

Abstract
Protein biogenesis and quality control are essential to maintaining a functional pool of proteins and involve numerous protein factors that dynamically and transiently interact with each other and with the substrate proteins in living cells. Conventional methods are hardly effective for studying dynamic, transient, and weak protein–protein interactions that occur in cells. Herein, we review how the site‐directed photocrosslinking approach, which relies on the genetic incorporation of a photoreactive unnatural amino acid into a protein of interest at selected individual amino acid residue positions and the covalent trapping of the interacting proteins upon ultraviolent irradiation, has become a highly efficient way to explore the aspects of protein contacts in living cells. For example, in the past decade, this approach has allowed the profiling of the in vivo substrate proteins of chaperones or proteases under both physiologically optimal and stressful (e.g., acidic) conditions, mapping residues located at protein interfaces, identifying new protein factors involved in the biogenesis of membrane proteins, trapping transiently formed protein complexes, and snapshotting different structural states of a protein. We anticipate that the site‐directed photocrosslinking approach will play a fundamental role in dissecting the detailed mechanisms of protein biogenesis, quality control, and dynamics in the future.
Keywords: membrane protein biogenesis, protein quality control, molecular chaperones, proteases, in vivo protein photocrosslinking, dynamics of proteins
Introduction
Biogenesis and quality control of proteins concern about how a functional pool of proteins are produced and maintained in living cells. A nascent polypeptide, after being synthesized from the ribosomes located in the cytosol, has to be targeted (delivered), often via translocation across membranes, to a proper subcellular location before it eventually folds/assembles into its native structure and becomes biologically functional.1, 2 During their biogenesis, the nascent proteins are often strictly monitored in cells by a network of quality control protein factors, including molecular chaperones, folding catalysts, and proteases. They either facilitate the proteins to fold/assemble by preventing the aggregation of the partially folded metastable intermediates or degrade the dramatically misfolded forms.3, 4 This trip of the nascent proteins involves many dynamic interactions either between the nascent polypeptides and the participating protein factors or between the latter proteins themselves. It would be highly desirable and important, although difficult, to explore this dynamic process as occurring in living cells, rather than under in vitro conditions or by genetic analysis. Understanding the mechanisms of such biogenesis and quality control of proteins in cells is not only biologically important but also clinically relevant, as protein misfolding has often been considered to account for a number of human disorders, such as the Alzheimer's and Parkinson's diseases, Type II diabetes, and a number of other so‐called folding diseases.3, 4, 5
Individual proteins are assumed to be in constant motion, that is, the coordinates of all the atoms of a protein are under constant time‐dependent fluctuations or, to put it in another way, a protein populates a large ensemble of many different conformations.6, 7 This concept has been perceived based on theoretical analysis8, 9, 10 and in vitro structural analysis, mainly via room temperature X‐ray crystallography and nuclear magnetic resonance (NMR) spectroscopy.11, 12, 13, 14 Nevertheless, the dynamic aspect of proteins has been far less studied in living cells, only with some preliminary in‐cell NMR spectroscopy analysis.15, 16
In this review, we do not intend to present a comprehensive discussion on the biogenesis, quality control, and dynamics of proteins. Rather, we will concentrate on more recent advances on how unnatural amino acid‐mediated photocrosslinking studies in living cells have advanced our understanding in these aspects. In the past decade, we and others have applied this approach to dissect the dynamic and transient interactions between the quality control factors and their substrate proteins or between the different quality control factors themselves, to map the interface residues involved in these interactions, or to capture and identify new proteins involved in protein biogenesis, as occurring in living cells under normal or stress conditions.
Site‐Directed Photocrosslinking Analysis Is a Powerful Technique to Explore Protein–Protein Interactions in Living Cells
The conventional chemical crosslinking of proteins in living cells
The conventional covalent crosslinking, via chemical agents containing at least two reactive (functional) groups, has been a powerful technique for studying protein–protein interactions in living cells.17 In this regard, proteins interacting through noncovalent interactions can be linked together by covalent bonds before being analyzed by electrophoresis, immunoblotting, mass spectrometry (after being purified), and other techniques. Although such crosslinking is able to capture weak and transient interactions, it apparently suffers from some limitations such as relying on the presence of pairs of proper active groups at the interface of two interacting proteins (no crosslinking if lacking them), crosslinking without protein selectivity, and cytotoxicity resulted from the uncaged reactivity of the agents.
Site‐directed photocrosslinking in living cells is mediated by genetically introduced unnatural amino acids
This technique has been mainly developed by Peter Schultz and his colleagues over the last two decades or so. The first photoreactive unnatural amino acid applied for this purpose was p‐benzoyl‐l‐phenylalanine (pBpa). This amino acid was initially developed for site‐specific incorporation into synthetic peptides, which could be covalently photocrosslinked to a target protein it specifically binds.18 It was then incorporated into specific residue positions of proteins by using the in vitro transcription/translation system19 before being eventually introduced into target proteins at the selected residue positions in living cells.20
Briefly speaking, the latest version of this methodology, as illustrated in Figure 1(a), relies on a specific orthogonal aminoacyl‐tRNA synthetase/tRNA pair. The orthogonal aminoacyl‐tRNA synthetase specifically recognizes the unnatural amino acid (but not any of the 20 natural amino acids) and adds it onto the orthogonal tRNA molecule (having the 5′ CUA 3′ anticodon), which in turn carries the unnatural amino acid to the ribosomes to incorporate the unnatural amino acid onto the target protein at a residue position where the original codon in the target gene was replaced by the amber codon TAG in the encoding gene, thus a 5′ UAG 3′ anticodon in the mRNA molecule. The method was first established in bacterial cells20 and more recently expanded to eukaryotic cells.23
Figure 1.
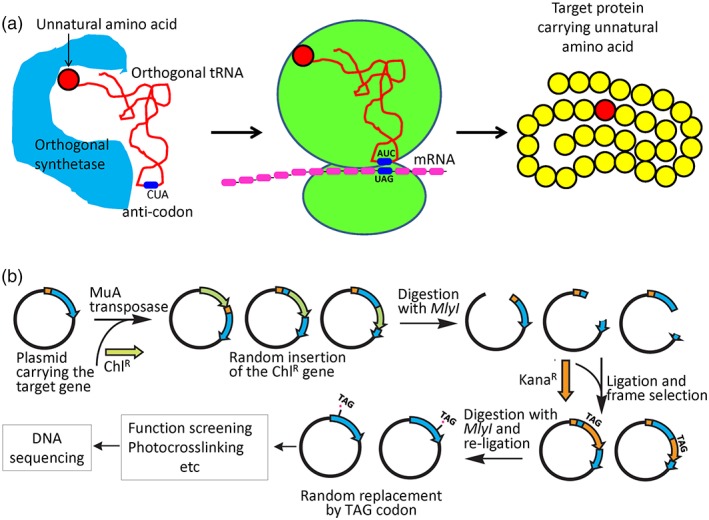
The general procedure for site‐specific (a) or random (b) incorporation of unnatural amino acid into a selected target protein in living cells. (a) The unnatural amino acid (the red ball) enters the cell that produces an orthogonal aminoacyl‐tRNA synthetase and an orthogonal tRNA (being usually expressed from a plasmid, but is expressed from the modified genome of the LY928 bacterial strain21) and is covalently linked to the orthogonal tRNA (carrying 5′ CUA 3′ as the anticodon) upon the catalysis of the orthogonal aminoacyl tRNA synthetase (left). The synthesized aminoacyl tRNA is then delivered to the ribosome to pair with the amber codon (5′ UAG 3′) on the mRNA transcribed from the genetically modified target gene (by site‐directed mutagenesis) that is usually carried by a plasmid (middle). A variant target protein in which an unnatural amino acid is incorporated at a selected position is produced (right). (b) Briefly,22 the chloramphenicol‐resistant gene (chlR) was randomly inserted into the target gene (usually carried by an expression plasmid) with the help of the MuA transposase. The plasmids were then digested with the restriction enzyme MlyI, which removes the chlR gene fragment by generating two blunt ends. Such linear plasmids were subsequently ligated with a kanamycin‐resistant gene (kanaR) carrying a TAG codon (the amber codon for unnatural amino acid incorporation) at one end, digested again with the MlyI restriction enzyme before being religated to form sealed plasmids. These manipulations randomly replace each codon of the target gene by a TAG amber codon (as indicated by the red asterisk), generating a library of plasmids each expressing a variant target protein having an unnatural amino acid introduced at a different residue position. This library was then applied to functional screening and photocrosslinking analysis. The nature of a variant (i.e., which residue is replaced by an unnatural amino acid) that forms photocrosslinked products is eventually characterized by a simple determination of its encoding DNA sequence.
The unnatural amino acid can be randomly introduced into a target protein
The conventional method for site‐directed photocrosslinking relies on the replacement of a specific codon on the target gene (carried either by a plasmid or by the genomic DNA), that encodes a unique amino acid residue, by the amber stop codon TAG via site‐directed mutagenesis. This is rather tedious and inefficient when many or all residue positions of a target protein need to be individually replaced. To overcome this, methods have been developed, as diagrammed in Figure 1(b), which allows each of the codons in a target gene to be randomly replaced by a TAG codon, thus generating a library of variants each incorporating an unnatural amino acid at a different residue position.22, 24 From such a library, we may first select the variants that are able to complement (i.e., to replace) the function of the wild‐type protein in the cells before examining whether these functional variants are able to form photocrosslinked products with itself or with other (prey) proteins. Using this approach, the exact residue position where the unnatural amino acid was incorporated for mediating a specific protein–protein interaction could be simply revealed at the end by determining the DNA sequence of the variant target gene.22 This strategy is somehow similar to that of directed evolution, where mutant enzymes of desired properties are selected from a random gene library, and the nature of the mutant enzyme could be similarly revealed by a DNA sequencing step at the very end.25, 26
Multiple unnatural amino acids carrying different photoreactive groups have been developed
One set of these unnatural amino acids have been developed, roughly as analogues of the phenylalanine and pyrrolysine amino acids [Fig. 2(a), left two panels], for the purpose of replacing residues at specifically selected positions of a target protein. For the incorporation of each of these unnatural amino acids, a specific pair of orthogonal aminoacyl‐tRNA synthetase and tRNA molecules have to be developed.27, 28, 29, 30, 31, 32, 36 The other set (e.g., photo‐Ile, photo‐Leu, photo‐Met, and photo‐Lys) have been developed for respectively replacing all the Ile, Leu, Met, and Lys residues nonspecifically in the target protein as well as in all other proteins [Fig. 2(a), right panel] in the living cells.27, 28 These unnatural amino acids, introduced into target proteins specifically or nonspecifically, could then be activated by irradiating the living cells with ultraviolet (UV) light, forming the highly reactive free radicals, which would react with a nearby hydrogen‐containing group from the interacting (prey) protein, as illustrated in Figure 2(b). These covalently crosslinked protein complexes could then be further characterized and identified via such analyses as immunoblotting and mass spectrometry, unveiling the nature of the proteins that interact with the target protein in living cells, under different environmental conditions.
Figure 2.
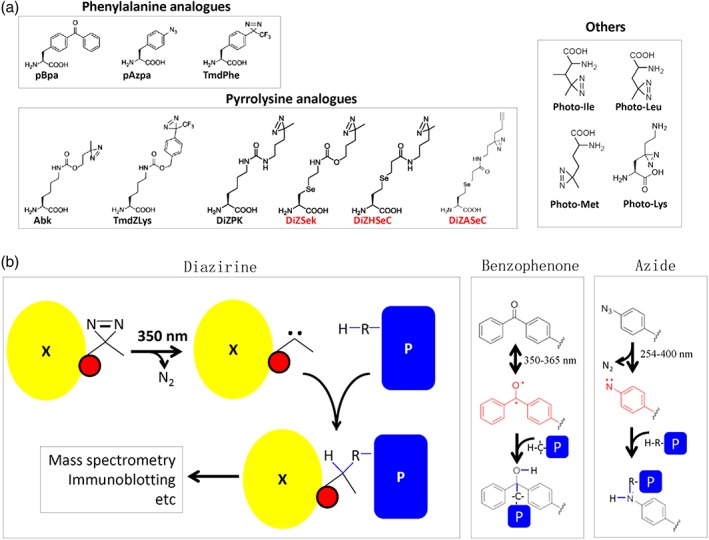
Chemical structures (a) and photocrosslinking mechanisms (b) of the common unnatural amino acids. (a) The common photoreactive unnatural amino acids that are designed mainly as analogues of phenylalanine (top) or pyrrolysine (bottom) are applied for site‐specific incorporations. The others (photo‐Leu, photo‐Ile, photo‐Met, and photo‐Lys) are used for nonspecific incorporations (right).27, 28 pBpa, p‐benzoyl‐l‐phenylalanine20; pAzpa, p‐azido‐l‐phenylalanine29; TmdPhe, 4′‐[3‐(trifluoromethyl)‐3H‐diazirin‐3‐yl]‐l‐phenylalanine30; Abk, 3′‐azibutyl‐N‐carbamoyllysine31; TmdZLys, N ε‐[((4‐(3‐(trifluoromethyl)‐3H‐diazirin‐3‐yl)‐benzyl)oxy)carbonyl]‐l‐lysine10; DiZPK, 3‐(3‐methyl‐3H‐diazirin‐3‐yl)‐propaminocarbonyl‐N ε‐l‐lysine32; DiZSek, N ε‐3‐(3‐methyl‐3H‐diazirin‐3‐yl)‐propaminocarbonyl‐γ‐seleno‐l‐lysine33; DiZHSeC, Se‐(N‐(3‐(3‐methyl‐3H‐diazirin‐3‐yl)propyl)propanamide)‐3‐ylhomoselenocysteine;34 and DiZASeC.35 Note that DiZSek, DiZHSeC, and DiZASeC (colored red) are all cleavable photocrosslinkers. (b) Shown here are the three common photoactivation mechanisms. A diazirine group incorporated into the target protein (X) is activated upon ultraviolet irradiation at 350 nm to form a carbine, which would be immediately covalently bonded with a neighboring XH group of the interacting protein (P), leading to an intermolecular covalent crosslinkage (left). The benzophenone will be activated to form a diradical (middle), while the azide group will be activated to form a nitrene (right) before covalently linked to an XH group nearby.
There are mainly three types of photoreactive groups in these unnatural amino acids. They are, as shown in Figure 2, the benzophenone (consisting of a carbonyl group linked to two benzene groups), the diazirine (consisting of a carbon bound to two nitrogen atoms) and the azide (consisting of the anion with the formula N−3). These photoreactive groups are not found in any molecules present in the cell per se and are kept inert without UV exposure. They will become highly reactive only after an irradiation with UV light [Fig. 2(b)]. The free radicals thus produced would effectively react with a nearby XH group (X being C, N, O, etc.). The diazirine could be activated upon exposure to the 350 nm UV light, producing a carbene (containing a neutral carbon atom and two unshared electrons) with a loss of N2. The carbenes are highly reactive and can insert into any neighboring C—H or heteroatom—H bond to form a covalent adduct [Fig. 2(b), left]. The benzophenone group can also be activated by UV light (350–365 nm) to produce a diradical (i.e., containing two radical centers), which can react with neighboring C—H bonds to form a covalent adduct [as shown in Fig. 2(b), middle]. The azide group could be activated by a light of broader wavelength (250–400 nm), depending on the nature of phenyl ring substitution, to form reactive nitrenes, accompanied by a loss of N2. Nitrenes are able to insert into neighboring C—H and heteroatom—H bonds, forming a new covalent adduct [as shown in Fig. 2(b), right]. The photocrosslinked products, with or without purification, can then be analyzed by mass spectrometry, immunoblotting, and other methods [Fig. 2(b), left].
The site‐directed photocrosslinking method exhibits multiple advantages in studying protein–protein interactions in living cells
The most remarkable advantage of this method is that it is based on a free radical reaction, which is largely independent of such cellular conditions as salt concentration, pH, and temperature.32, 37 In contrast, the conventional techniques for detecting protein–protein interactions, such as coimmunoprecipitation, yeast two‐hybrid system, fluorescence resonance energy transfer (FRET), and chemical crosslinking are highly affected, and thus are often made hardly feasible under these harsh conditions.38 This unique characteristic enables unnatural amino acid‐mediated photocrosslinking to be an efficient method for capturing protein–protein interactions in living cells grown either under normal or stressful conditions. For instance, via such site‐directed photocrosslinking analysis, we successfully examined the interacting substrate proteins of HdeA, a chaperone for acid resistance in bacteria, when the cells are placed in an extremely acidic (pH < 3) environment that mimicks the human stomach.32 None of the aforementioned conventional methods would be applicable under such a harsh condition.38
Due to the site‐specific nature of such crosslinking analysis, the amino acid residues of the target (bait) protein that are involved in a specific protein–protein interaction can be directly mapped out. In this regard, the unnatural amino acid‐incorporated variant protein often largely maintains the native structure of the wild‐type protein and remains functional, unless the unnatural amino acid replaces a residue critical for the function and folding of the target protein. Furthermore, amino acid residues of the interacting (prey) protein that are covalently linked via the unnatural amino acid residue to the target (bait) protein might be identified using a recently developed so‐called cleavable unnatural amino acid photocrosslinker, which carries a label that will be transferred to the crosslinking amino acid residue on the prey protein. This would allow the peptide containing the transferred label to be identified by mass spectrometry analysis (by measuring its exact molecular mass).34 This new approach may allow the interface of two interacting (bait and prey) proteins to be characterized at the residue resolution without relying on any beforehand‐supply of structural information of the interacting proteins. This would be particularly useful in characterizing the transient protein–protein interactions between the protein quality control factors and the not‐yet folded nascent or unfolded mature client proteins.
In addition, unnatural amino acid photocrosslinkers can be simultaneously introduced at different sites in one target protein or into different proteins that are coexpressed, which would allow transiently formed ternary protein complexes to be elegantly captured in living cells, as demonstrated in one of our recent work.21 Due to the short distance nature of such free radical reactions, false‐positive crosslinking reactions are much less frequent compared with the conventional methods.
Limitations of unnatural amino acid‐mediated photocrosslinking analysis
Similar to all other methods developed for exploring protein–protein interactions, the unnatural amino acid‐mediated photocrosslinking method has its own limitations. One of them is that, usually, the unnatural amino acid has to be introduced at the interface of two interacting proteins, otherwise crosslinking would not occur. In many cases, it is unknown what residues are located at the interface, thus many replacements (theoretically at all the residues) have to be individually tested before a conclusion can be made on whether two proteins interact or whether a target protein interacts with any other proteins. This would make the analysis tedious and time consuming. In addition, the incorporation of the unnatural amino acid may interfere with the folding, structure, protein expression, and/or biological functions of the target protein, making the subsequent analysis infeasible and unreliable. Furthermore, so far this technique has been effectively applied mainly to bacterial or cultured eukaryotic cells. But it remains ineffective on multicellular organisms, mainly due to the difficulties in having the unnatural amino acid to efficiently enter the host cells.
Site‐Directed Photocrosslinking Helps to Unveil the Molecular Events Occurring During the Biogenesis of Outer Membrane Proteins in Living Bacterial Cells
Biogenesis of outer membrane β‐barrel proteins involves a long and complex trip in bacterial cells
Outer membrane β‐barrel proteins (OMPs) are a type of proteins present not only in Gram‐negative bacteria but also in the mitochondria and chloroplasts of eukaryotes. They exhibit important functions in a variety of biological processes and are structurally characterized by adopting a cylindrical barrel‐like topology such that its N‐terminal and C‐terminal β‐strands, far from each other on the polypeptide chain, have to eventually pair with each other in forming the sealed barrel conformation.39 Their biogenesis in bacteria involves the following steps: after being synthesized by the ribosomes located in the cytosol, the nascent polypeptides have to be transported across the inner membrane (IM) and the periplasm before finally folded and inserted into the outer membrane40 (refer to Fig. 3). This probably represents one of the most complicated pathways in protein targeting.1, 2
Figure 3.
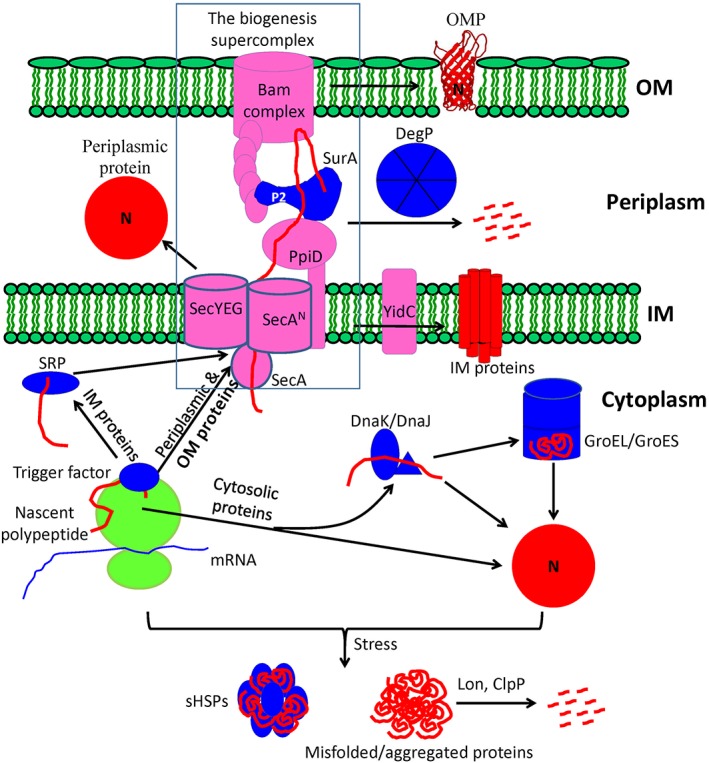
Major pathways of the biogenesis and quality control of proteins existing in Gram‐negative bacteria. They could be categorized into three major pathways according to the final subcellular destinations of the proteins. The first pathway deals with the secretory proteins. Their nascent polypeptides, after being synthesized by the cytosolic ribosomes, are destined to the protein‐conducting channels (the SecYEG or SecAN complexes) on the inner membrane (IM) for translocation, whose driving force is apparently provided by the SecA motor. Among the secretory proteins, the periplasmic proteins fold into their native conformations (N) after being translocated across the IM, while the outer membrane β‐barrel proteins (OMPs) (with the outer membrane lipoproteins being omitted for simplicity) are protected by such chaperones as SurA and delivered to the β‐barrel assembly machinery (BAM) complex for the final folding and integration into the outer membrane. A supercomplex for the biogenesis of OMPs seems to be formed in living cells (as shown with a dashed frame). The second pathway deals with the IM (i.e., cytoplasmic membrane) proteins. Their nascent polypeptides are probably also delivered to the SecYEG channel as guided by the signal recognition particle before being inserted into the IM with the help of the insertase YidC. The third pathway deals with the cytosolic proteins. Their nascent polypeptides fold into their native conformations, either cotranslationally under the assistance of such chaperones as the trigger factors and/or DnaK/DnaJ, or posttranslationally within the “Anfinsen” cage formed by the GroEL/GroES complex. Upon stress (e.g., heat shock), the unfolded proteins tend to aggregate unless they are protected by chaperones such as small heat shock proteins or degraded by proteases such as Lon/ClpP. Primary protein factors and complexes involved in protein biogenesis are diagrammed, mainly by referring to earlier review papers40, 41, 42 and our recent studies.21, 43
Numerous protein factors have been identified as participants of OMP biogenesis and quality control
Many protein factors have been revealed over the past few decades, mostly via genetic studies and partially via in vitro biochemical analysis, as participants of OMP biogenesis.41, 44 They mainly include the following: the SecA motor that may drive nascent chains of OMPs across the inner (i.e., cytoplasmic) membrane40, 41; the SecB that seems to function as a dedicated chaperone for the nascent polypeptides of the secretary proteins40, 41; the SecYEG complex that may function as the protein‐conducting channel; the chaperones such as SurA, PpiD, FkpA, and Skp that may facilitate the nascent OMP polypeptides to go through the hydrophilic periplasm40, 44; and the β‐barrel assembly machinery (BAM) complex composed of subunits BamA, BamB, BamC, BamD, and BamE that may mediate the β‐barrel structure formation and membrane insertion of OMPs at the outer membrane.40, 45 In addition, the potentially cytotoxic misfolded OMPs are removed by proteases such as DegP and DegQ.46, 47
Accordingly, the biogenesis process of OMPs relies on the dynamic and transient interactions between the aforementioned or other not‐yet identified protein factors and the nascent OMPs, as well as those between these quality control protein factors themselves. These interactions, though have been extensively studied via genetic, biophysical, biochemical, and structural approaches, had rarely been directly examined in living cells despite their utmost importance. This was apparently due to the lack of proper methodologies. In the past decade, site‐directed photocrosslinking mediated by genetically introduced unnatural amino acids has been effectively applied to probe such protein–protein interactions in living cells (as summarized in Table 1). These studies have greatly advanced our understanding of OMP biogenesis, whose complex nature, in comparison with that of cytosolic soluble proteins, makes living cell studies not only desired but indispensable.
Table 1.
Interactions of Protein Factors Involved in the Biogenesis and Quality Control of Proteins in Bacteria Revealed by Site‐Directed In Vivo Photocrosslinking Analysis
| Proteins | Assumed biological functions | Interacting proteins detected | References |
|---|---|---|---|
| SecA | ATPase motor that drives the translocation of secretory proteins across the membrane | SecY, SecG, and SecA (self‐interaction) and substrate proteins | 48, 49, 50, 51, 52 |
| SecY | A cytoplasmic membrane translocon essential for protein secretion | SecA, FtsY, YidC, SecF, and SecY (self‐interaction) and substrate proteins | 53, 54, 55, 56, 57, 58 |
| SurA/PpiD/Skp | Periplasmic chaperones assisting the transportation of nascent OMPs | SurA with PpiD, BamA, or substrate proteins; Skp with substrate proteins | 21, 59, 60 |
| BAM | A machinery essential for the β‐barrel formation and membrane integration of OMPs | BamA with SurA; BAM with substrate proteins | 21, 59, 60, 61, 62 |
| DegP | A protease quality control factor involved in OMP biogenesis and acid stress | HdeA, SurA, FkpA, and DegP (self‐assembly) and substrate proteins | 32, 35, 63, 64, 65 |
| HdeA/HdeB | chaperone for acid resistance | HdeA with DegP, SurA, and HdeA; HdeA/HdeB with substrate proteins | 32, 33, 34, 66 |
| Trigger factora | A chaperone assisting the folding of nascent polypeptides | nascent polypeptides | 67, 68, 69 |
| Hsp70/DnaKb | A critical chaperone assisting the folding of nascent polypeptides | substrate proteins | 70 |
| IbpB | A small heat shock protein involved in inclusion body formation | IbpB and substrate proteins | 37, 71 |
| Hsp18.1c | A small heat shock protein involved in protein refolding | model substrate proteins | 72 |
| GroEL/GroES, Hsp90d | Classic molecular chaperones for protein folding/refolding and assembly | No reports | |
| ClpP/Lond | Proteases degrading misfolded/aggregated proteins | No reports | |
| DsbA/DsbCd | Protein‐folding catalysts for disulfide bond formation | No reports | |
| Hsp33d | A critical chaperone for oxidative stress | No reports |
Explored by an in vitro translation system rather than in living cells.
The interaction of Hsp70 with substrate proteins in yeast cells rather than the interaction of DnaK with substrate proteins in bacteria was demonstrated.
Explored by purified bait proteins carrying unnatural amino acid under in vitro conditions.
Exhaustive literature search in the PubMed database reveals that these proteins have not been examined via site‐directed in vivo photocrosslinking analysis.
Site‐directed photocrosslinking analyses in living cells revealed new aspects of OMP biogenesis
The first such study was reported by Ito and Mori, who introduced the unnatural amino acid pBpa53 at a number of selected residue positions in the SecY protein. This allowed them to observe apparently two modes of interactions between the membrane‐integrated SecY and the cytosolic SecA proteins, with one region of SecY interacting with SecA in a continuous manner, while another region interacting with SecA only when a nascent polypeptide is being translocated.53 Oliver and Das then reciprocally introduced pBpa into SecA (rather than in SecY) and revealed that each molecule of the SecA protein seems to interact with two molecules of SecY, and also demonstrated a direct interaction between SecA and the SecY‐associated SecG.48 In addition, the SecA protein was confirmed to form dimers in living cells by such site‐directed photocrosslinking studies.49, 50
Recently, mainly via site‐directed photocrosslinking analysis in living cells, we revealed a shortened version of the SecA protein that consists of only the N‐terminal part of the SecA protein (thus designated as SecAN) that apparently function specifically as a protein‐conducting channel for nascent OMPs, but not for nascent periplasmic proteins.43 We initially conducted this study in an attempt to clarify whether or not nascent polypeptides of OMPs and periplasmic proteins, whose structures are dramatically different, are both translocated across the inner membrane through the SecYEG translocon, as commonly believed, although never directly demonstrated in living cells. However, our site‐directed in vivo photocrosslinking analysis failed to reveal any direct interaction between nascent OMPs and SecY, although it did reveal a direct interaction between nascent periplasmic proteins and SecY. We then accidentally observed that nascent OMPs, but not nascent periplasmic proteins, directly interact with this hitherto unreported shortened version of SecA, in living cells. We further characterized SecAN as follows.43 First, SecAN is almost exclusively present in the membrane fraction and apparently exists as homo‐oligomers. Second, bioinformatic analysis revealed that SecAN contains one putative transmembrane domain with a GXXXG motif known for mediating protein–protein interactions in biomembranes.73 Third, the processing of precursors of OMPs was severely retarded in cells producing assembly‐defective SecAN variants containing mutations in the GXXXG motif. Last but not least, the SecAN protein directly interacts with the BAM complex in living cells. It was only through such in vivo photocrosslinking analysis that this SecAN form was unveiled as a functional protein in the bacterial cells.
Detail interaction patterns between components of the protein translocon are revealed
Oliver and colleagues further utilized this strategy to thoroughly map the SecY regions (with pBpa individually introduced at 117 residue sites of SecY) interacting with SecA and surprisingly found that the SecA motor contacts most of the channel transmembrane helices and even the periplasmic regions of SecY,54 suggesting that SecA seems to be able to span the cytoplasmic membrane, conceivably via inserting into the heart of the SecY channel. Such a presumably dynamic interaction pattern would be difficult to demonstrate with any technique other than such in vivo photocrosslinking analysis. In addition, via this approach, the interaction of SecY with the signal recognition particle receptor FtsY55 or with the inner membrane protein insertase YidC,56 as well as the self‐dimerization of SecY,57 has been demonstrated to occur in living cells.
Intriguingly, when the unnatural amino acid pBpa was incorporated into OmpF, a typical OMP, at 21 randomly selected residue positions along the polypeptide, we detected no formation of photocrosslinked OmpF–SecY products in living cells.21, 43 Our study, though being not exhaustive, somehow challenges the current view that the SecYEG channel is responsible for the translocation of nascent OMPs across the cytoplasmic membrane. Instead, the newly identified membrane‐integrated SecAN protein seems to function as the channel for delivering nascent OMPs across the cytoplasmic membrane.43 In this regard, whether the SecAN oligomer translocates nascent OMPs by acting alone or by interacting with the SecYEG channel in living cells awaits further clarification.
Detail interactions among OMP nascent polypeptides, periplasmic chaperones, and the BAM complex components are analyzed in living cells
In the periplasm (the subcellular location between the inner and outer membranes), the nascent OMPs are believed to be protected by chaperones such as SurA, FkpA, Skp, and PpiD before being folded and inserted into the outer membrane of the Gram‐negative bacteria with the help of the BAM complex.21, 40, 74 Using site‐directed photocrosslinking, Ieva and Bernstein found that residues closer to the extracellular domain of the autotransporter EspP (an OMP) interact with SurA and Skp during its biogenesis.59 We recently demonstrated a unique pattern for the interaction between the nascent OMPs and SurA, with residues in the N‐terminal and C‐terminal regions, but not in their middle regions, both of OmpF and LamB (two OMPs), interacting with SurA,21 raising a possibility that SurA participates in sealing the β‐barrel structure during the biogenesis of OMPs. We also found that SurA interacts with nascent OMP substrates largely via its N‐terminal domain, while it interacts with the BamA protein mainly via a unique extended satellite P2 domain (as shown in Fig. 3). In addition, another inner membrane‐associated chaperone, PpiD, was also found to directly interact with the N‐terminal domain of SurA. Importantly, we demonstrated that the SurA–PpiD and SurA–BamA interactions, unlike the SurA–OmpF interaction, are barely affected when cellular protein synthesis was suppressed (by adding the antibiotic chloramphenicol), indicating that SurA interacts with OMPs as nascent substrates during their biogenesis but interacts with PpiD and BamA as a mature functional partner.21
The BAM complex, which is located partially on the outer membrane and partially in the periplasm, is known to be essential for OMP biogenesis. Despite the determination of its 3D structure,75, 76 the exact role of the BAM complex in living cells remains elusive. Site‐directed photocrosslinking analysis revealed an interaction of the discrete regions of the β‐barrel domain of nascent EspP (an OMP) with BamA, BamB, and BamD of the BAM complex,60 indicating that the BAM complex plays a direct role in the integration of nascent OMPs into the outer membrane. Further studies demonstrated that the BAM complex catalyzes the membrane integration of OMPs apparently in a multistep manner.61 In addition, by introducing pBpa into BamA and performing photocrosslinking analysis, we identified a few residues in its periplasmic POTRA‐2 domain that mediate an interaction with the SurA chaperone in living cells21 (refer to Fig. 3).
The fact that the SurA chaperone interacts with nascent OMPs largely via its N‐terminal domain but with BamA mainly via its satellite P2 domain guided us to design a dual photocrosslinking strategy to find out whether a transient OmpF–SurA–BamA complex is formed in living cells. For this, we first tried to simultaneously introduce the unnatural amino acid pBpa into two sites, respectively, located in the N‐terminal and P2 domains of SurA for capturing the potentially formed ternary OmpF–SurA–BamA complex, but we failed to capture it, probably due to the low expression level of the variant SurA protein carrying pBpa at two sites. We then tried to introduce pBpa into both OmpF (at a site known to interact with SurA) and BamA (also at a site known to interact with SurA) and successfully detected a formation of the OmpF–SurA–BamA ternary complex in living cells.21 This study for the first time demonstrated the formation of such a transient ternary complex in the cells during protein biogenesis, highlighting the power of the site‐directed photocrosslinking method. These observations, in combination with other genetic and biochemical studies, suggest the formation of a supercomplex, in which the components of SecYEG, BAM complex, SurA, and PpiD are assembled as an entity spanning the inner and outer membranes, thus functionally integrate the multiple discrete steps of OMP biogenesis, including translocation across the inner membrane, transportation through the periplasm, folding, and insertion into the outer membrane, all into one continuous and protected pathway21 (as diagrammed in Fig. 3).
The DegP protein primarily functions as a quality control protease for OMP biogenesis in living cells
In addition to the protein quality control factors mentioned above, we also systematically examined the role of the DegP protein in the quality control of OMP biogenesis by applying the site‐directed photocrosslinking analysis. After revealing an unusual activation mechanism of DegP, that is, by changing its oligomeric status,77 we then analyzed how DegP interacts with its substrate proteins in living cells by introducing pBpa into a protease‐deficient mutant DegP form (i.e., the DegP‐S210A variant, in which the critical serine residue at position 210 was replaced by an alanine residue) at multiple residue positions. We demonstrated that OMPs are the major natural substrate proteins of DegP in living cells while periplasmic proteins were only occasionally detected under both normal temperature and heat shock conditions.63 Furthermore, we demonstrated that DegP primarily functions as a protease, at both low and high temperatures (DegP was found to be essential for the bacterial cells to grow at high temperatures), to eliminate misfolded OMPs, with hardly any appreciable chaperone activity in living cells47, 63 (as illustrated in Fig. 3). Chen and colleagues developed a multifunctional photocrosslinker DiZASeC [Fig. 2(a)] that bears a bio‐orthogonal alkyne moiety (an unsaturated hydrocarbon containing one carbon–carbon triple bond) in addition to the diazirine photoactive group and a releasable linker based on selenium (Se).35 This photocrosslinker allows researchers to specifically enrich low‐abundance interacting proteins, for example, the substrate proteins of a fully active rather than of a protease‐deficient form of DegP. Taking advantage of this, they profiled the natural substrate proteins of the fully protease‐active DegP in cells and confirmed OMPs as the major substrate proteins,35 consistent with what we reported earlier.63
Unveiling the Protein Quality Control Mechanism in Bacterial Cells Living under Extremely Acidic Conditions
The extremely acidic human stomach juice (pH <3) serves as a natural barrier against many enteric bacteria, including pathogenic species, but some of them (e.g., the disease‐causing Escherichia coli strains O157:H7 and O104:H4) are able to survive for a few hours under such harsh conditions.78 Several anti‐acid mechanisms involving direct export and/or consumption of excessive protons are adopted by enteric bacteria to maintain the cytoplasmic pH at above 4.5 and thus effectively protect the cytosolic proteins.78
In contrast, proteins residing in the ATP‐deficient periplasm of Gram‐negative bacteria are more vulnerable to the extreme acidity of the stomach juice, primarily due to the highly permeable nature of their outer membranes. Although genetic analyses and in vitro biochemical studies have uncovered the critical role of the periplasmic chaperones HdeA and HdeB,78, 79, 80, 81 and the proteases DegP and DegQ64 for bacterial acid resistance by us and others, it is difficult to unveil how they bind to natural substrate proteins and what cellular proteins are protected in cells living under acidic conditions. For instance, our in vitro studies revealed that the HdeA protein exhibits chaperone activity only when its structure becomes largely disordered,80 but it was difficult to further explore how HdeA functions under such acidic conditions in living cells, mainly because the conventional techniques for studying protein–protein interactions are hardly applicable under such extremely acidic conditions.38
The functional mechanism of the HdeA chaperone is effectively explored in living cells
We applied site‐directed photocrosslinking analysis to probe protein–protein interactions in living cells that are placed under extremely acidic conditions. During this study, we developed a new orthogonal aminoacyl‐tRNA synthase/tRNA pair system for the incorporation of a new unnatural amino acid photocrosslinker, DiZPK, which is more flexible and more efficient than the commonly used unnatural amino acid pBpa [refer to Fig. 2(a)]. This enabled us to identify a total of 32 substrate proteins that bind to HdeA chaperone in living bacterial cells exposed to the acid stress condition (pH being <3), among which are two important quality control factors DegP and SurA.32 Both DegP and SurA have been known as quality control factors for the synthesis of OMPs40, 44 but neither has been known to play any role in resisting acid stress. We then further demonstrated that DegP and SurA assist the refolding or removal of the acid‐denatured substrate proteins after the bacterial cells are returned to neutral pH conditions.32, 78
In a following study, Chen and colleagues developed a selenium‐based cleavable unnatural amino acid photocrosslinker [DiZSek; refer to Fig. 2(a)] that allows the separation of the bait target (which initially carries the DiZSek) and the prey (interacting) proteins after the covalently crosslinked protein products (containing both the bait and the prey) were purified.33 This allows the captured prey proteins to be more efficiently identified by mass spectrometry analysis, eliminating the interference of the bait protein during this analysis. Recently, these authors applied this unnatural amino acid photocrosslinker in combination with the two‐dimensional (2D) difference gel electrophoresis technique to systematically profile the substrate proteins of HdeA and HdeB in living cells exposed to a wide range of acid stress conditions (of different pH values) or upon recovery to different pH conditions.66 As such, distinct substrate specificities between the HdeA and HdeB chaperones have been uncovered, apparently reflecting a fine‐tuned protein quality control strategy for bacterial cells to cope with the acid stress.
Furthermore, Chen and coworkers examined the interaction between HdeA and DegP in bacterial cells exposed to acidic conditions34 by applying another cleavable unnatural amino acid photocrosslinker, DiZHSeC [refer to Fig. 2(a)], which introduces an N‐(4, 4‐bis‐substituted‐pentyl)acrylamide moiety as a label (tag) on the prey protein, allowing the interaction surface on the prey protein to be identified with high confidence by mass spectrometry analysis. These analyses revealed that, interestingly, DegP (as the prey protein) uses its protease domain and PDZ1 domain, but not its PDZ2 domain, to directly contact the substrate‐binding regions of the HdeA chaperone (the bait protein) under acidic conditions. Apparently, this application represents a great advantage in comparison with the conventional unnatural amino acid photocrosslinkers and would be able to provide a residue‐by‐residue resolution dissection of the interfaces of two interacting proteins in living cells, without relying on any prior structural information of the two proteins.
The functional mechanism of DegP in acid resistance has been explored in living cells
Recently, we applied the site‐directed photocrosslinking method to capture the interacting substrate proteins of the DegP protease when the cells were placed under acidic conditions, as well as after returning to neutral pH conditions. This was performed after we demonstrated that the deletion of the degP gene dramatically impaired the viability of the E. coli cells against acid stress.64 Among the 13 DiZPK‐incorporated variants of DegP that we examined, the two variants having DiZPK [refer to Fig. 2(a)] introduced at the protease domain were highly efficient in forming photocrosslinked products with other proteins in living cells placed under either neutral or acidic pH conditions, reflecting the critical role of this domain in binding substrate proteins.
Interestingly, we observed that DegP interacts with HdeA and SurA only in cells placed in an acidic (pH being at 2.3) medium, but not in cells placed at or returned to neutral pH conditions.64 In addition to OMPs (e.g., OmpC and OmpF), several periplasmic proteins (e.g., OppA, MalE, and PhoA) were also found to be photocrosslinked with DegP in cells placed under acidic conditions and/or upon recovery to neutral pH condition, suggesting that the periplasmic proteins are also potential substrate proteins of the DegP protease in living cells exposed to acidic conditions. In a parallel study, Chen and coworkers combined the site‐directed photocrosslinking with 2D comparative proteomics analyses and demonstrated that DegP directly bind to diverse aggregation‐prone periplasmic proteins upon acid stress, and these premixed DegP‐substrate coaggregates are subsequently digested by the recovered DegP upon returning to neutral conditions.65 These data indicate that DegP represents an unprecedented protease that can survive the acid stress condition (by being protected by the acid chaperone HdeA32) and subsequently to function in maintaining the homeostasis of other proteins by effectively removing the acid‐denatured misfolded proteins, which otherwise might be toxic to cells, under recovered neutral pH conditions.47, 63, 64, 65
Certain Aspects of the Dynamic Structure of Proteins in Living Cells can be Effectively Probed via Site‐Directed Photocrosslinking
Protein structural dynamics are difficult to be explored in living cells
The dynamic structures have been believed to be central to protein functioning.6, 7 These aspects of proteins have been assessed under in vitro conditions using biophysical methods such as NMR spectroscopy,14, 82, 83 room temperature X‐ray crystallography,12, 13, 84 FRET,85 and hydrogen–deuterium exchange mass spectrometry.86 Nevertheless, it is far more technically challenging to study protein structural dynamics in living cells, partially due to the relatively small amount of a particular target protein that exists in a mixture of thousands of other cellular proteins. Since unnatural amino acids carrying a rationally designed functional group, as exemplified by a photoreactive group,20 a fluorescence probe,87 an infrared group,88 or a group for NMR detection,89 can be site‐specifically incorporated into a specific target protein, it is expected that unnatural amino acids may serve as an ideal tool for studying protein structural dynamics in living cells. Here, we summarize a few examples regarding the application of site‐directed photocrosslinking in exploring this biologically important but technically challenging issue.
The HdeA chaperone exhibits a dynamic structure in living cells
First, Chen and coworkers examined the pH‐dependent conformational change of the anti‐acid chaperone HdeA in living cells by fully exploiting the potential of the cleavable unnatural amino acid photocrosslinker DiZHSeC [refer to Fig. 2(a)], which allows an effective identification of the crosslinked peptide on the interacting (prey) proteins using mass spectrometry analysis.34 They found that the DiZHSeC replacing phenylalanine 35 in HdeA was able to form intramolecular crosslinking with tryptophan 82 in the peptide sequence 78‐VKGEWDK‐84 located at the C terminus of HdeA in living cells placed under neutral pH conditions, being consistent with the 3D structure of HdeA that was determined under in vitro neutral conditions.79, 90 However, this crosslinkage became no longer detectable in acid‐exposed cells (pH < 3), supporting a structural model such that acid treatment would trigger an opening of the C‐terminal region in HdeA. Furthermore, they observed that the crosslinking site (for DiZHSeC introduced at position phenylalanine 35) on the peptide 11‐KPVNSWTCEDFLAVDESFQPTAVGFAEALNNK‐42 is glutamate (E) 37 at pH 7 but more crosslinking sites are involved upon acid treatment,34 indicating a order‐to‐disorder transition within this region when the environmental pH drops from 7 to 2, in line with what we observed under in vitro conditions.80
Subunit interactions mediated by “noninterface” forbidden residues in living cells suggest a conformation far more dynamic than expected
Protein–protein interaction is usually assumed to occur through a specific interface that consists of a limited number of amino acid residues of the two interacting protein subunits. However, our systematic in vivo photocrosslinking analysis via genetically incorporated unnatural amino acids unexpectedly demonstrated that, under neutral pH conditions, the dimerization of HdeA is apparently mediated by residues along its whole polypeptide,91 including the “forbidden” residues that are far away from the dimerization interface as judged according to the reported 3D structure.79, 90 We provided evidence to show that such dimerization, though intriguing, is a result of neither protein overexpression nor any structural disturbance caused by the introduction of the unnatural amino acid.91
Dimerization mediated by similar “forbidden” noninterface residues was also observed for two other homo‐oligomeric proteins, IbpB (a member of the small heat shock protein family molecular chaperones and exists as polydispersed oligomers in vitro)92, 93 and DegP (a protease existing as hexamers in vitro).77, 94, 95 In contrast, dimerization of a few other oligomeric proteins (e.g., OmpF, LamB, SurA, FtsZ, and FkpA) that we similarly examined seems to be mediated only by specific residues.91 Collectively, these observations suggest that, for some oligomeric proteins (as represented by HdeA, IbpB, and DegP), their subunit interactions in living cells can also be mediated by residues other than those located at the interfaces as defined by in vitro structural determination, apparently indicating an extremely high dynamic structure of them in living cells (as schematically illustrated in Fig. 4).
Figure 4.
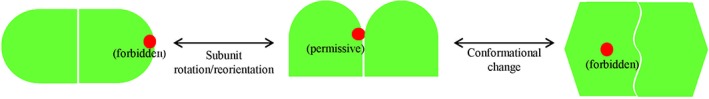
Certain proteins possess a highly dynamic structure in living cells. A “forbidden” residue (red ball) far away from the subunit “interfaces” (as revealed by in vitro structure determination) may become “permissive” to mediate the interaction of the protein subunits in living cells.91 This might happen through subunit rotation/reorientation (left arrow) and/or conformational change (right arrow) in living cells.
Consistent with the dynamic nature of the IbpB protein in living cells, we have demonstrated, also via site‐specific photocrosslinking analysis, that the IbpB protein acts as a robust chaperone in living cells by hierarchically activating its multiple‐type substrate‐binding residues.37 In addition, we have provided pieces of evidence to show that under in vitro conditions, IbpB93 and Hsp16.396, 97, another small heat shock protein exhibited a very dynamic oligomeric structure that allows them to regulate their chaperone activity in response to the environmental conditions.
The significantly shortened time needed for the photocrosslinking reaction may enable the capturing of more dynamic and transient protein–protein interactions
Recently, Akiyama and coworkers explored a high‐power UV irradiator, with which the UV exposure time of the living cells for an efficient photocrosslinking reaction is shortened from a couple of minutes as needed using the conventional UV irradiator to only 1 sec.58 This improvement could be of great significance for studying the kinetics of protein biogenesis in cells, because protein synthesis in bacterial cells growing at 37°C proceeds at a rate as high as 20 residues per second98 and thus the biogenesis of a protein would be usually finished within <1 min.58 The authors applied this new technique to study the folding kinetics of a cytoplasmic membrane protein SecD and its assembly with a partner protein SecF, and found that the folding of the periplasmic P1 domain of SecD occurs before the association of the membrane domains of SecD and SecF. In addition, an assembly intermediate between LptD (an OMP) and LptE (a lipoprotein), as well as two different conformational states for the LptD folding intermediate, was captured during the biogenesis of LptD, highlighting the power of this new technique (designated as PiXie, meaning “the pulse‐chase and in vivo photocrosslinking experiment”) in studying protein structure dynamics in living cells.58
Site‐directed photocrosslinking analysis revealed new assembly forms of the key cell division protein FtsZ in living cells
FtsZ is an essential protein for bacterial cell division. It polymerizes into protofilaments, which further assemble into higher‐order structures at the future division sites of the cell to form the dynamic Z‐ring structure.99 How the Z‐ring structure is assembled and disassembled in living cells has been poorly understood. We recently applied the site‐directed photocrosslinking technique, in combination with a strategy of random introduction of the unnatural amino acid pBpa into the FtsZ protein [as illustrated in Fig. 1(b)], to study the self‐assembly dynamics of FtsZ and identified four distinct residues that mediate the dimerization of FtsZ in living cells.22 Of particular interest is that three of these identified residues (R78, D82, and R85) make up a new lateral interface mediating the assembly of the FtsZ proteins (with the other one located at the known longitudinal interface). We further demonstrated, also by site‐directed photocrosslinking, that the preassembled protofilament (via longitudinal interactions) is a prerequisite for the lateral interactions to occur and that the latter are important for the FtsZ protofilaments to further assemble into the dynamic Z‐ring structure in cells. Such in vivo photocrosslinking analysis of the FtsZ protein also enabled us to unexpectedly unveil a new subcellular structure that we designate as regrowth‐delay body, which is formed only in the nongrowing bacterial cells and sequesters multiple key proteins including FtsZ, but dissolves to release the stored proteins for refunctioning when the cell resumes growth.100 The regrowth‐delay body is likely the most distinguishable subcellular structure hitherto identified to mark the multidrug‐tolerant persister (dormant) bacterial cells.
Concluding Remarks and Future Perspectives
The unnatural amino acid‐mediated site‐directed photocrosslinking technique, in combination with other analytic tools such as mass spectrometry, isotope‐labeled pulse‐chase, and immunoblotting, has greatly advanced our understanding on the biogenesis, quality control, and dynamics of proteins in living cells. Although those interactions related to OMP biogenesis have been extensively examined by this approach, many fundamental questions remain to be clarified. For instance, how are the newly synthesized polypeptides of the OMPs dynamically and transiently interact with all the participating protein factors, which are apparently assembled into a supercomplex21 (as shown in Fig. 3), on their way to the final destination? How the chaperone and protease factors recognize the not‐yet folded intermediates and the misfolded forms, respectively? Where and how the β‐barrel structures of the OMPs are formed and integrated into the outer membrane? Conceivably, the high‐power UV irradiator would be highly useful for these studies.58
To our surprise, protein–protein interactions related to the biogenesis and quality control of cytosolic proteins (as also diagrammed in the lower part of Fig. 3), although being extensively investigated via other techniques, have been rarely probed via site‐directed in vivo photocrosslinking analysis (as indicated in Table 1). For instance, the trigger factor, as an important chaperone binding to nascent polypeptides during the early stage of their biosynthesis on the ribosomes, has been studied only using in vitro site‐directed photocrosslinking analysis.67, 68, 69 Similarly, the interactions of many protein quality control factors with their substrate proteins have been extensively examined under in vitro conditions, but apparently have not yet been studied in living cells using this approach, according to our exhaustive literature search. These factors include the DnaK, GroEL, and Hsp90 chaperones,1, 3, 101 the Lon and ClpP proteases,102 and other chaperones/folding catalysts such as Hsp33103 and DsbA/DsbC.104 In addition, the protein quality control mechanism of extremophiles living in extremely harsh environments (e.g., thermophiles living in hot springs with a temperature of above 90°C) remains poorly understood. Although conventional methods are infeasible for probing protein–protein interactions under such conditions, the site‐directed photocrosslinking seems to be applicable and certainly worth testing.105
As regards the dynamics of protein structures in living cells, even less is known. For instance, how dynamic are proteins in general in living cells? How do protein conformations in living cells differ from what we usually perceive based on in vitro studies? How do the conformational ensembles of proteins correlate with their often multiple functions and interactions in living cells? Why some proteins seem to be more dynamic than others (as implicated by our observations)? Does the disease‐causing protein misfolding occurs due to problems of protein dynamics? Undoubtedly, answering these questions needs techniques beyond that represented by the site‐directed in vivo photocrosslinking analysis.
Acknowledgments
We thank Dr. Jiayu Yu and Mr. Yang Liu (both from Prof. Zengyi Chang's lab) for valuable discussions. This work was supported by research grants from the National Natural Science Foundation of China (Nos. 31670775 and 31470766 to ZYC, and Nos. 31770830 and 31570778 to XF), the Qidong‐SLS Innovation Fund (to ZYC), the Natural Science Foundation of Fujian Province (No. 2018J01725 to XF), and the Scientific Research Innovation Team Construction Program of Fujian Normal University (Z1707219021).
References
- 1. Blobel G (2000) Protein targeting. ChemBioChem 1:86–102. [DOI] [PubMed] [Google Scholar]
- 2. Chang Z. Biogenesis of secretory proteins: folding and quality control in the endoplasmic reticulum In: Bradshawa RA, Stahl P, Eds, 2016, Encyclopedia of cell biology. Waltham, MA: Academic Press; p. 535–544. [Google Scholar]
- 3. Balchin D, Hayer‐Hartl M, Hartl FU (2016) In vivo aspects of protein folding and quality control. Science 353:aac4354. [DOI] [PubMed] [Google Scholar]
- 4. Sontag EM, Samant RS, Frydman J (2017) Mechanisms and functions of spatial protein quality control. Annu Rev Biochem 86:97–122. [DOI] [PubMed] [Google Scholar]
- 5. Chiti F, Dobson CM (2017) Protein misfolding, amyloid formation, and human disease: a summary of progress over the last decade. Annu Rev Biochem 86:27–68. [DOI] [PubMed] [Google Scholar]
- 6. Henzler‐Wildman K, Kern D (2007) Dynamic personalities of proteins. Nature 450:964–972. [DOI] [PubMed] [Google Scholar]
- 7. Vinson VJ (2009) Proteins in motion. Introduction. Science 324:197. [DOI] [PubMed] [Google Scholar]
- 8. Suezaki Y, Go N (1975) Breathing mode of conformational fluctuations in globular proteins. Int J Pept Protein Res 7:333–334. [DOI] [PubMed] [Google Scholar]
- 9. McCammon JA, Gelin BR, Karplus M (1977) Dynamics of folded proteins. Nature 267:585–590. [DOI] [PubMed] [Google Scholar]
- 10. Frauenfelder H, Sligar SG, Wolynes PG (1991) The energy landscapes and motions of proteins. Science 254:1598–1603. [DOI] [PubMed] [Google Scholar]
- 11. Austin RH, Beeson KW, Eisenstein L, Frauenfelder H, Gunsalus IC (1975) Dynamics of ligand binding to myoglobin. Biochemistry 14:5355–5373. [DOI] [PubMed] [Google Scholar]
- 12. Frauenfelder H, Petsko GA, Tsernoglou D (1979) Temperature‐dependent X‐ray diffraction as a probe of protein structural dynamics. Nature 280:558–563. [DOI] [PubMed] [Google Scholar]
- 13. Fenwick RB, van den Bedem H, Fraser JS, Wright PE (2014) Integrated description of protein dynamics from room‐temperature X‐ray crystallography and NMR. Proc Natl Acad Sci U S A 111:E445–E454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Kovermann M, Rogne P, Wolf‐Watz M (2016) Protein dynamics and function from solution state NMR spectroscopy. Q Rev Biophys 49:e6. [DOI] [PubMed] [Google Scholar]
- 15. Freedberg DI, Selenko P (2014) Live cell NMR. Annu Rev Biophys 43:171–192. [DOI] [PubMed] [Google Scholar]
- 16. Luchinat E, Banci L (2016) A unique tool for cellular structural biology: in‐cell NMR. J Biol Chem 291:3776–3784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sinz A (2010) Investigation of protein‐protein interactions in living cells by chemical crosslinking and mass spectrometry. Anal Bioanal Chem 397:3433–3440. [DOI] [PubMed] [Google Scholar]
- 18. Kauer JC, Erickson‐Viitanen S, Wolfe HR Jr, DeGrado WF (1986) P‐benzoyl‐L‐phenylalanine, a new photoreactive amino acid. Photolabeling of calmodulin with a synthetic calmodulin‐binding peptide. J Biol Chem 261:10695–10700. [PubMed] [Google Scholar]
- 19. Cornish VW, Benson DR, Altenbach CA, Hideg K, Hubbell WL, Schultz PG (1994) Site‐specific incorporation of biophysical probes into proteins. Proc Natl Acad Sci U S A 91:2910–2914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chin JW, Martin AB, King DS, Wang L, Schultz PG (2002) Addition of a photocrosslinking amino acid to the genetic code of Escherichia coli . Proc Natl Acad Sci U S A 99:11020–11024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Wang Y, Wang R, Jin F, Liu Y, Yu J, Fu X, Chang Z (2016) A supercomplex spanning the inner and outer membranes mediates the biogenesis of beta‐barrel outer membrane proteins in bacteria. J Biol Chem 291:16720–16729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Guan F, Yu J, Liu Y, Li Y, Feng XH, Huang KC, Chang Z, Ye S (2018) Lateral interactions between protofilaments of the bacterial tubulin homolog FtsZ are essential for cell division. eLife 7:e35578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Liu CC, Schultz PG (2010) Adding new chemistries to the genetic code. Annu Rev Biochem 79:413–444. [DOI] [PubMed] [Google Scholar]
- 24. Daggett KA, Layer M, Cropp TA (2009) A general method for scanning unnatural amino acid mutagenesis. ACS Chem Biol 4:109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chen K, Arnold FH (1993) Tuning the activity of an enzyme for unusual environments: sequential random mutagenesis of subtilisin E for catalysis in dimethylformamide. Proc Natl Acad Sci U S A 90:5618–5622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Chang Z (2019) The 2018 Nobel prize in chemistry: engineering proteins (enzymes/peptide/antibodies) towards desired properties via the construction of random libraries. Sci China Life Sci 62:710–712. [DOI] [PubMed] [Google Scholar]
- 27. Suchanek M, Radzikowska A, Thiele C (2005) Photo‐leucine and photo‐methionine allow identification of protein‐protein interactions in living cells. Nat Methods 2:261–267. [DOI] [PubMed] [Google Scholar]
- 28. Yang T, Li XM, Bao X, Fung YM, Li XD (2016) Photo‐lysine captures proteins that bind lysine post‐translational modifications. Nat Chem Biol 12:70–72. [DOI] [PubMed] [Google Scholar]
- 29. Chin JW, Santoro SW, Martin AB, King DS, Wang L, Schultz PG (2002) Addition of p‐azido‐L‐phenylalanine to the genetic code of Escherichia coli . J Am Chem Soc 124:9026–9027. [DOI] [PubMed] [Google Scholar]
- 30. Tippmann EM, Liu W, Summerer D, Mack AV, Schultz PG (2007) A genetically encoded diazirine photocrosslinker in Escherichia coli . ChemBioChem 8:2210–2214. [DOI] [PubMed] [Google Scholar]
- 31. Ai HW, Shen W, Sagi A, Chen PR, Schultz PG (2011) Probing protein‐protein interactions with a genetically encoded photo‐crosslinking amino acid. ChemBioChem 12:1854–1857. [DOI] [PubMed] [Google Scholar]
- 32. Zhang M, Lin S, Song X, Liu J, Fu Y, Ge X, Fu X, Chang Z, Chen PR (2011) A genetically incorporated crosslinker reveals chaperone cooperation in acid resistance. Nat Chem Biol 7:671–677. [DOI] [PubMed] [Google Scholar]
- 33. Lin S, He D, Long T, Zhang S, Meng R, Chen PR (2014) Genetically encoded cleavable protein photo‐cross‐linker. J Am Chem Soc 136:11860–11863. [DOI] [PubMed] [Google Scholar]
- 34. Yang Y, Song H, He D, Zhang S, Dai S, Lin S, Meng R, Wang C, Chen PR (2016) Genetically encoded protein photocrosslinker with a transferable mass spectrometry‐identifiable label. Nat Commun 7:12299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. He D, Xie X, Yang F, Zhang H, Su H, Ge Y, Song H, Chen PR (2017) Quantitative and comparative profiling of protease substrates through a genetically encoded multifunctional photocrosslinker. Angew Chem Int Ed Engl 56:14521–14525. [DOI] [PubMed] [Google Scholar]
- 36. Yang Y, Song H, Chen PR (2016) Genetically encoded photocrosslinkers for identifying and mapping protein‐protein interactions in living cells. IUBMB Life 68:879–886. [DOI] [PubMed] [Google Scholar]
- 37. Fu X, Shi X, Yin L, Liu J, Joo K, Lee J, Chang Z (2013) Small heat shock protein IbpB acts as a robust chaperone in living cells by hierarchically activating its multi‐type substrate‐binding residues. J Biol Chem 288:11897–11906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Piehler J (2005) New methodologies for measuring protein interactions in vivo and in vitro. Curr Opin Struct Biol 15:4–14. [DOI] [PubMed] [Google Scholar]
- 39. Wimley WC (2003) The versatile beta‐barrel membrane protein. Curr Opin Struct Biol 13:404–411. [DOI] [PubMed] [Google Scholar]
- 40. Hagan CL, Silhavy TJ, Kahne D (2011) Beta‐barrel membrane protein assembly by the Bam complex. Annu Rev Biochem 80:189–210. [DOI] [PubMed] [Google Scholar]
- 41. Driessen AJ, Nouwen N (2008) Protein translocation across the bacterial cytoplasmic membrane. Annu Rev Biochem 77:643–667. [DOI] [PubMed] [Google Scholar]
- 42. Fu X (2014) Chaperone function and mechanism of small heat‐shock proteins. Acta Biochim Biophys Sin 46:347–356. [DOI] [PubMed] [Google Scholar]
- 43. Jin F, Chang Z (2017) Revelation of a novel protein translocon in bacterial plasma membrane. doi: 10.1101/121335. [DOI]
- 44. Rollauer SE, Sooreshjani MA, Noinaj N, Buchanan SK (2015) Outer membrane protein biogenesis in Gram‐negative bacteria. Philos Trans R Soc Lond B Biol Sci 370: 20150023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Hagan CL, Kim S, Kahne D (2010) Reconstitution of outer membrane protein assembly from purified components. Science 328:890–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Clausen T, Kaiser M, Huber R, Ehrmann M (2011) HTRA proteases: regulated proteolysis in protein quality control. Nat Rev Mol Cell Biol 12:152–162. [DOI] [PubMed] [Google Scholar]
- 47. Chang Z (2016) The function of the DegP (HtrA) protein: protease versus chaperone. IUBMB Life 68:904–907. [DOI] [PubMed] [Google Scholar]
- 48. Das S, Oliver DB (2011) Mapping of the SecA.SecY and SecA.SecG interfaces by site‐directed in vivo photocross‐linking. J Biol Chem 286:12371–12380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yu D, Wowor AJ, Cole JL, Kendall DA (2013) Defining the Escherichia coli SecA dimer interface residues through in vivo site‐specific photo‐cross‐linking. J Bacteriol 195:2817–2825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Banerjee T, Lindenthal C, Oliver D (2017) SecA functions in vivo as a discrete anti‐parallel dimer to promote protein transport. Mol Microbiol 103:439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Bensing BA, Yen YT, Seepersaud R, Sullam PM (2012) A specific interaction between SecA2 and a region of the preprotein adjacent to the signal peptide occurs during transport via the accessory sec system. J Biol Chem 287:24438–24447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Huber D, Jamshad M, Hanmer R, Schibich D, Doring K, Marcomini I, Kramer G, Bukau B (2017) SecA cotranslationally interacts with nascent substrate proteins in vivo. J Bacteriol 199:e00622–e00616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mori H, Ito K (2006) Different modes of SecY‐SecA interactions revealed by site‐directed in vivo photo‐cross‐linking. Proc Natl Acad Sci U S A 103:16159–16164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Banerjee T, Zheng Z, Abolafia J, Harper S, Oliver D (2017) The SecA protein deeply penetrates into the SecYEG channel during insertion, contacting most channel transmembrane helices and periplasmic regions. J Biol Chem 292:19693–19707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Kuhn P, Draycheva A, Vogt A, Petriman NA, Sturm L, Drepper F, Warscheid B, Wintermeyer W, Koch HG (2015) Ribosome binding induces repositioning of the signal recognition particle receptor on the translocon. J Cell Biol 211:91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Sachelaru I, Petriman NA, Kudva R, Kuhn P, Welte T, Knapp B, Drepper F, Warscheid B, Koch HG (2013) YidC occupies the lateral gate of the SecYEG translocon and is sequentially displaced by a nascent membrane protein. J Biol Chem 288:16295–16307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zheng Z, Blum A, Banerjee T, Wang Q, Dantis V, Oliver D (2016) Determination of the oligomeric state of SecYEG protein secretion channel complex using in vivo photo‐ and disulfide cross‐linking. J Biol Chem 291:5997–6010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Miyazaki R, Myougo N, Mori H, Akiyama Y (2018) A photo‐cross‐linking approach to monitor folding and assembly of newly synthesized proteins in a living cell. J Biol Chem 293:677–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Ieva R, Bernstein HD (2009) Interaction of an autotransporter passenger domain with BamA during its translocation across the bacterial outer membrane. Proc Natl Acad Sci U S A 106:19120–19125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ieva R, Tian P, Peterson JH, Bernstein HD (2011) Sequential and spatially restricted interactions of assembly factors with an autotransporter beta domain. Proc Natl Acad Sci U S A 108:E383–E391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Pavlova O, Peterson JH, Ieva R, Bernstein HD (2013) Mechanistic link between beta barrel assembly and the initiation of autotransporter secretion. Proc Natl Acad Sci U S A 110:E938–E947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Daimon Y, Iwama‐Masui C, Tanaka Y, Shiota T, Suzuki T, Miyazaki R, Sakurada H, Lithgow T, Dohmae N, Mori H, Tsukazaki T, Narita SI, Akiyama Y (2017) The TPR domain of BepA is required for productive interaction with substrate proteins and the beta‐barrel assembly machinery complex. Mol Microbiol 106:760–776. [DOI] [PubMed] [Google Scholar]
- 63. Ge X, Wang R, Ma J, Liu Y, Ezemaduka AN, Chen PR, Fu X, Chang Z (2014) DegP primarily functions as a protease for the biogenesis of beta‐barrel outer membrane proteins in the Gram‐negative bacterium Escherichia coli . FEBS J 281:1226–1240. [DOI] [PubMed] [Google Scholar]
- 64. Fu X, Wang Y, Shao H, Ma J, Song X, Zhang M, Chang Z (2018) DegP functions as a critical protease for bacterial acid resistance. FEBS J 285:3525–3538. [DOI] [PubMed] [Google Scholar]
- 65. He D, Zhang M, Liu S, Xie X, Chen PR (2019) Protease‐mediated protein quality control for bacterial acid resistance. Cell Chem Biol 26:144–150. [DOI] [PubMed] [Google Scholar]
- 66. Zhang S, He D, Yang Y, Lin S, Zhang M, Dai S, Chen PR (2016) Comparative proteomics reveal distinct chaperone‐client interactions in supporting bacterial acid resistance. Proc Natl Acad Sci U S A 113:10872–10877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ullers RS, Houben EN, Brunner J, Oudega B, Harms N, Luirink J (2006) Sequence‐specific interactions of nascent Escherichia coli polypeptides with trigger factor and signal recognition particle. J Biol Chem 281:13999–14005. [DOI] [PubMed] [Google Scholar]
- 68. Ullers RS, Houben EN, Raine A, ten Hagen‐Jongman CM, Ehrenberg M, Brunner J, Oudega B, Harms N, Luirink J (2003) Interplay of signal recognition particle and trigger factor at L23 near the nascent chain exit site on the Escherichia coli ribosome. J Cell Biol 161:679–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Jong WS, ten Hagen‐Jongman CM, Genevaux P, Brunner J, Oudega B, Luirink J (2004) Trigger factor interacts with the signal peptide of nascent Tat substrates but does not play a critical role in Tat‐mediated export. Eur J Biochem 271:4779–4787. [DOI] [PubMed] [Google Scholar]
- 70. Hoseini H, Pandey S, Jores T, Schmitt A, Franz‐Wachtel M, Macek B, Buchner J, Dimmer KS, Rapaport D (2016) The cytosolic cochaperone Sti1 is relevant for mitochondrial biogenesis and morphology. FEBS J 283:3338–3352. [DOI] [PubMed] [Google Scholar]
- 71. Fu X, Shi X, Yan L, Zhang H, Chang Z (2013) In vivo substrate diversity and preference of small heat shock protein IbpB as revealed by using a genetically incorporated photo‐cross‐linker. J Biol Chem 288:31646–31654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Jaya N, Garcia V, Vierling E (2009) Substrate binding site flexibility of the small heat shock protein molecular chaperones. Proc Natl Acad Sci U S A 106:15604–15609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Teese MG, Langosch D (2015) Role of GxxxG motifs in transmembrane domain interactions. Biochemistry 54:5125–5135. [DOI] [PubMed] [Google Scholar]
- 74. Ge X, Lyu ZX, Liu Y, Wang R, Zhao XS, Fu X, Chang Z (2014) Identification of FkpA as a key quality control factor for the biogenesis of outer membrane proteins under heat shock conditions. J Bacteriol 196:672–680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Han L, Zheng J, Wang Y, Yang X, Liu Y, Sun C, Cao B, Zhou H, Ni D, Lou J, Zhao Y, Huang Y (2016) Structure of the BAM complex and its implications for biogenesis of outer‐membrane proteins. Nat Struct Mol Biol 23:192–196. [DOI] [PubMed] [Google Scholar]
- 76. Gu Y, Li H, Dong H, Zeng Y, Zhang Z, Paterson NG, Stansfeld PJ, Wang Z, Zhang Y, Wang W, Dong C (2016) Structural basis of outer membrane protein insertion by the BAM complex. Nature 531:64–69. [DOI] [PubMed] [Google Scholar]
- 77. Jiang JS, Zhang XF, Chen Y, Wu Y, Zhou ZH, Chang Z, Sui SF (2008) Activation of DegP chaperone‐protease via formation of large cage‐like oligomers upon binding to substrate proteins. Proc Natl Acad Sci U S A 105:11939–11944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Hong W, Wu YE, Fu X, Chang Z (2012) Chaperone‐dependent mechanisms for acid resistance in enteric bacteria. Trends Microbiol 20:328–335. [DOI] [PubMed] [Google Scholar]
- 79. Gajiwala KS, Burley SK (2000) HDEA, a periplasmic protein that supports acid resistance in pathogenic enteric bacteria. J Mol Biol 295:605–612. [DOI] [PubMed] [Google Scholar]
- 80. Hong W, Jiao W, Hu J, Zhang J, Liu C, Fu X, Shen D, Xia B, Chang Z (2005) Periplasmic protein HdeA exhibits chaperone‐like activity exclusively within stomach pH range by transforming into disordered conformation. J Biol Chem 280:27029–27034. [DOI] [PubMed] [Google Scholar]
- 81. Kern R, Malki A, Abdallah J, Tagourti J, Richarme G (2007) Escherichia coli HdeB is an acid stress chaperone. J Bacteriol 189:603–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Kern D, Eisenmesser EZ, Wolf‐Watz M (2005) Enzyme dynamics during catalysis measured by NMR spectroscopy. Methods Enzymol 394:507–524. [DOI] [PubMed] [Google Scholar]
- 83. Mittermaier A, Kay LE (2006) New tools provide new insights in NMR studies of protein dynamics. Science 312:224–228. [DOI] [PubMed] [Google Scholar]
- 84. Fraser JS, van den Bedem H, Samelson AJ, Lang PT, Holton JM, Echols N, Alber T (2011) Accessing protein conformational ensembles using room‐temperature X‐ray crystallography. Proc Natl Acad Sci U S A 108:16247–16252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Ha TJ, Ting AY, Liang J, Caldwell WB, Deniz AA, Chemla DS, Schultz PG, Weiss S (1999) Single‐molecule fluorescence spectroscopy of enzyme conformational dynamics and cleavage mechanism. Proc Natl Acad Sci U S A 96:893–898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wales TE, Engen JR (2006) Hydrogen exchange mass spectrometry for the analysis of protein dynamics. Mass Spectrom Rev 25:158–170. [DOI] [PubMed] [Google Scholar]
- 87. Summerer D, Chen S, Wu N, Deiters A, Chin JW, Schultz PG (2006) A genetically encoded fluorescent amino acid. Proc Natl Acad Sci U S A 103:9785–9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Schultz KC, Supekova L, Ryu Y, Xie J, Perera R, Schultz PG (2006) A genetically encoded infrared probe. J Am Chem Soc 128:13984–13985. [DOI] [PubMed] [Google Scholar]
- 89. Deiters A, Geierstanger BH, Schultz PG (2005) Site‐specific in vivo labeling of proteins for NMR studies. ChemBioChem 6:55–58. [DOI] [PubMed] [Google Scholar]
- 90. Yang F, Gustafson KR, Boyd MR, Wlodawer A (1998) Crystal structure of Escherichia coli HdeA. Nat Struct Biol 5:763–764. [DOI] [PubMed] [Google Scholar]
- 91. Fu X, Wang Y, Song X, Shi X, Shao H, Liu Y, Zhang M, Chang Z (2019) Subunit interactions as mediated by “non‐interface” residues in living cells for multiple homo‐oligomeric proteins. Biochem Biophys Res Commun 512:100–105. [DOI] [PubMed] [Google Scholar]
- 92. Shearstone JR, Baneyx F (1999) Biochemical characterization of the small heat shock protein IbpB from Escherichia coli . J Biol Chem 274:9937–9945. [DOI] [PubMed] [Google Scholar]
- 93. Jiao W, Qian M, Li P, Zhao L, Chang Z (2005) The essential role of the flexible termini in the temperature‐responsiveness of the oligomeric state and chaperone‐like activity for the polydisperse small heat shock protein IbpB from Escherichia coli . J Mol Biol 347:871–884. [DOI] [PubMed] [Google Scholar]
- 94. Krojer T, Garrido‐Franco M, Huber R, Ehrmann M, Clausen T (2002) Crystal structure of DegP (HtrA) reveals a new protease‐chaperone machine. Nature 416:455–459. [DOI] [PubMed] [Google Scholar]
- 95. Krojer T, Sawa J, Schafer E, Saibil HR, Ehrmann M, Clausen T (2008) Structural basis for the regulated protease and chaperone function of DegP. Nature 453:885–890. [DOI] [PubMed] [Google Scholar]
- 96. Gu L, Abulimiti A, Li W, Chang Z (2002) Monodisperse Hsp16.3 nonamer exhibits dynamic dissociation and reassociation, with the nonamer dissociation prerequisite for chaperone‐like activity. J Mol Biol 319:517–526. [DOI] [PubMed] [Google Scholar]
- 97. Yang H, Huang S, Dai H, Gong Y, Zheng C, Chang Z (1999) The Mycobacterium tuberculosis small heat shock protein Hsp16.3 exposes hydrophobic surfaces at mild conditions: conformational flexibility and molecular chaperone activity. Protein Sci 8:174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Young R, Bremer H (1976) Polypeptide‐chain‐elongation rate in Escherichia coli B/r as a function of growth rate. Biochem J 160:185–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bi EF, Lutkenhaus J (1991) FtsZ ring structure associated with division in Escherichia coli . Nature 354:161–164. [DOI] [PubMed] [Google Scholar]
- 100. Yu J, Liu Y, Yin H, Chang Z (2019) Regrowth‐delay body as a bacterial subcellular structure marking multidrug‐tolerant persisters. Cell Discovery 5:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Hartl FU, Bracher A, Hayer‐Hartl M (2011) Molecular chaperones in protein folding and proteostasis. Nature 475:324–332. [DOI] [PubMed] [Google Scholar]
- 102. Laskowska E, Kuczynska‐Wisnik D, Skorko‐Glonek J, Taylor A (1996) Degradation by proteases Lon, Clp and HtrA, of Escherichia coli proteins aggregated in vivo by heat shock; HtrA protease action in vivo and in vitro. Mol Microbiol 22:555–571. [DOI] [PubMed] [Google Scholar]
- 103. Jakob U, Muse W, Eser M, Bardwell JC (1999) Chaperone activity with a redox switch. Cell 96:341–352. [DOI] [PubMed] [Google Scholar]
- 104. Inaba K, Ito K (2008) Structure and mechanisms of the DsbB‐DsbA disulfide bond generation machine. Biochim Biophys Acta 1783:520–529. [DOI] [PubMed] [Google Scholar]
- 105. Yanagisawa T, Hino N, Iraha F, Mukai T, Sakamoto K, Yokoyama S (2012) Wide‐range protein photo‐crosslinking achieved by a genetically encoded N(epsilon)‐(benzyloxycarbonyl)lysine derivative with a diazirinyl moiety. Mol Biosyst 8:1131–1135. [DOI] [PubMed] [Google Scholar]