

Abstract
Lipidomics is a rapidly growing field with numerous examples showing the importance of lipid molecules throughout biology. It has also shed light onto the vast and complex functions performed by many lipids that possess an immense diversity in molecular structures. Mass spectrometry (MS) is the tool of choice for analyzing lipids and has been the key catalyst driving the field forward. However, MS does not yet permit true molecular lipidomics wherein the identification and quantification of lipids having defined molecular structures can be routinely achieved. Here we describe recent advances in MS‐based lipidomics that allow access to higher levels of molecular information in lipidomics experiments. These advances will form a key piece of the puzzle as the field moves towards systems characterization of lipids at the molecular level.
Keywords: biochemistry, isomers, lipidomics, mass spectrometry, structural identification
1. Introduction
The appreciation of lipids as complex and critical molecular players has grown dramatically in the last two decades. No longer simply viewed as a means for energy storage and a structural scaffold for the cell membrane, it is now recognized that lipids play key and intricate roles in many cellular and metabolic functions,1 while specific alterations of the lipidome are implicated in the onset and progression of many diseases.2 By‐and‐large this understanding has been catalysed by the rapid emergence of mass spectrometry (MS)‐based lipidomics.3
The sensitivity and structural elucidation capabilities of modern MS make it the only analytical technology capable of even partially resolving the structural complexity of the lipidome, which can, in principle, consist of over 100 000 unique structures. As many of the mechanisms involved in lipid synthesis remain poorly understood,4 our knowledge of lipid composition comes almost entirely from MS measurements. The cornerstone of modern lipidomics is the analysis of tissue or cell extracts using electrospray ionization (ESI) coupled with MS using either direct infusion (shotgun lipidomics) or following chromatographic separation.49 However, with the rapid emergence of MS imaging (MSI) that enables the spatial distributions of lipids to be mapped within tissues, alternative ionization methods, such as matrix‐assisted laser desorption/ionization (MALDI), are finding more widespread use.5 In its various embodiments, conventional MS provides a wealth of structural information of detected lipid species (Figure 1). Within the context of a lipid structural hierarchy,6 ultra‐high mass resolution and accuracy (UHRAM) measurements enable the direct assignment of sum‐composition lipid species (i.e., the lipid class combined with the sum number of carbons and carbon–carbon double bonds (C=C bonds) in both fatty acyl chains).7 Beyond this, conventional tandem mass spectrometry (MS/MS) with low energy collision‐induced dissociation (CID) both confirms lipid class assignments and allows the identification of the length and degree of unsaturation of individual fatty acyls.9, 49 This is the current status of most lipidomics workflows, but it is still far from the unambiguous structural identification of lipids required to completely describe a lipid and its biochemical origins. Moreover, there is often insufficient information to reconcile whether the mass spectral signatures observed arise from a single molecule or an isomeric mixture. To address this challenge there has been great interest in recent years on the development of alterative ion‐activation methods providing greater structural detail and resolution of isomeric lipid populations.8
Figure 1.
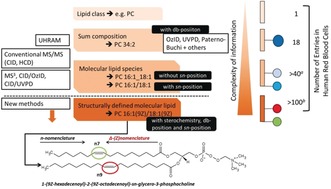
Hierarchy of lipid structure and MS methods required to reach each level. New methods (as discussed in this review) are needed to enable the identification of structurally defined molecular lipids. On the right, the number of unique lipid species at each level of identification expected in the red blood cell lipidome is shown(a and b indicate expected number of entries). Image adapted with permission from refs. 6, 13.
In this mini‐review we describe recent developments in MS‐based shotgun lipidomics that significantly add to the level of molecular information achieved in such experiments and pave the way for detection and quantitation of structurally defined lipid molecules, with an emphasis on glycero‐, glycerophospho‐, and sphingolipids. We focus on: 1) recently developed ion‐activation methods that provide greater structural details; 2) the benefit of combining the different ion‐activation methods with MSI to better localize defined lipid species in tissues; and 3) developments in time‐resolved (flux) lipidomics by incorporating and tracking stable isotopes (e.g., 13C‐, 15N‐, or 2H‐labeled precursors) to study lipid turnover. We also describe strategies for interpretation of lipid data, avoiding misinterpretation, and the need for standardization in the reporting of lipidomics data.
2. Mass Spectrometry Methods for the Detailed Structural Analysis of Lipids
The level of structural detail (and ambiguity) reached in MS‐based lipidomics depends on the methods employed. Consequently, so too does the ability to interpret the biochemical driving forces giving rise to the detected lipids. Prior to the widespread availability of UHRAM, low energy CID MS/MS was the primary approach used to identify lipids. It provides critical information relating to many structural features (see above)9 and is often coupled with triple quadrupole mass spectrometers employing structure‐specific precursor and neutral loss scans.10 In essence, CID enables the assignment of molecular lipid species (MLS) without sn‐positional information (Figure 1), although in some cases multi‐stage MS/MS (MSn) experiments using ion traps can move beyond this.11 A second advantage of MS/MS lies in the ability to selectively detect lipids not observed in the full‐scan spectrum, significantly increasing sensitivity and dynamic range. Employing precursor and neutral loss scanning on a q‐ToF analyzer, Ejsing et al. identified and quantified 250 molecular lipid species across 21 lipid classes in the yeast lipidome.12
The emergence of UHRAM instruments based on Fourier‐transform ion cyclotron resonance (FTICR) and Orbitrap has revolutionized shotgun lipidomics. Lipid analysis with a mass resolution equal to or greater than 150 000 (in the lipid m/z range) enables the resolution of many isobaric ion populations.3c, 13 When combined with accurate mass measurements (equal to or less than 3 ppm error) automatic sum‐composition assignment is directly possible. A second advantage arises when it is coupled with MS/MS. In the absence of sufficient resolving power, structural ambiguity may still arise in MS/MS analysis, especially when isobaric ions are co‐isolated and fragmented. In contrast, high resolution MS/MS facilitates the unambiguous assignment of individual fragment ions to co‐isolated isobaric precursors (provided they too are resolved). Almeida et al. have applied this approach in a data‐independent workflow to quantify 311 lipid species encompassing 20 classes and 202 unique molecular glycerophospholipids from a mouse brain extract.14 An important observation in this work was the requirement for resolving powers greater than 200 000 for the baseline separation of lipids containing 2×13C atoms from monoisotopic lipids containing one less double bond (e.g., triacylglycerides (TAGs) TAG 54:1 and 13C2‐TAG 54:2). Such resolution is only currently available using FTICR or high‐field Orbitraps allowing time‐domain signals of at least approximately 1 s in length to be recorded.
While possessing undisputed value in lipidomic workflows, UHRAM and CID are still far from offering the complete structural elucidation of lipids. They are blind to a variety of structural features, such as the positioning of C=C bonds and the esterification (sn) position of acyl chains on the glycerol backbone. This puts significant limitations on the ability to interpret measured lipid compositions and means identified MLS often encompass a population of unresolved isomers. In some cases the relative abundances of fatty acyl neutral loss fragments formed by CID can allow the identification and relative quantification of different sn‐isomers.11a While this can point to the major sn‐isomers present, such analysis does not exclude the possibility of low abundance, isomeric variants being present and is only applicable to a few phospholipid classes.
To address the increasingly recognized challenge of isomeric lipids and comprehensive structure elucidation, a variety of novel ion‐activation methods have been developed to provide access to alternative fragmentation channels and structural information. Several of these are discussed below. For a more in‐depth overview, the reader is referred to ref. 3c.
Ultraviolet photodissociation (UVPD) provides greater structural detail compared to CID alone. By enabling fragmentation following electronic excitation, fragmentation pathways not accessible with CID (vibrational excitation) are accessible. UVPD using 193 nm photons can cleave C=C bonds of both phospholipids and sphingolipids.8d,8f In the analysis of phospholipids Klein et al. observed the cleavage of the C−C bonds on either side of the double bond, providing a fragment ion pair separated by 24 Da for each double bond (db) position.8d Applying this method to the shotgun analysis of a bovine liver extract enabled a variety of phosphatidylcholine (PC) C=C isomers (db‐isomers) to be identified (Figure 2 a). Analogous cleavage of C=C bonds has been observed in a variety of unsaturated sphingolipid classes ionized as either lithiated or protonated adducts.8f Both fragment pairs, indicative of db‐position on the acyl chains, as well as the sphingosine C=C bond were observed. 193 nm UVPD has also been employed for MS3 fragmentation to resolve sn‐positional isomers of PC lipids. CID of sodiated PC lipids leads to the loss of the phosphocholine headgroup via a two‐step mechanism whereby the sn‐2 acyl chain becomes bonded to the backbone.8b This leaves behind a new C=C bond attached to the sn‐2 chain, the cleavage of which allows for the identification of the sn‐2 acyl chain.8e UVPD in this scenario also results in simultaneous cleavage of acyl chain C=C bonds, thus allowing fragment ions indicative of both PC sn‐ and db‐isomers to be produced in a single scan. UVPD may also be realized with photons not directly absorbed by the ionized lipid.15 Using radical‐directed dissociation (RDD) Pham et al. formed non‐covalent adducts of various phospholipids and TAGs with 4‐iodobenzoic acid (for the formation of anionic PC and SM ions) and 4‐iodoanaline(for the formation of cationic TAGs).15 UVPD of these adducts with 266 nm photons selectively cleaves the C−I bond, leaving behind a phenyl radical bound to the lipid. Further CID activation of this radical generates a radical site on the lipid that propagates RDD. In particular dissociation along unsaturated acyl chains was observed allowing the assignment of both C=C and chain‐branching positions.
Figure 2.
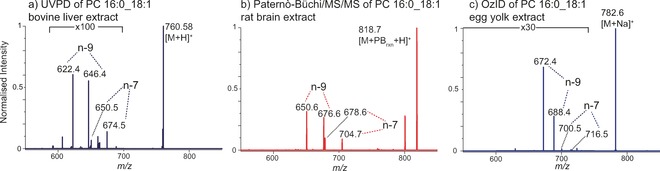
Differentiation of PC 16:0_18:1 double bond isomers using alterative ion‐activation strategies. a) UVPD of a bovine liver extract (data from ref. 8d), b) Paternò‐Büchi reactions followed by MS/MS of the reaction‐product ion acquired from a rat brain extract (data from ref. 16), and c) OzID from an egg yolk extract (unpublished data).
Paternò–Büchi (PB) reactions also allow the identification of db‐position.16 Here lipids are ionized using ESI from a solution containing acetone and irradiated using a 254 nm UV lamp through the nano‐ESI capillary. This induces a [2+2] photochemical reaction forming an oxetane ring. CID of the reaction product produces a pair of fragment ions 26 Da apart that are indicative of db‐positon. PB reactions have been employed online with shotgun lipidomics to reveal db‐isomers of both free fatty acids (FFAs) and phospholipids from a variety of tissues, including rat brain (Figure 2 b) and breast cancer tissue from mice.16 Here, alterations in db‐isomer populations that differentiated healthy and cancerous tissue were observed.16 This approach has the advantage of producing simple spectra and has been demonstrated for a variety of lipid families, including cholesterol esters.17 Moreover performing PB using carbonyl compounds that are themselves readily ionized allows simultaneous oxetane formation and ionization, including of non‐polar lipids not typically amenable to ESI. Using acetylpyridine that is readily protonated, the PB reaction and ionization has been demonstrated for a variety of lipids, including simple hydrocarbons.18 This also allows charge‐switching whereby lipids typically best analyzed in negative‐ion mode (such as FFAs) can be efficiently ionized in positive‐ion mode, greatly enhancing the depth of lipid coverage possible in a single analysis.18
Gas‐phase ozonolysis of mass‐selected lipid ions using ozone‐induced dissociation (OzID) permits the unambiguous identification of both db‐ and sn‐isomers.8a,8b Ozonolysis of C=C bonds generates a primary ozonide that rapidly decomposes into an aldehyde and the so‐called Criegee ion with their m/z values relative to the precursor indicating the position of the C=C bond (Figure 2 c). Like PB, OzID produces simple, easy to interpret spectra. Although OzID is applicable to many lipid classes, it has often been employed in positive‐mode analysis of sodiated PC and TAG lipids due to the greater reactivity of alkali adducts compared to the corresponding protonated and deprotonated ions. In its original linear ion‐trap implementation typical reaction times of 10 s are needed to give a reaction yield of several percent for db‐isomers.8a However recent adaptions to higher pressure compartments decoupled from mass analysis allow higher ozone concentrations and reduce this to the millisecond time scale, making extension to other lipid classes/ions and also coupling with fast chromatography possible.19 Analogous to the assignment of sn‐position with UVPD (see above), OzID of the fragment ions formed following loss of phosphocholine from alkali‐adducted PC lipids in an MS3 experiment (CID/OzID) enables selective cleavage of the sn‐2 acyl chain and thus assignment of sn‐position. The use of an analogous CID/OzID approach also makes it possible to assign the relative chain position in TAGs.20 Importantly the target C=C bond for CID/OzID is much more reactive, requiring an ion trap reaction time of only approximately 250 ms for high product‐ion yield. OzID can also be performed in parallel with CID enabling both conventional as well as OzID and CID/OzID fragments to be generated simultaneously.8b CID/OzID combined with later stages of CID and/or OzID can also allow the assignment of db‐position to individual acyl chain positions, a key step towards total structure elucidation.8b, 21 To our knowledge, OzID is the only method demonstrated to assign db‐positions to defined fatty acyl positions. It should be noted that the quantification of isomer populations with any of the methods described above will typically require the analysis of standards for each component to be quantified. This ensures isomer‐specific differences in product‐ion abundances or reactivities are accounted for. In the case of CID/OzID analysis of sn‐isomers, product‐ion abundances for monounsaturated PC lipids have been shown to reflect absolute molar fractions of each isomeric constituent.22
Ion mobility is another promising method to resolve MS signals originating from isobaric and isomeric lipids. In its various incarnations ion‐mobility spectrometry (IMS) has proven to be capable of at least partially resolving a broad selection of structural variations having different collision cross‐sections. This includes the separation of sn‐isomers, db‐isomers, stereoisomers, cis/trans isomers, sialic acid positional isomers of gangliosides (GD1a/GD1b), and sphingolipid isomers containing glucose or galactose.3c, 22, 23 For example, Maccarone et al., applied differential mobility spectrometry to the separation of PC sn‐isomers from a variety of biological extracts.22 Interestingly, the separation of PC 16:0/18:1 and PC 18:1/16:0 isomers was achieved only with the silver‐adducted lipid species. Applying this method to chick egg yolk revealed it to consist almost exclusively of PC 16:0/18:1, while analysis of bovine brain and kidney tissue revealed relative contributions for PC 18:1/16:0 of 19 % and 10 %, respectively. These results demonstrate that separation efficiency can be tuned by the choice of adduct ions. Similar adduct effects have also been demonstrated for the separation of db‐isomers and cis/trans isomers of PC lipids using drift‐tube IMS.23c In another study Kyle et al., combined liquid chromatography (LC) with drift‐tube IMS to identify a variety of isomeric lyso PC (LPC) lipids in different cell lines.23d While LC enabled the separation of cis/trans and sn‐isomers of LPC 16:1 from both embryonic epithelial kidney and Huh7.5.1 human hepatoma cell lines, the addition of IMS enabled the further observation of db‐isomers from the Huh7.5.1 cells that co‐eluted in the LC separation. IMS data showed the additional db‐isomers possessed a larger collision cross‐section, suggesting they contain the acyl‐chain double‐bond closer to the lipid head‐group, although the exact position was not determined. It is important to note that IMS alone does not allow the identification of the separated isomers. To achieve this, analysis of standards for each component in the mixture, or additional information acquired by orthogonal methods, is required. For example, this has been achieved by combination of OzID with IMS and LC to identify separated lipid isomers19b Nonetheless, by being able to rapidly resolve many isomeric lipid species, combined with its continued and rapid commercial development, IMS is expected to play a prominent role in future structural lipidomics and lipid biochemistry studies.
3. MS Imaging with Isomer Resolution
Over the last two decades MSI has rapidly evolved in biomedical and clinical research and shows much promise for tissue diagnostics, patient stratification, monitoring of therapy response, and prognosis prediction.24 In brief, MSI relies on desorption and ionization of molecules present on the surface of a solid, flat sample, such as a biological tissue section placed on a glass slide. Molecules are therefore visualized in situ within tissue sections and specific markers can be used to discriminate between healthy and diseased tissues. Lipids are the most common molecular class analyzed by MSI, largely due to the ease of detection of some key lipid classes. In many cases lipid profiles are analyzed that do not necessarily require the identification of the contributing lipids. For example MSI has been used to classify a variety of cancerous tissues based on localized lipid profiles.25 These profiles reflect different metabolic states and permit the differentiation of tissue and cell types within heterogeneous tissues. Ultimately, this information can be used in vivo and for real‐time tissue analysis and diagnosis during surgery to for example improve margin resection.26 However, when it comes to elucidating the biochemical processes occurring within tissues, robust identification and quantification of lipids is required. Here we outline recent work on methods to improve the structural specificity by which lipids can be visualized with MSI.
As outlined above, CID is the most popular MS/MS method used for lipid identification. In almost all cases MSI experiments are performed and MS/MS data only acquired in adjacent experiments (e.g., on‐tissue MS/MS or analysis of tissue extracts) after the ions of interest are determined. We note that selection of the ions of interest is typically based on the ion distributions observed in the MS1 analysis, which may already impose a bias on which lipid populations are further analyzed. To efficiently couple CID with MSI, data‐dependent acquisition (DDA)‐imaging has been introduced.27 DDA‐imaging uses a hybrid ion trap/Orbitrap instrument that enables simultaneous acquisition of UHRAM‐MSI data (using the Orbitrap) and MS/MS (using the ion trap). Due to this parallelization MS/MS data are acquired automatically, without extending the acquisition time compared to a conventional UHRAM‐MSI experiment of equivalent mass‐resolving power. Over 100 unique MLS (without sn‐positon) could be simultaneously imaged and identified in rat brain cerebellum by combining this method with an automated annotation routine.27 Even under these MS/MS conditions the identification of certain isomeric populations, such as at least two isomeric contributors to PS 36:1, namely PS 18:0_18:1 and PS 16:0_20:1, was achieved. However, while these isomeric populations were identified by MS/MS, they could not be distinguished in the full‐scan Orbitrap data used for MSI. In efforts to extend the utility of MSI for imaging lipids at higher structural specificity, a variety of the alternative ion activation methods described above have been recently applied to MSI. It is noteworthy that all of this work was reported in 2018, highlighting the rapidly growing demand for isomer‐resolved MSI.
UVPD has been coupled with desorption electrospray ionization (DESI)‐MSI to study the location of PC db‐isomers in brain and thyroid cancer tissues.28 Here, while the full scan DESI ion image displayed a lower abundance of the lipid ion of m/z 798 in the white matter compared to the gray matter (corresponding to the sum of all potassiated PC 16:0_18:1 isomers), DESI‐UVPD of this precursor revealed that the n‐9:n‐7 ratio of this isomeric population increases in white matter. Furthermore, DESI profiling for lymph node tissue sections with thyroid cancer metastasis revealed that the ratio of these sample isomers varies considerably between healthy and cancerous tissue.
PB reactions coupled with MSI and localized sampling have also been used to study the spatial distributions of db‐isomers. Tang et al., developed a rapid and online approach combining liquid microjunction surface sampling with PB reactions and MS/MS.29 Amongst other examples, relative quantitation, based on the n‐9:n‐7 isomeric ratio for fatty acid (FA) FA 18:1 and PC 16:0_18:1 in normal and cancerous mouse breast tissue, was performed and demonstrated significant diversity in the relative isomer populations across different tissues. Bednarik et al. extended the use of PB reactions to MALDI‐MSI.30 Here a PB derivatization protocol using benzaldehyde was developed that allowed efficient PB reactions to occur on tissue sections while preserving the spatial location of the lipids. After derivatization, the tissue was studied using post‐ionization‐enhanced MALDI‐MS/MS‐MSI.30 This enabled the localization and distinction of db‐isomers from a variety of lipid classes, including PC, PS, and hexosylceramides (HexCer) in mouse and pig brain tissues. The on‐tissue benzaldehyde reaction resulted in reaction yields of 5–30 % and produced analogous PB reaction products and MS/MS fragments as those observed using acetone. As an example, MS/MS of the PB product of PC 34:1 yielded two ion‐pairs at m/z 650.44/724.49 and 678.47/752.52 indicative of n‐9 and n‐7 db‐isomers, respectively, from porcine brain tissue. Similar changes in isomer populations throughout brain tissue to those seen with UVPD were observed. Unlike the differential images observed for PC 16:0_18:1 db‐isomers, the n‐9 and n‐7 double bond variants of the HexCer d18:1/24:1 both exhibited similar distributions, primarily in the myelin‐rich white matter.
Isomer‐resolved MALDI‐MSI has also been reported using OzID.31 This work provided the first example of the differentiation of both db‐ and sn‐isomers during MSI. When applied to identical lipid species OzID‐MSI gave similar db‐isomer distributions to those seen with UVPD‐ and PB‐based MSI. Figure 3 shows the differential imaging for the sn‐isomers of PC 34:1 and PC 36:1 in both a healthy rat brain (Figure 3 a) and a mouse brain containing a medulloblastoma tumor (Figure 3 e). In the case of PC 34:1, two sn‐isomers, namely PC 16:0/18:1 and PC 18:1/16:0, were observed. The fractional distribution image (FDI) for the PC 16:0/18:1 isomer (as a fraction of the sum signal from both isomers) is shown for both healthy brain (Figures 3 b) and the tumorous brain (Figure 3 f). Interestingly, changes in the relative isomer populations were observed between white and gray matter tissue regions. Heterogeneity in isomer populations within the tumor was also observed and potentially reflects the differing activities of phospholipases A1 and A2 throughout the tumor. A similar analysis of PC 36:1 sn‐isomers revealed four distinct isomers, namely a PC 18:0/18:1 and PC 18:1/18:0 pair (Figure 3 c,g), and a PC 16:0/20:1 and PC 20:1/16:0 pair (Figure 3 d,h) that again exhibit different populations in the tumor compared to the surrounding white and gray matter. To highlight the lipid diversity, both n‐9 and n‐7 db‐isomers were also observed for PC 36:1. This suggests the possible presence of at least eight distinct lipid structures contributing to the same sum‐composition level species, which are indistinguishable with conventional MSI approaches.
Figure 3.
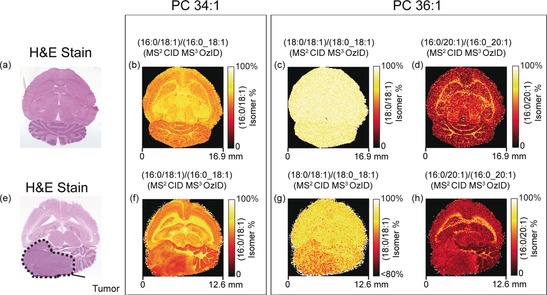
MALDI‐MSI of sn‐isomers in healthy rat brain (top) and mouse brain containing a medulloblastoma tumour (bottom) using CID/OzID. The corresponding H&E stains of the analyzed tissues are shown in (a) and (e), respectively. Two isomers, PC 16:0/18:1 and PC 18:1/16:0, are observed for the PC 34:1 sum‐composition species. The fractional distribution imaging (FDI) of these two isomers in the two tissues is shown in (b) and (f). Four isomers corresponding to a PC 18:0/18:1 PC 18:1/18:0 pair and a PC 16:0/20:1 and PC 20:1/16:0 pair are observed for the PC 36:1 sum‐composition species. (c), (d), (g), and (h) show the FDI of each isomeric pair from PC 36:1 throughout the tissues. Image adapted with permission from ref. 31.
4. Lipid‐Flux Analysis
It is well known that cellular lipidomes are complex, comprising of thousands of lipid species.1 However, the precise map of the lipidomes and how these are regulated at a detailed molecular level is still very limited. Several indications suggest that the distribution and content of lipids needs to be tightly regulated within the cell to maintain its proper functions. Addressing the function(s) of a single lipid becomes even more challenging as the current dogma is that similar lipid species can reside and act at multiple locations inside a cell simultaneously. This, and the dynamic mechanisms by which they are produced, still remain unanswered due to the lack of appropriate technologies for their precise determination.
The biosynthesis of lipids has, to this point, been mainly studied using radioactively labeled substances, fluorescent lipid analogues, and inhibitors/siRNAs against enzymes involved in lipid metabolism or cells lacking enzymes involved in lipid metabolism. Furthermore, such studies have primarily been restricted to lipid‐class analyses only, leaving the complexity of the detailed lipid structures underneath each class unexplored. While these investigations have added much to our present knowledge of lipid metabolism, these methods fail in delivering the precise information and functions of the involved molecular lipid species. How do we go about resolving this?
The use of stable mass isotopes, for example 13C‐, 15N‐ or 2H‐labeled precursor substances, or non‐endogenous substances thereof, offers an interesting possibility to study lipid turnover. Gas chromatography‐based tracer analyses have provided valuable insight to the fatty acid metabolism and turnover;32 however, due to its technological constraints determining the turnover of more complex lipids have been restricted. This has been overcome by applying lipidomics, where most of the studies have focused on adding labeled lipid precursors, such as FAs, long‐chain bases, and amino acids.33 Although cell‐culture systems have been the predominant experimental systems, some studies have also been performed in vivo.
Using FAs as labels allows the monitoring of the rate of FA uptake, the selectivity in the FA incorporation, and degradation rate of the labeled molecular lipid species. As a result, the FA turnover of MLS can be comprehensively determined, virtually on global lipidome scale. In contrast to FA‐ or long‐chain base (LCB)‐labeling, which labels any FA‐ or LCB‐containing molecular lipid species, head‐group labeling of phospholipids is more selective, targeting only the turnover of certain lipid classes at the sum‐composition level. Investigating de novo sphingolipid metabolism in mammalian cells has, for instance, been described by utilizing non‐endogenous sphingosine,34 labeled 1‐deoxy‐sphinganine,35 labeled serine,36 and labeled palmitate.37 Current lipidomics technologies allow the combination of several tracers to more comprehensively and precisely capture the metabolic flow of lipid metabolism. For instance, simultaneous labeling using stable isotopes of serine and glucose allows the contribution of de novo synthesis to the total synthesis of new glycosphingolipids (GSL) to be determined.36 These advancements further allowed the identification of lipid intermediates, other hitherto unknown metabolic routes, different lipid pools, and selectivity in molecular species, hence shedding new light on the complexity underlying the metabolic flow of endogenous GSL species in cells. Importantly, isotope‐labeling experiments have also successfully been performed in in vivo systems, permitting the investigation of complex lipid dynamics directly in living organisms. For instance, head‐group labeling of phospholipids has been applied for determining lung surfactant PC metabolism in transgenic mice38 and humans.39
To further determine the cellular location of the newly synthesized lipids, other types of tracers have been explored in conjunction with shotgun‐ and LC‐based lipidomics analyses. However, the prime challenge has been to develop precursors that can be both visualized and traced by MS simultaneously, and, importantly, closely mimic the physiological end products. An example is polyene fatty acids, which closely mimic the endogenous FAs and are visual under two‐photon excitation microscopy.40 However, care must also be taken to ensure that the behavior of fluorescently labeled lipids accurately mimics naturally occurring lipids, which may not always be the case due to their different chemical structures. As a promising alternative to visible tracers, MSI of stable mass isotope‐labeled samples has been demonstrated using both D2O and D3‐palmitate labeling.41 Combining stable isotope‐labeling and MSI provides not only an exciting technology for future location‐tracer experiments, but unlocks the possibilities for catching local metabolic events rather than a sum of many, which current extract‐based methodologies provides.
5. Interpreting Lipid Data
The interpretation of lipidomics data still remains challenging, as the data provided is much more detailed than the described classical lipid biosynthetic pathways that, in the context of more complex lipids, are only outlined at the lipid class level. Hence, the prime questions still remain; why do we have so many different lipids and what are their functions? To find the answers, we need to determine the structure and amount of each individual lipid species present. This is explicitly demonstrated in previous work by Ståhlman et al. showing that the metabolism of specific TAGs is explicitly associated with clinical dyslipidaemia.21 Using a combination of MS/MS and OzID, the almost complete structure of a TAG species, in this case TAG 16:0/18:1(n7)/16:0, could be determined. This permitted the identification of the active pathways associated with dyslipidaemic type‐2 diabetic patients that were not active under normolipidaemic conditions. Importantly, as the total TAG levels were unaffected, an obvious conclusion would have been that the TAG metabolism remains unchanged; however, performing sophisticated lipidomics measurements proved that changes do in fact occur at the lipid species level. These changes become increasingly apparent as more details regarding the lipid species are obtained.
As demonstrated, quantitatively capturing the complete lipid structures allows lipid metabolic networks to be more precisely mapped for a better understanding of their function and regulation. However, these analyses are still very challenging to conduct on a global lipidome scale and in a routine manner, due to incomplete technological and bioinformatics solutions. Major attempts to resolve the existing issues are ongoing, with a significant focus on the integration of ion‐mobility,22, 23 selective derivatization/chemical reactions, and alternative ion‐activation methods, or combinations of these (see above). Based on initial indications, it is most likely that the quantification of at least close to complete lipid structures will become a routine measurement in the near future.
As the lipidomics field still awaits the mainstream adoption of the next‐generation technologies mentioned above, the major routine lipidomics methodologies applied today, shotgun‐, chromatography‐, and imaging‐based MS,42 are still almost exclusively based on the conventional MS and MS/MS methods. These approaches yield lower structural information, typically sum lipid composition (e.g. PC 36:1), and molecular lipid species (e.g. PC 18:0_18:1). Despite this lower resolution, unprecedented insights into various lipidomes have still been gained, ranging from basic12, 43 to medical research,44 and clinical applications.45 However, the obtained partial lipid information still limits the biological interpretation as few metabolic pathways have been described in sufficient detail. To circumvent this issue, secondary analyses, such as correlation networks43 and correlation with gene expression data46 or clinical measurements,47 have been applied. Another successful approach is flux lipidomics, as outlined above.
6. The Need for Standardization and Proper Reporting of Lipidomics Data
There is major wave of interest in adopting lipidomics capabilities across research communities. However, the field is challenged by the large disparities in methodologies and technologies, resulting in an increasing number of publications of poor and/or inappropriate quality.48 The whole field urgently needs to establish its own standards to stop this destructive trend. This era has already begun with the recently established Lipidomics Standards Initiative (LSI) (https://lipidomics‐standards‐initiative.org) that aims to centralize lipidomics standards in all aspects of lipid analysis, covering pre‐analytics, extraction methods, mass spectrometry, data analysis, quality controls, and minimal reporting standards. LSI has taken on the role of facilitating the standardization of lipidomics through increased systematic and transparent reporting and engagement with the community.
A critical rule is to not over report lipidomics data, but rather report it according to the level of the information obtained, within the limits of the analysis performed.7 For instance, a precursor ion scan of m/z 184 on a low‐resolution instrument cannot differentiate isobaric ether PC O‐34:1 ([M+H]+ m/z 746.6) and diester PC 33:1 ([M+H]+ m/z 746.6), while these are readily resolved on a high‐resolution mass spectrometer with resolution greater than 40,000 full width at half maximum. Moreover, if both acyl anions, [FA 16:0‐H]− at m/z 255.2 and [FA 17:1‐H]‐ at m/z 267.2, are detected in negative ion MS/MS, then the annotation should be PC 16:0_17:1, rather than PC 33:1. The LSI works towards establishing the minimum rules for reporting lipid species data, which provides the common language highly needed for multi‐laboratory data comparison and exchange. Taken together, the guidelines on standards for lipidomics are needed and will trigger the awakening of the true potential of lipidomics. This sets the cornerstones to enter the regulatory environment where lipidomics is expected to have a highly valuable position in clinical research and diagnostics.
7. Conclusions and Outlook
The field of lipidomics is undergoing rapid evolution, both with respect to the technology and the applications. The large push in recent years towards more complete structural elucidation has resulted in a variety of new methods that should finally allow the study of structural‐defined lipid species. While still largely in the proof‐of‐concept stage, the ability to routinely execute such analyses will dramatically expand our knowledge in lipid biochemistry and biology. However, further development is needed to enable the total structural characterization with MS, e.g., assignment of db‐position to individual acyl chains, cis/trans and stereochemical assignments. To reach its full potential, these advances must be supplemented with parallel technologies permitting the precise monitoring of the kinetics and the localization of complete lipid structures, combined with a more complete understanding of the molecular machinery and mechanisms involved in lipid synthesis and conversions. With this in hand, the unlocking of the biological function of each individual lipid species becomes reachable, paving the way for true “molecular” lipidomics.
Conflict of interest
The authors declare no conflict of interest.
Biographical Information
Dr Tiffany Porta Siegel is an Assistant Professor at the Maastricht Multimodal Molecular Imaging Institute (M4I). Her research focuses on translation between the development of innovative MS‐based instrumentation and direct application to clinical research, with a special interest on intraoperative mass spectrometry. In particular, the use of molecular‐based diagnostics (mostly based on lipid signatures) is expected to improve patient management during surgery and overall outcome.

Biographical Information
Dr Kim Ekroos is the founder and CEO of Lipidomics Consulting Ltd. His expertise includes high‐throughput technologies for the precise assessment of lipidomes, enabled by advanced mass spectrometry, automation, and software tools, towards the discovery of biological architectures and of diagnostic biomarkers for clinical purpose. He is one of the pioneers in the field of lipidomics, with more than 20 years of experience in the academic, industry, and regulatory disciplines of lipidomics. He is a co‐founder of the Lipidomics Standards Initiative (LSI) and acts as scientific advisor for the European Lipidomics Meetings (ELM).

Biographical Information
Dr Shane R. Ellis is an Assistant Professor at the M4I Institute. His research focuses on the development and application of advanced mass spectrometry imaging methods to study, map, and eventually understand, localized biochemical processes occurring within tissues and cells. In particular, his group develops methods that greatly enhance the chemical information that can be obtained from MSI experiments. He is fascinated by the complex interplay between molecular structure, function, and biological implication and seeks to apply MSI techniques to study these relationships.

Acknowledgements
This work was supported by the Dutch Province of Limburg through the LINK program. S.R.E. acknowledges funding from InterregV‐EMR and the Netherlands Ministry of Economic Affairs within the “EURLIPIDS” project (project number EMR23). We are grateful to Prof. Jennifer Brodbelt (University of Texas at Austin, USA), Prof. Yu Xia (Tsinghua University, China), and Dr. Berwyck Poad/Prof. Stephen Blanksby (Queensland University of Technology, Australia) for providing the raw data used to prepare Figure 2.
T. Porta Siegel, K. Ekroos, S. R. Ellis, Angew. Chem. Int. Ed. 2019, 58, 6492.
References
- 1. van Meer G., Voelker D. R., Feigenson G. W., Nat. Rev. Mol. Cell Biol. 2008, 9, 112–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bandu R., Mok H. J., Kim K. P., Mass Spectrom. Rev. 2018, 37, 107–138. [DOI] [PubMed] [Google Scholar]
- 3.
- 3a. Wenk M. R., Cell 2010, 143, 888–895; [DOI] [PubMed] [Google Scholar]
- 3b. Blanksby S. J., Mitchell T. W., Annu. Rev. Anal. Chem. 2010, 3, 433–465; [DOI] [PubMed] [Google Scholar]
- 3c. Rustam Y. H., Reid G. E., Anal. Chem. 2018, 90, 374–397. [DOI] [PubMed] [Google Scholar]
- 4. Yetukuri L., Ekroos K., Vidal-Puig A., Oresic M., Mol. Biosyst. 2008, 4, 121–127. [DOI] [PubMed] [Google Scholar]
- 5.
- 5a. Zemski Berry K. A., Hankin J. A., Barkley R. M., Spraggins J. M., Caprioli R. M., Murphy R. C., Chem. Rev. 2011, 111, 6491–6512; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5b. Bowman A. P., Heeren R. M. A., Ellis S. R., TrAC Trends Anal. Chem. 2018. 10.1016/j.trac.2018.07.012. [DOI] [Google Scholar]
- 6. Ekroos K., Lipidomics: Technologies and Applications, Wiley, Hoboken, 2012. [Google Scholar]
- 7. Liebisch G., Vizcaino J. A., Kofeler H., Trotzmuller M., Griffiths W. J., Schmitz G., Spener F., Wakelam M. J., J. Lipid Res. 2013, 54, 1523–1530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.
- 8a. Thomas M. C., Mitchell T. W., Harman D. G., Deeley J. M., Nealon J. R., Blanksby S. J., Anal. Chem. 2008, 80, 303–311; [DOI] [PubMed] [Google Scholar]
- 8b. Pham H. T., Maccarone A. T., Thomas M. C., Campbell J. L., Mitchell T. W., Blanksby S. J., Analyst 2014, 139, 204–214; [DOI] [PubMed] [Google Scholar]
- 8c. Ma X., Xia Y., Angew. Chem. Int. Ed. 2014, 53, 2592–2596; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2630–2634; [Google Scholar]
- 8d. Klein D. R., Brodbelt J. S., Anal. Chem. 2017, 89, 1516–1522; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8e. Williams P. E., Klein D. R., Greer S. M., Brodbelt J. S., J. Am. Chem. Soc. 2017, 139, 15681–15690; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8f. Ryan E., Nguyen C. Q. N., Shiea C., Reid G. E., J. Am. Soc. Mass Spectrom. 2017, 28, 1406–1419; [DOI] [PubMed] [Google Scholar]
- 8g. Campbell J. L., Baba T., Anal. Chem. 2015, 87, 5837–5845. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Hsu F. F., Turk J., J. Chromatogr. B 2009, 877, 2673–2695; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9b. A. H. Merrill Jr , Sullards M. C., Allegood J. C., Kelly S., Wang E., Methods 2005, 36, 207–224. [DOI] [PubMed] [Google Scholar]
- 10. Brügger B., Erben G., Sandhoff R., Wieland F. T., Lehmann W. D., Proc. Natl. Acad. Sci. USA 1997, 94, 2339–2344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.
- 11a. Ekroos K., Ejsing C. S., Bahr U., Karas M., Simons K., Shevchenko A., J. Lipid Res. 2003, 44, 2181–2192; [DOI] [PubMed] [Google Scholar]
- 11b. Hsu F.-F., Turk J., J. Am. Soc. Mass Spectrom. 2010, 21, 657–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Ejsing C. S., Sampaio J. L., Surendranath V., Duchoslav E., Ekroos K., Klemm R. W., Simons K., Shevchenko A., Proc. Natl. Acad. Sci. USA 2009, 106, 2136–2141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ryan E., Reid G. E., Acc. Chem. Res. 2016, 49, 1596–1604. [DOI] [PubMed] [Google Scholar]
- 14. Almeida R., Pauling J. K., Sokol E., Hannibal-Bach H. K., Ejsing C. S., J. Am. Soc. Mass Spectrom. 2015, 26, 133–148. [DOI] [PubMed] [Google Scholar]
- 15. Pham H. T., Ly T., Trevitt A. J., Mitchell T. W., Blanksby S. J., Anal. Chem. 2012, 84, 7525–7532. [DOI] [PubMed] [Google Scholar]
- 16. Ma X., Chong L., Tian R., Shi R., Hu T. Y., Ouyang Z., Xia Y., Proc. Natl. Acad. Sci. USA 2016, 113, 2573–2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Ren J., Franklin E. T., Xia Y., J. Am. Soc. Mass Spectrom. 2017, 28, 1432–1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Esch P., Heiles S., J. Am. Soc. Mass Spectrom. 2018, 29, 1971–1980. [DOI] [PubMed] [Google Scholar]
- 19.
- 19a. Poad B. L. J., Green M. R., Kirk J. M., Tomczyk N., Mitchell T. W., Blanksby S. J., Anal. Chem. 2017, 89, 4223–4229; [DOI] [PubMed] [Google Scholar]
- 19b. Poad B. L. J., Zheng X., Mitchell T. W., Smith R. D., Baker E. S., Blanksby S. J., Anal. Chem. 2018, 90, 1292–1300; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19c. Vu N., Brown J., Giles K., Zhang Q., Rapid Commun. Mass Spectrom. 2017, 31, 1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Marshall D. L., Pham H. T., Bhujel M., Chin J. S. R., Yew J. Y., Mori K., Mitchell T. W., Blanksby S. J., Anal. Chem. 2016, 88, 2685–2692. [DOI] [PubMed] [Google Scholar]
- 21. Ståhlman M., Pham H. T., Adiels M., Mitchell T. W., Blanksby S. J., Fagerberg B., Ekroos K., Borén J., Diabetologia 2012, 55, 1156–1166. [DOI] [PubMed] [Google Scholar]
- 22. Maccarone A. T., Duldig J., Mitchell T. W., Blanksby S. J., Duchoslav E., Campbell J. L., J. Lipid Res. 2014, 55, 1668–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.
- 23a. Wojcik R., Webb I., Deng L., Garimella S., Prost S., Ibrahim Y., Baker E., Smith R., Int. J. Mol. Sci. 2017, 18, 183; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23b. Poad B. L. J., et al., Anal. Chem. 2018, 90, 5343–5351; [DOI] [PubMed] [Google Scholar]
- 23c. Groessl M., Graf S., Knochenmuss R., Analyst 2015, 140, 6904–6911; [DOI] [PubMed] [Google Scholar]
- 23d. Kyle J. E., et al., Analyst 2016, 141, 1649–1659; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23e. Bowman A. P., Abzalimov R. R., Shvartsburg A. A., J. Am. Soc. Mass Spectrom. 2017, 28, 1552–1561; [DOI] [PubMed] [Google Scholar]
- 23f. Hinz C., Liggi S., Griffin J. L., Curr. Opin. Chem. Biol. 2018, 42, 42–50. [DOI] [PubMed] [Google Scholar]
- 24. Vaysse P. M., Heeren R. M. A., Porta T., Balluff B., Analyst 2017, 142, 2690–2712. [DOI] [PubMed] [Google Scholar]
- 25.
- 25a. Sans M., et al., Cancer Res. 2017, 77, 2903–2913; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25b. Doria M. L., et al., Sci. Rep. 2016, 6, 39219; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25c. Mirnezami R. , et al., Molecular Oncology 2014, 8, 39–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Alexander J., et al., Surgical Endoscopy 2017, 31, 1361–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ellis S. R., Paine M. R. L., Eijkel G. B., Pauling J. K., Husen P., Jervelund M. W., Hermansson M., Ejsing C. S., Heeren R. M. A., Nat. Methods 2018, 15, 515–518. [DOI] [PubMed] [Google Scholar]
- 28. Klein D. R., Feider C. L., Garza K. Y., Lin J. Q., Eberlin L. S., Brodbelt J. S., Anal. Chem. 2018, 90, 10100–10104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tang F., et al., Anal. Chem. 2018, 90, 5612–5619. [DOI] [PubMed] [Google Scholar]
- 30. Bednařík A., Bölsker S., Soltwisch J., Dreisewerd K., Angew. Chem. Int. Ed. 2018, 57, 12092–12096; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 12268–12272. [Google Scholar]
- 31. Paine M. R. L., Poad B. L. J., Eijkel G. B., Marshall D. L., Blanksby S. J., Heeren R. M. A., Ellis S. R., Angew. Chem. Int. Ed. 2018, 57, 10530–10534; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 10690–10694. [Google Scholar]
- 32. Brenna J. T., Prostaglandins, Leukotrienes Essent. Fatty Acids 1997, 57, 467–472. [DOI] [PubMed] [Google Scholar]
- 33. Postle A. D., Curr. Opin. Clin. Nutr. Metab. Care 2012, 15, 127–133. [DOI] [PubMed] [Google Scholar]
- 34. Snider J. M., Snider A. J., Obeid L. M., Luberto C., Hannun Y. A., J. Lipid Res. 2018, 59, 1046–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Steiner R., Saied E. M., Othman A., Arenz C., Maccarone A. T., Poad B. L., Blanksby S. J., von Eckardstein A., Hornemann T., J. Lipid Res. 2016, 57, 1194–1203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Skotland T., Ekroos K., Kavaliauskiene S., Bergan J., Kauhanen D., Lintonen T., Sandvig K., J. Mol. Biol. 2016, 428, 4856–4866. [DOI] [PubMed] [Google Scholar]
- 37. Hu W., Bielawski J., Samad F., A. H. Merrill, Jr. , Cowart L. A., J. Lipid Res. 2009, 50, 1852–1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Postle A. D., Henderson N. G., Koster G., Clark H. W., Hunt A. N., Chem. Phys. Lipids 2011, 164, 549–555. [DOI] [PubMed] [Google Scholar]
- 39. Dushianthan A., Goss V., Cusack R., Grocott M. P., Postle A. D., Respir. Res. 2014, 15, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuerschner L., Ejsing C. S., Ekroos K., Shevchenko A., Anderson K. I., Thiele C., Nat. Methods 2005, 2, 39–45. [DOI] [PubMed] [Google Scholar]
- 41.
- 41a. Louie K. B., Bowen B. P., McAlhany S., Huang Y., Price J. C., Mao J.-h., Hellerstein M., Northen T. R., Sci. Rep. 2013, 3, 1656; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41b. Carson R. H., Lewis C. R., Erickson M. N., Zagieboylo A. P., Naylor B. C., Li K. W., Farnsworth P. B., Price J. C., J. Lipid Res. 2017, 58, 1884–1892; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41c. Sugiyama E., Yao I., Setou M., Biochem. Biophys. Res. Commun. 2018, 495, 1048–1054. [DOI] [PubMed] [Google Scholar]
- 42. Holčapek M., Liebisch G., Ekroos K., Anal. Chem. 2018, 90, 4249–4257. [DOI] [PubMed] [Google Scholar]
- 43.
- 43a. Sales S., et al., Sci. Rep. 2016, 6, 27710; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43b. Peng B., et al., Blood 2018, 132, e1-e12.29976776 [Google Scholar]
- 44.
- 44a. Vu T. M., et al., Nature 2017, 550, 524–528; [DOI] [PubMed] [Google Scholar]
- 44b. Ogasawara D., et al., Proc. Natl. Acad. Sci. USA 2016, 113, 26–33;26668358 [Google Scholar]
- 44c. Layre E., et al., Proc. Natl. Acad. Sci. USA 2014, 111, 2978–2983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.
- 45a. Laaksonen R., et al., Eur. Heart J. 2016, 37, 1967–1976; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45b. Wang Z., et al., Nature 2011, 472, 57–63; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45c. Hubbard W. C., et al., Mol. Genet. Metab. 2009, 97, 212–220. [DOI] [PubMed] [Google Scholar]
- 46. Perman J. C., et al., J. Clin. Invest. 2011, 121, 2625–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Hiukka A., et al., Diabetes 2009, 58, 2018–2026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.
- 48a. Liebisch G., Ejsing C. S., Ekroos K., Clin. Chem. 2015, 61, 1542–1544; [DOI] [PubMed] [Google Scholar]
- 48b. Bowden J. A., et al., J. Lipid Res. 2017, 58, 2275–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Blanksby S. J., Mitchell T. W., Annu. Rev. Anal. Chem. 2010, 3, 433–465. [DOI] [PubMed] [Google Scholar]