

We report the rational design of a polymer with unprecedented and robust efficiency for cytosolic delivery of native proteins.
Abstract
Cytosolic protein delivery is of central importance for the development of protein-based biotechnologies and therapeutics; however, efficient intracellular delivery of native proteins remains a challenge. Here, we reported a boronic acid–rich dendrimer with unprecedented efficiency for cytosolic delivery of native proteins. The dendrimer could bind with both negatively and positively charged proteins and efficiently delivered 13 cargo proteins into the cytosol of living cells. All the delivered proteins kept their bioactivities after cytosolic delivery. The dendrimer ensures efficient intracellular delivery of Cas9 protein into various cell lines and showed high efficiency in CRISPR-Cas9 genome editing. The rationally designed boronic acid–rich dendrimer permits the development of an efficient platform with high generality for the delivery of native proteins.
INTRODUCTION
Direct cytosolic delivery of proteins provides a promising strategy for specific and transient manipulation of cell function and treatment of various diseases, which avoids the limitations of gene delivery such as permanent or random genetic alteration, off-target effects due to sustained gene expression, stress response, and carcinogenesis (1–4). It is no surprise that approximately 20% of the drugs on the market are proteins, such as monoclonal antibodies, cytokines, growth factors, and insulin (1). However, all the clinically available protein-based therapeutics have been developed toward extracellular targets owing to the poor membrane permeability of most intracellular proteins (5–7). The lack of efficient approaches to translocate proteins into the cytosol of living cells remains a major challenge in expanding the family of proteins that could be identified as therapeutic candidates. During the last few years, researchers have proposed techniques including electroporation (8), microfluidic device (9), cell-penetrating peptides (10, 11), cell-derived vesicles (4, 6), synthetic virus-like nanoparticles (12), lipid nanoparticles (13, 14), polymers (2, 15, 16), and inorganic nanoparticles (17–20) for intracellular protein delivery. Among the delivery strategies, polymers have been one of the most promising candidates for cytosolic protein delivery both in vitro and in vivo due to ease of synthesis and functionalization, effectiveness in endosome escape, and material safety (2, 21, 22).
For polymer-mediated cytosolic protein delivery, the major concern is how to effectively bind proteins with the polymer and form stable complexes suitable for cell internalization. The protein molecules might be positively or negatively charged under physiological conditions depending on their isoelectric points (pIs). Therefore, it is difficult to develop a polymer with robust binding affinity with all sorts of proteins. To address this concern, researchers have proposed a protein supercharging strategy to increase the number of binding sites on proteins for cationic materials. They modified protein molecules with anionic species such as carboxyl-rich chemicals (23, 24), boronic acids (21), anionic proteins and peptides (17), anionic polymers (25), and small guide RNA or DNA nanoclews (2, 26), via covalent or noncovalent approaches. The engineered anionic species facilitate the formation of stable polymer/protein nanoparticles for intracellular protein delivery. In an alternative strategy, they directly conjugated the proteins with cationic polymers via dynamic covalent bonds for protein translocation (27). Though some of the modified species on protein surface could be removed by endolysosomal acidity/protease or reactive oxygen species, these strategies are involved with complicated syntheses or genetic engineering, alteration of protein structure or bioactivity after chemical modification, high costs, and unexpected safety concerns. Recent approaches to the cytosolic delivery of native proteins without the need for chemical modification have been reported, e.g., guanidinium-rich polymer to improve the binding affinity between polymer and proteins (22, 28–30) and fluoropolymers with excellent self-assembly behavior to improve the loading efficiency and capacity of polymers (15, 16, 31). Though these approaches achieved high efficiency for the translocation of proteins into cells, the generality of the technique toward different proteins might be problematic due to the distinct pIs and molecular sizes for the various proteins. As a result, there is still an urgent demand for an effective and robust approach for cytosolic delivery of proteins without the need for chemical modification.
The protein surface usually displays cationic groups such as amine, imidazole, and guanidinium, and anionic carboxylate groups. We hypothesized that the development of a polymer that is capable of binding with both the anionic and cationic groups on protein surface could provide a general approach for cytosolic protein delivery. Phenylboronic acid (PBA) is an electron-deficient group that can coordinate with cationic amine and imidazole groups on proteins via nitrogen-boronate complexation (32–34). The aromatic ring in PBA can bind with the guanidinium or ammonium groups on proteins via cation-π interactions, an unconventional force widely explored in protein-ligand interactions and protein structures (35, 36). On the other hand, anionic carboxylates on protein can efficiently bind with cationic species via ionic interactions. If the ligand PBA was conjugated onto a cationic polymer at a high grafting ratio, the yielding PBA-rich polymer may allow high binding affinity with different types of proteins via a combination of nitrogen-boronate complexation and cation-π and ionic interactions (Fig. 1). Besides the beneficial effect on protein binding, PBA moieties on polymer also provide opportunities for self-assembly and addressing the barriers existing in the cytosolic delivery of nucleic acids (37, 38). In this study, we proposed that the PBA-rich cationic polymer is a possible candidate to bind various proteins into stable nanoparticles and allow efficient cytosolic delivery. We investigated the behaviors of the PBA-rich polymer for intracellular protein delivery using different model proteins, including Cas9 ribonucleoprotein (RNP), which is widely explored as an efficient tool for CRISPR-Cas9 genome editing in a wide range of biomedical applications.
Fig. 1. Boronic acid–rich dendrimer with robust efficiency in cytosolic protein delivery.
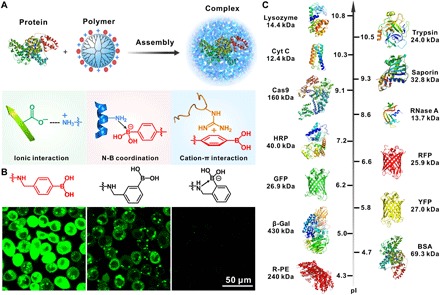
(A) Mechanism of boronate-rich dendrimer in complexation with protein complex formation. The dendrimer could bind with both negatively and positively charged proteins via a combination of nitrogen-boronate complexation and cation-π and ionic interactions between the two species. (B) Boronic acid–rich dendrimers with different chemical structures show distinct behaviors in the delivery of proteins into HeLa cells. Fluorescently labeled BSA was used as the model protein here. (C) Model proteins with different molecular weights and pIs in this study.
RESULTS
Boronic acid–rich dendrimer in cytosolic delivery
We used a generation 5 (G5) amine-terminated polyamidoamine (PAMAM) dendrimer with well-defined molecular size and surface functionality as the model cationic polymer. A series of PBA-modified dendrimers were synthesized by reacting 4-(bromomethyl)phenylboronic acid with the dendrimer at different feeding ratios. The conjugated number of PBA on each G5 dendrimer was 14 (P1), 24 (P2), 42 (P3), and 60 (P4), respectively, as calculated by a 1H NMR (nuclear magnetic resonance) method (fig. S1). Conjugation of more PBA moieties on the dendrimer yielded insoluble products in aqueous solutions. Therefore, we used the four PBA-modified dendrimers (P1 to P4) for further intracellular protein delivery. Bovine serum albumin (BSA) labeled with fluorescein isothiocyanate (FITC) was used as the model protein (BSA-FITC; 69.3 kDa, pI 4.7). We complexed the dendrimers with BSA-FITC via gentle mixing and treated the complexes with HeLa cells for 4 hours. As shown in Fig. 2A and fig. S2A, P1 to P3 and the non-modified dendrimer (P0) exhibit weak efficiency in the cytosolic delivery of BSA-FITC, while P4 shows markedly higher transduction efficiency, and the fluorescence intensity of cells treated with P4/BSA-FITC is more than one order of magnitude higher than that of P1 to P3 complexes. Dynamic light scattering (DLS) and transmission electron microscope (TEM) results show that P4 with the highest PBA density on dendrimer has a strong binding affinity with the protein to form stable and uniform nanoparticles around 100 nm (Fig. 2B). Fluorescence resonance energy transfer (FRET) analysis of the P4/BSA mixture also confirms the formation of condensed complexes (fig. S3A). We also compared the translocation efficiency of P4 with several analog materials (Fig. 2C and fig. S1), including G5 dendrimer modified with approximately 60 benzoic acid (P5), 3-(bromomethyl)phenylboronic acid (P6), 2-(bromomethyl)phenylboronic acid (P7), and 4-(bromobutyl)boronic acid (P8), respectively. As shown in Fig. 2 (D to F), P4 shows the highest efficiency among these dendrimers. Unexpectedly, P7 with a similar chemical composition to P4 shows extremely low efficiency for the delivery of BSA-FITC. This is due to the presence of intramolecular nitrogen-boronate coordination in the synthesized dendrimer P7, which reduces the efficient binding of dendrimer with proteins (Fig. 2F and fig. S3A). Similarly, P8 exhibits poor translocation efficiency in the delivery of BSA-FITC, which confirms the essential role of phenyl groups in protein binding and cytosolic delivery (Fig. 2F and fig. S3A). The boronic acid–rich dendrimer P4 is even more efficient than commercial protein transduction reagents such as PULSin and TransEx. Incubation of the P4-transduced cells with trypan blue, a membrane-impermeable fluorescence quencher, scarcely inhibits the fluorescence intensity of cells (fig. S2B), suggesting the efficient translocation of BSA-FITC into the cytosol rather than physical adsorption on cell membrane (15). The translocation efficiency of P4/BSA-FITC depends on the incubation time (Fig. 2G), and the internalization is energy dependent, probably mediated by micropinocytosis-, clathrin-, and lipid raft–dependent pathways (fig. S2C). The internalized BSA-FITC is generally not localized with acidic compartments such as endosomes and lysosomes stained by LysoTracker Red, and the dispersion of BSA-FITC in the cytosol is observed as early as 2 hours after complex incubation (fig. S4). These results suggest fast endosomal escape from the acidic compartments and efficient intracellular protein release into the cytosols probably triggered by abundant proteins in the cytosol or polyanionic species (fig. S3B). The tested dendrimers show minimal toxicity on the treated HeLa cells (Fig. 2H) as well as normal cell lines such as NIH3T3 cells (fig. S2D).
Fig. 2. Boronic acid–rich dendrimer in cytosolic delivery.
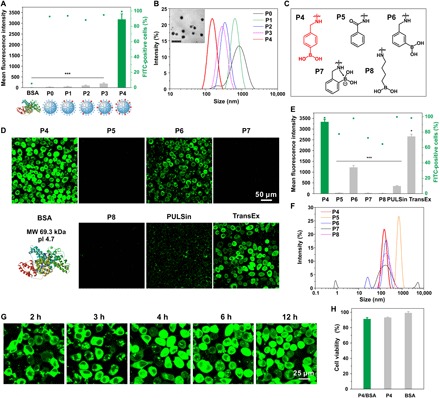
(A) Cytosolic delivery of BSA-FITC into HeLa cells by P0 to P4 for 4 hours. The doses of protein and dendrimer in each well were 6 and 8 μg, respectively. (B) DLS analysis of P0, P1, P2, P3, and P4/BSA complexes, and TEM of P4/BSA nanoparticles. Scale bar, 200 nm. (C) Structure of the PBA analog materials. (D) Confocal images and (E) flow cytometry analysis of HeLa cells treated with P4/BSA-FITC or analog complexes of P5 to P8 for 4 hours. The doses of protein and dendrimer were 6 and 8 μg in each well, respectively. Commercial reagents PULSin and TransEx were used as positive controls. (F) DLS analysis of P4, P5, P6, P7, and P8/BSA-FITC complexes. (G) Time-dependent internalization of P4/BSA-FITC by HeLa cells. (H) Viability of HeLa cells treated with P4/BSA, P4, and BSA complexes. The concentrations of materials were equal to those in cytosolic protein delivery.
Cytosolic delivery of fluorescent proteins
We further tested the transduction efficiency of P4 in the delivery of other fluorescent proteins. Phycoerythrin (R-PE) is a red fluorescent protein isolated from red algae. It is a relatively large protein with a molecular mass of 240 kDa and is negatively charged under physiological conditions (pI 4.3). P4 and phycoerythrin form uniform and positively charged nanoparticles (fig. S5A). As shown in Fig. 3 (A and B), P4 shows efficient cytosolic delivery of phycoerythrin into HeLa cells, and red fluorescence is dispersed in the cytosol after 4 hours of incubation with P4/phycoerythrin. Similarly, P4 efficiently delivers other fluorescent proteins including green fluorescent protein (GFP; 26.9 kDa, pI 6.2), yellow fluorescent protein (YFP; 27.0 kDa, pI 5.8), and red fluorescent protein (RFP; 25.9 kDa, pI 6.6) into HeLa cells (Fig. 3C). Besides negatively charged proteins, P4 is also efficient in the cytosolic delivery of cationic proteins (Fig. 3D and fig. S5, B to D), such as cytochrome c (Cyt C; 12.4 kDa, pI 10.3), lysozyme (14.4 kDa, pI 10.8), and trypsin (24.0 kDa, pI 10.5).
Fig. 3. Cytosolic delivery of fluorescent proteins.
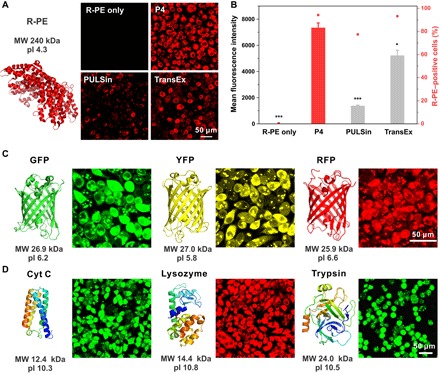
(A) P4 in the cytosolic delivery of R-PE into HeLa cells for 4 hours. Commercial reagents PULSin and TransEx were used as positive controls. (B) Quantitative analysis of the protein internalization in (A) by flow cytometry. The dose of R-PE in each well was 0.5 μg. P4 in the cytosolic delivery of (C) negatively charged GFP, YFP, and RFP into HeLa cells for 4 hours. The dose of proteins in each well was 3 μg. P4 in the cytosolic delivery of (D) positively charged Cyt C, lysosome, and trypsin into HeLa cells for 4 hours. The doses of proteins in each well were 6, 6, and 3 μg, respectively, and the dose of P4 for all the experiments was 8 μg.
Cytosolic delivery of native enzymes
Besides high transduction efficiency, we need to maintain the bioactivity of proteins after cytosolic delivery. In this case, we used a bioactive protein, β-galactosidase (β-Gal), as the model protein to confirm whether the intracellular released β-Gal keeps its bioactivity in catalytic hydrolysis of β-Gal–containing compounds (Fig. 4A). P4 and β-Gal form uniform and positively charged nanoparticles (fig. S5E), and the binding of β-Gal with P4 partially decreases its enzymatic activity. However, the addition of heparin sulfate into the solution fully recovers the enzyme bioactivity due to efficient release of β-Gal from the complex nanoparticles (fig. S6A). We then treated HeLa cells with the P4/β-Gal complex and further added the transduced cells with 5-bromo-4-chloro-3-indolyl-β-d-galactoside (X-Gal), the substrate for β-Gal. The enzyme catalyzes the hydrolysis of X-Gal into an insoluble blue dye, which can be used to reveal the amount of β-Gal in the treated cells. As shown in Fig. 4B, P4 shows high accumulation of blue dyes in the cells after X-Gal staining, and its translocation efficiency is superior to the control materials. The relative β-Gal activity of cells treated with P4/β-Gal determined by a β-Gal activity assay kit reveals that more than 80% of the enzyme activity is maintained after intracellular delivery (Fig. 4C). Horseradish peroxidase (HRP; 40.0 kDa, pI 7.2) is an enzyme that catalyzes the oxidation of a nonfluorescent substrate, Amplex Red, into a red fluorescent and membrane-impermeable product in the presence of hydrogen peroxide (Fig. 4D) (39). Complexation of HRP with P4 (fig. S5F) slightly reduces its catalytic activity, while the addition of other proteins such as BSA or heparin sulfate could recover the enzymatic activity of HRP (fig. S6B). In addition, P4 shows high activity in the cytosolic delivery of HRP as revealed by the red fluorescence signals observed in the treated cells (Fig. 4E). Similarly, HRP catalyzes the reaction of a colorless substrate, tetramethylbenzidine (TMB), into a blue dye in the presence of hydrogen peroxide. The cells treated with P4/HRP show the highest HRP activity after cytosolic delivery, as determined by the TMB assay (Fig. 4F).
Fig. 4. Cytosolic delivery of native enzymes.
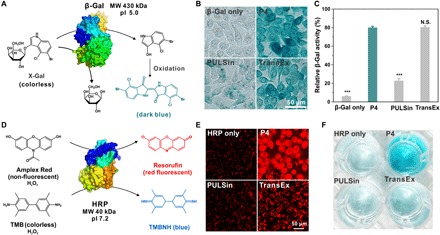
(A) β-Gal catalyzes the hydrolysis of colorless substrate X-Gal into a blue-colored product. (B) X-Gal staining of the cells treated with β-Gal/P4 for 4 hours. (C) Enzymatic activity of the β-Gal after cytosolic delivery determined by a kit. The dose of β-Gal in each well was 5 μg. (D) HRP catalyzes colorless substrates Amplex Red or TMB into a red fluorescent product, resorufin, or a blue dye, respectively, in the presence of hydrogen peroxide. (E) Confocal microscopy images of HeLa cells treated with P4/HRP and Amplex Red. Commercial reagents PULSin and TransEx were used as positive controls. (F) HRP enzymatic activity in the transduced cell analysis by TMB assay. The dose of HRP in each well was 6 μg. The dose of P4 in the experiments was 8 μg in each well.
Cytosolic delivery of toxic proteins
We also tested the efficiencies of P4 in the delivery of toxic proteins such as saporin (32.8 kDa, pI 9.3), ribonuclease A (RNase A; 13.7 kDa, pI 8.6), and trypsin. Saporin is a ribosome-inactivating protein that irreversibly blocks protein synthesis in mammalian cells via the depurination of a specific nucleotide on the 28S subunit (Fig. 5A). P4 is efficient in binding with saporin and forms uniform and positively charged nanoparticles (fig. S5G). Saporin only showed minimal toxicity on breast cancer cells MDA-MB-231 due to limited membrane penetration; however, the presence of P4 efficiently translocates saporin into the cytosol (Fig. 5B), where the protein inactivates the ribosomes. This is evident by the significantly increased cytotoxicity on the cells treated with the P4/saporin complex, and the increased toxicity is not caused by the dendrimer P4 (Fig. 5B). RNase A is a positively charged enzyme that can catalyze the degradation of RNA chains (Fig. 5A). RNase A could keep its enzymatic activity after intracellular release from the dendrimer P4 (figs. S5H and S6C), and the dendrimer efficiently delivers RNase A into the cells and decreases the viability of treated cells (Fig. 5C). Similarly, the efficiently delivered trypsin by P4 also leads to efficient cell death due to the degradation of intracellular proteins by the internalized protease (Fig. 5D and fig. S5D). These results together suggest that P4 is a potent and versatile dendrimer for cytosolic delivery of native proteins.
Fig. 5. Cytosolic delivery of toxic proteins.
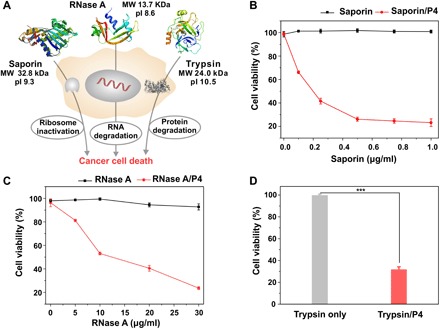
(A) Cytosolic delivery of saporin, RNase A, and trypsin into cancer cells leads to ribosome inactivity, RNA degradation, and intracellular protein hydrolysis, respectively, and cell death. Viability of MDA-MB-231 cells treated with (B) saporin/P4 and (C) RNase A/P4, and (D) HeLa cells treated with trypsin/P4 complexes determined by MTT assay. The concentration of trypsin was 40 μg/ml. Free saporin, RNase A, and trypsin were tested as controls. The dose of P4 in the experiments of cytosolic delivery of saporin, RNase A, and trypsin were 0.6, 0.6, and 2.5 μg in each well, respectively.
Cytosolic delivery of Cas9 RNP for CRISPR-Cas9 gene editing
We lastly investigated the potential of P4 to deliver Streptococcus pyogenes Cas9 RNP for genome editing (Fig. 6A). Cas9 is widely explored in a variety of biomedical applications and is capable of inducing site-specific cleavage at endogenous genomic loci directed by single guide RNAs (sgRNAs) in mammalian cells (40). At present, the shortage of delivery materials that can deliver Cas9 RNP still represents one of the major challenges for successful CRISPR-Cas–based genome editing. As shown in Fig. 6B, P4 and Cas9 RNP form spherical and monodisperse nanoparticles around 300 nm. Subsequently, we used 293T cells stably expressing enhanced green fluorescence protein (EGFP) as a model to illustrate the potential of P4-mediated Cas9 delivery for the disruption of the EGFP gene (Fig. 6A). To this end, a single guide RNA targeting enhanced green fluorescence protein (sgEGFP) that is designed to target the EGFP gene was complexed with Cas9 to form RNP. As shown in Fig. 6 (C and D), the intensity of EGFP in the 293T-EGFP cell line is significantly decreased upon the intracellular delivery of P4/RNP, due to the loss-of-function effect of EGFP. Flow cytometry analysis indicated that approximately 40% of the 293T-EGFP cells treated with P4/RNP exhibited EGFP knockout (Fig. 6E), whereas treatment with RNP alone had negligible knockout effects. Note that the ability of P4 to mediate the disruption of the EGFP gene was greater over the commercially available RNP delivery agent CRISPRMAX (CMAX). To further validate the genome editing efficiency mediated by P4, we investigated the intracellular delivery of RNP targeting different genome loci in the 293T cell line (Fig. 6, F to H). Indels (insertion and deletion) detected by T7 endonuclease I (T7E1) digestion assays were studied to investigate whether the genome editing occurred at the targeted genome locus. After the intracellular delivery, the digestion bands (cuts), which were derived from the digestion products of T7E1 recognizing indels in the double-stranded DNA, were observed from the uncut bands in both adeno-associated virus integration site 1 (AAVS1) and hemoglobin subunit beta (HBB). As expected, the delivery of both P4/RNP resulted in efficient genome editing in these two loci. AAVS1 genome editing mediated by P4/RNP and CMAX/RNP resulted in indel rates of 23.1 and 17.5%, respectively. Likewise, HBB genome editing by P4/RNP and CMAX resulted in indel rates of 21.1 and 9.7%, respectively. To confirm the robustness of P4 in the delivery of Cas9 RNP, we lastly validated the genome editing capability in HCT-116 and HT-29 cell lines. As evident from Fig. 6 (I and J), indel studies suggested that the transduction of P4/RNP also resulted in efficient genome editing at the AAVS1 and HBB genome loci in HCT-116 cells. In this study, we also used P4/RNP to knock out the catenin beta-1 (CTNNB1) gene, which plays an important functional role in the canonical Wnt signaling pathway in HCT-116 and HT-29 cell lines. As shown in fig. S7, T7E1 cleavage assay demonstrated that HCT-116 and HT-29 cells transfected with P4/RNP induced efficient DNA mutation. Owing to the simple chemical structure of P4, further modifications to render additional functional moieties such as targeting ligands and protective antifouling layers could be easily achievable to meet the specified genome editing applications in vivo.
Fig. 6. Cytosolic delivery of Cas9 RNP for CRISPR-Cas9 gene editing.
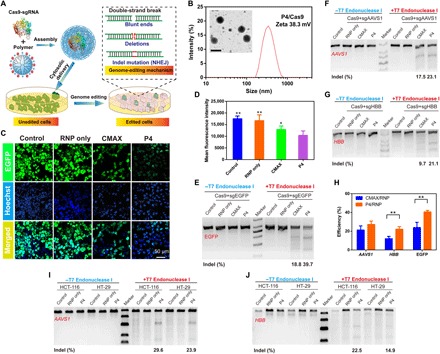
(A) P4-mediated Cas9/sgEGFP delivery for efficient EGFP genome editing. (B) DLS analysis of P4/Cas9 RNP nanoparticles. The inset is the TEM image of P4/Cas9 RNP nanoparticles. Scale bar, 500 nm. (C) Confocal images of 293T-EGFP cells treated with P4/RNP for 4 hours. Scale bar, 50 μm. (D) Flow cytometry analysis of 293T-EGFP cells treated with P4/RNP for 4 hours by means of mean fluorescence intensity. (E) Indel analysis from samples treated by P4/RNP. Commercial reagent CMAX was used as a positive control. T7E1 assay results from the intracellular delivery of P4/RNP targeting (F) AAVS1 and (G) HBB genes in 293T cells. (H) Genome editing efficiency, as revealed by the grayscale density of T7E1 results, after the intracellular delivery of RNP targeting EGFP locus (for 293T-EGFP cells), AAVS1 locus, and HBB locus (for 293T cells). Data represent mean ± SD (n = 3, Student’s t test, **P < 0.01). T7E1 assay results from the intracellular delivery of P4/RNP targeting (I) AAVS1 and (J) HBB genes in HCT-116 and HT-29 cells.
DISCUSSION
In summary, we reported the rational design of a boronic acid–rich dendrimer with unprecedented efficiency for cytosolic delivery of proteins with different pIs and sizes. The dendrimer efficiently assembles various proteins into nanoparticles, and the delivered proteins could maintain their bioactivity after intracellular release. This dendrimer also allows the efficient delivery of Cas9 RNP targeting multiple genome loci of different types of cell lines, which defines a useful material for the delivery of genome editing tools in a wide range of biomedical applications in the future.
MATERIALS AND METHODS
Materials
Amine-terminated and ethylenediamine-cored G5 PAMAM (theoretical molecular mass: 28,826 Da) was purchased from Dendritech (Midland, MI). Benzoic acid, dicyclohexylcarbodiimide, N-hydroxysuccinimide, rhodamine B isothiocyanate (RBITC), Cyt C from equine heart, saporin, and lysozyme from chicken egg white were purchased from Sigma-Aldrich (St. Louis, MO). 4-(Bromomethyl)phenylboronic acid was purchased from Meryer Chem. (Shanghai, China). β-Gal and RNase A were purchased from J&K Scientific (Shanghai, China). FITC, trimethylamine, trypsin from porcine pancreas, and Amplex Red were purchased from Macklin (Shanghai, China). BSA and hydrogen peroxide were purchased from Aladdin (Shanghai, China). CRISPR-Cas9 protein was purchased from GenScript (Nanjing, China). T7E1 enzyme was obtained from New England Biolabs (Beijing, China). PULSin was purchased from Polyplus-transfection (France). TransEx was obtained from Cenji Biotech (Shanghai, China). Lipofectamine CMAX Transfection Reagent was bought from Thermo Fisher Scientific (Germany). R-phycoerythrin (R-PE) was purchased from Cayman Chemical (Michigan, USA). GFP, YFP, and RFP were gifts from N. Chen in CAS. HRP and TMB were purchased from Yuanye Biotechnology (Shanghai, China). LysoTracker Red (DND-99, Invitrogen) was purchased from Thermo Fisher Scientific (Germany).
Synthesis and characterization of PBA-modified dendrimers
G5 PAMAM dendrimer was reacted with 4-(bromomethyl)phenylboronic acid at various molar ratios in anhydrous methanol for 24 hours. The yielding products were intensively dialyzed against methanol and distilled water. Then, the products were ultrafiltrated for concentration or lyophilized directly to obtain PBA-decorated dendrimers. PBA analog materials including 3-(bromomethyl)phenylboronic acid–, 2-(bromomethyl)phenylboronic acid–, and 4-(bromobutyl)boronic acid–modified dendrimer were synthesized by a similar procedure. For benzoic acid–modified dendrimer, dicyclohexylcarbodiimide, N-hydroxysuccinimide, and benzoic acid were dissolved in dimethylformamide. The solution was stirred for 6 hours at room temperature. Then, G5 dendrimer and trimethylamine dissolved in dimethyl sulfoxide were added, and the reaction was continued for 7 days. The product was extensively dialyzed against dimethyl sulfoxide and distilled water and followed by lyophilization to obtain white powders. The purified materials were further characterized by 1H NMR spectroscopy (Varian, 699.804 MHz) to determine the average conjugated numbers of ligands on each G5 dendrimer. RBITC-labeled dendrimers were synthesized by stirring the mixture of dendrimers and RBITC in the dark at an RBITC/dendrimer molar ratio of 3:1 for 24 hours. The products were intensively dialyzed against distilled water and lyophilized to obtain RBITC-labeled dendrimers.
Synthesis of fluorescent dye–labeled proteins
Briefly, BSA, Cyt C, and trypsin were dissolved in phosphate-buffered saline (PBS) buffer (pH 7.4), respectively. The protein solutions were mixed with FITC at a FITC/protein molar ratio of 3:1. The reaction solutions were stirred for 24 hours at room temperature in the dark. The yielding products were intensively dialyzed against PBS and distilled water, respectively. The purified products were lyophilized to obtain FITC-labeled proteins BSA-FITC, Cyt C–FITC, and trypsin-FITC, respectively. RBITC-labeled lysozyme was synthesized with a similar method. The molar ratio of RBITC to lysozyme was 2:1. The fluorescent dye–labeled proteins were stored at −20°C before further experiments.
Protein/dendrimer complex formation and characterization
The boronic acid–decorated dendrimers were mixed with model proteins and incubated for 30 min, respectively. Then, the mixtures were diluted with deionized water for further characterization. The size and zeta potential of the formed complexes were characterized by DLS using Malvern Zetasizer (Nano ZS 90, Malvern, UK) at 25°C. The morphology of the nanoparticles was observed by TEM (HT7700, Hitachi Ltd., Japan). FRET assay was conducted to evaluate the formation of the protein/dendrimer complex and in vitro protein release. RBITC-labeled dendrimer was used to quench the fluorescence of BSA-FITC in the formed complex. Heparin sodium or native BSA was added into the complex solution to trigger the release of proteins. The fluorescence spectra were recorded by a Hitachi F-4500 fluorescence spectrophotometer (Hitachi Ltd., Japan) at excitation and emission wavelengths of λex = 470 nm and λem = 500 – 700 nm, respectively.
Cell culture and cytosolic protein delivery
HeLa cells [a human cervical carcinoma cell line, American Type Culture Collection (ATCC)] and NIH3T3 cells (a mouse embryo fibroblast cell line, ATCC) were cultured in Dulbecco’s modified Eagle’s medium (DMEM, Gibco). MDA-MB-231 cells (a human mammary cancer cell line, ATCC) were cultured in minimum essential medium (MEM, GIBO). The cell culture media contain 10% fetal bovine serum (FBS, Gemini), penicillin (100 μg/ml), and streptomycin (100 μg/ml) at 37°C under 5% CO2.
HeLa cells were seeded in 48-well plates before cytosolic protein delivery. The protein solutions were mixed with dendrimer at various weight ratios. The complex solutions were diluted with 100 μl of serum-free DMEM and incubated for 30 min at room temperature and further diluted with 150 μl of serum-free DMEM. The culture media were removed, and the cells were washed twice with PBS, before adding the protein complex solutions into the wells. After incubation for 4 hours, the culture media containing proteins were removed, and the cells were washed twice with PBS. The fluorescence intensity of the treated cells was determined by flow cytometry (BD FACSCalibur, San Jose). The cells were also observed by laser scanning confocal microscopy (LSCM, Leica SP5, Germany). The commercial protein delivery reagents such as PULSin and TransEx were used as positive controls according to the manufacturers’ protocols. Naked proteins without complexation with the dendrimers were used as negative controls. In a separate study, trypan blue (0.04%) was added to the treated cells before flow cytometry analysis to quench the fluorescence of proteins physically adsorbed on cell membrane. The time-dependent intracellular delivery of dendrimer/protein complexes into HeLa cells was observed by LCSM. The cells were incubated with dendrimer/BSA-FITC complexes for 1, 2, 3, 4, 6, and 12 hours, respectively. The acidic organelles in the treated cells were stained by LysoTracker Red (DND-99, Invitrogen) before confocal imaging. To investigate the internalization mechanism of protein/dendrimer complexes, we used BSA-FITC as the model protein. HeLa cells were preincubated with endocytosis inhibitors such as cytochalasin D (10 μM), chlorpromazine (20 μM), genistein (700 μM), and methyl-β-cyclodextrin (10 mM) for 2 hours before protein transduction experiments, respectively, or incubated at 4°C during cytosolic protein delivery. The cells treated without any inhibitors and transduced at 37°C were tested as a control.
Cell viability assay
The viability of cells incubated with dendrimer/protein complexes was evaluated by a standard 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay. Briefly, HeLa cells or NIH3T3 cells were seeded in a 96-well plate overnight. Then, the culture medium in each well was replaced by 100 μl of serum-free DMEM containing dendrimer/protein complexes, native proteins, or dendrimers. The concentrations of proteins and dendrimers were equal to the optimal concentrations during protein delivery. The cells were incubated for 4 hours, and the media were replaced by 100 μl of fresh DMEM containing 10% FBS. After another 20 hours of incubation, the cells were further tested by MTT assay to determine the cell viability. Five repeats were performed for each sample.
β-Gal staining and activity assay
Cytosolic delivery of enzymes such as β-Gal into HeLa cells was determined by intracellular β-Gal enzymatic activity measurements. In situ X-Gal staining kit and a quantitative β-Gal enzyme activity assay kit (Beyotime Biotech) were conducted according to the manufacturer’s protocols. Briefly, HeLa cells were treated with dendrimer/β-Gal complexes for 4 hours with a similar procedure as described above. Then, the treated cells were washed twice with PBS and fixed for 10 min at room temperature. The cells were further washed three times with PBS and incubated with the working solution containing 5% X-Gal for 2 hours at 37°C in the absence of CO2. The treated cells were further washed with PBS and observed by an optical microscope (Olympus, Japan). The intracellular β-Gal activity of the treated cells was quantitatively analyzed using a β-Gal enzyme activity assay kit. Generally, the treated cells were lysed with cell lysis buffer. Then, 50 μl of cell lysates was mixed with 50 μl of working solution containing enzyme substrate O-nitrophenyl-β-d-galactopyranoside and incubated for 30 min at 37°C. After incubation, 150 μl of stop reaction solution was added in each well. The enzyme activity was determined by measurement of the absorbance of the solution at 420 nm using a microplate reader (Thermo Fisher Scientific, Germany). The enzyme activity of native β-Gal at the same concentration was measured as control and defined as 100%. For in vitro β-Gal activity assay, the enzyme activity was measured by X-Gal staining assay. Generally, the enzyme/P4 complex solutions were incubated with substrate solution containing X-Gal at 37°C for 1 hour. Dimethyl sulfoxide was added to dissolve the yielding product, and the absorbance of the solutions at 633 nm was recorded by a microplate reader. Native β-Gal activity was measured as control. The recovery of enzyme activity was conducted by the addition of BSA (0.1 mg/ml) or heparin sodium (0.1 mg/ml) into the complex solutions.
HRP staining and activity
The cytosolic HRP delivery was determined by intracellular HRP enzymatic activity assay. Intracellularly delivered HRP catalyzed a nonfluorescent substrate, Amplex Red, to a fluorescent product, resorufin, in the presence of hydrogen peroxide. Briefly, HeLa cells were treated with dendrimer/HRP complexes for 4 hours with a similar procedure. Then, the cells were washed three times with PBS and incubated in PBS containing Amplex Red (50 μM) and hydrogen peroxide (500 μM). After 30 min of incubation at room temperature, the treated cells were washed three times and observed by LSCM. In another typical intracellular enzyme activity assay, TMB was used as a colorless substrate and turned into a blue product in the presence of HRP. Generally, cytosolic HRP delivery was performed with a similar procedure, and the treated cells were washed five times with PBS. TMB substrate (0.5 ml; 10 μg/ml) dissolved in acetate buffer (pH 5) with 3 mM hydrogen peroxide was added into each well. After 10 min of incubation at room temperature, the mixture solution in each well was imaged by an optical camera. Cytosolic enzyme activity of native HRP without dendrimer complexion was measured as a control. The in vitro HRP enzymatic activity was measured by TMB staining assay. Generally, the native HRP and HRP/P4 complex solutions were centrifuged at 13,000 rpm for 30 min. The supernatants were diluted with distilled water and incubated with TMB solution. The enzyme activity of HRP was determined by measurement of the absorbance of the solution at 630 nm by a microplate reader at 30-s intervals for 11 min. The recovery of enzyme activity was conducted by the addition of BSA (0.1 mg/ml) or heparin sodium (0.1 mg/ml) into the complex solutions.
Cytosolic delivery of saporin, RNase A, and trypsin
The cytosolic delivery of toxic proteins was tested on MDA-MB-231 cells (saporin and RNase A) and HeLa cells (trypsin). The cells were cultured in 96-well plates overnight. Saporin, RNase A, and trypsin complexes with P4 were prepared as described above. The dendrimer/protein complexes were diluted with 100 μl of serum-free MEM and incubated for 30 min. Then, the mixtures were further diluted with 500 μl of serum-free MEM. The cell culture media were removed, and the cells were washed with PBS. Protein complexes (100 μl) were added into each well. After 6 hours of incubation, the media were replaced with 100 μl of fresh MEM containing 10% FBS, and the cells were further incubated for 42 hours. The cytotoxicity of cells treated with saporin, RNase A, and trypsin complexes was evaluated by MTT assay. Five repeats were tested for each sample. Proteins were tested at various concentrations. Naked saporin, RNase A, trypsin, and dendrimers at equal concentrations were tested as controls. For in vitro RNase A activity assay, the enzyme activity was measured by an RNaseAlert Kit according to the manufacturer’s protocol. Briefly, RNaseAlert substrate dissolved in assay buffer was incubated with native enzyme or the RNase A/P4 complex in RNase-free buffer. The recovery of enzyme activity was conducted by the addition of BSA (0.1 mg/ml) or heparin sodium (0.1 mg/ml) into the complex solutions. The fluorescence intensity was recorded at 5-min intervals for 30 min. The excitation and emission wavelengths were λex = 490 nm and λem = 520 nm, respectively.
sgRNA design and synthesis
Transcription templates were designed by online tools (http://crispr.mit.edu/ and http://chopchop.cbu.uib.no/). A 65-nucleotiode (nt) specific Cas9 forward primer containing the T7 promoter and the 20-nt AAVS1, HBB, EGFP, and CTNNB1 targeting sequence and an 83-nt universal reverse primer (table S1) containing the 3′ tail sequence of sgRNA were mixed for the polymerase chain reaction (PCR). The PCR was set up in the thermal cycler under the following conditions: 97°C for 3 min; 40 cycles of 97°C for 30 s, 55°C for 30 s, and 68°C for 10 s; and 68°C for 10 s. After the PCR program was completed, the PCR sample was purified by the MiniElute PCR Purification Kit (Qiagen) and then used as a DNA template for in vitro transcription reaction. The in vitro transcription reaction to generate sgRNA was performed with DNA template for 12 hours at a 37°C water bath using the MEGAscript T7 Transcription Kit (Invitrogen). The sgRNA product was then purified by the RNeasy Mini Kit (Qiagen). After the purification, the sample was ran on a polyacrylamide gel electrophoresis gel to visualize the sgRNA products and quantified with NanoDrop (Thermo Fisher Scientific, Germany).
AAVS1, HBB, and CTNNB1 gene disruption assay
The 293T cells, 293T-EGFP cells, HCT-116 cells, and HT-29 cells were cultured in DMEM (Gibco). The cell culture medium contains 10% FBS (Gemini), penicillin (100 μg/ml), and streptomycin (100 μg/ml) at 37°C under 5% CO2. P4/RNP-mediated transfection was conducted by seeding into a six-well plate at a density of 2 × 105 cells per well the day before transfection. The doses of Cas9 protein, sgRNA, and P4 in each well were 4, 2, and 8 μg, respectively. Six hours after transfection, transfection mixtures were replaced with DMEM containing 10% FBS and further incubated for 48 hours.
EGFP gene disruption assay
293T-EGFP cells were established by infecting 293T cells with lentivirus harboring an EGFP-expressing cassette. P4/RNP-mediated transfection was conducted by seeding into a glass-bottom cell well at a density of 104 cells per well the day before transfection. The doses of Cas9 protein, sgRNA, and P4 in each well were 2, 1, and 4 μg, respectively. After 4 hours of incubation, the Cas9-containing medium was replaced with fresh DMEM containing 10% FBS and incubated for 48 hours before analyzed by LSCM (ZEISS LSM 880, Germany). For flow cytometer analysis, 293T-EGFP cells were seeded into a six-well plate at a density of 2 × 105 cells per well. The doses of Cas9 protein, sgRNA, and P4 in each well were 4, 2, and 8 μg, respectively. After 48 hours of incubation, the cells were rinsed three times with 1 × PBS and trypsinized with 0.25% trypsin. Subsequently, the cells were resuspended in 200 μl of 1 × PBS and subjected to flow cytometry analysis (Beckman Coulter DxFLEX, USA).
In vitro T7E1 assay
After the treatment of 293T cells, 293T-EGFP cells, HCT-116 cells, and HT-29 cells for 48 hours, genomic DNA of cells was isolated using a DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer’s recommended protocol. The specific PCR amplicons were generated using the primers listed in table S2 and were purified with the QIAquick PCR Purification Kit (Qiagen). The PCR was set up in the thermal cycler with the following conditions: 97°C for 5 min; 40 cycles of 97°C for 30 s, 62°C for 15 s, and 68°C for 30 s. Afterward, a total of 200 ng of PCR products were mixed in 20 μl of nuclease-free ddH2O with 1 × NEBuffer 2, and the mixtures underwent a denature/annealing process to form a heteroduplex: 95°C for 5 min, −2°C s−1 to 85°C, and −0.1°C s−1 to 25°C. Two hundred nanograms of reannealed PCR products was mixed with 0.3 μl of T7E1 in the 200-μl PCR tubes and incubated at 37°C for 30 min following the recommended protocol. Digestion PCR products were analyzed on 2% agarose gels. The gray levels of the undigested band and the digested band were quantified with ImageJ. Indel percentage analysis was calculated on the basis of the following formula: 100 × (1 − (1 − fraction cleaved)1/2), where fraction cleaved = band intensity of each digested band/(band intensity of each digested band + band intensity of undigested band).
Statistical analysis
The data for mean fluorescence intensity, enzyme activity, and cell viability were presented as means ± SD. Statistically significant difference was analyzed by Student’s t test. P < 0.05 was considered significant. N.S., P > 0.05; *P < 0.05, **P < 0.01, and ***P < 0.001.
Supplementary Material
Acknowledgments
We thank N. Chen from Shanghai Institute of Applied Physics, CAS, for the gifts of GFP, YFP, and RFP used in this study. Funding: We appreciate the grants from the National Natural Science Foundation of China (21725402, 21474030, and 81872807), the Shanghai Municipal Science and Technology Commission (17XD1401600), Fundamental Research Funds for the Central Universities, and Thousand Talents Plan. Author contributions: C.L. and H.W. synthesized and characterized the materials. C.L. performed cytosolic protein delivery experiments, analyzed the data, and wrote the manuscript. T.W. and Y.P. conducted and analyzed the CRISPR-Cas9 genome editing experiments. S.Z. investigated the mechanism of complex uptake and enzyme activity assay. Y.C. designed and supervised the study and wrote the manuscript. All authors read and approved the manuscript. Competing interests: Y.C., C.L., and H.W. are inventors on a patent application related to this work filed by East China Normal University (no. 201910187501.8, filed 13 March 2019). Y.C., Y.P., T.W., and C.L. have applied for an additional patent application related to this work, filed by East China Normal University (filed 10 April 2019). The authors declare no other competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaaw8922/DC1
Fig. S1. Characterization of the modified dendrimers by 1H NMR.
Fig. S2. Cytosolic delivery of BSA.
Fig. S3. Characterization of the BSA-FITC/dendrimer-RBITC complex by FRET.
Fig. S4. Confocal images of HeLa cells treated with the BSA-FITC/P4 complex at different times.
Fig. S5. Characterization of the protein/dendrimer complexes by DLS and TEM.
Fig. S6. Enzyme activity of β-Gal, HRP, and RNase A.
Fig. S7. CRISPR-Cas9 gene editing in different cells.
Table S1. Sequences of sgRNA used in this study.
Table S2. Primer sequences for PCR amplification of target genes.
REFERENCES AND NOTES
- 1.Mix K. A., Lomax J. E., Raines R. T., Cytosolic delivery of proteins by bioreversible esterification. J. Am. Chem. Soc. 139, 14396–14398 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lee K., Conboy M., Park H. M., Jiang F., Kim H. J., Dewitt M. A., Mackley V. A., Chang K., Rao A., Skinner C., Shobha T., Mehdipour M., Liu H., Huang W.-c., Lan F., Bray N. L., Li S., Corn J. E., Kataoka K., Doudna J. A., Conboy I., Murthy N., Nanoparticle delivery of Cas9 ribonucleoprotein and donor DNA in vivo induces homology-directed DNA repair. Nat. Biomed. Eng. 1, 889–901 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rouet R., Thuma B. A., Roy M. D., Lintner N. G., Rubitski D. M., Finley J. E., Wisniewska H. M., Mendonsa R., Hirsh A., de Oñate L., Barrón J. C., McLellan T. J., Bellenger J., Feng X., Varghese A., Chrunyk B. A., Borzilleri K., Hesp K. D., Zhou K., Ma N., Tu M., Dullea R., McClure K. F., Wilson R. C., Liras S., Mascitti V., Doudna J. A., Receptor-mediated delivery of CRISPR-Cas9 endonuclease for cell-type-specific gene editing. J. Am. Chem. Soc. 140, 6596–6603 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cheng G., Li W., Ha L., Han X., Hao S., Wan Y., Wang Z., Dong F., Zou X., Mao Y., Zheng S.-Y., Self-assembly of extracellular vesicle-like metal–organic framework nanoparticles for protection and intracellular delivery of biofunctional proteins. J. Am. Chem. Soc. 140, 7282–7291 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Zelikin A. N., Ehrhardt C., Healy A. M., Materials and methods for delivery of biological drugs. Nat. Chem. 8, 997–1007 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Yim N., Ryu S.-W., Choi K., Lee K. R., Lee S., Choi H., Kim J., Shaker M. R., Sun W., Park J.-H., Kim D., Do Heo W., Choi C., Exosome engineering for efficient intracellular delivery of soluble proteins using optically reversible protein–protein interaction module. Nat. Commun. 7, 12277 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Postupalenko V., Desplancq D., Orlov I., Arntz Y., Spehner D., Mely Y., Klaholz B. P., Schultz P., Weiss E., Zuber G., Protein delivery system containing a nickel-immobilized polymer for multimerization of affinity-purified his-tagged proteins enhances cytosolic transfer. Angew. Chem. Int. Ed. 54, 10583–10586 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Schmidt S., Adjobo-Hermans M. J., Wallbrecher R., Verdurmen W. P., Bovée-Geurts P. H., van Oostrum J., Milletti F., Enderle T., Brock R., Detecting cytosolic peptide delivery with the GFP complementation assay in the low micromolar range. Angew. Chem. Int. Ed. 54, 15105–15108 (2015). [DOI] [PubMed] [Google Scholar]
- 9.Sharei A., Zoldan J., Adamo A., Sim W. Y., Cho N., Jackson E., Mao S., Schneider S., Han M.-J., Lytton-Jean A., Basto P. A., Jhunjhunwala S., Lee J., Heller D. A., Kang J. W., Hartoularos G. C., Kim K. S., Anderson D. G., Langer R., Jensen K. F., A vector-free microfluidic platform for intracellular delivery. Proc. Natl. Acad. Sci. U.S.A. 110, 2082–2087 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Akishiba M., Takeuchi T., Kawaguchi Y., Sakamoto K., Yu H.-H., Nakase I., Takatani-Nakase T., Madani F., Gräslund A., Futaki S., Cytosolic antibody delivery by lipid-sensitive endosomolytic peptide. Nat. Chem. 9, 751–761 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Nischan N., Herce H. D., Natale F., Bohlke N., Budisa N., Cardoso M. C., Hackenberger C. P. R., Covalent attachment of cyclic TAT peptides to GFP results in protein delivery into live cells with immediate bioavailability. Angew. Chem. Int. Ed. 54, 1950–1953 (2015). [DOI] [PubMed] [Google Scholar]
- 12.Dashti N. H., Abidin R. S., Sainsbury F., Programmable in vitro coencapsidation of guest proteins for intracellular delivery by virus-like particles. ACS Nano 12, 4615–4623 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Zuris J. A., Thompson D. B., Shu Y., Guilinger J. P., Bessen J. L., Hu J. H., Maeder M. L., Joung J. K., Chen Z.-Y., Liu D. R., Cationic lipid-mediated delivery of proteins enables efficient protein-based genome editing in vitro and in vivo. Nat. Biotechnol. 33, 73–80 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wang M., Zuris J. A., Meng F. T., Rees H., Sun S., Deng P., Han Y., Gao X., Pouli D., Wu Q., Efficient delivery of genome-editing proteins using bioreducible lipid nanoparticles. Proc. Natl. Acad. Sci. U.S.A. 113, 2868–2873 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang Z., Shen W., Ling J., Yan Y., Hu J., Cheng Y., The fluorination effect of fluoroamphiphiles in cytosolic protein delivery. Nat. Commun. 9, 1377 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng Y. Y., Fluorinated polymers in gene delivery. Acta Polym. Sin. 8, 1234–1245 (2017). [Google Scholar]
- 17.Mout R., Ray M., Yesilbag Tonga G., Lee Y.-W., Tay T., Sasaki K., Rotello V. M., Direct cytosolic delivery of CRISPR/Cas9-ribonucleoprotein for efficient gene editing. ACS Nano 11, 2452–2458 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mout R., Ray M., Tay T., Sasaki K., Yesilbag Tonga G., Rotello V. M., General strategy for direct cytosolic protein delivery via protein–nanoparticle co-engineering. ACS Nano 11, 6416–6421 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tang R., Wang M., Ray M., Jiang Y., Jiang Z., Xu Q., Rotello V. M., Active targeting of the nucleus using nonpeptidic boronate tags. J. Am. Chem. Soc. 139, 8547–8551 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shao D., Li M., Wang Z., Zheng X., Lao Y.-H., Chang Z. M., Zhang F., Lu M., Yue J., Hu H., Yan H., Chen L., Dong W.-f., Leong K. W., Bioinspired diselenide-bridged mesoporous silica nanoparticles for dual-responsive protein delivery. Adv. Mater. 30, 1801198 (2018). [DOI] [PubMed] [Google Scholar]
- 21.He H., Chen Y., Li Y., Song Z., Zhong Y., Zhu R., Cheng J., Yin L., Effective and selective anti-cancer protein delivery via all-functions-in-one nanocarriers coupled with visible light-responsive, reversible protein engineering. Adv. Funct. Mater. 28, 1706710 (2018). [Google Scholar]
- 22.Chang H., Lv J., Gao X., Wang X., Wang H., Chen H., He X., Li L., Cheng Y., Rational design of a polymer with robust efficacy for intracellular protein and peptide delivery. Nano Lett. 17, 1678–1684 (2017). [DOI] [PubMed] [Google Scholar]
- 23.Lee Y., Ishii T., Cabral H., Kim H. J., Seo J.-H., Nishiyama N., Oshima H., Osada K., Kataoka K., Charge-conversional polyionic complex micelles—Efficient nanocarriers for protein delivery into cytoplasm. Angew. Chem. Int. Ed. 48, 5309–5312 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Lee Y., Ishii T., Kim H. J., Nishiyama N., Hayakawa Y., Itaka K., Kataoka K., Efficient delivery of bioactive antibodies into the cytoplasm of living cells by charge-conversional polyion complex micelles. Angew. Chem. Int. Ed. 49, 2552–2555 (2010). [DOI] [PubMed] [Google Scholar]
- 25.Postupalenko V., Sibler A. P., Desplancq D., Nomine Y., Spehner D., Schultz P., Weiss E., Zuber G., Intracellular delivery of functionally active proteins using self-assembling pyridylthiourea-polyethylenimine. J. Control. Release 178, 86–94 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Sun W. J., Ji W. Y., Hall J. M., Hu Q. Y., Wang C., Beisel C. L., Gu Z., Self-assembled DNA nanoclews for the efficient delivery of CRISPR-Cas9 for genome editing. Angew. Chem. Int. Ed. 127, 12197–12201 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ng D. Y. W., Arzt M., Wu Y., Kuan S. L., Lamla M., Weil T., Constructing hybrid protein zymogens through protective dendritic assembly. Angew. Chem. Int. Ed. 53, 324–328 (2014). [DOI] [PubMed] [Google Scholar]
- 28.Sgolastra F., deRonde B. M., Sarapas J. M., Som A., Tew G. N., Designing mimics of membrane active proteins. Acc. Chem. Res. 46, 2977–2987 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tezgel A. Ö., Jacobs P., Backlund C. M., Telfer J. C., Tew G. N., Synthetic protein mimics for functional protein delivery. Biomacromolecules 18, 819–825 (2017). [DOI] [PubMed] [Google Scholar]
- 30.Posey N. D., Hango C. R., Minter L. M., Tew G. N., The role of cargo binding strength in polymer-mediated intracellular protein delivery. Bioconjug. Chem. 29, 2679–2690 (2018). [DOI] [PubMed] [Google Scholar]
- 31.Lv J., He B. W., Yu J. W., Wang Y. T., Wang C. P., Zhang S., Wang H., Hu J. J., Zhang Q., Cheng Y. Y., Fluoropolymers for intracellular and in vivo protein delivery. Biomaterials 182, 167–175 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Lv S., Wu Y., Cai K., He H., Li Y., Lan M., Chen X., Cheng J., Yin L., High drug loading and sub-quantitative loading efficiency of polymeric micelles driven by donor–receptor coordination interactions. J. Am. Chem. Soc. 140, 1235–1238 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Wang J., Wu W., Zhang Y. J., Wang X., Qian H. Q., Liu B. R., Jiang X. Q., The combined effects of size and surface chemistry on the accumulation of boronic acid-rich protein nanoparticles in tumors. Biomaterials 35, 866–878 (2014). [DOI] [PubMed] [Google Scholar]
- 34.Wang X., Zhen X., Wang J., Zhang J. L., Wu W., Jiang X. Q., Doxorubicin delivery to 3D multicellular spheroids and tumors based on boronic acid-rich chitosan nanoparticles. Biomaterials 34, 4667–4679 (2013). [DOI] [PubMed] [Google Scholar]
- 35.Mahadevi A. S., Sastry G. N., Cation-π interaction: Its role and relevance in chemistry, biology, and material science. Chem. Rev. 113, 2100–2138 (2012). [DOI] [PubMed] [Google Scholar]
- 36.Kennedy C. R., Lin S., Jacobsen E. N., The cation-π interaction in small-molecule catalysis. Angew. Chem. Int. Ed. 55, 12596–12624 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Liu C., Shao N., Wang Y., Cheng Y., Clustering small dendrimers into nanoaggregates for efficient DNA and siRNA delivery with minimal toxicity. Adv. Healthc. Mater. 5, 584–592 (2016). [DOI] [PubMed] [Google Scholar]
- 38.Piest M., Engbersen J. F. J., Role of boronic acid moieties in poly(amido amine)s for gene delivery. J. Control. Release 155, 331–340 (2011). [DOI] [PubMed] [Google Scholar]
- 39.Sun L., Gao Y., Wang Y., Wei Q., Shi J., Chen N., Li D., Fan C., Guiding protein delivery into live cells using DNA-programmed membrane fusion. Chem. Sci. 9, 5967–5975 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cong L., Ran F. A., Cox D., Lin S. L., Barretto R., Habib N., Hsu P. D., Wu X. B., Jiang W. Y., Marraffini L., Zhang F., Multiplex genome engineering using CRISPR/Cas systems. Science 339, 819–823 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaaw8922/DC1
Fig. S1. Characterization of the modified dendrimers by 1H NMR.
Fig. S2. Cytosolic delivery of BSA.
Fig. S3. Characterization of the BSA-FITC/dendrimer-RBITC complex by FRET.
Fig. S4. Confocal images of HeLa cells treated with the BSA-FITC/P4 complex at different times.
Fig. S5. Characterization of the protein/dendrimer complexes by DLS and TEM.
Fig. S6. Enzyme activity of β-Gal, HRP, and RNase A.
Fig. S7. CRISPR-Cas9 gene editing in different cells.
Table S1. Sequences of sgRNA used in this study.
Table S2. Primer sequences for PCR amplification of target genes.