

Abstract
Chediak-Higashi syndrome is a genetic disorder caused by mutations in a gene encoding a protein named LYST in humans (“lysosomal trafficking regulator”) or Beige in mice. A prominent feature of this disease is the accumulation of enlarged lysosome-related granules in a variety of cells. The genome of Dictyostelium discoideum contains six genes encoding proteins that are related to LYST/Beige in amino acid sequence, and disruption of one of these genes, lvsA (large volume sphere), results in profound defects in cytokinesis. To better understand the function of this family of proteins in membrane trafficking, we have analyzed mutants disrupted in lvsA, lvsB, lvsC, lvsD, lvsE, and lvsF. Of all these, only lvsA and lvsB mutants displayed interesting phenotypes in our assays. lvsA-null cells exhibited defects in phagocytosis and contained abnormal looking contractile vacuole membranes. Loss of LvsB, the Dictyostelium protein most similar to LYST/Beige, resulted in the formation of enlarged vesicles that by multiple criteria appeared to be acidic lysosomes. The rates of endocytosis, phagocytosis, and fluid phase exocytosis were normal in lvsB-null cells. Also, the rates of processing and the efficiency of targeting of lysosomal α-mannosidase were normal, although lvsB mutants inefficiently retained α-mannosidase, as well as two other lysosomal cysteine proteinases. Finally, results of pulse-chase experiments indicated that an increase in fusion rates accounted for the enlarged lysosomes in lvsB-null cells, suggesting that LvsB acts as a negative regulator of fusion. Our results support the notion that LvsB/LYST/Beige function in a similar manner to regulate lysosome biogenesis.
INTRODUCTION
Chediak-Higashi syndrome (CHS) is a rare autosomal recessive genetic disorder of humans that also occurs in other mammals, including cattle (Padgett, 1967), mice (Lutzner et al., 1967), rats (Nishimura et al., 1989), minks (Padgett, 1967), and killer whales (Ridgway, 1979). Patients with this disorder suffer from partial albinism, excessive bleeding, and recurrent bacterial infections.
The defining clinical manifestation of CHS is the presence in a wide variety of cell types of enlarged lysosomes or granules that form as the result of a mutation in the LYST gene in humans (chromosome 1) or the beige gene in the murine model (chromosome 13; Dufourcq-Lagelouse et al., 1999; Introne et al., 1999). A few models have been proposed to explain the role of LYST in the formation of enlarged lysosomes. The first model, based primarily on electron microscope studies, hypothesizes that LYST normally acts as a negative regulator of homotypic and heterotypic lysosome fusion, thus accounting for the increase in size of lysosomes in cells lacking LYST (Oliver and Essner, 1975). In addition, other studies have demonstrated that secretory lysosomes fuse to form giant lysosomes in maturing CHS cytotoxic T lymphocytes (Stinchcombe et al., 2000). The second model proposes that LYST is a positive regulator of fission, and in the absence of LYST, the balance is tilted in favor of fusion, and large lysosomes accumulate (Burkhardt et al., 1993; Perou et al., 1997). In support of this model, it was observed that overexpression of LYST induced a more peripheral redistribution of smaller lysosomes. Finally, a third model suggests that LYST may regulate protein transport to late endosomes; thus, trafficking defects could account for the morphological changes observed in lysosomes and lysosome-related organelles (Faigle et al., 1998).
Although the CHS gene family is conserved in a wide variety of species, the amino acid sequence of the encoded gene products predicts little regarding the role of LYST/Beige in lysosome biogenesis or membrane trafficking. LYST/Beige comprises three distinct domains. The amino-terminal 80% of the protein has limited homology to other proteins in the database. This portion of the protein is followed by the BEACH (Beige and Chediak-Higashi) domain, the defining consensus sequence for CHS-related proteins, and several WD-40 repeats, which are thought to be important in protein-protein interaction. Several other mammalian gene products contain the BEACH and WD-40 domains, including FAN (factor associated with sphingomyelinase activation; Adam-Klages et al., 1996), neurobeachin (Wang et al., 2000), and CDC4L (Feuchter et al., 1992).
Dictyostelium discoideum will be a useful system to study the function of BEACH/WD-40 domain-containing proteins related to LYST/Beige. First, a wide range of genetic and biochemical tools are available to explore the function of this haploid organism (reviewed in a special issue of Biochimica et Biophysica Acta (2001, Vol 1525). Second, the Dictyostelium genome contains six genes, termed lvs (large volume sphere) A through F, that encode proteins containing the BEACH and WD-40 domains that are related to the LYST/Beige family of proteins (Wang, N., Wu, W., and DeLozanne, A., unpublished data ). LvsA is the first member of this family to be characterized and has been found to play an important role in cytokinesis and osmoregulation (Kwak et al., 1999; Gerald et al., 2001). Third, the endolysosomal/phagosomal pathways in Dictyostelium are relatively well characterized and comparable to related pathways in mammalian cells (reviewed by Cardelli, 2001). Fluid phase enters the endosomal pathway primarily through macropinocytosis, although clathrin-mediated micropinocytic internalization also occurs. Endosomes derived from the fission of macropinosomes, fuse to form acidic lysosomes that, in turn, fuse to form nonacidic secretory vesicles, termed postlysosomes (reviewed by Maniak, 2001). A number of proteins have been identified that regulate fusion of lysosomes with each other and with newly formed phagosomes (reviewed in Rupper and Cardelli, 2001). In addition, it has been proposed that vesicles, containing membrane and soluble luminal proteins, form by fission and recycle from late endosomal to earlier endocytic compartments or the plasma membrane. Proteins that regulate this process include DdRab7, which may recycle membrane components from postlysosomes back to early endosomes and lysosomes (Buczynski et al., 1997), and myosin I, which recycles membrane from early endosomes back to plasma membrane (Neuhaus and Soldati, 2000).
Of the six known lvs genes in Dictyostelium, the lvsB gene encodes a protein most closely related in amino acid sequence to the LYST/Beige protein. To determine whether LvsB functions like LYST/Beige proteins, we biochemically and microscopically analyzed lvsA-null through lvsF-null mutants. This report demonstrates that of the six proteins only LvsB appears to function like the LYST/Beige protein. Notably, lvsB-null mutants contain enlarged acidic lysosomes that retain most, but not all, of the proteins found in normal lysosomes. Furthermore, these enlarged lysosomes appear to form as a result of an increase in the rate of vesicle fusion.
MATERIALS AND METHODS
Cell Lines
Dictyostelium strain NC4A2 was used as the control parental strain (kind gift of David Knecht). lvsA-null cell strain AD-63 (alias VIG9) was described previously (Kwak et al., 1999). All other Lvs-related knockout strains were made in NC4A2 as described by Wang, N., Wu, W. and DeLozanne, A. (unpublished data). Briefly, knockout constructs were made using a blasticidin-resistance cassette bounded by two segments of each gene. Each construct was introduced into NC4A2 cells by electroporation, and clonal transformants were selected in 96-well dishes. Individual clones were screened for double-crossover insertion of the knockout construct by PCR, and the appropriate insertion was confirmed by Southern blot analysis.
Microscopy
To visualize the entire endocytic pathway by fluorescence microscopy, cells grown in T-25 tissue culture flasks were allowed to settle on 22- × 22-mm coverslips (Fisherbrand, Fisher Scientific, Hampton, NH) in a six-well dish and were incubated for 2 h in HL-5 growth medium (10.0 g proteose peptone [Oxide LTD., Basingstoke, Hampshire, United Kingdom], 10.0 g of glucose, 5.0 g yeast extract, 0.19 g Na2HPO4, 0.35 g KH2PO4) containing 1.0 mg/ml fluorescein isothiocyanate (FITC)-conjugated dextran (FD-70S, Sigma, St. Louis, MO). To view a subset of vesicles in the endocytic pathway, cells were pulsed with FITC-dextran for 5 min, washed, and chased for 0 min (to visualize macropinosomes), 20 min (lysosomes), or 60 min (postlysosomes). After the chase periods, cells were washed with fresh HL-5, fixed in 1% formaldehyde in HL-5 for 3 min, and viewed using a fluorescence microscope (Olympus, Tokyo, Japan).
To assess acidification of vesicles, cells were allowed to settle on coverslips in HL-5 and incubated with a 1:100 dilution of DND-189 Lysosensor (Molecular Probes, Eugene, OR) in HL-5 for 2–3 min. Lysosensor fluoresces only in acidic compartments (5.2 pKa), and live cells were visualized in the fluorescein channel.
For immunofluorescence microscopy, cells grown in T-25 flasks in log phase were allowed to settle on 22- × 22-mm coverslips in six-well plates for 20 min, fixed, and permeabilized as described by Bush et al. (1994). Cells were incubated with the primary (B832, anti–100-kDa pump) and secondary antibodies (Texas Red goat anti-mouse; Jackson Laboratories, Bar Harbor, ME), each for 1 h at 4°C. Coverslips were washed and mounted on slides and visualized using an Olympus BX-50 fluorescence microscope.
Endocytosis, Phagocytosis, and Exocytosis Assays
Fluid phase endocytosis and exocytosis rates were measured using FITC-dextran, as described by Temesvari et al. (1996a). Phagocytosis rates were measured using fluorescent latex beads as described by Temesvari et al. (2000).
Purification of Lysosomes
Lysosomes were purified as described before with some modifications (Temesvari et al., 1994). Cells were pulsed for 15 min with an equal volume of HL-5 media containing 2.0 mg/ml iron dextran, washed twice in cold HL-5, and chased in fresh media for 15 min. Harvested cells were resuspended to 2.0 × 108 cells/ml in sucrose buffer (5.0 mM glycine, 100 mM sucrose, pH 8.5) with a protease inhibitor cocktail (10 μg/ml leupeptin, 20 μg/ml chymostatin, 20 μg/ml pepstatin, 5 mg/ml aprotinin, 100 mM Na-p-tosyl-l-lysine chloromethyl ketone) and were broken by passage through two 5-μm pore polycarbonate filters (Poretics, Livermore, CA). Postnuclear supernatants were pumped through the column of fine mesh wire (25 μm diameter; Goodfellow, Malvern, PA) at 70 ml/h. After washing, lysosomes were recovered from the wire mesh using a 5-ml pipette and centrifuged in 35-ml Oakridge tubes at 39,000 × g at 4°C for 30 min. The sucrose buffer was aspirated and the lysosomes were resuspended in Laemmli buffer (Laemmli, 1970), heated to 65°C for 5 min, and subjected to SDS-PAGE or stored at −80°C.
Western Blot Analysis
SDS-PAGE and Western blot analysis were done as described by Buczynski et al. (1997). The membranes were blocked overnight in TBSTG (10 mM Tris base, 150 mM NaCl, 0.1% gelatin (Knox), 0.1% Tween-20, pH 7.5) and exposed to the following antibodies that were diluted in TBSTG: mouse anti-100-kDa subunit of the vacuolar H+-ATPase (B832, 1:200; Fok et al., 1993); rabbit anti-41-kDa subunit of the vacuolar H+-ATPase (LSU 18-2, 1:2000; Temesvari et al., 1996b); rabbit anti–α-mannosidase (LSU 27-2, 1:5000; J. Cardelli, unpublished results); rabbit anti-D. discoideum Rab7 (LSU 7-2-4, 1:200; Buczynski et al., 1997); affinity pure rabbit anti-RabB (LSU11 25-1-2, 1:200; J. Cardelli, unpublished results,); rabbit anti-RabD (Rab14 homolog, LSU 4-7-2, 1:200; Bush et al., 1994); anti-cathepsin D (1:500; Journet et al., 1999); anti-CPp36, which recognizes cysteine proteinase p36, (1:1000; a kind gift from J. Garin); anti-LmpA, which recognizes D. discoideum LmpA, (1:1000; Karakesisoglou et al., 1999); AD7.5, which recognizes N-acetylglucose 1-phosphate–modified cysteine proteinases (1:1000; kind gift of Hudson Freeze); anti-acid phosphatase (5E1, 1:250); anti–β-glucosidase (AP8, 1:250). All blots were exposed to the same mixture of alkaline phosphatase-conjugated secondary antibodies diluted in TBSTG (both goat anti-mouse, 1:3000; catalog no. 170-6520, Bio-Rad, Hercules, CA; and goat anti-rabbit, 1:30,000; catalog no. A-3687, Sigma). The blots were developed in NBT buffer (100 mM Tris base, 100 mM NaCl, 5 mM MgCl2) containing 0.6 mM nitroblue tetrazolium (Sigma) and 1.2 mM 5-bromo-4-choro-3-indolyl phosphate (ICN Biomedicals, Cleveland, OH). Densitometry was calculated using a Bio-Rad imager with Quantity One Quantitation software, version 4.1.0 (Bio-Rad Laboratories, Hercules, CA).
α-Mannosidase Processing
Radiolabeling of cells, immunoprecipitation of α-mannosidase, SDS-PAGE, and fluorography were done as described by Buczynski et al. (1997).
Endosome/Phagosome Fusion Assay
Two methods were used to determine the fusion of either endosomes or phagosomes. To determine the rate of fusion of endosomes, cells were allowed to settle on coverslips placed in six-well plates and incubated for 5 min with 1.5 mg/ml rhodamine isothiocyanate (RITC)-conjugated dextran (catalog no. R-9379, Sigma) in HL-5. Cells were washed twice with fresh HL-5 and incubated with 1.5 mg/ml FITC-dextran for 5 min. Coverslips were washed once with fresh HL-5 and chased for 10 min in HL-5, and cells were fixed with 1% formaldehyde in HL-5 for 3 min. Coverslips were mounted on slides and visualized using both the fluorescein and rhodamine channels. Images of 50–100 cells were electronically captured using an Olympus upright fluorescence microscope equipped with a digital camera (Sensy, Brussels, Belgium). Images were analyzed using MetaView software (Universal Imaging, Dowingtown, PA).
For phagosome fusion, we used FITC-labeled bacteria (fl-bacteria) to visualize individual phagosomes. Cells were placed in shaking suspension and allowed to phagocytose fl-bacteria for 10 min and then washed three times with fresh HL-5. Cells were allowed to settle on coverslips in HL-5 to initiate the 30-min chase period. Coverslips were gently immersed in fresh HL-5 to wash the remaining fl-bacteria off, and then the cells were fixed in 1% formaldehyde in HL-5. Coverslips were immersed again in HL-5 to wash off the remaining fl-bacteria and mounted on coverslips to be examined with the fluorescent microscope. Cells (50–100) with ∼10 fl-bacteria per cell were examined using a 100× oil objective, and the number of fl-bacteria in each phagosome was assessed using both phase-contrast and fluorescent filters. Fusion events are typically easy to observe because the phagosome contains some soluble fluorescent FITC that clearly marks the inner edge of the vacuole and demonstrates that a single compartment contains multiple bacteria.
RESULTS
Dictyostelium Has Six Proteins Related to the Mammalian LYST/Beige Proteins
A search of the nearly complete Dictyostelium genome database identified six genes encoding BEACH domain-containing proteins (Wang, N., Wu, W. and DeLozanne, A., unpublished data). Of these, lvsA was already characterized for its essential role in cytokinesis and contractile vacuole (CV) function (Kwak et al., 1999; Gerald et al., 2001). Thus, the family of Dictyostelium genes was named lvsA through lvsF. All of the proteins encoded by these six genes share a high degree of similarity to each other but only in the BEACH and WD domain regions (Wang, N., Wu, W. and DeLozanne, A., unpublished data).
The BEACH and WD domains from the Dictyostelium LvsA–F proteins were compared with those from related proteins from many other species. Blast scores and sequence comparison by the Clustal method consistently indicated the sequence similarities illustrated in Figure 1. LvsB is the Dictyostelium protein most closely related to the mammalian LYST/Beige proteins. LvsF is related to the mammalian protein FAN and the other Lvs proteins are related to predicted proteins of unknown function in other species.
Figure 1.

LvsB is the Dictyostelium protein most similar to mammalian LYST/Beige. Sequence comparison of the BEACH and WD domains of the six Dictyostelium Lvs proteins and proteins from other organisms. The BEACH and WD domains of the indicated proteins were aligned using DNAStar with the ClustalW algorithm. The position of LvsB is indicated with an asterisk. Note its close relationship to mammalian LYST proteins. Accession codes are indicated for each protein. A.t., Arabidopsis thaliana; C.e., Caenorhabditis elegans; D.d., Dictyostelium discoideum; D.m., Drosophila melanogaster.
To understand the function of this family of proteins, we disrupted each of the Dictyostelium lvsB–F genes by homologous recombination. Initial analysis of the phenotype of each mutant indicated that, other than lvsA-null mutants, none of them are essential for cytokinesis, cell growth, osmoregulation, or development (Wang, N., Wu, W. and DeLozanne, A., unpublished data). Interestingly, the lvsB mutants displayed morphological differences that stimulated further study.
lvsB-Null Cells Accumulate Enlarged Endosomal Vesicles
Wild-type cells, and lvsA through lvsF-null mutants were examined using an inverted microscope equipped with phase-contrast optics. The most striking difference in morphology observed was an increase in the number of enlarged vacuolar structures observed in lvsB-null cells (Figure 2, compare E with A and C). These vacuoles were observed in the lvsB-null mutants at all growth densities in axenic media and over a culturing period of several months. The majority of the enlarged vacuoles in lvsB-null cells were typically >2 μm in size; vacuoles of this size were seldom seen in control and lvsA-null cells. The doubling times for all three strains in tissue culture flasks were comparable, indicating that the enlarged vacuoles did not influence division rate of cells.
Figure 2.

The enlarged vesicles in the lvsB-null mutant are endocytic. NC4A2 control (A and B), lvsA-null (C and D), and lvsB-null (E and F) cells were incubated in HL-5 medium containing FITC-dextran for 1 h to illuminate the endocytic vesicles. Cells were visualized using a fluorescence microscope. The arrows point to enlarged endocytic vesicles in lvsB-null cells, and the arrowheads point to vesicles the approximate size of normal lysosomes. Bar, 1 μm.
The enlarged vacuoles could be derived from the endosomal pathway or from the CV system of membranes. To distinguish between these two possibilities, we incubated cells in growth medium with the fluid phase marker FITC-dextran for 1 h to label the entire endosomal pathway, including macropinosomes, endosomes, lysosomes, and postlysosomes; internalized FITC-dextran is not trafficked into the CV compartments (Gabriel et al., 1999). Fluorescence microscopy indicated that all of the enlarged vacuoles in the lvsB-null mutant contained FITC-dextran, confirming that these vacuoles were part of the endosomal pathway (Figure 2F). Control (Figure 2B) and lvsA-null cells (Figure 2D) contained the predicted normal range in size of fluorescent endosomal vesicles (0.2–1.0 μm).
The Enlarged Vacuoles in the lvsB-Null Mutants Are Lysosomes
To determine whether the enlarged vacuoles in the lvsB-null cells were morphologically modified lysosomes, we performed a pulse-chase experiment using the fluid phase marker, FITC-dextran. Cells in growth medium were pulsed with FITC-dextran for 10 min followed by a 15-min chase in fresh medium, a time frame previously demonstrated to be sufficient to transport fluid to lysosomes (Temesvari et al., 1996a). As demonstrated in Figure 3, most of the enlarged vacuoles in lvsB-null cells (G) contained fluorescent fluid (H), whereas control cells (A and B) and lvsA-null cells (D and E) contained fluorescent vesicles the size of normal lysosomes (0.5 μm). Consistent with these large vacuoles being lysosomes, shorter pulses with FITC-dextran (to label macropinosomes) and longer chases (to label postlysosomes) inefficiently labeled the large vacuoles (Harris, Wang, Wu, Weatherford, De Lozanne, and Cardelli, unpublished results).
Figure 3.

The enlarged vesicles in the lvsB-null cells are acidic lysosomes. To visualize lysosomes, NC4A2 control (A and B), lvsA-null (D and E), and lvsB-null (G and H) cells were subjected to a 15-min pulse with FITC-dextran, washed, and chased for 15 min in fresh growth medium. Cells were examined with phase-contrast optics (A, D, and G) or by fluorescence microscopy (B, E, and H). The control (B) and lvsA-null (E) cells contained lysosomes of normal size, whereas the lvsB-null cells (H) contained enlarged lysosomes. Control (C), lvsA-null (F), and lvsB-null (I) cells were incubated with the acidophilic dye LysoSensor DND-189 (Molecular Probes) in HL-5 growth medium and visualized using a fluorescence microscope. The arrows point to enlarged acidic lysosomes, and the arrowheads point to normal size lysosomes. Bar, 2 μm.
To demonstrate that these large vesicles were acidic, consistent with their being lysosomes, cells were incubated with Lysosensor DND-189 (Molecular Probes), a probe that fluoresces only under acidic conditions. Figure 3 indicates that only lvsB-null cells (I) contained enlarged acidic compartments, whereas the wild-type cells (C) and the lvsA-null mutant (F) contained acidic vesicles the size of normal lysosomes.
To determine whether enlarged acidic lysosomes formed only in lvsB-null mutants, we pulsed wild-type cells and lvsA-, lvsB-, lvsC-, lvsD-, lvsE-, and lvsF-null mutants with FITC-dextran for 1 h to label the endosomal compartments. Fluorescence microscopy indicated that enlarged FITC-dextran–positive vesicles did not form in any mutant other than the lvsB-null strain (Figure 4; Harris, Wang, Wu, Weatherford, De Lozanne, and Cardelli, unpublished results for lvsF-null cells). Together with the results presented above, these data indicate that only the disruption of a gene encoding the protein closest in homology to LYST/Beige resulted in the accumulation of large acidic lysosomes, the phenotype associated with fibroblasts and other cells from patients with CHS.
Figure 4.

Only the lvsB-null cells exhibit the CHS cellular morphology. To examine the size of the endocytic vesicles, control (A), lvsA-null (B), lvsB-null (C), lvsC-null (D), lvsD-null (E), and lvsE-null (F) cells were pulsed in HL-5 medium with FITC-dextran for 2 h, washed, and fixed (lvsF-null not shown). Vesicle morphology was examined with a fluorescent microscope at 400× magnification. Bar, 2 μm.
Lysosomes from lvsB-Null Cells Contain Most but Not All of the Proteins Found Enriched in Normal Lysosomes
To determine whether the presence of enlarged lysosomes in lvsB-null cells disrupted trafficking or retention of lysosomal proteins, we used a previously described magnetic fractionation procedure (Temesvari et al., 1994) to purify lysosomes from control cells and lvsB-null cells. Proteins in cell lysates and lysosomes, prepared from the parental and lvsB-null cells, were separated by SDS-PAGE and silver stained or blotted to nitrocellulose. Blots were incubated with antibodies that have all been previously determined to recognize luminal or membrane proteins enriched in lysosomes (see MATERIALS AND METHODS and the references therein). The qualitative pattern of proteins detected by silver stain was comparable between the various fractions prepared from lvsB-null and control cells with a few exceptions. Lysosomes from the control cells contained higher levels of a 40- and 42-kDa protein as compared with lysosomes from lvsB-null cells (Figure 5, top, marked with an arrow). The relative enriched levels of the 100-kDa vacuolar H+-ATPase subunit (100 kDa su), the 41-kDa vacuolar H+-ATPase subunit (41 kDa su), RabB, RabD, Rab7, cathepsin D (Cath. D), the 36-kDa cysteine proteinase (CP36), eight different cysteine proteinases (GlcNac 1-P containing), β-glucosidase (β-glu), acid phosphatase (Acid Phos.), and LmpA were similar in lysosomes prepared from control and lvsB-null cells (Figure 5). In contrast, the relative amount of mature α-mannosidase (α-mann) in the lysosomes from lvsB-null cells was reduced by 50% as compared with control lysosomes (Figure 5). In addition, two proteins detectable in control lysosomes by an antibody (anti-GlcNac 1-P) that recognizes N-acetylglucosamine 1-phosphate (Souza et al., 1997) were not detected in blots of the lysosomes from lvsB-null cells (Figure 5).
Figure 5.

Lysosomes from the lvsB-null mutant reduced levels of α-mannosidase and several cysteine proteinases. NC4A2 control and lvsB-null cells were fed iron-dextran for 15 min and then were chased for 15 min in fresh medium to load up lysosomes. Iron-dextran–filled vesicles (lysosomes) were collected on a magnetic column, concentrated, and subjected to SDS-PAGE (see MATERIALS AND METHODS). Control cell lysates (lane 1), control lysosomes (lane 2), and lvsB-null lysosomes (lane 3) were subjected to SDS-PAGE and silver stained (top) or incubated with a host of antibodies against lysosomal proteins. The antibodies reacted with the 100-kDa subunit of the vacuolar H+-ATPase (100 kDa su), the 41-kDa subunit of the vacuolar H+-ATPase (41 kDa su), RabB (human Rab21 homolog), RabD (human Rab14 homolog), Rab7, cathepsin D (Cath. D), cysteine proteinase 36 (CP36), GlcNac 1-P-sulfate (lysosomal cysteine proteinases), α-mannosidase (α-mann), β-glucosidase (β-glu), acid phosphatase (Acid Phos.), and lysosomal membrane protein A (LmpA).
To determine whether the reduction in the levels of α-mannosidase in lysosomes from lvsB-null cells was the result of defects in processing or sorting of this enzyme, we performed a radiolabel pulse-chase experiment. Control, lvsA-null, and lvsB-null cells were pulsed with [35S]methionine for 20 min and chased in fresh HL-5 medium for 15, 60, 120, and 240 min. For each time point, α-mannosidase was immunoprecipitated from the medium (to detect secreted enzyme) and from cell lysates (to detect synthesis and processing rate of the enzyme). Lysosomal α-mannosidase is synthesized as a 140 kDa precursor that is, first, proteolytically cleaved to an immature form of 80 kDa, followed by further processing to the mature subunits of 58 and 60 kDa that are secreted slowly over time into the medium (Mierendorf et al., 1985). As indicated in the fluorographs shown in Figure 6, the rate of processing of the precursor form of α-mannosidase was similar for all strains examined. All of the precursor polypeptide was proteolytically processed in all strains 60 min into the chase with a half-time of processing of 10–15 min, and very little of the precursor was missorted and secreted from cells. In contrast, secreted radiolabeled mature forms of α-mannosidase were first detected at 120 min in the medium from lvsB-null cells, and by 240 min of chase, >50% of the mature enzyme had been secreted. In contrast, <25% of the mature forms had been released from control cells and lvsA-null cells at this chase time point. These results suggest that the processing and targeting of α-mannosidase were normal in the three strains examined but that lvsB-null cells may be defective in the retention of the mature form of this enzyme. This result was confirmed by demonstrating that the rate of secretion of the mature enzyme was twice as fast from lvsB-null cells as compared with control cells (Harris, Wang, Wu, Weatherford, De Lozanne, and Cardelli, unpublished results).
Figure 6.

lvsB-null cells process and target lysosomal α-mannosidase normally but oversecrete the mature enzyme. Wild-type control, lvsA-null, and lvsB-null cells were pulsed with [35S]methionine, washed, and chased in fresh HL-5. At each time point during the chase period, α-mannosidase was immunoprecipitated from both the medium, to detect secreted α-mannosidase, and cell lysates, to detect intracellular α-mannosidase. Immunoprecipitates were subjected to SDS-PAGE, followed by fluorography.
lvsB-Null Cells Are Not Defective in Endocytosis, Phagocytosis, or Endosomal Efflux
Because LvsB appears to play a role in the regulation of fusion and/or fission of lysosomes in the endocytic pathway, we wanted to determine whether this protein also regulated other pathways relevant to the biogenesis and/or function of lysosomes, such as endocytosis, phagocytosis, and endosomal efflux. Therefore, the rates of endocytosis and release of fluid (using FITC-dextran as a fluid phase marker) and phagocytosis (using latex beads as a marker) were measured in control, lvsA-null, and lvsB-null cell lines. Figure 7 indicates that the rates of endocytosis (A) and exocytosis (B) were statistically similar in the control and both of the lvs mutant cell lines. The rates of phagocytosis (C) were also comparable in both the control and lvsB-null cells. In contrast, the rate of phagocytosis was reduced by 40% in the lvsA-null cells as compared with control cells. These data suggest that LvsB does not play a significant role in the uptake of either fluid phase or particles, nor does this protein regulate fluid phase movement along the endosomal pathway. Instead, it appears that LvsA may regulate internalization of particles but not of fluid.
Figure 7.

The rates of endocytosis, phagocytosis, and fluid exocytosis are comparable between lvsB-null and control cells; lvsA-null cells are defective in phagocytosis. Endocytosis rates (A), exocytosis rates (B), and phagocytosis rates (C) were measured for control cells and the lvsA-null and lvsB-null mutants as described in MATERIALS AND METHODS. lvsA-null cells were significantly reduced in the rate of phagocytosis (unpaired, two-tailed Student's t test, p < 0.01) compared with control cells, whereas, lvsB-null and control cells were not significantly different.
The CV System Is Morphologically Normal in lvsB-Null Cells but Is Altered in lvsA-Null Cells
The CV is an osmoregulatory organelle consisting of a reticulum network (the collecting ducts that run throughout the cytoplasm) and the bladder, a contracting vacuole that empties the collecting ducts and expels water from the cell (Heuser et al., 1993). The membrane of the CV and the endocytic pathway are distinct, and no apparent trafficking of the bulk flow of membranes and protein occurs between these organelles (Gabriel et al., 1999). Despite this, these two pathways share some common proteins, such as RabD (Bush et al., 1994) and the vacuolar H+-ATPase (Heuser et al., 1993), and we have hypothesized that RabD regulates trafficking of a subset of proteins between lysosomes and the CV system (Bush et al., 1996). Therefore, because LvsB regulated the morphology of the lysosomes, we asked whether this protein also regulated the morphology of the CV. Immunofluorescence microscopy using antibodies directed against the membrane-inserted 100-kDa subunit of the H+-ATPase revealed that the morphology of the CV appeared normal in both the parent strain (Figure 8, A and B) and lvsB-null strain (Figure 8, E and F). In contrast, as described in more detail elsewhere (Gerald et al., 2001), the CV network in the lvsA-null cells appeared more filamentous and finer in morphology, suggesting that LvsA plays a role in maintaining CV morphology and function (Figure 8, C and D).
Figure 8.

The CV system of membranes appears normal in the lvsB-null cells in contrast to the altered morphology seen for CV membranes in lvsA-null cells. Control NC4A2 (A and B), lvsA-null (C and D), and lvsB-null (E and F) were fixed, permeabilized, and incubated with an antibody against the 100-kDa subunit of the proton pump, a marker for the CV membranes. CV reticular network membranes were visualized with a fluorescent microscope at 1000×.
The Enlarged Lysosomes in the lvsB-Null Cells May Result from an Increase in Vesicle Fusion
Overexpression of LYST in both normal and CHS fibroblasts results in numerous smaller-than-normal lysosomes that distribute near the periphery, interpreted to mean that LYST acts as a positive regulator of fission (Perou et al., 1997). However, this result is also consistent with LYST being a negative regulator of fusion, and therefore a greater amount of LYST in the cytoplasm would more efficiently inhibit fusion, resulting in the accumulation of smaller lysosomes. To further investigate the role of LvsB, we developed a fusion assay designed to distinguish whether the large lysosomes in the lvsB-null cells accumulate as a result of an increase in fusion or as a result of a decrease in fission. Cells were first pulsed with RITC-dextran for 5 min, washed for 2 min, pulsed with FITC-dextran for 5 min, chased for 10 min, and then fixed in formaldehyde. This pulse-chase format will label endosomes and lysosomes. Fluorescent microscopic images were collected in both the fluorescein and rhodamine channels, and the number of red, green, and red/green-merged vesicles were counted in several pools of cells. The fluorescence micrograph shown in Figure 9 reveals that the relative number of red/green-merged vesicles appeared greater in the lvsB-null cells (B) versus the number observed in control cells (A). The percentage of fused vesicles is graphically shown in the bar graph in Figure 11A and demonstrates that the number of fused endolysosomal vesicles was threefold higher in the lvsB-null cells as compared with fusion observed for the lvsA-null and control cells. If cells were examined immediately after the pulse period with the second fluor, no merged vesicles were found, suggesting that the merged vesicles arise from fusion following internalization (Figure 9C).
Figure 9.
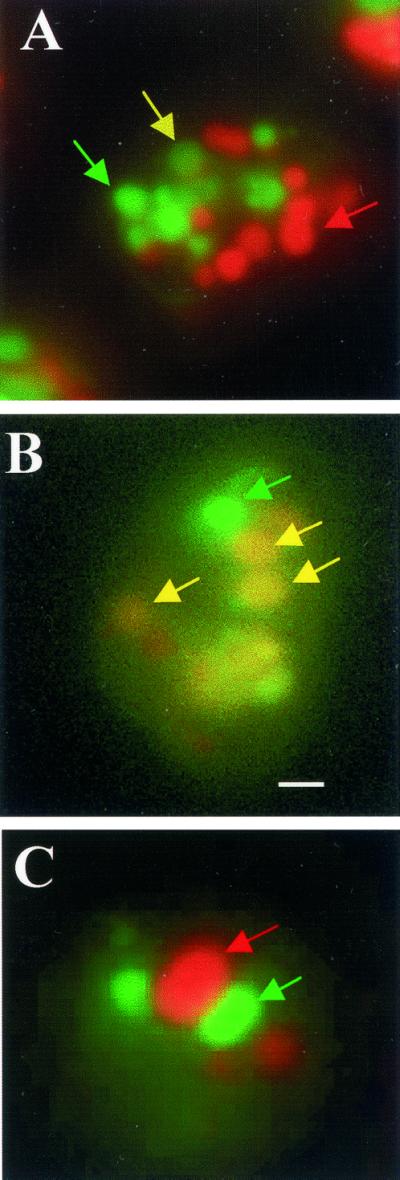
Large endolysosomes in the lvsB-null cells are a result of an increase in fusion. Control NC4A2, lvsA-null, and lvsB-null cells were pulsed with RITC-dextran, washed, pulsed with FITC-dextran, washed, chased for 5 min, fixed, and examined under a fluorescence microscope. Most of the red and green vesicles were distinct and separate from each other in the control (A), whereas, a higher percentage of vesicles in the lvsB-null (B) fused with each other. Examination of cells at the end of the double-pulse period revealed that little colocalization was observed in the mutant, suggesting that separately internalized vesicles fuse over time (C). Bar, 2 μm.
Figure 11.

The rate of endolysosomal and phagosomal fusion was increased in lvsB-null cells. A enumerates the number of red/green-merged endosomal vesicles and reveals that this number is significantly higher in the lvsB-null cells relative to control (wt) and lvsA-null cells (unpaired, two-tailed Student's t test, p = 0.0016). B illustrates that the number of fusion events per cell is significantly higher at the 30-min chase point in the lvsB-null mutant as compared with lvsA-null and control cells (wt) when using FITC-labeled bacteria (see MATERIALS AND METHODS for details) as a marker for phagolysosomes (unpaired, two-tailed Student's t test, p < 0.0001). These experiments were repeated three times each and for each experiment between 50 and 100 cells in random fields were analyzed.
The merged vesicles observed are presumed to be the result of the fusion between red and green vesicles; unfortunately, this method cannot quantitatively measure multiple fusion events, especially those that occur between vesicles with the same fluorescent substance. Also, these merged vesicles may result from trafficking of smaller vesicles between these large vesicles or a “kiss and run” process (Storrie and Desjardins, 1996). Therefore, to more accurately measure the number of fusion events for each merged vesicle and to determine whether fusion was complete, we used a previously developed phagosomal fusion assay (Rupper et al., 2001). Cells were allowed to internalize fluorescent bacteria for 10 min in shaking suspension, washed, and chased in fresh HL-5 on coverslips for 30 min. Microscopic images were collected, and the total number of bacteria per cell and bacteria per phagosome were enumerated as described by Rupper et al. (2001). Fusion rates are linear between 30 and 90 min of chase in cells (Rupper et al., 2001). Figure 10, A–D, indicates that after the pulse period only phagosomes containing single bacteria are observed in control and mutant cells. At the 30-min chase point, a greater number of multiparticle phagosomes are found in lvsB-null cells (Figure 10, G and H) as compared with control cells (Figure 10, E and F). A quantitative analysis of fusion events indicates that the number of phagosomal fusion events in the lvsB-null cells was threefold higher after a 30-min chase as compared with fusion in control and lvsA-null cells ( Figure 11). This result strongly suggests that the absence of LvsB led to an increase in endolysosomal fusion and phagosome-phagosome fusion.
Figure 10.

The rate of phagosome-phagosome fusion was increased in lvsB-null cells. Control cells were pulsed with FITC-labeled Escherichia coli for 10 min, washed, and chased on coverslips for 30 min. At this time point indicated, cells were fixed and examined microscopically to determine the number of fusion events. The assay is linear between 30 and 90 min of chase. A, C, E, and G are phase contrast images, whereas B, D, F, and H are fluorescent images. A and B (control) and C and D (mutant) indicate that after a short pulse period only single-particle–containing phagosomes are observed in cells (marked with arrowheads). E and F (control) and G and H (mutant) indicate that at the 30-min chase point at greater number of phagosomes contain multiparticles in the mutant cells as compared with control cells (marked with arrows). Bar, 2 μm.
DISCUSSION
We report here that disruption of the Dictyostelium lvsB gene, encoding a protein related in amino acid sequence to LYST/Beige proteins, particularly in the BEACH and WD-40 domains, resulted in a phenotype similar to that observed in cells from patients with CHS. The cellular changes observed in lvsB-null mutants included the presence of large acidic proteinase- and glycosidase-rich vesicles that appeared to be lysosomes. Enlarged lysosomes were not detected in lvsA-, lvsC-, lvsD-, lvsE-, or lvsF-null mutants. In addition, a small subset of proteins was decreased in abundance in lysosomes prepared from lvsB-null cells, although the sorting and processing of one of these proteins, a lysosomal glycosidase, appeared normal. Finally, the studies described here indicate that LvsB appeared to function as a negative regulator of lysosome biogenesis, because in the absence of this protein the rate of homotypic vesicle fusion increased. Because of the availability of biochemical and genetic approaches to study Dictyostelium and the fact that its endolysosomal system has been well characterized, our studies suggest that this organism will be a useful system to explore the molecular and biochemical nature of CHS.
It appears significant then that, among all six Dictyostelium Lvs proteins, LvsB is the most closely related to LYST/Beige in protein sequence. Intriguingly, this similarity is borne only in the BEACH and WD domains and not in the rest (80%) of the protein. Further studies are required to determine whether this portion of the protein contributes in any way to the function of LvsB/LYST/Beige proteins.
The main diagnostic feature of CHS disease at the cellular level is the striking presence of enlarged lysosomes and lysosome-related organelles in different cells from CHS patients. Three models have been developed to explain the formation of these enlarged structures. One model proposes that LYST/Beige acts as a negative regulator of fusion (Oliver and Essner, 1975), another suggests that this protein acts as a positive regulator of fission (Burkhardt et al., 1993; Perou et al., 1997), and a third model hypothesizes that LYST/Beige regulates vesicle trafficking (Faigle et al., 1998), a process that contributes to lysosome morphology and size.
Early morphological and biochemical studies supported the first model and suggested that these large lysosomes formed from the homotypic fusion of lysosomes or the fusion of lysosomes with prelysosomal endocytic vesicles (Irimajiri et al., 1992; Jones et al., 1992). Furthermore, fusion can account for identical and nonidentical membrane-enclosed organelles in neutrophils, suggesting that vesicles along different stages of maturation in the endolysosomal pathway were fusing together. Finally, homotypic fusion of lysosomes and phagosomes is not a process confined to CHS or Beige cells but in fact occurs in normal cells (Tjelle et al., 2000). Together, these data can be interpreted to support a role for LYST/Beige as a negative regulator of endolysosomal fusion processes that occur normally in cells.
More recently, it has been proposed that the formation of large lysosomes in CHS cells may be the result of a reduction in the rate of fission or budding of vesicles from maturing lysosomes that have undergone fusion. First, it was demonstrated that lysosomes and late endosomes fuse to form a hybrid organelle in normal cells and that fission reactions restore both late endosomes and lysosomes (Luzio et al., 2000). Second, when the LYST protein is overexpressed in CHS fibroblasts, large lysosomes disappear with the emergence of smaller-than-normal granules (Perou et al., 1997).
The experiments described here support a role for the LvsB protein as a negative regulator of fusion of lysosomes. First, our results demonstrate that the enlarged vesicles in lvsB-null mutants were lysosomes. These enlarged structures contained fluid phase markers (Figure 2), they were acidic (Figure 3), and they contained a host of proteins previously demonstrated to be enriched in normal lysosomes (Figure 5). In addition, these large vesicles received internalized fluid with the same kinetics as normal lysosomes (Figure 3). Second, we found that the rate of homotypic fusion of phagosomes and endolysosomes was significantly increased in lvsB-null cells relative to control cells and lvsA-, lvsC-, lvsD-, lvsE-, and lvsF-null mutants (Figure 9). Although our studies do not exclude the possibility that LvsB also acts to regulate fission, they more strongly support the idea that LvsB, (and by analogy, LYST/Beige), normally acts to negatively regulate the rate of fusion of endolysosomes.
How might LvsB and LYST/Beige proteins act to down-regulate fusion of endolysosomal vesicles? One possibility is that LYST-like proteins may inhibit the formation of tethering/docking complexes, necessary to bring vesicles close together, or SNARE complexes, necessary for membrane fusion. Nothing currently known about the BEACH or WD-40 domains suggests an interaction with docking or fusion-promoting proteins, although this needs to be investigated.
Another possible function for LYST/Beige/LvsB proteins could be to act as a molecular scaffold to regulate a signaling pathway that modifies the vesicle membrane to regulate fusion (Ward et al., 2000). One possible signaling pathway may involve the formation of ceramide. Ceramide levels are known to increase in Beige cells, and ceramide has been demonstrated to regulate endosome fusion. Intriguingly, FAN has BEACH and WD-40 domains related to LYST/Beige and is known to interact with a neutral sphingomyelinase to regulate the production of ceramide. Thus, it is possible that LYST/Beige interacts with sphingomyelinase to generate ceramide that in turn regulates vesicle fusion. LvsB/LYST/Beige might also alter the levels of other membrane-associated lipids, including phosphoinositides, which could alter vesicle fusion rates.
LvsB and LYST/Beige proteins may also regulate the trafficking of lysosomes and endosomes along cytoskeletal structures such as microtubules and F-actin. Movement of lysosomes from the periphery of cells to a more juxtanuclear position may increase the likelihood of docking and fusion. In fact, overexpression of the Beige protein in human fibroblasts results in smaller-than-normal lysosomes and a more peripheral distribution of lysosomes (Perou et al., 1997). The LYST/Beige proteins could negatively regulate proteins like Rab7, a small GTPase implicated in the trafficking and fusion of lysosomes in a juxtanuclear position in the cell (Bucci et al., 2000; Caplan et al., 2001). Alternatively, LYST/Beige-like proteins could positively regulate proteins like Rab27a (Bahadoran et al., 2001;Hume et al., 2001) or myosin V (Wu et al., 2001), both implicated in the F-actin- and microtubular-dependent trafficking of melanosomes to the periphery of melanocytes. Dictyostelium RabD, a Rab14-like GTPase (Harris, and Cardelli, unpublished results), and two phosphoinositide 3-kinases (Rupper et al., 2001) have been implicated in the regulation of lysosome-lysosome and phagosome-phagosome fusion and potentially could be targets for the activity of LvsB.
The study presented here also revealed only minor defects in protein trafficking in lvsB-null cells, even although this mutant contained enlarged lysosomes. Of >20 proteins assayed, only mature lysosomal α-mannosidase and two cysteine proteinases were inefficiently retained in mutant cells. The oversecretion of α-mannosidase was not the result of missorting, because the newly synthesized protein was processed and sorted correctly in lvsB-null cells. Finally, the rates of endocytosis, phagocytosis and fluid phase exocytosis were also normal in lvsB-null cells, consistent with only minor defects in trafficking of membrane and protein. Changes in the rate of secretion of α-mannosidase but not other hydrolases that reside in the same organelle have been observed before in clathrin-null mutants (Ruscetti et al., 1994). Lysosomal enzymes are probably retained in Dictyostelium by a process that involves the recycling of these enzymes from lysosomes and postlysosomes (a secretory compartment) back to early endosomes (Buczynski et al., 1997). In contrast, 100% of the internalized fluid is released from postlysosomes. The large size of lysosomes may inhibit the efficient recycling and retention of some lysosomal enzymes.
The observations of only subtle defects in retention or trafficking of lysosomal proteins in lvsB-null mutants are similar to the conclusions reached by others in the study of CHS or Beige cells. For instance, it has been reported that acidic α-mannosidase levels are lower in CHS cells as compared with controls (Tanaka, 1980) and that only two lysosomal proteinases, elastase and cathepsin G, were deficient in Beige neutrophils (Takeuchi et al., 1986). Furthermore, it was demonstrated that the processing and targeting of lysosomal cathepsin D and the delivery of proteins to lysosomes were normal in cytotoxic T cells from patients with CHS, even though these cells contained enlarged vacuoles and were defective in cytotoxic T-lymphocyte–mediated functions (Stinchcombe et al., 2000).
Minor trafficking defects can, however, have profound effects. For instance, peptide loading onto the major histocompatibility complex type II molecules and antigen presentation is delayed in the B cells of CHS patients (Faigle et al., 1998). In addition, CTLA-4, a negative regulator of T-cell activation (Karandikar et al., 1996) is normally enriched in endocytic vesicles and the secretory granules, whereas it is found in the large granules of the T cells from patients with CHS. Conformationally altered CTLA-4 is found on the surface of cells, leading to a disruption of T-cell homeostasis and possibly contributing to the accelerated phase in CHS patients (Barrat et al., 1999). The altered conformation of CTLA-4 may be the result of decreased processing of this protein in lysosomes because of a missorted processing enzyme.
LvsA, also closely related to LYST/Beige, has been proposed to regulate membrane trafficking to the contractile ring, a process important during cytokinesis (Kwak et al., 1999). As reported here, lvsA-null cells, but not lvsB-null cells, were defective in phagocytosis, although lvsA-null cells were normal in other endosomal processes, including lysosome biogenesis. LvsA is thus added to a growing list of Dictyostelium proteins that regulate phagocytosis (Cardelli, 2001).
Another known role of LvsA is in the function of the CV during osmoregulation (Gerald et al., 2001). The CV network of membranes was also altered in lvsA-null mutants and appeared finer and more vesicular in structure as compared with the more coarse tubular-vesicular structures observed in control cells or lvsB-null mutants. The possible connection between the morphology of the CV network and the regulation of phagocytosis is intriguing, because we have observed that overexpression of two different dominant negative GTPases, RabDN121I (Bush et al., 1996) and Rab11N126I (Harris et al., 2001), alters the CV network and affects phagocytosis. Rates of phagocytosis are increased in cells expressing Rab11N126I, and the CV network appears “thicker” in morphology, perhaps because of an increase in the amount of CV membrane present. In contrast, expression of RabDN121I results in decreases in the rates of phagocytosis and the formation of a reduced patch or cluster of vesicular CV structures next to the plasma membrane (Harris et al., 2001 ).
We have proposed that the CV network in Dictyostelium may function like the recycling endosomal compartment functions in mammalian cells. Also, as proposed for recycling endosomal compartments, we hypothesize that the CV network of membranes may regulate internalization of particles by supplying membrane to help generate the phagocytic cup (Cardelli, 2001). The CV network and phagocytosis have also been linked in Tetrahymena pyriformis. A 71-kDa protein, associated with the actin-binding proteins, localizes to both the CV and oral apparatus responsible for food uptake, suggesting that a connection may exist between the membranes involved for internalization and osmoregularity in this single-celled organism (Watanabe et al., 1998). We are currently investigating possible trafficking pathways between the CV membranes and the phagosomes in Dictyostelium.
In conclusion, we have demonstrated that LvsA and LvsB, two proteins that are related to LYST/Beige and contain BEACH and WD-40 domains, play a role in the regulation of phagocytosis and lysosome biogenesis, respectively. In contrast, null mutants of LvsC, LvsD, LvsE, and LvsF contained normal size lysosomes, and preliminary results suggest that endosomal processes also function normally. Future studies will focus on defining the molecular functions of LvsB and LvsA and on investigating the possible membrane-trafficking defects that exist in these other lvs mutants.
ACKNOWLEDGMENTS
The authors would like to thank members of the DeLozanne and Cardelli laboratories for careful reading of the manuscript. This research was supported by National Institutes of Health grant DK-39232 (to J.C.).
Footnotes
DOI:10.1091/mbc.01–09-0454.
REFERENCES
- Adam-Klages S, Adam D, Wiegmann K, Struve S, Kolanus W, Schneider-Mergener J, Kronke M. FAN, a novel WD-repeat protein, couples the p55 TNF-receptor to neutral sphingomyelinase. Cell. 1996;86:937–947. doi: 10.1016/s0092-8674(00)80169-5. [DOI] [PubMed] [Google Scholar]
- Bahadoran P, Aberdam E, Mantoux F, Busca R, Bille K, Yalman N, de Saint-Basile G, Casaroli-Marano R, Ortonne JP, Ballotti R. Rab27a: a key to melanosome transport in human melanocytes. J Cell Biol. 2001;152:843–850. doi: 10.1083/jcb.152.4.843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrat FJ, Le Deist F, Benkerrou M, Bousso P, Feldmann J, Fischer A, de Saint B. Defective CTLA-4 cycling pathway in Chediak-Higashi syndrome: a possible mechanism for deregulation of T lymphocyte activation. Proc Natl Acad Sci USA. 1999;96:8645–8650. doi: 10.1073/pnas.96.15.8645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bucci C, Thomsen P, Nicoziani P, McCarthy J, van Deurs B. Rab7: a key to lysosome biogenesis. Mol Biol Cell. 2000;11:467–480. doi: 10.1091/mbc.11.2.467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buczynski G, Bush J, Zhang L, Rodriguez-Paris J, Cardelli J. Evidence for a recycling role for Rab7 in regulating a late step in endocytosis and in retention of lysosomal enzymes in Dictyostelium discoideum. Mol Biol Cell. 1997;8:1343–1360. doi: 10.1091/mbc.8.7.1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burkhardt JK, Wiebel FA, Hester S, Argon Y. The giant organelles in beige and Chediak-Higashi fibroblasts are derived from late endosomes and mature lysosomes. J Exp Med. 1993;178:1845–1856. doi: 10.1084/jem.178.6.1845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bush J, Nolta K, Rodriguez-Paris J, Kaufmann N, O'Halloran T, Ruscetti T, Temesvari L, Steck T, Cardelli J. A Rab4-like GTPase in Dictyostelium discoideum colocalizes with V-H(+)-ATPases in reticular membranes of the contractile vacuole complex and in lysosomes. J Cell Sci. 1994;107:2801–2812. doi: 10.1242/jcs.107.10.2801. [DOI] [PubMed] [Google Scholar]
- Bush J, Temesvari L, Rodriguez-Paris J, Buczynski G, Cardelli J. A role for a Rab4-like GTPase in endocytosis and in regulation of contractile vacuole structure and function in Dictyostelium discoideum. Mol Biol Cell. 1996;7:1623–1638. doi: 10.1091/mbc.7.10.1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caplan S, Hartnell LM, Aguilar RC, Naslavsky N, Bonifacino JS. Human Vam6p promotes lysosome clustering and fusion in vivo. J Cell Biol. 2001;154:109–122. doi: 10.1083/jcb.200102142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cardelli J. Phagocytosis and macropinocytosis in Dictyostelium: phosphoinositide-based processes, biochemically distinct. Traffic. 2001;2:311–320. doi: 10.1034/j.1600-0854.2001.002005311.x. [DOI] [PubMed] [Google Scholar]
- Dufourcq-Lagelouse R, Lambert N, Duval M, Viot G, Vilmer E, Fischer A, Prieur M, de Saint B. Chediak-Higashi syndrome associated with maternal uniparental isodisomy of chromosome 1. Eur J Hum Genet. 1999;7:633–637. doi: 10.1038/sj.ejhg.5200355. [DOI] [PubMed] [Google Scholar]
- Faigle W, Raposo G, Tenza D, Pinet V, Vogt AB, Kropshofer H, Fischer A, de Saint-Basile G, Amigorena S. Deficient peptide loading and MHC class II endosomal sorting in a human genetic immunodeficiency disease: the Chediak-Higashi syndrome. J Cell Biol. 1998;141:1121–1134. doi: 10.1083/jcb.141.5.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feuchter AE, Freeman JD, Mager DL. Strategy for detecting cellular transcripts promoted by human endogenous long terminal repeats: identification of a novel gene (CDC4L) with homology to yeast CDC4. Genomics. 1992;13:1237–1246. doi: 10.1016/0888-7543(92)90041-p. [DOI] [PubMed] [Google Scholar]
- Fok AK, Clarke M, Ma L, Allen RD. Vacuolar H(+)-ATPase of Dictyostelium discoideum: a monoclonal antibody study. J Cell Sci. 1993;106:1103–1113. doi: 10.1242/jcs.106.4.1103. [DOI] [PubMed] [Google Scholar]
- Gabriel D, Hacker U, Kohler J, Muller-Taubenberger A, Schwartz JM, Westphal M, Gerisch G. The contractile vacuole network of Dictyostelium as a distinct organelle: its dynamics visualized by a GFP marker protein. J Cell Sci. 1999;112:3995–4005. doi: 10.1242/jcs.112.22.3995. [DOI] [PubMed] [Google Scholar]
- Gerald N, Siano M, DeLozanne A. The Dictyostelium LusA protein is localized on the contractile vacuole and is required for osmoregulation. Traffic. 2002;3:50–60. doi: 10.1034/j.1600-0854.2002.30107.x. [DOI] [PubMed] [Google Scholar]
- Harris E, Yoshida K, Cardelli J, Bush J. Rab11-like GTPase associates with and regulates the structure and function of the contractile vacuole system in Dictyostelium. J Cell Sci. 2001;114:3035–3045. doi: 10.1242/jcs.114.16.3035. [DOI] [PubMed] [Google Scholar]
- Heuser J, Zhu Q, Clarke M. Proton pumps populate the contractile vacuoles of Dictyostelium amoebae. J Cell Biol. 1993;121:1311–1327. doi: 10.1083/jcb.121.6.1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hume AN, Collinson LM, Rapak A, Gomes AQ, Hopkins CR, Seabra MC. Rab27a regulates the peripheral distribution of melanosomes in melanocytes. J Cell Biol. 2001;152:795–808. doi: 10.1083/jcb.152.4.795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Introne W, Boissy RE, Gahl WA. Clinical, molecular, and cell biological aspects of Chediak-Higashi syndrome. Mol Genet Metab. 1999;68:283–303. doi: 10.1006/mgme.1999.2927. [DOI] [PubMed] [Google Scholar]
- Irimajiri K, Iwamoto I, Kawanishi K, Tsuji K, Morita S, Koyama A, Hamazaki H, Horiuchi F, Horiuchi A, Akiyama T. Studies on pseudo-Chediak-Higashi granules formation in acute promyelocytic leukemia. Rinsho Ketsueki. 1992;33:1057–1065. [PubMed] [Google Scholar]
- Jones K, Steward R, Fowler M, Fukuda M, Holcombe R. Chediak-Higashi lymphoblastoid cell lines: granule characteristics and expression of lysosome-associated membrane proteins. Clin Immunol Immunopathol. 1992;65:219–226. doi: 10.1016/0090-1229(92)90150-m. [DOI] [PubMed] [Google Scholar]
- Journet A, Chapel A, Jehan S, Adessi C, Freeze H, Klein G, Garin J. Characterization of Dictyostelium discoideum cathepsin D. J Cell Sci. 1999;112:3833–3843. doi: 10.1242/jcs.112.21.3833. [DOI] [PubMed] [Google Scholar]
- Karakesisoglou I, Janssen KP, Eichinger L, Noegel AA, Schleicher M. Identification of a suppressor of the Dictyostelium profilin-minus phenotype as a CD36/LIMP-II homologue. J Cell Biol. 1999;145:167–181. doi: 10.1083/jcb.145.1.167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karandikar NJ, Vanderlugt CL, Walunas TL, Miller SD, Bluestone JA. CTLA-4: a negative regulator of autoimmune disease. J Exp Med. 1996;184:783–788. doi: 10.1084/jem.184.2.783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwak E, Gerald N, Larochelle DA, Vithalani KK, Niswonger ML, Maready M, De Lozanne A. LvsA, a protein related to the mouse beige protein, is required for cytokinesis in Dictyostelium. Mol Biol Cell. 1999;10:4429–4439. doi: 10.1091/mbc.10.12.4429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laemmli UK. Cleavage of structural proteins during the assembly of the head of bacteriophage T4. Nature. 1970;227:680–685. doi: 10.1038/227680a0. [DOI] [PubMed] [Google Scholar]
- Lutzner MA, Lowrie CT, Jordan HW. Giant granules in leukocytes of the beige mouse. J Hered. 1967;58:299–300. doi: 10.1093/oxfordjournals.jhered.a107620. [DOI] [PubMed] [Google Scholar]
- Luzio JP, Rous BA, Bright NA, Pryor PR, Mullock BM, Piper RC. Lysosome-endosome fusion and lysosome biogenesis. J Cell Sci. 2000;113:1515–1524. doi: 10.1242/jcs.113.9.1515. [DOI] [PubMed] [Google Scholar]
- Maniak M. Fluid-phase uptake and transit in axenic Dictyostelium cells. Biochim Biophys Acta. 2001;1525:197–204. doi: 10.1016/s0304-4165(01)00105-2. [DOI] [PubMed] [Google Scholar]
- Mierendorf RCJ, Cardelli JA, Dimond RL. Pathways involved in targeting and secretion of a lysosomal enzyme in Dictyostelium discoideum. J Cell Biol. 1985;100:1777–1787. doi: 10.1083/jcb.100.5.1777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neuhaus EM, Soldati T. A myosin I is involved in membrane recycling from early endosomes. J Cell Biol. 2000;150:1013–1026. doi: 10.1083/jcb.150.5.1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimura M, Inoue M, Nakano T, Nishikawa T, Miyamoto M, Kobayashi T, Kitamura Y. Beige rat: a new animal model of Chediak-Higashi syndrome. Blood. 1989;74:270–273. [PubMed] [Google Scholar]
- Oliver C, Essner E. Formation of anomalous lysosomes in monocytes, neutrophils, and eosinophils from bone marrow of mice with Chediak-Higashi syndrome. Lab Invest. 1975;32:17–27. [PubMed] [Google Scholar]
- Padgett GA. Neutrophilic function in animals with the Chediak-Higashi syndrome. Blood. 1967;29:906–915. [PubMed] [Google Scholar]
- Perou CM, Leslie JD, Green W, Li L, Ward DM, Kaplan J. The Beige/Chediak-Higashi syndrome gene encodes a widely expressed cytosolic protein. J Biol Chem. 1997;272:29790–29794. doi: 10.1074/jbc.272.47.29790. [DOI] [PubMed] [Google Scholar]
- Ridgway SH. Reported causes of death of captive killer whales (Orcinus orca) J Wildl Dis. 1979;15:99–104. doi: 10.7589/0090-3558-15.1.99. [DOI] [PubMed] [Google Scholar]
- Rupper A, Cardelli J. Regulation of phagocytosis and endo-phagosomal trafficking pathways in Dictyostelium discoideum. Biochim Biophys Acta. 2001;1525:205–216. doi: 10.1016/s0304-4165(01)00106-4. [DOI] [PubMed] [Google Scholar]
- Rupper AC, Rodriguez-Paris JM, Grove BD, Cardelli JA. p110-related PI 3-kinases regulate phagosome-phagosome fusion, and phagosomal pH through a PKB/Akt dependent pathway in Dictyostelium. J Cell Sci. 2001;114:1283–1295. doi: 10.1242/jcs.114.7.1283. [DOI] [PubMed] [Google Scholar]
- Ruscetti T, Cardelli JA, Niswonger ML, O'Halloran TJ. Clathrin heavy chain functions in sorting and secretion of lysosomal enzymes in Dictyostelium discoideum. J Cell Biol. 1994;126:343–352. doi: 10.1083/jcb.126.2.343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Souza GM, Mehta DP, Lammertz M, Rodriguez-Paris J, Wu R, Cardelli JA, Freeze HH. Dictyostelium lysosomal proteins with different sugar modifications sort to functionally distinct compartments. J Cell Sci. 1997;110:2239–2248. doi: 10.1242/jcs.110.18.2239. [DOI] [PubMed] [Google Scholar]
- Stinchcombe JC, Page LJ, Griffiths GM. Secretory lysosome biogenesis in cytotoxic T lymphocytes from normal and Chediak Higashi syndrome patients. Traffic. 2000;1:435–444. doi: 10.1034/j.1600-0854.2000.010508.x. [DOI] [PubMed] [Google Scholar]
- Storrie B, Desjardins M. The biogenesis of lysosomes: is it a kiss and run, continuous fusion and fission process? Bioessays. 1996;18:895–903. doi: 10.1002/bies.950181108. [DOI] [PubMed] [Google Scholar]
- Takeuchi K, Wood H, Swank RT. Lysosomal elastase and cathepsin G in beige mice: neutrophils of beige (Chediak-Higashi) mice selectively lack lysosomal elastase and cathepsin G. J Exp Med. 1986;163:665–677. doi: 10.1084/jem.163.3.665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka T. Chediak-Higashi syndrome: abnormal lysosomal enzyme levels in granulocytes of patients and family members. Pediatr Res. 1980;14:901–904. doi: 10.1203/00006450-198008000-00001. [DOI] [PubMed] [Google Scholar]
- Temesvari L, Rodriguez-Paris J, Bush J, Steck TL, Cardelli J. Characterization of lysosomal membrane proteins of Dictyostelium discoideum: a complex population of acidic integral membrane glycoproteins, Rab GTP-binding proteins and vacuolar ATPase subunits. J Biol Chem. 1994;269:25719–25727. [PubMed] [Google Scholar]
- Temesvari L, Zhang L, Fodera B, Janssen KP, Schleicher M, Cardelli JA. Inactivation of lmpA, encoding a LIMPII-related endosomal protein, suppresses the internalization and endosomal trafficking defects in profilin-null mutants. Mol Biol Cell. 2000;11:2019–2031. doi: 10.1091/mbc.11.6.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Temesvari LA, Bush JM, Peterson MD, Novak KD, Titus MA, Cardelli JA. Examination of the endosomal and lysosomal pathways in Dictyostelium discoideum myosin I mutants. J Cell Sci. 1996a;109:663–673. doi: 10.1242/jcs.109.3.663. [DOI] [PubMed] [Google Scholar]
- Temesvari LA, Rodriguez-Paris JM, Bush JM, Zhang L, Cardelli JA. Involvement of the vacuolar proton-translocating ATPase in multiple steps of the endo-lysosomal system and in the contractile vacuole system of Dictyostelium discoideum. J Cell Sci. 1996b;109:1479–1495. doi: 10.1242/jcs.109.6.1479. [DOI] [PubMed] [Google Scholar]
- Tjelle TE, Lovdal T, Berg T. Phagosome dynamics and function. Bioessays. 2000;22:255–263. doi: 10.1002/(SICI)1521-1878(200003)22:3<255::AID-BIES7>3.0.CO;2-R. [DOI] [PubMed] [Google Scholar]
- Wang X, Herberg FW, Laue MM, Wullner C, Hu B, Petrasch-Parwez E, Kilimann MW. Neurobeachin: a protein kinase A-anchoring, beige/Chediak-Higashi protein homolog implicated in neuronal membrane traffic. J Neurosci. 2000;20:8551–8565. doi: 10.1523/JNEUROSCI.20-23-08551.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward DM, Griffiths GM, Stinchcombe JC, Kaplan J. Analysis of the lysosomal storage disease Chediak-Higashi syndrome. Traffic. 2000;1:816–822. doi: 10.1034/j.1600-0854.2000.011102.x. [DOI] [PubMed] [Google Scholar]
- Watanabe A, Kurasawa Y, Watanabe Y, Numata O. A new Tetrahymena actin-binding protein is localized in the division furrow. J Biochem (Tokyo) 1998;123:607–613. doi: 10.1093/oxfordjournals.jbchem.a021980. [DOI] [PubMed] [Google Scholar]
- Wu X, Rao K, Bowers MB, Copeland NG, Jenkins NA, Hammer JA. Rab27a enables myosin Va-dependent melanosome capture by recruiting the myosin to the organelle. J Cell Sci. 2001;114:1091–1100. doi: 10.1242/jcs.114.6.1091. [DOI] [PubMed] [Google Scholar]