

Abstract
Biotechnological processes are typically perceived to be greener than chemical processes. A life cycle assessment (LCA) was performed to compare the chemical and biochemical synthesis of lactones obtained by Baeyer–Villiger oxidation. The LCA is prospective (based on experiments at a small scale with primary data) because the process is at an early stage. The results show that the synthesis route has no significant effect on the climate change impact [(1.65±0.59) kg gproduct −1 vs. (1.64±0.67) kg gproduct −1]. Key process performance metrics affecting the environmental impact were evaluated by performing a sensitivity analysis. Recycling of solvents and enzyme were shown to provide an advantage to the enzymatic synthesis. Additionally, the climate change impact was decreased by 71 % if renewable electricity was used. The study shows that comparative LCAs can be used to usefully support decisions at an early stage of process development.
Keywords: biocatalysis, life cycle assessment, oxidative chemistry, process metrics, sustainable chemistry
Introduction
Enzymatic reactions are typically perceived, and sometimes claimed, to be greener than the corresponding chemical conversions.1 Their assumed environmental benefits are an incentive for the development of such processes. This contributes to the growing importance of industrial biocatalysis for the synthesis of molecules used as pharmaceuticals, flavors and fragrances, and bulk chemicals.2 Typical green features associated with the use of enzymes are correlated with the green chemistry principles.3 Enzymatic reactions are usually performed under mild reaction conditions, at low reaction temperatures, and at ambient pressure. Enzymes operate by using water as the reaction medium, thereby reducing the use of organic solvents. Additionally, enzymes are biobased, because they are produced from renewable resources in fermentation processes, and biodegradable. More importantly, the major advantages of enzymes are their regio‐ and enantioselectivity, which results in more straightforward synthetic routes and avoids multiple protection/deprotection steps, which generate waste.4
There is, however, some criticism concerning the environmental benefits of enzymatic reactions.5 The use of water as the reaction medium typically leads to dilute reaction streams because many compounds are poorly water‐soluble and may also be inhibitory at higher concentrations. This results in an increased solvent use for product isolation during downstream processing (DSP).6 Because water is the solvent, this also comes with costly technology for concentration on account of its high boiling point. Another concern is the high energy consumption associated with the production of enzymes, particularly if they are further purified and immobilized.7 Additionally, although oxidative biocatalysis allows the replacement of toxic oxidants by molecular oxygen, it raises other concerns such as the generation of waste associated with the use of co‐substrates to regenerate co‐factors, especially if isolated enzymes are applied.5
To quantify and compare the environmental impact of chemical reactions, some green process metrics have been developed. The most commonly used is the E‐factor, which is a measure of the amount of waste generated by a reaction (mass of waste/mass of product).8 However, the comparison of the environmental impact of two processes, for example, chemical and enzymatic, is limited owing to the fact that the E‐factor does not take into consideration the type of waste that is generated, nor the energy consumption of each process.5
To obtain a more accurate quantification of the environmental impact, life cycle assessments (LCAs) are being more widely performed by the scientific community.9 A LCA is a standardized and internationally recognized tool specifically designed to quantify the environmental impact of a product or service, taking into account its entire life cycle (ISO 14044:2006).10 Several LCAs have been applied to chemical processes (both with biobased and fossil‐based processes),11 and some of them have demonstrated unexpected results. For example, some biobased polymers and biobased solvents have a higher environmental impact than their fossil‐based counterparts.12 Such counter‐intuitive results demonstrate the need to perform an LCA before claiming the sustainability or green aspects of a given reaction or product synthesis. Comparative LCAs are especially useful to identify the most sustainable synthetic route to a given product if several routes are potentially possible. The sustainability of enzymatic reactions, as opposed to chemical routes, could, for example, be claimed for the industrial synthesis of a bulk chemical13 and a pharmaceutical molecule.14
The quality and comprehensive character of an LCA depends on the reliability of the data, which should also be as representative and as complete as possible.15 Conventionally, LCAs have been based on retrospective data from existing processes. To provide a quantitative analysis of the environmental impact of a process that has not yet been implemented at the industrial scale, a prospective approach has been developed in recent years.16 Prospective LCAs, which are performed on different development stages of processes such as laboratory scale, simulations, and pilot plants, enable the identification of key steps in the process that require focus for improvement and provide guidance regarding process upscaling.16 Although the limitation of prospective LCAs is that they cannot provide an absolute quantification of the environmental impact, owing to the type of data on which they are based, they are particularly relevant for the comparison of early‐stage processes to identify the most advantageous route for upscaling from an environmental perspective. For example, comparative early‐stage LCAs have proven useful for the selection of raw chemicals.17 It appears that accounting for the energy consumption may be crucial because it plays an important role in LCA and is very dependent on the scale of operation.18
The goal of the current study is to compare the environmental impact of two early‐stage synthetic routes for the preparation of lactones. These cyclic esters were obtained by Baeyer–Villiger oxidation, inserting an oxygen atom in the C−C bond of a ketone.19 This reaction is, for example, of interest for the synthesis of lactones as monomers for polymer applications20 and in the pharmaceutical industry.21 Although it has been known for many years that organic peracids are efficient oxidants for the Baeyer–Villiger oxidation,22 recent research focuses on the development of greener oxidants.23
The desire to replace toxic oxidants by molecular oxygen is one of the most important motivations for the development of industrial oxidative biocatalysis. Biocatalytic Baeyer–Villiger oxidations are enabled by the use of Baeyer–Villiger monooxygenases, which catalyze the reaction by using molecular oxygen as the oxidant. Although the range of esters and lactones that can be synthesized by using Baeyer–Villiger monooxygenases is increasing, including for example (substituted) lactones of various ring sizes, steroids, and bicyclic compounds,24 these biocatalysts have not yet been reported on a truly industrial scale. Consequently, these reactions are still performed at laboratory or pilot‐plant scale, with a few exceptions.25
The enzymatic synthesis of functionalized lactones, β,δ‐trimethyl‐ϵ‐caprolactones (TMCL), used as a monomer for polymeric applications, has previously been established (Figure 1).20b, 26 This product can be synthesized through two synthetic routes from the same cyclic ketone substrate 3,3,5‐trimethylcyclohexanone (TMCH). The first route is a chemical Baeyer–Villiger oxidation that uses the peracid m‐chloroperbenzoic acid (m‐CPBA) as a chemical oxidant.20a The second route is an enzymatic oxidation with oxygen as the oxidant, catalyzed by a Baeyer–Villiger monooxygenase from Thermocrispum municipale (TmCHMO).27 Ring‐opening polymerization of TMCL yields amorphous polyesters, with potential applications as plasticizers or dispersants for inks and coatings (Figure 1).20a, 28 Because the product can be used for several types of polymers that have different applications, the end‐of‐life of the product is not included in this LCA. The studied boundaries of this assessment are therefore cradle‐to‐gate as shown in Figure 1. This LCA is based on primary data for the two syntheses, which comprises the synthesis of the product by oxidation and the product isolation procedure. The substrate, the chemical oxidant, and the enzymes were also modeled and included in the study.
Figure 1.
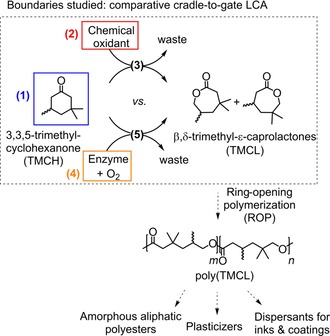
Boundaries studied for the synthesis of the product (TMCL): comparative cradle‐to‐gate assessment comprising (1) the synthesis of the substrate (TMCH), (2) the synthesis of the chemical oxidant, (3) the synthesis of the product by using a chemical oxidant, (4) the enzyme preparation, and (5) the enzymatic synthesis of the product by using a Baeyer–Villiger monooxygenase (BVMO). All reactions include DSP.
It is worth noting that this comparative LCA is a worst‐case scenario for the comparison of this type of enzymatic reaction with its chemical counterpart because the main advantage of biocatalysts and Baeyer–Villiger monooxygenases in particular, namely their regio‐ and enantioselectivity, is not exploited in this case. This LCA evaluates the synthesis of a monomer that can be considered as a bulk chemical, with high production volume and low price, for which enzymatic reactions need to be particularly efficient to be competitive. This means that the enzymatic reaction has been further developed to improve its process performance metrics to achieve higher space‐time yield by increasing the product concentration and lowering the reaction time.26b These improvements are also expected to contribute to reducing the environmental impact of the biocatalytic reaction.
After presenting the methodology and the results, a sensitivity analysis is performed. The source of electricity, the effect of recycling of several components including enzymes, solvents, and co‐product, as well the type of chemical oxidant used were evaluated. Lastly, two key process metrics influencing the environmental impact of the reactions were identified, thereby demonstrating the potential of early‐stage LCAs as a tool for the improvement of enzymatic reactions.
Methodology
The present LCA was performed in accordance with the ISO standard (ISO 14044:2006). The methodology followed the LCA framework and is described below.
Goal, scope definition, function, and functional unit
This study is a comparative LCA between two synthetic routes for the same lactone product, TMCL. Both syntheses were performed at the laboratory scale starting from the same substrate, TMCH. The function of the study is the synthesis and purification of the TMCL product. The functional unit (FU) was selected according to the product category rules (PCR) of the environmental product declaration (EDP), which suggests using a physical reference.29 An FU of 1 g of TMCL product was chosen because it is representative of the laboratory‐scale experiments.
System description and boundaries
As mentioned previously, the boundaries of this comparative LCA are cradle‐to‐gate because there are several applications for the evaluated product. The study includes the synthesis and purification of the product TMCL, which is synthesized either chemically by using m‐CPBA as the chemical oxidant (Figure 2), or enzymatically with a Baeyer–Villiger monooxygenase, which uses molecular oxygen as the oxidant (Figure 3). In total, the life cycle inventory consists of five parts:
Figure 2.
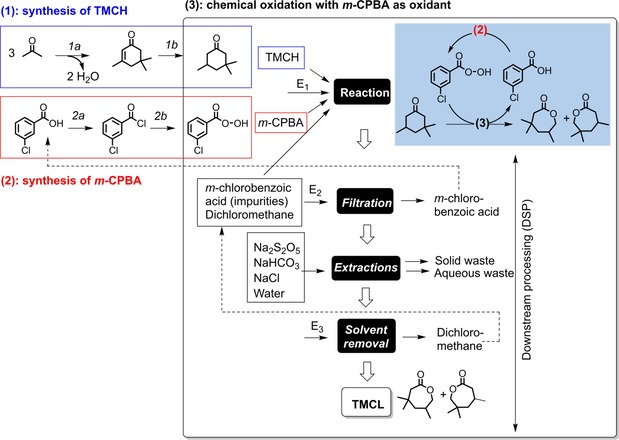
Process flowsheet for the chemical synthesis of the product TMCL (3), describing the synthesis of the substrate (1) and the synthesis of the chemical oxidant m‐CPBA (2). Reaction conditions: 1 a) base‐catalyzed aldol condensation (KOH, 90 °C, 20 h), 1 b) Pd‐catalyzed hydrogenation (supercritical CO2, 104–116 °C), 2 a) chlorination (SOCl2, 70 °C, 4 h), 2 b) nucleophilic reaction with hydrogen peroxide (H2O2, aq. NaOH, dioxane, MgSO4, dioxane, 15 min), 3) chemical Baeyer–Villiger oxidation (RT, 72 h). Electricity consumptions are indicated as Ei (see Table S1 in the Supporting Information for details). Dotted arrows indicate potential recycled streams (in the sensitivity analysis only).
Figure 3.
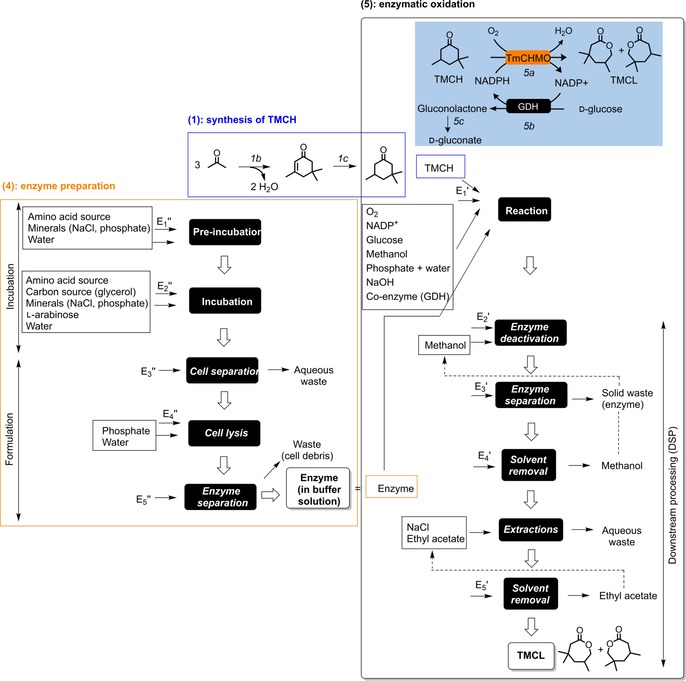
Process flowsheet for the enzymatic synthesis of the product TMCL (5), describing the synthesis of the substrate TMCH (1) and the preparation of the enzyme (4). Reaction conditions: 1 a) base‐catalyzed aldol condensation (KOH, 90 °C, 20 h), 1 b) Pd‐catalyzed hydrogenation (supercritical CO2, 104–116 °C), 5) enzymatic Baeyer–Villiger oxidation (30 °C, 28 h) with 5 a) oxidation, 5 b) co‐factor regeneration, and 5 c) spontaneous hydrolysis of the co‐product. Electricity consumptions are indicated as Ei′ and Ei′′ (see Table S1 in the Supporting Information for details). Dotted arrows indicate potential recycled streams (in the sensitivity analysis only).
The synthesis of the substrate TMCH is common to the two synthetic routes (Figures 2 and 3). The base‐catalyzed aldol condensation of acetone gives isophorone (step 1 a), which is then hydrogenated in supercritical CO2 with a Pd catalyst to produce TMCH (step 1 b). Many catalysts and experimental procedures have been reported for the preparation of isophorone from acetone.30 Although some procedures report better selectivity, the procedure selected gave the most complete information concerning the mass balance.31
The synthesis of the chemical oxidant m‐CPBA is performed in two steps (Figure 2).32 In step 2 a, m‐chlorobenzoic acid is chlorinated by using thionyl chloride. The corresponding peracid m‐CPBA is then formed in step 2 b by nucleophilic substitution with hydrogen peroxide.
The synthesis of the product TMCL by chemical oxidation is performed by using m‐CPBA (Figure 2).20a The reaction is performed in dichloromethane with an excess of peracid, with m‐chlorobenzoic formed as co‐product. Because this acid is also the precursor of the synthesis of m‐CPBA, this chemical is isolated by filtration and recycled. Because of the excess oxidant, the DSP steps require its neutralization by extraction with a sulfite, followed by base extraction to remove the remaining acid co‐product. Finally, the organic phase is washed with brine, and the solvent is removed under rotary evaporation. The product is recovered as an oil.
The preparation of the oxidizing enzyme, TmCHMO, is included in this LCA (Figure 3).26 In this study, the enzyme is used as a cell‐free extract, prepared in phosphate buffer. Enzyme preparation consists of a pre‐incubation and an incubation step with a source of amino acids, minerals, glycerol as carbon source, and water for the fermentation broth. The resultant E. coli K12 derivative cells are then separated from the broth by centrifugation and resuspended in phosphate buffer, after which they are subjected to cell lysis to create a cell‐free extract. The cell debris are separated by centrifugation to obtain the enzymes in buffer solution.
The synthesis of the product TMCL by enzymatic oxidation is performed by using TmCHMO as an oxidizing enzyme in phosphate buffer.26b Methanol is used as the co‐solvent to aid the solubility of the substrate TMCH. The reaction requires the use of nicotinamide adenine dinucleotide phosphate (NADPH) as a co‐factor (step 5 a). Because of its price and relatively low stability, this co‐factor is used in low amounts and is regenerated by using a glucose dehydrogenase (GDH) as co‐enzyme with d‐glucose as co‐substrate (step 5 b). This reaction leads to the formation of gluconolactone, which spontaneously hydrolyzes in the aqueous medium to form d‐gluconic acid, resulting in a decreased pH of the reaction (step 5 c). To maintain constant pH, the reaction is auto‐titrated with a base solution, thereby forming d‐gluconate as a co‐product. In the DSP, the enzymes are first deactivated by addition of methanol and then separated by centrifugation from the reaction mixture, which is then concentrated by rotary evaporation to remove the methanol. The product is isolated from the aqueous solution by extraction with ethyl acetate after saturation with sodium chloride to afford an oil. Both solvents (methanol and ethyl acetate) are recycled.
Data collection
In this LCA, priority was given to primary data from laboratory‐scale experiments. The data used are divided in several categories, in order of importance:
Primary data from laboratory‐scale experiments, which were performed in our group. This is the case for the chemical and enzymatic syntheses of the product as well as the enzyme preparation.20a, 26b Given the importance of accurate energy consumption data,33 the electricity consumption was measured with laboratory equipment used in the syntheses (see Table S1 in the Supporting Information for the details of the electricity consumption).
Secondary data modeled based on the literature were used for chemicals specific to this study: the substrate TMCH and the oxidant m‐CPBA. All these data were based on laboratory‐scale experiments, except for step 1 b. In this step, the data were based on experiments performed at pilot‐plant scale for which the electricity consumption was estimated.31, 32, 34
Secondary data from the Ecoinvent v3.2 database (Ecoinvent Center, St‐Gallen, Switzerland) were used for basic chemicals such as solvents, acids, bases, inorganic salts, and so on. These data were obtained from the global market, or from the European market if not available, to make sure the data are more representative of the context of this study. Likewise, the data from the electricity production were taken as an average of the European energy grid mix with low voltage.
Data from alternative chemicals were used for chemicals that were not available in the Ecoinvent database and were replaced by chemicals with equivalent functions (l‐arabinose: glucose; tryptone: soy bean meal; yeast extract: fodder yeast; m‐chlorobenzoic acid: benzoic acid; potassium sulfate: sodium sulfate; sodium metabisulfite: sodium sulfite; sodium bicarbonate: sodium carbonate; GDH enzyme: enzyme production from potato starch). The impact of these replacement chemicals is discussed in the results.
-
Suppressed data: a cut‐off rule was applied to chemicals that were present in negligible weight percentages compared with the total chemical input of a given reaction
(<0.05 % for NADP+, <0.004 % for ampicillin, 0.0002 % for supported Pd catalyst).
Data quality
The pedigree matrix is representative of the quality of the data, in particular its geographical and temporal correlation as well as completeness and technological level (Table S2 in the Supporting Information).35 The matrix allows determination of the uncertainty for data following a log‐normal distribution. The distribution of emissions in the environment is often log‐normal.36
Methods and environmental impacts assessed
The two synthetic routes were modeled in Simapro V8 (PRé consultant, NL). Environmental impacts were calculated with the IMPACT 2002+ V2.14 method, which covers the major environmental effects.37 Only the end‐point categories are presented in the Results and Discussion section, but mid‐points were used for the life cycle impact assessment (LCIA) interpretations. The climate change impact was calculated with the IPCC GWP 100a mid‐point method according to the updated method from the Intergovernmental Panel on Climate Change (IPCC).38 The water intensity was calculated based on the mass balance of the synthesis and product isolation because all data were available.
Results and Discussion
The potential environmental impacts of the chemical and enzymatic syntheses were compared for five different end‐point categories relative to the FU (which is 1 g of TMCL product): 1) carbon emissions or climate change impact, which is a measure of the increasing temperature in the lower atmosphere as a result of the emission of greenhouse gases such as carbon dioxide, methane, and nitrogen dioxide into the air. Emissions are converted in CO2 equivalents by using the global warming potential (GWP) provided by the IPCC as a conversion factor, expressed in kg of CO2 equivalents; 2) water intensity, which measures the amount of water used for the synthesis and product isolation, expressed in g of water; 3) damage to human health, which is an aggregation of toxicity to humans, respiratory effects, ionizing radiation, ozone‐layer depletion, and photochemical oxidation. It is expressed in disability‐adjusted life year (DALY); 4) resources, which combines the energy from mineral extraction and non‐renewable resources, expressed in MJ primary; 5) ecosystem quality, which includes several factors such as ecotoxicity, acidification, eutrophication, and land use. It is expressed in potentially disappeared fraction of species per m2 per year (PDF m−2 yr−1).
The relative environmental impact of the chemical and enzymatic synthesis for four impact categories is shown in Figure 4. The uncertainties were calculated in Simapro by using a Monte‐Carlo simulation method and are represented by the error bars. The absolute values for the environmental impact and the uncertainties are given in Table S3 (in the Supporting Information). The uncertainty is quite high with relative standard deviations of approximately 50 % on average. Because the LCA is based on data from laboratory experiments performed at one place, the uncertainty is higher than for an LCA performed on industrially implemented processes running at several sites over a long time period.
Figure 4.
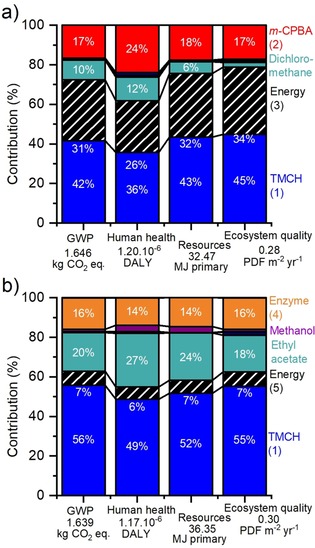
Contribution distribution for four environmental impact categories for a) chemical synthesis and b) enzymatic synthesis. The energy contribution is the electricity used for the oxidation synthesis only. The percentages of contributions lower than 3 % are not indicated. The total values for each impact category are indicated below the x axis.
The contribution inputs of the chemicals and energy (electricity) consumption are quite similar for all impact categories considered (Figure 4). It should be noted that the electricity consumption shown in Figure 4 represents only the electricity required for the oxidation synthesis, and that the impact of the chemicals (chemical oxidant, enzyme, substrate) takes into account the electricity required for their synthesis. Chemical and enzymatic syntheses have an almost identical environmental impact if no chemical is recycled (less than 0.4 % difference in favor of the enzymatic synthesis). The potential impact of the use of alternative chemicals [from data type 4)] on climate change is negligible because these chemicals represent a very small fraction of the total climate change (Figure S1 in the Supporting Information).
The contributions of the substrate, electricity, chemical oxidant, and enzyme preparation are quite constant independently of the environmental category that is considered. The only exception is the increased impact made on human health, which is mainly owing to the respiratory organics. This is attributed to the use of organic solvents, which is associated with some health risks such as dizziness (ethyl acetate), toxicity upon ingestion and inhalation (methanol), and potential carcinogenic effects (dichloromethane). Additionally, the synthesis of m‐CPBA requires the use of thionyl chloride, which is toxic and can cause severe burns and eye damage, and dioxane, which is a suspected carcinogenic solvent. Although 10 % of the climate change impact is caused by the use of organic solvent for the chemical synthesis, this impact is doubled for the enzymatic synthesis owing to the use of ethyl acetate for DSP. This is one of the disadvantages of enzymatic reactions, in which the product often displays limited water solubility, thereby requiring larger amounts of organic solvent for extraction. As such, careful solvent selection combined with solvent recycling is crucial for industrial processes.39 The effect of solvent recycling is investigated in the sensitivity analysis in the next section.
The rest of the study focuses on the comparison of climate change impacts of both oxidation syntheses of the product because climate change is in general the main environmental concern. Moreover, the contribution of each input to the GWP is quite representative of the distribution of the other impact categories (Figure 4). Because this study was based on laboratory‐scale experiments, the energy consumption is from electricity only. It should be noted that this differs from reactions performed at industrial scale, for which the energy source is not exclusively electricity (e.g., with heat exchangers). Energy consumption in lab‐scale processes is usually higher than the corresponding optimized reactions performed at industrial scale (per unit of product).18 Consequently, the contributions to climate change of both reactions were also detailed further, grouping the total electricity required for the entire process (Figure 5 a).
Figure 5.
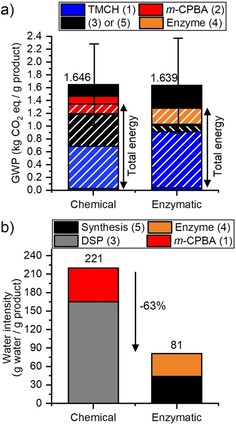
Performances of the chemical and enzymatic reactions with a) GWP and b) water intensity. The values on top of the columns indicate the total GWP and total water intensity, respectively. Stripped bars indicate contributions owing to electricity consumption. The error bars indicate the standard deviations for the GWP.
In both the chemical and enzymatic oxidation routes, the largest impact is made by the total electricity consumption for the oxidation step itself but also for the synthesis of the chemicals required for this reaction. The electricity consumption contributes up to 80 % of the GWP from the chemical synthesis and up to 76 % of the GWP from the enzymatic synthesis. This difference in electricity consumption for the same product synthesized chemically and enzymatically is caused by three aspects. The first is the increased electricity consumption of the oxidation step itself, which is almost four times higher for the chemical reaction, which is directly related to the increased reaction time (72 h compared with 28 h for the enzymatic reaction). The second aspect is that the electricity needed to synthesize the TMCH substrate is higher for the enzymatic reaction because, although both reactions have a substrate conversion higher than 99 %, the isolated yield of the enzymatic reaction is lower (68 % compared with 90 % for the chemical synthesis). Consequently, more substrate is needed to obtain the same quantity of product after DSP. Lastly, the contribution of the synthesis of the chemical oxidant, m‐CPBA, is similar to that of the enzyme preparation (less than 17 % of the total for each synthesis considering the climate change impact category). However, although half of the impact made by the chemical oxidant is caused by the chemicals needed for its preparation, the electricity consumption required for the preparation of the enzymes represents almost all of its impact, with only 2 % attributed to the chemicals. More specifically, the electricity consumption for (pre)‐incubation represented the largest contribution, whereas 16 % of the GWP was caused by formulation of the enzyme.
The prominent contribution of electricity in the GWP of the synthesis is quite typical of an early‐stage LCA.17b Clearly, the electricity consumption of laboratory data, on which early‐stage LCAs are based, is not representative of the energy consumption of the same optimized process at industrial scale.18 They are several reasons for this difference in scaling: 1) the electricity consumption of laboratory equipment is usually not a factor that is taken into account when developing a reaction at laboratory scale, thereby resulting in a higher contribution of the energy consumption for early‐stage LCAs; 2) such equipment usually suffers from an increased energy consumption at start‐up, which increases the average energy consumption of the process;40 and 3) most importantly, the equipment used for large‐scale processes differs from that used at laboratory scale and is usually more energy efficient. This shows the importance of scale in LCAs, especially regarding equipment and their energy efficiencies. It also demonstrates the need to take the electricity consumption into account for early‐stage LCAs. Several ways are possible to account for the electricity consumption: it can be measured from the actual equipment, which is time‐consuming but the most accurate, or it can also be estimated from similar equipment or calculated based on thermodynamic properties. For laboratory‐scale LCAs, it appears that measuring the actual electricity consumption of apparatus used becomes crucial because the energy consumption will typically represent an important contribution to the environmental impact.
Another important environmental impact that is particularly relevant for enzymatic reactions is the water intensity,41 that is, the quantity of water needed for the synthesis and DSP. Typically, water is the reaction medium for enzymatic reactions. Compared with the corresponding chemical reactions, biocatalytic reactions are therefore expected to have a higher water intensity. However, it is important to take into account DSP because the use of m‐CPBA as the chemical oxidant in the corresponding chemical synthesis induces several washing steps to neutralize the remaining oxidant and remove any acid co‐product. As a result, the water intensity of the chemical synthesis is in reality higher than that of the enzymatic synthesis, despite the use of an organic solvent as the reaction medium, in contrast to the enzymatic synthesis for which water is the reaction medium (Figure 5 b). The water intensity of the enzymatic reaction is 63 % lower than that of the chemical reaction. Approximately half of this water intensity is contributed by the preparation of the enzymes, and the other half represents the water used as the reaction medium. Surprisingly, the synthesis of the chemical oxidant is more water intensive than the preparation of the enzymes, although the latter are obtained through fermentation in aqueous medium. Because no water is used for the synthesis and purification of the TMCH substrate, the water intensity values are independent of the conversion and isolated yield of both oxidation reactions.
Sensitivity analysis
In the base‐case scenario, the results of which are presented above, the electricity was sourced from Europe, the peracid m‐CPBA was selected as the chemical oxidant for the chemical synthesis, and the enzymes used in the biocatalyzed reactions were cell‐free extracts used for one reaction only. In both cases, no chemical was recycled (Figures 2 and 3). A sensitivity analysis was performed on these parameters to evaluate their influence on the environmental impact and to test the robustness of the assumptions made in the methodology.
Effect of electricity source
Electricity consumption was shown to be the largest contributor to the synthesis of the product in both cases, with the current average EU electricity mix as the electricity source. As such, different scenarios regarding the source of electricity were also evaluated (Figure 6). The GWP for both the chemical and the enzymatic reactions was compared with electricity sourced from the Netherlands (mostly fossil‐based) and with electricity sourced from Norway (mostly from renewable resources; Ecoinvent v3.2).
Figure 6.
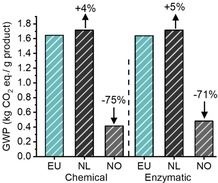
Evolution of GWP for the chemical and enzymatic synthesis as a function of the electricity source, with average from Europe (EU, GWP=0.49 kg kWh−1), the Netherlands (NL, GWP=0.55 kg kWh−1), and Norway (NO, GWP=0.04 kg kWh−1). The percentages on top of the columns indicate the difference of GWP compared with the EU electricity source.
Changing the electricity source from EU to the Netherlands resulted in an increase to GWP of 4–5 %. This is owing to the increased carbon intensity of the Dutch electricity grid mix, which is mainly produced by using natural gas and coal, compared with the average electricity from Europe, which is a mix of combustible fuels, nuclear energy, and renewable energy such as wind and hydro‐electric power. A dramatic decrease in environmental impact was observed if the electricity source was replaced by greener electricity from Norway, which is mainly from hydroelectric power. In this case, the GWP of the enzymatic and chemical reactions could be decreased by 71 and 75 %, respectively. This is of course correlated with the high contribution of electricity consumption to the climate change impact of the chemical and enzymatic reactions. It furthermore indicates that the geographical location of industrial (bio)chemical reactions also contributes to its environmental impact.
Effect of recycling efficiency of solvents and co‐product streams
In both oxidation reactions, several streams can be recycled: solvents, the co‐product of the chemical synthesis, and m‐chlorobenzoic acid, which is the precursor of the chemical oxidant m‐CPBA. This co‐product can be recycled for the synthesis of m‐CPBA because it is isolated during the filtration step of product purification (Figure 2). Similarly, dichloromethane can be recycled as the reaction medium after the solvent removal step during product isolation. In the enzymatic synthesis, two solvent streams can be recycled: methanol and ethyl acetate, which are both used during DSP and evaporated (Figure 3).
Considering solvents, dichloromethane production results in the highest GWP contribution (3.50 kg kg−1 vs. 2.56 kg kg−1 for ethyl acetate). However, the enzymatic synthesis requires at least twice the amount of solvent for the DSP. Consequently, recycling the solvent stream has a more drastic effect on the GWP of the enzymatic synthesis (Figure 7 a). If dichloromethane and m‐chlorobenzoic acid were recycled with 90 % efficiency, the GWP of the chemical synthesis decreased by approximately 9 % compared with if no recycling was implemented. Further improving the recycling efficiency to 95 % resulted in a slight improvement, with 9.6 % decrease in GWP compared with no recycling. Interestingly, it is the recycling of the solvents that allows the enzymatic synthesis to be of lower environmental impact than the chemical synthesis. The environmental impact could be reduced by almost 20 % with a recycling efficiency of 95 %. Overall, recycling of solvents has a significant impact on the syntheses because they have a high environmental impact (see Figure S1 in the Supporting Information for details). For the rest of the sensitivity analysis, a recycling efficiency of 90 % was applied.
Figure 7.

a) GWP as a function of the recycling efficiency of co‐product (m‐chlorobenzoic acid) and solvent for the chemical and enzymatic syntheses, and b) comparison of the total GWP of the syntheses with 90 % recycling efficiency of solvents with the replacement with peracetic acid (chemical synthesis) and reutilization of the enzyme (enzymatic synthesis) with either whole‐cells in buffer (total GWP=1.080 kg gproduct −1) or whole‐cells in fermentation broth (total GWP=1.079 kg gproduct −1). The values on top of the bars indicate the total GWP.
Effect of enzyme recycling
Another industrially relevant alternative to decrease the GWP of the enzymatic reaction is to reuse the enzyme over several reaction cycles. For established industrial processes, biocatalysts can be recycled up to 200 times.42 A reusability of ten times with 2 % of loss of biocatalytic activity per reuse was assumed. This is a realistic estimation of the performance of this particular enzyme, based on direct experimental studies on its stability and reusability if immobilized.43 To model the reuse of the enzyme, the enzyme preparation procedure and the product isolation step must be adapted appropriately because the standard procedure includes the deactivation of all remaining enzyme by addition of methanol. The enzyme can potentially be recycled if used in the form of whole‐cells, which are separated from the reaction mixture by ultrafiltration, after which the product can be directly extracted from the reaction mixture. The electricity consumption for ultrafiltration was calculated based on data from the literature.44 Two whole‐cell preparation procedures were tested: in buffer solution or in fermentation broth. The latter option has the advantage higher energy efficiency because one centrifugation step can be omitted, although this biocatalyst format may ultimately prove more challenging owing to possible side‐reactions. The alternative flowsheet of the enzymatic reaction with recycled whole‐cells is shown in Figure S2 (in the Supporting Information).
Reusing the enzymes reduces the GWP by approximately 20 % (Figure 7 b). In this case, the contribution of the enzyme to the total GWP of the enzymatic synthesis drops to less than 3 % whereas it accounts for approximately 19 % of the total GWP if it is not recycled. It should also be noted that the reusability of the enzymes induces lower electricity consumption owing to the simplification of the DSP steps.
Effect of oxidant type
Although m‐CPBA is an effective chemical oxidant, it displays several disadvantages such as safety issues related to its flammability and shock sensitivity, making it potentially explosive.22 Additionally, its use generates one equivalent of co‐product, m‐chlorobenzoic acid, which can be recycled as detailed above. It is typically used in excess, which requires quenching of any unreacted oxidant, leading to a more complex DSP. Consequently, greener alternatives such as hydrogen peroxide, peracetic acid, and of course oxygen, which require the use of oxidative biocatalysts, have been identified.23a If m‐CPBA was replaced with peracetic acid, no recycling of the co‐product (acetic acid in this case) was modeled because its isolation from the aqueous medium would require additional steps after extraction with ethyl acetate (Figure S3 in the Supporting Information). The synthesis of peracetic acid was modeled based on data from the literature.45 The use of peracetic acid instead of m‐CPBA leads to a decrease of approximately 18 % of the GWP of the chemical synthesis (Figure 7 b). Despite the lack of recycling for peracetic acid, the contribution of this oxidant to the total GWP of the chemical synthesis is negligible (less than 1 %). This is because the synthesis of peracetic acid is more efficient than that of m‐CPBA because it is synthesized in a single step, uses fewer chemicals (water, hydrogen peroxide, acetic acid, and sulfuric acid), and the production of these chemicals has a low impact on the GWP contribution.
Key process performance metrics influencing the environmental impact
The electricity consumption, as well as the amount of substrate, are crucial parameters for the environmental impact of the synthesis of the product. These two parameters are directly correlated with the reaction time, the substrate conversion, and the yield of the reaction, which are typical process metrics investigated for process intensification of biocatalytic reactions.42 The enzymatic reaction has a lower isolated yield than the chemical reaction (65 vs. 90 %), which results in a higher amount of substrate needed for the synthesis of the same amount of product. However, the chemical synthesis is less efficient and requires 3 days to reach full conversion (see Figure S4 in the Supporting Information for the kinetics), whereas the enzymatic oxidation is completed within 28 h. To use laboratory‐scale LCA as a tool to help improve reactions while keeping their environmental impact in mind, the GWP of both reactions was calculated as a function of the isolated yield and the reaction time (Figure 8). The isolated yield is calculated by assuming full substrate conversion.
Figure 8.
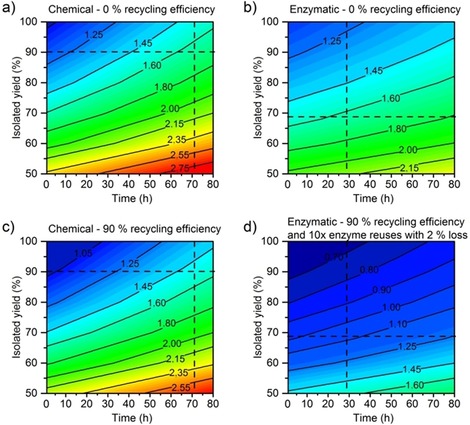
Evolution of GWP (kg gproduct −1) as a function of the reaction time and the isolated yield for a) the chemical synthesis with m‐CPBA without recycling, b) the enzymatic synthesis without recycling, c) the chemical synthesis with m‐CPBA with 90 % recycling efficiency of solvents and co‐product, and d) the enzymatic synthesis with 90 % recycling efficiency of solvents and reuse of enzyme (10 cycles with 2 % loss). The intersection of the dotted lines indicates the current isolated yield and reaction time.
If both reactions have the same process metrics (reaction time, conversion, and isolated yield) and the same recycling efficiency, the environmental impact of the enzymatic reaction is lower than that of the chemical reaction independent of the efficiency of the reaction (Figure 8 a, b). The reaction time is the most important factor to consider for reducing the environmental impact of the chemical reaction, whereas the most important factor for the enzymatic reaction is the isolated yield. Interestingly, the GWP of the chemical reaction would equal that of the enzymatic reaction if the reaction time of the chemical reaction was more than halved and the isolated yield of the enzymatic reaction was increased to approximately 80 %, which can easily be achieved (Figure 8 a, b). This decrease in reaction time, however, seems hardly feasible if full substrate conversion of the chemical reaction is to be achieved.
An optimistic scenario in which the solvents, co‐product, and enzymes were recycled was considered. This scenario does not consider the use of energy from more renewable resources, which would drastically decrease the environmental impact of both reactions as shown above, because the energy contribution is overrepresented at the laboratory scale. If both reactions are optimized in terms of recycling, the enzymatic synthesis clearly has the lower environmental impact (Figure 8 c, d). If the reaction time of the chemical synthesis could be reduced with an improvement in the isolated yield, a GWP lower than 1.25 kg of CO2 equivalents could be obtained. However, the isolated yield of the enzymatic synthesis can easily be improved to a level similar to that of the chemical reaction, which would result in a GWP below 0.8 kg of CO2 equivalents. This shows that the improvement of the environmental impact of the reactions is different depending on the reaction. Whereas a reduction of the reaction time impacts the chemical reaction, recycling of solvents and enzyme is crucial for the enzymatic reaction.
Conclusions
The environmental impact of an enzymatic oxidation reaction was evaluated and compared with its chemical equivalent in an early‐stage cradle‐to‐gate life cycle assessment (LCA). This comparative LCA demonstrates that, at laboratory scale, the enzymatic and chemical synthesis have very similar environmental impacts if no recycling of solvents or co‐product is considered. It is in fact the recycling of solvents and enzyme that makes the enzymatic synthesis of lower environmental impact than the chemical synthesis. This is owing to the higher recycling potential of the enzymatic synthesis, which is more solvent‐intensive because the product isolation requires organic extractions from the aqueous phase. It should be noted that although recycling can be advantageous it can also be limited in some cases by economic considerations owing to the potential increase of the complexity of the process or energy requirements to achieve increased recycling efficiencies. The enzymatic reaction was, however, more competitive considering the water intensity because it resulted in less than half the water consumption of the chemical reaction, despite the use of water as the reaction medium for enzyme preparation and the oxidative reaction itself.
A sensitivity analysis performed on the source of the electricity showed that the total climate change impact of the chemical and biocatalyzed reactions can be decreased by up to 71–75 %, respectively, by using renewable electricity. This drastic decrease shows the importance of the electricity consumption to the environmental impact of both reactions and to early‐stage LCAs in general. Several factors contributed to decreasing the environmental impact of the reactions still further, namely recycling of solvent and co‐product streams, recycling of the enzyme as whole‐cells, and replacing the chemical oxidant with peracetic acid. In the two latter cases, the contribution of the oxidant (chemical or enzymatic) was almost negligible with less than 1 % contribution to the total GWP contribution of the oxidation.
Prospective LCA based on laboratory‐scale data can also be used as a tool for the improvement of enzymatic reactions. The reduction of the reaction time (correlated with the amount of electricity consumed) and the improvement of the substrate conversion and isolated yield have a greater influence on the environmental impact of the chemical reactions than the recycling of solvent streams. For the enzymatic synthesis, the recycling of solvents and enzymes influences its environmental impact the most. These parameters help to evaluate the improvement of key process performance metrics, such as conversion, isolated yield, and space‐time yield, which are necessary for the commercialization of enzymatic reactions. This optimistic scenario did not take into account replacing the electricity with renewable electricity because of the overrepresentation of electricity in the data based on laboratory‐scale experiments. Such an analysis performed on data from pilot‐plant scale or simulated data at industrial scale would help still further in identifying crucial parameters influencing the environmental impact.
The key learnings from this comparative cradle‐to‐gate LCA at laboratory scale are 1) the importance of scale in LCA, and its impact of the energy consumption, and 2) the use of such LCAs as learning tools to compare the environmental performances of chemical and enzymatic reactions even at a prospective scale. This tool should enable targeting of the key process performance metrics, which can make an important difference for optimizing enzymatic reactions. We hope that this work can inspire early‐stage comparative cradle‐to‐gate LCAs for the development of greener processes and in particular biocatalytic processes.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
The research for this work has received funding from the European Union (EU) project ROBOX (grant agreement No. 635734) under the EU′s Horizon 2020 Programme Research and Innovation actions H2020‐LEIT BIO‐2014‐1. The views and opinions expressed in this article are only those of the authors, and do not necessarily reflect those of the European Union Research Agency. The European Union is not liable for any use that may be made of the information contained herein.
M. A. F. Delgove, A.-B. Laurent, J. M. Woodley, S. M. A. De Wildeman, K. V. Bernaerts, Y. van der Meer, ChemSusChem 2019, 12, 1349.
References
- 1. Wenda S., Illner S., Mell A., Kragl U., Green Chem. 2011, 13, 3007–3047. [Google Scholar]
- 2.
- 2a. Industrial Biotransformations (Eds.: A. Liese, K. Seelbach, C. Wandrey), Wiley-VCH, Weinheim, 2006; [Google Scholar]
- 2b. Straathof A. J. J., Panke S., Schmid A., Curr. Opin. Biotechnol. 2002, 13, 548–556. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Anastas P. T., Warner J. C., Green Chemistry: Theory and Practice, Oxford University Press, Oxford, 2000; [Google Scholar]
- 3b. Sheldon R. A., Woodley J. M., Chem. Rev. 2018, 118, 801–838. [DOI] [PubMed] [Google Scholar]
- 4.
- 4a. Woodley J. M., Trends Biotechnol. 2008, 26, 321–327; [DOI] [PubMed] [Google Scholar]
- 4b. Ran N., Zhao L., Chen Z., Tao J., Green Chem. 2008, 10, 361–372. [Google Scholar]
- 5. Ni Y., Holtmann D., Hollmann F., ChemCatChem 2014, 6, 930–943. [Google Scholar]
- 6. Clouthier C. M., Pelletier J. N., Chem. Soc. Rev. 2012, 41, 1585–1605. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Kim S., Jiménez-González C., Dale B. E., Int. J. Life Cycle Assess. 2009, 14, 392–400; [Google Scholar]
- 7b. Nielsen P. H., Oxenbøll K. M., Wenzel H., Int. J. Life Cycle Assess. 2007, 12, 432. [Google Scholar]
- 8.
- 8a. Constable D. J. C., Curzons A. D., Cunningham V. L., Green Chem. 2002, 4, 521–527; [Google Scholar]
- 8b. Sheldon R. A., Green Chem. 2017, 19, 18–43. [Google Scholar]
- 9. McManus M. C., Taylor C. M., Biomass Bioenergy 2015, 82, 13–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.ISO 14044:2006, Environmental Management – Life Cycle Assessment – Requirements and Guidelines, International Organization for Standardization, 2006.
- 11. Burgess A. A., Brennan D. J., Chem. Eng. Sci. 2001, 56, 2589–2604. [Google Scholar]
- 12.
- 12a. Tabone M. D., Cregg J. J., Beckman E. J., Landis A. E., Environ. Sci. Technol. 2010, 44, 8264–8269; [DOI] [PubMed] [Google Scholar]
- 12b. Khoo H. H., Ee W. L., Isoni V., Green Chem. 2016, 18, 1912–1922. [Google Scholar]
- 13. Thum O., Oxenbøll K., SOFW-J. 2008, 134, 44–47. [Google Scholar]
- 14. Henderson R. K., Jiménez-González C., Preston C., Constable D. J., Woodley J. M., Ind. Biotechnol. 2008, 4, 180–192. [Google Scholar]
- 15. Khoo H. H., Isoni V., Sharratt P. N., Sustainable Prod. Consumption 2018, 16, 68–87. [Google Scholar]
- 16. Arvidsson R., Tillman A.-M., Sandén B. A., Janssen M., Nordelöf A., Kushnir D., Molander S., J. Ind. Ecol. 2018, 22, 1286–1294. [Google Scholar]
- 17.
- 17a. Hischier R., Kwon N. H., Brog J.-P., Fromm K. M., ChemSusChem 2018, 11, 2068–2076; [DOI] [PubMed] [Google Scholar]
- 17b. Cossutta M., McKechnie J., Pickering S. J., Green Chem. 2017, 19, 5874–5884. [Google Scholar]
- 18. Piccinno F., Hischier R., Seeger S., Som C., J. Cleaner Prod. 2016, 135, 1085–1097. [Google Scholar]
- 19. Baeyer A., Villiger V., Ber. Dtsch. Chem. Ges. 1899, 32, 3625–3633. [Google Scholar]
- 20.
- 20a. Delgove M. A. F., Luchies J., Wauters I., Deroover G. G. P., De Wildeman S. M. A., Bernaerts K. V., Polym. Chem. 2017, 8, 4696–4706; [Google Scholar]
- 20b. Delgove M. A. F., Fürst M. J. L. J., Fraaije M. W., Bernaerts K. V., De Wildeman S. M. A., ChemBioChem 2018, 19, 354–360. [DOI] [PubMed] [Google Scholar]
- 21. Caron S., Dugger R. W., Ruggeri S. G., Ragan J. A., Ripin D. H. B., Chem. Rev. 2006, 106, 2943–2989. [DOI] [PubMed] [Google Scholar]
- 22. Hussain H., Al-Harrasi A., Green I. R., Ahmed I., Abbas G., Rehman N. U., RSC Adv. 2014, 4, 12882–12917. [Google Scholar]
- 23.
- 23a. ten Brink G. J., Arends I. W. C. E., Sheldon R. A., Chem. Rev. 2004, 104, 4105–4124; [DOI] [PubMed] [Google Scholar]
- 23b. Yakabi K., Mathieux T., Milne K., López-Vidal E. M., Buchard A., Hammond C., ChemSusChem 2017, 10, 3652–3659; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23c. Ríos M. Y., Salazar E., Olivo H. F., Green Chem. 2007, 9, 459–462. [Google Scholar]
- 24. Alphand V., Wohlgemuth R., Curr. Org. Chem. 2010, 14, 1928–1965. [Google Scholar]
- 25. Baldwin C. V. F., Wohlgemuth R., Woodley J. M., Org. Process Res. Dev. 2008, 12, 660–665. [Google Scholar]
- 26.
- 26a. Delgove M. A. F., Elford M. T., Bernaerts K. V., De Wildeman S. M. A., J. Chem. Technol. Biotechnol. 2018, 93, 2131–2140; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26b. Delgove M., Elford M., Bernaerts K., De Wildeman S., Org. Process Res. Dev. 2018, 22, 803–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Romero E., Castellanos J. R. G., Mattevi A., Fraaije M. W., Angew. Chem. Int. Ed. 2016, 55, 15852–15855; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 16084–16087. [Google Scholar]
- 28.
- 28a. Zhou J., Ritter H., Macromolecules 2008, 41, 1663–1666; [Google Scholar]
- 28b. Zhou J., Ritter H., Polym. Int. 2011, 60, 1158–1161. [Google Scholar]
- 29.Environmental Product Declaration (EPD)—PCR Basic Module—Basic Chemicals CPC 34 (v3.0), 2017.
- 30.
- 30a. Grebinoski M. C., Glassman D., Elias C. L., Schutz A. A. (Aristech Chemical Corporation, Philadelphia, PA) US5352839A, 1994;
- 30b. Mei J., Chen Z., Yuan S., Mao J., Li H., Yin H., Chem. Eng. Technol. 2016, 39, 1867–1874; [Google Scholar]
- 30c. Koklin A. E., Hasyanova G. M., Glukhov L. M., Bogdan V. I., Russ. Chem. Bull. 2017, 66, 488–490. [Google Scholar]
- 31. Li Y., Meng H., Lu Y., Li C., Ind. Eng. Chem. Res. 2016, 55, 5257–5262. [Google Scholar]
- 32.
- 32a. Zhao X.-J., Gong D.-M., Jiang Y.-R., Guo D., Zhu Y., Deng Y.-C., Eur. J. Med. Chem. 2017, 138, 738–747; [DOI] [PubMed] [Google Scholar]
- 32b. McDonald R. N., Steppel R. N., Dorsey J. E., Org. Synth. 1970, 50, 15. [Google Scholar]
- 33.D. Beloin-Saint-Pierre, in Society of Environmental Toxicology and Chemistry (SETAC) Europe 24th LCA Symposium, Vienna, Austria, 24–28 September 2018.
- 34. Licence P., Ke J., Sokolova M., Ross S. K., Poliakoff M., Green Chem. 2003, 5, 99–104. [Google Scholar]
- 35. Weidema B. P., Wesnæs M. S., J. Cleaner Prod. 1996, 4, 167–174. [Google Scholar]
- 36. Limpert E., Stahel W. A., Abbt M., BioScience 2001, 51, 341–352. [Google Scholar]
- 37. Jolliet O., Margni M., Charles R., Humbert S., Payet J., Rebitzer G., Rosenbaum R., Int. J. Life Cycle Assess. 2003, 8, 324. [Google Scholar]
- 38. Fifth Assessment Report, The Physical Science Basis, Intergovernmental Panel on Climate Change, 2013.
- 39. Jiménez-González C., Poechlauer P., Broxterman Q. B., Yang B.-S., am Ende D., Baird J., Bertsch C., Hannah R. E., Dell′Orco P., Noorman H., Yee S., Reintjens R., Wells A., Massonneau V., Manley J., Org. Process Res. Dev. 2011, 15, 900–911. [Google Scholar]
- 40. Hetherington A. C., Borrion A. L., Griffiths O. G., McManus M. C., Int. J. Life Cycle Assess. 2014, 19, 130–143. [Google Scholar]
- 41. Pfister S., Boulay A.-M., Berger M., Hadjikakou M., Motoshita M., Hess T., Ridoutt B., Weinzettel J., Scherer L., Döll P., Manzardo A., Núñez M., Verones F., Humbert S., Buxmann K., Harding K., Benini L., Oki T., Finkbeiner M., Henderson A., Ecol. Indic. 2017, 72, 352–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tufvesson P., Fu W., Jensen J. S., Woodley J. M., Food Bioprod. Process. 2010, 88, 3–11. [Google Scholar]
- 43. Delgove M. A. F., Valencia D., Solé J., Bernaerts K. V., De Wildeman S. M. A., Guillén M., Álvaro G., Appl. Catal. A 2019, 572, 134–141. [Google Scholar]
- 44. Bahnasawy A. H., Shenana M. E., Aust. J. Agric. Eng. 2010, 1, 54–65. [Google Scholar]
- 45. Greenspan F. P., Ind. Eng. Chem. 1947, 39, 847–848. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary