

Abstract
Background
Gene duplications have a major role in the evolution of new biological functions. Theoretical studies often assume that a duplication per se is selectively neutral and that, following a duplication, one of the gene copies is freed from purifying (stabilizing) selection, which creates the potential for evolution of a new function.
Results
In search of systematic evidence of accelerated evolution after duplication, we used data from 26 bacterial, six archaeal, and seven eukaryotic genomes to compare the mode and strength of selection acting on recently duplicated genes (paralogs) and on similarly diverged, unduplicated orthologous genes in different species. We find that the ratio of nonsynonymous to synonymous substitutions (Kn/Ks) in most paralogous pairs is <<1 and that paralogs typically evolve at similar rates, without significant asymmetry, indicating that both paralogs produced by a duplication are subject to purifying selection. This selection is, however, substantially weaker than the purifying selection affecting unduplicated orthologs that have diverged to the same extent as the analyzed paralogs. Most of the recently duplicated genes appear to be involved in various forms of environmental response; in particular, many of them encode membrane and secreted proteins.
Conclusions
The results of this analysis indicate that recently duplicated paralogs evolve faster than orthologs with the same level of divergence and similar functions, but apparently do not experience a phase of neutral evolution. We hypothesize that gene duplications that persist in an evolving lineage are beneficial from the time of their origin, due primarily to a protein dosage effect in response to variable environmental conditions; duplications are likely to give rise to new functions at a later phase of their evolution once a higher level of divergence is reached.
Background
Gene duplications are traditionally considered to be a major evolutionary source of new protein functions. The conventional view, pioneered by Susumu Ohno, holds that a gene duplication produces two functionally redundant, paralogous genes and thereby frees one of them from selective constraints. This unconstrained paralog is then free to accumulate neutral mutations that would have been deleterious in a unique gene [1]. Although the most likely outcome of such neutral evolution is for one of the paralogs to fix a null mutation and become a pseudogene, there is also the possibility of fixation of mutations that lead to a new function [2,3,4,5,6].
One of the predictions of the conventional model of evolution of duplicated genes is the rapid loss of paralogs due to null mutations [2,3,5,7]. However, this prediction was not supported by studies on isozyme spectra of polyploids in a number of organisms (reviewed in [8]). Furthermore, a study of 17 pairs of duplicated genes in the tetraploid frog Xenopus laevis has shown that both copies were subject to purifying selection [9], contrary to the notion of neutrality of one of the copies [1]. The failure of empirical research to support Ohno's model has led to the proposal of two alternative hypotheses.
The 'subfunctionalization' hypothesis is based on the same assumption as Ohno's model, namely that duplicated genes are redundant in function and, accordingly, a duplication event is selectively neutral [6,10]. However, it was argued that, as natural selection does not 'know' which duplicated gene should be under selection and which is free of selective constraint, both paralogs experience a period of relaxed selection and accelerated evolution. During this period, both genes might accumulate mutations that impair different functions of the ancestral gene, so that, after a certain point, none of the paralogs is capable of substituting for the ancestor [6,10].
The second hypothesis holds that a gene duplication that leads to a new function is preceded by a period of 'gene sharing', such that the original, unduplicated gene encodes two distinct functions. When two paralogs of such a bifunctional gene are produced by duplication, each may be driven by positive selection to specialize in one of these functions, which it performs more efficiently than the ancestor gene, leading to the creation of two indispensable genes with distinct functions [11].
The strong interest in the evolution of gene duplications stems from the notion that duplication leads to new functions [1,12,13]. With the increasing availability of genomic data, it became clear that numerous gene families have diverged in function through series of duplications [14,15], including many lineage-specific expansions identified in each of the genomes sequenced [16,17,18,19]. Such creation of novel gene functions obviously provides a long-term, but not a short-term [20] advantage for gene duplication.
Duplicated genes have also been observed to affect fitness immediately after duplication (see Discussion), providing a short-term advantage for duplication. Although these observations remained largely unnoticed by evolutionary biologists investigating the evolution of gene duplications, such short-term advantage is crucial to the long-term fate of a duplication as group selection favoring a duplication because of its long-term advantage cannot overcome the individual selection that depends exclusively on short-term effects [20]. A recent genome-wide analysis of duplicated genes of eukaryotes has suggested that they evolve under purifying selection, with an apparent early phase of relaxed constraint or even near-neutrality [21]. Here we show that the same pattern of purifying selection is observed among bacterial and archaeal paralogs and demonstrate that amino-acid substitution rate is greater in paralogs than in orthologs with the same level of divergence at synonymous sites. We also present evidence in support of genome-wide short-term advantage of duplicated genes, and discuss its relevance to the evolution of new gene functions.
Results
We identified recent gene duplications in 26 bacterial, six archaeal and three eukaryotic complete genomes, two completely sequenced chromosomes from Arabidopsis, and among the available sequences from three mammalian species - human, mouse and rat (Table 1). Because the duplications were identified through the similarity of full-length protein sequences (see Materials and methods), this procedure primarily, if not exclusively, detected functional genes rather than pseudogenes. For comparison, orthologs with no apparent recent duplications were identified in pairs of closely related organisms, namely the bacteria Chlamydia trachomatis and C. muridarum (603 orthologous pairs)and two strains of Helicobacter pylori (1,202 orthologous pairs), and the three mammals (3,428 orthologous pairs).
Table 1.
Ratio of nonsynonymous to synonymous substitution rates (Kn/Ks) for recently diverged paralogs (0.05 < Ks < 0.5)
| Archaea | Bacteria | S. cerevisiae | A. thaliana | C. elegans | D. melanogaster | Mammalia | |
| Total number of paralogous clusters | 43 | 330 | 166 | 325 | 318 | 103 | 2,948 |
| Average ± standard error (standard deviation) | 0.266 ± 0.062 (0.231) | 0.346 ± 0.033 (0.301) | 0.104 ± 0.020 (0.171) | 0.293 ± 0.013 (0.149) | 0.244 ± 0.015 (0.162) | 0.233 ± 0.041 (0.180) | 0.451 ± 0.016 (0.257) |
Selection was measured in terms of the ratio of the rate of nonsynonymous substitutions (Kn), which are usually subject to selective pressure, to the rate of synonymous substitutions (Ks), which are assumed to be (nearly) neutral. A Kn/Ks ratio of 1 is assumed to indicate neutrality, Kn/Ks > 1 is a signature of positive selection at the amino-acid level, and Kn/Ks < 1 is indicative of purifying selection, which is the most common mode of selection [14,15]. Such analysis is applicable to homologs with a relatively low level of divergence, in which synonymous substitutions have not yet reached saturation.
In all genomes analyzed, the nonsynonymous substitution rate was significantly lower than the synonymous substitution rate, such that Kn/Ks << 1 (Figure 1a,b and Table 1), in agreement with the recent genome-wide study of paralogs [21]. Pairwise comparisons of the Kn/Ks values for different clades revealed that the strength of selection was generally similar in bacterial, archaeal and eukaryotic sets of paralogous genes, although apparently stronger selection was observed among the yeast paralogs and weaker selection was seen in mammalian paralogs (Tables 1,2).
Figure 1.
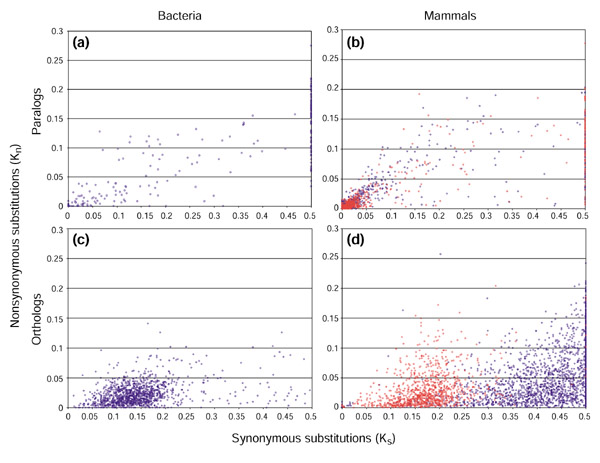
Synonymous and nonsynonymous substitution rates for (a,b) paralogs and (c,d) orthologs from (a,c) bacteria and (b,d) mammals. All points with Ks > 0.5 (approaching saturation) were assigned a Ks value of 0.5. In (b), the blue circles represent human duplicated genes and the red circles show duplicated genes from mouse and rat. In (d), the blue circles are human-rodent orthologous comparisons, and the red circles are the mouse-rat comparisons.
Table 2.
p-values of pairwise Student's t-tests of the Kn/Ks(0.05 < Ks < 0.5) ratios from Table 1 for different lineages
| Bacteria | S. cerevisiae | A. thaliana | C. elegans | D. melanogaster | Mammalia | |
| Archaea | 0.359 | 0.0706 | 0.746 | 0.895 | 0.708 | 0.0456 |
| Bacteria | 3.65e-10 | 0.128 | 0.00538 | 0.0370 | 0.0513 | |
| S. cerevisiae | 1.47e-12 | 1.07e-7 | 0.00915 | 5.74e-28 | ||
| A. thaliana | 0.0141 | 0.181 | 7.57e-14 | |||
| C. elegans | 0.789 | 4.81e-19 | ||||
| D. melanogaster | 5.71e-5 |
Significant p-values (< 0.05) are in bold.
The observation that Kn << Ks for most of the recently diverged paralogs is, in itself, not sufficient to conclude that both paralogs in each pair are subject to purifying selection. The same result would have been obtained if, in accordance with Ohno's original model, one of the paralogs was free from selective constraints, whereas the other one was maintained by purifying selection. Therefore, we carried out a relative rate test on amino-acid substitutions [15] on those pairs of paralogs, for which an ortholog was available that was more distant from either of the paralogs than the paralogs were from each other (orthologs that were too distant to be used for the relative rate test were discarded; see Materials and methods). A comparison of the evolutionary distances between each of the paralogs and the ortholog used as an outgroup showed that, of a total of 101 analyzed pairs, only five evolve at significantly (p < 0.05) different rate. The proportions of paralogous pairs that evolved at significantly different rates in each of the analyzed clades are shown in Table 3. Thus, in the majority of cases at least, both paralogs evolved under similar levels of purifying selection, in agreement with the results of an early analysis of 17 pairs of paralogs from X. laevis and a more recent analysis of 19 pairs of paralogs in human and teleost lineages [9,22].
Table 3.
Results of the relative rate test for recently diverged paralogs
| Archaea | Bacteria | S. cerevisiae | C. elegans | D. melanogaster | Mammalia | |
| Ndiff/Niden | 2/7 | 1/10 | 0/15 | 0/2 | 2/11 | 2/49 |
Ndiff, number of paralogous genes pairs that evolved at significantly (p < 0.05) different rates; Niden, number of paralogous pairs that evolved at (approximately) the same rate.
We next sought to compare the strength of purifying selection acting on paralogs to that acting on orthologs with the same level of sequence divergence, which were identified as bidirectional best hits [23] between two pairs of bacterial genomes (C. trachomatis-C. muridarum and two strains of H. pylori) and three pairs of mammalian species (human-mouse, human-rat and mouse-rat). The results of this comparison indicated that paralogs were subject to significantly weaker purifying selection than similarly diverged orthologs (compare Figure 1a and c; b and d). Specifically, the Kn/Ks ratio for orthologs was approximately twice the ratio for paralogs in bacteria, and approximately three times the ratio for paralogs in mammals (Table 4).
Table 4.
A comparison of the Kn/Ks ratios for paralogs and orthologs
| Bacteria | Mammals | |
| All proteins | ||
| Paralogs | 0.346 ± 0.033 (0.301) | 0.451 ± 0.016 (0.257) |
| Orthologs | 0.164 ± 0.004 (0.128) | 0.131 ± 0.003 (0.131) |
| Proteins with predicted signal peptides (probably secreted) | ||
| Paralogs | 0.352 ± 0.079 (0.137) | 0.476 ± 0.038 (0.239) |
| Orthologs | 0.184 ± 0.014 (0.118) | 0.197 ± 0.014 (0.174) |
| Predicted membrane proteins | ||
| Paralogs | 0.342 ± 0.036 (0.185) | 0.448 ± 0.030 (0.246) |
| Orthologs | 0.162 ± 0.004 (0.107) | 0.142 ± 0.004 (0.132) |
The numbers are the average Kn/Ks ratio ± standard error (standard deviation), 0.05 < Ks < 0.5.
Because we restricted our gene comparisons to cases in which the synonymous divergence rate (Ks) did not exceed 0.5 substitutions per site, a substantial part of the human-rodent orthologous comparisons was excluded from the analysis (Figure 1d). The use of the orthologous comparisons with a lower Ks, and thus with potentially stronger selection in the synonymous sites, could, in principle, bias our comparison of the Kn/Ks ratio. To rule out this possibility, we calculated the Kn/Ks ratio for the center of the rodent orthologous comparisons (0.15 < Ks < 0.25) and compared it to the Kn/Ks ratio of paralogs within the same range of Ks. The difference in the Kn/Ks ratio in this range of Ks (0.448 in paralogs versus 0.175 in orthologs) was not substantially dif ferent from the difference between the Kn/Ks ratios for 0.05 < Ks < 0.5 (Table 4). Extrapolating this observation to the human-rodent comparisons, it appears most likely that the exclusion of the human-rodent orthologous pairs with Ks > 0.5 did not substantially bias the results.
Weak selection acting on synonymous sites or differences in mutation bias among related species might potentially result in homogeneity of codon usage in genes within one genome, which would translate into a much greater heterogeneity of codon vocabulary in orthologous genes compared to paralogous genes. Under this scenario, synonymous divergence could be substantially underestimated for paralogous, but not for orthologous genes in our dataset, falsely suggesting an acceleration of evolution of amino-acid substitutions (larger Kn/Ks values) in paralogous genes. If there was such homogeneity of codon usage in paralogous genes, paralogs should show a greater correlation between codon bias and Ks values than orthologs. This prediction is not supported by linear least-square regressions, which fail to detect any substantial differences in the correlation between Ks and codon bias in paralogous versus orthologous genes (data not shown), suggesting that the observation of a higher Kn/Ks ratio in paralogous genes cannot be explained by an underestimate of synonymous divergence in paralogs.
Because genes with different functions evolve at different rates [24,25], the observed relaxation of selection in paralogs could potentially be due to an over-representation of fast-evolving genes in the paralogous gene set. In an attempt to isolate the effect of duplication per se on purifying selection, we analyzed paralogs and orthologs that appeared to encode proteins with similar functions, namely secreted and membrane proteins that were identified by signal peptide and transmembrane helix prediction. The difference in the Kn/Ks ratios for paralogous versus orthologous genes was approximately the same for the predicted secreted and membrane proteins as it was for the complete analyzed gene sets (Table 4), indicating that the acceleration of evolution probably is an inherent feature of duplicated genes.
Discussion
The present analysis confirms the earlier observations that paralogs evolve under purifying selection [21], which typically acts with similar strength on both duplicated copies of genes [9,22]. Additionally, however, we found that paralogs evolve significantly faster than unduplicated genes with a similar level of divergence, which is compatible with the notion that gene duplications are a source of new protein functions.
The inconsistency of empirical evidence with Ohno's model prompts questions on the validity of some of the assumptions underlying this model. One major assumption, which was inherited by the subfunctionalization model, is that one gene copy is sufficient to perform the respective function, so that a gene duplication is redundant and has no effect on fitness [1,10]. This notion has been widely accepted, and often becomes one of the central postulates of models of duplicated gene evolution [3,7,26,27]. Should this be the case, however, a duplication event would only very rarely achieve fixation [28,29]; moreover, in the event that a duplication is slightly deleterious, it would be effectively prevented from achieving fixation [30].
Although the notion of duplication producing redundant genes is central to current theories of duplicated gene evolution, the short-term benefits of gene duplications are well known. This is illustrated by the numerous observations of adaptive gene amplifications in response to antibiotics [31,32,33], anticancer drug treatments and exposure to various toxins [34,35,36,37,38,39] or heavy metals [40,41,42,43,44], nutrient limitations [32,33,45,46,47,48,49,50], pesticide treatments [51,52,53], extreme temperatures [54,55] and symbiotic and parasitic interactions [56,57]. Combining this information with the observations that recently duplicated genes evolve under purifying selection ([21] and our present work), it seems reasonable to hypothesize that a majority of duplicated genes that achieve fixation in a population increase fitness when present in two or more copies in a genome and thus are subject to purifying selection from the moment of duplication.
Recently duplicated paralogs appear to be a nonrandom group enriched in genes coding for proteins involved in different aspects of the organisms' interaction with the environment (see Additional data files). In particular, a substantial fraction of these paralogs encode (predicted) membrane or secreted proteins. The prevalent functions of duplicated genes varied among different organisms. In bacteria, the majority of these genes encoded different types of surface molecules, which, in pathogens, are involved in interaction with substrate cells. In yeast, there was an emphasis on membrane transporters as well as on genes involved in stress response (for example, heat shock). In multicellular eukaryotes, receptors (for example, olfactory receptors) and (predicted) secreted signaling molecules were predominant. Proteins with a predicted signal peptide were more prevalent in Arabidopsis thaliana (35% of all paralogs versus 17% of all unduplicated genes from the proteome), Caenorhabditis elegans (41% among paralogs versus 31% among unduplicated genes) and Drosophila melanogaster (39% in paralogs versus 23% in orthologs) and predicted membrane proteins were more numerous in mammals (56% in paralogs versus 49% in unduplicated genes). Each of these differences in the functional composition of the sets of paralogs and unduplicated genes were statistically highly significant (p < 0.0001, χ2 test).
A number of genes for which amplification has been shown to have a role in adaptation to various conditions were among the recent duplicates in our dataset. Examples include genes for the yeast hexose transporter, whose amplification has been shown to increase fitness in a low-glucose environment [49], the nematode P-glycoprotein gene, the classical eukaryotic multidrug-resistance gene [36], and the fruit fly copper-binding metallothionein gene whose duplication is implicated in copper tolerance [40,41]. Many genes that are known to amplify in response to selection, primarily during anticancer drug treatments, were found to be present in multiple copies in mammalian genomes as well, in particular, aminoacyl-tRNA synthetases, glutamine synthetase and other anabolic enzymes, adenosine deaminase, ornithine decarboxylase, AMP deaminase, and N-acetyl glucosaminyltransferase [34,46]. Permeases, transporters, synthetases and various detoxification enzymes, which, in general, tend to amplify as a response to stressful conditions [32,33,34,46,48,51], also showed a substantial presence among the recently duplicated genes in all species (see Additional data files). Duplication of genes belonging to these functional groups is thought to be adaptive because increase in the production of the corresponding proteins may either increase the efficiency of transport of a nutrient into the cell or of toxins out of the cell, increase the rate of catabolism of toxic substances, or allow a greater rate of synthesis of metabolites, for example amino acids, that are required in increased amounts under the given conditions [32,33,34,46,48,51].
Thus, the observation that purifying selection appears to act on all recent duplicates and examination of the functions of recently duplicated genes do not support the notion that gene duplication results in true functional redundancy and duplications may achieve fixation despite being redundant [26]. The alternative hypothesis - that gene duplications are fixed in a population by positive selection in all organisms - is supported by a combination of evidence of adaptive duplications from many types of living organisms: prokaryotes [31,33,45,46,48,50,55,56], protists [35,58,59], plants [39,44], fungi [43,49], invertebrates [40,41,51,52,53], non-mammalian vertebrates [54], as well as mammalian somatic tissues [34,36,37,38]. Combining these observations with the suggestion that gene duplication may be a general mechanism of adaptation to various conditions of environmental stress [32,33,46,48,49,50,52,53,55,60], we suggest that, in both prokaryotes and eukaryotes, most paralogs that are fixed in a population have a direct effect on fitness from the moment of duplication, and aid in the adaptation to various environmental conditions, primarily through a protein dosage effect.
That the short-term benefit of a gene duplication is a direct effect on protein dosage also stems from a variety of experimental observations in a number of organisms, prokaryotic and eukaryotic. Gene duplication may be a temporary mechanism to increase protein or RNA dosage, as in the case of rRNA genes in amphibian oocytes and ciliate macronuclei, the chorion genes in some dipterans, actin genes in chicken as well as drug transporters in somatic tissues (see [34,37] for reviews). Protein dosage effects have also been demonstrated in a number of other studies of inheritable adaptive gene duplications [32,34,35,43,44,46,49,51,53,61]. Furthermore, there is evidence from the analysis of the yeast genome that duplicated genes tend to be from those sets of functions that are more highly expressed [62], supporting a general role for selection on protein dosage in duplicated genes.
We show here a substantial acceleration of evolution in recently diverged paralogs compared to orthologs with the same level of synonymous sequence divergence. This acceleration may be explained by positive selection or by a relaxation of purifying selection or by a combination of the two. A few cases of differentiation of paralogs driven by positive selection have been described [63,64,65,66]; however, such cases are notoriously difficult to uncover. Thus, we make no attempt here to examine in greater detail the selective forces that act on duplicated genes. In general, however, only a small fraction of sites in some genes appears to evolve under positive selection [15], and relaxation of purifying selection is likely to be the main mechanism behind the acceleration of evolution after duplication.
Our claim of the prevalence of the protein dosage effect as the short-term advantage of duplicated genes is not incompatible with the possibility of a long-term advantage in terms of new functions or the observation that duplicated genes evolve at a faster rate. Indeed, evolution of a new beneficial function in one of the duplicates, once a relatively high level of divergence (which will be different for different genes, depending on the specific function involved) is reached, appears to be common among duplicated genes as demonstrated for several paralogous gene families [33,52,54,67]. Inasmuch as this happens, positive selection might contribute to the observed acceleration of evolution in duplicated genes.
However, assuming that gene duplications primarily evolve under purifying selection, at least soon after duplication, the observed acceleration of evolution may be explained by epistatic interaction between gene copies. In the absence of epistasis, a fitness function is an exponential function that depends on a single additive variable called fitness potential [68,69,70,71]. Assuming that the acceleration of evolution observed in paralogs is primarily due to a relaxation of purifying selection, the fitness potential variable describing the fitness function of diverging gene copies can be thought of as the effective amount of functional protein, which depends both on gene duplication and point mutations. Assuming that all point mutations are equally deleterious, such that it takes, for example, 10 point mutations to eliminate the increase in the amount of functional protein resulting from one duplication, p = d - m/10, where p is the fitness potential, d is the number of gene copies and m is the number of deleterious point mutations. By definition, in the absence of epistasis, selection against a deleterious mutation is constant for all values of the fitness potential.
However, if a fitness function grows at a less than exponential rate, then a relaxation of selection against deleterious mutations is expected [68,69,70,71]. For example, consider a linear fitness function f(p) = 2p. In this case, the fitness of an organism with one gene copy is f(p) = 2p = 2(1) = 2, and the fitness of the same organism with one point mutation is f(p) = 2p = 2(d - m/10) = 2(1 - 1/10) = 1.8; thus the mutation reduced fitness by 10% (1 - 1.8/2). The fitness of an organism with two gene copies is f(p) = 2p = 2(2) = 4, the fitness of the same organism with two gene copies and one point mutation is f(p) = 2(2 - 1/10) = 3.8; in this case, the same mutation will cause only a 5% reduction in fitness (1-3.8/4). Thus, the observed acceleration of evolution in paralogous genes implies that either their divergence occurs under positive selection or that the fitness function for gene duplications grows at a slower than exponential rate.
Experimental evidence from several organisms supports the notion that the increase of fitness due to gene duplication is far from being exponential; more specifically, under static environmental conditions, there is an optimal gene copy number [49,50,53]. As duplication events are known to incur a fitness cost [50,53], the fitness function of duplicated genes most probably resembles an inverted parabola, such that it contains a clearly defined maximum [28,53]. Numerous observations that gene duplications that increase protein dosage can be pathogenic [61,72,73,74,75] further support the model of optimum copy number for each gene.
Thus, it seems reasonable to hypothesize that, for each gene, there is an optimum number of copies per genome that may vary depending on environmental conditions. Assuming that the environmental condition that determines the optimum gene number fluctuates, positive selection would regulate the gene copy number in the genome accordingly. Under this model, if environmental conditions, and therefore the number of gene copies, fluctuate frequently, then most paralogs will be closely related, such that an excess of (nearly) identical genes will be found within a genome, as indeed has been observed for many genomes ([21] and data not shown).
The present observation that duplicated genes experience a substantial relaxation of selection compared to unduplicated genes is compatible with the traditional view that gene duplications make a major contribution to the evolution of new gene functions. Additionally, the repertoire of protein functions among recent duplicates suggests that many gene duplications contribute to adaptation of the organism to various forms of environmental stress. The results of the present analysis of recent duplications suggest a two-stage evolutionary model of gene duplication: in the first stage, immediately after duplication and during the early phase of their evolution, paralogs are retained and are subject to purifying selection because of the short-term advantage of protein dosage regulation; at a later stage in their evolution, gene duplications are likely to provide a long-term advantage by enabling the creation of new functions.
Materials and methods
Genome sequence data
The complete genome sequences of bacteria and archaea, the yeast Saccharomyces cerevisiae, the nematode C. elegans and the fruit fly D. melanogaster, and the sequences of chromosomes II and IV of the thale cress A. thaliana were extracted from the Genomes division of the Entrez retrieval system [76]. The complete sequences of cDNAs from mouse, rat and humans were extracted from GenBank.
Data analysis
Clusters of paralogs were identified by comparing the protein sequences encoded by all genes from each genome using the BLASTP program [77] followed by single-linkage clustering using the BLASTCLUST program (I. Dondoshan-sky, Y.I.W. and E.V.K., unpublished results; [78]). Each cluster included sequences that aligned over at least 95% of their lengths with a score density of 1.5 bits per position, which approximately corresponds to 75% identity. Probable orthologs were operationally defined as bidirectional best hits between proteins from two genomes [23]. A subset of probable orthologous pairs that met the criteria used for identification of paralogs (score density of 1.5 bit/position and aligned over at least 95% of their lengths) was selected. Orthologous pairs, in which one or both of the members was identified as part of a paralogous cluster, were excluded from further analysis to ensure a set of unique genes in the orthologous dataset.
Protein sequences were aligned using the CLUSTAL W program [79] and the corresponding nucleotide-sequence alignments were derived, by substituting the respective coding sequences for the protein sequences. The number of synonymous nucleotide substitutions per synonymous site (Ks), and the number of nonsynonymous nucleotide substitutions per nonsynonymous site (Kn) were estimatedusing the Pamilo-Bianchi-Li method [80,81]. Families of paralogs that included more than two genes were separated into all possible pairs, and each pair was subjected to the same analysis, after which an average of both Ks and Kn was taken and used as a single point. However, only paralogous clusters with two paralogs were used for the relative rate test. For the substitution rate calculations, the data from 26 bacterial genomes, six archaeal genomes and three mammalian genomes were combined into three kingdom-specific datasets; the remaining eukaryotic genomes were analyzed independently. The substitution rates for orthologous gene pairs were calculated using the same approach. Pairs of paralogs and orthologs with Ks > 0.5, which approach saturation in synonymous sites, and those with Ks < 0.05, which are subject to statistical and database-annotation errors, were excluded from all calculations of the Kn/Ks ratios.
For the relative rate test, the closest ortholog from the same taxon was identified for each pair of paralogs. To ensure correct identification, those ortholog candidates that were closer to either of the paralogs than the paralogs were to each other were discarded. In the remaining triplets, which consisted of an ortholog and two paralogs, p-distances POP1, POP2 and PPP for ortholog-paralog 1, ortholog-paralog 2, and paralog-paralog pairs, respectively, were calculated from the multiple alignment. The conversion between p-distances and corresponding evolutionary distances, DOP1, DOP2 and DPP, was carried out using the gamma-distance correction [82,83] with an α parameter of 1.0. The significance of the difference between POP1 and POP2 was estimated using the normalized deviation |POP1 - POP2|/(POP1(1 - POP1)/LP1 + POP2(1 - POP2)/LP2)1/2, where LP1 and LP2 are the lengths of the paralogs [84]. To estimate the significance of the maximum possible difference between the paralogs, evolutionary rates, DOPmax and DOPmin were calculated as (DOP1 + DOP2 ± DPP)/2 (as follows from the triangle inequality, |DOP1 - DOP2| ≤ DPP). DOPmax and DOPmin were converted to the respective p-distances POPmax and POPmin; the significance of the maximum possible difference was calculated in the same manner as for the real POP1 and POP2. Triplets that failed to yield significant difference between POPmax and POPmin (because of paralogs being too similar) were discarded because no amount of evolutionary pressure (resulting in different rates of evolution in paralogs 1 and 2) would be sufficient to produce a significant difference in POP1 and POP2 on such short evolutionary distances.
For prediction of protein function, detailed sequence analysis was carried out wherever needed using the PSI-BLAST program [78] and the SMART system for protein domain identification [85]. Signal peptides in protein sequences were predicted using the SignalP program [86] and membrane proteins using the MEMSAT program [87]. Codon bias was estimated using the effective number of codons measure [88].
Additional data file
The following plain text additional data files are available with this article and at [89].
Lists of pairs of orthologs (in the form of gis, which are unique gene identifiers) between human and mouse (File 1), human and rat (File 2), rat and mouse (File 3), C. trachomatis and C. muridarum (File 4) and Helicobacter pylori 26695 and J99 (File 5).
Lists of paralogs for archaea (File 6), bacteria (File 7), mammalia (File 8), C. elegans (File 9), D. melanogaster (File 10), A. thaliana (File 11) and S. cerevisiae (File 12). Each file includes annotated lists of clusters of recently duplicated paralogs (see text for the definitions) from the corresponding genome or, in the case of archaea and bacteria, from the set of all available complete genomes. Clusters marked with '#' were excluded from the calculations described in the paper, typically because they consist of proteins encoded by mobile elements.
Supplementary Material
Pairs of orthologs between human and mouse
Pairs of orthologs between human and rat
Pairs of orthologs between rat and mouse
Pairs of orthologs between C. trachomatis and C. muridarum
Pairs of orthologs between Helicobacter pylori 26695 and J99
Lists of paralogs for archaea
Lists of paralogs for bacteria
Lists of paralogs for mammalia
Lists of paralogs for C. elegans
Lists of paralogs for D. melanogaster
Lists of paralogs for A. thaliana
Lists of paralogs for S. cerevisiae
Acknowledgments
Acknowledgements
We are grateful to A.S. Kondrashov for numerous helpful suggestions, to I. King Jordan, M.A. Roytberg, J.L. Spouge and D.A. Kondrashov for useful discussions and to A.S. Kondrashov, I. King Jordan and D.J. Lipman for critical reading of the manuscript.
References
- Ohno S. Evolution by Gene Duplication Berlin-Heidelberg-New York: Springer-Verlag, 1970.
- Kimura M, King JL. Fixation of a deleterious allele at one of two "duplicate" loci by mutation pressure and random drift. Proc Natl Acad Sci USA. 1979;76:2858–2861. doi: 10.1073/pnas.76.6.2858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh JB. How often do duplicated genes evolve new functions? Genetics. 1995;139:421–428. doi: 10.1093/genetics/139.1.421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner A. The fate of duplicated genes: loss or new function? BioEssays. 1998;20:785–788. doi: 10.1002/(SICI)1521-1878(199810)20:10<785::AID-BIES2>3.0.CO;2-M. [DOI] [PubMed] [Google Scholar]
- Stoltzfus A. On the possibility of constructive neutral evolution. J Mol Evol. 1999;49:169–181. doi: 10.1007/pl00006540. [DOI] [PubMed] [Google Scholar]
- Lynch M, Force A. The probability of duplicate gene preservation by subfunctionalization. Genetics. 2000;154:459–473. doi: 10.1093/genetics/154.1.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T. How gene families evolve. Theor Popul Biol. 1990;37:213–219. doi: 10.1016/0040-5809(90)90036-u. [DOI] [PubMed] [Google Scholar]
- Li WH. Rate of gene silencing at duplicate loci: a theoretical study and interpretation of data from tetraploid fishes. Genetics. 1980;95:237–258. doi: 10.1093/genetics/95.1.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes MK, Hughes AL. Evolution of duplicate genes in a tetraploid animal, Xenopus laevis. Mol Biol Evol. 1993;10:1360–1369. doi: 10.1093/oxfordjournals.molbev.a040080. [DOI] [PubMed] [Google Scholar]
- Force A, Lynch M, Pickett FB, Amores A, Yan YL, Postlethwait J. Preservation of duplicate genes by complementary, degenerative mutations. Genetics. 1999;151:1531–1545. doi: 10.1093/genetics/151.4.1531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes AL. The evolution of functionally novel proteins after gene duplication. Proc R Soc Lond B Biol Sci. 1994;256:119–124. doi: 10.1098/rspb.1994.0058. [DOI] [PubMed] [Google Scholar]
- Bridges CA. Salivary chromosome maps. J Hered. 1935;26:60–64. [Google Scholar]
- Lewis EB. Pseudoallelism and gene evolution. Cold Spring Harbor Symp Quant Biol. 1951;16:159–174. doi: 10.1101/sqb.1951.016.01.014. [DOI] [PubMed] [Google Scholar]
- Li WH. Molecular Evolution Sunderland, MA: Sinauer, 1997.
- Hughes AL. Adaptive Evolution of Genes and Genomes New York-Oxford: Oxford University Press, 1999.
- Chervitz SA, Aravind L, Sherlock G, Ball CA, Koonin EV, Dwight SS, Harris MA, Dolinski K, Mohr S, Smith T, et al. Comparison of the complete protein sets of worm and yeast: orthology and divergence. Science. 1998;282:2022–2028. doi: 10.1126/science.282.5396.2022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, et al. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- International Human Genome Consortium Initial sequencing and analysis of the human genome. Nature. 2001;409:860–921. doi: 10.1038/35057062. [DOI] [PubMed] [Google Scholar]
- Jordan IK, Makarova KS, Spouge JL, Wolf YI, Koonin EV. Lineage-specific gene expansions in bacterial and archaeal genomes. Genome Res. 2001;11:555–565. doi: 10.1101/gr.166001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maynard Smith J. The Evolution of Sex. 1978.
- Lynch M, Conery JS. The evolutionary fate and consequences of duplicate genes. Science. 2000;290:1151–1155. doi: 10.1126/science.290.5494.1151. [DOI] [PubMed] [Google Scholar]
- Robinson-Rechavi M, Laudet V. Evolutionary rates of duplicate genes in fish and mammals. Mol Biol Evol. 2001;18:681–683. doi: 10.1093/oxfordjournals.molbev.a003849. [DOI] [PubMed] [Google Scholar]
- Tatusov RL, Koonin EV, Lipman DJ. A genomic perspective on protein families. Science. 1997;278:631–637. doi: 10.1126/science.278.5338.631. [DOI] [PubMed] [Google Scholar]
- Kimura M. The Neutral Theory of Molecular Evolution. 1983. [DOI] [PubMed]
- Grishin NV, Wolf YI, Koonin EV. From complete genomes to measures of substitution rate variability within and between proteins. Genome Res. 2000;10:991–1000. doi: 10.1101/gr.10.7.991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nowak MA, Boerlijst MC, Cooke J, Smith JM. Evolution of genetic redundancy. Nature. 1997;388:167–171. doi: 10.1038/40618. [DOI] [PubMed] [Google Scholar]
- Wagner A. Redundant gene functions and natural selection. J Evol Biol. 1999;12:1–16. [Google Scholar]
- Koch AL. Selection and recombination in populations containing tandem multiplet genes. J Mol Evol. 1979;14:273–285. doi: 10.1007/BF01732495. [DOI] [PubMed] [Google Scholar]
- Clark AG. Invasion and maintenance of a gene duplication. Proc Natl Acad Sci USA. 1994;91:2950–2954. doi: 10.1073/pnas.91.8.2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JF, Kimura M. An Introduction to Population Genetics Theory New York: Harper & Row, 1970.
- Koch AL. Evolution of antibiotic resistance gene function. Microbiol Rev. 1981;45:355–378. doi: 10.1128/mr.45.2.355-378.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Velkov VV. Gene amplification in prokaryotic and eukaryotic systems. Genetika. 1982;18:529–543. [Google Scholar]
- Romero D, Palacios R. Gene amplification and genomic plasticity in prokaryotes. Annu Rev Genet. 1997;31:91–111. doi: 10.1146/annurev.genet.31.1.91. [DOI] [PubMed] [Google Scholar]
- Stark GR, Wahl GM. Gene amplification. Annu Rev Biochem. 1984;53:447–491. doi: 10.1146/annurev.bi.53.070184.002311. [DOI] [PubMed] [Google Scholar]
- Reinbothe S, Ortel B, Parthier B. Overproduction by gene amplification of the multifunctional arom protein confers glyphosate tolerance to a plastid-free mutant of Euglena gracilis. Mol Gen Genet. 1993;239:416–424. doi: 10.1007/BF00276940. [DOI] [PubMed] [Google Scholar]
- Gottesman MM, Hrycyna CA, Schoenlein PV, Germann UA, Pastan I. Genetic analysis of the multidrug transporter. Annu Rev Genet. 1995;29:607–649. doi: 10.1146/annurev.ge.29.120195.003135. [DOI] [PubMed] [Google Scholar]
- Schwab M. Oncogene amplification in solid tumors. Semin Cancer Biol. 1999;9:319–325. doi: 10.1006/scbi.1999.0126. [DOI] [PubMed] [Google Scholar]
- Montgomery JS, Price DK, Figg WD. The androgen receptor gene and its influence on the development and progression of prostate cancer. J Pathol. 2001;195:138–146. doi: 10.1002/1096-9896(200109)195:2<138::AID-PATH961>3.0.CO;2-Y. [DOI] [PubMed] [Google Scholar]
- Widholm JM, Chinnala AR, Ryu JH, Song HS, Eggett T, Brotherton JE. Glyphosate selection of gene amplification in suspension cultures of three plant species. Physiol Plant. 2001;112:540–545. doi: 10.1034/j.1399-3054.2001.1120411.x. [DOI] [PubMed] [Google Scholar]
- Otto E, Young JE, Maroni G. Structure and expression of a tandem duplication of the Drosophila metallothionein gene. Proc Natl Acad Sci USA. 1986;83:6025–6029. doi: 10.1073/pnas.83.16.6025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maroni G, Wise J, Young JE, Otto E. Metallothionein gene duplications and metal tolerance in natural populations of Drosophila melanogaster. Genetics. 1987;117:739–744. doi: 10.1093/genetics/117.4.739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratyeva TF, Muntyan LN, Karvaiko GI. Zinc-resistant and arsenic-resistant strains of Thiobacillus ferrooxidans have increased copy numbers of chromosomal resistance genes. Microbiology. 1995;141:1157–1162. doi: 10.1099/13500872-141-5-1157. [DOI] [PubMed] [Google Scholar]
- Tohoyama H, Shiraishi E, Amano S, Inouhe M, Joho M, Murayama T. Amplification of a gene for metallothionein by tandem repeat in a strain of cadmium-resistant yeast cells. FEMS Microbiol Lett. 1996;136:269–273. [Google Scholar]
- van Hoof NA, Hassinen VH, Hakvoort HW, Ballintijn KF, Schat H, Verkleij JA, Ernst WH, Karenlampi SO, Tervahauta AI. Enhanced copper tolerance in Silene vulgaris (Moench) Garcke populations from copper mines is associated with increased transcript levels of a 2b-type metallothionein gene. Plant Physiol. 2001;126:1519–1526. doi: 10.1104/pp.126.4.1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horiuchi T, Horiuchi S, Novick A. The genetic basis of hypersynthesis of β-galactosidase. Genetics. 1963;48:157–169. doi: 10.1093/genetics/48.2.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson RP, Roth JR. Tandem genetic duplications in phage and bacteria. Annu Rev Microbiol. 1977;31:473–505. doi: 10.1146/annurev.mi.31.100177.002353. [DOI] [PubMed] [Google Scholar]
- Hartley BS. In: In Microorganisms as Model Systems for Studying Evolution. Mortlock RP, editor. New York: Plenum Press,; 1984. pp. 23–54. [Google Scholar]
- Sonti RV, Roth JR. Role of gene duplications in the adaptation of Salmonella typhimurium to growth on limiting carbon sources. Genetics. 1989;123:19–28. doi: 10.1093/genetics/123.1.19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown CJ, Todd KM, Rosenzweig RF. Multiple duplications of yeast hexose transport genes in response to selection in a glucose-limited environment. Mol Biol Evol. 1998;15:931–942. doi: 10.1093/oxfordjournals.molbev.a026009. [DOI] [PubMed] [Google Scholar]
- Hastings PJ, Bull HJ, Klump JR, Rosenberg SM. Adaptive amplification: an inducible chromosomal instability mechanism. Cell. 2000;103:723–731. doi: 10.1016/s0092-8674(00)00176-8. [DOI] [PubMed] [Google Scholar]
- Tabashnik BE. Implications of gene amplification for evolution and management of insecticide resistance. J Econ Entomol. 1990;83:1170–1176. doi: 10.1093/jee/83.4.1170. [DOI] [PubMed] [Google Scholar]
- Lenormand T, Guillemaud T, Bourguet D, Raymond M. Appearance and sweep of a gene duplication: adaptive response and potential for new functions in the mosquito Culex pipiens. Evolution. 1998;52:1705–1712. doi: 10.1111/j.1558-5646.1998.tb02250.x. [DOI] [PubMed] [Google Scholar]
- Guillemaud T, Raymond M, Tsagkarakou A, Bernard C, Rochard P, Pasteur N. Quantitative variation and selection of esterase gene amplification in Culex pipiens. Heredity. 1999;83:87–99. doi: 10.1038/sj.hdy.6885370. [DOI] [PubMed] [Google Scholar]
- Chen L, DeVries AL, Cheng CH. Evolution of antifreeze glycoprotein gene from a trypsinogen gene in Antarctic notothenioid fish. Proc Natl Acad Sci USA. 1997;94:3811–3816. doi: 10.1073/pnas.94.8.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riehle MM, Bennett AF, Long AD. Genetic architecture of thermal adaptation in Escherichia coli. Proc Natl Acad Sci USA. 2001;98:525–530. doi: 10.1073/pnas.021448998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai CY, Baumann L, Baumann P. Amplification of trpEG: adaptation of Buchnera aphidicola to an endosymbiotic association with aphids. Proc Natl Acad Sci USA. 1994;91:3819–3823. doi: 10.1073/pnas.91.9.3819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero D, Davila G, Palacios R. The dynamic genome of Rhizobium. In: de Bruijn FJ, Lupski G, Weinstock G, editor. In Bacterial Genomes: Physical Structure and Analysis. London: Chapman and Hall,; 1997. pp. 153–161. [Google Scholar]
- Kaufmann J, Klein A. Gene dosage as a possible major determinant for equal expression levels of genes encoding RNA polymerase subunits in the hypotrichous ciliate Euplotes octocarinatus. Nucleic Acids Res. 1992;20:4445–4450. doi: 10.1093/nar/20.17.4445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segovia M. Leishmania gene amplification: a mechanism of drug resistance. Ann Trop Med Parasitol. 1994;88:123–130. doi: 10.1080/00034983.1994.11812849. [DOI] [PubMed] [Google Scholar]
- Tlsty TD, Albertini AM, Miller JH. Gene amplification in the lac region of E. coli. Cell. 1984;37:217–224. doi: 10.1016/0092-8674(84)90317-9. [DOI] [PubMed] [Google Scholar]
- Lupski JR, Roth JR, Weinstock GM. Chromosomal duplications in bacteria, fruit flies, and humans. Am J Hum Genet. 1996;58:21–27. [PMC free article] [PubMed] [Google Scholar]
- Seoighe C, Wolfe KH. Yeast genome evolution in the post-genome era. Curr Opin Microbiol. 1999;2:548–554. doi: 10.1016/s1369-5274(99)00015-6. [DOI] [PubMed] [Google Scholar]
- Zhang J, Rosenberg HF, Nei M. Positive Darwinian selection after gene duplication in primate ribonuclease genes. Proc Natl Acad Sci USA. 1998;95:3708–3713. doi: 10.1073/pnas.95.7.3708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson ME, Viggiano L, Bailey JA, Abdul-Rauf M, Goodwin G, Rocchi M, Eichler EE. Positive selection of a gene family during the emergence of humans and African apes. Nature. 2001;413:514–519. doi: 10.1038/35097067. [DOI] [PubMed] [Google Scholar]
- Merritt TJ, Quattro JM. Evidence for a period of directional selection following gene duplication in a neurally expressed locus of triosephosphate isomerase. Genetics. 2001;159:689–697. doi: 10.1093/genetics/159.2.689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van De Peer Y, Taylor JS, Braasch I, Meyer A. The ghost of selection past: rates of evolution and functional divergence of anciently duplicated genes. J Mol Evol. 2001;53:436–446. doi: 10.1007/s002390010233. [DOI] [PubMed] [Google Scholar]
- Alm RA, Guerry P, Trust TJ. Significance of duplicated flagellin genes in Campylobacter. J Mol Biol. 1993;230:359–363. doi: 10.1006/jmbi.1993.1151. [DOI] [PubMed] [Google Scholar]
- Milkman R. Selection differentials and selection coefficients. Genetics. 1978;88:391–403. doi: 10.1093/genetics/88.2.391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura M, Crow JF. Effect of overall phenotypic selection on genetic change at individual loci. Proc Natl Acad Sci USA. 1978;75:6168–6171. doi: 10.1073/pnas.75.12.6168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crow JF, Kimura M. Efficiency of truncation selection. Proc Natl Acad Sci USA. 1979;76:396–399. doi: 10.1073/pnas.76.1.396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shnol EE, Kondrashov AS. The effect of selection on the phenotypic variance. Genetics. 1993;134:995–996. doi: 10.1093/genetics/134.3.995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lupski JR. Charcot-Marie-Tooth disease: a gene-dosage effect. Hosp Pract (Off Ed) 1997;32:83–112. doi: 10.1080/21548331.1997.11443485. [DOI] [PubMed] [Google Scholar]
- Pratt VM, Roberson JR, Weiss L, Van Dyke DL. Duplication 6q21q23 in two unrelated patients. Am J Med Genet. 1998;80:112–114. doi: 10.1002/(sici)1096-8628(19981102)80:2<112::aid-ajmg4>3.0.co;2-u. [DOI] [PubMed] [Google Scholar]
- Inoue K, Osaka H, Imaizumi K, Nezu A, Takanashi J, Arii J, Murayama K, Ono J, Kikawa Y, Mito T, et al. Proteolipid protein gene duplications causing Pelizaeus-Merzbacher disease: molecular mechanism and phenotypic manifestations. Ann Neurol. 1999;45:624–632. [PubMed] [Google Scholar]
- Fan YS, Siu VM. Molecular cytogenetic characterization of a derivative chromosome 8 with an inverted duplication of 8p21.3→p23.3 and a rearranged duplication of 8q24.13→qter. Am J Med Genet. 2001;102:266–271. doi: 10.1002/ajmg.1460. [DOI] [PubMed] [Google Scholar]
- Tatusova TA, Karsch-Mizrachi I, Ostell JA. Complete genomes in WWW Entrez: data representation and analysis. Bioinformatics. 1999;15:536–543. doi: 10.1093/bioinformatics/15.7.536. [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 1997;25:3389–3402. doi: 10.1093/nar/25.17.3389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stand-alone BLAST executables ftp://ncbi.nlm.nih.gov/blast/executables
- Thompson JD, Higgins DG, Gibson TJ. CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 1994;22:4673–4680. doi: 10.1093/nar/22.22.4673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pamilo P, Bianchi NO. Evolution of the Zfx and Zfy genes: rates and interdependence between the genes. Mol Biol Evol. 1993;10:271–281. doi: 10.1093/oxfordjournals.molbev.a040003. [DOI] [PubMed] [Google Scholar]
- Li WH. Unbiased estimation of the rates of synonymous and nonsynonymous substitution. J Mol Evol. 1993;36:96–99. doi: 10.1007/BF02407308. [DOI] [PubMed] [Google Scholar]
- Ota T, Nei M. Estimation of the number of amino-acid substitutions per site when the substitution rate varies among sites. J Mol Evol. 1994;38:642–643. doi: 10.1007/BF00175826. [DOI] [PubMed] [Google Scholar]
- Grishin NV. Estimation of the number of amino-acid substitutions per site when the substitution rate varies among sites. J Mol Evol. 1995;41:675–679. doi: 10.1007/BF00175826. [DOI] [PubMed] [Google Scholar]
- Nei M, Kumar S. Molecular Evolution and Phylogenetics Oxford: Oxford University Press, 2000.
- Schultz J, Milpetz F, Bork P, Ponting CP. SMART, a simple modular architecture research tool: identification of signaling domains. Proc Natl Acad Sci USA. 1998;95:5857–5864. doi: 10.1073/pnas.95.11.5857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H, Engelbrecht J, Brunak S, von Heijne G. A neural network method for identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Int J Neural Syst. 1997;8:581–599. doi: 10.1142/s0129065797000537. [DOI] [PubMed] [Google Scholar]
- Jones DT, Taylor WR, Thornton JM. A model recognition approach to the prediction of all-helical membrane protein structure and topology. Biochemistry. 1994;33:3038–3049. doi: 10.1021/bi00176a037. [DOI] [PubMed] [Google Scholar]
- Wright F. The 'effective number of codons' used in a gene. Gene. 1990;87:23–29. doi: 10.1016/0378-1119(90)90491-9. [DOI] [PubMed] [Google Scholar]
- Annotated lists of orthologs and paralogs ftp://ncbi.nlm.nih.gov/pub/koonin/duplication
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Pairs of orthologs between human and mouse
Pairs of orthologs between human and rat
Pairs of orthologs between rat and mouse
Pairs of orthologs between C. trachomatis and C. muridarum
Pairs of orthologs between Helicobacter pylori 26695 and J99
Lists of paralogs for archaea
Lists of paralogs for bacteria
Lists of paralogs for mammalia
Lists of paralogs for C. elegans
Lists of paralogs for D. melanogaster
Lists of paralogs for A. thaliana
Lists of paralogs for S. cerevisiae