

Abstract
The underlying molecular basis for neurodevelopmental or neuropsychiatric disorders is not known. In contrast, mechanistic understanding of other brain disorders including neurodegeneration has advanced considerably. Yet, these do not approach the knowledge accrued for many cancers with precision therapeutics acting on well‐characterized targets. Although the identification of genes responsible for neurodevelopmental and neuropsychiatric disorders remains a major obstacle, the few causally associated genes are ripe for discovery by focusing efforts to dissect their mechanisms. Here, we make a case for delving into mechanisms of the poorly characterized human KCTD gene family. Varying levels of evidence support their roles in neurocognitive disorders (KCTD3), neurodevelopmental disease (KCTD7), bipolar disorder (KCTD12), autism and schizophrenia (KCTD13), movement disorders (KCTD17), cancer (KCTD11), and obesity (KCTD15). Collective knowledge about these genes adds enhanced value, and critical insights into potential disease mechanisms have come from unexpected sources. Translation of basic research on the KCTD‐related yeast protein Whi2 has revealed roles in nutrient signaling to mTORC1 (KCTD11) and an autophagy‐lysosome pathway affecting mitochondria (KCTD7). Recent biochemical and structure‐based studies (KCTD12, KCTD13, KCTD16) reveal mechanisms of regulating membrane channel activities through modulation of distinct GTPases. We explore how these seemingly varied functions may be disease related.
Keywords: KCTD11, KCTD13, KCTD7, Neurodegeneration, Neurodevelopmental disorders
1. INTRODUCTION
Understanding the molecular basis of neurodevelopmental and neuropsychiatric disorders has many obstacles inherent to disease complexities and the lack of tractable model systems analogous to cancer biology. However, remarkable advancements in genomics have identified many potential candidate genes, some with additional compelling evidence for causal involvement in developmental and psychiatric brain disorders. Most gene candidates are relatively uncharacterized compared to the decades of accumulated knowledge for some tumor suppressors and oncogenes. New exploratory research is needed to decipher the mechanistic details and organismal functions of those genes contributing to neurodevelopmental and neuropsychiatric disorders.
A prime candidate for focused attention is the understudied 25‐member KCTD gene family. Several human KCTD genes have emerged in association with neurodevelopmental, neuropsychiatric, and neurodegenerative disorders. Additional KCTD family members are associated with several types of cancer and other disorders, providing additional perspectives. While mutations in individual KCTD genes are found in a limited number of patients, collectively they provide a compelling basis to justify interrogation of their molecular and cellular functions to understand disease mechanisms. The biochemical and biological functions of KCTD proteins have not been deciphered, but progress is underway (Table 1). Some KCTD disease associations will need further validation, and likely many others are not yet identified. Deciphering the shared and distinct functions of multiple KCTD family members will provide a wealth of knowledge toward understanding neurodevelopmental, neuropsychiatric, and degenerative processes that were previously impermeable to interrogation.
Table 1.
Disease associations, protein functions and structure determinations for all human KCTD family proteins and yeast Whi2.
| Clade Figure 2 | Protein | BTB structure | Binding partners | Biological functions | Disease relevance |
|---|---|---|---|---|---|
| E | KCTD17 | closed pentamer (X‐ray7) | Cul3 2, 32 (5:5 SAXS 5, 7) | Promotes ciliogenesis by degrading trichoplein32, 106 | Gen vars associated with dystonia79, 83 |
| KCTD5 | closed pentamer (EM,107 X‐ray3) | Cul32 (5:5 ITC37) | Inhibits GPCR signal, degrades Gβγ18; monoubiquitination of ΔNp63α108 | Involved in sleep regulation86, 109 | |
| KCTD2 | ND | Clu329 |
Degrades c‐Myc29
Regulates sleep86 |
Low in patient‐derived glioma stem cells 29
Gen vars assoc. with Alzheimer's risk (GWAS)110, 111 |
|
| KCTD9 | Closed pentamer (X‐ray2) | Cul3 (5:5 cryo‐EM2) | ND | ND | |
| D | SHKBP1 | monomer (X‐ray5) |
Cul3 (5:5 SAXS5) CIN85112 SETA113 |
Promotes EGFR pathway by disrupting c‐Cbl‐CIN85 complex112 |
Mutated in cervical cancer114
Mutated in leukemia115 Biomarker in small intestinal neuroendocrine tumors116 |
| KCTD3 | ND | HCN391 | Up‐regulation of HCN391 |
Biallelic mutations in epileptic encephalopathy90
Gen vars in intellectual disability/ seizures (WES)88, 89 |
|
| C | KCTD10 | tetramer (X‐ray5) |
Cul331, 35, 117
PCNA6 TNFAIP1118 |
Degrades RhoB31, 35
Promotes cilium, degrades CEP97117 DNA synthesis, cell proliferation6 Inhibits NF‐κB and AP‐1118 |
Tumor suppressor in gastrointestinal stromal tumor119 |
| TNFAIP1 | ND |
Cul333, 76
RhoB120 PCNA8 KCTD10118 |
Degrades RhoA33, 76
Regulates apoptosis120 Inhibits NF‐κB and AP‐1 118 |
Aa a tumor suppressor in nonsmall cell lung cancer121
Poor prognosis if overexpressed in breast cancer122 Overexpressed in osteosarcoma123 |
|
| KCTD13 |
tetra‐ (X‐ray5) pentamer (EM107) |
Cul311, 33, 76 (5:5 SAXS5) SAXS5
PCNA7 |
Degrades RhoA11, 33, 34, 76 |
Copy‐number var associated with autism11, 52, 76
Mutations associated with schizophrenia124 Overexpression: microcephaly in zebrafish, mouse34 |
|
| H | KCTD14 | ND | ND | ND | ND |
| KCTD7 | ND | Cul313, 36 | Regulates neuronal autophagy,13 Gln transport SAT2,23 K+ conductance20 | Bi‐allelic mutations cause severe early onset progressive disorder with epilepsy13, 38, 39, 40, 41, 125 | |
| B | KCTD6 | pentamer (EM107) | Cul32 (4:4 gel filtration16) |
Suppresses Hh pathway by degrading HDAC30 and USP21126
Degrades small ankyrin‐1127 |
ND |
| KCTD21 | ND | Cul330 | Inhibits Hh by degrading HDAC30 | Gen vars associated with autism (WES)128 | |
| KCTD11 |
tetramer (gel filtration),96
pentamer (EM107) |
Cul3 (4:4 gel filtration16, 96) |
Inhibits mTORC1 activity14
Inhibits Hh pathway by degrading HDAC12 |
Deletion/ reduced expression in medulloblastoma94
Loss of heterozygosity in prostate adenocarcinoma129 Reduced expression in hepatocellular carcinoma130 |
|
| Other | KCTD4 | ND | ND | ND | ND |
| A | KCTD15 | pentamer (EM107) | AP‐2α10 | Inhibits neural crest formation by inhibiting AP‐2α10 & Wnt pathway99 | Genetic variants associated with obesity97, 98 |
| KCTD1 | closed/open pentamer (EM,107 X‐ray2) | AP‐2α transcription factor9 | Inhibits transcription factor AP‐2α9 and Wnt signaling by degrading β‐catenin131 |
I27N mutation caused kidney dysfunction in mice132
Missense mutations associated with scalp‐ear‐nipple syndrome133 |
|
| Other | KCTD19 | ND | ND | ND | ND |
| F | KCTD12 | pentamer (EM107; X‐ray19) |
GABAB2
17
Gβγ19 CDC25B134 |
Regulates GABAB2 receptor signaling17, 19, 135, 136
Suppresses Wnt‐Notch pathway137 Promotes G2/M transition134 |
Emotionality, neuronal excitability (mice)65
KCTD12 increases 5‐y survival in GI stromal tumor138 Increased KCTD12 in cervical and lung cancers134 Bipolar disorder (GWAS)62 |
| KCTD16 | open pentamer (X‐ray5, 19) |
GABAB2
17
Gβγ19 |
Regulates GABAB2 receptor signaling17, 19, 135, 136 | ND | |
| KCTD8 | ND | GABAB2 17 | Regulates GABAB2 signaling17, 135, 136 | ND | |
| Other | KCNRG | ND | Kv channel139, 140 | Suppresses K + channel activity139 | Deleted in B‐cell chronic lymphocytic leukemia,140, 141, 142 prostate cancer140 and multiple myeloma142 |
| Other | KCTD18 | ND | ND | ND |
Duplication of 2q33 in one patient with epilepsy, devel. delay, autistic behavior143
Haplotype associated with restless legs syndrome144 |
| G | KCTD20 | ND | ND | Activates Akt145, 146 | Gen var associated with insulin resistance (GWAS)147 |
| BTBD10 | ND | Akt1‐3148 | Inhibits apoptosis, activates Akt149, 150 | Sporadic amyotrophic lateral sclerosis151 | |
| Sc | Whi2 | ND | Psr113 | Suppresses TORC1, promotes autophagy induction13 | Plant pathogen CoWhi2 has suggested role in pathogenesis during infection152 |
5:5/4:4, pentameric or tetrameric symmetry when bound to binding partners; X‐ray/EM/gel‐filtration/SAXS‐small angle X‐ray scattering SAXS, structure determination methods.
Abbreviations: Gen vars, genetic variants associated with disease; ND, not determined; GWAS, genome‐wide association study; Sc, Saccharomyces cerevisiae (baker's yeast); WES, whole exome sequencing.
Bold type: KCTD proteins discussed in separate sections of this article.
Human KCTD family proteins (KCTD1‐21, TNFAIP1, KCNRG, SHKBP1, and BTBD10) can localize in the cytoplasm or the nucleus, and range in size from 26‐kDa KCTD5 (234 amino acids) to 105‐kDa KCTD19 (926 amino acids). Mice encode an additional KCTD protein, Kctd12b (chromosome X), which is highly similar to mouse Kctd12 (chromosome 14). The distinguishing feature of KCTD proteins is a single N‐terminal BTB/POZ (bric‐a‐brac, tramtrak, and broad complex/poxvirus zinc finger) domain, the exception being KCTD19 with three separate BTBs that may reflect tandem gene amplification (Figure 1).1 BLAST searches readily reveal that the BTB domains of KCTD family proteins are most similar in amino acid sequence to the T1/BTB domains that mediate tetramerization of voltage‐gated potassium channel subunits to form functional channels.1, 2 This sequence similarity to Kv channels explains how KCTDs acquired their official name (potassium channel tetramerization domain). However, KCTD family proteins lack predicted transmembrane domains.3
Figure 1.
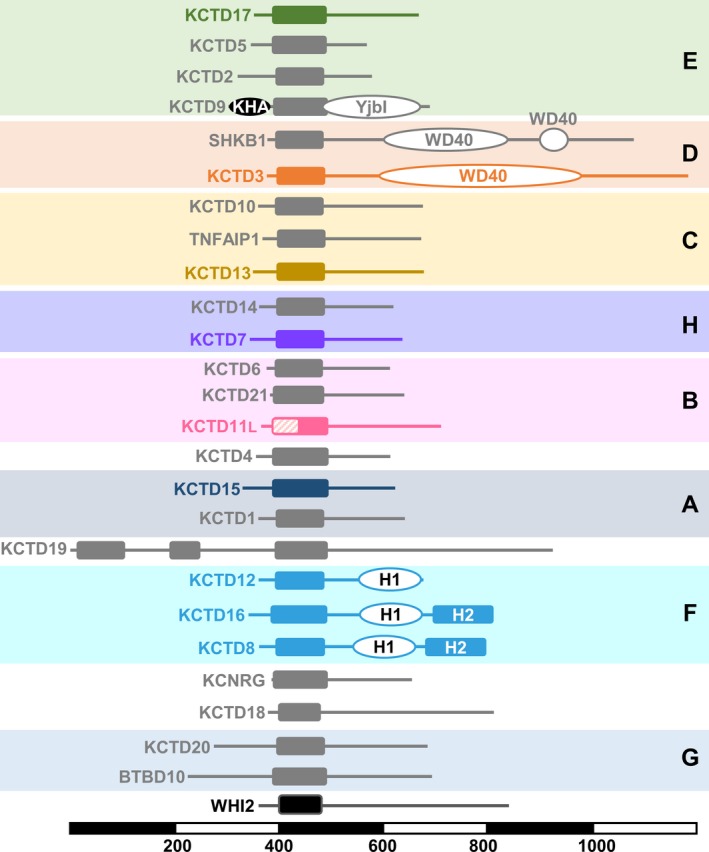
The diverse human KCTD protein family and yeast Whi2. Line diagrams of the 25 human KCTD family proteins and Saccharomyces cerevisiae Whi2 are drawn to scale, grouped in color‐coded clades (A‐H), ordered as in Figure 2, and aligned with respect to their BTB domain (solid rectangles). Additional protein domains with known or inferred structures (KHA, YjbI, WD40, H1) and similarity region H2 are also represented. KCTD11L starts at an AUU start codon adding 39 N‐terminal residues (hashed box) before the first in‐frame AUG translate start. Gray line diagrams indicate proteins not discussed in detail. Scale bar indicates protein length in amino acid residues
An unusual feature of this gene family is their diversity outside the BTB domain. Except within subgroups of closely related family members, KCTD proteins lack obvious sequence similarity in their highly variable C‐terminal regions.1 This feature is consistent with their proposed roles as adaptor molecules that use their C‐termini to bind and recruit diverse cellular proteins destined for degradation. In this model, KCTDs are responsible for selecting protein substrates for ubiquitination by cullin‐RING ubiquitin ligases (CRLs) that bind to the BTB domains of KCTD proteins.2, 4, 5 Thus, disrupted proteostasis required for the delicate balance between protein function versus degradation could potentially underlie neurological disorders now associated with the brain‐enriched proteins KCTD3, KCTD7, KCTD13, and KCTD17. This proposed adaptor function for KCTD proteins could potentially underlie the diverse biological processes reported for KCTD proteins, including DNA replication (KCTD10 and TNFAIP1),6, 7, 8 transcription inhibition (KCTD1 and KCTD15),9, 10 regulation of Rho GTPases in brain development (KCTD13),11 suppression of hedgehog signaling (KCTD11),12 autophagy induction and amino acid signaling to mTORC1 (KCTD11),13, 14, 15 and more (Table 1). However, other KCTDs appear to lack the ability to bind cullin‐3, implying distinct biochemical mechanisms.16 For example, several KCTD family proteins (e.g., KCTD12 and KCTD13) may alter neuronal activity and other signaling pathways by regulating diverse types of GTPases 11, 17, 18, 19. Here, we convey our current understanding of KCTD protein structure and function and how this may relate to disease mechanisms, focusing on a subset of KCTD family members implicated in disorders originating from the neural crest.
2. STRUCTURE AND FUNCTION OF KCTD FAMILY PROTEINS
KCTD family members likely represent paralogs that arose by gene duplication from a common ancestral gene followed by divergence, which led to diversification of the current KCTD protein family in the animal kingdom (Figure 2). Solved structures are available for the N‐terminal BTB domain of several KCTD family proteins primarily revealing pentameric homo‐oligomers. This fivefold symmetry appears to extend through the C‐terminus.1, 2, 3, 19 The BTB domains of KCTD proteins also mediate other protein‐protein interactions, leading to three main hypotheses for general KCTD mechanisms. The first of these is not favored currently. Reasoning that KCTD‐BTB domains might directly bind to their closest cousins, the T1/BTB (tetramerization) domains of voltage‐gated potassium channels, KCTD proteins could potentially regulate channel assembly or activity. KCTD5 was tested for the ability to bind or affect the functions of Kv1.2, Kv2.1, Kv3.4, and Kv4.2 channels but without success.3 In a second model, several KCTD family members are reported to indirectly influence channel activity by mechanisms not yet delineated.20, 21, 22, 23 However, recent advancements in this direction stem for the finding that the BTB domain of a different subset of KCTDs (clade F) interacts with the cytoplasmic tail of membrane‐embedded GABAB neurotransmitter receptors allowing the KCTD C‐terminus to transmit a signal that modulates channels in close proximity.19 The third proposed biochemical mechanisms for KCTD family proteins potentially apply more broadly to the KCTD protein family. The BTB domain of many KCTD family proteins are reported to bind to the cullin‐3 ubiquitin ligase, potentially serving as adaptor molecules to recruit substrates for ubiquitination.5 However, exactly how the KCTD BTB domain of KCTD proteins would fit onto cullin‐3 is a matter of speculation. Furthermore, little is known about the structure or function of most KCTD C‐termini.
Figure 2.
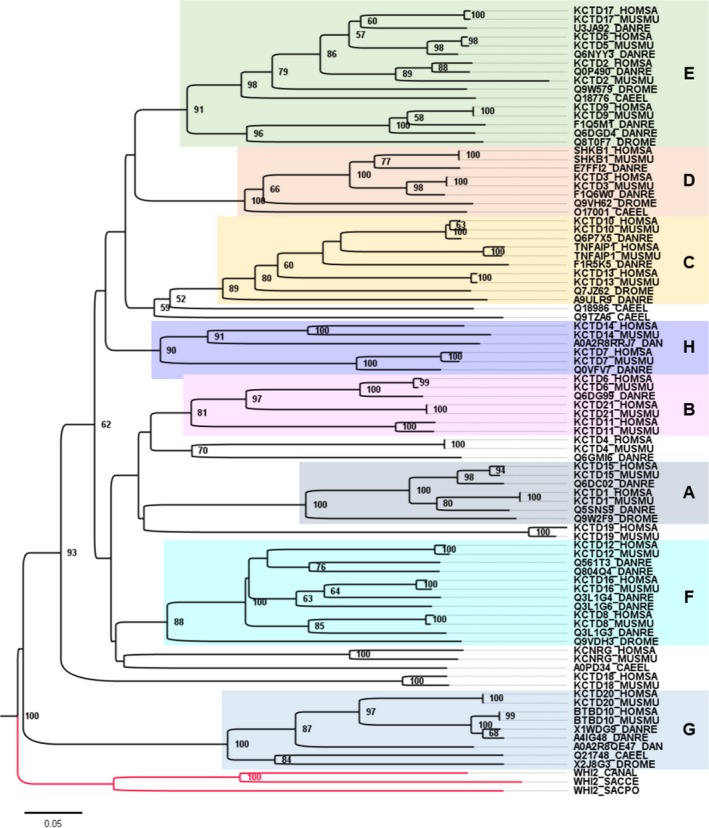
Phylogenetic tree of isolated BTB domains from KCTD family homologs. Amino acid sequences of KCTD family proteins from human (Homo sapiens, HOMSA), mouse (Mus musculus, MUSMU), zebrafish (Danio rerio, DANRE), Drosophila melanogaster (DROME), Caenorhabditis elegans (CAEEL), and three yeast species (Saccharomyces cerevisiae, SACCE; Schizosaccharomyces pombe, SACPO; Candida albicans, CANAL) were collected from UniProt (release 2019_02) or after searches using the DELTA‐BLAST algorithm on the NCBI website. Sequences were aligned using MAFFT (version 7), and a neighbor‐joining (NJ) analysis was performed with 1000 bootstrap replicates. Bootstrap support values above 50 are shown at each node. The tree was rooted using Whi2p from S pombe. Yeast sequences were represented as an outgroup (red branches). The arbitrary cluster designations for groups A‐G were assigned to match those reported by Skoblov et al.1 The new H group is deduced from this analysis. Compared to Skoblov et al,1 we found that KCTD9 segregates within group E. Amino acid sequences (Table S1) and alignment results (Table S2) for this analysis are found in Supporting information
KCTD family proteins were previously classified into seven phylogenetic clades based on the amino acid sequences of the BTB regions alone or of the full‐length proteins.1 Our analysis based on the minimal BTB domains is in agreement with the previous study and suggests the existence of an additional 8th clade that we termed H comprised of KCTD7 and KCTD14 (Figures 1, 2). In addition, based on our analysis, we propose to include the BTB of KCTD9 in the E group. Like the related tetrameric T1/BTB domains of voltage‐gated potassium (Kv) channels, the BTB domains of KCTD10 and KCTD13 are capable of forming tetramers. However, the BTB domains of most KCTD proteins form pentamers (KCTD1, −5, −6, −9, −11, −12, −15, −16, −17) based on crystal structures, cryo‐EM, or other methods (Table 1). The exception is the available structure of SHKBP1‐BTB, which is a monomer.5 The only available full‐length KCTD protein structure reveals a fivefold symmetry extending through the C‐terminus of KCTD5,3 consistent with the pentameric structure of the C‐terminal H1 domain of KCTD12.19
BTB domains are found in other well‐known proteins, such as Skip1, an adaptor for cullin‐1 ubiquitin ligase complexes, and KEAP1, which regulates localization of NRF2 in a redox‐responsive manner.24 The identification of BTB domains from other protein families as cullin‐binding partners by mass spectrometry, including several KCTD family members,25, 26, 27 fuels the search for biological roles for KCTD family proteins as exchangeable adaptors of cullin‐3‐RING E3 ubiquitin ligase complexes (CRLs). In this model, cullin‐3 interacts with the BTB domains of exchangeable adaptor proteins that serve to recruit different protein substrates for ubiquitination by the RBX‐RING protein bound to the C‐terminus of cullin‐3 (Figure 3).27, 28 Thus, KCTD family proteins could function like a multihead screwdriver to recruit different cellular proteins for selective degradation by the proteasome or the lysosome to fine‐tune many cellular processes.27
Figure 3.
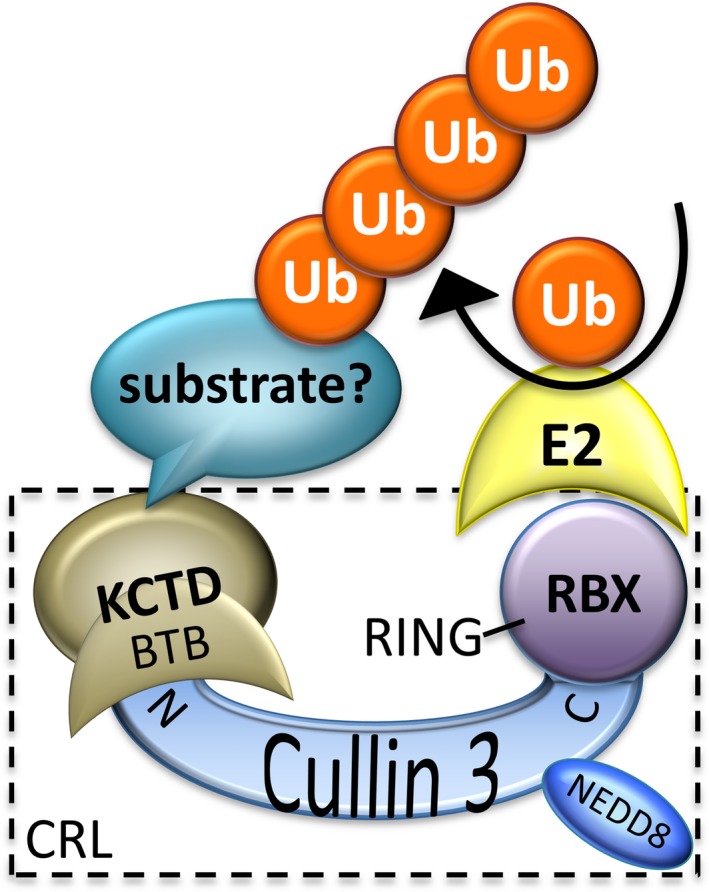
Proposed role for a subset of KCTD family proteins as adaptors for cullin‐3 ubiquitin ligase complexes (CRLs)
Several KCTD family proteins (KCTD2, −5, −6, −10, −11, −13, −17, −21, TNFAIP1) have been reported to interact with cullin‐3 and to mediate ubiquitination and degradation of specific target proteins.11, 12, 18, 29, 30, 31, 32, 33 Cullin‐3 binding to KCTD13, TNFAIP1, and KCTD10 may regulate actin organization and other cell functions by degrading Rho GTPases RhoA or RhoB.31, 33, 34, 35 KCTD6, KCTD11, and KCTD21 can each assemble with cullin‐3 and are reported to suppress hedgehog signaling by degrading Gli deacetylase HDAC1.12, 30 KCTD7, KCTD9, and SHKBP1 are capable of binding cullin‐3, but their substrates have not yet been identified.2, 5, 36 Intriguingly, KCTD5, −9, −13, −17, and SHKBP1 can form 5:5 heterodecameric complexes with cullin‐3 based on biochemical experiments, even though their purified BTB domains may adopt geometries of tetramers or monomers.2, 5, 37 This suggests that cullin‐3 may drive assembly of KCTD protein structures. Validation of this adaptor function by in vitro reconstitution of KCTD‐cullin3‐RBX‐E2 ubiquitination reactions is challenged by the apparent need to identify and include the specific target substrate in these reactions. Detailed biochemistry and structure determinations are also needed to confirm the biological evidence that KCTDs function as cullin‐3 adaptors. Additional binding partners of KCTD proteins also have been identified. KCTD1 and KCTD15 were reported to bind and inhibit the activity of transcription factor AP‐2α.9, 10 KCTD10, KCTD13, and TNFAIP1 were reported to regulate DNA replication by interacting with PCNA (proliferating cell nuclear antigen).6, 8 Whether these KCTD functions involve cullin‐3 or unrelated mechanisms is not yet established.
3. KCTD GENES ASSOCIATED WITH NEURODEVELOPMENTAL AND NEUROPSYCHIATRIC DISORDERS
3.1. KCTD7 mutations cause a severe neurodevelopmental disorder
Our recent genetic analysis confirms that mutations in KCTD7 cause a rare early‐onset, autosomal recessive disorder (progressive myoclonic epilepsy/PME3, also called EPM3).13 Over 50 patients with over 40 unique variants in KCTD7 have been identified to date, though many more likely remain unidentified.13, 38, 39, 40, 41, 42, 43, 44, 45 In all cases, patients have homozygous or compound heterozygous mutations (missense, stop‐gain, frameshifts, or large deletions), while all heterozygous family members are unaffected.13 Thus far, the highest density of patient variants occurs within the N‐terminal BTB domain. A cluster of mutations also occurs in the last 30 residues and in an ~100‐residue middle region, both of unknown function.13 Patients appear to develop normally and achieve early childhood milestones. However, between 10 and 20 months of age these children develop refractory myoclonic seizures, movement disorders, and/or developmental delays.13 This younger age of onset with KCTD7 mutations (average 16 months) distinguishes these patients from other types of progressive myoclonic epilepsies (PME). All patients subsequently progress, exhibiting severe cognitive decline, motor deficits, and seizures.13 Most patients become nonverbal and wheelchair‐bound within 2 years of diagnosis, but the few ambulatory patients now in their teens and early twenties have diagnoses of autism or schizophrenia. This disorder is also designated as neuronal ceroid lipofuscinosis type 14 (CLN14), primarily on the basis of two patients with subcellular inclusions and lysosome storage material.36, 45 However, most studies concluded that the subcellular pathologies of KCTD7 patients are distinct from previously described CLN pathologies and other lysosomal storage disorders.13, 41
Persistent difficulties with diagnosing this disorder have been attributed to earlier onset ages than expected for PME disorders, negative biopsy tests for CLN‐related pathologies, negative brain MRI findings (with a few exceptions), and the fact that KCTD7 was not confirmed as a disease gene until relatively recently. These challenges have been partially overcome by including KCTD7 on diagnostic sequencing panels for epilepsy. However, at least 25% of patients develop movement disorders (ataxia, tremors, dyskinesia, choreoathetosis, dystonia) or regression of milestones before the onset of seizures, though all eventually develop myoclonic epilepsy. Electrical activity in the brain detected by EEG tests is generally positive, and brain biopsies are predicted to be diagnostic based on the prevalence of lipofuscin/lysosome‐like structures in patient brain.13 One patient underwent callosotomy with reported benefit.13 A number of patients with heterozygous KCTD7 mutations and some overlapping neurological symptoms have also been identified, although any causal role for KCTD7 in these cases is unknown.13 Undiagnosed bi‐allelic KCTD7 gene mutations have contributed to the misassignment of disease symptoms to unrelated events such as vaccinations routinely administered around the expected age of disease onset. In addition, the gene name for KCTDs has evoked assumptions that patients could be treated for a channelopathy,46 though currently available evidence does not justify this therapeutic approach.
Although the molecular mechanisms underlying disease in patients with KCTD7 mutations are not known, research efforts have begun to dissect some biological functions. KCTD7 protein expression was reported in hippocampal neurons and Purkinje cells of mouse brain.20, 41 Expressed wild‐type KCTD7 protein in cultured mouse neurons or Xenopus oocytes was reported to hyperpolarize the resting cell membrane potential, and some patient mutations inhibited the K+ flux observed with wild‐type KCTD7.20, 23 How KCTD7 might influence potassium currents is not known but may be indirect as compelling evidence of a direct interaction with K+ channels is currently lacking. Cerebrospinal fluid from some patients was reported to have higher levels of glutamine and lower levels of glutamate, which was suggested to result from impaired regulation of the neuronal glutamine transporter SAT2 by mutant KCTD7.23
Our biochemical studies indicate that KCTD7 protein interacts with cullin‐3,13, 20 raising the possibility that KCTD7 may serve as an adaptor for the cullin‐3 E3 ubiquitin ligase to mediate protein degradation of targeted substrates. However, no KCTD7‐recruited substrate proteins have been identified. One study implied that the C‐terminus of KCTD7 may be involved in binding cullin‐3.36 More recent evidence indicates that the BTB‐containing N‐terminus of KCTD7 is required and sufficient for cullin‐3 interactions based on co‐immunoprecipitation assays and subcellular localization of expressed proteins.13 Furthermore, this interaction with cullin‐3 is partially impaired by BTB domain mutations found in patients (R70W, R84Q, L108M).13 Thus, a role for KCTD7 in proteostasis could conceivably contribute to progressive disease by causing the accumulation of undegraded proteins, correlating with the prevalence of abnormal lysosome‐like structures observed by electron microscopy in neurons of a patient brain biopsy.13 Consistent with these findings, electron microscopy analysis of low‐passage skin fibroblasts derived from two additional KCTD7 patients exhibits abnormal mitochondrial cristae morphologies, lipid droplet accumulation around mitochondria, and phagolysosomes containing partially degraded material, features that were absent from matched control cells.13
One potential mechanism to explain these observations arose from yeast genetic studies. Fungal Whi2 protein sequences and metazoan KCTD family sequences share a homologous BTB domain (IPR011333) with significant sequence similarity (Figures 1, 2).47 It is not known whether both yeast and mammalian KCTD proteins descended from a common ancestral gene or whether they evolved as a result of BTB domain insertion into unrelated ancestral genes (therefore, they share sequence homology but are not referred to as homologs). The yeast WHI2 gene from Saccharomyces cerevisiae was originally discovered when a spontaneous inactivating mutation in WHI2 was identified as the cause for a cell growth phenotype.48, 49 Yeast WHI2 was later rediscovered for similar reasons, because spontaneous WHI2 mutations caused cells to continue growing inappropriately after switching cells to medium with low levels of amino acids.50 This is because Whi2 is required to suppress TORC1 kinase, the master regulator of cellular responses to nutrient status.14, 15, 51 Interestingly, knockdown of Kctd13 in neuro2A cells was reported to increase cell proliferation.52 Whether KCTD13 or KCTD7 regulates TORC1, or whether this involves cullin‐3‐dependent protein degradation is not known. However, given that TORC1 is well known to actively suppress autophagy in yeast and mammals, it is not surprising that whi2 ‐mutant yeast, which have sustained TORC1 activity in low amino acid conditions, fail to induce autophagy.13 Interestingly, KCTD7 patient fibroblasts were found to have defective autophagy induction when starved.13 Consistent with a role for BTB‐containing, cullin‐interacting proteins in autophagy regulation, the BTB‐kelch‐repeat protein KLHL20 regulates autophagy by functioning as a cullin‐3 adaptor to degrade the mTORC1‐inhibited ULK1 protein kinase and the lipid kinase VPS34, both important for early steps of autophagosome formation.53 Similarly, the F‐box protein and associated BTB/POZ protein Skp1 can mediate cullin‐1‐dependent degradation of VPS34 to regulate autophagy.54
Interestingly, a spontaneous whi2 mutation in yeast partially rescues defective mitochondrial respiratory function (petite phenotype) caused by loss of the mitochondrial fission factor Fis1, also conserved in humans.47, 50 By promoting mitochondrial organelle fission, Fis1 is thought to promote turnover of mitochondria by generating small organelles that can be engulfed by autophagosomes.55, 56 Perhaps sustained TORC1 activity in fis1whi2 double mutants helps compensate for mitochondrial insufficiency without Fis1, explaining why most FIS1‐deletion strains develop a secondary WHI2 mutation.50 While the role of Whi2 versus Fis1 in mitochondrial turnover via mitophagy is debated,57, 58 the profound defect in autophagy observed in knockouts lacking Whi2 (yeast KCTD) provided the first clue about the function of human KCTD7 in autophagy. This model is consistent with the prevalence of mitochondria containing defective cristae membrane structures 13 and the altered branching patterns of mitochondrial organelles observed in KCTD7 patient fibroblasts (Figure 4). In the future, animal models will likely be needed to understand the physiological and pathological consequences of Kctd7 deficiency before grasping the organismal and behavioral consequences relevant to human disease mechanisms.
Figure 4.
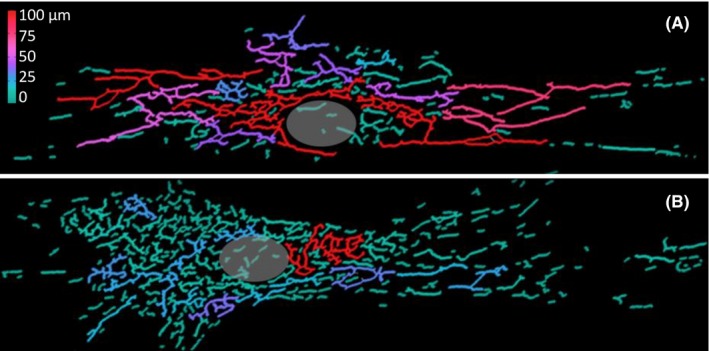
Altered mitochondrial morphology in KCTD7 mutant patient fibroblasts. Primary passage‐matched human fibroblasts from (A) an age‐matched control and (B) a patient with compound heterozygous R84W/D106fs mutations in KCTD7 were confirmed by Sanger sequencing and qRT‐PCR analysis as described.13 To visualize mitochondrial organelles, cells grown on round 12‐mm‐diameter glass coverslips (FisherBrand) were fixed (10 min in cold 4% paraformaldehyde), permeabilized (5 min with 0.2% Triton X‐100) and immunostained 1 h with anti‐Tom20 antibody and Alexa Fluor® secondary antibodies (Santa Cruz), mounted in Prolong Gold, and 0.5 μmol/L Z‐stack images were captured on a Nikon 90i at 40x or 60x magnification using Volocity software for deconvolution. (For quantification, mitochondria in some experiments were labeled instead with 100 nmol/L Mitotracker Red for 15 min prior to fixation.) Double‐blinded images were converted to 8‐bit grayscale, binarized and skeletonized using a custom ImageJ plug‐in, and mitochondrial structure parameters (including length, size, branching, degree of clustering, circularity) were quantified using “Analyze Skeleton 2D/3D” ImageJ plug‐in for 2‐3 independent experiments. The total mitochondrial network per cells was significantly reduced in long‐branch frequency in KCTD7 mutant fibroblast compared to control fibroblast. Individual mitochondrial subnetworks (skeletons) are rainbow colored according to total length (red longest, blue shortest). Position of the nucleus in each cell is marked by a gray circle
3.2. KCTD8, KCTD12, and KCTD16 in neurotransmitter receptor signaling
Mouse Kctd8, Kctd12, Kctd12b (not found in humans), and Kctd16 belong to clade F of the KCTD protein family (Figure 2, Table 1)1 and are considered auxiliary subunits of the inhibitory neurotransmitter receptor complex GABAB1/2 (G‐protein‐coupled receptor/GPCR 3 family) present on both inhibitory and excitatory neurons.17, 59 Supported by studies in GABAB‐deficient mice, GABAB receptor aberrations are implicated in neurodegenerative and neuropsychiatric disorders, including seizure disorders, depression, schizophrenia, addiction, and several neurodevelopmental disorders.59, 60, 61 Therefore, disruption of the auxiliary subunits KCTD8, KCTD12, and KCTD16 may cause related conditions. A mutation in the promoter region of human KCTD12 was reported to contribute to bipolar I disorder.62 Similarly, elevated protein levels of human KCTD12 were associated with depression63 and schizophrenia.64 Consistent with these findings, kctd12 ‐knockout mice exhibit related phenotypes including altered emotional behaviors and increased neuronal excitability,65 supporting a potential role for KCTD12 in neuropsychiatric disorders. KCTD12 has also been implicated in several cancers not discussed here.
Insights into the molecular mechanisms involved are at the forefront of understanding KCTD family protein functions, and recent structure determinations further advance the field overall. The homologous GABAB1 and GABAB2 receptors (GABBR1 and GABBR2) function as heterodimers. GABAB1 binds the inhibitory neurotransmitter gamma‐aminobutyric acid (GABA), and the GABAB2 subunit interacts with G‐proteins for signaling. GABAB1/2 receptors modulate synaptic transmission by indirectly regulating specific Ca2+ and K+ channels through trimeric G‐proteins.66 KCTD8, KCTD12, and KCTD16 can increase the activation rate of GABAB responses, and KCTD12 can cause fast desensitization of GABAB receptor responses.17, 21, 67 A new crystal structure containing the C‐terminus of GABAB2 (amino acid residues 876‐913) reveals how the pentameric BTB domain of KCTD16 enwraps the cytoplasmic tail of the neurotransmitter receptor GABAB2.19 The interaction with KCTD16 and also with KCTD12 is abolished by the BTB mutation Phe80Ala in KCTD16 and Phe87Ala in KCTD12, further validating the crystal structure.19
The same study also connects KCTD proteins with trimeric G‐protein complexes, providing a model for how clade F KCTDs may transmit a signal to regulate potassium flux across the cell membrane. The conserved H1 and H2 homology regions were previously recognized in the C‐terminus of clade F proteins, except H2 is not present in the shorter KCTD12 C‐terminus (Figure 1).21 The KCTD12 H1 region was previously shown to be responsible for desensitization of GABAB receptor responses, whereas H2 domains of KCTD8 and KCTD16 have auto‐inhibitory effects on their H1 region.21 A new crystal structure of KCTD12 H1 bound to Gβ1γ2 reveals an H1 pentamer surrounded by five Gβ1γ2 dimers.19 Taking the evidence together, the proposed model is that a KCTD12 pentamer dangles from the extended cytoplasmic tail of GABAB2, which is anchored in the cell membrane with GABAB1.19 Upon GABAB1 receptor stimulation, KCTD12 expels Gα from the inhibited trimeric G‐protein complex Gαβ1γ2, and membrane‐associated Gβ1γ2 can rapidly activate the associated GIRK (G‐protein‐coupled inwardly rectifying K+ channel). Then, rapid deactivation/desensitization of GIRK channels would subsequently occur when KCTD12 H1 sequesters Gβγ away from these channels.19 A role for KCTD12 in GABAB‐Gβγ signaling to regulate potassium channel activity is not mutually exclusive with a role as a cullin‐3 adaptor (e.g., to degrade Gα), except that the α2β3 loop of KCTD12 is predicted to interfere with cullin‐3 interactions.16 Therefore, any congruency between the cullin‐binding KCTDs and GTPase signaling KCTDs is currently unresolved but may represent independent functions of the same or different KCTD family proteins. However, it is tempting to consider that these functions could be present in the same KCTD protein to coordinate cellular functions.
3.3. KCTD13 association with autism and schizophrenia
Recent genetic studies have revealed copy‐number variations (CNV) in many genes in association with developmental brain disorders, intellectual disability, epilepsy, autism spectrum disorder, and schizophrenia.68 KCTD13 (also known as BACURD1 or POLDIP1) is located in the 16p11.2 locus, which is known to contribute to risk of multiple neuropsychiatric disorders. Deletions of 16p11.2 are associated with epilepsy, autism, and autism spectrum disorder (ASD),69 while 16p11.2 duplications are associated with autism and schizophrenia.70 Interestingly, dosage effects of 16p11.2 appear to affect head size in humans, with deletions observed in macrocephaly and duplication observed in microcephaly.70
Zebrafish and mouse model systems have helped to overcome the major challenge of dissecting the individual contributions to disease of the many genes present 16p11.2 duplications/deletions. Zebrafish have been useful in other studies to investigate human dosage‐sensitive genes and can reflect anatomical phenotypes observed in early human development.71 Therefore, zebrafish were used to dissect human 16p11.2, which encompasses 29 genes that when deleted in humans can confer susceptibility to neurocognitive defects.52, 69 Results from overexpression of each of these 29 genes individually in zebrafish embryos identified a single gene, KCTD13, capable of inducing microcephaly, a phenotype of patients with 16p11.2 duplication.52 Conversely, transient suppression of the orthologous Zebrafish kctd13 locus resulted in the reciprocal macrocephaly phenotype.52, 72, 73 The importance of Kctd13 for cellular proliferation was confirmed in developing mouse brains. In contrast to these studies, others failed to detect increased brain size or increased neurogenesis in mice or zebrafish when the entire Kctd13 locus was deleted in zebrafish or mice.11 The discrepancy between both lines of data may be due to different compensation mechanisms between knockdown approaches and genetic deletion, possible phenotypic differences between Kctd13 knockdown in only a subset of neural progenitors versus complete genetic deletion of the Kctd13 locus, or alternatively, contributions from other genes located in 16p11.2.
Other studies demonstrate that in mouse models, Kctd13 epistatically affects anatomical phenotypes in combination with Mvp (major vault protein) or Lat (linker for activation of T cells), genes, which are also located in 16p11.2 loci52, 74. However, recent studies reported no robust abnormalities in brain structure of mice with genetic ablation of Kctd13, and instead observed sex‐specific differences in brain volume of double heterozygous mice lacking one copy of Kctd13 and one copy of either Lat or Mvp, also located in 16p11.2.75 These results suggest that altered dosage of Kctd13, and Mvp or Lat may have epistatic effects on brain size.
KCTD13 has been reported to function as a cullin‐3 adaptor for ubiquitination and degradation of RhoA, a small GTPase protein that is a key regulator of actin cytoskeleton and plays critical roles in neuronal development and synaptic function.33, 76 Consistently, genetic deletion of the entire Kctd13 gene has resulted in increased RhoA expression, the loss of dendritic spines and reduced synaptic activity in the CA1 region of the hippocampus.11, 76 Reduced synaptic transmission is normalized by pharmacological inhibition of RhoA. These results suggest that KCTD13 may recruit RhoA for modulating its turnover via the cullin‐3 ubiquitin ligase, thereby regulating synaptic function, consistent with spatiotemporal network analysis of brain subregions.34 This Kctd13‐knockout mouse (entire Kctd13 gene deleted) lacked detectable memory deficits.11 However, an independently constructed Kctd13‐deficient mouse with an out‐of‐frame exon 2 deletion (expected to fully ablate Kctd13) exhibited deficits in short‐term recognition memory, but lacked detectable changes in expression levels of RhoA, the candidate KCTD13‐cullin target substrate.75 However, RNA‐seq analyses of gene expression profiles from the cortex and hippocampus of Kctd13 ‐deficient (exon 2‐deleted) mice revealed altered signaling pathways critical for neurodevelopment, including synaptic formation, and both knockout mouse lines exhibited reduced spine density in the hippocampus.11, 75 Thus, further studies are required to understand the mechanistic complexities by which Kctd13 copy number modulates brain development beyond RhoA signaling. It would also be of great interest to investigate how multiple genes in 16p11.2 loci interplay to regulate brain development and contribute to neurodevelopmental abnormalities and psychiatric disorders such as schizophrenia and autism.
3.4. KCTD17 in myoclonus‐dystonia
Myoclonus‐dystonia syndrome (MDS) is a rare movement disorder characterized by nonepileptic spontaneous muscle contractions and dystonia.77 Approximately 25‐50% of myoclonus‐dystonia cases are caused by autosomal dominant mutations in the SGCE gene, coding for ε‐sarcoglycan.78 Thus, there are additional genetic variants responsible for this disease, and several candidate genes have been identified, some of which have been confirmed.79, 80 KCTD17 variant c.434G > A, p.Arg145His was identified by combining genome‐wide linkage analysis and whole‐exome sequencing of a large British pedigree and of a second German family with autosomal dominant myoclonus‐dystonia but lacking SGCE gene mutations.79 Additional tests confirmed the lack of a common ancestor between these two families. KCTD17 (c.434G > A, p.Arg145His) was the only segregating variant among seven candidates from affected myoclonus‐dystonia patients in the British family.79 Very recently, two additional KCTD17 mutations affecting the same splice acceptor site (c.508‐2A > T and c.508‐1G > T) were identified in two independent studies.81, 82 It has been pointed out that the clinical features of the KCTD17 patients are phenotypically distinguishable from MDS due to SGCE mutations.80 However, the evidence is reasonably compelling that KCTD17 mutations are responsible for a subset of myoclonus‐dystonia. Although autosomal dominant KCTD17 mutations cause less severe disease than bi‐allelic KCTD7 mutations discussed in section 3.1, both KCTD7 and KCTD17 disorders have some overlapping clinical features including difficulty swallowing, impaired verbal skills, cognitive impairment, difficulties with fine motor skills, and their disease is progressive, unlike SGCE mutations.
The biological functions of KCTD17 are not yet clear. KCTD17 mRNA was shown to be broadly expressed across the brain but particularly in the putamen, consistent with dystonia being caused by dysfunction of basal ganglia circuits.79 Fibroblasts derived from a KCTD17 patient (p.Arg145His) exhibit defective ER calcium signaling, which is suggested to underlie myoclonus‐dystonia linked to mutations in other genes (e.g., HPCA, CACNA1A, ANO3).79, 83
KCTD17 has also been reported to function as an adaptor of the cullin‐3 ubiquitin ligase to mediate ubiquitination and degradation of trichoplein, which is a negative regulator of ciliogenesis.32 Both neurons and astrocytes contain a primary cilium, and ciliogenesis has been reported to play an important role in brain development.84 This raises the possibility that defective ciliogenesis caused by KCTD17 mutations contributes to the pathology of myoclonus‐dystonia.
The BTB domain of mammalian KCTD17 is most similar in sequence to KCTD2, KCTD5, and KCTD9, which together constitute clade E (Figure 2, Table 1).1 Mammalian KCTD2, KCTD5, and KCTD17 are homologs of Drosophila Insomniac protein (Inc), a regulator of sleep homeostasis and synaptic function in flies.85 Insomniac was reported to be a substrate adaptor of Drosophila cullin‐3 and may regulate turnover of yet unknown neuronal targets to regulate sleep and synaptic functions.85 Both insomniac and its mammalian homologs are expressed in the nervous system and localize to synapses.86 Mouse KCTD2, KCTD5, and KCTD17 can each heteromultimerize with Drosophila Insomniac and also bind to Drosophila cullin‐3 in vitro, suggesting conserved functions.86 Although only mouse KCTD2 and KCTD5, but not KCTD17, were able to rescue the sleep phenotype in flies lacking Insomniac, the inability of KCTD17 to restore sleep in insomniac mutant flies was suggested to be due to its low expression in transgenic flies.86 Drosophila Insomniac and cullin‐3 also regulate dopaminergic signaling.85 Dysfunction of dopaminergic pathways has been associated with myoclonus‐dystonia.87 Currently, the molecular links between KCTD17‐cullin3‐dependent protein degradation, synaptic function, dopaminergic signaling, and pathology of myoclonus‐dystonia remain unclear.
3.5. KCTD3 in neurocognitive disease
KCTD3, also known as NY‐REN‐45, has been identified in several genome‐wide screens for disease variants. A bi‐allelic frameshift mutation in KCTD3 (c.1036_1073del, p.P346Tfs*4) was first identified in one family by whole exon sequencing of 143 multiplex families with neurocognitive disorders.88 This same KCTD3 mutation was later reported in a 2.5‐year‐old patient.89 Homozygous KCTD3 mutations were identified in three additional families, one harboring the same frameshift mutation (c.1036_1073del, p.P346Tfs*4), and the other two harboring a missense mutation (c.166C > T, p.Arg56*).90 KCTD3 patients exhibit global developmental delay, seizures, and cerebellar hypoplasia.88, 90
The biological function of KCTD3 protein has not been investigated in‐depth, but one study provides some evidence that links KCTD3 with the nervous system. Mouse Kctd3 was identified as a binding partner of Hcn3 (hyperpolarization‐activated cyclic nucleotide‐gated channel) in a yeast two‐hybrid screen.91 Immunoprecipitation from mouse brain lysates suggests that Kctd3 specifically binds to Hcn3, but not to the other Hcn channels (Hcn1, Hcn2, and Hcn4).91 Immunostaining confirmed that Kctd3 and Hcn3 colocalize in several brain regions including hypothalamus, midbrain and cerebellum. Kctd3 increases Hcn3 current density by promoting trafficking of Hcn3 protein to the cell membrane.91 Human HCN channels are widely expressed in the brain and are reported to control cellular excitability and synaptic transmission.92, 93 Whether KCTD3 also regulates these neuronal functions is not yet known.
4. RELEVANCE OF OTHER KCTD FAMILY MEMBERS TO THE NERVOUS SYSTEM
4.1. KCTD11 in cancer
KCTD11 has been implicated as a tumor suppressor in several cancers, most notably in medulloblastoma, a primary brain tumor of childhood.94 Suggested mechanisms include KCTD11‐mediated suppression of the hedgehog signaling pathway by interacting with cullin‐3 via the BTB domain of KCTD11, while the KCTD11 C‐terminus recruits the Gli deacetylase HDAC1 for degradation.12 KCTD11 is located on 17p13.1 near TP53. In an in vivo mouse screen to test the effect of haploinsufficiency of TP53‐linked genes, mouse Kctd11 was identified as a tumor suppressor gene.95 However, neither of these findings has been confirmed by follow‐up investigations.
Until very recently, human KCTD11 was annotated in the NCBI and UniProt databases as a 232 amino acid protein with a truncated BTB domain (currently annotated at NCBI as KCTD11s, NP_001002914). However, coding sequencings for the missing N‐terminal segment of the BTB domain are present in‐frame immediately preceding the most 5‐prime ATG start of translation. In vitro studies suggest that KCTD11 is translated from an upstream non‐AUG (AUU) start codon (which may occur more commonly than appreciated), adding 39 amino acids to the N‐terminus of human KCTD11.96 NCBI recently revised the annotation of human KCTD11 as a 271 amino acid protein including a full BTB domain (KCTD11l, NP_001350571). A recent study showed that yeast Whi2 sharing a BTB domain homologous to that of human KCTD proteins is capable of inhibiting TORC1 under low amino acid conditions.14 Remarkably, human KCTD11 but not other KCTD proteins tested (KCTD7, KCTD8, KCTD11, KCTD12, and KCTD16) could suppress TORC1 activity when expressed in whi2 ‐deficient yeast and in mammalian cell lines under low amino acid conditions.14 Furthermore, knockdown of KCTD11 in HEK293 cells confirmed that KCTD11 is required to suppress mTORC1 during amino acid deprivation.14 The detailed molecular mechanism of how KCTD11 regulates mTORC1 activity is still unknown. One speculation is that there is crosstalk between mTORC1 and the hedgehog signaling pathways through KCTD11 in cancer.
4.2. KCTD15 in neural crest formation and obesity
Genome‐wide association studies (GWAS) have identified KCTD15 variants in association with increased risk of obesity.97, 98 Although the detailed molecular mechanisms are not known, several lines of evidence suggest a potential role for KCTD15 in obesity through inhibition of Wnt signaling. KCTD15 was reported to control/limit neural crest formation in zebrafish and frog embryos by attenuating the Wnt‐β‐catenin signaling pathway, as overexpression of KCTD15 decreased neural crest formation while KCTD15 knockdown caused neural crest size to increase.99 Follow‐up studies carried out by the same group showed that in zebrafish embryos and in human cells, KCTD15 directly inhibits the transcription factor AP‐2α, a target of Wnt signaling, consistent with a role for KCTD15 in neural crest development.10 The proposed mechanism is that KCTD15 binds to the proline‐rich activation domain of AP‐2α to prevent transcriptional activation by AP‐2α and that SUMO modification in the C‐terminus of zebrafish Kctd15 on Lys252 (human K278) inhibits the ability of KCTD15 to suppress transcription and inhibit neural crest formation.10, 100 Given that mesenchymal stem cells and some adipocytes are derived from the neural crest,101 and that AP‐2 regulates the expression of genes important for adipogenesis, such as C/EBPα and IRS‐ 1,102, 103 it is conceivable that KCTD15 may regulate adipogenesis by regulating AP‐2 transcription activity in neural crest during development.
5. PERSPECTIVES
Several human KCTD family genes are expressed predominantly in the brain. Genetic alternations in KCTD family members have been associated with neurodevelopmental disorders, epilepsy, autism, schizophrenia, movement disorders, obesity, and several cancers, but little is understood about the biological functions of KCTD family proteins (Table 1). The original expectation that BTB domains of KCTD family members might directly partner with their nearest homologs, the T1/BTB domains of voltage‐gated potassium channels, currently lacks confirmation. However, new evidence supports roles for KCTDs in signaling pathways to indirectly modulate potassium channel activity. KCTDs appear to be involved in other processes, including nutrient sensing and autophagy, based on work in yeast showing that the yeast KCTD‐like protein Whi2 interacts with yeast phosphatases Psr1/Psr2 to regulate TORC1 activity.14 The plethora of effects of KCTD family proteins may reflect an adaptor function of KCTDs that recruits substrates for ubiquitination by cullin‐3 and subsequent degradation, though evidence also supports additional mechanisms. The highly variable C‐terminal regions of KCTD proteins could potentially reflect their roles in recruiting diverse substrates for cullin‐3‐mediated ubiquitination and degradation, though this is only one possibility. A binding site of the heterotrimeric G‐protein subunits Gβγ has been mapped to the C‐terminus of KCTD12 and KCTD16. Though it is not known whether this interaction serves only to desensitize the G‐protein coupled inwardly rectifying potassium channel GIRK or whether KCTD‐mediated protein turnover or other functions are involved. Defects in ubiquitination‐dependent protein function and/or GTPase modulated ion flux may underlie the neurodevelopmental and neuropsychiatric disorders associated with mutations affecting KCTD family proteins. In addition, more animal models are needed to understand pathophysiological roles of KCTDs before comprehending the detailed mechanisms of human disease.
Difficulties in diagnosing children with rare disease mutations in KCTD proteins have contributed to the misassignment of symptoms to other causes. The early‐onset age for autism and several other KCTD‐associated disorders coincide with the timing of childhood vaccinations. Consequently, patient narratives and social media indicate that these families spent years without an explanation for their child's illness. Patients with undiagnosed disease mutations seek explanations from circumstantial evidence. For example, until 2012 there was only a single publication implicating KCTD7 mutations in disease.38 Similarly, a separate cohort of patients with encephalopathies attributed to vaccine reactions were instead due to de novo mutations in SCN1A.104, 105 Thus, from a policy perspective as well as from a therapeutic perspective, new knowledge about KCTD family protein functions is urgently needed.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
Supporting information
Table S1. BTB amino acid sequences used to generate figures 1 and 2.
Table S2. BTB alignment used for the analysis presented in figure 2.
ACKNOWLEDGMENTS
Supported by the National Natural Science Foundation of China 31401197, Jiangsu Key Laboratory of Neuropsychiatric Diseases BM2013003, and the Priority Academic Program Development of the Jiangsu Higher Education Institutes (PAPD) to X.T., by the Fondation ARC and the Ligue Contre le Cancer Comité du Gard to A.A., by the National Institutes of Health USA grants P50MH094268 and R01DA041208 to A.K., R01NS083373 and R01GM077875 to J.M.H., and the Wendy Klag Center for Autism and Developmental Disabilities to J.M.H.
Teng X, Aouacheria A, Lionnard L, et al. KCTD: A new gene family involved in neurodevelopmental and neuropsychiatric disorders. CNS Neurosci Ther. 2019;25:887–902. 10.1111/cns.13156
REFERENCES
- 1. Skoblov M, Marakhonov A, Marakasova E, et al. Protein partners of KCTD proteins provide insights about their functional roles in cell differentiation and vertebrate development. BioEssays. 2013;35(7):586‐596. [DOI] [PubMed] [Google Scholar]
- 2. Ji AX, Chu A, Nielsen TK, Benlekbir S, Rubinstein JL, Prive GG. Structural insights into KCTD protein assembly and cullin3 recognition. J Mol Biol. 2016;428(1):92‐107. [DOI] [PubMed] [Google Scholar]
- 3. Dementieva IS, Tereshko V, McCrossan ZA, et al. Pentameric assembly of potassium channel tetramerization domain‐containing protein 5. J Mol Biol. 2009;387(1):175‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bayón Y, Trinidad AG, de la Puerta ML, et al. KCTD5, a putative substrate adaptor for cullin3 ubiquitin ligases. FEBS J. 2008;275(15):3900‐3910. [DOI] [PubMed] [Google Scholar]
- 5. Pinkas DM, Sanvitale CE, Bufton JC, et al. Structural complexity in the KCTD family of Cullin3‐dependent E3 ubiquitin ligases. Biochem J. 2017;474(22):3747‐3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Wang Y, Zheng YI, Luo F, et al. KCTD10 interacts with proliferating cell nuclear antigen and its down‐regulation could inhibit cell proliferation. J Cell Biochem. 2009;106(3):409‐413. [DOI] [PubMed] [Google Scholar]
- 7. He H, Tan CK, Downey KM, So AG. A tumor necrosis factor alpha‐ and interleukin 6‐inducible protein that interacts with the small subunit of DNA polymerase delta and proliferating cell nuclear antigen. Proc Natl Acad Sci USA. 2001;98(21):11979‐11984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Yang L, Liu N, Hu X, et al. CK2 phosphorylates TNFAIP1 to affect its subcellular localization and interaction with PCNA. Mol Biol Rep. 2010;37(6):2967‐2973. [DOI] [PubMed] [Google Scholar]
- 9. Ding X, Luo C, Zhou J, et al. The interaction of KCTD1 with transcription factor AP‐2alpha inhibits its transactivation. J Cell Biochem. 2009;106(2):285‐295. [DOI] [PubMed] [Google Scholar]
- 10. Zarelli VE, Dawid IB. Inhibition of neural crest formation by Kctd15 involves regulation of transcription factor AP‐2. Proc Natl Acad Sci USA. 2013;110(8):2870‐2875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Escamilla CO, Filonova I, Walker AK, et al. Kctd13 deletion reduces synaptic transmission via increased RhoA. Nature. 2017;551:227‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Canettieri G, Di Marcotullio L, Greco A, et al. Histone deacetylase and Cullin3‐REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat Cell Biol. 2010;12(2):132‐142. [DOI] [PubMed] [Google Scholar]
- 13. Metz KA, Teng X, Coppens I, et al. KCTD7 deficiency defines a distinct neurodegenerative disorder with a conserved autophagy‐lysosome defect. Ann Neurol. 2018;84(5):766‐780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chen X, Wang G, Zhang YU, et al. Whi2 is a conserved negative regulator of TORC1 in response to low amino acids. PLoS Genet. 2018;14(8):e1007592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Teng X, Hardwick JM. Whi2: a new player in amino acid sensing. Curr Genet. 2019;65(3):701‐709. [DOI] [PubMed] [Google Scholar]
- 16. Smaldone G, Pirone L, Balasco N, Di Gaetano S, Pedone EM, Vitagliano L. Cullin 3 recognition is not a universal property among KCTD proteins. PLoS ONE. 2015;10(5):e0126808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Schwenk J, Metz M, Zolles G, et al. Native GABA(B) receptors are heteromultimers with a family of auxiliary subunits. Nature. 2010;465(7295):231‐235. [DOI] [PubMed] [Google Scholar]
- 18. Brockmann M, Blomen VA, Nieuwenhuis J, et al. Genetic wiring maps of single‐cell protein states reveal an off‐switch for GPCR signalling. Nature. 2017;546(7657):307‐311. [DOI] [PubMed] [Google Scholar]
- 19. Zheng S, Abreu N, Levitz J, Kruse AC. Structural basis for KCTD‐mediated rapid desensitization of GABAB signalling. Nature. 2019;567(7746):127‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Azizieh R, Orduz D, Van Bogaert P, et al. Progressive myoclonic epilepsy‐associated gene KCTD7 is a regulator of potassium conductance in neurons. Mol Neurobiol. 2011;44(1):111‐121. [DOI] [PubMed] [Google Scholar]
- 21. Seddik R, Jungblut SP, Silander OK, et al. Opposite effects of KCTD subunit domains on GABA(B) receptor‐mediated desensitization. J Biol Chem. 2012;287(47):39869‐39877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Turecek R, Schwenk J, Fritzius T, et al. Auxiliary GABAB receptor subunits uncouple G protein betagamma subunits from effector channels to induce desensitization. Neuron. 2014;82(5):1032‐1044. [DOI] [PubMed] [Google Scholar]
- 23. Moen MN, Fjaer R, Hamdani EH, et al. Pathogenic variants in KCTD7 perturb neuronal K+ fluxes and glutamine transport. Brain. 2016;139(Pt 12):3109‐3120. [DOI] [PubMed] [Google Scholar]
- 24. Holmstrom KM, Kostov RV, Dinkova‐Kostova AT. The multifaceted role of Nrf2 in mitochondrial function. Curr Opin Toxicol. 2016;1:80‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Zheng N, Schulman BA, Song L, et al. Structure of the Cul1‐Rbx1‐Skp1‐F boxSkp2 SCF ubiquitin ligase complex. Nature. 2002;416(6882):703‐709. [DOI] [PubMed] [Google Scholar]
- 26. Zhuang M, Calabrese MF, Liu J, et al. Structures of SPOP‐substrate complexes: insights into molecular architectures of BTB‐Cul3 ubiquitin ligases. Mol Cell. 2009;36(1):39‐50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Scott DC, Rhee DY, Duda DM, et al. Two distinct types of E3 ligases work in unison to regulate substrate ubiquitylation. Cell. 2016;166(5): 1198‐1214 e1124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Cheng JI, Guo J, Wang Z, et al. Functional analysis of Cullin 3 E3 ligases in tumorigenesis. Biochim Biophys Acta Rev Cancer. 2018;1869(1):11‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kim EJ, Kim SH, Jin X, Jin X, Kim H. KCTD2, an adaptor of Cullin3 E3 ubiquitin ligase, suppresses gliomagenesis by destabilizing c‐Myc. Cell Death Differ. 2017;24(4):649‐659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. De Smaele E, Di Marcotullio L, Moretti M, et al. Identification and characterization of KCASH2 and KCASH3, 2 novel Cullin3 adaptors suppressing histone deacetylase and Hedgehog activity in medulloblastoma. Neoplasia. 2011;13(4):374‐385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kovačević I, Sakaue T, Majoleé J, et al. The Cullin‐3‐Rbx1‐KCTD10 complex controls endothelial barrier function via K63 ubiquitination of RhoB. J Cell Biol. 2018;217(3):1015‐1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kasahara K, Kawakami Y, Kiyono T, et al. Ubiquitin‐proteasome system controls ciliogenesis at the initial step of axoneme extension. Nat Commun. 2014;5:5081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Chen Y, Yang Z, Meng M, et al. Cullin mediates degradation of RhoA through evolutionarily conserved BTB adaptors to control actin cytoskeleton structure and cell movement. Mol Cell. 2009;35(6):841‐855. [DOI] [PubMed] [Google Scholar]
- 34. Lin GN, Corominas R, Lemmens I, et al. Spatiotemporal 16p11.2 protein network implicates cortical late mid‐fetal brain development and KCTD13‐Cul3‐RhoA pathway in psychiatric diseases. Neuron. 2015;85(4):742‐754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Murakami A, Maekawa M, Kawai K, et al. Cullin‐3/KCTD10 E3 complex is essential for Rac1 activation through RhoB degradation in human epidermal growth factor receptor 2‐positive breast cancer cells. Cancer Sci. 2019;110(2):650‐661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Staropoli JF, Karaa A, Lim ET, et al. A Homozygous mutation in KCTD7 links neuronal ceroid lipofuscinosis to the ubiquitin‐proteasome system. Am J Hum Genet. 2012;91(1):202‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Balasco N, Pirone L, Smaldone G, et al. Molecular recognition of Cullin3 by KCTDs: insights from experimental and computational investigations. Biochim Biophys Acta. 2014;1844(7):1289‐1298. [DOI] [PubMed] [Google Scholar]
- 38. Van Bogaert P, Azizieh R, Désir J, et al. Mutation of a potassium channel‐related gene in progressive myoclonic epilepsy. Ann Neurol. 2007;61(6):579‐586. [DOI] [PubMed] [Google Scholar]
- 39. Farhan SM, Murphy LM, Robinson JF, et al. Linkage analysis and exome sequencing identify a novel mutation in KCTD7 in patients with progressive myoclonus epilepsy with ataxia. Epilepsia. 2014;55(9):e106‐111. [DOI] [PubMed] [Google Scholar]
- 40. Krabichler B, Rostasy K, Baumann M, et al. Novel mutation in potassium channel related gene KCTD7 and progressive myoclonic epilepsy. Ann Hum Genet. 2012;76(4):326‐331. [DOI] [PubMed] [Google Scholar]
- 41. Kousi M, Anttila V, Schulz A, et al. Novel mutations consolidate KCTD7 as a progressive myoclonus epilepsy gene. J Med Genet. 2012;49(6):391‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vairo FP, Boczek NJ, Cousin MA, et al. The prevalence of diseases caused by lysosome‐related genes in a cohort of undiagnosed patients. Mol Genet Metab Rep. 2017;13:46‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ebrahimi‐Fakhari D, Hildebrandt C, Davis PE, Rodan LH, Anselm I, Bodamer O. The spectrum of movement disorders in childhood‐onset lysosomal storage diseases. Mov Disord Clin Pract. 2018;5(2):149‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lindy AS, Stosser MB, Butler E, et al. Diagnostic outcomes for genetic testing of 70 genes in 8565 patients with epilepsy and neurodevelopmental disorders. Epilepsia. 2018;59(5):1062‐1071. [DOI] [PubMed] [Google Scholar]
- 45. Mastrangelo M, Sartori S, Simonati A, et al. Progressive myoclonus epilepsy and ceroidolipofuscinosis 14: The multifaceted phenotypic spectrum of KCTD7‐related disorders. Eur J Med Genet. 2018. [DOI] [PubMed] [Google Scholar]
- 46. Oyrer J, Maljevic S, Scheffer IE, Berkovic SF, Petrou S, Reid CA. Ion channels in genetic epilepsy: from genes and mechanisms to disease‐targeted therapies. Pharmacol Rev. 2018;70(1):142‐173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Teng X, Dayhoff‐Brannigan M, Cheng W‐C, et al. Genome‐wide consequences of deleting any single gene. Mol Cell. 2013;52(4):485‐494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sudbery PE, Goodey AR, Carter BL. Genes which control cell proliferation in the yeast Saccharomyces cerevisiae . Nature. 1980;288(5789):401‐404. [DOI] [PubMed] [Google Scholar]
- 49. Saul DJ, Walton EF, Sudbery PE, Carter B. Saccharomyces cerevisiae whi2 mutants in stationary phase retain the properties of exponentially growing cells. J Gen Microbiol. 1985;131:2245‐2251. [Google Scholar]
- 50. Cheng WC, Teng X, Park HK, Tucker CM, Dunham MJ, Hardwick JM. Fis1 deficiency selects for compensatory mutations responsible for cell death and growth control defects. Cell Death Differ. 2008;15(12):1838‐1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Teng X, Yau E, Sing C, Hardwick JM. Whi2 signals low leucine availability to halt yeast growth and cell death. FEMS Yeast Res. 2018;18(8):887‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Golzio C, Willer J, Talkowski ME, et al. KCTD13 is a major driver of mirrored neuroanatomical phenotypes of the 16p11.2 copy number variant. Nature. 2012;485(7398):363‐367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Liu CC, Lin YC, Chen YH, et al. Cul3‐KLHL20 ubiquitin ligase governs the turnover of ULK1 and VPS34 complexes to control autophagy termination. Mol Cell. 2016;61(1):84‐97. [DOI] [PubMed] [Google Scholar]
- 54. Xiao J, Zhang T, Xu D, et al. FBXL20‐mediated Vps34 ubiquitination as a p53 controlled checkpoint in regulating autophagy and receptor degradation. Genes Dev. 2015;29(2):184‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Mao K, Klionsky DJ. Participation of mitochondrial fission during mitophagy. Cell Cycle. 2013;12(19):3131‐3132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Mao K, Klionsky DJ. Mitochondrial fission facilitates mitophagy in Saccharomyces cerevisiae . Autophagy. 2013;9(11):1900‐1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Mendl N, Occhipinti A, Muller M, Wild P, Dikic I, Reichert AS. Mitophagy in yeast is independent of mitochondrial fission and requires the stress response gene WHI2. J Cell Sci. 2011;124(Pt 8):1339‐1350. [DOI] [PubMed] [Google Scholar]
- 58. Mao K, Wang K, Liu X, Klionsky DJ. The scaffold protein Atg11 recruits fission machinery to drive selective mitochondria degradation by autophagy. Dev Cell. 2013;26(1):9‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Gassmann M, Bettler B. Regulation of neuronal GABA(B) receptor functions by subunit composition. Nat Rev Neurosci. 2012;13(6):380‐394. [DOI] [PubMed] [Google Scholar]
- 60. Schuler V, Lüscher C, Blanchet C, et al. Epilepsy, hyperalgesia, impaired memory, and loss of pre‐ and postsynaptic GABA(B) responses in mice lacking GABA(B(1)). Neuron. 2001;31(1):47‐58. [DOI] [PubMed] [Google Scholar]
- 61. Yoo Y, Jung J, Lee YN, et al. GABBR2 mutations determine phenotype in rett syndrome and epileptic encephalopathy. Ann Neurol. 2017;82(3):466‐478. [DOI] [PubMed] [Google Scholar]
- 62. Lee MTM, Chen CH, Lee CS, et al. Genome‐wide association study of bipolar I disorder in the Han Chinese population. Mol Psychiatry. 2011;16(5):548‐556. [DOI] [PubMed] [Google Scholar]
- 63. Sibille E, Wang Y, Joeyen‐Waldorf J, et al. A molecular signature of depression in the amygdala. Am J Psychiatry. 2009;166(9):1011‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Benes FM. Amygdalocortical circuitry in schizophrenia: from circuits to molecules. Neuropsychopharmacology. 2010;35(1):239‐257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Cathomas F, Stegen M, Sigrist H, et al. Altered emotionality and neuronal excitability in mice lacking KCTD12, an auxiliary subunit of GABAB receptors associated with mood disorders. Transl Psychiatry. 2015;5:e510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Chalifoux JR, Carter AG. GABAB receptor modulation of synaptic function. Curr Opin Neurobiol. 2011;21(2):339‐344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Adelfinger L, Turecek R, Ivankova K, et al. GABAB receptor phosphorylation regulates KCTD12‐induced K(+) current desensitization. Biochem Pharmacol. 2014;91(3):369‐379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Michaelson JJ, Shi Y, Gujral M, et al. Whole‐genome sequencing in autism identifies hot spots for de novo germline mutation. Cell. 2012;151(7):1431‐1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Weiss LA, Shen Y, Korn JM, et al. Association between microdeletion and microduplication at 16p11.2 and autism. N Engl J Med. 2008;358(7):667‐675. [DOI] [PubMed] [Google Scholar]
- 70. McCarthy SE, Makarov V, Kirov G, et al. Microduplications of 16p11.2 are associated with schizophrenia. Nat Genet. 2009;41(11):1223‐1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Ceol CJ, Houvras Y, Jane‐Valbuena J, et al. The histone methyltransferase SETDB1 is recurrently amplified in melanoma and accelerates its onset. Nature. 2011;471(7339):513‐517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Walters RG, Jacquemont S, Valsesia A, et al. A new highly penetrant form of obesity due to deletions on chromosome 16p11.2. Nature. 2010;463(7281):671‐675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Kumar RA, KaraMohamed S, Sudi J, et al. Recurrent 16p11.2 microdeletions in autism. Hum Mol Genet. 2008;17(4):628‐638. [DOI] [PubMed] [Google Scholar]
- 74. Loviglio MN, Arbogast T, Jønch AE, et al. The immune signaling adaptor LAT contributes to the neuroanatomical phenotype of 16p11.2 BP2‐BP3 CNVs. Am J Hum Genet. 2017;101(4):564‐577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Arbogast T, Razaz P, Ellegood J, et al. Kctd13‐deficient mice display short‐term memory impairment and sex‐dependent genetic interactions. Hum Mol Genet. 2019;28(9):1474–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Gladwyn‐Ng I, Huang L, Ngo L, et al. Bacurd1/Kctd13 and Bacurd2/Tnfaip1 are interacting partners to Rnd proteins which influence the long‐term positioning and dendritic maturation of cerebral cortical neurons. Neural Dev. 2016;11:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Albanese A, Bhatia K, Bressman SB, et al. Phenomenology and classification of dystonia: a consensus update. Mov Disord. 2013;28(7):863‐873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Zimprich A, Grabowski M, Asmus F, et al. Mutations in the gene encoding epsilon‐sarcoglycan cause myoclonus‐dystonia syndrome. Nat Genet. 2001;29(1):66‐69. [DOI] [PubMed] [Google Scholar]
- 79. Mencacci NE, Rubio‐Agusti I, Zdebik A, et al. A missense mutation in KCTD17 causes autosomal dominant myoclonus‐dystonia. Am J Hum Genet. 2015;96(6):938‐947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Mencacci NE, Bruggemann N. KCTD17 is a confirmed new gene for dystonia, but is it responsible for SGCE‐negative myoclonus‐dystonia? Parkinsonism Relat Disord. 2019;61:887‐3. [DOI] [PubMed] [Google Scholar]
- 81. Graziola F, Stregapede F, Travaglini L, et al. A novel KCTD17 mutation is associated with childhood early‐onset hyperkinetic movement disorder. Parkinsonism Relat Disord. 2019;61:4‐6. [DOI] [PubMed] [Google Scholar]
- 82. Marcé‐Grau A, Correa M, Vanegas MI, et al. Childhood onset progressive myoclonic dystonia due to a de novo KCTD17 splicing mutation. Parkinsonism Relat Disord. 2019;61:7‐9. [DOI] [PubMed] [Google Scholar]
- 83. Domingo A, Erro R, Lohmann K. Novel dystonia genes: Clues on disease mechanisms and the complexities of high‐throughput sequencing. Mov Disord. 2016;31(4):471‐477. [DOI] [PubMed] [Google Scholar]
- 84. Metin C, Pedraza M. Cilia: traffic directors along the road of cortical development. Neuroscientist. 2014;20(5):468‐482. [DOI] [PubMed] [Google Scholar]
- 85. Pfeiffenberger C, Allada R. Cul3 and the BTB adaptor insomniac are key regulators of sleep homeostasis and a dopamine arousal pathway in Drosophila. PLoS Genet. 2012;8(10):e1003003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Li Q, Kellner DA, Hatch HAM, et al. Conserved properties of Drosophila Insomniac link sleep regulation and synaptic function. PLoS Genet. 2017;13(5):e1006815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Karimi M, Perlmutter JS. The role of dopamine and dopaminergic pathways in dystonia: insights from neuroimaging. Tremor Other Hyperkinet Mov (N Y). 2015;5:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Alazami AM, Patel N, Shamseldin HE, et al. Accelerating novel candidate gene discovery in neurogenetic disorders via whole‐exome sequencing of prescreened multiplex consanguineous families. Cell Rep. 2015;10(2):148‐161. [DOI] [PubMed] [Google Scholar]
- 89. Trujillano D, Bertoli‐Avella AM, Kumar Kandaswamy K, et al. Clinical exome sequencing: results from 2819 samples reflecting 1000 families. Eur J Hum Genet. 2017;25(2):176‐182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Faqeih EA, Almannai M, Saleh MM, AlWadei AH, Samman MM, Alkuraya FS. Phenotypic characterization of KCTD3‐related developmental epileptic encephalopathy. Clin Genet. 2018;93(5):1081‐1086. [DOI] [PubMed] [Google Scholar]
- 91. Cao‐Ehlker X, Zong X, Hammelmann V, et al. Up‐regulation of hyperpolarization‐activated cyclic nucleotide‐gated channel 3 (HCN3) by specific interaction with K+ channel tetramerization domain‐containing protein 3 (KCTD3). J Biol Chem. 2013;288(11):7580‐7589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Biel M, Wahl‐Schott C, Michalakis S, Zong X. Hyperpolarization‐activated cation channels: from genes to function. Physiol Rev. 2009;89(3):847‐885. [DOI] [PubMed] [Google Scholar]
- 93. Huang Z, Lujan R, Kadurin I, et al. Presynaptic HCN1 channels regulate Cav3.2 activity and neurotransmission at select cortical synapses. Nat Neurosci. 2011;14(4):478‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Di Marcotullio L, Ferretti E, De Smaele E, et al. REN(KCTD11) is a suppressor of Hedgehog signaling and is deleted in human medulloblastoma. Proc Natl Acad Sci USA. 2004;101(29):10833‐10838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Scuoppo C, Miething C, Lindqvist L, et al. A tumour suppressor network relying on the polyamine‐hypusine axis. Nature. 2012;487(7406):244‐248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Correale S, Pirone L, Di Marcotullio L, et al. Molecular organization of the cullin E3 ligase adaptor KCTD11. Biochimie. 2011;93(4):715‐724. [DOI] [PubMed] [Google Scholar]
- 97. Willer CJ, Speliotes EK, Loos RJ, et al. Six new loci associated with body mass index highlight a neuronal influence on body weight regulation. Nat Genet. 2009;41(1):25‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Baranski TJ, Kraja AT, Fink JL, et al. A high throughput, functional screen of human Body Mass Index GWAS loci using tissue‐specific RNAi Drosophila melanogaster crosses. PLoS Genet. 2018;14(4):e1007222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Dutta S, Dawid IB. Kctd15 inhibits neural crest formation by attenuating Wnt/beta‐catenin signaling output. Development. 2010;137(18):3013‐3018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Zarelli VE, Dawid IB. The BTB‐containing protein Kctd15 is SUMOylated in vivo. PLoS ONE. 2013;8(9):e75016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Billon N, Monteiro MC, Dani C. Developmental origin of adipocytes: new insights into a pending question. Biol Cell. 2008;100(10):563‐575. [DOI] [PubMed] [Google Scholar]
- 102. Jiang MS, Tang QQ, McLenithan J, et al. Derepression of the C/EBPalpha gene during adipogenesis: identification of AP‐2alpha as a repressor. Proc Natl Acad Sci USA. 1998;95(7):3467‐3471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Meng X, Kondo M, Morino K, et al. Transcription factor AP‐2beta: a negative regulator of IRS‐1 gene expression. Biochem Biophys Res Commun. 2010;392(4):526‐532. [DOI] [PubMed] [Google Scholar]
- 104. Novy J, Catarino CB, Chinthapalli K, et al. Another cause of vaccine encephalopathy: a case of Angelman syndrome. Eur J Med Genet. 2012;55(5):338‐341. [DOI] [PubMed] [Google Scholar]
- 105. Zamponi N, Passamonti C, Petrelli C, et al. Vaccination and occurrence of seizures in SCN1A mutation‐positive patients: a multicenter Italian study. Pediatr Neurol. 2014;50(3):228‐232. [DOI] [PubMed] [Google Scholar]
- 106. Inaba H, Goto H, Kasahara K, et al. Ndel1 suppresses ciliogenesis in proliferating cells by regulating the trichoplein‐Aurora A pathway. J Cell Biol. 2016;212(4):409‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Smaldone G, Pirone L, Pedone E, Marlovits T, Vitagliano L, Ciccarelli L. The BTB domains of the potassium channel tetramerization domain proteins prevalently assume pentameric states. FEBS Lett. 2016;590(11):1663‐1671. [DOI] [PubMed] [Google Scholar]
- 108. Vlasenko DO, Novosylna OV, Negrutskii BS, El'skaya AV. Truncation of the A, A(*), A' helices segment impairs the actin bundling activity of mammalian eEF1A1. FEBS Lett. 2015;589(11):1187‐1193. [DOI] [PubMed] [Google Scholar]
- 109. Pirone L, Correale S, de Paola I, et al. Design, synthesis and characterization of a peptide able to bind proteins of the KCTD family: implications for KCTD‐cullin 3 recognition. J Pept Sci. 2011;17(5):373‐376. [DOI] [PubMed] [Google Scholar]
- 110. Traylor M, Adib‐Samii P, Harold D, et al. Shared genetic contribution to ischaemic stroke and Alzheimer's Disease. Ann Neurol. 2016;79(5):739‐747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Boada M, Antúnez C, Ramírez‐Lorca R, et al. ATP5H/KCTD2 locus is associated with Alzheimer's disease risk. Mol Psychiatry. 2014;19(6):682‐687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Feng L, Wang JT, Jin H, Qian K, Geng JG. SH3KBP1‐binding protein 1 prevents epidermal growth factor receptor degradation by the interruption of c‐Cbl‐CIN85 complex. Cell Biochem Funct. 2011;29(7):589‐596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Borinstein SC, Hyatt MA, Sykes VW, et al. SETA is a multifunctional adapter protein with three SH3 domains that binds Grb2, Cbl, and the novel SB1 proteins. Cell Signal. 2000;12(11–12):769‐779. [DOI] [PubMed] [Google Scholar]
- 114. Cancer Genome Atlas Research N , Albert Einstein College of M , Analytical Biological S , et al. Integrated genomic and molecular characterization of cervical cancer. Nature. 2017;543(7645):378‐384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Greif PA, Eck SH, Konstandin NP, et al. Identification of recurring tumor‐specific somatic mutations in acute myeloid leukemia by transcriptome sequencing. Leukemia. 2011;25(5):821‐827. [DOI] [PubMed] [Google Scholar]
- 116. Darmanis S, Cui T, Drobin K, et al. Identification of candidate serum proteins for classifying well‐differentiated small intestinal neuroendocrine tumors. PLoS ONE. 2013;8(11):e81712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Nagai T, Mukoyama S, Kagiwada H, Goshima N, Mizuno K. Cullin‐3‐KCTD10‐mediated CEP97 degradation promotes primary cilium formation. J Cell Sci. 2018;131(24):jcs219527. [DOI] [PubMed] [Google Scholar]
- 118. Hu X, Yan F, Wang F, et al. TNFAIP1 interacts with KCTD10 to promote the degradation of KCTD10 proteins and inhibit the transcriptional activities of NF‐kappaB and AP‐1. Mol Biol Rep. 2012;39(11):9911‐9919. [DOI] [PubMed] [Google Scholar]
- 119. Kubota D, Yoshida A, Tsuda H, et al. Gene expression network analysis of ETV1 reveals KCTD10 as a novel prognostic biomarker in gastrointestinal stromal tumor. PLoS ONE. 2013;8(8):e73896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Kim DM, Chung KS, Choi SJ, et al. RhoB induces apoptosis via direct interaction with TNFAIP1 in HeLa cells. Int J Cancer. 2009;125(11):2520‐2527. [DOI] [PubMed] [Google Scholar]
- 121. Cui RI, Meng W, Sun H‐L, et al. MicroRNA‐224 promotes tumor progression in nonsmall cell lung cancer. Proc Natl Acad Sci USA. 2015;112(31):E4288‐4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Grinchuk OV, Motakis E, Kuznetsov VA. Complex sense‐antisense architecture of TNFAIP1/POLDIP2 on 17q11.2 represents a novel transcriptional structural‐functional gene module involved in breast cancer progression. BMC Genom. 2010;11(Suppl 1):S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Zhang CL, Wang C, Yan WJ, Gao R, Li YH, Zhou XH. Knockdown of TNFAIP1 inhibits growth and induces apoptosis in osteosarcoma cells through inhibition of the nuclear factor‐kappaB pathway. Oncol Rep. 2014;32(3):1149‐1155. [DOI] [PubMed] [Google Scholar]
- 124. Degenhardt F, Heinemann B, Strohmaier J, et al. Identification of rare variants in KCTD13 at the schizophrenia risk locus 16p11.2. Psychiatr Genet. 2016;26(6):293‐296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Blumkin L, Kivity S, Lev D, et al. A compound heterozygous missense mutation and a large deletion in the KCTD7 gene presenting as an opsoclonus‐myoclonus ataxia‐like syndrome. J Neurol. 2012;259:2590‐2598. [DOI] [PubMed] [Google Scholar]
- 126. Heride C, Rigden DJ, Bertsoulaki E, et al. The centrosomal deubiquitylase USP21 regulates Gli1 transcriptional activity and stability. J Cell Sci. 2016;129(21):4001‐4013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lange S, Perera S, Teh P, Chen J. Obscurin and KCTD6 regulate cullin‐dependent small ankyrin‐1 (sAnk1.5) protein turnover. Mol Biol Cell. 2012;23(13):2490‐2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Al‐Mubarak B, Abouelhoda M, Omar A, et al. Whole exome sequencing reveals inherited and de novo variants in autism spectrum disorder: a trio study from Saudi families. Sci Rep. 2017;7(1):5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Zazzeroni F, Nicosia D, Tessitore A, et al. KCTD11 tumor suppressor gene expression is reduced in prostate adenocarcinoma. Biomed Res Int. 2014;2014:380398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Tong R, Yang B, Xiao H, et al. KCTD11 inhibits growth and metastasis of hepatocellular carcinoma through activating Hippo signaling. Oncotarget. 2017;8(23):37717‐37729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Li X, Chen C, Wang F, et al. KCTD1 suppresses canonical Wnt signaling pathway by enhancing beta‐catenin degradation. PLoS ONE. 2014;9(4):e94343. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 132. Kumar S, Rathkolb B, Sabrautzki S, et al. Standardized, systemic phenotypic analysis reveals kidney dysfunction as main alteration of Kctd1 (I27N) mutant mice. J Biomed Sci. 2017;24(1):57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. Marneros AG, Beck AE, Turner EH, et al. Mutations in KCTD1 cause scalp‐ear‐nipple syndrome. Am J Hum Genet. 2013;92(4):621‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134. Zhong Y, Yang J, Xu WW, et al. KCTD12 promotes tumorigenesis by facilitating CDC25B/CDK1/Aurora A‐dependent G2/M transition. Oncogene. 2017;36(44):6177‐6189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Ivankova K, Turecek R, Fritzius T, et al. Up‐regulation of GABA(B) receptor signaling by constitutive assembly with the K+ channel tetramerization domain‐containing protein 12 (KCTD12). J Biol Chem. 2013;288(34):24848‐24856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Fritzius T, Turecek R, Seddik R, et al. KCTD hetero‐oligomers confer unique kinetic properties on hippocampal GABAB receptor‐induced K+ currents. J Neurosci. 2017;37(5):1162‐1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137. Abbaszadegan MR, Taghehchian N, Li L, Aarabi A, Moghbeli M. Contribution of KCTD12 to esophageal squamous cell carcinoma. BMC Cancer. 2018;18(1):853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138. Hasegawa T, Asanuma H, Ogino J, et al. Use of potassium channel tetramerization domain‐containing 12 as a biomarker for diagnosis and prognosis of gastrointestinal stromal tumor. Hum Pathol. 2013;44(7):1271‐1277. [DOI] [PubMed] [Google Scholar]
- 139. Usman H, Mathew MK. Potassium channel regulator KCNRG regulates surface expression of Shaker‐type potassium channels. Biochem Biophys Res Commun. 2010;391(3):1301‐1305. [DOI] [PubMed] [Google Scholar]
- 140. Ivanov DV, Tyazhelova TV, Lemonnier L, et al. A new human gene KCNRG encoding potassium channel regulating protein is a cancer suppressor gene candidate located in 13q14.3. FEBS Lett. 2003;539(1–3):156‐160. [DOI] [PubMed] [Google Scholar]
- 141. Tyybakinoja A, Vilpo J, Knuutila S. High‐resolution oligonucleotide array‐CGH pinpoints genes involved in cryptic losses in chronic lymphocytic leukemia. Cytogenet Genome Res. 2007;118(1):8‐12. [DOI] [PubMed] [Google Scholar]
- 142. Birerdinc A, Nohelty E, Marakhonov A, et al. Pro‐apoptotic and antiproliferative activity of human KCNRG, a putative tumor suppressor in 13q14 region. Tumour Biol. 2010;31(1):33‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Usui D, Shimada S, Shimojima K, et al. Interstitial duplication of 2q32.1‐q33.3 in a patient with epilepsy, developmental delay, and autistic behavior. Am J Med Genet A. 2013;161A(5):1078‐1084. [DOI] [PubMed] [Google Scholar]
- 144. Pichler I, Schwienbacher C, Zanon A, et al. Fine‐mapping of restless legs locus 4 (RLS4) identifies a haplotype over the SPATS2L and KCTD18 genes. J Mol Neurosci. 2013;49(3):600‐605. [DOI] [PubMed] [Google Scholar]
- 145. Nawa M, Matsuoka M. KCTD20, a relative of BTBD10, is a positive regulator of Akt. BMC Biochem. 2013;14:27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Zhang X, Zhou H, Cai L, et al. Kctd20 promotes the development of non‐small cell lung cancer through activating Fak/AKT pathway and predicts poor overall survival of patients. Mol Carcinog. 2017;56(9):2058‐2065. [DOI] [PubMed] [Google Scholar]
- 147. Rich SS, Goodarzi MO, Palmer ND, et al. A genome‐wide association scan for acute insulin response to glucose in Hispanic‐Americans: the Insulin Resistance Atherosclerosis Family Study (IRAS FS). Diabetologia. 2009;52(7):1326‐1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Nawa M, Kanekura K, Hashimoto Y, Aiso S, Matsuoka M. A novel Akt/PKB‐interacting protein promotes cell adhesion and inhibits familial amyotrophic lateral sclerosis‐linked mutant SOD1‐induced neuronal death via inhibition of PP2A‐mediated dephosphorylation of Akt/PKB. Cell Signal. 2008;20(3):493‐505. [DOI] [PubMed] [Google Scholar]
- 149. Nawa M, Kage‐Nakadai E, Aiso S, Okamoto K, Mitani S, Matsuoka M. Reduced expression of BTBD10, an Akt activator, leads to motor neuron death. Cell Death Differ. 2012;19(8):1398‐1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150. Zheng M, Zhu H, Gong Y, et al. Involvement of GMRP1, a novel mediator of Akt pathway, in brain damage after intracerebral hemorrhage. Int J Clin Exp Pathol. 2013;6(2):224‐229. [PMC free article] [PubMed] [Google Scholar]
- 151. Furuta N, Makioka K, Fujita Y, et al. Reduced expression of BTBD10 in anterior horn cells with Golgi fragmentation and pTDP‐43‐positive inclusions in patients with sporadic amyotrophic lateral sclerosis. Neuropathology. 2013;33(4):397‐404. [DOI] [PubMed] [Google Scholar]
- 152. Harata K, Nishiuchi T, Kubo Y. Colletotrichum orbiculare WHI2, a yeast stress‐response regulator homolog, controls the biotrophic stage of hemibiotrophic infection through TOR signaling. Mol Plant Microbe Interact. 2016;29(6):468‐483. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. BTB amino acid sequences used to generate figures 1 and 2.
Table S2. BTB alignment used for the analysis presented in figure 2.