

Superhydrophilic zwitterionic polymers derived from trimethylamine N-oxide are reported as a new class of nonfouling materials.
Abstract
Materials that resist nonspecific protein adsorption are needed for many applications. However, few are able to achieve ultralow fouling in complex biological milieu. Zwitterionic polymers emerge as a class of highly effective ultralow fouling materials due to their superhydrophilicity, outperforming other hydrophilic materials such as poly(ethylene glycol). Unfortunately, there are only three major classes of zwitterionic materials based on poly(phosphorylcholine), poly(sulfobetaine), and poly(carboxybetaine) currently available. Inspired by trimethylamine N-oxide (TMAO), a zwitterionic osmolyte and the most effective protein stabilizer, we here report TMAO-derived zwitterionic polymers (PTMAO) as a new class of ultralow fouling biomaterials. The nonfouling properties of PTMAO were demonstrated under highly challenging conditions. The mechanism accounting for the extraordinary hydration of PTMAO was elucidated by molecular dynamics simulations. The discovery of PTMAO polymers demonstrates the power of molecular understanding in the design of new biomimetic materials and provides the biomaterials community with another class of nonfouling zwitterionic materials.
INTRODUCTION
Unwanted adsorption of biomolecules, cells, and microorganisms represents a great challenge in many applications from medical devices to drug delivery systems (1, 2). For example, biomaterials tend to be covered with a layer of host proteins shortly after implantation, which thereafter will provoke an irrevocable foreign body reaction (FBR) (2, 3). As a result, a variety of iatrogenic complications including inflammation, infection, tissue fibrosis, and capsulate formation will be triggered, leading to the failure of biomaterials. Likewise, drug-delivering carriers are also susceptible to the nonspecific attachment of proteins and cells, which may result in the rapid clearance of therapeutic drugs and adverse immune responses (4, 5). Therefore, reducing or eliminating undesirable “biofouling” is of paramount importance to the safety and function of medical devices or drugs. To achieve a nonfouling surface, a number of hydrophilic materials have been used, where the surface hydration plays a pivotal role (6). These materials have been shown to create a hydration shell that repels biomolecules or cells from contacting surfaces, making the underlying substrates “stealthy” (7). However, the hydration capability of most hydrophilic materials is often not sufficient to produce a nonfouling surface. At present, there are few highly hydrophilic materials that can meet the nonfouling demand of practical applications, particularly in complex biological media.
Poly(ethylene glycol) (PEG) is the most widely used stealth material. The attachment of PEG to surfaces, known as “PEGylation,” has been a “gold standard” strategy to resist nonspecific protein adsorption. Although commonly considered hydrophilic, PEG containing a hydrophobic C─C backbone along with a hydrophobic −O(CH3) terminal group is amphiphilic, as evidenced by its good solubility in many organic solvents in addition to water. As any nonspecific interaction moieties can be detected by the immune system, the hydrophobic character of PEG would be amplified under in vivo conditions, particularly when it is conjugated with highly immunogenic proteins, generating PEG-specific antibodies (8, 9). In recent decades, zwitterionic materials that contain an equal number of oppositely charged ions (zwitterions) have been emerging as a new series of hydrophilic biomaterials for nonfouling purposes as they can strongly hold water molecules via electrostatically induced hydration (10, 11). Poly(phosphorylcholine) (PPC), which mimics zwitterionic phosphorylcholine moieties outside cell membranes, has been extensively studied and used as a biomimetic fouling-resistant material in many applications over the past decades (12). Another two classes of zwitterionic materials, poly(sulfobetaine) (PSB) derived from taurine and poly(carboxybetaine) (PCB) derived from glycine betaine, have been demonstrated for their superior hydration capability and fouling resistance (13–17). Typically, PCB polymer-coated surfaces have been shown to reduce nonspecific protein adsorption to an ultralow level, i.e., <0.3 ng/cm2 in undiluted human serum or plasma, the lowest detection limit of a surface plasmon resonance (SPR) biosensor (10, 18). Furthermore, such an excellent nonfouling property has been shown to persist in vivo: PCB hydrogels implanted in mice could prevent the occurrence of FBRs such as capsule formation for at least 3 months, while PCB-coated proteins display enhanced circulation but much lessened immunogenicity in mice and rats (19–23). With these attractive benefits, zwitterionic materials are receiving increasingly high attention. Unfortunately, only three classes of zwitterionic materials (PPC, PSB, and PCB) are currently available.
Previous studies have shown that the hydration capacity along with the nonfouling property of zwitterionic polymers increases as the intramolecular distance between the positively and negatively charged sites of the zwitterionic headgroups decreases (24, 25). On the basis of this rule of thumb, we have scouted naturally occurring zwitterionic molecules and found that trimethylamine N-oxide (TMAO), a small organic osmolyte present in saltwater fishes, might be an excellent zwitterionic headgroup for nonfouling materials, as its opposing charge moieties are directly connected (Me3N+–O−) with no spacer between two charges (26). In comparison, the zwitterionic moiety of PCB that derives from glycine contains at least a one-carbon separation (RMe2N+−CH2CH2−COO−). Moreover, TMAO is renowned to stabilize the folded structures of proteins by counteracting the effects of protein denaturants (e.g., urea), heat, and pressure in the most effective way (27–30). Inspired by the superhydrophilic and protein-stabilizing nature of TMAO, we here report a TMAO-derived polymer (PTMAO) as a new-generation zwitterionic material with fully characterized properties (Fig. 1A). The exceptional nonfouling performance of PTMAO was demonstrated under harsh in vitro and in vivo conditions, and the mechanism accounting for the extraordinary hydration of PTMAO was elucidated by molecular dynamics (MD) simulations. The discovery of PTMAO represents the fourth class of nonfouling zwitterionic material after PPC, PSB, and PCB, enriching the arsenal of nonfouling materials, and is expected to benefit a wide range of applications.
Fig. 1. Scheme illustration of PTMAO.
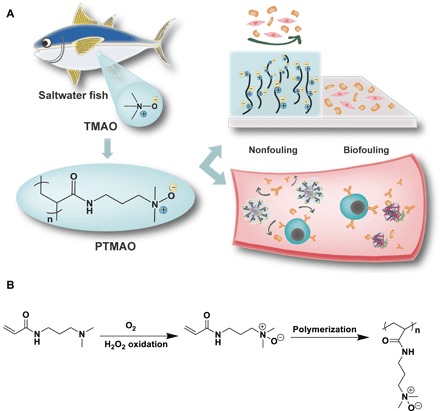
(A) The design of PTMAO is derived from TMAO, a zwitterionic osmolyte in saltwater fishes. The nonfouling property of PTMAO could effectively prevent a surface from biofouling both in vitro and in vivo. (B) Scheme of TMAO monomer and polymer synthesis.
RESULTS AND DISCUSSION
Synthesis of TMAO-analog monomer
Given the superhydrophilic and protein-stabilizing nature of TMAO, a TMAO-derived polymer is envisioned to provide a new class of nonfouling zwitterionic materials. In this work, a zwitterionic TMAO-derived monomer was synthesized by oxidizing N,N-dimethylaminopropyl acrylamide (DMAPA) (Fig. 1B). Initially, several mild oxidizing agents were used to oxidize the tertiary amine. However, these oxidizing agents resulted in incomplete reactions along with side products. The use of stronger oxidizing agents such as 50% hydrogen peroxide resulted in the complete conversion of DMAPA with no side products. After further purification, we obtained the final product of TMAO monomer as a colorless and viscous liquid. The resulting monomer was characterized using nuclear magnetic resonance (NMR; fig. S1) and high-resolution mass spectrometry (HRMS; fig. S2). With a single-step reaction, an ecofriendly solvent (water), high-conversion efficiency and ease in purification, high-purity TMAO monomer can be easily scaled up for large production.
In vitro fouling tests of TMAO polymers
The adsorption of plasma proteins onto a typical synthetic surface occurs rapidly upon contact between the surface and the blood. Among numerous plasma proteins, fibrinogen plays a major role in determining the hemocompatibility of a particular material due to its abundance in the blood, its essential role in coagulation, and its ability to promote platelet adhesion. Therefore, we first polymerized TMAO monomers into a hydrogel, which was then punched into disks and exposed to a highly concentrated fibrinogen solution (10 mg/ml). In parallel, polypropylene (PP) disks with the same size were prepared and incubated with fibrinogen under the same condition as the positive control. After a 2-hour exposure, the amount of fibrinogen adhered onto PTMAO and PP disks was quantitatively measured by enzyme-linked immunosorbent assay (ELISA). As shown in Fig. 2A, PTMAO disks exhibited an exceptional nonfouling property after 2-hour incubation in a highly concentrated fibrinogen solution (10 mg/ml) by reducing 97.6% of adsorbed fibrinogen with respect to that of PP disks. Considering that the fibrinogen concentration in human plasma (1.5 to 4 mg/ml) is much lower than the test solution (10 mg/ml), PTMAO is anticipated to maintain an ultralow fibrinogen adsorption in the real blood environment. Furthermore, the potential of PTMAO in activating complement proteins was also studied. The unexpected activation of complement systems would evoke the propagation of protein adsorption on material surfaces, enhancing the vulnerability of materials to immune recognition. In this work, the level of C5b-9, a typical marker of complement activation in human serum, was assessed after serum had been incubated with PTMAO hydrogels for 3 hours (Fig. 2B). In contrast to the control hydrogel composed of another hydrophilic polymer, poly(vinyl alcohol) (PVA), PTMAO displayed minimal impact on C5b-9 level. Such a low complement activation by PTMAO may be attributed to the hydration layer surrounding PTMAO surfaces.
Fig. 2. In vitro tests of the nonfouling property of PTMAO.
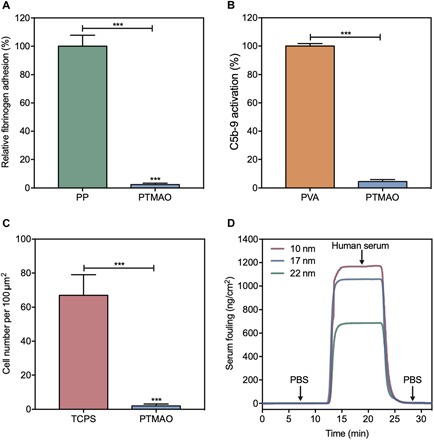
(A) Fibrinogen adsorbed to PP and PTMAO hydrogel surfaces measured by ELISA. (B) Complement C5b-9 activation by poly(vinyl alcohol) (PVA) and PTMAO hydrogels. (C) NIH 3T3 cells adhered to tissue culture polystyrene (TCPS) and PTMAO hydrogel surfaces after 3 days of culture. (D) SPR sensorgram of PTMAO films at different thicknesses (10, 17, and 22 nm) in human blood serum. All statistical analyses were performed using Student’s t test. ***P < 0.001.
In addition to protein adsorption, cell adhesion represents another major type of biofouling, which may directly contribute to the generation of FBR. For instance, fibroblasts that adhered to the surface of medical implants could mediate the formation of fibrous avascular capsules, which sequester the implant from its target tissues and wall off its effects from the rest of the body. Thus, to evaluate the resistance of PTMAO against cell adhesion, we seeded NIH 3T3 fibroblasts onto PTMAO hydrogel disks, and the number of adhered cells was analyzed after a 3-day cell culture (Fig. 2C and fig. S3). In contrast to the hydrogel disks made by tissue culture polystyrene (TCPS), wherein a large number of aggregated fibroblasts were observed (e.g., more than 60 cells per 100 μm by 100 μm), the PTMAO hydrogel disks manifested clean surfaces as the number of adhered fibroblasts was markedly reduced (e.g., less than two cells per 100 μm by 100 μm).
Moreover, the nonfouling property of PTMAO was further tested in undiluted human blood serum, the most challenging system in vitro that mimics the complex biological environment. Briefly, gold chips coated with uniform PTMAO at three different thicknesses (10, 17, and 22 nm) were achieved via a controlled surface-initiated atom transfer radical polymerization (SI-ATRP). Then, undiluted human blood serum was flowed through PTMAO-coated gold surfaces, during which any adsorption of proteins or other components in the serum would be detected by an ultrasensitive SPR binding analysis. Surfaces with adsorbed proteins less than 5 ng/cm2 in undiluted blood plasma or serum are defined as ultralow fouling. Results from SPR experiments (Fig. 2D) showed that gold chips coated with PTMAO films at different thicknesses (10, 17, and 22 nm) all displayed adsorbed proteins less than 3 ng/cm2 in human blood serum. This further demonstrates the ultralow fouling property of PTMAO in complex biological media, suggesting the promising potential of PTMAO for medical applications.
Minimal immunogenic potential of TMAO polymers
Zwitterionic polymers that are entirely superhydrophilic are anticipated to have excellent nonfouling properties and bear little nonspecific interaction with the biological environment, thus inducing minimal immune response (10). In contrast, the existence of hydrophobic domains in polymers such as PEG could meditate the immune recognition and evoke the generation of polymer-specific antibody (Ab) responses. Hence, the immunogenicity of PTMAO is of particular relevance to understand the nonfouling properties of PTMAO in a challenging in vivo system.
The attachment of a polymer to carrier proteins has been shown to facilitate the exhibition of its immunogenicity. A typical example is PEG: Although free PEG exhibits little or no immunogenicity, it would become highly immunogenic once being attached to large carriers such as liposomes and proteins, eliciting PEG-specific Ab responses much like a hapten. Therefore, attaching PTMAO to immunogenic proteins would allow us to amplify its potential immunogenicity and thus better understand its nonfouling property in vivo. Hence, we conjugated PTMAO onto the surface of keyhole limpet hemocyanin (KLH) by a free radical nanoemulsion method. Note that KLH has an extremely high immunogenicity in mammalian hosts and is the most commonly used to promote the induction of immune responses to haptens because of its remarkable immune-stimulating properties, large size, and numerous sites for conjugation. The hydrodynamic size of the PTMAO-KLH conjugates prepared was approximately the same as that of PEG10k-KLH conjugates (fig. S4), as determined by size exclusion chromatography (SEC). Then, four immunizations of PTMAO-KLH conjugates were conducted on C57BL/6J mice (n = 5) via subcutaneous injection for one dose per week, while another cohort of mice were immunized with PEG-KLH conjugates as the control group at the same dose and schedule. At first to fifth weeks after the first immunization, mice sera were collected for Ab tests through ELISA tests (Fig. 3A). Results indicated that the PEG polymers grafted on KLH ultimately elicited a significant level of PEG-specific Abs [average immunoglobulin M (IgM) titer, >4000; average IgG titer, >1000], which was consistent with the clinical findings, and validated the haptenic character of PEG (Fig. 3, B and C). In stark contrast, TMAO was invisible to the immune recognition as no detectable TMAO-specific Abs (average IgM and IgG titers, <200) were developed after five weekly immunizations of TMAO-KLH conjugates. The KLH immunization study suggests that TMAO could maintain its superior nonfouling property even under extremely challenging in vivo conditions.
Fig. 3. Evaluation of PTMAO immunogenicity.
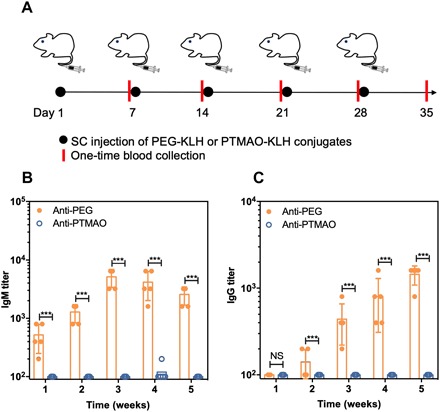
C57BL/6J mice were subcutaneously injected with either PEG-KLH or PTMAO-KLH (n = 5) for five doses (one dose per week). SC, subcutaneous. Mice blood was collected on day 7, 14, 21, 28, and 35 (A). The titer of polymer-specific IgM (B) and IgG (C) in mice sera was detected with ELISA tests. All statistical analyses were performed using Student’s t test. NS, no significant difference. ***P < 0.001.
Improved circulation and efficacy of PTMAO-conjugated proteins
Attachment of hydrophilic materials onto the surfaces of nanoparticles and proteins can afford strong hydration layers that can not only deviate them from immune surveillance but also delay their clearance by the mononuclear phagocyte system and the kidneys, thus contributing to enhanced circulation times. Therefore, the capability of extending the circulation time of a substrate represents another standard to gauge the nonfouling property of a hydrophilic polymer in vivo. Hence, we investigated the impact of PTMAO on the in vivo behavior of uricase, a highly immunogenic enzyme, to further demonstrate its superior hydration effect and nonfouling property. PTMAO polymers were attached onto uricase by the same means of PTMAO-KLH. The hydrodynamic size of the PTMAO-conjugated uricase (PTMAO-uricase) conjugates prepared was approximately the same as that of PEG10k-uricase conjugates, as determined by SEC (fig. S5). In parallel, PEGylated uricase (PEG-uricase) with the similar hydrodynamic size as confirmed by SEC was also prepared as the control (fig. S5). The retained conjugates in the blood could be easily determined by measuring the activity of its associated uricase with AmplexTM Red Uric Acid/Uricase Assay kit.
Before in vivo studies, the stability of native uricase, PEG-uricase, and PTMAO-uricase was first tested. Uricase samples were stressed in urea, a chemical known to destabilize the structure of proteins and to inhibit enzyme activity (fig. S6A). With the increase of urea concentration, both native uricase and PEG-uricase exhibited a marked decrease in bioactivity, whereas that of uricase conjugated with PTMAO was well maintained. Furthermore, a thermal stability test was also performed, measuring the effect of temperature on enzyme activity (fig. S6B). It was also observed that PTMAO conjugation significantly enhanced the stability of uricase at high temperature. The stabilizing effect of PTMAO may be ascribed to the protein-stabilizing effect of TMAO, which is a protective osmolyte long recognized to offset the deleterious effects of urea and to increase the melting temperature and the unfolding free energy of proteins.
Then, three intravenous injections of native uricase, PEG-uricase, and PTMAO-uricase were performed on C57BL/6J mice (n = 5), respectively (one dose per week). After the first and third administrations of uricase samples, mice sera were collected at various time points from each cohort for pharmacokinetic (PK) and pharmacodynamic (PD) studies (Fig. 4A). As shown in Fig. 4 (B and C), native uricase displayed a half-life (T1/2) as short as 3.9 hours after the first injection, which further decreased to 1.9 hours after five repeated administrations. This is a phenomenal accelerated blood clearance (ABC) phenomenon caused by as anti-uricase Abs. On the other hand, although PEG conjugation extended the circulation (T1/2 = 16.2 hours) of uricase after a single injection, an ABC phenomenon was still observed upon PEG-uricase as its T1/2 shrunk to 10.1 hours after five administrations. In contrast, PTMAO-uricase exhibited a persistently superb circulation half-life as long as 19.1 hours (first dose) and 18.2 hours (third dose) after repeated administrations. The extended presence of PTMAO-uricase in systemic circulation also promotes the efficacy of uricase in catalyzing urate metabolism. As shown in Fig. 4 (D and E), PTMAO-uricase reduced approximately 80% of urate in mice blood at 72 hours after the injection, and this remarkable urate-eliminating ability was well maintained after three injections, demonstrating the sustained and enhanced PD profiles of PTMAO-uricase. In contrast, native uricase and PEG-uricase, although could also to some extend reduce the urate level in the blood, lost more than 50 and 30% of their original urate-eliminating ability, respectively, after three injections. On day 21, mice sera were collected for the evaluation of anti-uricase and anticonjugate titers with ELISA test (Fig. 4, F and G). Compared to native uricase, PEGylation of uricase decreased anti-uricase IgM and IgG titers by 16- and 8-folds, respectively. However, a high level of anticonjugate Abs (average IgM titers, >1000; average IgG titer, >500) was still observed, which was proved to mainly consist of anti-PEG Abs and has been recognized as the main culprit for the efficacy loss of PEG-uricase (Krystexxa) in the clinic. In contrast, the level of anti-uricase and anticonjugate Abs was also significantly lower in the group treated with PTMAO-uricase. These results confirm the nonfouling property PTMAO, including prolonging circulation time of substrates and preventing them from the immune recognition.
Fig. 4. In vivo nonfouling performance of PTMAO.
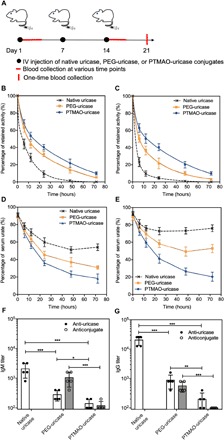
C57BL/6J mice were intravenously injected with native uricase, PEG-uricase, or PTMAO-uricase (n = 5) for three doses (one dose per week). Mice blood at various time points (0.1, 6, 12, 24, 48, and 72 hours) were collected after the first and third injections for the characterization of circulation and efficacy, and mice blood on day 21 after the first injection was also collected for antibody detection (A). PK profiles of each uricase sample after the first (B) and third intravenous (IV) injection (C) were determined by measuring the retained uricase activity in mice sera. PD profiles of each uricase sample after the first (D) and third intravenous (IV) injections (E) were determined by measuring the urate level in mice sera. IgM (F) and IgG (G) against uricase or conjugates were detected by direct ELISAs. All statistical analyses were performed using Student’s t test. *P < 0.05, **P < 0.01 and ***P < 0.001).
Nonfouling mechanism of TMAO at molecular level
To study the mechanism accounting for the extraordinary hydration of PTMAO, we performed MD simulations of a TMAO small molecule and a 10-residue PTMAO oligomer in an aqueous solution. Simulations of a three-residue PEG oligomer (OEG) were conducted for comparison. Simulation results showed that the TMAO oxygen accepts an average of 2.5 hydrogen bonds from water, accepting either two or three hydrogens bonds with approximately equal probability, while OEG oxygen atoms typically accept only one hydrogen bond from water, as shown in Fig. 5A. In addition, hydrogen bond lifetime (τHB) for TMAO water was observed to be longer than that for OEG water (Fig. 5B). The persistent binding of multiple water molecules to TMAO molecule suggested very strong interactions with water. The radial distribution functions (RDFs) for water oxygen atoms with respect to the heavy atoms of the TMAO small molecule (OTMAO, NTMAO, and CTMAO) suggested a near-contiguous sphere of hydration centered on the quaternary nitrogen (Fig. 5C). The first peak of the NTMAO RDF is composed of a polar hydration peak contributed by hydration around the quaternary amine cation and a shoulder contributed by the tightly bound water at the oxygen anion. Figure 5D shows a single frame from the TMAO monomer simulation, including the water molecules with a NTMAO-water oxygen distance less than the first minimum in the NTMAO RDF (0.6 nm). This snapshot reveals that the first hydration shell, with respect to NTMAO, covers the whole TMAO molecule and includes the strongly hydrogen-bonded water at the TMAO oxygen (high opacity). The contiguous hydration shell observed in the simulation of the TMAO small molecule was also observed in the PTMAO simulation (Fig. 5E), which indicates that water is similarly ordered near the PTMAO headgroups. Together, MD simulations suggest that PTMAO retains the superhydrophilicity observed for the TMAO small molecule. The strong hydrogen bonding with water and a contiguous hydration shell around the PTMAO headgroups could be responsible for its superhydrophilic properties.
Fig. 5. MD simulation on the nonfouling mechanism of PTMAO at molecular level.
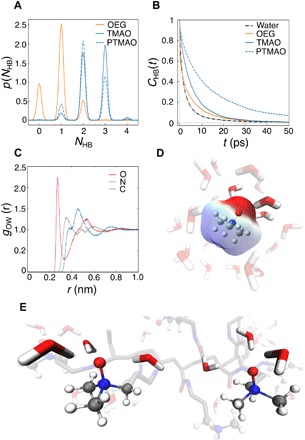
(A) The probability distributions for the number of hydrogen bonds, NHB, between water and the TMAO oxygen (blue line), water and the PTMAO oxygen (blue dashed line), and water and each OEG oxygen (orange line). (B) Autocorrelation function for τHB, CHB(t), for water-TMAO oxygen (blue line), water-PTMAO oxygen (blue dashed line), water-OEG oxygen (orange line), and water-water hydrogen bonds (black dashed line). (C) RDF of water oxygen with respect to O (red), N (blue), and C (gray) atoms of TMAO. (D) Snapshot of aqueous TMAO with opaque and semitransparent water corresponding to the first shoulder and main peak in the RDF of water oxygen with respect to N atoms of TMAO, respectively. (E) Snapshot of PTMAO in aqueous solution, highlighting the tightly bound waters near two of the TMAO headgroup.
CONCLUSIONS
In summary, a TMAO-derived polymer, PTMAO, was successfully obtained as a new-generation zwitterionic material. The superhydrophilicity and nonfouling properties of PTMAO were demonstrated from different angles under challenging conditions, both in vitro and in vivo. Results showed that PTMAO-coated surfaces could achieved an ultralow protein adsorption in undiluted blood serum. Furthermore, PTMAO exhibited minimal immunogenicity and extended circulation after being conjugated to highly immunogenic protein carriers. The molecular-level nonfouling mechanism of PTMAO stemming from strong hydration was elucidated from MD simulations. The development of PTMAO as a broadly applicable nonfouling material for many applications represents an important milestone in the development of biomaterials.
MATERIALS AND METHODS
Materials
PP sheet was purchased from TAP Plastics. Fibrinogen, KLH, recombinant uricase from Candida sp., and all chemicals were purchased from Sigma-Aldrich unless otherwise noted and were used as received. Methoxy-PEG– N-hydroxysuccinimide (mPEG-NHS) (10 kDa, 95%) was purchased from Nanocs Corporation. Goat anti-mouse IgM Ab and goat anti-mouse IgG Ab was purchased from Bethyl Laboratories. Pierce Protein Concentrators (100,000 molecular weight cutoff) were purchased from Thermo Fisher Scientific (Waltham, MA). A 3,3′,5,5′-tetramethylbenzidine substrate solution was purchased from eBioscience (San Diego, CA). High-resolution SEC resin (Sephacryl S-500HR) was purchased from GE Healthcare Life Sciences. The C5b-9 assay ELISA kit was purchased from BD Bioscience.
Synthesis of TMAO monomer
Diethylenetriaminepentaacetic acid (800 mg) was added to 30 ml of Milli-Q water and mixed vigorously until the white powder dissolved. Then, hydrogen peroxide (50% solution, 2.87 g) was slowly added, and reaction contents were heated to 60°C. Oxygen gas was then slowly purged into solution (31). Dimethylaminopropylacrylamide (14.4 g) in 10 ml of Milli-Q water was added dropwise for 30 min. The reaction was carried out for 6 hours at 60°C. After completion of reaction, the reaction contents were cooled. NMR analysis of the product confirmed the formation of TMAO monomer. The pH of the final product was about 7.5, and the solid content was about 32.9%. TMAO monomer was then precipitated by an organic solvent. The final product is colorless and viscous liquid. NMR result is shown in fig. S1. 1H NMR (500 MHz, D2O): δ = 6.07 to 5.93 (m, 2H), 5.55 (d, J = 9.9 Hz, 1H), 3.12 (m, 4H), 3.01 (s, 6H), 1.90 to 1.74 (m, 2H). HRMS analysis was carried out by using a Thermo Fisher Scientific Exactive Plus Orbitrap spectrometer, and the result is shown in fig. S2. HRMS (mass/charge ratio): calculated for C8H17N2O2, 173.1284 ([M + H]+); found 173.1282.
Preparation of TMAO hydrogel
TMAO hydrogel was fabricated by bulk photopolymerization with a hydrogel aqueous solution containing TMAO monomer (0.67 g of Milli-Q water and 330 mg of TMAO monomer), cross-linker N,N′-methylenebis(acrylamide) [1 weight % (wt %), 3.3 mg], and photoinitiator 2-hydroxy-2-methylpropiophenone (0.33 mg). The hydrogel aqueous solution was placed between two glass slides separated by a 0.5-mm-thick polytetrafluoroethylene spacer and was then photopolymerized at room temperature for 30 min. After polymerization, hydrogels were removed from the casts and soaked in phosphate-buffered saline (PBS) for 3 days to remove unreacted chemicals and to reach the fully hydrated hydrogel network. PBS was refreshed every 12 hours.
Fibrinogen adsorption test
Biopsy punches were used to punch the hydrated TMAO hydrogel and the PP sheet into 5-mm-diameter disks. The sample disks were then placed into a 24-well plate and incubated with 1 ml of fibrinogen (10 mg/ml) in PBS buffer for 2 hours, followed by five washes with pure PBS buffer. Subsequently, the sample disks were then transferred to new wells and incubated with 1 ml of horseradish peroxidase (HRP)–conjugated antifibrinogen (1 μg/ml) in PBS buffer for 1 hour. All sample disks were then transferred to new wells after five washes with pure PBS buffer. Next, 1 ml of o-phenylenediamine citrate phosphate solution [1 mg/ml; 0.1 M (pH 5.0)] containing 0.03% hydrogen peroxide was added. After the 15-min incubation, the enzymatic reaction was stopped by adding an equal volume of 1 M HCl. The same procedure was conducted on PP disks with the same surface area as the control. An absorbance value at 492 nm was recorded by a plate reader and was normalized to that of the PP sample. Average data were acquired from three specimens.
Cell adhesion test
TMAO hydrogel disks with 1 wt % cross-linker were soaked in 70% ethanol for 2 hours for sterilization and soaked in sterilized PBS until equilibrium. NIH 3T3 fibroblasts were respectively seeded onto TMAO hydrogel disks at a concentration of 105 cells/ml into 24-well plates. The same procedure was conducted on TCPS hydrogel disks with the same surface area as the control. The cells were cultured at 37°C, 5% CO2, and 100% humidity for 72 hours and then were observed and photographed on a Nikon Eclipse TE2000-U microscope at ×100 magnification.
Complement activation test
PTMAO and PVA were made into highly cross-linked cylindrical hydrogel bowls (6 mm in diameter and 8 mm in depth) with 50 wt % of solid fraction using a home-made mold. Then, 300 μl of pooled complement human serum (Innovative Research, Novi, MI) was added into replicates of each polymer bowl (n = 3) and incubated at 37°C for 90 min, and followed by quenching with 30 μl of 10 mM EDTA solution. The complement activation degree was measured by quantifying a complement degradation fragment C5b-9 in serum using a standard ELISA kit (BD Bioscience, San Diego, CA) following the manufacturer’s protocol. The level of preexisting C5b-9 in the serum was measured and subtracted as the background.
Preparation of TMAO-coated surface by SI-ATRP
Clean, gold-coated glass substrates were first soaked in ω-mercaptoundecyl bromoisobutyrate solution (0.2 mM in ethanol) for overnight to get initiator-coated substrates. The initiator-coated substrates, together with copper(I) bromide (14.35 mg and 0.1 mmol), were then placed into a Schlenk tube and deoxygenated via pump vacuum for 10 cycles. TMAO monomer (1.72 g and 10 mmol), Me6TREN (23 mg and 0.1 mmol), methanol (3.6 ml), and H2O (0.4 ml) were added into Schlenk tube and deoxygenated via the same method. After full deoxygenation, the mixed aqueous solution of TMAO monomer and Me6TREN were transferred to the tube, which held the substrate and copper bromide. The reaction mixture was placed at ambient temperature overnight. The substrate was then taken out from the mixture and washed with alternating ethanol and water, three times each, and air-dried before being used as SPR sample. The thickness of the coated TMAO polymer film was measured by ellipsometer.
SPR test in human serum
The nonspecific protein adsorption of the TMAO polymer films was determined with a SPR biosensor using a flow rate of 40 μl/min at 25°C. After first establishing a baseline using PBS, undiluted human serum was flowed for 10 min, followed by buffer to reestablish the baseline. Protein adsorption was quantified as the difference between buffer baselines and converted to a surface coverage using the appropriate sensitivity factor.
Preparation of PTMAO-KLH conjugate
AOT [sodium bis(2-ethylhexyl) sulfosuccinate, 120 mg] and Brij 30 [PEG dodecyl ether, 230 mg] were added to a glass vial (20 ml) to which a stir bar was added. The vial was sealed with a Teflon-lined septum cap and purged with dry nitrogen for 10 min. Nitrogen-deoxygenated hexane (5 ml) was then added to the vial under vigorous stirring. For the aqueous phase, KLH (premodified with acryloyl group, 1 mg) was dissolved in Hepes buffer [150 μl (pH 8.5)] to which TMAO monomer (100 mg) was added and dissolved. Dry nitrogen was bubbled through the monomer/protein solution for 2 min, after which the aqueous phase was slowly added to the organic continuous phase dropwise. The vial was sonicated to form a stable microemulsion. A 20% (w/v) solution of ammonium persulfate (APS) in deionized water (20 μl) was then added to the emulsion. After 5 min, polymerization was initiated by the addition of tetramethylethylenediamine (TEMED; 12 μl) and maintained at 4°C under rapid magnetic stirring. After the 2-hour reaction, the organic solvent was removed by rotary evaporator, and the PTMAO-KLH conjugate was precipitated and washed with tetrahydrofuran (THF) for three times. The PTMAO-KLH conjugate was resuspended in PBS buffer and purified with high-resolution SEC (Sephacryl S-500HR) to remove the free KLH. Lastly, the conjugates were washed and concentrated with PBS (pH 7.4) for three times using a 100-kDa molecular mass cutoff centrifugal filter. The protein-polymer conjugates were characterized by gel permeation chromatogram (GPC; Wyatt technology).
Preparation of PEGylated KLH
KLH (1 mg/ml) and mPEG-NHS (10 kDa, 20 mg/ml) were mixed in 50 mM Hepes buffer (pH 8.5). The reaction was stirred at 4°C overnight. Then, the PEG-KLH conjugates were purified with high-resolution SEC (Sephacryl S-500HR) to remove the free KLH. Last, the conjugates were washed and concentrated with PBS (pH 7.4) for three times using a 100-kDa molecular mass cutoff centrifugal filter. The protein-polymer conjugates were characterized by GPC (Wyatt technology).
Preparation of PTMAO-uricase conjugate
AOT (120 mg) and Brij 30 (230 mg) were added to a glass vial (20 ml) to which a stir bar was added. The vial was sealed with a Teflon-lined septum cap and purged with dry nitrogen for 10 min. Nitrogen-deoxygenated hexane (5 ml) was then added to the vial under vigorous stirring. For the aqueous phase, uricase (premodified with acryloyl group, 1 mg) was dissolved in Hepes buffer (125 μl, pH 8.5) to which TMAO monomer (50 mg) was added and dissolved. Dry nitrogen was bubbled through the monomer/protein solution for 2 min, after which the aqueous phase was slowly added to the organic continuous phase dropwise. The vial was sonicated to form a stable microemulsion. A 20% (w/v) solution of APS (10 μl) in Milli-Q water was then added to the emulsion. After 5 min, polymerization was initiated by the addition of TEMED (6 μl) and maintained at 4°C under rapid magnetic stirring. After the 2-hour reaction, the organic solvent was removed by rotary evaporator, and the PTMAO-uricase conjugate was precipitated and washed with THF three times. The PTMAO-uricase conjugate was resuspended in PBS buffer and purified with high-resolution SEC (Sephacryl S-500HR) to remove the free uricase. Lastly, the conjugates were washed and concentrated with PBS (pH 7.4) three times using a 100-kDa molecular mass cutoff centrifugal filter. The protein-polymer conjugates were characterized by GPC (Wyatt technology).
Preparation of PEGylated uricase
Uricase (1 mg/ml) and mPEG-NHS (10 kDa, 10 mg/ml) were mixed in 50 mM Hepes buffer (pH 8.5). The reaction was stirred at 4°C overnight. Then, the PEG-uricase conjugates were purified with high-resolution SEC (Sephacryl S-500HR) to remove the free uricase. Lastly, the conjugates were washed and concentrated with PBS (pH 7.4) for three times using a 100-kDa molecular mass cutoff centrifugal filter. The protein-polymer conjugates were characterized by GPC.
Stability test
Native uricase, PEG-uricase, and PTMAO-uricase conjugates were incubated in PBS solution (pH 7.4) at 10 μg/ml for 6 hours with and without urea (0.4, 0.8, 1.6, and 3.2 M), after which the activities of the retained uricase were tested. Likewise, the thermal stability of native uricase, PEG-uricase, and PTMAO-uricase conjugates were tested after incubation at different temperatures (40°, 50°, 60°, and 70°C) for 30 min. The activity of uricase was measured by AmplexTM Red Uric Acid/Uricase Assay kit following the manufacturer’s protocol. Measurements were performed in triplicate.
Animal studies
The University of Washington Institutional Animal Care and Use Committee approved all animal experiments under protocol no. 4203-01. Male C57BL/6J mice of ∼20 g were randomly divided.
Immunogenicity study of PTMAO
To compare the immunogenicity of PEG and PTMAO, two groups of male C57BL/6J mice (n = 5) were administered the PEG-KLH and PTMAO-KLH conjugates, respectively, via subcutaneous injection (2 mg protein/kg weight) for 5 weeks (one dose per week). Mice sera were collected on day 7, 14, 21, 28, and 35 during the immunization study, and the anti-PEG or anti-PTMAO antibodies (IgM and IgG) in mice sera were detected using ELISA tests.
For ELISA tests, 100 μl of antigen solution (10 μg/ml of protein concentration), prepared in 0.1 M sodium carbonate buffer (pH 10.5), was used to coat each well of the 96-well plates. Antigens used in direct ELISAs for plate coating consisted of PEG–bovine serum albumin (BSA) (for the mice sera immunized with PEG-KLH) or PTMAO-BSA conjugates (for mice sera immunized with PTMAO-KLH). PEG-BSA and PTMA-BSAO conjugates were prepared, following the same preparation protocol as the PEG-KLH and PTMAO-KLH conjugates as described above. During coating procedure, plates were incubated at 4°C overnight. After removing antigen solutions, the plates were washed five times using PBS (pH 7.4) and then filled with blocking buffer [1% nonfat milk solution in 0.1 M tris buffer (pH 8.0)]. It is important to avoid using any buffer that contains PEG-like detergents, e.g., Tween 20 and Tween 80. After incubation at room temperature for 1 hour, blocking buffer was removed, and all wells were washed by PBS for another five times. Serial dilutions of monoclonal anti-PEG Abs in PBS containing 1% nonfat milk were added to the plates (100 μl per well), which were incubated for 1 hour at 37°C. The plates were then washed five times with PBS, followed by adding secondary antibody HRP conjugates (100 μl per well, 1:50,000 dilution; Bethyl Laboratories). After adding the secondary antibody, plates were incubated at room temperature for 1 hour and then washed five times using PBS before the addition of HRP substrate TMB (100 μl per well). The plates were shaken for 15 min, and 100 μl of stop solution (0.2 M H2SO4) was added to each well. Absorbance at 450 (signal) and 570 nm (background) was recorded by a microplate reader.
In vivo nonfouling performance of PTMAO
Three groups of male C57BL/6J mice (n = 5) were administered the native uricase, PEG-uricase, and PTMAO-uricase conjugates, respectively, via intravenous injection (25 U/kg weight) for 3 weeks (one dose per week). The mice sera were collected at various time points (0.1, 6, 12, 24, 48, and 72 hours) after the first and the third injection of uricase samples for evaluating the level of uricase/uric acid. The uricase concentration in plasma was estimated on the basis of the enzyme activity measured by AmplexTM Red Uric Acid/Uricase Assay kit. The circulation time of each uricase sample was calculated using PKSolver software following the instructions. The urate concentration in the mice sera was also measured by AmplexTM Red Uric Acid/Uricase Assay kit. All the mice were sacrificed on day 21, and their sera were harvested for antibody detection via ELISA tests as described above. For ELISA tests, uricase was used as the coated antigen for the detection of anti-uricase Ab, while PEG-uricase or PTMAO-uricase conjugate was used as the coated antigen for the detection of anticonjugate Ab.
MD simulations
Partial charges for TMAO, OEG, and PTMAO atoms were assigned using the restrained electrostatic potential (RESP) method (32). Quantum mechanical calculations for RESP were performed in Gaussian 09 using the B3LYP hybrid functional with the 6-31G(d) basis set (33, 34). For PTMAO, the quantum calculations were performed for a monomer with a methyl-capped backbone, and the partial charge of the backbone atoms was adjusted to neutralize the net charge of the monomer. Water molecules were described by the TIP3P water model.
The initial configuration for each simulation was generated with GROMACS 5.1.2 utilities (35). For the small-molecule simulations, one solute molecule (TMAO or OEG) was centered in a cubic box with 2.5 nm sides. The box was then solvated with 504 randomly placed water molecules. A two-step equilibration procedure was followed to provide reasonable starting positions and velocities for production simulations. Energy minimization was performed with the solvated configuration using the steepest descent algorithm for 10,000 steps. Energy minimization was followed by a simulation (1 ns) in the number of atoms, pressure and temperature (NPT) ensemble with the Bussi-Donadio-Parrinello (V-rescale) thermostat and Berendsen barostat for temperature and pressure control, respectively (36, 37). The output atomic coordinates and velocities for each system were used as the initial coordinates and velocities for the production simulations.
For the production phase, each system was simulated for 4 ns in the NPT ensemble with the Bussi-Donadio-Parrinello (V-rescale) thermostat and Parrinello-Rahman barostat for temperature and pressure control, respectively (36, 38). Frames were saved every 100 fs (50 steps) during the production simulation, and trajectories were analyzed with GROMACS utilities. The hydrogen bond count for every frame was calculated with the “gmx hbond” utility using the default geometric definition of the hydrogen bond: a donor-acceptor distance of less than 0.35 nm and a hydrogen donor-acceptor angle of less than 30°. The probability distribution of the number of hydrogen bonds accepted by OEG and TMAO oxygens was generated with kernel density estimation, essentially a technique to smooth the hydrogen bond histogram with Gaussians. The autocorrelation function for τHB was calculated using the method of Luzar and Chandler (39), as implemented in the GROMACS utility gmx hbond. RDFs were calculated using the “gmx rdf” utility.
Supplementary Material
Acknowledgments
Funding: This work was supported by the Defense Threat Reduction Agency (HDTRA1-13-1-0044) the Office of Navel Research (N00014-19-1-2063) and the University of Washington. Author contributions: B.L., P.J., and S.J. designed experiments. B.L, P.J., J.M., J.K.S., Z.Y., H.-C.H., Y.H., X.L., and K.W. performed experiments. J.K.S. and J.P. conducted simulations. B.L. wrote the manuscript. B.L., J.K.S., and S.J. edited the manuscript. Competing interests: B.L., P.J., J.M., and S.J. are authors on a patent application related to this work filed by the University of Washington (patent application no. WO2019006398A3, published on 28 February 2019). All other authors declare that they have no competing interests. S.J. is the cofounder of Taproot Medical Technologies, which licensed this patent. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaaw9562/DC1
Fig. S1. 1H NMR spectrum of TMAO monomer.
Fig. S2. HRMS spectrum of TMAO monomer.
Fig. S3. Cell adhesion test.
Fig. S4. GPC graph of KLH, PEG10k-KLH, and TMAO-KLH conjugates.
Fig. S5. GPC graph of native uricase, PEG10k-uricase, and TMAO-uricase conjugates.
Fig. S6. Protein stability test.
Table S1. Circulation time of uricase samples after repeated intravenous injections.
REFERENCES AND NOTES
- 1.Meyers S. R., Grinstaff M. W., Biocompatible and bioactive surface modifications for prolonged in vivo efficacy. Chem. Rev. 112, 1615–1632 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Williams D. F., On the mechanisms of biocompatibility. Biomaterials 29, 2941–2953 (2008). [DOI] [PubMed] [Google Scholar]
- 3.Ratner B. D., Replacing and renewing: Synthetic materials, biomimetics, and tissue engineering in implant dentistry. J. Dent. Educ. 65, 1340–1347 (2001). [PubMed] [Google Scholar]
- 4.van der Giessen W. J., Lincoff A. M., Schwartz R. S., van Beusekom H. M. M., Serruys P. W., Holmes D. R., Ellis S. G., Topol E. J., Marked inflammatory sequelae to implantation of biodegradable and nonbiodegradable polymers in porcine coronary arteries. Circulation 94, 1690–1697 (1996). [DOI] [PubMed] [Google Scholar]
- 5.Grabbe S., Landfester K., Schuppan D., Barz M., Zentel R., Nanoparticles and the immune system: Challenges and opportunities. Nanomedicine 11, 2621–2624 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Paterlini T. T., Nogueira L. F. B., Tovani C. B., Cruz M. A. E., Derradi R., Ramos A. P., The role played by modified bioinspired surfaces in interfacial properties of biomaterials. Biophys. Rev. 9, 683–698 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leng C., Huang H., Zhang K., Hung H.-C., Xu Y., Li Y., Jiang S., Chen Z., Effect of surface hydration on antifouling properties of mixed charged polymers. Langmuir 34, 6538–6545 (2018). [DOI] [PubMed] [Google Scholar]
- 8.Li B., Yuan Z., Hung H.-C., Ma J., Jain P., Tsao C., Xie J., Zhang P., Lin X., Wu K., Jiang S., Revealing the immunogenic risk of polymers. Angew. Chem. Int. Ed. 57, 13873–13876 (2018). [DOI] [PubMed] [Google Scholar]
- 9.Yang Q., Lai S. K., Anti-PEG immunity: Emergence, characteristics, and unaddressed questions. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 7, 655–677 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jiang S., Cao Z., Ultralow-fouling, functionalizable, and hydrolyzable zwitterionic materials and their derivatives for biological applications. Adv. Mater. 22, 920–932 (2010). [DOI] [PubMed] [Google Scholar]
- 11.Shao Q., White A. D., Jiang S., Difference of carboxybetaine and oligo(ethylene glycol) moieties in altering hydrophobic interactions: A molecular simulation study. J. Phys. Chem. B 118, 189–194 (2014). [DOI] [PubMed] [Google Scholar]
- 12.Lee I., Kobayashi K., Sun H. Y., Takatani S., Zhong L. G., Biomembrane mimetic polymer poly (2-methacryloyloxyethyl phosphorylcholine-co-n-butyl methacrylate) at the interface of polyurethane surfaces. J. Biomed. Mater. Res. A 82, 316–322 (2007). [DOI] [PubMed] [Google Scholar]
- 13.Chen S., Yu Q., Li L., Boozer C. L., Homola J., Yee S. S., Jiang S., Detecting the adsorption of dye molecules in homogeneous poly(propylene imine) dendrimer monolayers by surface plasmon resonance sensor. J. Am. Chem. Soc. 124, 3395–3401 (2002). [DOI] [PubMed] [Google Scholar]
- 14.Leng C., Sun S., Zhang K., Jiang S., Chen Z., Molecular level studies on interfacial hydration of zwitterionic and other antifouling polymers in situ. Acta Biomater. 40, 6–15 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Chien H.-W., Cheng P.-H., Chen S.-Y., Yu J., Tsai W.-B., Low-fouling and functional poly(carboxybetaine) coating via a photo-crosslinking process. Biomater. Sci. 5, 523–531 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Zhang Z., Chao T., Chen S., Jiang S., Superlow fouling sulfobetaine and carboxybetaine polymers on glass slides. Langmuir 22, 10072–10077 (2006). [DOI] [PubMed] [Google Scholar]
- 17.Li B., Xie J., Yuan Z., Jain P., Lin X., Wu K., Jiang S., Mitigation of inflammatory immune responses with hydrophilic nanoparticles. Angew. Chem. Int. Ed. 57, 4527–4531 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Ladd J., Zhang Z., Chen S., Hower J. C., Jiang S., Zwitterionic polymers exhibiting high resistance to nonspecific protein adsorption from human serum and plasma. Biomacromolecules 9, 1357–1361 (2008). [DOI] [PubMed] [Google Scholar]
- 19.Zhang L., Cao Z., Bai T., Carr L., Ella-Menye J.-R., Irvin C., Ratner B. D., Jiang S., Zwitterionic hydrogels implanted in mice resist the foreign-body reaction. Nat. Biotechnol. 31, 553–556 (2013). [DOI] [PubMed] [Google Scholar]
- 20.Li B., Yuan Z., Zhang P., Sinclair A., Jain P., Wu K., Tsao C., Xie J., Hung H.-C., Lin X., Bai T., Jiang S., Zwitterionic nanocages overcome the efficacy loss of biologic drugs. Adv. Mater. 30, e1705728 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Liu S., Liu J., Esker A. R., Edgar K. J., An Efficient, Regioselective Pathway to Cationic and Zwitterionic N-Heterocyclic Cellulose Ionomers. Biomacromolecules 17, 503–513 (2016). [DOI] [PubMed] [Google Scholar]
- 22.Keefe A. J., Jiang S., Poly(zwitterionic)protein conjugates offer increased stability without sacrificing binding affinity or bioactivity. Nat. Chem. 4, 59–63 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li Y., Zhang X., Zwitterionic poly(carboxybetaine) modified liposomes enhancing tumor therapy without accelerated blood clearance phenomenon. J. Control. Release 213, e125 (2015). [DOI] [PubMed] [Google Scholar]
- 24.Shao Q., Jiang S., Effect of carbon spacer length on zwitterionic carboxybetaines. J. Phys. Chem. B 117, 1357–1366 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Shao Q., Jiang S., Molecular understanding and design of zwitterionic materials. Adv. Mater. 27, 15–26 (2015). [DOI] [PubMed] [Google Scholar]
- 26.Hunger J., Ottosson N., Mazur K., Bonn M., Bakker H. J., Water-mediated interactions between trimethylamine-N-oxide and urea. Phys. Chem. Chem. Phys. 17, 298–306 (2015). [DOI] [PubMed] [Google Scholar]
- 27.Liao Y.-T., Manson A. C., DeLyser M. R., Noid W. G., Cremer P. S., Trimethylamine N-oxide stabilizes proteins via a distinct mechanism compared with betaine and glycine. Proc. Natl. Acad. Sci. U.S.A. 114, 2479–2484 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gluick T. C., Yadav S., Trimethylamine N-oxide stabilizes RNA tertiary structure and attenuates the denaturating effects of urea. J. Am. Chem. Soc. 125, 4418–4419 (2003). [DOI] [PubMed] [Google Scholar]
- 29.Ma J., Pazos I. M., Gai F., Microscopic insights into the protein-stabilizing effect of trimethylamine N-oxide (TMAO). Proc. Natl. Acad. Sci. U.S.A. 111, 8476–8481 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rani A., Jayaraj A., Jayaram B., Pannuru V., Trimethylamine-N-oxide switches from stabilizing nature: A mechanistic outlook through experimental techniques and molecular dynamics simulation. Sci. Rep. 6, 23656 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.M. R. Sivik, J.-F. Bodet, B. W. Kluesener, W. M. Scheper, D. W.-K. Yeung, V. Bergeron, Compositions and methods for using amine oxide monomeric unit-containing polymeric suds enhancers, U.S. Patent 20050026803 (2005).
- 32.Bayly C. I., Cieplak P., Cornell W., Kollman P. A., A well-behaved electrostatic potential based method using charge restraints for deriving atomic charges: The RESP model. J. Phys. Chem. 97, 10269–10280 (1993). [Google Scholar]
- 33.Becke A. D., Density-functional thermochemistry. III. The role of exact exchange. J. Chem. Phys. 98, 5648–5652 (1993). [Google Scholar]
- 34.Petersson G. A., Bennett A., Tensfeldt T. G., Al-Laham M. A., Shirley W. A., Mantzaris J., A complete basis set model chemistry. I. The total energies of closed-shell atoms and hydrides of the first-row elements. J. Chem. Phys. 89, 2193–2218 (1988). [Google Scholar]
- 35.Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., Lindahl E., GROMACS: High performance molecular simulations through multi-level parallelism from laptops to supercomputers. SoftwareX 1–2, 19–25 (2015). [Google Scholar]
- 36.Bussi G., Donadio D., Parrinello M., Canonical sampling through velocity rescaling. J. Chem. Phys. 126, 014101 (2007). [DOI] [PubMed] [Google Scholar]
- 37.Berendsen H. J. C., Postma J. P. M., van Gunsteren W. F., DiNola A., Haak J. R., Molecular dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 (1984). [Google Scholar]
- 38.Parrinello M., Rahman A., Polymorphic transitions in single crystals: A new molecular dynamics method. J. Appl. Phys. 52, 7182–7190 (1981). [Google Scholar]
- 39.Luzar A., Chandler D., Hydrogen-bond kinetics in liquid water. Nature 379, 55–57 (1996). [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaaw9562/DC1
Fig. S1. 1H NMR spectrum of TMAO monomer.
Fig. S2. HRMS spectrum of TMAO monomer.
Fig. S3. Cell adhesion test.
Fig. S4. GPC graph of KLH, PEG10k-KLH, and TMAO-KLH conjugates.
Fig. S5. GPC graph of native uricase, PEG10k-uricase, and TMAO-uricase conjugates.
Fig. S6. Protein stability test.
Table S1. Circulation time of uricase samples after repeated intravenous injections.