

Abstract
SIRT1 is an NAD+-dependent lysine deacetylase that promotes healthy aging and longevity in diverse organisms. Small molecule allosteric activators of SIRT1 such as resveratrol and SRT2104 directly bind to the N-terminus of SIRT1 and lower the Km for the protein substrate. In rodents, sirtuin-activating compounds (STACs) protect from age-related diseases and extend life span. In human clinical trials, STACs have a high safety profile and anti-inflammatory activities. Here, we describe methods for identifying and characterizing STACs, including production of recombinant protein, in vitro assays with recombinant protein, and cellular assays based on mitochondrial dynamics. The methods described in this chapter will facilitate this discovery of improved STACs, natural and synthetic, in the pursuit of interventions to treat age-related diseases.
Keywords: Sirtuin, SIRT1, Histone deacetylase (HDAC), Deacylase, Deacetylase, NAD+, Nicotinamide, ADP-ribose, Aging, Longevity, Metabolism, Epigenetics, Histone, p53, Mitochondria, Membrane potential, Reactive oxygen species (ROS), Sirtuin-activating compound (STAC), Resveratrol, Allosteric activator, Recombinant protein
1. Introduction
Since the discovery of the yeast longevity gene Silent Information Regulator 2 (SIR2), much attention has been given to its seven mammalian homologs, the sirtuins [1]. Of these, SIRT1 is the closest homolog to its yeast counterpart and, along with SIRT6, is one of only two sirtuins shown to extend life span in mammals [2]. SIRT1, an NAD+-dependent lysine deacetylase, serves as an NAD+-sensor that promotes efficient energy utilization and cellular defenses in response to changes in the environment such a decline in nutrient availability. SIRT1 has numerous deacetylation targets involved in key biological processes, including histones [3], transcription factors like p53 [4, 5], PGC-1α [6], and NF-κB [7], signaling proteins such as the Notch intracellular domain (NICD) [8], and insulin receptor substrate-2 (IRS2) [9], and enzymes such as LKB1 [10]. The net effect of increased SIRT1 activity is to send a cell into “survival mode,” with improved DNA repair, epigenetic stability, and metabolic efficiency. SIRT1 promotes healthy aging and longevity, while its dysregulation accelerates many age-related diseases such as Alzheimer’s disease, cancer, cardiovascular disease, and diabetes [11].
Because of SIRT1’s salutary effects, it is an enticing target for pharmacological activation. In comparison to enzyme inhibition, discovering an activator is a rarity; it is easier to throw a wrench in the works than to enhance a machine’s function. Luckily, SIRT1 is the rare enzyme with activation potential. It possesses a large, loosely structured N-terminus called the STAC binding domain (SBD), the only mammalian sirtuin with such a domain. While not required for the enzyme’s activity, the SBD increase SIRT1 activity by stabilizing the interaction of the domain with its acetylated protein targets. STACs allosterically bind to this N-terminus, further stabilizing the interaction between SIRT1 and its targets beyond normal levels, in effect lowering the apparent Km of the enzyme [12]. It is not known if this opportunity for allosteric activation is an accident of biochemistry or a product of natural selection. If an endogenous small molecule SIRT1 activator exists in mammals, it has yet to be discovered.
The first discovery of SIRT1 allosteric activators came in 2003 with the identification of resveratrol and similarly structured polyphenols [13]. The first SIRT1-mediated activities described for these molecules were increased mitochondrial function and protection from the negative effects of a high fat diet [10, 14]. Since 2003, synthetic molecules have been discovered with much higher affinity for SIRT1. Two such molecules, SRT1720 and SRT2104, extend healthspan and longevity in mice [15, 16]. In humans, over 50 clinical trials have been carried out with resveratrol or SRT2104, with a focus on neuropathies, cardiovascular disease, inflammation, and diabetes. Considering resveratrol’s less-than ideal pharmacokinetic properties and the variability of human patients, clinical results have been mixed. A recent meta-analysis concluded that resveratrol supplementation has positive effects on multiple health parameters in type 2 diabetes patients, with no adverse effects over placebo [17]. Similarly, SRT2104 has been shown to reduce disease severity in a subset of psoriasis patients when compared to placebo in a randomized double-blind trial [18]. New SIRT1 activating compounds with improved pharmacokinetic and pharmacodynamic profiles are an area of intense research interest.
Here, we describe methods for the discovery and characterization of STACs. First, we describe a method for producing recombinant SIRT1 enzyme for biochemical assays [19]. In addition to producing the wild-type enzyme, it is also possible to produce mutant versions of SIRT1. For example, SIRT1-H363Y is enzymatically dead and SIRT1-E230K is resistant to allosteric activation [12]. This protocol is also used to produce recombinant yPNC1, which is required for a subsequent assay.
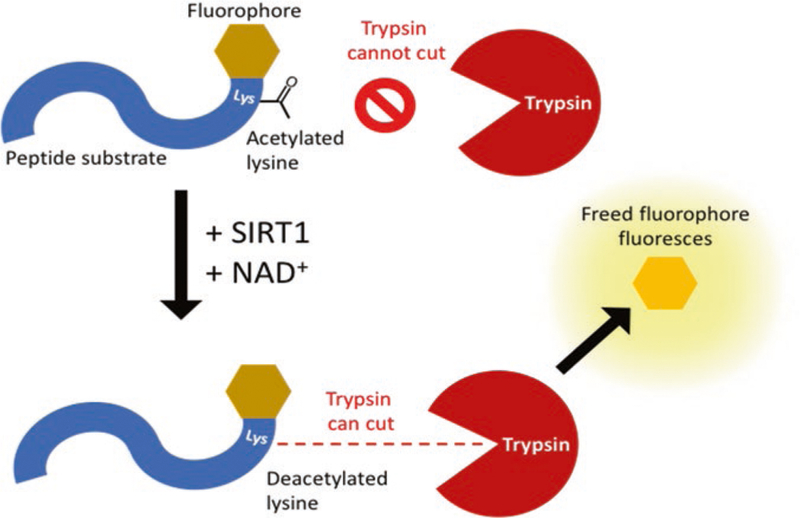
Next, we describe the Fluor-de-Lys assay for measuring SIRT1 activity in vitro [20]. SIRT1 is incubated with an acetylated sub-strate, NAD+, and the test STAC. The substrate has a quenched fluorophore proximal to its acetylated lysine such that the removal of the acetyl group by SIRT1 allows the lysine to be cleaved by trypsin in the subsequent development stage, thereby freeing and un-quenching the fluorophore (Fig. 1). Fluorescence is therefore proportional to the amount of deacetylated substrate due to SIRT1 activity. This assay has been a focus of controversy, as some have argued that the activity of SIRT1 on a fluorophore-tagged sub-strate does not reflect its activity on endogenous substrates [21]. However, further investigation has shown that the hydrophobic fluorophore in this assay mimics natural, bulky, hydrophobic residues such as tryptophan and phenylalanine that are present in endogenous substrates [12]. STACs identified in this assay have proved robust in other in vitro and in vivo contexts.
Fig. 1. Schematic of the Fluor-de-Lys assay.
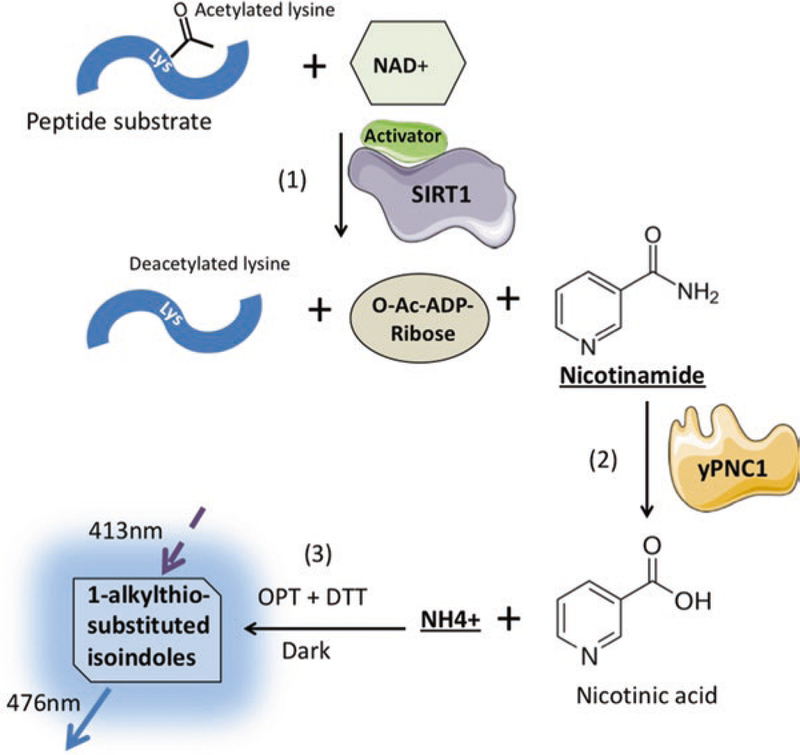
Another assay for measuring SIRT1 activity in vitro is the PNC1-OPT assay [22] (Fig. 2). This assay is designed for using a native peptide (without a fluorophore) as the SIRT1 substrate. It measures the amount of nicotinamide, another product produced in the SIRT1 deacetylation reaction. SIRT1 activators will increase the production of nicotinamide in step (1). yPnc1, an enzyme from budding yeast, will then convert the nicotinamide into nicotinic acid and NH4+ (free ammonia) in step (2). Finally in step (3), the ammonia will react with ortho-phthalaldehyde (OPT) and dithiothreitol (DTT) to generate a fluorescent product—1-alkylthio-substituted isoindoles, which can emit fluorescence near 476 nm once excited. Therefore, the fluorescence detected is proportional to the amount of nicotinamide produced and is thereby representative of SIRT1 activity.
Fig. 2. Schematic of the PNC1-OPT assay. A potential SIRT1 activator is added in step (1) and its effect on the SIRT1 deacetylation reaction will be revealed in step (3).
Finally, we describe methods for measuring STACs’ effects on mitochondrial activity in cells. Resveratrol, SRT1720, and SRT2104 have all been shown to promote mitochondrial biogenesis and function and to suppress the production of reactive oxygen species (ROS) [10, 23, 24]. As mitochondrial content and activity vary with diet, exercise, aging, and disease status, these methods are a strategic starting place for assessing novel STACs’ efficacy in vivo. Here, we present relatively simple methods for quantifying mitochondrial mass, membrane potential, and ROS production based on the use of fluorescent dyes and flow cytometry. Four dyes are utilized in this assay: mitotracker deep red (MitoTracker® Deep Red FM) and nonyl-acridine orange (NAO) for measuring mitochondrial mass; tetramethylrhodamine methyl ester (TMRM) for measuring mitochondrial membrane potential; and dihydroethidium (DHE) measuring ROS.
2. Materials
2.1. Expression and Purification of Recombinant SIRT1
pET-based His-tagged SIRT1 plasmid.
BL21 pLysS(DE3), Rosetta, or other competent bacteria for protein expression.
LB media and LB-agar plates with antibiotic.
Bacterial incubator.
Centrifuge for spinning down large volumes of bacterial cultures.
Protease inhibitors (EDTA free).
Isopropyl beta-D-1-thiogalactopyranoside (IPTG).
Sonicator.
Ni-NTA agarose beads.
-
Lysis buffer
-
(a)
1% Triton X-100.
-
(b)
50 mM Tris pH 8.0.
-
(c)
150 mM NaCl.
-
(d)
20 mM imidazole.
-
(e)
3 mM beta-mercaptoethanol (BME).
-
(a)
- Wash buffer
-
(a)1% Triton X-100.
-
(b)50 mM Tris pH 8.0.
-
(c)300 mM NaCl.
-
(d)20 mM imidazole.
-
(e)3 mM BME.
-
(a)
- Elution buffer
-
(a)50 mM Tris pH 8.0.
-
(b)250 mM imidazole.
-
(c)3 mM BME.
-
(a)
Spectrophotometer for measuring absorbance.
Optional: Bio-Rad Poly-Prep® columns.
Optional: Millipore Microcon® dialysis columns.
2.2. Fluor de Lys Assay for SIRT1 Activators In Vitro
All of the following materials are available in the SIRT1 Fluorometric Drug Discovery Kit (Enzo® Life Sciences)
SIRT1 (Sirtuin 1, hSir2SIRT1) (human, recombinant) in 25 mM Tris, pH 7.5, 100 mM sodium chloride, 5 mM dithiothreitol, and 10% glycerol. Store at –80 °C.
Fluro de Lys® SIRT1, Deacetylase Substrate (100 μL; 5 mM solution in 50 mM Tris/Cl, pH 8.0, 137 mM sodium chloride, 2.7 mM potassium chloride, 1 mM magnesium chloride). Store at –80 °C.
Fluor-de-Lys® Developer II Concentrate (5×) (5 × 250 μL; 5× Stock Solution; Dilute in Assay Buffer before use). Store at –80 °C.
NAD+ (Sirtuin Substrate) (500 μL; 50 mM β-Nicotinamide adenine dinucleotide (oxidized form) in 50 mM Tris/Cl, pH 8.0, 137 mM sodium chloride, 2.7 mM potassium chloride, 1 mM magnesium chloride). Store at –80 °C.
Nicotinamide (Sirtuin Inhibitor) (500 μL; 50 mM Nicotinamide in 50 mM Tris/Cl, pH 8.0, 137 mM sodium chloride, 2.7 mM potassium chloride, 1 mM magnesium chloride). Store at –80 °C.
Resveratrol (Sirtuin Activator) (10 mg; Solid MW: 228.2, soluble in DMSO or 100% ethanol (to 100 mM)). Store at –80 °C.
Suramin sodium (Sirtuin Inhibitor) (10 mg; Solid MW: 1429.2, soluble in water or assay buffer (to 25 mM)). Store at –80 °C.
Fluor-de-Lys® Deacetylated Standard (30 μL; 10 mM in DMSO). Store at –80 °C.
Sirtuin Assay Buffer (50 mM Tris/Cl, pH 8.0, 137 mM sodium chloride, 2.7 mM potassium chloride, 1 mM magnesium chloride, 1 mg/mL bovine serum albumin) (20 mL). Store at –80 °C.
96-well white ½ area plates for fluorometry.
Spectrophotometer with fluorescence capabilities and appropriate filters (excitation 350–380 nm and emission 450–480 nm).
2.3. PNC1-OPTAssay for SIRT1 Activators In Vitro
Purified recombinant SIRT1 and yPnc1 enzymes should be expressed by using the protocol in the same chapter [18].
SIRT1 requires acetylated peptide substrates, the peptide length is usually 5–15 amino acids and it has acetylated lysine near the middle of the sequence (e.g., Ac-TARK(ac)STG-NH2) (see Note 1). Peptides with hydrophobic groups adjacent to the acetyl-lysine are recommended for STAC-mediated SIRT1 activation [12].
Reaction Buffer: PBS (pH 7.4) or Tris buffer (pH = 8.0) supplemented with 1 mM DTT (Thermo Fisher, R0861).
OPT Developer Reagent: To make this 30% ethanol/70% PBS (pH 7.4) solution supplemented with 10 mM OPT and 10 mM DTT, first dissolve OPT (Sigma, P0657) in pure ethanol (33.3 mM OPT) and DTT in PBS (14.28 mM DTT), respectively, then mix 3 volumes of ethanol (with OPT) to 7 volumes of PBS (with DTT). Protect from light and store at –20 °C until use.
NAD+ (Sigma, N7004) should be dissolved in distilled water to make 100 mM aliquots and stored at –20 °C. Avoid freezethaw cycles.
Nicotinamide (Sigma, 72340) is used to make standard curve, prepare its 100 mM stock solution in distilled water, aliquoted and stored at –20 °C.
96-well black polystyrene plates (Corning, 3915) suitable for fluorometry.
Plate reader capable of reading fluorescence (Ex/ Em = 413/476 nm).
Aluminum foil.
37 °C incubator.
Orbital shaker.
2.4. Mitochondrial Assays for SIRT1 Activators in Cells
Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum, 2 mM L-glutamine, and 1% antibiotic: penicillin/streptomycin.
Phosphate-buffered saline (PBS) (GIBCO).
Dimethyl sulfoxide (DMSO).
6-Well Cell Culture plate (2 mL volume per well).
Mouse Embryonic Fibroblasts (MEFs) and/or C2C12 cells.
12 × 75 mm round bottom polystyrene tubes.
Tetramethylrhodamine methyl ester (TMRM perchlorate, Invitrogen T-668), a light-sensitive cationic, mitochondrial membrane-permeable fluorescent dye, dissolved in DMSO at 100–500 times greater concentration than the final concentration used in the assay. The concentration used in the assay varies depending on the mode used (see Note 2). Cells are equilibrated with the dye in the incubation media for 45–60 min. TMRM displays excitation/emission spectra of 552/575 nm.
Dihydroethidium (DHE, Sigma D7008), a cell-permeable fluorescent dye, is extensively used in tissue culture experiments to evaluate ROS. DHE is dissolved in DMSO at 100–500 times greater concentration than the final concentration used in the assay, protected from the light. Cells are equilibrated with the dye in the incubation media for 30–60 min. It displays excitation/emission spectra of 490/590 nm.
Nonyl acridine orange (NAO, Acridine Orange 10-Nonyl Bromide, Invitrogen A1372), a light-sensitive metachromatic dye which binds to cardiolipin, a phospholipid specifically present on the mitochondrial membrane, should be dissolved in DMSO at 100–500 times greater concentration than the working concentration (see Note 3). Cells are equilibrated with the dye in the incubation media for 30–60 min and its excitation/ emission spectra of 495/538 nm.
MitoTracker® Deep Red FM (Invitrogen M22426), a light-sensitive dye that passively diffuses across membranes and accumulates in active mitochondria, and is well retained after aldehyde fixation or after permeabilization with detergents in subsequent steps. This dye, which is not easily washed out of cells once the mitochondria experience a loss in membrane potential, is dissolved in DMSO at 100–500 times greater concentration than the final concentration used in the assay. Cells are equilibrated with the dye in the incubation media for 30–60 min. It displays excitation/emission spectra of 644/665 nm.
Carbonyl cyanide p-(trifluoromethoxy) phenylhydrazone (FCCP) (Sigma C2920), dissolved in ethanol, 1 mM stock solution.
Antimycin A (Sigma A8674), dissolved in ethanol, 1 mM stock solution.
Hydrogen peroxide (Sigma H1009), 30% w/w.
3. Methods
3.1. Expression and Purification of Recombinant SIRT1
Day 1: Transform competent bacterial cells with the His-tagged SIRT1 plasmid, according to manufacturer’s instructions.
Grow cells on LB-agar plates with appropriate antibiotic overnight at 37 °C.
Day 2: Pick a colony from the plate and inoculate a tube with 5 mL of LB media with antibiotic. Grow overnight at 37 °C shaking at 225 rpm.
Day 3: Add the 5 mL culture to 2 L of LB media with antibiotic. Grow at 37 °C shaking at 225 rpm, taking optical density (OD) reading occasionally using the spectrophotometer, until an OD600 of 0.6 is reached.
Add IPTG to a final concentration of 1 mM to induce recombinant protein expression.
Shake culture at 16 °C at 225 rpm overnight.
Day 4: Spin down bacterial culture in a centrifuge (e.g., 4633 × g for 20 min at 4 °C), and pour off the supernatant.
Add protease inhibitors (without EDTA) to lysis and wash buffers (see Note 4). Keep the solutions on ice.
Add 50–200 mL of lysis buffer to the cell pellet. Resuspend the pellet thorough by pipetting up and down.
Let the solution sit on ice for about 30 min until it becomes viscous, which can be monitored by pipetting a small amount of the mixture.
Sonicate the solution on ice for 30 s, 5 times, with 1-min cooldowns on ice in between sonication steps.
Centrifuge the samples at 30,000 × g (16,000 rpm using a Sorval SS34 rotor) at 4 °C for 30 min to pellet the cell debris.
Meanwhile, prepare the Ni-NTA agarose beads. Use 1.5 mL of slurry (50% packed beads) per liter of bacteria, for approximately 3 mL slurry. Briefly spin down the slurry at a low speed (~100 × g) and aspirate off the supernatant. Wash the beads 2× with 10 mL cold lysis buffer, briefly spinning and removing the supernatant between washes.
Add the sample supernatant to the washed beads. Rotate at 4 °C for 1–2 h.
Centrifuge the sample at 100 × g for 1 min to pellet the beads. Aspirate and discard the supernatant.
Add twice the bead volume of cold wash buffer. Resuspend, spin, and remove the supernatant. Repeat this for a total of at least five wash steps.
Add 1.5 times the bead volume of elution buffer. Resuspend and rotate for 1 h at 4 °C.
Remove the beads by spinning at (100 × g). Keep the supernatant.
Optional: Transfer the supernatant to a Polyprep column. Filter the supernatant by gravity through the column into a collector tube, according to manufacturer’s instructions.
Optional: Dialysis with a Millipore Microcon column may be performed according to manufacturer’s instructions to remove imidazole.
Dilute the protein 1:1 with glycerol, aliquot, and freeze at –20 °C.
3.2. Fluor de-Lys Assay for SIRT1 Activators In Vitro
3.2.1 Preparing Reagents
All kit components, as well as all dilutions of components, should be defrosted on ice until use. Aliquot the volume of needed reagent so the reagents and plates can be pre-warmed to 37 °C prior to the first incubation (see Note 5).
Include an extra number of wells when calculating the volume of reagents needed to account for pipetting error or bubbles. You will need a minimum 10 μL of each reagent per well.
A typical experiment will have each reagent with a working concentration at 3× since the wells will contain three separate parts (SIRT1, compound of interest and NAD+/Substrate) at equal volumes (30 μL total).
Dilute the SIRT1 in assay buffer to 3× (0.06 U/μL).
Prepare 500 μL dilutions in assay buffer of 3× (300 μM) resve-ratrol and suramin and aliquot five 100 μL tubes to be used for up to three freeze-thaw cycles each.
NAD+/Substrate should be combined into 1 master mix of assay buffer at 3× (75 μM) each for ease of measuring and pipetting (see Note 6).
Final concentrations of water, DMSO, or ethanol >1% can affect the SIRT1 activity, so all compounds should be resus-pended in assay buffer, when possible.
Within 15 min of use, dilute the 5× developer into assay buffer with nicotinamide. The 1 mL of working concentration developer will contain 760 μL assay buffer, 200 μL 5× developer, and 40 μL of 50 mM (stock concentration) nicotinamide. You will need 30 ϑL of developer per well.
3.2.2. Performing the Assay
Table 1 is a scheme of what a typical experiment will look like with the Fluor de Lys assay.
Prepare the solutions as described above and put on ice.
Place the plate and solutions of SIRT1, resveratrol, suramin, NAD+/Substrate, and test compounds in a 37 °C incubator to pre-warm for 10 min (see Note 7).
Add 10–20 μL of assay buffer to the appropriate wells.
Add 10 μL of the 3× resveratrol, suramin, or the test compounds to their respective wells.
Add 10 μL of the 3× SIRT1 to all of the wells, except for the No Enzyme wells (see Note 8).
At this step, you can either immediately move on, or wait 5–10 min for the compounds to interact with the SIRT1.
Add 10 μL of the 3× NAD+/Substrate to all wells. This must be done last, as it will start the reaction.
Cover the plate with foil and incubate at 37 °C for the desired length of time (30–60 min).
Once there is ~10 min in the incubation left, quickly thaw the 5× developer and nicotinamide and make the 1× developer mixture.
Add 30 μL of the 1× developer to all wells (see Note 9).
Recover the plate with foil and leave at RT for at least 15 min before reading. (30–60 min will have the reaction fully stopped and at max signal.)
Read the plate in a top-reading microplate fluorometer with an excitation of 360 nm and emission reading of 460 nm (see Note 10).
Table 1. Example of assay mixtures (per well volume).
| Sample | Assay buffer (μL) |
SIRT1 (3×) (μL) |
Amount of sample (3×) (μL) |
NAD+/substrate (3×) (μL) |
|---|---|---|---|---|
| No enzyme | 20 | 0 | 0 | 10 |
| Control | 10 | 10 | 0 | 10 |
| Resveratrol | 0 | 10 | 10 | 10 |
| Suramin | 0 | 10 | 10 | 10 |
| Test compound | 0 | 10 | 10 | 10 |
3.3. PNC1-OPTAssay for SIRT1 Activators In Vitro
3.3.1. Setup and Reaction (Involves Steps 1 and 2)
Dilute 100 mM nicotinamide stock solution to make 0, 5, 10, 20, 30, 40, and 50 μM standards. Pipette 1 μL of each standard into its respective well of a 96-well plate. (The final nicotinamide concentrations will be 100-fold further diluted.)
Add 1 μg yPNC1 with 100 μL of the reaction buffer to each standard well and mix by pipetting.
Prepare a master-mix on ice. Since an extra set of reactions will be used to account for background fluorescence, the matermix volume should be enough for assaying two times the number of samples in triplicates. Each reaction needs the following: 10–30 μM acetylated peptide substrate, ~1–2 μg yPnc1 enzyme, ~1–2 μg SIRT1 enzyme (see Note 11) and reaction buffer (100 μL minus the volume of other components). Mix by gentle vortexing.
Split the master-mix into two tubes. Add 75–200 μM final concentration of NAD+ to one tube and add an equal volume of water to another tube (see Note 12). Mix again by gentle vortexing.
For each compound sample to be tested, apply 100 μL of master-mix without NAD+ to three wells, and 100 μL mastermix with NAD+ to three wells (all performed in triplicate). Then the same amount of test compound, such as a potential SIRT1 activator, can be added into each well. Test compounds must not alter yPnc1 activity or diminish the fluorescence signal. This can be discerned by incubating compound with a nicotinamide standard curve in this assay. Add equal volume of solvent (e.g. DMSO, the solvent for resveratrol) into control wells (see Note 13). Mix by pipetting.
Gently tap the plate to mix the solutions, and cover with a plate sealer. Incubate at 37 °C on a plate shaker with gentle agitation for 1 h. Longer incubation times may be needed if: (1) the enzyme has lower activity; (2) the activity on the assayed substrate is weak; or (3) the nicotinamide standard curve doses are very high.
3.3.2. Development (Step 3)
OPT Developer Reagent should be warm at 37 or 42 °C for 15 min (avoid light) before developing. Vortex if precipitation of DTT is observed.
Following the incubation period, add 100 μL of OPT Developer Reagent into each well quickly under dimly lit or dark conditions with a multichannel pipette. Gently tap to mix the solutions, then use plate sealer to cover the 96-wells and wrap it with aluminum foil. Put the plate on a plate shaker in room temperature for 1 h with gentle agitation.
Read the fluorescence on a spectrophotometer under dim light. A 0.1–1 s read time is recommended. Filters set to excitation ~420 nm (±10), and emission ~460 nm (±20).
3.3.3. Analysis
Use the nicotinamide standards to plot a standard curve of fluorescence intensity versus nicotinamide concentration.
Calculate the net fluorescence for each reaction condition by subtracting the mean fluorescence of the background control reactions (no NAD+) from the experimental reaction (with NAD+), F-corrected = F + NAD–F — NAD control (mean value). If high dose of NAD+ is used, performing parallel reactions with and without SIRT1 enzyme will be more appropriate.
Using the linear equation generated from the standard curve, net fluorescence can be converted into amounts of nicotinamide. More nicotinamide produced during the reaction represents higher enzyme activity.
3.4. Mitochondrial Assays for SIRT1 Activators in Cells
3.4.1. Method Introduction
While for many years, isolated mitochondria were the preferred choice to investigate the role of the organelle in bioenergetic tissues, several techniques have emerged to analyze mitochondria within the cell. As with any technique, these methods have advantages and disadvantages. As the cellular environment of the mitochondria are maintained, in situ methods may be more physiologically relevant. However, many substrates and reagents that work on isolated mitochondria cannot be used within intact cells due the restricted permeability of the plasma membrane. While it is generally difficult to culture primary cells from adult animals for in situ comparisons, isolation of mitochondria can be used to compare animals of different ages, genotypes, or treatments.
Finally, while we frequently analyze mitochondrial function to study cellular bioenergetics, it is important to remember that although the primary function of mitochondria is to fuel ATP to the cell, additional processes are related to mitochondria, such as removal of ROS, ion transport, calcium buffering, and metabolism of fatty acids, amino acids, and nucleotides. Mitochondrial dysfunction can be observed when a failure in any one of these processes is present. Conclusions must take into account that altered bioenergetic behavior may be a cause or an effect of cellular dysfunction.
In this section of the chapter, we describe methodology for the investigation of in situ mitochondrial bioenergetics in response to SIRT1 activators using fluorescent dyes (Table 2).
Table 2. Mitochondrial selective dyes.
| Dye | Conc. | Measurement | Wavelength excitation/ emission |
FACS channel |
Reference |
|---|---|---|---|---|---|
| TMRM | 2–100 nM | Mitochondrial membrane potential | 552/575 | PE | Invitrogen T-668 |
| DHE | 10 μM | Reactive oxygen species | 490/590 | PE | Sigma D7008 |
| NAO | 10 nM | Mitochondrial mass | 495/538 | GFP | Invitrogen A1372 |
| MitoTracker® Deep Red FM | 10 nM | Mitochondrial mass | 644/665 | APC | Invitrogen M22426 |
3.4.2. Performing the Assay
Plate MEFs and/or C2C12 cells at 60–70% cell confluence (see Note 14).
On the following day treat cell with STACs (see Note 15).
Remove the media and wash the cell with PBS.
Incubate cells with new fresh media supplemented with either 10 nM TMRM for 45–60 min, 10 μM DHE for 30–60 min, 10 nM MitoTracker® Deep Red FM for 30–60 min, or 10 nM NAO for 30–60 min at 37 °C in a 5% CO2 incubator (see Note 16). NAO and MitoTracker® Deep Red FM may be incubated and analyzed together, as their spectra do not interfere with one another.
Remove the media and wash the cell with PBS.
Add trypsin to the cells and wait for them to detach.
Collect media and cells in a 15 mL conical tube.
Pellet cells by centrifugation at 300 × g for 3 min.
Remove the supernatant and add 300 μL of PBS without the probe and transfer all cells to a FACS tube.
Keep cells on ice.
Read in the fluorescence-activated cell sorting (FACS) machine (10,000 or more events) using appropriate channel for the fluorescence intensity of the dye. Analyze by taking median or mean fluorescence of a sample.
Acknowledgments
We are grateful for support from the Paul F. Glenn Foundation for Medical Research. This work was supported by NIH R37 AG028730, R01AG019719, R01 DK100263 and R21 DE027490. J.A.A. is recipient of scholarship from the Portuguese national funds via FCT – Fundação para a Ciência e a Tecnologia (PD/ BD/114173/2016).
4 Notes
To reduce background fluorescence, shorter peptides or peptides with fewer aromatic groups are preferred.
In situ experiments on any type of cells loaded with membrane potential dyes require a careful interpretation [25, 26]. Typically, as we select the best dye to use in our experiments, it is necessary to consider if the dye can be used in either quenching or non-quenching modes [26, 27]. Quenching mode approaches are a sensitive way to monitor real-time effects on mitochondrial membrane potential (ΔΨm) in response to acute application of experimental treatment strategies (i.e., pharmacologic or toxic treatments) [25, 26, 28]. They require higher dye concentrations (50–100 nM to several micromolar), so that the dye accumulates within mitochondrial matrix in a sufficient concentration to form aggregates, thus quenching some fluorescent emissions of the aggregated dye (a phenomenon called autoquenching). Consequently, quenching mode is a nonlinear event [27], which does not detect preexisting differences in ΔΨm between two different populations (i.e., two different cell lines). On the other hand, in non-quenching mode, a lower dye concentration is used (2–30 nM), thus circumventing dye aggregation and quenching in the mitochondrial matrix. Experiments involving dye loading after experimental treatments affecting ΔΨm (i.e., STACs treatment before the dye loading to compare preexisting relative mitochondrial polarization) should typically utilize dyes in non-quenching mode [25, 26, 28], since at very low concentrations, the fluorescent signal shows a linear relationship with the concentration of the dye. Thus, the concentration of the dye reflects the Nernstian distribution of the dye between compartments in response to local changes in potential [27]. The use of more permeant dyes, like TMRM, is therefore advantageous for this experiment. Importantly, empirical determination of the right concentration of TMRM for non-quenching experiments should be the first step in such a study.
The mitochondrial uptake of NAO has been shown to be independent of ΔΨm, unlike many other mitochondrial dyes. At a high concentration shows toxicity and can bind to cardiolipin in all mitochondria, regardless of their energetic state.
EDTA must not be used, as it can interfere with the Ni beads.
The assay undergoes two incubation steps. The first at 37 °C and the second, which uses the developer, at room temperature. It is best to keep the developer and nicotinamide for the second incubation frozen until just prior to use. Do not refreeze the assay buffer between the incubations.
The concentration of NAD+ can be changed to increase or decrease the amount of signal produced by SIRT1 activity. This allows for better optimization for signal vs. reagent use.
Using a white plate can give ~5× higher signal than that of an transparent plate, which when read in a top-reading fluorometer can greatly increase the sensitivity of the assay.
Having a negative control of no SIRT1 added is recommended to ensure there is no increased background and/or interactions with the compounds of interest.
Taking a measurement of the plate prior to adding developer can be useful as another background measurement.
Due to the fluorescence, this assay has a good tolerance for extracts that may discolor the wells. However, proper background readings should still be taken to ensure accuracy.
For enzymes that have lower activity than SIRT1, the protein amount added should be increased (e.g., SIRT6), and longer incubations performed.
NAD+ could exhibit fluorescence at high concentrations (>200 μM). If high NAD+ concentrations are required, the background reaction formulation can be altered. Instead of not adding NAD+, background reactions can include NAD+ but exclude the enzyme or use the corresponding non-acetylated peptide.
DMSO may inhibit the enzyme activity and lower the signal. Therefore, adding equal amount of DMSO in the same concentration to control wells is important. Typically, <4% DMSO in the final reaction is desirable.
Each cell line should be assessed individually to determine the optimal cell density.
Empirical determination of the concentration of STACs, as well as determination of the time points to be used, is one of the first steps in an experiment.
For a complete understanding of the results, appropriate controls should be done. In addition to the group of cells treated with only vehicle when measuring (ΔΨm, a protonophore such as FCCP or CCCP (widely used to depolarize mitochondria and collapse (ΔΨm, 2–10 μM at 37 °C, 5% CO2 incubator for 15–30 min) serves as a positive control. Titration of the FCCP or CCCP may be necessary for optimal results across different cell lines. When measuring ROS levels, antimycin A at 5 μM or H2O2 at 100–300 μM, at 37 °C, 5% CO2 incubator for 15–30 min, should be used as a positive control.
Disclosure: D.A.S. is a founder, equity owner, board member, advisor to, director of, consultant to, investor in and/or inventor on patents licensed to Vium, Jupiter Orphan Therapeutics, Cohbar, Galilei Biosciences, GlaxoSmithKline, OvaScience, EMD Millipore, Wellomics, Inside Tracker, Caudalie, Bayer Crop Science, Longwood Fund, Zymo Research, EdenRoc Sciences (and affiliates Arc-Bio, Dovetail Genomics, Claret Bioscience, Revere Biosensors, UpRNA and MetroBiotech (an NAD booster company), Liberty Biosecurity), Life Biosciences (and affiliates Selphagy, Senolytic Therapeutics, Spotlight Biosciences, Animal Biosciences, Iduna, Immetas, Prana, Continuum Biosciences, Jumpstart Fertility (an NAD booster company), and Lua Communications). D.A.S. sits on the board of directors of both companies. D.A.S. is an inventor on a patent application filed by Mayo Clinic and Harvard Medical School that has been licensed to Elysium Health; his personal royalty share is directed to the Sinclair lab. For more information see https://genetics.med.harvard.edu/sinclair-test/people/sinclair-other.php. Y.L. is an equity owner of Iduna.
References
- 1.Kaeberlein M, McVey M, Guarente L (1999) The SIR2/3/4 complex and SIR2 alone promote longevity in Saccharomyces cerevisiae by two different mechanisms. Genes Dev 13:2570–2580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Satoh A, Brace CS, Rensing N et al. (2013) Sirtl extends life span and delays aging in mice through the regulation of Nk2 homeobox 1 in the DMH and LH. Cell Metab 18:416–430. 10.1016/j.cmet.2013.07.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Imai S, Armstrong CM, Kaeberlein M, Guarente L (2000) Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 403:795–800. 10.1038/35001622 [DOI] [PubMed] [Google Scholar]
- 4.Luo J, Nikolaev AY, Imai S et al. (2001) Negative control of p53 by Sir2alpha promotes cell survival under stress. Cell 107:137–148. 10.1016/S0092-8674(01)00524-4 [DOI] [PubMed] [Google Scholar]
- 5.Vaziri H, Dessain SK, Ng Eaton E et al. (2001) hSIR2(SIRT1) functions as an NAD-dependent p53 deacetylase. Cell 107:149–159 [DOI] [PubMed] [Google Scholar]
- 6.Rodgers JT, Lerin C, Haas W et al. (2005) Nutrient control of glucose homeostasis through a complex of PGC-lalpha and SIRT1. Nature 434:113–118. 10.1038/nature03354 [DOI] [PubMed] [Google Scholar]
- 7.Yeung F, Hoberg JE, Ramsey CS et al. (2004) Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 23:2369–2380. 10.1038/sj.emboj.7600244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Guarani V, Deflorian G, Franco CA et al. (2011) Acetylation-dependent regulation of endothelial Notch signalling by the SIRT1 deacetylase. Nature 473:234–238. 10.1038/nature09917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhang J (2007) The direct involvement of SirT1 in insulin-induced insulin receptor substrate-2 tyrosine phosphorylation. J Biol Chem 282:34356–34364. 10.1074/jbc.M706644200 [DOI] [PubMed] [Google Scholar]
- 10.Price NL, Gomes AP, Ling AJ et al. (2012) SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell Metab 15:675–690. 10.1016/j.cmet.2012.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bonkowski MS, Sinclair DA (2016) Slowing ageing by design: the rise of NAD+ and sirtuin-activating compounds. Nat Rev Mol Cell Biol 17:679–690. 10.1038/nrm.2016.93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hubbard BP, Gomes AP, Dai H et al. (2013) Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 339:1216–1219. 10.1126/science.1231097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Howitz KT, Bitterman KJ, Cohen HY et al. (2003) Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 425:191–196. 10.1038/nature01960 [DOI] [PubMed] [Google Scholar]
- 14.Baur JA, Pearson KJ, Price NL et al. (2006) Resveratrol improves health and survival of mice on a high-calorie diet. Nature 444:337–342. 10.1038/nature05354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mitchell SJ, Martin-Montalvo A, Mercken EM et al. (2014) The SIRT1 activator SRT1720 extends lifespan and improves health of mice fed a standard diet. Cell Rep 6:836–843. 10.1016/j.celrep.2014.01.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mercken EM, Mitchell SJ, Martin-Montalvo A et al. (2014) SRT2104 extends survival of male mice on a standard diet and preserves bone and muscle mass. Aging Cell 13:787–796. 10.1111/acel.12220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hausenblas HA, Schoulda JA, Smoliga JM (2015) Resveratrol treatment as an adjunct to pharmacological management in type 2 diabetes mellitus—systematic review and meta-analysis. Mol Nutr Food Res 59:147–159. 10.1002/mnfr.201400173 [DOI] [PubMed] [Google Scholar]
- 18.Krueger JG, Suárez-Fariñas M, Cueto I et al. (2018) A randomized, placebo-controlled study of SRT2104, a SIRT1 activator, in patients with moderate to severe psoriasis. PLoS One 10:e0142081. 10.1371/journal.pone.0142081 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dai H, Ellis JL, Sinclair DA, Hubbard BP (2016) Synthesis and assay of SIRT1-activating compounds. Meth Enzymol 574:213–244. 10.1016/bs.mie.2016.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wegener D, Wirsching F, Riester D, Schwienhorst A (2003) A fluorogenic histone deacetylase assay well suited for high-throughput activity screening. Chem Biol 10:61–68 [DOI] [PubMed] [Google Scholar]
- 21.Pacholec M, Bleasdale JE, Chrunyk B et al. (2010) SRT1720, SRT2183, SRT1460, and resveratrol are not direct activators of SIRT1. J Biol Chem 285:8340–8351. 10.1074/jbc.M109.088682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hubbard BP, Sinclair DA (2013) Measurement of sirtuin enzyme activity using a substrate-agnostic fluorometric nicotinamide assay. Methods Mol Biol 1077:167–177. 10.1007/978-1-62703-637-5_11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gomes AP, Price NL, Ling AJ et al. (2013) Declining NAD( + ) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155:1624–1638. 10.1016/j.cell.2013.11.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Funk JA, Odejinmi S, Schnellmann RG (2010) SRT1720 induces mitochondrial biogenesis and rescues mitochondrial function after oxidant injury in renal proximal tubule cells. J Pharmacol Exp Ther 333:593–601. 10.1124/jpet.109.161992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ward MW, Rego AC, Frenguelli BG, Nicholls DG (2000) Mitochondrial membrane potential and glutamate excitotoxicity in cultured cerebellar granule cells. J Neurosci 20: 7208–7219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicholls DG, Ward MW (2000) Mitochondrial membrane potential and neuronal glutamate excitotoxicity: mortality and millivolts. Trends Neurosci 23:166–174 [DOI] [PubMed] [Google Scholar]
- 27.Duchen MR (2004) Mitochondria in health and disease: perspectives on a new mitochondrial biology. Mol Asp Med 25:365–451. 10.1016/j.mam.2004.03.001 [DOI] [PubMed] [Google Scholar]
- 28.Nicholls DG (2006) Simultaneous monitoring of ionophore-and inhibitor-mediated plasma and mitochondrial membrane potential changes in cultured neurons. J Biol Chem 281: 14864–14874. 10.1074/jbc.M510916200 [DOI] [PubMed] [Google Scholar]