

Summary
Small heat-shock proteins (sHSPs) are molecular chaperones that bind partially and globally unfolded states of their client proteins. Previously, we discovered that the archaeal Hsp16.5, which forms ordered and symmetric 24-subunit oligomers, can be engineered to transition to an ordered and symmetric 48-subunit oligomer by insertion of a peptide from human HspB1 (Hsp27). Here, we uncovered the existence of an array of oligomeric states (30 to 38 subunits) that can be populated as a consequence of altering the sequence and length of the inserted peptide. Polydisperse Hsp16.5 oligomers displayed higher affinity to a model client protein consistent with a general mechanism for recognition and binding that involves increased access of the hydrophobic N-terminal region. Our findings, which integrate structural and functional analyses from evolutionarily-distant sHSPs, support a model wherein the modular architecture of these proteins encodes motifs of oligomer polydispersity, dissociation and expansion to achieve functional diversity and regulation.
Introduction
Small heat-shock proteins (sHSPs) are a ubiquitous class of molecular chaperones that play a central role in the stress resistance of all organisms (Carra et al., 2017; Jakob et al., 1993). While they share the general property of binding non-native and unfolded proteins with other heat shock proteins, sHSP chaperone activity does not involve the direct consumption of ATP (McHaourab et al., 2009; Van Montfort et al., 2002). They bind their client proteins in stable complexes thereby inhibiting aggregation and as such provide an energy-efficient, first line defense against protein aggregation (Carra et al., 2017; Haslbeck and Buchner, 2002; Haslbeck et al., 2005; Haslbeck and Vierling, 2015). Humans express 10 distinct sHSPs that have critical physiological roles including maintenance of lens transparency and the integrity of cardiac and skeletal muscles (Clark et al., 2012; Garrido et al., 2012; Kappe et al., 2003; Kappé et al., 2010; Sun and MacRae, 2005). Mutations in human sHSPs have been associated with inherited diseases, most notably cataract and cardiomyopathy (Andley, 2007; Andley et al., 2011; Ecroyd and Carver, 2009; Strauch and Haslbeck, 2016; Treweek et al., 2015; Vicart et al., 1998).
The molecular architecture of sHSPs is built around a conserved 90-amino acid domain, the α-crystallin domain (de Jong et al., 1998; Kappé et al., 2010). This domain forms a dimeric building block which assembles into oligomers through interactions in the sequence-variable N-terminal domain and the C-terminal tail (Berengian et al., 1997; Berengian et al., 1999; Braun et al., 2011; Jehle et al., 2010; Jehle et al., 2011; Koteiche et al., 1998; Laganowsky et al., 2010; Laganowsky and Eisenberg, 2010; McHaourab et al., 1997; van Montfort et al., 2001). Crystal structures and spectroscopic analyses have defined the architecture of this building block across the evolutionary spectrum, identifying two motifs of dimerization so far (Jehle et al., 2010; Kim et al., 1998; Kim et al., 2003b; Koteiche et al., 1998; Koteiche and McHaourab, 1999; van Montfort et al., 2001). The packing of these dimers dictates the structure of sHSP oligomers which display remarkable diversity in size and symmetry (Hochberg and Benesch, 2015).
Far from being static “sponges” for unfolded proteins, sHSP oligomeric structures, particularly from eukaryotes, are characterized by a fascinating spectrum of conformational flexibility (Carra et al., 2017; Haslbeck and Vierling, 2015). Multiple studies have demonstrated that polydispersity of sHSP oligomers is a mechanism for regulation of the binding affinity and capacity for client proteins (Koteiche et al., 2015; Shashidharamurthy et al., 2005; Sobott et al., 2002; Stengel et al., 2010). Dynamic oligomer dissociation, manifested by subunit exchange between oligomers, exposes a flexible N-terminal region containing putative interaction sites that are otherwise inaccessible (White et al., 2006). Shifts in the oligomeric equilibrium from large oligomers to dimers, e.g. by phosphorylation of Hsp27 and αB-crystallin, enhance the affinity to client proteins and the binding capacity (Ecroyd et al., 2007; Rogalla et al., 1999; Shashidharamurthy et al., 2005; Shi et al., 2013; Thériault et al., 2004). Alternatively, oligomer expansion, resulting from an increase in the number of subunits per oligomer, can expose the N-terminal region (Bova et al., 1999; McHaourab et al., 2012). Initially observed in cataract-linked mutants of α-crystallins (Koteiche and McHaourab, 2006; Kumar et al., 1999; Shroff et al., 2000), the structural basis of expansion was uncovered by an engineered variant of Methanococcus jannaschii Hsp16.5 (Shi et al., 2013; Shi et al., 2006). Insertion of a 14-amino acid peptide (hereafter referred to as the P1 peptide), found at the junction of the N-terminal and α-crystallin domain of human Hsp27 (HspB1), at the equivalent position in Hsp16.5 resulted in the expansion of the oligomer from 24 subunits to 48 subunits (McHaourab et al., 2012; Shi et al., 2013; Shi et al., 2006). P1 plays an important role in the dynamics of Hsp27 concentration-dependent equilibrium dissociation. Shashidharamurthy et al. demonstrated that the deletion of this peptide shifts the equilibrium towards the larger oligomers and concomitantly reduce the apparent affinity of Hsp27 towards destabilized mutants of the model client protein T4 lysozyme (T4L) (Shashidharamurthy et al., 2005). A high resolution structure of the expanded Hsp16.5 variant revealed that the changes in the sequence of the N-terminal region lead to two different orientations of the C-terminal tail which in turn enable two different packing modes between α-crystallin dimers within the same oligomer (McHaourab et al., 2012). This lead to the appearance of a large window in the expanded oligomer while preserving the octahedral symmetry of the assembly.
The expansion of the P1-containing Hsp16.5 variant was unexpected considering the lack of polydispersity of Wild Type (WT) Hsp16.5, and raised the question of whether oligomeric plasticity is an intrinsic property of sHSPs. That is, are all sHSPs capable of displaying a broad spectrum of oligomeric structures? A related question is whether the P1 peptide sequence encodes specific structural and dynamic properties that are essential to oligomer expansion. However, the P1 peptide was not resolved in the crystal structure, hindering an understanding of its conformational properties. While P1 is not native to Hsp16.5, its role in modulating oligomer dissociation of human Hsp27 implies evolutionary optimization of its sequence motifs (McDonald et al., 2012; Shashidharamurthy et al., 2005; Shi et al., 2013). Indeed, sequence alignment of Hsp27 reveals that only humans (Homo sapiens), chimpanzee (Pan troglodytes) and rhesus macaque (Macaca mulatta) have identical P1 sequences (Fig S1). Even in the evolutionarily-close mouse (Mus musculus), P1 has undergone substantial sequence changes even in comparison with the less conserved N-terminal domain (Table S1).
The work reported here follows up on structural and functional studies of the role of P1 in Hsp27 equilibrium dissociation and in the expansion of the Hsp16.5 oligomer, hereafter referred to as Hsp16.5-P1. Hsp16.5 offers a more tractable model for this investigation given its amenability to biophysical and EM analyses than the heterogeneous Hsp27 (Haley et al., 2000). For this purpose, the amino acid composition and/or length of the P1 peptide sequence were manipulated and the resulting variants of Hsp16.5-P1 were characterized for the size of the oligomer and its polydispersity by size exclusion chromatography (SEC), multi-angle light scattering (MALS), and blue native gel electrophoresis (BN-PAGE). Furthermore, we visualized the various populated oligomeric assemblies using negative stain electron microscopy (EM) and analyzed oligomer distributions by native mass spectrometry (MS) (Benesch et al., 2006). Finally, the changes in the oligomeric assembly of the variants were correlated with their affinities to a destabilized mutant of T4L.
A number of general themes emerge from this systematic investigation that extends the current understanding of the structural basis of sHSPs polydispersity and activation. We find that changes in the amino acid sequence of P1 lead to previously unobserved oligomeric assemblies of Hsp16.5-P1 characterized by numbers of subunits distinct from the WT and P1 forms. Two of the engineered variants can form concurrently 24- and 36-subunit oligomers, suggesting that the latter are intermediate states on the assembly pathway from 24 to 48 subunits. Moreover, analysis of substrate binding affinities of the Hsp16.5-P1 variants further reinforces the relationship between oligomer size, polydispersity and chaperone efficiency. Finally, a sequence motif in P1 is identified as critical to the dynamic oligomer dissociation of human Hsp27 and alters its apparent chaperone activity.
Results and discussion
Changes in the sequence of P1 induce polydispersity of Hsp16.5-P1.
Inspection of the P1 peptide reveals three distinct sequence characteristics: a PLPP sequence at the N-terminus and an alanine rich AVAA sequence at the C-terminus interrupted by an IESP sequence (Fig. 1A). To address the question of whether the exact sequence of P1 is required for oligomer expansion and to assess the role of the various motifs, we generated nine variants (referred to as Var-1 to −9, Fig. 1A) that were expressed in E. Coli (Fig. S2) and analyzed for oligomer size distribution by BN-PAGE (Fig. 1B) and SEC in conjunction with MALS (Fig. 1C). Substitution of amino acids or changes to the length of P1 led to profound alterations of oligomer size and heterogeneity. Therefore, the variants are grouped and discussed based on the properties of the resulting oligomer.
Figure 1. Characterization of the oligomers formed by insertion of peptides in Hsp16.5.

A, Sequences of the peptides inserted in the Hsp16.5 sequence between residues 33-34. The variants are grouped by color according to oligomeric properties shown in panels B and C. B, BN-PAGE. Left panel, 5% acrylamide gel; right panel, 7.5% acrylamide gel (STAR Methods). The approximate number of subunits corresponding to band mobility is shown with small arrows on the left side of the left panel. The molecular weight marker is shown in the first lane of the right panel. Large arrows highlight mobility differences of the P1-insert and WT constructs in the 7.5% acrylamide gel. Lanes have been rearranged according to variants after documenting the gels. C, SEC-MALS analysis of purified variants. Variants are grouped according to similar characteristics. The molecular mass range that is sampled across the elution peak is indicated by the slope of the line for each variant. The vertical lines benchmark the WT (dotted red) and the P1-insert (dotted black) elution peak. (See also Figure S2 and Table S2)
A subset of P1 variants, color coded in black in Fig. 1, maintained the 48 subunit oligomer as the main assembly but appeared to increase its heterogeneity. Var-1, where two N-terminal residues of P1 were substituted by acidic residues, and Var-2, where P1 was extended by two Ala residues at the C-terminus, displayed a wide range of molecular masses by SEC-MALS (Fig. 1B-C and Table S2). In addition, BN-PAGE analysis indicated the presence of a 24-subunit oligomer more prominently for Var-1. For reference, Fig. 1 shows the corresponding analysis for the insertion of native P1, which in accordance with previous results led to a well-defined expanded 48-subunit oligomer with an average molar mass of ~0.8 MDa as observed by SEC-MALS (Fig. 1B-C and Table S2) (McHaourab et al., 2012; Shi et al., 2013; Shi et al., 2006).
Perhaps the most remarkable effect on the assembly was observed for a set of variants, color coded green in Fig. 1A, generated by diverse manipulations of the P1 sequence. These manipulations ranged from increasing the redundancy of the peptide sequence by complete substitution with an isometric poly-Ala insert (Var-3) to truncation of 4 residues from either its N-terminus (Var-5) or the C-terminus (Var-6). A previously unobserved ensemble of oligomer sizes centered around 36 subunits was invariably detected by SEC-MALS. However, despite the comparable average molar mass, the oligomeric populations of Var-5 and Var-6 were more polydisperse than Var-3, as reflected by larger spreads in molar mass distributions across their SEC profiles (Fig. 1C).
Shortening the P1 peptide stabilizes a 36-subunit oligomeric cluster.
Further truncation of the P1 peptide by an additional four residues from either the N-terminus (Var-7) or C-terminus (Var-8) leads to two distinct populations corresponding to a 36-subunit cluster and WT-like 24-subunit oligomers (Fig. 1C, blue traces). The concurrent assembly into two forms suggests that they are of similar free energy or that one of them is kinetically trapped. The relative population of the 36-subunit oligomer was greater in Var-8 where the N-terminal half of P1 remained intact. Similarly, a shift toward formation of larger oligomeric species was observed in Var-6 where the N-terminus of P1 remains intact relative to Var-5 where the first 4 amino acids where truncated (Table S2).
Comparative analysis by BN-PAGE (Fig. 1B) complemented the SEC-MALS results underscoring the relative polydispersity within the oligomeric ensemble of the variants. WT Hsp16.5 and Hsp16.5-P1 resolved as near homogenous 24- and 48-subunit entities, respectively, on a 7.5% acrylamide gel. In contrast, Var-3 through Var-6 demonstrated broad heterogeneity around the putative 36-subunit cluster. Importantly, BN-PAGE exposed a discrete oligomer species for Var-3 which appears to consist of about 30 subunits.
These observations demonstrate that while the chemical properties and length of P1 modulate the oligomer size distribution of Hsp16.5, there were no simple correlations between the P1 sequence details and the changes in oligomer assembly. In contrast, a strong dependence on the length of the insertion suggests that the intrinsic structural flexibility of Hsp16.5 was more of a determining factor of oligomer size and heterogeneity than the exact sequence inserted. Only the N-terminus of the P1 sequence may have a structural role in promoting formation of larger oligomers as insertion of only the four leading N-terminal PLPP residues (Var-9) was sufficient to promote formation of a 36-subunit cluster.
EM analysis of the 36-subunit cluster.
While SEC-MALS and BN-PAGE revealed changes in oligomer size and distribution, they are inherently low resolution indicators of heterogeneity. Therefore, to visualize the structural features of the 30/36-subunit oligomers, we characterized the variants by negative stain EM as shown in Fig. 2 and Fig. S3. The particles were classified broadly into three classes: a WT-like 24-subunit oligomer, a 36-subunit oligomeric cluster, and the 48-subunit expanded form (Table 1). A fourth minor class is that of a 30-subunit oligomer. The overall trends in populations of the assemblies were consistent with the SEC-MALS and BN-PAGE analysis, emphasizing that short insertions promote expansion and confirming that the only the P1 sequence lead to a homogenous population of 48-subunit assembly.
Figure 2. Three dimensional negative stain-EM reconstructions of representative oligomers from peptide insertion variants of Hsp16.5:

24-subunit (WT-like), 30-subunit, 36-subunit, and 48-subunit (P1-like). The top panel shows a surface representation and the middle panel is a clipped view revealing internal structure. Unlike the 36-subunit oligomer, the internal anatomy of the intermediate 30 subunit class structure lacks density. However, the circumference for these two types of particles is similar. In contrast to the well-ordered arrangement of the 24- and 48-subunit oligomers, the intermediate size oligomers demonstrate enhanced heterogeneity and lack of symmetry. The relative particle distribution for each variant is given in Table 1. The bottom panel correlates the EM analysis with the BN-PAGE results (in cartoon representation) of the oligomer distribution and identifies the predominant species for P1 variants. (See also Figure S3)
Table 1.
Particle distribution of HSP variants determined by negative stain EM
| HSP variant | 24 subunit | 36 subunit | 48 subunit |
|---|---|---|---|
| P1 | - | 7.5 | 92.5 |
| Var-1 | - | 15.1 | 84.7 |
| Var-2 | 3.1 | 57.9 | 39.0 |
| Var-3 | 8.5 | 91.5 | - |
| Var-4 | - | 100 | - |
| Var-5 | 12.0 | 86.2 | 3.8 |
| Var-6 | 1.0 | 97.4 | 1.6 |
| Var-7 | 53.8 | 41.7 | 6.6 |
| Var-8 | 32.9 | 53.1 | 7.4 |
The Hsp16.5 mutant datasets were refined against three representative structures of 24, 36, and 48 subunits with the EMAN multi-refine subroutine in order to partition individual particle images into one of three classes. For each Hsp16.5 mutant the percentage of particles placed into each of the three classes is shown.
To gain insight into the structure of the 36-subunit assembly, we analyzed Var-4, which promoted the most homogeneous assembly of this intermediate (Fig. 1B and Table S2). While classified Var-4 particles revealed a size consistent with 36 subunits, the outer shell appears to have both WT and P1 dimer-dimer arrangements. The 3D reconstruction from 9824 negative stained particles achieved a modest resolution of 28Å (Fig 2: 36-mer panel), which is indicative of structural heterogeneity despite the classification by SEC as consisting primarily of 36 subunits.
Similarly, 3D reconstruction of the 30-subunit assembly, attempted for particle images from Var-8, yielded limited resolution (>25Å) even after class-based sorting, suggesting that this intermediate-sized assembly is structurally heterogeneous. The average structures of 30/36 subunit assemblies from Var-8 and Var-4 reveal gaps both in the internal assembly of dimers as well as the outer shell that has regions of octahedral-like symmetry. Overall, both the 30-subunit and 36-subunit reconstructions resembled that of the 24-subunit and 48-subunit assemblies, but without complete octahedral symmetry and with regions of incomplete outer shells.
Thus, while Hsp16.5-P1 variants can expand into intermediate size oligomers, the reconstructions are suggestive of incomplete outer shells and variable interfaces. An understanding of how such assemblies are populated invokes the principles inferred from the crystal structure of Hsp16.5-P1. Introduction of P1 facilitates changes in the C-terminal tail orientation to accommodate oligomer expansion while leaving the α-crystallin dimer unaffected (McHaourab et al., 2012). In the context of this model, the EM analysis of Var-8 and −4 suggests that either the two orientations of the C-terminal tail corresponding to the WT and P1 oligomer co-exist in the intermediate size assemblies or that this tail adopts a new orientation that is incompatible with either of the two ordered and symmetric 24 and 48-subunit oligomers.
High resolution oligomer binning by native state mass spectrometry.
In light of the structural heterogeneity of the 36-subunit oligomeric cluster and the consequent low resolution of the EM reconstruction, we analyzed a set of variants, selected to represent the three groups of oligomers, by native state mass spectrometry to determine the exact subunit stoichiometries populated by the Hsp16.5-P1 variants. All variants examined returned high quality mass spectra featuring signal >8000 m/z, indicative of high-mass oligomers spanning multiple stoichiometries; such dispersity in structure is shown for Var-3 in Figure 3. To compare with EM particle-binning, we extracted the ion counts for the different oligomers (grouped according to 24-, 48-subunits, and stoichiometries in between) by integrating the signal recorded in defined regions of the spectra (Fig S4). The abundances we obtained in this way showed a remarkable correspondence with the single-particle analysis from EM data (Table 2), despite the simplifications in this analysis.
Figure 3. Oligomer binning of Var-3 by native state mass spectrometry.
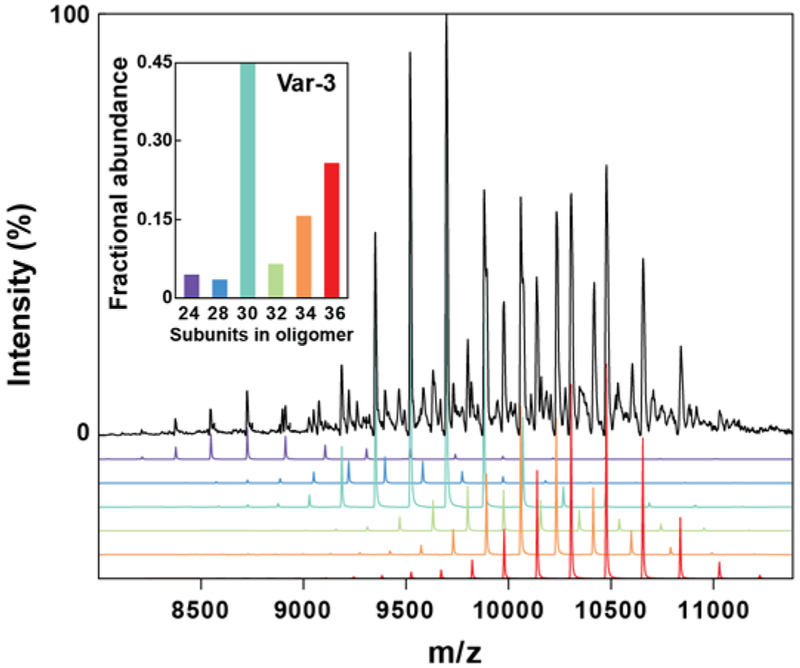
Native mass spectrum of Var-3 revealed the co-population of multiple oligomeric states. The relative abundances of the different states (inset) were extracted from the signal intensity. (See also Table S3)
Table 2.
Relative abundances of HSP variants determined by native MS
| HSP variant |
24 subunit |
28-38 subunits |
48 subunit |
|---|---|---|---|
| P1 | 0.04 | 0.07 | 0.89 |
| Var-2 | 0.11 | 0.52 | 0.37 |
| Var-3 | 0.05 | 0.91 | 0.04 |
| Var-6 | 0.11 | 0.81 | 0.08 |
| Var-7 | 0.44 | 0.39 | 0.17 |
| Var-8 | 0.69 | 0.24 | 0.07 |
The relative abundances of clustered oligomeric states of HSP16.5 variants determined by native mass spectrometry are shown. The relative abundance of each oligomeric subset was determined by integration of the native mass spectrum across the m/z ranges shown in Figure S4.
The high resolution afforded by the MS data allowed us to identify individual stoichiometries. For all investigated variants, we observed assemblies composed of 28 to 38 subunits, which were separated by dimeric intervals. Using a combination of MS and tandem-MS data, in which the ions were collisionally-activated to cause their dissociation and corresponding charge-reduction, we were able to obtain abundances for each stoichiometry offering an additional level of detail with good agreement to all methods presented (Figures S5, S6 and Table S3) (Benesch et al., 2006). This data demonstrated that the 24- and 48- subunit assemblies are well defined entities, with no 26 or 40-46-subunit assemblies observed. In contrast, the 36-subunit assembly was flanked by numerous other assemblies of varying subunit stoichiometries. This observation suggests that the 24- and 48-subunit oligomers are much more favourable symmetries while the 36-subunit oligomer is of marginal stability relative to neighbouring stoichiometries. The size heterogeneity of the oligomers is reminiscent of eukaryotic sHSPs (Aquilina et al., 2003; Basha et al., 2012; Benesch et al., 2008; Haslbeck and Vierling, 2015). The difference between oligomers by two subunits is consistent with the α-crystallin dimer being the building block of all oligomers and the monomer of Hsp16.5 being highly unstable.
Stability of the 36-subunit oligomeric cluster.
The EM and MS results indicate that while Hsp16.5 oligomer heterogeneity likely arises from the flexibility of α-crystallin dimer-dimer interactions coupled with reorientation of the C-terminal tail, the intermediate size oligomers may be less stable. Previous studies have shown that Hsp16.5 is thermally stable up to 85 °C and maintains a native 24-subunit quaternary structure, even in the presence of rapid subunit exchange at elevated temperatures (Bova et al., 2002; Kim et al., 2003a; Kim et al., 2003b; Quinlan et al., 2013). Consistent with these results, exposure to high temperature did not alter the oligomeric state of WT Hsp16.5 or Hsp16.5-P1 as evident from SEC-MALS profiles (Fig. 4A). Similarly, the Var-5 and Var-6 oligomers, which mostly occupy the 36-subunit intermediate, were largely unaffected by exposure to higher temperature (Fig. 4B).
Figure 4. Temperature-induced oligomer rearrangement in Hsp16.5 variants.

Variants were analyzed by SEC-MALS before (continuous line) and after incubation at 68 °C for 30 min (dashed line). A-B, the 24-subunit WT, the 48-subunit P1-insert, and the intermediate-size Var-5 and Var-6 oligomers were not susceptible to temperature-induced structural rearrangement. C, In contrast, Var-3 oligomers reassembled as a smaller oligomer upon heat treatment. D, incubation of Var-8 induced an increase of the WT-like 24-subunit arrangement and a reduction of the intermediate-size oligomer. The dotted vertical lines in C and D benchmark the WT 24-subunit elution peak.
In contrast, the intermediate oligomers formed by the poly-Ala Var-3 eluted from the SEC column with a larger retention volume (Fig. 4C). The temperature-induced change in average molar mass of Var-3 is consistent with a rearrangement into a smaller oligomer. Reduced oligomeric stability was exacerbated for the highly-truncated P1 constructs of Var-7 and Var-8, which readily partition between two discrete populations. Incubation at high temperature resulted in depletion of the 36-subunit cluster while simultaneously increasing the population of 24-subunit oligomers (Fig. 4D). Presumably the free energy of transition between the 36-subunit and 24-subunit oligomers at ambient temperature is sufficiently large to enable isolation of the two forms by SEC. Upon re-injection, the isolated 36-subunit cluster and the 24-subunit assembly of Var-8 eluted as distinct symmetrical peaks that did not scramble during extended storage at 4 °C (Fig. 5A and Fig. S8) and their molar masses agreed with their equilibrium populations (Fig. 5A). However, after incubation at 68 °C, the 36-subunit oligomeric pool of Var-8 shifted and converged upon the 24-subunit form according to SEC-MALS. This 24-subunit oligomer did not dissociate further with heat treatment, indicating that fundamental subunit interactions were preserved (Fig. 5B).
Figure 5. Isolation, characterization, and rearrangement of Var-8 oligomer subpopulations.

A, 24- (gray trace) and 36-subunit (dotted trace) oligomers of Var-8 demonstrated similar MALS size distribution properties as the equilibrium populations observed prior to isolation by SEC. B, Incubation of the isolated 36-subunit oligomer (dotted trace) at 68 °C for 30 min induced collapse of the oligomer to a more thermostable 24-subunit arrangement (gray trace). (See also Figure S8)
To identify the distribution of smaller oligomers induced by exposure to high temperature, we analyzed the size distribution of four P1 variants by BN-PAGE following heating at four different exposure times (Fig. 6). Var-7 and Var-8 showed a pattern consistent with that observed by SEC-MALS favoring a 24-subunit, WT-like oligomer. Remarkably, both Var-3 and Var-4 showed preferences for a species larger than the WT after heating, suggesting that full-length insertion is less compatible energetically with the 24-subunit assembly. BN-PAGE revealed the population of oligomers with an apparent composition of approximately 30 subunits. While the resolution of this technique does not allow for accurate determination of the number of subunits, the data showed that Hsp16.5-P1 variants can occupy a ladder of different size oligomers consistent with the results from native state MS.
Figure 6. Temperature-induced subunit-rearrangement in Hsp16.5-P1 variants.

30μg proteins were incubated at 68° C for 0, 30, 60, and 120 min, and were resolved at pH 7.0 on 7.5% blue native polyacrylamide gels.
Together, these results suggest that although intermediate-sized oligomers are likely populated in the expansion to the ordered 48-subunit oligomer, they have lower stability than the 24 or 48 subunit oligomers, neither of which dissociate to a measurable extent. Moreover, the stability of the 36-subunit cluster is dependent on the length of the insertion. Whereas Var-5 and 6 (10 amino acids) are not sensitive to incubation at 68 °C, Var-7 and −8 (6 amino acids) rearranged into the 24-subunit oligomer. Interpreted in the context of the two-orientation model of the C-terminal tail, the striking differences in behavior between the variants upon heat treatment likely reflect the stability of the interactions of the C-terminal tail with the α-crystallin domain of a neighboring subunit. This observation suggests that there may be intermediate orientations of this tail that are less optimal than the two corresponding to the 24- and 48-subunits assemblies.
Heterogenous expansion enhances client protein binding
A hallmark of the expanded Hsp16.5-P1 48-subunit oligomer is an increase in binding of client proteins relative to WT Hsp16.5, presumably due to greater accessibility of the high affinity binding sites within the N-terminal domain (Shi et al., 2006). Likewise, a measurable increase in binding affinity would be anticipated for P1 variants that shift the oligomeric population toward larger structures relative to WT. To characterize the binding efficiency of Hsp16.5 WT and P1 variants, we employed a well-established assay wherein thermodynamically-destabilized mutants of the model client protein T4L (for example T4 lysozyme L99A, T4L-L99A) are titrated with increasing concentrations of the chaperone (Claxton et al., 2008; Sathish et al., 2003). Steady state binding of sHSPs to T4L has been used as a surrogate to their chaperone activity. Recognition and binding by sHSPs depend on the free energy of unfolding of the T4L mutants and display two modes of binding (Koteiche and McHaourab, 2003; McDonald et al., 2012; McHaourab et al., 2002; Sathish et al., 2003; Shashidharamurthy et al., 2005).
To monitor formation of a stable chaperone-substrate complex, T4L-L99A was labeled with the Cys-specific probe monobromobimane at residue T151C. Changes in bimane fluorescence anisotropy were observed upon addition of various molar ratios of Hsp16.5-P1 variants, indicating the formation of a complex between Hsp16.5 and T4L (Fig. 7) (Koteiche et al., 2015). Previous applications of this assay that bimane monitor fluorescence intensity revealed the two-mode nature of chaperone binding characterized by low and high affinity interactions each manifested by a distinct number of binding sites (Claxton et al., 2008; Koteiche and McHaourab, 2003; McHaourab et al., 2002; Sathish et al., 2003; Shashidharamurthy et al., 2005). Since bimane anisotropy is similar in both low and high affinity binding modes, we performed the assay under conditions in which the high affinity mode dominates to reduce convolution with low affinity binding parameters (Koteiche et al., 2015). The experimental binding curves were fit using a non-linear least squares approach assuming a single-mode binding model to obtain an apparent KD. The number of binding sites was set to 0.5 T4L molecules per chaperone subunit to account for contribution by the low affinity mode for some of the variants.
Figure 7. Binding isotherms of selected Hsp16.5 variants to bimane-labeled T4L-L99A.
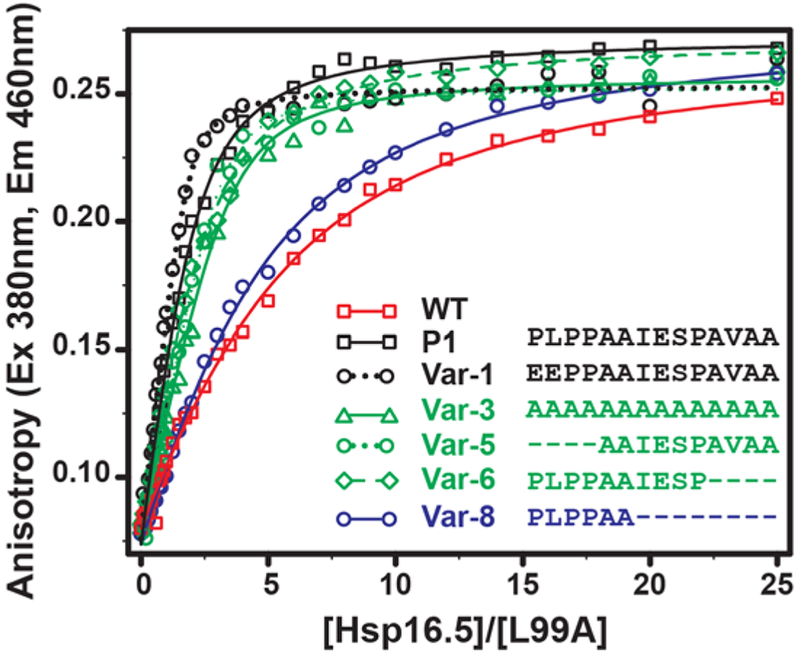
An increase in binding affinity correlates with variants that form large oligomers relative to WT. T4L (5μM) was incubated with increasing molar concentrations (X-axis) of sHSP variants for 2hrs at 37 °C in pH 7.2 buffer, and the fluorescence anisotropy (Y-axis) of the labeled substrate T4L-L99A was measured. The solid lines are non-linear least-squares fits to a single-mode binding model. The KD for each variant is reported in Table S4.
For binding assays, we selected variants that represent the spectrum of oligomer sizes and polydispersity. Binding isotherms of WT Hsp16.5 and P1 variants to T4L-L99A are compared in Fig. 7 and the parameters are shown in Table S4. For all constructs, a monotonic increase in bimane anisotropy was observed followed by a region of saturation at high Hsp16.5 concentrations as expected. Consistent with previous studies, insertion of the P1 peptide increases binding affinity by an order of magnitude (Shi et al., 2006). Furthermore, in most cases, T4L-L99A was bound to a higher level by Hsp16.5-P1 variants than by the WT, revealing a correlation between apparent KD and the propensity to form larger oligomers. For instance, Var-5 and Var-6, which form oligomeric ensembles clustered around 36-subunit assemblies, demonstrated a similar KD to Hsp16.5-P1. In contrast, Var-8, which possesses a substantial fraction of 24-subunit oligomers, binds with an affinity similar to Hsp16.5 WT. Together with the structural analyses, these results reinforce the direct link between oligomer expansion and chaperone activity. The finding that Var-3, −5 and −6 have similar binding characteristics to Hsp16.5-P1 suggests equivalence between polydispersity and expansion in increasing affinity to substrates.
Role of the PLPP motif in modulating the polydispersity of human Hsp27 oligomers.
While the crystal structure of the expanded Hsp16.5-P1 oligomer did not resolve the P1 peptide, the N-terminus of the P1 peptide contains a low complexity sequence, PLPP, that occurs in proteins at higher frequency than expected and generally adopts a conserved rigid loop structural motif (Zielenkiewicz and Plochocka, 1995). Given the results above suggesting a role of the PLPP motif in conferring the properties of the P1 peptide, we explored its importance in modulating the equilibrium dissociation of human Hsp27 oligomers. Hsp27 undergoes concentration-dependent dissociation from a polydisperse oligomer to a functionally-active dimer, and the two oligomeric states of Hsp27 exist in equilibrium at physiological pH (Jovcevski et al., 2015; McDonald et al., 2012; Shashidharamurthy et al., 2005). This equilibrium is illustrated for the C137A mutant of Hsp27 (hereafter referred Hsp27*) by a series of analytical SEC of the purified protein at progressively lower concentrations (Fig. 8A). Injected at or above 500μg/ml concentration, Hsp27* eluted from the SEC column at retention volumes expected for the heterogeneous large oligomer. Progressive dilution of the protein led to the appearance of a peak corresponding to the dimer, which predominates at 10μg/ml injected concentration. Stabilization of the dissociated dimer was observed at all concentrations for the Hsp27*-D3 mutant (S15D/S78D/S82D), which mimics the fully-phosphorylated state (Fig. S7) (Hayes et al., 2009; Kato et al., 1994). Deletion of the P1 peptide in both the Hsp27 and Hsp27-D3 has been shown to favor the larger oligomeric assembly, suggesting that the P1 peptide is important for the structural rearrangements mediating dynamic disassembly of larger oligomers (Shashidharamurthy et al., 2005).
Figure 8. Impact of the PLPP motif on the assembly of Hsp27* oligomers and client protein binding.

A, Concentration-dependent dissociation of the Hsp27* oligomer into the functionally-active dimer is disrupted when the PLPP sequence is substituted with a tetra-alanine peptide. 100μl of the proteins, at the indicated concentrations, were eluted at room temperature from the SEC column at pH 7.2. B, Binding to bimane-labeled T4L-L99A is highly attenuated for Hsp27*-PLPP/AAAA mutant, which correlates with reduced propensity to form the dimeric species. T4L-L99A (5μM) was incubated with increasing molar excess of Hsp27* (X-axis) for 2hrs at 37 °C in pH 7.2 buffer, and the fluorescence anisotropy (Y-axis) of the labeled substrate T4L-L99A was measured. (See also Figure S7)
To probe the importance of the PLPP structural unit in the dissociation equilibrium of Hsp27*, we substituted the sequence with tetra-alanine (AAAA). The mutant Hsp27*-PLPP/AAAA eluted from the SEC column at the retention volume identical to that of Hsp27* when injected at 1mg/ml and 500μg/ml concentrations (Fig. 8A). However at less than 100μg/ml injected concentrations, the peak corresponding to the dimer was less populated in the substitution mutant than in the WT. Thus replacement of PLPP mimics to a large extent the deletion of P1, consistent with a critical role in the modulation of oligomer dissociation. At the level of chaperone activity, impaired transition to the dimeric species correlated with a loss of high affinity substrate binding in the PLPP/AAAA mutant (Fig. 8B). Minimal changes in bimane anisotropy of Hsp27*-PLPP/AAAA indicated marginal binding under the conditions of the assay. In conjunction with the Hsp16.5 expansion analysis where only the exact sequence of P1 supported an ordered 48-subunit oligomer, the analysis of Hsp27 oligomer equilibrium and T4L binding suggests that P1 confers unique structural and dynamic characteristics. P1 sequences are less conserved than the N-terminal domain predicting diversity in the dynamics of oligomer dissociation across the evolutionary spectrum.
Concluding remarks
The discovery that Hsp16.5 can expand to a continuum of assemblies from its ordered and symmetric 24-subunit oligomer demonstrates that polydispersity is intrinsically encoded in the architecture of the sHSPs. Furthermore, the lack of stringent dependence on the exact sequence of the insertion (for example, the poly Ala Var-3) suggests that the expansion can be triggered by perturbation of the structure and interactions of the N-terminal domain which is located within the oligomer interior. In support of this notion, we have previously demonstrated by cryoEM that binding of T4L to the N-terminal domain of Hsp16.5 leads to heterogenous oligomer expansion (Shi et al., 2013). Together these findings suggest that the modular architecture of sHSPs, wherein the conserved modules of the α-crystallin domain and C-terminal tail are paired with a sequence variable N-terminal domain, enables diversity in the size and dynamics of sHSP oligomers. Crystal and cryoEM structures of Hsp16.5-P1 revealed that the conformation of the C-terminal tail is structurally coupled to the sequence of the N-terminal domain (McHaourab et al., 2012) thus enabling the propagation of sequence changes in this domain to the oligomeric structure. The conservation of this structural mechanism is supported by the structure of Triticum aestivum Hsp16.9 where the change in orientation of the C-terminal tail led to alternative packing of α-crystallin domain dimers within the same oligomer (van Montfort et al., 2001).
The flexibility of the sHSPs quaternary structure underpins the tuning and regulation of the binding affinity and capacity to client proteins. Comparison of the structural and functional properties of archaeal Hsp16.5 and mammalian Hsp27 underscores conserved features of chaperone-like activity mediated by two distinct mechanisms. Dynamic dissociation of Hsp27 to dimers, characteristic of a number of eukaryotic proteins, is tightly coupled to activation of binding to destabilized T4L. Oligomer expansion of Hsp16.5 is an alternative mechanism of activation triggered by repacking of the N-terminal domain and propagated by the consequent reorientation of the C-terminal tail. We surmise that the heterogeneity of client proteins and the need for tight regulation of chaperone activity necessitated the evolution of a complement of sHSPs tailored for the specific demands of cells and tissues. An example of this diversity is the emergence of 10 distinct human sHSPs or species-specific forms such as mouse αAins-crystallin where a peptide insertion leads to an isoform of αA with high affinity to client proteins (Koteiche et al., 2015). The interplay of these evolutionary forces likely accounts for the lack of strict conservation of sHSPs oligomeric structure and the observed diversity in their size, symmetry and chaperone activity.
STAR★Methods
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Hassane S. Mchaourab (hassane.mchaourab@vanderbilt.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All proteins were expressed in vitro using pET-20b(+) vector in Escherichia coli BL21(DE3) cells grown in lysogeny broth (LB) supplemented with Ampicillin, in conditions further elaborated in the method details.
METHOD DETAILS
Cloning and mutagenesis:
Site-directed mutagenesis of T4L-L99A and insertion of Hsp27 P1 peptide (the 14-amino acid peptide from residues 57-70 of human Hsp27) in the Hsp16.5 gene between residues 33 and 34 is described previously (Koteiche et al., 2005; Shi et al., 2013; Shi et al., 2006). The tridecamer alanine insert Var-3 was generated by overlap extension PCR as detailed elsewhere (Koteiche and McHaourab, 2002). Other variants of Hsp16.5-P1 with varying lengths of the insertion peptide based on the P1 sequence were generated by the QuikChange method (Stratagene) on the Hsp16.5-P1 template. Hsp27 was cloned and the cysteine-less Hsp27, referred hereinafter as Hsp27*, was generated by site directed mutagenesis of the native cysteine at position 137 to alanine, as described previously (McDonald et al., 2012). It has previously been shown that the cysteine to alanine substitution does not affect the chaperone efficiency of Hsp27* in aggregation assays and equilibrium binding to T4L (McHaourab et al., 1997). The Hsp27*-PLPP/AAAA mutant was generated by substituting proline – lysine – proline – proline residues at positions 57-60 to alanines by QuikChange. The phosphorylation mimic Hsp27*-D3 was generated by site-directed mutagenesis S15D, S78D, and S82D on the cysteine-free Hsp27* as described previously (Shashidharamurthy et al., 2005). The plasmid constructs were confirmed by DNA sequencing.
Expression, purification, and labelling of T4L:
The T4L-L99A mutant was expressed, purified, and labelled with monobromobimane at residue T151C as described previously (Claxton et al., 2008). Briefly, E. coli cultures inoculated from overnight seeds were grown in LB broth by shaking at 30°C until OD600 ~ 1 and induced by 0.4 mM isopropyl-β-D-thiogalactopyranoside (IPTG). After 3 hr induction at 30°C with continued shaking, the cells were harvested by centrifugation, resuspended into the lysis buffer (25mM 4-morpholinepropanesulfonic acid (MOPS), 25mM 2-Amino-2-(hydroxymethyl)-1,3-propanediol (Tris), pH 7.6; 1mM Ethylenediaminetetraacetic acid (EDTA), 0.02% (w/v) NaN3, 10 mM Dithiothreitol (DTT)), disrupted by sonication and centrifuged (15000 xg). T4L-L99A was purified by cation exchange on Resource S column by eluting with linear gradient of 1M NaCl. The eluted protein was incubated with 10-fold molar excess of monobromobimane for two hours at room temperature, followed by overnight incubation at 4°C. Unbound label was removed in the subsequent size exclusion chromatography (SEC) on a Superdex 75 10/300 GL column (GE Healthcare Life Sciences).
Expression and purification of sHSPs:
Hsp16.5 and the variants of Hsp16.5-P1 were expressed and purified by anion exchange, hydrophobic interaction, and size exclusion chromatography (SEC) as described previously (Koteiche et al., 2005; Shi et al., 2013; Shi et al., 2006). Briefly, E. coli cultures inoculated from overnight seeds were grown by shaking at 37°C until OD600 ~ 0.9 and induced by 0.4 mM IPTG. After 3 hr induction at 37°C with continued shaking, the cells were harvested by centrifugation (15000 xg), resuspended into the lysis buffer (20 mM Tris, pH 8.0; 100mM NaCl, 0.1 mM EDTA, 0.02% NaN3), disrupted by sonication, and the nucleic acids were precipitated by polyethyleneimine (0.06% w/v). After removing cell debris by centrifugation, Hsp16.5 variants were purified by anion exchange on a source Q column by eluting with linear gradient of 1M NaCl. Ammonium sulfate was added to a final concentration of 1 M, and the samples were loaded on a phenyl sepharose column and eluted with reverse gradient of 1 to 0 M ammonium sulfate. Proteins were finally purified by size exclusion (SEC) chromatography on a Superdex 200 10/300 GL or a Superose 6, 10/300 GL (GE Healthcare Life Sciences) and stored on ice.
Hsp27* and Hsp27*-PLPP/AAAA were purified by sequential anion exchange and size exclusion chromatography as described previously (McDonald et al., 2012). Briefly, E. coli cells were expressed by autoinduction for 12 h at 30°C (Studier, 2005). The cells were harvested by centrifugation and resuspended in lysis buffer (20 mM Tris, pH 8.0; 75 mM NaCl, 1 mM EDTA, and 10 mM DTT). The resuspension was sonicated and DNA was precipitated with polyethyleneimine (0.05% w/v). The lysate was cleared by centrifugation (15000 xg) and purified by anion exchange chromatography on Source Q column. Proteins were finally purified by size exclusion (SEC) chromatography on a Superose 6, 10/300 GL column (GE Healthcare Life Sciences) and kept stored on ice.
E. coli cells expressing Hsp27*-D3 by autoinduction for 12 h at 30 °C were lysed in the buffer containing 20 mM Tris, 1 mM EDTA, and 10 mM DTT and the supernatant as above was loaded on diethylaminoethanol (DEAE) Sepharose column. Ammonium sulfate was added to the final concentration of 0.5 M, and protein was further purified from a phenyl sepharose column with reverse gradient of 0.5 to 0 M ammonium sulfate. Protein was finally purified by size exclusion (SEC) chromatography on a Superose 6, 10/300 GL column (GE Healthcare Life Sciences) and kept stored on ice.
All proteins were eluted from SEC columns at room temperature in the SEC buffer containing 9mM MOPS, 6mM Tris, 50mM NaCl, 0.1mM EDTA, and 0.02% (w/v) NaN3 at pH 7.2. Protein concentrations were determined from absorbance at 280nm based on the extinction coefficients calculated by the ProtParam tool at the ExPASy Webserver (Wilkins et al., 1999). The homogeneity of all purified proteins was confirmed by SDS-PAGE and Coomassie staining.
Analytical Size Exclusion Chromatography:
Analytical SEC on purified HSPs was performed at room temperature on Superose 6, 10/300 GL column (GE Healthcare Life Sciences) equilibrated in the SEC buffer described above in an Agilent HP1100 HPLC system. Elution profiles of the proteins were monitored by absorption at 280nm wavelength using a UV detector and by the tryptophan fluorescence (Excitation: 295nm; Emission 330nm) using a fluorescence detector. Proteins were injected from 100μl loop at the indicated concentrations at an isocratic flow rate of 0.5ml/min.
Molar Mass Determination:
Molar mass of the proteins was determined by the multi-angle laser light-scattering detector (Wyatt Technologies) connected in-tandem to a refractive Index (RI) detector (Agilent). 100μl of each protein at 1mg/ml concentrations was injected at 25°C by an Agilent HP1100 HPLC system on a Superose 6 column equilibrated in the SEC buffer at the isocratic flow rate of 0.5ml/min. The elution of the proteins was also monitored by an absorbance detector. The average molar mass and the average hydrodynamic radius of the proteins were calculated by the ASTRA software (Wyatt) from the concentration of proteins derived from the RI signal based on constant dn/dc (0.185 ml/g).
Blue-Native PAGE:
The oligomers (30μg) were resolved at pH 7.0 by electrophoresis on hand casted imidazole / 6-aminocaproic acid polyacrylamide gels of the indicated acrylamide concentration in presence of Coomasie Blue G-250 (Wittig et al., 2006). Electrophoresis was performed in imidazole / tricine buffers (Cathode buffer: 50mM Tricine, 7.5mM Imidazole, 0.02% Coomasie Blue G-250; Anode buffer: 25mM Imidazole, pH 7) in a cold room (BioRad MiniProtean 3 cell) at 100V until the samples entered the resolving gel, followed by constant 150V (current limited to 15mA) for ~2 hours. The gels were visualized by standard Coomasie staining.
Subunit rearrangement:
Proteins diluted to 1mg/ml in the SEC buffer were incubated in Eppendorf tubes at 68 °C for 30min, unless specified otherwise, in a water bath to stimulate subunit rearrangement. The thermally induced subunit rearrangement was quenched by transferring sample tubes to ice-water slush, and the proteins were kept on ice until used in the subsequent analyses.
Binding of T4L to Hsp16.5 and Hsp27* variants:
Solutions of monobromobimane-labelled T4L-L99A at final concentration of 5μM with increasing molar excess (0-25 fold molar excess) of Hsp16.5 or Hsp27* variants diluted in the SEC buffer were incubated at 37 °C for 2 h. The fluorescence anisotropy was measured by a SynergyH4 microplate reader (BioTek) at 37 °C. The monobromobimane label was excited by 380nm filter (bandpass 20nm) and the emission fluorescence intensities parallel (I∥) and perpendicular (I⊥) to the direction of polarized light were recorded at 460 nm (band-pass 40 nm). Steady state anisotropy (r) was calculated from these intensities by the equation: r = (I∥ - I⊥)/(I∥ - 2I⊥). The binding isotherms were generated by the nonlinear least-squares fits using the Levenberg–Marquart method from the calculated bimane anisotropy plotted as a function of the [sHSP]/[T4L] ratio by Origin 8 software (OriginLab Corporation, Northampton, MA). The maximum anisotropy was fixed to 0.3, and the number of binding site binding site parameter, n, was constrained to 0.5 in all fits.
Electron Microscopy:
10 nM droplets of proteins were applied for two minute to freshly glow discharged solid carbon support on copper 400 mesh TEM grids. Excess water was blotted away and replaced with pH neutralized 0.75% uranyl formate solution, then allowed to air dry. Prepared grids were visualized at a nominal magnification of 67,000X on an FEI Tecnai T12 operating at 120 KeV. Digital images were captured using the Script Assisted Microscopy (SAM) package (Shi et al., 2008) on a Gatan US1000 CCD camera. Images were normalized and individual HSP particles were selected from micrographs of each mutant variant using the EMAN Boxer routine (Tang et al., 2007). The total number of particles for each sample is as follows: P1 = 6853, Var-1 = 2747, Var-2 = 4987, Var-3 = 2550, Var-4 = 9834, Var-5 = 5770, Var-6 = 22040, Var-7 = 4756, and Var-8 = 2846 particles. Previously determined Hsp16.5 24mer, 36mer and 48mer complexes were used as starting models for each mutant variant in multirefine.py subroutine from EMAN. Each particle set was individually refined against the three starting structures to determine the relative distribution of assembly states for each mutant. Post-multirefine.py bins for each of the three resulting structures were reclassified in EMAN refine2d.py to confirm the sorting process. Molecular graphics images were produced using the UCSF Chimera (Pettersen et al., 2004)
Native mass spectrometry:
Native mass spectrometry was performed as described elsewhere for oligomeric identification and quantification (Kondrat et al., 2015). Ions were generated in positive polarity from a 200 mM NH4OAc solution (Sigma-Aldrich, Gillingham, UK), at room temperature, using a nanospray source and gold-coated borosilicate capillaries. Capillaries were prepared in-house using a model P-97 (Sutter Instruments) capillary puller and a sputter coater (Polaron, Newhaven, United Kingdom). Ions were introduced into a Q Exactive Plus hybrid quadrupole-Orbitrap mass spectrometer (Thermo Fisher Scientific, Bremen Germany) modified for the transmission, selection and detection of high mass ions (van de Waterbeemd et al., 2017). All spectra were acquired in “EMR Mode” and a pulser mode of 50. Ions were generated in positive polarity from a nanospray source using gold-coated capillaries prepared in-house. Transfer capillary temperature was (100-120 °C). The injection flatapole was modified for in-source trapping and a desolvation voltage of −100 V applied. No in-source activation was used. Ions were trapped in the HCD cell before being transferred into the C-trap and Orbitrap mass analyser for detection. Transient times were 64 ms and AGC target was 1×106. The collision gas was nitrogen and UHV pressure was maintained at approximately 1×10−9 mbar. Spectra were acquired with 10 microscans, averaged over a minimum of 100 scans and with a noise level parameter set to 3. For MS/MS experiments the HCD activation voltage was increased to between 150-250 V to achieve full or near-complete dissociation of the selected precursor. Tandem MS was performed across the whole oligomeric range where sufficient ion intensity allowed.
Intact oligomeric distributions were fitted to spectra, where possible, using mass spectrometry deconvolution software, Unidec, as described previously (Marty et al., 2015). All spectra underwent minimal curved background subtraction with a value of 100, except variant 2 which is shown with a value of 20 (Morgner and Robinson, 2012). CID spectra were assigned using Unidec software and the precursor identity determined manually. Estimates for the relative abundance of each oligomeric cluster (24, 28-38, 48) were achieved through integration of the intact MS spectrum over the m/z regions shown in Fig.S4. Relative abundances for each individual oligomeric state, including discrimination by truncation number, were determined for variants 2, 3, and 6 by direct fitting of oligomeric species to the intact spectrum using Unidec. Resulting fit distributions were consistent with CID spectra for the corresponding range. The relative abundance for the 28-38 subunit region for variants 7 and 8 was calculated from the reconstruction of tandem MS species weighted by the integral of the spectrum across the quadrupole selection window. This was then scaled relative to the well resolved 24 subunit region in each case. The 36 subunit cluster was then generated for comparison through summation of the oligomeric states 28-38. All deconvoluted data was scaled according to the reciprocal of the charge state to account for the magnitude of charge on the detector signal.
QUANTIFICATION AND STATISTICAL ANALYSIS
Where appropriate, statistical details are given in the Method Details section.
DATA AND SOFTWARE AVAILABILITY
Data on Native MS stoichiometry determination, 3D negative stain-EM reconstruction, and Molecular Mass Determination by Light Scattering is included in the paper. The raw data of this study are available upon reasonable request.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health Grant R01-EY12018 to H.S.M. S.A.C. and J.L.P.B. thank the Biotechnology and Biological Sciences Research Council (BB/L017067/1) and Waters Corp for an iCASE studentship. We thank Joseph Gault (Oxford) for useful discussions and advice.
Footnotes
Declaration of Interests
The authors declare no competing interests.
References
- Andley UP (2007). Crystallins in the eye: Function and pathology. Progress in Retinal and Eye Research 26, 78–98. [DOI] [PubMed] [Google Scholar]
- Andley UP, Hamilton PD, Ravi N, and Weihl CC (2011). A Knock-In Mouse Model for the R120G Mutation of αB-Crystallin Recapitulates Human Hereditary Myopathy and Cataracts. PLoS ONE 6, e17671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aquilina JA, Benesch JL, Bateman OA, Slingsby C, and Robinson CV (2003). Polydispersity of a mammalian chaperone: mass spectrometry reveals the population of oligomers in alphaB-crystallin. Proc Natl Acad Sci U S A 100, 10611–10616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basha E, O'Neill H, and Vierling E (2012). Small heat shock proteins and α-crystallins: dynamic proteins with flexible functions. Trends Biochem Sci 37, 106–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benesch JL, Aquilina JA, Ruotolo BT, Sobott F, and Robinson CV (2006). Tandem mass spectrometry reveals the quaternary organization of macromolecular assemblies. Chem Biol 13, 597–605. [DOI] [PubMed] [Google Scholar]
- Benesch JLP, Ayoub M, Robinson CV, and Aquilina JA (2008). Small heat shock protein activity is regulated by variable oligomeric substructure. Journal of Biological Chemistry 283, 28513–28517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berengian AR, Bova MP, and McHaourab HS (1997). Structure and Function of the Conserved Domain in αA-Crystallin. Site-Directed Spin Labeling Identifies a β-Strand Located near a Subunit Interface†. Biochemistry 36, 9951–9957. [DOI] [PubMed] [Google Scholar]
- Berengian AR, Parfenova M, and McHaourab HS (1999). Site-directed Spin Labeling Study of Subunit Interactions in the α-Crystallin Domain of Small Heat-shock Proteins: COMPARISON OF THE OLIGOMER SYMMETRY IN αA-CRYSTALLIN, HSP 27, and HSP 16.3. Journal of Biological Chemistry 274, 6305–6314. [DOI] [PubMed] [Google Scholar]
- Bova MP, Huang Q, Ding L, and Horwitz J (2002). Subunit Exchange, Conformational Stability, and Chaperone-like Function of the Small Heat Shock Protein 16.5 from Methanococcus jannaschii. Journal of Biological Chemistry 277, 38468–38475. [DOI] [PubMed] [Google Scholar]
- Bova MP, Yaron O, Huang Q, Ding L, Haley DA, Stewart PL, and Horwitz J (1999). Mutation R120G in alphaB-crystallin, which is linked to a desmin-related myopathy, results in an irregular structure and defective chaperone-like function. Proc Natl Acad Sci U S A 96, 6137–6142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun N, Zacharias M, Peschek J, Kastenmiiller A, Zou J, Hanzlik M, Haslbeck M, Rappsilber J, Buchner J, and Weinkauf S (2011). Multiple molecular architectures of the eye lens chaperone αB-crystallin elucidated by a triple hybrid approach. Proceedings of the National Academy of Sciences 108, 20491–20496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carra S, Alberti S, Arrigo PA, Benesch JL, Benjamin IJ, Boelens W, Bartelt-Kirbach B, Brundel B, Buchner J, Bukau B, et al. (2017). The growing world of small heat shock proteins: from structure to functions. Cell Stress & Chaperones 22, 601–611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark AR, Lubsen NH, and Slingsby C (2012). sHSP in the eye lens: Crystallin mutations, cataract and proteostasis. International Journal of Biochemistry & Cell Biology 44, 1687–1697. [DOI] [PubMed] [Google Scholar]
- Claxton DP, Zou P, and Mchaourab HS (2008). Structure and orientation of T4 lysozyme bound to the small heat shock protein alphα-crystallin. J Mol Biol 375, 1026–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Jong WW, Caspers GJ, and Leunissen JAM (1998). Genealogy of the alphα-crystallin – small heat-shock protein superfamily. International Journal of Biological Macromolecules 22, 151–162. [DOI] [PubMed] [Google Scholar]
- Ecroyd H, and Carver J (2009). Crystallin proteins and amyloid fibrils. Cellular and Molecular Life Sciences 66, 62–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ecroyd H, Meehan S, Horwitz J, Aquilina JA, Benesch JLP, Robinson CV, Macphee CE, and Carver JA (2007). Mimicking phosphorylation of alpha B-crystallin affects its chaperone activity. Biochemical Journal 401, 129–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido C, Paul C, Seigneuric R, and Kampinga HH (2012). The small heat shock proteins family: The long forgotten chaperones. International Journal of Biochemistry & Cell Biology 44, 1588–1592. [DOI] [PubMed] [Google Scholar]
- Haley DA, Bova MP, Huang QL, McHaourab HS, and Stewart PL (2000). Small heat-shock protein structures reveal a continuum from symmetric to variable assemblies. Journal of Molecular Biology 298, 261–272. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, and Buchner J (2002). Chaperone function of sHsps. Progress in Molecular and Subcellular Biology. Small stress proteins 28, 37–59. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, Franzmann T, Weinfurtner D, and Buchner J (2005). Some like it hot: the structure and function of small heat-shock proteins. Nat Struct Mol Biol 12, 842–846. [DOI] [PubMed] [Google Scholar]
- Haslbeck M, and Vierling E (2015). A first line of stress defense: small heat shock proteins and their function in protein homeostasis. J Mol Biol 427, 1537–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes D, Napoli V, Mazurkie A, Stafford WF, and Graceffa P (2009). Phosphorylation dependence of hsp27 multimeric size and molecular chaperone function. J Biol Chem 284, 18801–18807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hochberg GKA, and Benesch JLP (2015). Dynamics-Function Relationships of the Small Heat-Shock Proteins In The Big Book on Small Heat Shock Proteins, Tanguay RM, and Hightower LE, eds. (Cham: Springer International Publishing; ), pp. 87–100. [Google Scholar]
- Jakob U, Gaestel M, Engel K, and Buchner J (1993). SMALL HEAT-SHOCK PROTEINS ARE MOLECULAR CHAPERONES. Journal of Biological Chemistry 268, 1517–1520. [PubMed] [Google Scholar]
- Jehle S, Rajagopal P, Bardiaux B, Markovic S, Kuhne R, Stout JR, Higman VA, Klevit RE, van Rossum B-J, and Oschkinat H (2010). Solid-state NMR and SAXS studies provide a structural basis for the activation of [alpha]B-crystallin oligomers. Nat Struct Mol Biol 17, 1037–1042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jehle S, Vollmar BS, Bardiaux B, Dove KK, Rajagopal P, Gonen T, Oschkinat H, and Klevit RE (2011). N-terminal domain of αΒ-crystallin provides a conformational switch for multimerization and structural heterogeneity. Proceedings of the National Academy of Sciences 108, 6409–6414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jovcevski B, Kelly MA, Rote AP, Berg T, Gastall HY, Benesch JL, Aquilina JA, and Ecroyd H (2015). Phosphomimics destabilize Hsp27 oligomeric assemblies and enhance chaperone activity. Chem Biol 22, 186–195. [DOI] [PubMed] [Google Scholar]
- Kappe G, Franck E, Verschuure P, Boelens WC, Leunissen JAM, and de Jong WW (2003). The human genome encodes 10 alphα-crystallin-related small heat shock proteins: HspBl-10. Cell Stress & Chaperones 8, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kappe G, Boelens W, and Jong W (2010). Why proteins without an α-crystallin domain should not be included in the human small heat shock protein family HSPB. Cell Stress and Chaperones 15, 457–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kato K, Hasegawa K, Goto S, and Inaguma Y (1994). Dissociation as a result of phosphorylation of an aggregated form of the small stress protein, hsp27. J Biol Chem 269, 11274–11278. [PubMed] [Google Scholar]
- Kim DR, Lee I, Ha SC, and Kim KK (2003a). Activation mechanism of HSP16.5 from Methanococcus jannaschii. Biochemical and Biophysical Research Communications 307, 991–998. [DOI] [PubMed] [Google Scholar]
- Kim KK, Kim R, and Kim SH (1998). Crystal structure of a small heat-shock protein. Nature 394, 595–599. [DOI] [PubMed] [Google Scholar]
- Kim R, Lai L, Lee H-H, Cheong G-W, Kim KK, Wu Z, Yokota H, Marqusee S, and Kim S-H (2003b). On the mechanism of chaperone activity of the small heat-shock protein of Methanococcus jannaschii. Proceedings of the National Academy of Sciences 100, 8151–8155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrat FD, Struwe WB, and Benesch JL (2015). Native mass spectrometry: towards high-throughput structural proteomics. Methods Mol Biol 1261, 349–371. [DOI] [PubMed] [Google Scholar]
- Koteiche HA, Berengian AR, and Mchaourab HS (1998). Identification of protein folding patterns using site-directed spin labeling. Structural characterization of a beta-sheet and putative substrate binding regions in the conserved domain of alpha A-crystallin. Biochemistry 37, 12681–12688. [DOI] [PubMed] [Google Scholar]
- Koteiche HA, Chiu S, Majdoch RL, Stewart PL, and McHaourab HS (2005). Atomic models by cryo-EM and site-directed spin labeling: Application to the N-terminal region of Hsp16.5. Structure 13, 1165–1171. [DOI] [PubMed] [Google Scholar]
- Koteiche HA, Claxton DP, Mishra S, Stein RA, McDonald ET, and McHaourab HS (2015). Species-Specific Structural and Functional Divergence of alpha-crystallins: Zebrafish alpha Ba- and Rodent alpha A(ins)-Crystallin Encode Activated Chaperones. Biochemistry 54, 5949–5958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koteiche HA, and McHaourab HS (1999). Folding pattern of the alpha-crystallin domain in alphaA-crystallin determined by site-directed spin labeling. J Mol Biol 294, 561–577. [DOI] [PubMed] [Google Scholar]
- Koteiche HA, and McHaourab HS (2002). The determinants of the oligomeric structure in Hsp16.5 are encoded in the alpha-crystallin domain. FEBS Lett 519, 16–22. [DOI] [PubMed] [Google Scholar]
- Koteiche HA, and McHaourab HS (2003). Mechanism of chaperone function in small heat-shock proteins. Phosphorylation-induced activation of two-mode binding in alphaB-crystallin. J Biol Chem 278, 10361–10367. [DOI] [PubMed] [Google Scholar]
- Koteiche HA, and McHaourab HS (2006). Mechanism of a hereditary cataract phenotype. Mutations in alphaA-crystallin activate substrate binding. J Biol Chem 281, 14273–14279. [DOI] [PubMed] [Google Scholar]
- Kumar LVS, Ramakrishna T, and Rao CM (1999). Structural and Functional Consequences of the Mutation of a Conserved Arginine Residue in αA and αB Crystallins. Journal of Biological Chemistry 274, 24137–24141. [DOI] [PubMed] [Google Scholar]
- Laganowsky A, Benesch JLP, Landau M, Ding L, Sawaya MR, Cascio D, Huang Q, Robinson CV, Horwitz J, and Eisenberg D (2010). Crystal structures of truncated alphaA and alphaB crystallins reveal structural mechanisms of polydispersity important for eye lens function. Protein Science 19, 1031–1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laganowsky A, and Eisenberg D (2010). Non-3D domain swapped crystal structure of truncated zebrafish alphaA crystallin. Protein Science 19, 1978–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marty MT, Baldwin AJ, Marklund EG, Hochberg GK, Benesch JL, and Robinson CV (2015). Bayesian deconvolution of mass and ion mobility spectra: from binary interactions to polydisperse ensembles. Anal Chem 87, 4370–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ET, Bortolus M, Koteiche HA, and McHaourab HS (2012). Sequence, Structure, and Dynamic Determinants of Hsp27 (HspB1) Equilibrium Dissociation Are Encoded by the N-Terminal Domain. Biochemistry 51, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHaourab HS, Berengian AR, and Koteiche HA (1997). Site-directed spin-labeling study of the structure and subunit interactions along a conserved sequence in the alpha-crystallin domain of heat-shock protein .27. Evidence of a conserved subunit interface. Biochemistry 36, 14627–14634. [DOI] [PubMed] [Google Scholar]
- McHaourab HS, Dodson EK, and Koteiche HA (2002). Mechanism of chaperone function in small heat shock proteins. Two-mode binding of the excited states of T4 lysozyme mutants by alphaA-crystallin. J Biol Chem 277, 40557–40566. [DOI] [PubMed] [Google Scholar]
- McHaourab HS, Godar JA, and Stewart PL (2009). Structure and Mechanism of Protein Stability Sensors: Chaperone Activity of Small Heat Shock Proteins. Biochemistry 48, 3828–3837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McHaourab HS, Lin Y-L, and Spiller BW (2012). Crystal Structure of an Activated Variant of Small Heat Shock Protein Hsp16.5. Biochemistry 51, 5105–5112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgner N, and Robinson CV (2012). Massign: an assignment strategy for maximizing information from the mass spectra of heterogeneous protein assemblies. Anal Chem 84, 2939–2948. [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, and Ferrin TE (2004). UCSF chimera - A visualization system for exploratory research and analysis. Journal of Computational Chemistry 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Quinlan RA, Zhang Y, Lansbury A, Williamson I, Pohl E, and Sun F (2013). Changes in the quaternary structure and function of MjHSP16.5 attributable to deletion of the IXI motif and introduction of the substitution, R107G, in the alpha-crystallin domain. Philosophical Transactions of the Royal Society B-Biological Sciences 368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogalla T, Ehrnsperger M, Preville X, Kotlyarov A, Lutsch G, Ducasse C, Paul C, Wieske M, Arrigo AP, Buchner J, et al. (1999). Regulation of Hsp27 oligomerization, chaperone function, and protective activity against oxidative stress/tumor necrosis factor alpha by phosphorylation. J Biol Chem 274, 18947–18956. [DOI] [PubMed] [Google Scholar]
- Sathish HA, Stein RA, Yang GY, and McHaourab HS (2003). Mechanism of chaperone function in small heat-shock proteins - Fluorescence studies of the conformations of T4 lysozyme bound to alpha B-crystallin. Journal of Biological Chemistry 278, 44214–44221. [DOI] [PubMed] [Google Scholar]
- Shashidharamurthy R, Koteiche HA, Dong J, and McHaourab HS (2005). Mechanism of chaperone function in small heat shock proteins: dissociation of the HSP27 oligomer is required for recognition and binding of destabilized T4 lysozyme. J Biol Chem 280, 5281–5289. [DOI] [PubMed] [Google Scholar]
- Shi J, Koteiche HA, McDonald ET, Fox TL, Stewart PL, and McHaourab HS (2013). Cryoelectron microscopy analysis of small heat shock protein 16.5 (Hsp16.5) complexes with T4 lysozyme reveals the structural basis of multimode binding. J Biol Chem 288, 4819–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi J, Koteiche HA, McHaourab HS, and Stewart PL (2006). Cryoelectron microscopy and EPR analysis of engineered symmetric and polydisperse Hsp16.5 assemblies reveals determinants of polydispersity and substrate binding. J Biol Chem 281, 40420–40428. [DOI] [PubMed] [Google Scholar]
- Shi J, Williams DR, and Stewart PL (2008). A Script-Assisted Microscopy (SAM) package to improve data acquisition rates on FEI Tecnai electron microscopes equipped with Gatan CCD cameras. J Struct Biol 164, 166–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shroff NP, Cherian-Shaw M, Bera S, and Abraham EC (2000). Mutation of R116C Results in Highly Oligomerized αA-Crystallin with Modified Structure and Defective Chaperone-like Function. Biochemistry 39, 1420–1426. [DOI] [PubMed] [Google Scholar]
- Sobott F, Benesch JL, Vierling E, and Robinson CV (2002). Subunit exchange of multimeric protein complexes. Real-time monitoring of subunit exchange between small heat shock proteins by using electrospray mass spectrometry. J Biol Chem 277, 38921–38929. [DOI] [PubMed] [Google Scholar]
- Stengel F, Baldwin AJ, Painter AJ, Jaya N, Basha E, Kay LE, Vierling E, Robinson CV, and Benesch JLP (2010). Quaternary dynamics and plasticity underlie small heat shock protein chaperone function. Proceedings of the National Academy of Sciences of the United States of America 107, 2007–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strauch A, and Haslbeck M (2016). The function of small heat-shock proteins and their implication in proteostasis. Essays Biochem 60, 163–172. [DOI] [PubMed] [Google Scholar]
- Studier FW (2005). Protein production by auto-induction in high density shaking cultures. Protein Expr Purif 41, 207–234. [DOI] [PubMed] [Google Scholar]
- Sun Y, and MacRae TH (2005). The small heat shock proteins and their role in human disease. Febs Journal 272, 2613–2627. [DOI] [PubMed] [Google Scholar]
- Tang G, Peng L, Baldwin PR, Mann DS, Jiang W, Rees I, and Ludtke SJ (2007). EMAN2: an extensible image processing suite for electron microscopy. J Struct Biol 157, 38–46. [DOI] [PubMed] [Google Scholar]
- Thériault JR, Lambert H, Chávez-Zobel AT, Charest G, Lavigne P, and Landry J (2004). Essential role of the NH2-terminal WD/EPF motif in the phosphorylation-activated protective function of mammalian Hsp27. J Biol Chem 279, 23463–23471. [DOI] [PubMed] [Google Scholar]
- Treweek TM, Meehan S, Ecroyd H, and Carver JA (2015). Small heat-shock proteins: important players in regulating cellular proteostasis. Cell Mol Life Sci 72, 429–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Waterbeemd M, Fort KL, Boll D, Reinhardt-Szyba M, Routh A, Makarov A, and Heck AJ (2017). High-fidelity mass analysis unveils heterogeneity in intact ribosomal particles. Nat Methods 14, 283–286. [DOI] [PubMed] [Google Scholar]
- Van Montfort R, Slingsby C, and Vierling E (2002). Structure and function of the small heat shock protein/alpha-crystallin family of molecular chaperones. Protein Folding in the Cell 59, 105–156. [DOI] [PubMed] [Google Scholar]
- van Montfort RLM, Basha E, Friedrich KL, Slingsby C, and Vierling E (2001). Crystal structure and assembly of a eukaryotic small heat shock protein. Nature Structural Biology 8, 1025–1030. [DOI] [PubMed] [Google Scholar]
- Vicart P, Caron A, Guicheney P, Li ZL, Prevost MC, Faure A, Chateau D, Chapon F, Tome F, Dupret JM, et al. (1998). A missense mutation in the alpha B-crystallin chaperone gene causes a desmin-related myopathy. Nature Genetics 20, 92–95. [DOI] [PubMed] [Google Scholar]
- White HE, Orlova EV, Chen SX, Wang LC, Ignatiou A, Gowen B, Stromer T, Franzmann TM, Haslbeck M, Buchner J, et al. (2006). Multiple distinct assemblies reveal conformational flexibility in the small heat shock protein Hsp26. Structure 14, 1197–1204. [DOI] [PubMed] [Google Scholar]
- Wilkins MR, Gasteiger E, Bairoch A, Sanchez JC, Williams KL, Appel RD, and Hochstrasser DF (1999). Protein identification and analysis tools in the ExPASy server. Methods Mol Biol 112, 531–552. [DOI] [PubMed] [Google Scholar]
- Wittig I, Braun H-P, and Schagger H (2006). Blue native PAGE. Nat. Protocols 1, 418–428. [DOI] [PubMed] [Google Scholar]
- Zielenkiewicz P, and Plochocka D (1995). Abundant PLPP sequence has the same conformation in unrelated proteins. Protein and Peptide Letters 2, 299–304. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data on Native MS stoichiometry determination, 3D negative stain-EM reconstruction, and Molecular Mass Determination by Light Scattering is included in the paper. The raw data of this study are available upon reasonable request.