technology | Cat# 9255; RRID: AB_2307321 |
| p-PLCγ2 (Tyr759), Clone K86-689.37 | BD Biosciences | Cat# 558490; RRID: AB_647226 |
| GFP, Clone FM264G | BioLegend | Cat# 338002; RRID: AB_1279414 |
| p-HH3 (Ser28), Clone HTA28 | BioLegend | Cat# 641002; RRID: AB_1227659 |
| p-S6 (Ser235/Ser236), Clone N7-548 | BD Biosciences | Custom made |
| cleaved PARP, Clone F21-852 | BD Biosciences | Cat# 552596; RRID: AB_394437 |
| p-mTOR (Ser2448), Clone D9C2 | Cell Signaling Technology | Cat# 5536; RRID: AB_10691552 |
| p-c-RAF (Ser259), Clone Polyclonal | Thermo Fisher Scientific | Cat# 44-502; RRID: AB_2533669 |
| p-RB (Ser807/811), Clone D20B12 | Cell Signaling Technology | Cat# 8516; RRID: AB_11178658 |
| p-4EBP1 (Thr37/46), Clone 236B4 | Cell Signaling Technology | Cat# 2855; RRID: AB_560835 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15710 |
| Maleimido mono amide DOTA (mDOTA) | Macrocyclics | Cat# B-272 |
| Iridium | Fluidigm | Cat# 201192A |
| Maxpar X8 Multimetal labeling kit | Fluidigm | Cat# 201300 |
| Lanthanide (III) metal isotopes as chloride salts | Fluidigm | N/A |
| jetPRIME | PolyPlus | Cat# 114-15 |
| X-treme GENE HP | Roche | Cat# 6366236001 |
| Recombinant Murine EGF | Peprotech | Cat# 315-09 |
| vemurafenib | Selleckchem | Cat# S1267 |
| CI1040 | Selleckchem | Cat# S1020 |
| Deposited Data | ||
| Integrated raw data and preprocessed data | This paper | https://doi.org/10.17632/3kh7ypz232.1 |
| Experimental Models: Cell Lines | ||
| HEK293T | ATCC | Cat# CRL-3216, RRID:CVCL_0063 |
| A375 | A gift from Dr. Lukas Sommer | N/A |
| Recombinant DNA | ||
| The human kinase library plasmid kit | Addgene | Kit# 1000000014 |
| ORFeome Human Entry Collection Phosphatase | Dharmacon | Cat# OHS4941 |
| pDEST pcDNA5 FRT TO-eGFP | A gift from Dr. Anne-Claude Gingras | N/A |
| pDEST 3′ Triple Flag pcDNA5 FRT TO | A gift from Dr. Anne-Claude Gingras | N/A |
| pFLAG-CMV-hErk1 (K71R) | Addgene | Plasmid # 49329 |
| pCIG AKT3 (K177M) | Addgene | Plasmid # 73051 |
| pMCL-HA-MAPKK1-8E (K97M) | Addgene | Plasmid # 40811 |
| IRES-GFP-AXL-KD (K567R) | Addgene | Plasmid # 65498 |
| FLAG.PKCepsilon.K/W (K437W) | Addgene | Plasmid # 10796 |
| Software and Algorithms | ||
| Cytobank | Cytobank | https://www.cytobank.org/ |
| Concatenation tool | Cytobank | https://support.cytobank.org/hc/en-us/articles/206336147-FCS-file-concatenation-tool |
| Normalizer | Finck et al., 2013 | https://github.com/nolanlab/bead-normalization/releases |
| Single cell debarcoder | Zunder et al., 2015 | https://github.com/nolanlab/single-cell-debarcoder |
| BP-R2 analysis | Lun et al., 2017 | https://github.com/BodenmillerGroup/Adnet |
| t-SNE | van der Maaten and Hinton, 2008 | https://github.com/jkrijthe/Rtsne |
| STRING | Szklarczyk et al., 2017 | https://string-db.org/ |
| OmniPath | Türei et al., 2016 | http://omnipathdb.org/ |
| Shape-based clustering | Genolini et al., 2015 | R package ‘kml’ |
Contact for Reagent and Resource Sharing
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Bernd Bodenmiller (bernd.bodenmiller@imls.uzh.ch).
Experimental Model and Subject Details
HEK293T cells, obtained from ATCC, were cultured in DMEM (D5671, SIGMA), supplemented with 10% FBS, 2 mM L-glutamine, 100 U/ml penicillin, and 100 μg/ml streptomycin. A375 cells, a gift from Dr. Lukas Sommer at University of Zürich, were cultured in RPMI Medium 1640 (21875-034, GIBCO) with 10% FBS, 100 U/ml penicillin, and 100 μg/ml streptomycin. For cell passaging or harvesting, HEK293T cells and A375 cells were incubated with 1X TrypLE Express (Life Technologies) at 37°C for 2 min and 5 min, respectively.
Method Details
Cloning
The human kinase library plasmid kit, containing open reading frames (ORFs) in Gateway Entry vectors, was provided by William Hahn and David Root (Johannessen et al., 2010, Yang et al., 2011) via Addgene (Kit # 1000000014). The human phosphatase library was obtained from Dharmacon (OHS4941, ORFeome Human Entry Collection Phosphatase). Destination vectors, including pDEST pcDNA5 FRT TO-eGFP and pDEST 3′ Triple Flag pcDNA5 FRT TO, were kindly provided by Dr. Anne-Claude Gingras at Mount Sinai Hospital, Toronto, Canada (Couzens et al., 2013). Expression vectors encoding the FLAG- or GFP-tagged fusion proteins were generated via Gateway Cloning and sequenced before transfection. Vectors for kinase-dead mutants, including pFLAG-CMV-hErk1 (K71R) (Addgene plasmid # 49329), pCIG AKT3 (K177M) (Addgene plasmid # 73051), pMCL-HA-MAPKK1-8E (K97M) (Addgene plasmid # 40811), IRES-GFP-AXL-KD (K567R) (Addgene plasmid # 65498), and FLAG.PKCepsilon.K/W (K437W) (Addgene plasmid # 10796) were gifts from Melanie Cobb, Joseph Gleeson, Natalie Ahn, Aaron Meyer, and Alex Toker, respectively (Baek et al., 2015, Cenni et al., 2002, Meyer et al., 2015, O’Neil et al., 1990).
HEK293T cell transfection and stimulation
HEK293T cells were seeded at the density of 0.7 million per well in 6-well plates. After 24 h, cells were transfected with 2 μg plasmid and 4 μL of jetPRIME (PolyPlus) per well with the standard protocol provided by the manufacturer. For kinase and phosphatase double transfection experiments, cells were transfected with plasmids encoding GFP-tagged kinases and FLAG-tagged phosphatases 16 h and 24 h after seeding, respectively. Half the amounts of plasmid and jetPRIME were used in each round for co-overexpression experiments. At 18 h after transfection, EGF (Peprotech) was added to a final concentration of 100 ng/ml. At 20 min before a given EGF stimulation time point, 5-iodo-deoxycytidine (IdU) was added to the medium at the final concentration of 10 μM. At 2 min before a given EGF stimulation time point, medium was replaced by 1X TrypLE to induce cell detachment. At the time point, paraformaldehyde (PFA, from Electron Microscopy Sciences) was added to the cell suspension to a final percentage of 1.6%, and cells were incubated at room temperature for 10 min. If EGF stimulation was not necessary in the experiment, cells were directly harvested and crosslinked with PFA.
A375 cell transfection
A375 cells were seeded at the density of 0.15 million per well in 6-well plates. At 24 h after seeding, transfection was performed using 2 μg plasmid and 4 μL of X-treme GENE HP reagent (06 366 236 001, Roche) per well with the standard protocol provided by the manufacturer.
Kinase inhibition with small molecules
BRAFV600E inhibitor vemurafenib and MEK inhibitor CI1040 (stock solutions of 10 mM/mL in DMSO, Selleckchem) were added to the cells 18 h after transfection independently or in combination at final concentrations of 1 μM and 10 μM, respectively. The same volume of DMSO was added to the control samples. Cells were treated either for 3 h or for 2 days. At the end of the treatment, cells were labeled with IdU during 20-minute incubation and subsequently harvested by 5-minute TrypLE digestion and 10-minute PFA crosslinking as described above.
Methanol permeabilization
Crosslinked cells were washed twice with cell staining media (CSM, PBS with 0.5% BSA). After centrifugation, ice-cold methanol was used to resuspend the cells, followed by 10-minute permeabilization on ice or for long-term storage at −80°C.
Antibody conjugation
The MaxPAR antibody conjugation kit (Fluidigm) was used to generate isotope-labeled antibodies using the manufacturer’s standard protocol. After conjugation, the antibody yield was determined based on absorbance of 280 nm. Candor PBS Antibody Stabilization solution (Candor Bioscience GmbH) was used to dilute antibodies for long-term storage at 4°C.
Barcoding and staining protocol
Formalin-crosslinked and methanol-permeabilized cells were washed three times with CSM and once with PBS. Cells were incubated in PBS containing barcoding reagents of 89Y (100 nM), 103Rh (2 μM), 105Pd (100 nM), 106Pd (100 nM), 108Pd (100 nM), 110Pd (100 nM), 113In (200 nM), 115In (100 nM), and 209Bi (20 nM) for 30 min at room temperature and then washed three times with CSM. Barcoded cells were then pooled and stained with the metal-conjugated antibody mix (Table S2) at room temperature for 1 h. The antibody mix was removed by washing cells three times with CSM and once with PBS. For DNA staining, iridium-containing intercalator (Fluidigm) diluted in PBS with 1.6% PFA was incubated with the cells at 4°C overnight. On the day of the measurement, the intercalator solution was removed, and cells were washed with CSM, PBS, and ddH2O. After the last washing step, cells were resuspended in ddH2O and filtered through a 70-μm strainer.
Mass cytometry analysis
EQ Four Element Calibration Beads (Fluidigm) were added to cell suspensions in a 1:10 ratio (v/v). Samples were analyzed on a CyTOF2 (Fluidigm). The manufacturer’s standard operation procedures were used for acquisition at a cell rate of ∼500 cells per second. After the acquisition, all FCS files from the same barcoded sample were concatenated (Bodenmiller et al., 2012). Data were then normalized, and bead events were removed (Finck et al., 2013) before doublet removal and de-barcoding of cells into their corresponding wells using a doublet-filtering scheme and single-cell deconvolution algorithm (Zunder et al., 2015). Subsequently, data were processed using Cytobank (https://www.cytobank.org). Additional gating on the DNA channels (191Ir and 193Ir) and 139La/141Pr was used to remove remained doublets, debris and contaminating particulate.
Quantification and Statistical Analysis
Data preprocessing and BP-R2 analysis
Data preprocessing
Raw data were transformed using the inverse hyperbolic sine transform with a cofactor of 5:
Except where use of raw data values is specifically noted, all visualizations and analyses were performed using transformed data.
Data binning
For data binning, the range between the lower and upper 2.5% of observations was divided into ten equal bins bin1,…,bin10. The observations in the lower and upper 2.5% were assigned to the lowest and highest bins, respectively. In order to be able to compare expression levels between samples within a time course experiment, all observations of the time course were pooled to determine the binning.
BP-R2
BP-R2 analysis is described in Lun et al., 2017 (https://github.com/BodenmillerGroup/Adnet). In brief, the median of a measured marker was calculated for each bin . Additionally, the overall mean of the medians of all the 10 bins () was calculated (bins with less than 25 cells were discarded). Then, for each bin, we computed the sum of squared deviations from the bin medians and the sum of squared deviations from the overall mean of medians. These values were summed over all bins and the BP-R2 was defined as one minus the ratio between them:
Threshold determination
Following the method described in Lun et al., 2017, we chose the maximum BP-R2 among all the 108 control samples (FLAG-GFP overexpression and untransfected cells) as a cutoff. Relationships that had a BP-R2 higher than this threshold were considered as sufficiently strong to be of interest.
Signed-BP-R2
The relationship strengths calculated as BP-R2 were mostly positive, with a few exceptions of negative BP-R2 values mostly from the cell cycle marker IdU, due to bimodality. These rare and weak negative BP-R2 values were considered as negligible and were therefore assigned to 0. This allowed the integration of signaling relationship directionalities, determined by Spearman correlation of bin medians (), with the relationship strengths (). The signed-BP-R2 score was calculated as:
Hierarchical clustering
Hierarchical clustering was performed for kinases and phosphatases on their abundance-dependent signaling relationships, as signed-BP-R2, to all measured phosphorylation sites with and without 10-minute EGF stimulation. Ward’s method and Euclidean distances (Ward, 1963) were used for the clustering, and the hierarchical tree was cut at the height of 5 to obtain 10 clusters of kinases and phosphatases as shown in Figure S2A.
t-SNE analysis
t-SNE analysis was performed with the Package ‘Rtsne’ in R.
Functional enrichment and association analysis using STRING database
The functional enrichment and interaction enrichment analyses were performed using the STRING database v10.5 (Szklarczyk et al., 2017). All the kinases and phosphates tested were mapped to STRING protein name-space establishing the background protein set for the further analysis. The functional enrichment p values were corrected using Benjamini and Hochberg method (Benjamini and Hochberg, 1995) (the detailed description of the statistical methods can be found in Franceschini et al., 2013). To test whether the functional signal within the clusters arises exclusively from a homology between the proteins, the homologous proteins were grouped together into one node, and, therefore, the proteins that exhibited high or medium homology did not contribute independently to the enrichment functional term count. In order to form the grouped representation of the STRING network the single-linkage clustering method was applied to the homology relationships between the proteins in which neighbors were defined as having a self-normalized bit score (BLAST bit score of alignment between the two proteins divided by the bit score of self-alignment of shorter of the two proteins) equal to or higher than 0.2. For each functional term the grouped node contributed to the enrichment count when one or more of the proteins forming the group were annotated with the term in question. This process was applied to both the clusters and the background separately to ensure that for the groups in which proteins were shared between the cluster and the background the functional term was counted in both sets.
Shortest signed directed path analysis using OmniPath
The pathway analysis was performed for all signaling relationships between overexpressed POIs and measured phosphorylation sites using OmniPath (http://omnipathdb.org/), a collection of literature curated human signaling pathways integrated from 25 databases (pathway databases: TRIP, SPIKE, SignaLink3, Guide2Pharma, CA1, ARN, NRF2ome, Macrophage, DeathDomain, PDZBase, Signor; interaction databases: BioGRID, CancerCellMap, MPPI, DIP, InnateDB, MatrixDB; PTM databases: PhosphoSite, DEPOD, LMPID, phosphoELM, ELM, DOMINO, dbPTM, HPRD-phos) (Türei et al., 2016). The shortest path was determined based on based on Breadth-First Search methods, computed through a Python module called pyPath (Türei et al., 2016).
Shape-based clustering
For each signaling relationship between a phosphorylation site and an overexpressed protein, the median phosphorylation abundance in each pre-defined bin was calculated using arcsinh transformed data. K-means shape-based clustering was performed with the package ‘kml’ (Genolini et al., 2015) in R for all strong POI abundance-dependent signaling relationships after 0-1 normalization on phosphorylation abundance (in Figure 1D), or for signaling trajectories over 1-hour EGF stimulation time course without data normalization (in Figure S6). Euclidean distance was used as similarity measure.
Selection of strong signaling dynamic influencing POIs
For each pair of signaling relationships between an overexpressed POI and a measured phosphorylation site, the delta BP-R2 score was calculated as the signed-BP-R2 value with 10-minute EGF stimulation minus the signed-BP-R2 value in unstimulated cells. We selected the 10 POIs with the largest positive differences in signed-BP-R2, the 10 POIs with the largest negative difference in signed-BP-R2, the 20 POIs with the most signaling relationships in the 99th percentile of the difference in signed-BP-R2, and the 10 central signaling dynamic regulators in the MAPK/ERK and AKT pathways known from the literature (Steelman et al., 2011). Some POIs were in more than one set, so this resulted in 39 kinases and phosphatases.
Signaling amplitudes analysis
The signaling amplitudes analysis was adapted from our previous methods (Lun et al., 2017). The fold change of median phosphorylation abundance for each bin in EGF-stimulated samples over the corresponding bin of the unstimulated sample (EGF 0 min) was calculated using the raw count. The amplitude for each bin was identified as the maximal fold change over all time points. Amplitude ratios between the second highest and the second lowest bin amplitudes were computed for all samples, and the highest amplitude ratio in all FLAG-GFP overexpression and untransfected controls was used to determine the cutoff for robust and strong abundance-dependent changes.
Data and Software Availability
All raw data and pre-analyzed data are available at Mendeley Data under the following link:
Acknowledgments
We would like to thank the Bodenmiller lab for support and fruitful discussions, the Sommer lab for sharing experimental materials, the Lehner lab and the Mosimann lab for sharing equipment, and Dr. Vinko Tosevski and Dr. Tess Brodie at the Mass Cytometry Facility, University of Zürich, for support and troubleshooting help. We would especially like to thank Dr. A.-C. Gingras, Lunenfeld-Tanenbaum Research Institute, for sharing the pDEST vectors used in this study. This work was supported by a Swiss National Science Foundation (SNSF) R’Equip grant, an SNSF Assistant Professorship grant (PP00P3-144874), the European Research Council (ERC) under the European Union’s Seventh Framework Program (FP/2007-2013)/ERC Grant Agreement no. 336921, and an NIH grant (UC4 DK108132).
Author Contributions
X.-K.L. and B.B. conceived the study. X.-K.L. performed the experiments, the data processing, and the data analysis. D.S. performed the functional enrichment and functional association analysis. A.G. performed the OmniPath analysis. N.D. cloned the phosphatase expression library. V.R.T.Z. implemented the BP-R2 platform and helped with the data analysis. X.-K.L., D.S., A.G., N.D., V.R.T.Z., J.S.-R., C.v.M., and B.B. performed the biological interpretation. X.-K.L. and B.B. wrote the manuscript with input from all of the authors.
Declaration of Interests
The authors declare no competing interests.
Published: May 14, 2019
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.molcel.2019.04.021.
Supplemental Information
References
- Abbas T., Keaton M.A., Dutta A. Genomic instability in cancer. Cold Spring Harb. Perspect. Biol. 2013;5:a012914. doi: 10.1101/cshperspect.a012914. [DOI] [PMC free article] [PubMed] [Google Scholar]; Abbas, T., Keaton, M.A., and Dutta, A. (2013). Genomic instability in cancer. Cold Spring Harb. Perspect. Biol. 5, a012914. [DOI] [PMC free article] [PubMed]
- Allen L.F., Sebolt-Leopold J., Meyer M.B. CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK) Semin. Oncol. 2003;30(5, Suppl 16):105–116. doi: 10.1053/j.seminoncol.2003.08.012. [DOI] [PubMed] [Google Scholar]; Allen, L.F., Sebolt-Leopold, J., and Meyer, M.B. (2003). CI-1040 (PD184352), a targeted signal transduction inhibitor of MEK (MAPKK). Semin. Oncol. 30(5, Suppl 16) 105-116. [DOI] [PubMed]
- Baek S.T., Copeland B., Yun E.-J., Kwon S.-K., Guemez-Gamboa A., Schaffer A.E., Kim S., Kang H.-C., Song S., Mathern G.W., Gleeson J.G. An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development. Nat. Med. 2015;21:1445–1454. doi: 10.1038/nm.3982. [DOI] [PMC free article] [PubMed] [Google Scholar]; Baek, S.T., Copeland, B., Yun, E.-J., Kwon, S.-K., Guemez-Gamboa, A., Schaffer, A.E., Kim, S., Kang, H.-C., Song, S., Mathern, G.W., and Gleeson, J.G. (2015). An AKT3-FOXG1-reelin network underlies defective migration in human focal malformations of cortical development. Nat. Med. 21, 1445-1454. [DOI] [PMC free article] [PubMed]
- Barrios-Rodiles M., Brown K.R., Ozdamar B., Bose R., Liu Z., Donovan R.S., Shinjo F., Liu Y., Dembowy J., Taylor I.W. High-Throughput Mapping of a Dynamic Signaling Network in Mammalian Cells. Science. 2005;307:1621–1625. doi: 10.1126/science.1105776. [DOI] [PubMed] [Google Scholar]; Barrios-Rodiles, M., Brown, K.R., Ozdamar, B., Bose, R., Liu, Z., Donovan, R.S., Shinjo, F., Liu, Y., Dembowy, J., Taylor, I.W., et al. (2005). High-Throughput Mapping of a Dynamic Signaling Network in Mammalian Cells. Science 307, 1621-1625. [DOI] [PubMed]
- Bendall S.C., Simonds E.F., Qiu P., Amir A.D., Krutzik P.O., Finck R., Bruggner R.V., Melamed R., Trejo A., Ornatsky O.I. Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science. 2011;332:687–696. doi: 10.1126/science.1198704. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bendall, S.C., Simonds, E.F., Qiu, P., Amir, A.D., Krutzik, P.O., Finck, R., Bruggner, R.V., Melamed, R., Trejo, A., Ornatsky, O.I., et al. (2011). Single-cell mass cytometry of differential immune and drug responses across a human hematopoietic continuum. Science 332, 687-696. [DOI] [PMC free article] [PubMed]
- Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B. 1995;57:289–300. [Google Scholar]; Benjamini, Y., and Hochberg, Y. (1995). Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. B 57, 289-300.
- Bodenmiller B., Wanka S., Kraft C., Urban J., Campbell D., Pedrioli P.G., Gerrits B., Picotti P., Lam H., Vitek O. Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci. Signal. 2010;3:rs4. doi: 10.1126/scisignal.2001182. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bodenmiller, B., Wanka, S., Kraft, C., Urban, J., Campbell, D., Pedrioli, P.G., Gerrits, B., Picotti, P., Lam, H., Vitek, O., et al. (2010). Phosphoproteomic analysis reveals interconnected system-wide responses to perturbations of kinases and phosphatases in yeast. Sci. Signal. 3, rs4. [DOI] [PMC free article] [PubMed]
- Bodenmiller B., Zunder E.R., Finck R., Chen T.J., Savig E.S., Bruggner R.V., Simonds E.F., Bendall S.C., Sachs K., Krutzik P.O., Nolan G.P. Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat. Biotechnol. 2012;30:858–867. doi: 10.1038/nbt.2317. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bodenmiller, B., Zunder, E.R., Finck, R., Chen, T.J., Savig, E.S., Bruggner, R.V., Simonds, E.F., Bendall, S.C., Sachs, K., Krutzik, P.O., and Nolan, G.P. (2012). Multiplexed mass cytometry profiling of cellular states perturbed by small-molecule regulators. Nat. Biotechnol. 30, 858-867. [DOI] [PMC free article] [PubMed]
- Bollu L.R., Mazumdar A., Savage M.I., Brown P.H. Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clin. Cancer Res. 2017;23:2136–2142. doi: 10.1158/1078-0432.CCR-16-0934. [DOI] [PMC free article] [PubMed] [Google Scholar]; Bollu, L.R., Mazumdar, A., Savage, M.I., and Brown, P.H. (2017). Molecular Pathways: Targeting Protein Tyrosine Phosphatases in Cancer. Clin. Cancer Res. 23, 2136-2142. [DOI] [PMC free article] [PubMed]
- Breitkreutz A., Choi H., Sharom J.R., Boucher L., Neduva V., Larsen B., Lin Z.-Y., Breitkreutz B.-J., Stark C., Liu G. A global protein kinase and phosphatase interaction network in yeast. Science. 2010;328:1043–1046. doi: 10.1126/science.1176495. [DOI] [PMC free article] [PubMed] [Google Scholar]; Breitkreutz, A., Choi, H., Sharom, J.R., Boucher, L., Neduva, V., Larsen, B., Lin, Z.-Y., Breitkreutz, B.-J., Stark, C., Liu, G., et al. (2010). A global protein kinase and phosphatase interaction network in yeast. Science 328, 1043-1046. [DOI] [PMC free article] [PubMed]
- Burgermeister E., Seger R. PPARgamma and MEK Interactions in Cancer. PPAR Res. 2008;2008:309469. doi: 10.1155/2008/309469. [DOI] [PMC free article] [PubMed] [Google Scholar]; Burgermeister, E., and Seger, R. (2008). PPARgamma and MEK Interactions in Cancer. PPAR Res. 2008, 309469. [DOI] [PMC free article] [PubMed]
- Carlino M.S., Todd J.R., Gowrishankar K., Mijatov B., Pupo G.M., Fung C., Snoyman S., Hersey P., Long G.V., Kefford R.F., Rizos H. Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol. Oncol. 2014;8:544–554. doi: 10.1016/j.molonc.2014.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]; Carlino, M.S., Todd, J.R., Gowrishankar, K., Mijatov, B., Pupo, G.M., Fung, C., Snoyman, S., Hersey, P., Long, G.V., Kefford, R.F., and Rizos, H. (2014). Differential activity of MEK and ERK inhibitors in BRAF inhibitor resistant melanoma. Mol. Oncol. 8, 544-554. [DOI] [PMC free article] [PubMed]
- Cenni V., Döppler H., Sonnenburg E.D., Maraldi N., Newton A.C., Toker A. Regulation of novel protein kinase C epsilon by phosphorylation. Biochem. J. 2002;363:537–545. doi: 10.1042/0264-6021:3630537. [DOI] [PMC free article] [PubMed] [Google Scholar]; Cenni, V., Doppler, H., Sonnenburg, E.D., Maraldi, N., Newton, A.C., and Toker, A. (2002). Regulation of novel protein kinase C epsilon by phosphorylation. Biochem. J. 363, 537-545. [DOI] [PMC free article] [PubMed]
- Chaube B., Malvi P., Singh S.V., Mohammad N., Viollet B., Bhat M.K. AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discov. 2015;1:15063. doi: 10.1038/cddiscovery.2015.63. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chaube, B., Malvi, P., Singh, S.V., Mohammad, N., Viollet, B., and Bhat, M.K. (2015). AMPK maintains energy homeostasis and survival in cancer cells via regulating p38/PGC-1α-mediated mitochondrial biogenesis. Cell Death Discov. 1, 15063. [DOI] [PMC free article] [PubMed]
- Chen M.J., Dixon J.E., Manning G. Genomics and evolution of protein phosphatases. Sci. Signal. 2017;10:eaag1796. doi: 10.1126/scisignal.aag1796. [DOI] [PubMed] [Google Scholar]; Chen, M.J., Dixon, J.E., and Manning, G. (2017). Genomics and evolution of protein phosphatases. Sci. Signal. 10, eaag1796. [DOI] [PubMed]
- Chevrier S., Levine J.H., Zanotelli V.R.T., Silina K., Schulz D., Bacac M., Ries C.H., Ailles L., Jewett M.A.S., Moch H. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell. 2017;169:736–749.e18. doi: 10.1016/j.cell.2017.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]; Chevrier, S., Levine, J.H., Zanotelli, V.R.T., Silina, K., Schulz, D., Bacac, M., Ries, C.H., Ailles, L., Jewett, M.A.S., Moch, H., et al. (2017). An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell 169, 736-749.e18. [DOI] [PMC free article] [PubMed]
- Couzens A.L., Knight J.D.R., Kean M.J., Teo G., Weiss A., Dunham W.H., Lin Z.-Y., Bagshaw R.D., Sicheri F., Pawson T. Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal. 2013;6:rs15. doi: 10.1126/scisignal.2004712. [DOI] [PubMed] [Google Scholar]; Couzens, A.L., Knight, J.D.R., Kean, M.J., Teo, G., Weiss, A., Dunham, W.H., Lin, Z.-Y., Bagshaw, R.D., Sicheri, F., Pawson, T., et al. (2013). Protein interaction network of the mammalian Hippo pathway reveals mechanisms of kinase-phosphatase interactions. Sci. Signal. 6, rs15. [DOI] [PubMed]
- Creixell P., Schoof E.M., Simpson C.D., Longden J., Miller C.J., Lou H.J., Perryman L., Cox T.R., Zivanovic N., Palmeri A. Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell. 2015;163:202–217. doi: 10.1016/j.cell.2015.08.056. [DOI] [PMC free article] [PubMed] [Google Scholar]; Creixell, P., Schoof, E.M., Simpson, C.D., Longden, J., Miller, C.J., Lou, H.J., Perryman, L., Cox, T.R., Zivanovic, N., Palmeri, A., et al. (2015). Kinome-wide decoding of network-attacking mutations rewiring cancer signaling. Cell 163, 202-217. [DOI] [PMC free article] [PubMed]
- De Vriendt V., De Roock W., Di Narzo A.F., Tian S., Biesmans B., Jacobs B., Budinska E., Sagaert X., Rossi S., D’Ario G. DUSP 4 expression identifies a subset of colorectal cancer tumors that differ in MAPK activation, regardless of the genotype. Biomarkers. 2013;18:516–524. doi: 10.3109/1354750X.2013.819038. [DOI] [PubMed] [Google Scholar]; De Vriendt, V., De Roock, W., Di Narzo, A.F., Tian, S., Biesmans, B., Jacobs, B., Budinska, E., Sagaert, X., Rossi, S., D’Ario, G., et al. (2013). DUSP 4 expression identifies a subset of colorectal cancer tumors that differ in MAPK activation, regardless of the genotype. Biomarkers 18, 516-524. [DOI] [PubMed]
- Du W., Elemento O. Cancer systems biology: embracing complexity to develop better anticancer therapeutic strategies. Oncogene. 2015;34:3215–3225. doi: 10.1038/onc.2014.291. [DOI] [PubMed] [Google Scholar]; Du, W., and Elemento, O. (2015). Cancer systems biology: embracing complexity to develop better anticancer therapeutic strategies. Oncogene 34, 3215-3225. [DOI] [PubMed]
- Duan Y., Wong W., Chua S.C., Wee H.L., Lim S.G., Chua B.T., Ho H.K. Overexpression of Tyro3 and its implications on hepatocellular carcinoma progression. Int. J. Oncol. 2016;48:358–366. doi: 10.3892/ijo.2015.3244. [DOI] [PubMed] [Google Scholar]; Duan, Y., Wong, W., Chua, S.C., Wee, H.L., Lim, S.G., Chua, B.T., and Ho, H.K. (2016). Overexpression of Tyro3 and its implications on hepatocellular carcinoma progression. Int. J. Oncol. 48, 358-366. [DOI] [PubMed]
- Eid S., Turk S., Volkamer A., Rippmann F., Fulle S. KinMap: a web-based tool for interactive navigation through human kinome data. BMC Bioinformatics. 2017;18:16. doi: 10.1186/s12859-016-1433-7. [DOI] [PMC free article] [PubMed] [Google Scholar]; Eid, S., Turk, S., Volkamer, A., Rippmann, F., and Fulle, S. (2017). KinMap: a web-based tool for interactive navigation through human kinome data. BMC Bioinformatics 18, 16. [DOI] [PMC free article] [PubMed]
- Eralp Y., Derin D., Ozluk Y., Yavuz E., Guney N., Saip P., Muslumanoglu M., Igci A., Kücücük S., Dincer M. MAPK overexpression is associated with anthracycline resistance and increased risk for recurrence in patients with triple-negative breast cancer. Ann. Oncol. 2008;19:669–674. doi: 10.1093/annonc/mdm522. [DOI] [PubMed] [Google Scholar]; Eralp, Y., Derin, D., Ozluk, Y., Yavuz, E., Guney, N., Saip, P., Muslumanoglu, M., Igci, A., Kucucuk, S., Dincer, M., et al. (2008). MAPK overexpression is associated with anthracycline resistance and increased risk for recurrence in patients with triple-negative breast cancer. Ann. Oncol. 19, 669-674. [DOI] [PubMed]
- Eroglu Z., Ribas A. Combination therapy with BRAF and MEK inhibitors for melanoma: latest evidence and place in therapy. Ther. Adv. Med. Oncol. 2016;8:48–56. doi: 10.1177/1758834015616934. [DOI] [PMC free article] [PubMed] [Google Scholar]; Eroglu, Z., and Ribas, A. (2016). Combination therapy with BRAF and MEK inhibitors for melanoma: latest evidence and place in therapy. Ther. Adv. Med. Oncol. 8, 48-56. [DOI] [PMC free article] [PubMed]
- Finch A.R., Caunt C.J., Perrett R.M., Tsaneva-Atanasova K., McArdle C.A. Dual specificity phosphatases 10 and 16 are positive regulators of EGF-stimulated ERK activity: indirect regulation of ERK signals by JNK/p38 selective MAPK phosphatases. Cell. Signal. 2012;24:1002–1011. doi: 10.1016/j.cellsig.2011.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]; Finch, A.R., Caunt, C.J., Perrett, R.M., Tsaneva-Atanasova, K., and McArdle, C.A. (2012). Dual specificity phosphatases 10 and 16 are positive regulators of EGF-stimulated ERK activity: indirect regulation of ERK signals by JNK/p38 selective MAPK phosphatases. Cell. Signal. 24, 1002-1011. [DOI] [PMC free article] [PubMed]
- Finck R., Simonds E.F., Jager A., Krishnaswamy S., Sachs K., Fantl W., Pe’er D., Nolan G.P., Bendall S.C. Normalization of mass cytometry data with bead standards. Cytometry A. 2013;83:483–494. doi: 10.1002/cyto.a.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]; Finck, R., Simonds, E.F., Jager, A., Krishnaswamy, S., Sachs, K., Fantl, W., Pe’er, D., Nolan, G.P., and Bendall, S.C. (2013). Normalization of mass cytometry data with bead standards. Cytometry A 83, 483-494. [DOI] [PMC free article] [PubMed]
- Fleuren E.D.G., Zhang L., Wu J., Daly R.J. The kinome ‘at large’ in cancer. Nat. Rev. Cancer. 2016;16:83–98. doi: 10.1038/nrc.2015.18. [DOI] [PubMed] [Google Scholar]; Fleuren, E.D.G., Zhang, L., Wu, J., and Daly, R.J. (2016). The kinome ‘at large’ in cancer. Nat. Rev. Cancer 16, 83-98. [DOI] [PubMed]
- Franceschini A., Szklarczyk D., Frankild S., Kuhn M., Simonovic M., Roth A., Lin J., Minguez P., Bork P., von Mering C., Jensen L.J. STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 2013;41:D808–D815. doi: 10.1093/nar/gks1094. [DOI] [PMC free article] [PubMed] [Google Scholar]; Franceschini, A., Szklarczyk, D., Frankild, S., Kuhn, M., Simonovic, M., Roth, A., Lin, J., Minguez, P., Bork, P., von Mering, C., and Jensen, L.J. (2013). STRING v9.1: protein-protein interaction networks, with increased coverage and integration. Nucleic Acids Res. 41, D808-D815. [DOI] [PMC free article] [PubMed]
- Genolini C., Alacoque X., Sentenac M., Arnaud C. kml and kml3d: R Packages to Cluster Longitudinal Data. J. Stat. Softw. 2015;65:1–34. [Google Scholar]; Genolini, C., Alacoque, X., Sentenac, M., and Arnaud, C. (2015). kml and kml3d: R Packages to Cluster Longitudinal Data. J. Stat. Softw. 65, 1-34.
- Guan K.L., Butch E. Isolation and characterization of a novel dual specific phosphatase, HVH2, which selectively dephosphorylates the mitogen-activated protein kinase. J. Biol. Chem. 1995;270:7197–7203. doi: 10.1074/jbc.270.13.7197. [DOI] [PubMed] [Google Scholar]; Guan, K.L., and Butch, E. (1995). Isolation and characterization of a novel dual specific phosphatase, HVH2, which selectively dephosphorylates the mitogen-activated protein kinase. J. Biol. Chem. 270, 7197-7203. [DOI] [PubMed]
- Guo G., Gong K., Wohlfeld B., Hatanpaa K.J., Zhao D., Habib A.A. Ligand-Independent EGFR Signaling. Cancer Res. 2015;75:3436–3441. doi: 10.1158/0008-5472.CAN-15-0989. [DOI] [PMC free article] [PubMed] [Google Scholar]; Guo, G., Gong, K., Wohlfeld, B., Hatanpaa, K.J., Zhao, D., and Habib, A.A. (2015). Ligand-Independent EGFR Signaling. Cancer Res. 75, 3436-3441. [DOI] [PMC free article] [PubMed]
- Hennessy B.T., Smith D.L., Ram P.T., Lu Y., Mills G.B. Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 2005;4:988–1004. doi: 10.1038/nrd1902. [DOI] [PubMed] [Google Scholar]; Hennessy, B.T., Smith, D.L., Ram, P.T., Lu, Y., and Mills, G.B. (2005). Exploiting the PI3K/AKT pathway for cancer drug discovery. Nat. Rev. Drug Discov. 4, 988-1004. [DOI] [PubMed]
- Horn T., Sandmann T., Fischer B., Axelsson E., Huber W., Boutros M. Mapping of signaling networks through synthetic genetic interaction analysis by RNAi. Nat. Methods. 2011;8:341–346. doi: 10.1038/nmeth.1581. [DOI] [PubMed] [Google Scholar]; Horn, T., Sandmann, T., Fischer, B., Axelsson, E., Huber, W., and Boutros, M. (2011). Mapping of signaling networks through synthetic genetic interaction analysis by RNAi. Nat. Methods 8, 341-346. [DOI] [PubMed]
- Hughey J.J., Lee T.K., Covert M.W. Computational modeling of mammalian signaling networks. Wiley Interdiscip. Rev. Syst. Biol. Med. 2010;2:194–209. doi: 10.1002/wsbm.52. [DOI] [PMC free article] [PubMed] [Google Scholar]; Hughey, J.J., Lee, T.K., and Covert, M.W. (2010). Computational modeling of mammalian signaling networks. Wiley Interdiscip. Rev. Syst. Biol. Med. 2, 194-209. [DOI] [PMC free article] [PubMed]
- Johannessen C.M., Boehm J.S., Kim S.Y., Thomas S.R., Wardwell L., Johnson L.A., Emery C.M., Stransky N., Cogdill A.P., Barretina J. COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature. 2010;468:968–972. doi: 10.1038/nature09627. [DOI] [PMC free article] [PubMed] [Google Scholar]; Johannessen, C.M., Boehm, J.S., Kim, S.Y., Thomas, S.R., Wardwell, L., Johnson, L.A., Emery, C.M., Stransky, N., Cogdill, A.P., Barretina, J., et al. (2010). COT drives resistance to RAF inhibition through MAP kinase pathway reactivation. Nature 468, 968-972. [DOI] [PMC free article] [PubMed]
- Johannessen C.M., Johnson L.A., Piccioni F., Townes A., Frederick D.T., Donahue M.K., Narayan R., Flaherty K.T., Wargo J.A., Root D.E., Garraway L.A. A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature. 2013;504:138–142. doi: 10.1038/nature12688. [DOI] [PMC free article] [PubMed] [Google Scholar]; Johannessen, C.M., Johnson, L.A., Piccioni, F., Townes, A., Frederick, D.T., Donahue, M.K., Narayan, R., Flaherty, K.T., Wargo, J.A., Root, D.E., and Garraway, L.A. (2013). A melanocyte lineage program confers resistance to MAP kinase pathway inhibition. Nature 504, 138-142. [DOI] [PMC free article] [PubMed]
- Julien S.G., Dubé N., Read M., Penney J., Paquet M., Han Y., Kennedy B.P., Muller W.J., Tremblay M.L. Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 2007;39:338–346. doi: 10.1038/ng1963. [DOI] [PubMed] [Google Scholar]; Julien, S.G., Dube, N., Read, M., Penney, J., Paquet, M., Han, Y., Kennedy, B.P., Muller, W.J., and Tremblay, M.L. (2007). Protein tyrosine phosphatase 1B deficiency or inhibition delays ErbB2-induced mammary tumorigenesis and protects from lung metastasis. Nat. Genet. 39, 338-346. [DOI] [PubMed]
- Julien S.G., Dubé N., Hardy S., Tremblay M.L. Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer. 2011;11:35–49. doi: 10.1038/nrc2980. [DOI] [PubMed] [Google Scholar]; Julien, S.G., Dube, N., Hardy, S., and Tremblay, M.L. (2011). Inside the human cancer tyrosine phosphatome. Nat. Rev. Cancer 11, 35-49. [DOI] [PubMed]
- Koboldt D.C., Fulton R.S., McLellan M.D., Schmidt H., Kalicki-Veizer J., McMichael J.F., Fulton L.L., Dooling D.J., Ding L., Mardis E.R., Cancer Genome Atlas Network Comprehensive molecular portraits of human breast tumours. Nature. 2012;490:61–70. doi: 10.1038/nature11412. [DOI] [PMC free article] [PubMed] [Google Scholar]; Koboldt, D.C., Fulton, R.S., McLellan, M.D., Schmidt, H., Kalicki-Veizer, J., McMichael, J.F., Fulton, L.L., Dooling, D.J., Ding, L., Mardis, E.R., et al.; Cancer Genome Atlas Network (2012). Comprehensive molecular portraits of human breast tumours. Nature 490, 61-70. [DOI] [PMC free article] [PubMed]
- Koseska A., Bastiaens P.I. Cell signaling as a cognitive process. EMBO J. 2017;36:568–582. doi: 10.15252/embj.201695383. [DOI] [PMC free article] [PubMed] [Google Scholar]; Koseska, A., and Bastiaens, P.I. (2017). Cell signaling as a cognitive process. EMBO J. 36, 568-582. [DOI] [PMC free article] [PubMed]
- Krumsiek J., Suhre K., Illig T., Adamski J., Theis F.J. Gaussian graphical modeling reconstructs pathway reactions from high-throughput metabolomics data. BMC Syst. Biol. 2011;5:21. doi: 10.1186/1752-0509-5-21. [DOI] [PMC free article] [PubMed] [Google Scholar]; Krumsiek, J., Suhre, K., Illig, T., Adamski, J., and Theis, F.J. (2011). Gaussian graphical modeling reconstructs pathway reactions from high-throughput metabolomics data. BMC Syst. Biol. 5, 21. [DOI] [PMC free article] [PubMed]
- Krzywinski M., Schein J., Birol I., Connors J., Gascoyne R., Horsman D., Jones S.J., Marra M.A. Circos: an information aesthetic for comparative genomics. Genome Res. 2009;19:1639–1645. doi: 10.1101/gr.092759.109. [DOI] [PMC free article] [PubMed] [Google Scholar]; Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., Jones, S.J., and Marra, M.A. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639-1645. [DOI] [PMC free article] [PubMed]
- Kung J.E., Jura N. Structural Basis for the Non-catalytic Functions of Protein Kinases. Structure. 2016;24:7–24. doi: 10.1016/j.str.2015.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; Kung, J.E., and Jura, N. (2016). Structural Basis for the Non-catalytic Functions of Protein Kinases. Structure 24, 7-24. [DOI] [PMC free article] [PubMed]
- Lavoie H., Therrien M. Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 2015;16:281–298. doi: 10.1038/nrm3979. [DOI] [PubMed] [Google Scholar]; Lavoie, H., and Therrien, M. (2015). Regulation of RAF protein kinases in ERK signalling. Nat. Rev. Mol. Cell Biol. 16, 281-298. [DOI] [PubMed]
- Levine J.H., Simonds E.F., Bendall S.C., Davis K.L., Amir A.D., Tadmor M.D., Litvin O., Fienberg H.G., Jager A., Zunder E.R. Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell. 2015;162:184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]; Levine, J.H., Simonds, E.F., Bendall, S.C., Davis, K.L., Amir, A.D., Tadmor, M.D., Litvin, O., Fienberg, H.G., Jager, A., Zunder, E.R., et al. (2015). Data-Driven Phenotypic Dissection of AML Reveals Progenitor-like Cells that Correlate with Prognosis. Cell 162, 184-197. [DOI] [PMC free article] [PubMed]
- Linding R., Jensen L.J., Ostheimer G.J., van Vugt M.A.T.M., Jørgensen C., Miron I.M., Diella F., Colwill K., Taylor L., Elder K. Systematic discovery of in vivo phosphorylation networks. Cell. 2007;129:1415–1426. doi: 10.1016/j.cell.2007.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]; Linding, R., Jensen, L.J., Ostheimer, G.J., van Vugt, M.A.T.M., Jorgensen, C., Miron, I.M., Diella, F., Colwill, K., Taylor, L., Elder, K., et al. (2007). Systematic discovery of in vivo phosphorylation networks. Cell 129, 1415-1426. [DOI] [PMC free article] [PubMed]
- Liu K., Chen H.-L., Wang S., Gu M.-M., Chen X.-M., Zhang S.-L., Yu K.-J., You Q.-S. High Expression of RIOK2 and NOB1 Predict Human Non-small Cell Lung Cancer Outcomes. Sci. Rep. 2016;6:28666. doi: 10.1038/srep28666. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]; Liu, K., Chen, H.-L., Wang, S., Gu, M.-M., Chen, X.-M., Zhang, S.-L., Yu, K.-J., and You, Q.-S. (2016). High Expression of RIOK2 and NOB1 Predict Human Non-small Cell Lung Cancer Outcomes. Sci. Rep. 6, 28666. [DOI] [PMC free article] [PubMed] [Retracted]
- Logue J.S., Morrison D.K. Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 2012;26:641–650. doi: 10.1101/gad.186965.112. [DOI] [PMC free article] [PubMed] [Google Scholar]; Logue, J.S., and Morrison, D.K. (2012). Complexity in the signaling network: insights from the use of targeted inhibitors in cancer therapy. Genes Dev. 26, 641-650. [DOI] [PMC free article] [PubMed]
- Low H.B., Zhang Y. Regulatory Roles of MAPK Phosphatases in Cancer. Immune Netw. 2016;16:85–98. doi: 10.4110/in.2016.16.2.85. [DOI] [PMC free article] [PubMed] [Google Scholar]; Low, H.B., and Zhang, Y. (2016). Regulatory Roles of MAPK Phosphatases in Cancer. Immune Netw. 16, 85-98. [DOI] [PMC free article] [PubMed]
- Lun X.-K., Zanotelli V.R.T., Wade J.D., Schapiro D., Tognetti M., Dobberstein N., Bodenmiller B. Influence of node abundance on signaling network state and dynamics analyzed by mass cytometry. Nat. Biotechnol. 2017;35:164–172. doi: 10.1038/nbt.3770. [DOI] [PMC free article] [PubMed] [Google Scholar]; Lun, X.-K., Zanotelli, V.R.T., Wade, J.D., Schapiro, D., Tognetti, M., Dobberstein, N., and Bodenmiller, B. (2017). Influence of node abundance on signaling network state and dynamics analyzed by mass cytometry. Nat. Biotechnol. 35, 164-172. [DOI] [PMC free article] [PubMed]
- Manning G., Whyte D.B., Martinez R., Hunter T., Sudarsanam S. The protein kinase complement of the human genome. Science. 2002;298:1912–1934. doi: 10.1126/science.1075762. [DOI] [PubMed] [Google Scholar]; Manning, G., Whyte, D.B., Martinez, R., Hunter, T., and Sudarsanam, S. (2002). The protein kinase complement of the human genome. Science 298, 1912-1934. [DOI] [PubMed]
- Masuda K., Shima H., Katagiri C., Kikuchi K. Activation of ERK induces phosphorylation of MAPK phosphatase-7, a JNK specific phosphatase, at Ser-446. J. Biol. Chem. 2003;278:32448–32456. doi: 10.1074/jbc.M213254200. [DOI] [PubMed] [Google Scholar]; Masuda, K., Shima, H., Katagiri, C., and Kikuchi, K. (2003). Activation of ERK induces phosphorylation of MAPK phosphatase-7, a JNK specific phosphatase, at Ser-446. J. Biol. Chem. 278, 32448-32456. [DOI] [PubMed]
- Mattila E., Pellinen T., Nevo J., Vuoriluoto K., Arjonen A., Ivaska J. Negative regulation of EGFR signalling through integrin-α1β1-mediated activation of protein tyrosine phosphatase TCPTP. Nat. Cell Biol. 2005;7:78–85. doi: 10.1038/ncb1209. [DOI] [PubMed] [Google Scholar]; Mattila, E., Pellinen, T., Nevo, J., Vuoriluoto, K., Arjonen, A., and Ivaska, J. (2005). Negative regulation of EGFR signalling through integrin-α1β1-mediated activation of protein tyrosine phosphatase TCPTP. Nat. Cell Biol. 7, 78-85. [DOI] [PubMed]
- Mertins P., Mani D.R., Ruggles K.V., Gillette M.A., Clauser K.R., Wang P., Wang X., Qiao J.W., Cao S., Petralia F., NCI CPTAC Proteogenomics connects somatic mutations to signalling in breast cancer. Nature. 2016;534:55–62. doi: 10.1038/nature18003. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mertins, P., Mani, D.R., Ruggles, K.V., Gillette, M.A., Clauser, K.R., Wang, P., Wang, X., Qiao, J.W., Cao, S., Petralia, F., et al.; NCI CPTAC (2016). Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55-62. [DOI] [PMC free article] [PubMed]
- Meyer A.S., Zweemer A.J.M., Lauffenburger D.A. The AXL Receptor is a Sensor of Ligand Spatial Heterogeneity. Cell Syst. 2015;1:25–36. doi: 10.1016/j.cels.2015.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]; Meyer, A.S., Zweemer, A.J.M., and Lauffenburger, D.A. (2015). The AXL Receptor is a Sensor of Ligand Spatial Heterogeneity. Cell Syst. 1, 25-36. [DOI] [PMC free article] [PubMed]
- Mok J., Kim P.M., Lam H.Y.K., Piccirillo S., Zhou X., Jeschke G.R., Sheridan D.L., Parker S.A., Desai V., Jwa M. Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signal. 2010;3:ra12. doi: 10.1126/scisignal.2000482. [DOI] [PMC free article] [PubMed] [Google Scholar]; Mok, J., Kim, P.M., Lam, H.Y.K., Piccirillo, S., Zhou, X., Jeschke, G.R., Sheridan, D.L., Parker, S.A., Desai, V., Jwa, M., et al. (2010). Deciphering protein kinase specificity through large-scale analysis of yeast phosphorylation site motifs. Sci. Signal. 3, ra12. [DOI] [PMC free article] [PubMed]
- Morandell S., Reinhardt H.C., Cannell I.G., Kim J.S., Ruf D.M., Mitra T., Couvillon A.D., Jacks T., Yaffe M.B. A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep. 2013;5:868–877. doi: 10.1016/j.celrep.2013.10.025. [DOI] [PMC free article] [PubMed] [Google Scholar]; Morandell, S., Reinhardt, H.C., Cannell, I.G., Kim, J.S., Ruf, D.M., Mitra, T., Couvillon, A.D., Jacks, T., and Yaffe, M.B. (2013). A reversible gene-targeting strategy identifies synthetic lethal interactions between MK2 and p53 in the DNA damage response in vivo. Cell Rep. 5, 868-877. [DOI] [PMC free article] [PubMed]
- O’Neil K., DeGrado W., Hermann A., Candia J., Rong S., Fukasawa K., Woude G., Vande, Ahn N. A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science. 1990;250:646–651. doi: 10.1126/science.2237415. [DOI] [PubMed] [Google Scholar]; O’Neil, K., DeGrado, W., Hermann, A., Candia, J., Rong, S., Fukasawa, K., Woude, G. Vande, and Ahn, N. (1990). A thermodynamic scale for the helix-forming tendencies of the commonly occurring amino acids. Science 250, 646-651. [DOI] [PubMed]
- Ochoa D., Jonikas M., Lawrence R.T., El Debs B., Selkrig J., Typas A., Villén J., Santos S.D., Beltrao P. An atlas of human kinase regulation. Mol. Syst. Biol. 2016;12:888. doi: 10.15252/msb.20167295. [DOI] [PMC free article] [PubMed] [Google Scholar]; Ochoa, D., Jonikas, M., Lawrence, R.T., El Debs, B., Selkrig, J., Typas, A., Villen, J., Santos, S.D., and Beltrao, P. (2016). An atlas of human kinase regulation. Mol. Syst. Biol. 12, 888. [DOI] [PMC free article] [PubMed]
- Parsons S.J., Parsons J.T. Src family kinases, key regulators of signal transduction. Oncogene. 2004;23:7906–7909. doi: 10.1038/sj.onc.1208160. [DOI] [PubMed] [Google Scholar]; Parsons, S.J., and Parsons, J.T. (2004). Src family kinases, key regulators of signal transduction. Oncogene 23, 7906-7909. [DOI] [PubMed]
- Patel A.P., Tirosh I., Trombetta J.J., Shalek A.K., Gillespie S.M., Wakimoto H., Cahill D.P., Nahed B.V., Curry W.T., Martuza R.L. Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science. 2014;344:1396–1401. doi: 10.1126/science.1254257. [DOI] [PMC free article] [PubMed] [Google Scholar]; Patel, A.P., Tirosh, I., Trombetta, J.J., Shalek, A.K., Gillespie, S.M., Wakimoto, H., Cahill, D.P., Nahed, B.V., Curry, W.T., Martuza, R.L., et al. (2014). Single-cell RNA-seq highlights intratumoral heterogeneity in primary glioblastoma. Science 344, 1396-1401. [DOI] [PMC free article] [PubMed]
- Paul M.K., Mukhopadhyay A.K. Tyrosine kinase - Role and significance in Cancer. Int. J. Med. Sci. 2004;1:101–115. doi: 10.7150/ijms.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]; Paul, M.K., and Mukhopadhyay, A.K. (2004). Tyrosine kinase - Role and significance in Cancer. Int. J. Med. Sci. 1, 101-115. [DOI] [PMC free article] [PubMed]
- Pawson T., Warner N. Oncogenic re-wiring of cellular signaling pathways. Oncogene. 2007;26:1268–1275. doi: 10.1038/sj.onc.1210255. [DOI] [PubMed] [Google Scholar]; Pawson, T., and Warner, N. (2007). Oncogenic re-wiring of cellular signaling pathways. Oncogene 26, 1268-1275. [DOI] [PubMed]
- Perfetto L., Briganti L., Calderone A., Cerquone Perpetuini A., Iannuccelli M., Langone F., Licata L., Marinkovic M., Mattioni A., Pavlidou T. SIGNOR: a database of causal relationships between biological entities. Nucleic Acids Res. 2016;44(D1):D548–D554. doi: 10.1093/nar/gkv1048. [DOI] [PMC free article] [PubMed] [Google Scholar]; Perfetto, L., Briganti, L., Calderone, A., Cerquone Perpetuini, A., Iannuccelli, M., Langone, F., Licata, L., Marinkovic, M., Mattioni, A., Pavlidou, T., et al. (2016). SIGNOR: a database of causal relationships between biological entities. Nucleic Acids Res. 44 (D1), D548-D554. [DOI] [PMC free article] [PubMed]
- Petryszak R., Keays M., Tang Y.A., Fonseca N.A., Barrera E., Burdett T., Füllgrabe A., Fuentes A.M.-P., Jupp S., Koskinen S. Expression Atlas update--an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 2016;44(D1):D746–D752. doi: 10.1093/nar/gkv1045. [DOI] [PMC free article] [PubMed] [Google Scholar]; Petryszak, R., Keays, M., Tang, Y.A., Fonseca, N.A., Barrera, E., Burdett, T., Fullgrabe, A., Fuentes, A.M.-P., Jupp, S., Koskinen, S., et al. (2016). Expression Atlas update--an integrated database of gene and protein expression in humans, animals and plants. Nucleic Acids Res. 44 (D1), D746-D752. [DOI] [PMC free article] [PubMed]
- Roth A., McPherson A., Laks E., Biele J., Yap D., Wan A., Smith M.A., Nielsen C.B., McAlpine J.N., Aparicio S. Clonal genotype and population structure inference from single-cell tumor sequencing. Nat. Methods. 2016;13:573–576. doi: 10.1038/nmeth.3867. [DOI] [PubMed] [Google Scholar]; Roth, A., McPherson, A., Laks, E., Biele, J., Yap, D., Wan, A., Smith, M.A., Nielsen, C.B., McAlpine, J.N., Aparicio, S., et al. (2016). Clonal genotype and population structure inference from single-cell tumor sequencing. Nat. Methods 13, 573-576. [DOI] [PubMed]
- Sacco F., Gherardini P.F., Paoluzi S., Saez-Rodriguez J., Helmer-Citterich M., Ragnini-Wilson A., Castagnoli L., Cesareni G. Mapping the human phosphatome on growth pathways. Mol. Syst. Biol. 2012;8:603. doi: 10.1038/msb.2012.36. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sacco, F., Gherardini, P.F., Paoluzi, S., Saez-Rodriguez, J., Helmer-Citterich, M., Ragnini-Wilson, A., Castagnoli, L., and Cesareni, G. (2012a). Mapping the human phosphatome on growth pathways. Mol. Syst. Biol. 8, 603. [DOI] [PMC free article] [PubMed]
- Sacco F., Perfetto L., Castagnoli L., Cesareni G. The human phosphatase interactome: an intricate family portrait. FEBS Lett. 2012;586:2732–2739. doi: 10.1016/j.febslet.2012.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]; Sacco, F., Perfetto, L., Castagnoli, L., and Cesareni, G. (2012b). The human phosphatase interactome: an intricate family portrait. FEBS Lett. 586, 2732-2739. [DOI] [PMC free article] [PubMed]
- Salgia R. MET in Lung Cancer: Biomarker Selection Based on Scientific Rationale. Mol. Cancer Ther. 2017;16:555–565. doi: 10.1158/1535-7163.MCT-16-0472. [DOI] [PubMed] [Google Scholar]; Salgia, R. (2017). MET in Lung Cancer: Biomarker Selection Based on Scientific Rationale. Mol. Cancer Ther. 16, 555-565. [DOI] [PubMed]
- Santarius T., Shipley J., Brewer D., Stratton M.R., Cooper C.S. A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer. 2010;10:59–64. doi: 10.1038/nrc2771. [DOI] [PubMed] [Google Scholar]; Santarius, T., Shipley, J., Brewer, D., Stratton, M.R., and Cooper, C.S. (2010). A census of amplified and overexpressed human cancer genes. Nat. Rev. Cancer 10, 59-64. [DOI] [PubMed]
- Santos S.D.M., Verveer P.J., Bastiaens P.I.H. Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat. Cell Biol. 2007;9:324–330. doi: 10.1038/ncb1543. [DOI] [PubMed] [Google Scholar]; Santos, S.D.M., Verveer, P.J., and Bastiaens, P.I.H. (2007). Growth factor-induced MAPK network topology shapes Erk response determining PC-12 cell fate. Nat. Cell Biol. 9 324-330. [DOI] [PubMed]
- Shaffer S.M., Dunagin M.C., Torborg S.R., Torre E.A., Emert B., Krepler C., Beqiri M., Sproesser K., Brafford P.A., Xiao M. Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature. 2017;546:431–435. doi: 10.1038/nature22794. [DOI] [PMC free article] [PubMed] [Google Scholar]; Shaffer, S.M., Dunagin, M.C., Torborg, S.R., Torre, E.A., Emert, B., Krepler, C., Beqiri, M., Sproesser, K., Brafford, P.A., Xiao, M., et al. (2017). Rare cell variability and drug-induced reprogramming as a mode of cancer drug resistance. Nature 546, 431-435. [DOI] [PMC free article] [PubMed]
- Steelman L., Chappell W., Abrams S.L., Kempf R.C., Long J., Laidler P., Mijatovic S., Maksimovic-Ivanic D., Stivala F., Mazzarino M.C. Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY) 2011;3:192–222. doi: 10.18632/aging.100296. [DOI] [PMC free article] [PubMed] [Google Scholar]; Steelman, L., Chappell, W., Abrams, S.L., Kempf, R.C., Long, J., Laidler, P., Mijatovic, S., Maksimovic-Ivanic, D., Stivala, F., Mazzarino, M.C., et al. (2011). Roles of the Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR pathways in controlling growth and sensitivity to therapy-implications for cancer and aging. Aging (Albany NY) 3, 192-222. [DOI] [PMC free article] [PubMed]
- Szklarczyk D., Morris J.H., Cook H., Kuhn M., Wyder S., Simonovic M., Santos A., Doncheva N.T., Roth A., Bork P. The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 2017;45(D1):D362–D368. doi: 10.1093/nar/gkw937. [DOI] [PMC free article] [PubMed] [Google Scholar]; Szklarczyk, D., Morris, J.H., Cook, H., Kuhn, M., Wyder, S., Simonovic, M., Santos, A., Doncheva, N.T., Roth, A., Bork, P., et al. (2017). The STRING database in 2017: quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45 (D1), D362-D368. [DOI] [PMC free article] [PubMed]
- Takahashi-Yanaga F., Shiraishi F., Hirata M., Miwa Y., Morimoto S., Sasaguri T. Glycogen synthase kinase-3β is tyrosine-phosphorylated by MEK1 in human skin fibroblasts. Biochem. Biophys. Res. Commun. 2004;316:411–415. doi: 10.1016/j.bbrc.2004.02.061. [DOI] [PubMed] [Google Scholar]; Takahashi-Yanaga, F., Shiraishi, F., Hirata, M., Miwa, Y., Morimoto, S., and Sasaguri, T. (2004). Glycogen synthase kinase-3β is tyrosine-phosphorylated by MEK1 in human skin fibroblasts. Biochem. Biophys. Res. Commun. 316, 411-415. [DOI] [PubMed]
- Türei D., Korcsmáros T., Saez-Rodriguez J. OmniPath: guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods. 2016;13:966–967. doi: 10.1038/nmeth.4077. [DOI] [PubMed] [Google Scholar]; Turei, D., Korcsmaros, T., and Saez-Rodriguez, J. (2016). OmniPath: guidelines and gateway for literature-curated signaling pathway resources. Nat. Methods 13, 966-967. [DOI] [PubMed]
- Uhlen M., Zhang C., Lee S., Sjöstedt E., Fagerberg L., Bidkhori G., Benfeitas R., Arif M., Liu Z., Edfors F. A pathology atlas of the human cancer transcriptome. Science. 2017;357:eaan2507. doi: 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]; Uhlen, M., Zhang, C., Lee, S., Sjostedt, E., Fagerberg, L., Bidkhori, G., Benfeitas, R., Arif, M., Liu, Z., Edfors, F., et al. (2017). A pathology atlas of the human cancer transcriptome. Science 357, eaan2507. [DOI] [PubMed]
- van der Maaten L., Hinton G. Visualizing Data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2605. [Google Scholar]; van der Maaten, L., and Hinton, G. (2008). Visualizing Data using t-SNE. J. Mach. Learn. Res. 9, 2579-2605.
- Wang M., Herrmann C.J., Simonovic M., Szklarczyk D., von Mering C. Version 4.0 of PaxDb: protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics. 2015;15:3163–3168. doi: 10.1002/pmic.201400441. [DOI] [PMC free article] [PubMed] [Google Scholar]; Wang, M., Herrmann, C.J., Simonovic, M., Szklarczyk, D., and von Mering, C. (2015). Version 4.0 of PaxDb: protein abundance data, integrated across model organisms, tissues, and cell-lines. Proteomics 15, 3163-3168. [DOI] [PMC free article] [PubMed]
- Ward J.H. Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 1963;58:236–244. [Google Scholar]; Ward, J.H. (1963). Hierarchical Grouping to Optimize an Objective Function. J. Am. Stat. Assoc. 58, 236-244.
- Yang X., Boehm J.S., Yang X., Salehi-Ashtiani K., Hao T., Shen Y., Lubonja R., Thomas S.R., Alkan O., Bhimdi T. A public genome-scale lentiviral expression library of human ORFs. Nat. Methods. 2011;8:659–661. doi: 10.1038/nmeth.1638. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yang, X., Boehm, J.S., Yang, X., Salehi-Ashtiani, K., Hao, T., Shen, Y., Lubonja, R., Thomas, S.R., Alkan, O., Bhimdi, T., et al. (2011). A public genome-scale lentiviral expression library of human ORFs. Nat. Methods 8, 659-661. [DOI] [PMC free article] [PubMed]
- Yu Y., Anjum R., Kubota K., Rush J., Villen J., Gygi S.P. A site-specific, multiplexed kinase activity assay using stable-isotope dilution and high-resolution mass spectrometry. Proc. Natl. Acad. Sci. USA. 2009;106:11606–11611. doi: 10.1073/pnas.0905165106. [DOI] [PMC free article] [PubMed] [Google Scholar]; Yu, Y., Anjum, R., Kubota, K., Rush, J., Villen, J., and Gygi, S.P. (2009). A site-specific, multiplexed kinase activity assay using stable-isotope dilution and high-resolution mass spectrometry. Proc. Natl. Acad. Sci. USA 106, 11606-11611. [DOI] [PMC free article] [PubMed]
- Zadra G., Batista J.L., Loda M. Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol. Cancer Res. 2015;13:1059–1072. doi: 10.1158/1541-7786.MCR-15-0068. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zadra, G., Batista, J.L., and Loda, M. (2015). Dissecting the Dual Role of AMPK in Cancer: From Experimental to Human Studies. Mol. Cancer Res. 13, 1059-1072. [DOI] [PMC free article] [PubMed]
- Zhang H., Liu T., Zhang Z., Payne S.H., Zhang B., McDermott J.E., Zhou J.-Y., Petyuk V.A., Chen L., Ray D., CPTAC Investigators Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell. 2016;166:755–765. doi: 10.1016/j.cell.2016.05.069. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zhang, H., Liu, T., Zhang, Z., Payne, S.H., Zhang, B., McDermott, J.E., Zhou, J.-Y., Petyuk, V.A., Chen, L., Ray, D., et al.; CPTAC Investigators (2016). Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 166, 755-765. [DOI] [PMC free article] [PubMed]
- Zunder E.R., Finck R., Behbehani G.K., Amir A.D., Krishnaswamy S., Gonzalez V.D., Lorang C.G., Bjornson Z., Spitzer M.H., Bodenmiller B. Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat. Protoc. 2015;10:316–333. doi: 10.1038/nprot.2015.020. [DOI] [PMC free article] [PubMed] [Google Scholar]; Zunder, E.R., Finck, R., Behbehani, G.K., Amir, A.D., Krishnaswamy, S., Gonzalez, V.D., Lorang, C.G., Bjornson, Z., Spitzer, M.H., Bodenmiller, B., et al. (2015). Palladium-based mass tag cell barcoding with a doublet-filtering scheme and single-cell deconvolution algorithm. Nat. Protoc. 10, 316-333. [DOI] [PMC free article] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data and pre-analyzed data are available at Mendeley Data under the following link: