

Abstract
Background
Bacillus subtilis is developed to be an attractive expression host to produce both secreted and cytoplasmic proteins owing to its prominent biological characteristics. Chromosomal integration is a stable expression strategy while the expression level is not ideal compared with plasmid expression. Thus, to meet the requirement of protein overexpression, promoter, as one of the key elements, is important. It is necessary to obtain an ideal promoter for overproduction of foreign proteins from a single copy expression cassette.
Results
The activity of promoter Pylb was further enhanced by optimizing the − 35, − 10 core region and upstream sequence (UP) by substituting both sequences with consensus sequences. The final engineered promoter exhibited almost 26-fold in β-galactosidase (BgaB) activity and 195-fold in super-folded green fluorescent protein (sfGFP) intensity than that of WT. The two proteins account for 43% and 30% of intracellular proteins, respectively. The promoter was eventually tested by successful extracellular overproduction of Methyl Parathion Hydrolase (MPH) and Chlorothalonil hydrolytic dehalogenase (Chd) to a level of 0.3 g/L (144 U/mL) and 0.27 g/L (4.4 U/mL) on shake-flask culture condition.
Conclusions
A strong promoter was engineered for efficient chromosomally integrated expression of heterologous proteins.
Electronic supplementary material
The online version of this article (10.1186/s12934-019-1159-0) contains supplementary material, which is available to authorized users.
Keywords: Bacillus subtilis, Promoter engineering, Chromosomal integration, Highly expression
Background
Bacillus subtilis, a species of Gram-positive aerobic soil bacteria, is an attractive industrial workhorse for production of various enzymes and industrial recombinant proteins due to its GRAS (generally recognized as safe) status, well-characterized protein secretion mechanisms and large-scale fermentation processes [1–5]. In addition, the bacterium has no significant bias in codon usage and efficient genetic manipulation is available [6, 7]. Thus, more attention has been paid to its expression systems for the purpose of the commercial application and basic research.
Plasmid-mediated recombinant production of proteins in bacteria is unstable during the late stage of fermentation [8]. Moreover, the safety concerns and legal requirements surrounding the use of antibiotic is another bottleneck in food industry. Chromosomal integration offers a more stable alternative to maintenance of foreign inserted expression cassette. However, the expression level may not meet the requirement when compared with that of multi-copy plasmid expression. Protein production was mainly determined by transcription, translation and post-translation level. To realize efficient chromosomally integrated protein expression, promoter is of great importance in transcription level because it directly affects the efficient synthesis of fundamental transcripts. Therefore, a powerful promoter is desirable to drive gene overexpression.
Nowadays, existing promoters can be generally divided into three major groups: constitutive promoters [9–13], inducible promoters [14–25] and stationary phase promoters [26–28]. Recently, a stationary phase promoter Pylb [29] was identified. Based on the Pylb promoter, in this work, a new strong promoter was developed to realize high level protein expression through single-copy expression cassette integration.
Results
Assessment of the WT promoter Pylb
To begin with, WT promoter was compared with commonly used constitutive promoter P43 [9], inducible promoter PxylA [14], and stationary phase promoter PsrfA [27]. The expression level of reporter protein BgaB was used to reflect the strength of the promoters. Promoter Pylb was prominent for its strength and stationary phase (Fig. 1b, c and Additional file 1: Figures S1, S2). During the lag phase and the early exponential phase, there was little BgaB activity detected. The reporter protein began to emerge at the mid-exponential phase; the activity sharply increased to the peak value during the transition to stationary phase, and remained constant during the followed stationary phase (Fig. 1c). Thus, the WT Pylb is deserved to be further engineered for overexpression of proteins in B. subtilis.
Fig. 1.

a Construction of the integrative plasmid pYBGB. b Comparison of the maximum yield of BgaB under the control of different promoters. All cultures were grown in triplicate, and each experiment was performed at least twice. Error bars indicate standard deviations. c The expression level and pattern of BgaB measured in strain WBBgaB. During 24 h of cultivation, cells were sampled periodically and analyzed by examining the biomass and BgaB activity
Engineering the core regions to improve transcription
− 35 and − 10 regions were the most important sequences in promoter strength and the regions were widely engineered to enhance promoter transcription [30]. Thus, the − 10 and − 35 regions of Pylb were changed into the corresponding consensus sequence separately or in combination (Fig. 2a). The reporter protein BgaB was used to assess the strength of the engineered promoters. A promoter with the consensus − 10 hexamer (P10) was 0.5-fold stronger than Pylb while the BgaB expression level controlled by the P35 promoter resulted in a sevenfold increase (Fig. 2b, c). When both changes were combined into one single promoter (P3510), its activity was enhanced about ninefold (Fig. 2b, c and Additional file 1: Figures S3, S4). In addition to − 35 and − 10 regions, − 16 and − 22 region will also influence promoter strength [30, 31]. Next, − 16 region was also changed into the corresponding consensus sequence TRTG (where R stands for A or G) based on P3510, generating P351016 (Fig. 2a). However, BgaB activity could not be detected under the control of P351016 (Fig. 2b, c and Additional file 1: Figures S3, S4). The activity of promoter with mutation in the − 22 region (Fig. 2a) decreased slightly under the control of P351022 (Fig. 2b, c and Additional file 1: Figures S3, S4).
Fig. 2.
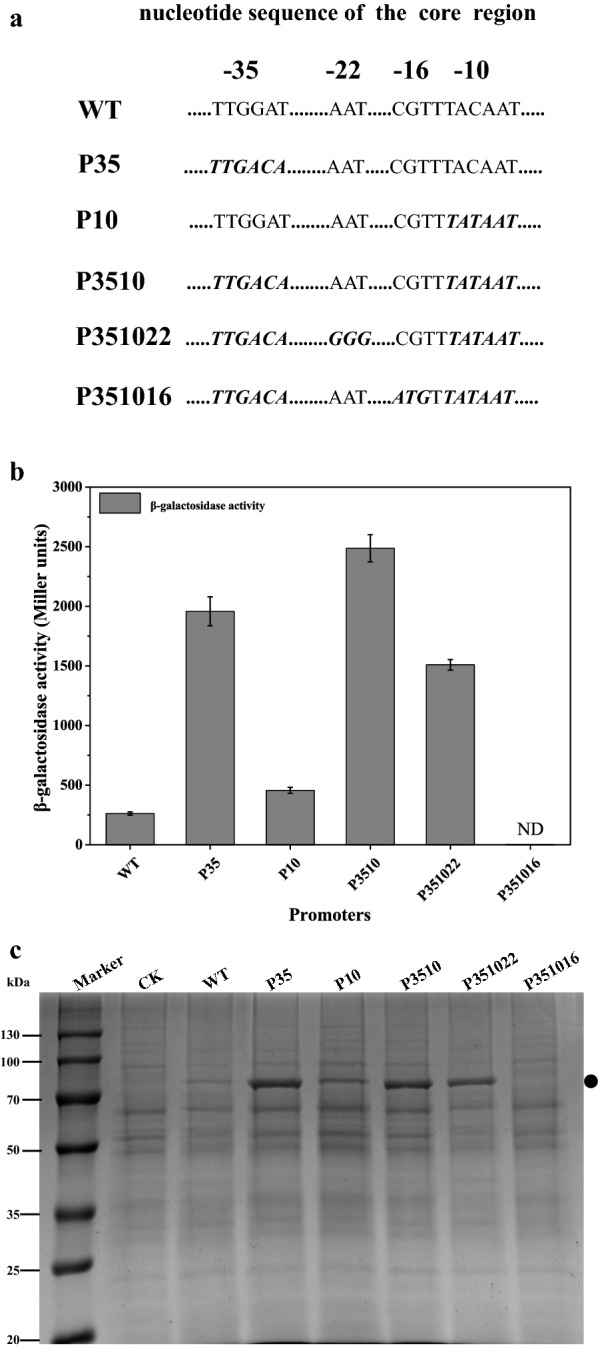
Optimization of the core region of the Pylb promoter. a the core regions of the Pylb promoter were changed to corresponding consensus sequence. The nucleotides in bold italic indicate mutated sequences. b The BgaB expression level under the control of Pylb derivatives. c SDS-PAGE analysis of the BgaB expression. Equal amounts (30 μg) of total protein were loaded into each lane. The band corresponding to BgaB was marked. All cultures were grown in triplicate, and each experiment was performed at least twice. Error bars indicate standard deviations. CK represents the intracellular protein of strain WB800
Engineering the upstream sequence to improve transcription
Since upstream elements could enhance transcription initiation in B. subtilis [31, 32], to further enhance the promoter P3510, putative UP elements (− 59 to − 38) of four rrn operons (rrnO, rrnJ, rrnD, rrnB) controlled by tandem promoters (P1 and P2) [33] were introduced to replace the native region (Fig. 3a). As shown in Fig. 3b, four UP elements showed a distinct activation of the transcription as compared to the native UP element. The engineered BP3510 and JP3510 was approximately one fold stronger than that of the P3510 promoter, demonstrating the strong stimulation of the promoter activity. When the UP element of rrnB was engineered to a consensus sequence [34], − 59 nnAAA(A/T)(A/T)T(A/T)TTTTnnAAAAnnn − 38, the new UP element was mutated to TTAAAAATTTTTTTTAAAAAAA (Fig. 3a). The mutant promoter NBP3510 showed superior activity to all of the other promoters and the BgaB expression was twofold higher than that of the P3510 (Fig. 3b, c and Additional file 1: Figures S5, S6) and enhanced by 26-fold compared with that of the WT promoter. In addition, the native UP region of P3510 was also changed to a consensus sequence (Fig. 3a) and the resulting promoter NP3510 was enhanced up to 1.7-fold (Fig. 3b, c and Additional file 1: Figures S5, S6). In summary, the strongest promoter NBP3510 allowed intracellular accumulation up to about 43% of the total cellular protein. In addition, promoter engineering had no significant effect on bacterial growth (Additional file 1: Figures S14, S15 and Table S2). Thus, the increased production was mainly due to the improved transcription. Western blot results showed that BgaB was expressed (Additional file 1: Figures S11 and S13). Furthermore, qRT-PCR was used to verify the transcription level of NBP3510. RNA was extracted after 4, 8, 12, 16 h. The highest transcription level of NBP3510 was 340-fold stronger than promoter Pylb (Fig. 3d).
Fig. 3.

Optimization of the upstream region of the P3510 promoter. a the upstream regions of the P3510 promoter were changed to corresponding sequence. The nucleotides in bold italic indicate mutated sequences. b The BgaB expression level under the control of P3510 derivatives. c SDS-PAGE analysis of the BgaB expression. Equal amounts (30 μg) of total protein were loaded into each lane. The band corresponding to BgaB was marked. d Comparative analysis of the transcription levels of two promoters at different time points. The transcription level of Pylb promoter at the time point 4 h was set as 1. The reference gene was 16 s rRNA. All cultures were grown in triplicate, and each experiment was performed at least twice. Error bars indicate standard deviations
Although the strength of NBP3510 was dramatically enhanced at both log phage and stationary phase after engineering, it still exhibits the property of “stationary phase” (Fig. 1c and Additional file 1: Figures S5, S6). Since target gene was also transcribed in the early stage of cell growth under the control of stationary-phase promoter. Thus, the kinetics of BgaB production were not fit very well with Luedeking and Piret [35] (Additional file 1: Figure S17 and Table S3).
Intracellular expression of the sfGFP protein
Another reporter protein sfGFP [36] was used to verify if the strong promoter NBP3510 was suitable for highly efficient intracellular expression. The NBP3510 showed prominent fluorescence by naked-eye detection (Fig. 4a) and the fluorescence intensity was enhanced up to 195-fold than that of the WT promoter (Fig. 4b and Additional file 1: Figures S7, S8). The sfGFP expression reached 30% of total cellular protein in SDS-PAGE (Fig. 4c). Western blot results showed that sfGFP was expressed (Additional file 1: Figures S11 and S13). Together with the BgaB expression, these results revealed that the engineered promoter NBP3510 was sufficient for efficient chromosomally integrated intracellular expression.
Fig. 4.

Intracellular expression of sfGFP. a The fluorescence imagines of strains WBGFP (Pylb) and WBSGFP (NBP3510). Strains were cultured for 16 h in LB medium and imagines were taken. b The fluorescence intensity controlled by different promoters. c The accumulative sfGFP protein in different strains. M, Marker. Lane 1, Strain WB800. Lane 2, Strain WBGFP. Lane 3, Strain WBSGFP. Equal amounts (30 μg) of total protein were loaded into each lane. The band corresponding to sfGFP was marked. All cultures were grown in triplicate, and each experiment was performed at least twice. Error bars indicate standard deviations
Extracellular expression of Methyl Parathion Hydrolase (MPH) and Chlorothalonil hydrolytic dehalogenase (Chd) by promoter NBP3510
To test whether the engineered promoter was suitable for overproduction of extracellular protein, strains WBSMPH and WBSChd were cultured in 2 × SR [37] medium. The cell growth, MPH activity, Chd activity and protein overproduction were measured throughout cultivation (Fig. 5 and Additional file 1: Figures S9, S10). Mutant strains showed no significant difference on bacterial growth compared with strain WB800 (Additional file 1: Figure S16 and Table S2). The extracellular protein displayed stationary phase-dependent pattern and the activity was significantly increased from the mid-exponential phase to stationary phase (Fig. 5a, c). The activity of MPH measured from the supernatant was as high as 144 U/ml (Fig. 5a, b) which was 5.3-fold of that of plasmid pP43NMK-mediated expression (P43-mpd cassette) [38]. The yield of MPH was increased to 0.3 g/L on shake-flask culture condition. The activity of Chd measured from the supernatant was as high as 4.4 U/mL (Fig. 5c, d) which was 300-fold higher than that of plasmid pP43Chd-mediated expression (P43-chd cassette) [39]. The yield of Chd was increased to 0.27 g/L on shake-flask culture condition. Western blot results showed that MPH and Chd were expressed (Additional file 1: Figures S12, S13).
Fig. 5.

Overproduction of Methyl Parathion Hydrolase (MPH) and Chlorothalonil hydrolytic dehalogenase (Chd) using the promoter NBP3510. a The activities of MPH in the supernatant (solid rectangle) and cell density (empty circle) were determined at different times. b The expression of MPH was analyzed by SDS-PAGE. Equal amounts (20 μL) of culture supernatant were loaded into each lane. c The activities of Chd in the supernatant (solid rectangle) and cell density (empty circle) were determined at different times. d The expression of Chd was analyzed by SDS-PAGE. Equal amounts (10 μL) of culture supernatant were loaded into each lane. The bands indicating to the target proteins were marked. All cultures were grown in triplicate, and each experiment was performed at least twice. Error bars indicate standard deviations. CK represents the extracellular protein of strain WB800
Discussion
So far, most promoters reported in B. subtilis were ligated to plasmid for protein production [19, 22, 27, 40–43]. Only four promoters (cry3Aa [26], amyQ [44], aprE [45] and a hybrid promoter [46] consist of P43 [9], Pylb [29] and Prha [47] were integrated to express single-copy gene. However, detailed protein production level was not mentioned except promoter aprE (10% intracellular total protein). Since single copy of expression cassette was not comparable to plasmid-expression system, it is necessary to choose an ideal promoter for stable and efficient protein production.
To date, promoters could be obtained by three fundamental approaches: (1) screening from the microbial genome and identification of the promoter [48–52]; (2) generating libraries of artificial promoters [36, 53–56]; (3) engineering the core region of known promoters [26, 28, 31, 45]. In this work, existing stationary phase-dependent promoter Pylb was engineered to achieve protein overproduction in a single copy cassette.
It is widely recognized that core region and the UP element were the key elements that contribute most to promoter activity. By engineering the core region to consensus sequence, the results indicated − 35 region played a pivotal role in promoter activity, which was in accordance with previous findings that the promoter activity of PaprN and PgroES-groEL were improved by modifying the − 35 region [31, 57]. It is presumably the poorer homology of the − 35 motifs than the − 10 region in Pylb to δA-dependent consensuses that resulted in the major contribution of the − 35 region. The − 16 region was reported to have a pronounced effect on transcription [31] when changed to consensus sequence TRTG and the TRTG motif was shown to stabilizes the transcription initiation open complex [58]. However, our result was opposed to the report and no BgaB activity was detected while engineering the − 16 region. It is probably that the native − 16 region is a key sequence involved in transcriptional activity. This result may be the most difference compared with other promoters. The − 22 region was also a potential target [31]. The phenomenon that the activity of promoter decreased slightly when changed from AAT to GGG indicated that “AT base” maybe suitable for “G” at positions − 22 region.
The UP element, a component of bacterial promoters located upstream of the − 35 hexamer, increases transcription by interacting with the RNA polymerase α-subunit [34]. It has been reported that UP element could stimulate transcription initiation in B. subtilis although UP elements are not crucial for transcription of all promoters [32]. Since the major products of all cellular transcription in B. subtilis are rRNA and tRNA, which constitute more than 95% of the total RNA [59], the UP elements from rrn operator may further stimulate promoter activity. There are 10 rrn operons in B. subtilis, controlled by tandem (P1 and P2) promoters. According to previously reported rrn operons [33], we chose UPs from four strong rrn operons controlled by P1 promoter. While replacing the UP sequence with that of rrnB operon, the promoter activity showed the best performance compared to other three. This is consistent with the report that strong rrnB P1 UP element increased the rate of RNAP binding [60]. To further engineering the promoter, the UP of rrnB was changed to consensus sequence, generating a new UP element consisting of A and T only. The UP engineering was verified again that the activity of a natural UP element could be improved by making it more AT-rich [34].
In this study, two reporter proteins BgaB and sfGFP were highly expressed. The intracellular expression level (43%) of BgaB was higher than that driven by promoter Pgrac in plasmid pHT100 (30%) [31] and the intracellular expression level (30%) of sfGFP was comparable to that driven by promoter PsrfA in plasmid pBSG3x (28.4%) [28]. The expression of two intracellular single-copy reporter proteins demonstrated that promoter NBP3510 was suitable for intracellular protein expression. To further exploit the application of NBP3510 promoter in B. subtilis, MPH and Chd proteins were extracellular expressed. The activity of MPH (144 U/mL) was higher than that driven by promoter P43 in plasmid pP43NMK (27.1 U/mL) and the expression level (0.3 g/L) was also superior to pP43NMK-mediated expression (53 mg/L) [9]. The activity of Chd (4.4 U/mL) was higher than that driven by promoter P43 in plasmid pP43Chd (14.5 U/L) and the expression level (0.27 g/L) was also superior to pP43Chd-mediated expression (5.65 mg/L) [39]. All in all, all these results indicate that single copy of promoter NBP3510 could be used to overexpress foreign proteins in B. subtilis.
Aside from promoter strength, there are still many variables affecting expression levels, including the stability of the mRNA [61], protein translation [62, 63], the culture conditions [64, 65] and so on. Thus, protein production can be further improved by optimizing the variables above.
Conclusions
Bacillus subtilis, a versatile microorganism, has been used to overexpress various recombinant proteins owing to its outstanding biological characteristics. In this study, highly efficient promoter NBP3510 was generated and two intracellular proteins (BgaB, sfGFP) and extracellular proteins (MPH, Chd) were overexpressed from a single copy expression cassette.
Methods
Strains, plasmids and growth conditions
Bacterial strains and plasmids used in this study are listed in Table 1. Plasmid pAX01 was a gift from the Bacillus Genetic Stock Center (BGSC). Escherichia coli Top 10 were used as the host for gene cloning. B. subtilis WB800 was used for gene expression and integration. Unless otherwise indicated, the final concentrations of antibiotics were as follows, mg/L: ampicillin (Amp), 100 for E. coli; erythromycin (Em), 5 for Bacillus. Strains were cultivated in Luria–Bertani (LB) medium or 2 × Super-Rich (SR) [37] medium. LB medium consisted of 1% tryptone, 0.5% yeast extract and 0.5% NaCl while 2 × SR medium consisted of 3% tryptone, 5% yeast extract and 0.6% K2HPO4, pH 7.2. Strains were cultivated at 37 °C in shaking flasks (SHUNIU, GG-17, Sichuan SHUBO Co., LTD, China) on an incubator shaker (IS-RDV1, Crystal, China) operating at 200 rpm. To express recombinant protein, 0.3 mL of preculture was transferred into 250-mL shaking flasks that were loaded with 30 mL of LB or 2 × SR liquid medium. The percentages of the produced intracellular proteins were calculated by Imagine J software. The cell density was determined by measuring the OD600 with a UV-1800/PC spectrophotometer (Shanghai, MAPADA Instrument Co., Ltd., China).
Table 1.
Strains and plasmids used in this study
| Strain or plasmid | Characteristicsa | Source or referenceb |
|---|---|---|
| Plasmids | ||
| pAX01 | PxylA, Apr, Emr, lacA | [15] |
| pYBGB | pAX01 containing Pylb-bgaB cassette | This work |
| pLJ-2 | CmR; E. coli-Bacillus shuttle vector | [19] |
| pUS20 | Spcr, unstable in B. subtilis, E. coli-Bacillus shuttle vector | [71] |
| Strains | ||
| E. coli Top10 | F− mcrA Δ(mrr-hsdRMS-mcrBC) ϕ80lacZΔM15 ΔlacX74 recA1 araD139 Δ(ara leu)7697 galU galK endA1 nupG | TransGene Biotech (Beijing, China) |
| B. subtilis | ||
| 168 | trpC2 | BGSC 1A1 |
| WB800 | (168) ΔnprE, Δ aprA, Δepr, Δ bpr, Δmpr, ΔnprB, Δvpr, ΔwprA | [72] |
| WBEmBgaB | Strain WB800 derivate, lacA::Pylb-bgaB, Emr | This work |
| WBBgaB | Strain WB800 derivate, lacA::Pylb-bgaB | This work |
| BS43 | Strain WB800 derivate, lacA::P43-bgaB | This work |
| BSxylA | Strain WB800 derivate, lacA::PxylA-bgaB | This work |
| BSsrfA | Strain WB800 derivate, lacA::PsrfA-bgaB | This work |
| 35BgaB | Strain WB800 derivate, lacA::P35-bgaB | This work |
| 10BgaB | Strain WB800 derivate, lacA::P10-bgaB | This work |
| 3510BgaB | Strain WB800 derivate, lacA::P3510-bgaB | This work |
| 351016BgaB | Strain WB800 derivate, lacA::P351016-bgaB | This work |
| 351022BgaB | Strain WB800 derivate, lacA::P351022-bgaB | This work |
| OBgaB | Strain WB800 derivate, lacA::OP3510-bgaB | This work |
| JBgaB | Strain WB800 derivate, lacA::JP3510-bgaB | This work |
| DBgaB | Strain WB800 derivate, lacA::DP3510-bgaB | This work |
| BBgaB | Strain WB800 derivate, lacA::BP3510-bgaB | This work |
| WBSBgaB | Strain WB800 derivate, lacA::NBP3510-bgaB | This work |
| WBGFP | Strain WB800 derivate, lacA:: Pylb -sfGFP | This work |
| WBSGFP | Strain WB800 derivate, lacA::NBP3510-bgaB | This work |
| WBSMPH | Strain WB800 derivate, lacA::NBP3510-mpd | This work |
| WBSChd | Strain WB800 derivate, lacA::NBP3510-chd | This work |
aApR: ampicillin resistance; CmR: chloramphenicol resistance; EmR: erythromycin resistance; Spcr: spectinomycin resistance
bBGSC: Bacillus Genetic Stock Center
DNA manipulation techniques
Oligonucleotides synthesis (Additional file 1: Table S1) and DNA sequencing were performed by Sangon Biotech Co., Ltd. (Shanghai, China). The isolation and manipulation of recombinant DNA was performed using standard techniques. All enzymes were commercial preparations. Phusion DNA high-fidelity polymerase was purchased from NEB (Shanghai, China). The transformation of B. subtilis was carried out as previously described [66].
Comparison of the WT promoter Pylb with other promoters in BagB expression
To construct Pylb-driven bagB expression strain, Fragment 1, a 2.0-kb fragment, comprising the bgaB encoded β-galactosidase activity was cloned from plasmid pLJ-2 [19] using the primer pair P1/P2. Concomitantly, Fragment 2, carrying the Pylb promoter from B. subtilis 168 genome was generated using the primer pair P3/P4. The third fragment, the pAX01 plasmid backbone was amplified using the primer pair P5/P6. The terminuses of three fragments were flanked by 30-bp homology in order using the Sequence and Ligation Independent Cloning (SLIC) method, yielding integrating plasmid pYBGB (Fig. 1a). The plasmid was then transformed into B. subtilis WB800, resulting in strain WBEmBgaB.
To eliminate the Em resistance, left flanking region (LF) and the right flanking region of erm was fused together using the primer pairs P7/P8 and P9/P10. The fragment was transformed to B. subtilis WBEmBgaB with the temperature-sensitive plasmid pUS20 by nature co-transformation. The temperature-sensitive plasmid pUS20 was subsequently cured by overnight growth without selection, generating the marker-free strain WBBgaB.
As for other promoter-driven expression strains, co-transformation was applied to replace the promoters with the Pylb promoters in strain WBBgaB. First, the promoters P43, PxylA and PsrfA were amplified from B. subtilis 168 genome using primer pairs P11/P12, P13/P14 and P15/P16. Next, the corresponding LF region and RF region were amplified from strain WBBgaB using the primer pairs P7/P17, P7/P19, P7/P21 and P18/P10, P20/P10, P22/P10, respectively. The promoters were flanked by the corresponding LF and RF region by overlapping-extension PCR. Three fused fragments with promoter-driven bgaB expression cassettes were separately transformed to B. subtilis WB800 by nature co-transformation described above. The resulting strains were designed as BS43, BSxylA and BSsrfA. All of the target mutation was confirmed by PCR amplification followed by DNA sequencing.
Site-mutation of the WT Pylb promoter
To mutate the core region of the Pylb promoter, for instance, the − 35 region, the strain WBBgaB containing Pylb-bgaB cassette was used as template. Primer pairs P7/P23 and P24/P10 were used to amplify the LF region and RF region, respectively. The mutation was introduced into the primers P23 and P24. Primers P23 and P24 were reverse complementation. The LF and RF fragments were fused by overlapping PCR and co-transformed with the plasmid pUS20 followed by elimination of pUS20, generating the strain 35bgaB. Other mutant promoters, P10, P3510, P351016 and P351022 were also constructed using the method described above and the corresponding strains (10BgaB, 3510BgaB, 351016BgaB and 351022BgaB) are also constructed using the primers listed in Additional file 1: Table S1.
As for mutation of the upstream sequence of P3510, strain 3510bgaB was used as template. The native UP was replaced by four upstream sequences of rrn operon (rrnO, rrnJ, rrnD, rrnB) P1 promoter [33]. New promoters OP3510, JP3510, DP3510, BP3510 and NBP3510 were generated the same as above. The corresponding strains (OBgaB, JBgaB, DBgaB, BBgaB and WBSBgaB) and the primers are listed in Additional file 1: Table S1.
Measurement of BgaB encoded β-galactosidase activity
Bacillus subtilis WB800 containing each engineered promoter with the bgaB reporter gene was cultured at 37 °C in an orbital shaker at 200 rpm in LB medium without antibiotic. After incubation for 16 h, samples were taken for determination of β-galactosidase activities. The β-galactosidase specific activities were converted to Miller units, as described previously [67]. The values shown are the average of three independent experiments.
Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE)
The protein samples were mixed with 5 × SDS-PAGE sample buffer (125 mM Tris–HCl pH 6.8, 4% SDS, 20% glycerol, 10% β-mercaptoethanol and 0.004% bromophenol) and heated at 100 °C for 10 min. The sample was centrifuged at 12,000 rpm for 5 min and the supernatant was used for SDS-PAGE. The electrophoresis was performed at 80 V through the stacking gel (5%) and at 120 V through the separation gel (9%) until the bromophenol blue marker dye reached to within 0.5 cm of the bottom of the gel. Then, the protein bands were stained with Coomassie Brilliant Blue R-250. After staining, gels were destained overnight in a solution containing 5% ethanol and 10% acetic acid.
Construction of the GFP, Methyl Parathion Hydrolase (MPH) and Chlorothalonil hydrolytic dehalogenase (Chd) expression strains
To construct integrative strain for intracellular GFP expression and extracellular MPH and Chd expression, sfgfp [36] was synthesized from Genescript Company (Nanjing, China). Gene mpd encoding methyl parathion hydrolase was amplified from the plasmid pP43NMK [38] using primer pairs P43/P44. Gene chd encoding chlorothalonil hydrolytic dehalogenase was amplified from the plasmid pP43Chd using primer pairs P45/P46 [39]. For extracellular expression of MPH and Chd, the single peptide of aprE was assembled to mpd and chd using primer pairs P47/P48 and P49/P50. The corresponding LF (including the promoter Pylb or NBP3510) region was cloned from the strain WBBgaB and WBSBgaB. The common RF region was amplified from the strain WB800. Then the LF (Pylb or NBP3510), sfGFP and the RF fragment were fused together and transformed to WB800, generating the strains WBGFP and WBSGFP. Strains harboring NBP3510-mpd cassette and NBP3510-chd were also constructed as described above, namely WBSMPH and WBSChd, respectively.
Flow cytometric analysis
Bacillus subtilis WB800 with the sfGFP reporter gene was cultured at 37 °C in an orbital shaker at 200 rpm in LB medium without antibiotic. Cells were taken and washed twice with phosphate-buffered saline (PBS, pH 7.5), diluted tenfold in PBS and then analyzed with a BD Accuri C6 flow cytometer (BD, Oxford, UK) using an argon laser at 488 nm as described previously [68]. For each sample, at least 5 × 104 cells were analyzed. Standard deviations are based on a minimum of three statistically independent experiments. Data were obtained using FlowJo V10 software (http://www.flowjochina.com/).
Expression of MPH and Chd using promoter NBP3510
A fresh overnight culture of the recombinant strain containing mpd or chd cassette was inoculated into 250-mL shake flasks containing 30 mL 2 × SR [37] liquid medium, cultivated 24 h and periodically sampled. A cell-free supernatant was obtained by centrifugation (5 min, 10,000×g). MPH activity measurements were performed as previously described [38, 69]. One unit of MPH activity was defined as the amount of enzyme required to hydrolyze 1 μmol methyl parathion in 1 min at 35 °C. Chd activity measurements were performed as previously described [39, 70]. One unit of Chd activity was defined as the amount of enzyme needed to decrease 1 mmol chlorothalonil per minute under optimal conditions.
Additional file
Additional file 1. Fig. S1–S2 The expression pattern of BgaB in mutant strains with different promoters. Fig. S3–S4 The expression pattern of BgaB controlled by mutant Pylb. Fig. S5-S6 The expression pattern of BgaB controlled by mutant P3510. Fig. S7–S8 The expression pattern of sfGFP in mutant strains. Fig. S9–S10 The control of MPH (a) and Chd (b) expression in strain WB800. Fig. S11 Purification of BgaB and sfGFP. Fig. S12 Purification of MPH and Chd. Fig. S13 Verification of BgaB, sfGFP, MPH and Chd by western blot. Fig. S14-S16 The kinetics of bacterial growth. Fig. S17 The kinetic model for BgaB production in mutant strains. Table S1 Primers used in this study. Table S2 the parameters in the kinetics of bacterial growth. Table S3 the parameters in the kinetics of BgaB production.
Acknowledgements
Not applicable.
Abbreviations
- BGSC
Bacillus Genetic Stock Center
- SLIC
sequence and ligation independent cloning
- MPH
Methyl Parathion Hydrolase
- Chd
chlorothalonil hydrolytic dehalogenase
- sfGFP
super-folded green fluorescent protein
- UP
upstream sequence
Authors’ contributions
ZC constructed the plasmids, the variant strain, accomplished the determination of the recombinant expression system, analyzed the data and wrote the manuscript. YB conceived the idea, designed this study. CS was participated in the determination of expression pattern using reporter gene. ZL and LY analyzed the data. YX and JJ conceived of the study, participated in its design, and coordination. All authors read and approved the final manuscript.
Funding
This work was supported by the National Natural Science Foundation (31770125 and 31800095), the 863 Plan of China (2014AA020543) and the Natural Science Foundation of Jiangsu Province (BK20180541).
Availability of data and materials
All data generated or analyzed during this study are included in this published article.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Chaoyang Zhou, Email: 2015216019@njau.edu.cn.
Bin Ye, Email: 2016216017@njau.edu.cn.
Shan Cheng, Email: 2017116088@njau.edu.cn.
Leizhen Zhao, Email: 2016116081@njau.edu.cn.
Yuanxin Liu, Email: 2017816155@njau.edu.cn.
Jiandong Jiang, Email: jiang_jjd@njau.edu.cn.
Xin Yan, Phone: 86 25 84396685, Email: yanxin@njau.edu.cn.
References
- 1.Terpe K. Overview of bacterial expression systems for heterologous protein production: from molecular and biochemical fundamentals to commercial systems. Appl Microbiol Biotechnol. 2006;72:211–222. doi: 10.1007/s00253-006-0465-8. [DOI] [PubMed] [Google Scholar]
- 2.Schumann W. Production of recombinant proteins in Bacillus subtilis. Adv Appl Microbiol. 2007;62:137–189. doi: 10.1016/S0065-2164(07)62006-1. [DOI] [PubMed] [Google Scholar]
- 3.Commichau FM, Alzinger A, Sande R, Bretzel W, Meyer FM, Chevreux B, Wyss M, Hohmann HP, Pragai Z. Overexpression of a non-native deoxyxylulose-dependent vitamin B6 pathway in Bacillus subtilis for the production of pyridoxine. Metab Eng. 2014;25:38–49. doi: 10.1016/j.ymben.2014.06.007. [DOI] [PubMed] [Google Scholar]
- 4.Yang H, Liu L, Li J, Du G, Chen J. Heterologous expression, biochemical characterization, and overproduction of alkaline alpha-amylase from Bacillus alcalophilus in Bacillus subtilis. Microb Cell Fact. 2011;10:77. doi: 10.1186/1475-2859-10-77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marcus S, Ajay S, Ward OP. Developments in the use of Bacillus species for industrial production. Can J Microbiol. 2004;50:1. doi: 10.1139/w03-076. [DOI] [PubMed] [Google Scholar]
- 6.Vavrová Ľ, Muchová K, Barák I. Comparison of different Bacillus subtilis expression systems. Res Microbiol. 2010;161:791–797. doi: 10.1016/j.resmic.2010.09.004. [DOI] [PubMed] [Google Scholar]
- 7.Dong H, Zhang D. Current development in genetic engineering strategies of Bacillus species. Microb Cell Fact. 2014;13:63. doi: 10.1186/1475-2859-13-63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bron S, Holsappel S, Venema G, Peeters BPH. Plasmid deletion formation between short direct repeats in Bacillus subtilis is stimulated by single-stranded rolling-circle replication intermediates. Mol Gen Genet. 1991;226:88–96. doi: 10.1007/BF00273591. [DOI] [PubMed] [Google Scholar]
- 9.Wang PZ, Doi RH. Overlapping promoters transcribed by Bacillus subtilis sigma 55 and sigma 37 RNA polymerase holoenzymes during growth and stationary phases. J Biol Chem. 1984;259:8619–8625. [PubMed] [Google Scholar]
- 10.Herbort M, Klein M, Manting EH, Driessen AJ, Freudl R. Temporal expression of the Bacillus subtilis secA gene, encoding a central component of the preprotein translocase. J Bacteriol. 1999;181:493–500. doi: 10.1128/jb.181.2.493-500.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang AL, Liu H, Yang MM, Gong YS, Chen H. Assay and characterization of a strong promoter element from B. subtilis. Biochem Biophys Res Commun. 2007;354:90–95. doi: 10.1016/j.bbrc.2006.12.137. [DOI] [PubMed] [Google Scholar]
- 12.Zyprian E, Matzura H. Characterization of signals promoting gene expression on the Staphylococcus aureus plasmid pUB110 and development of a gram-positive expression vector system. DNA. 1986;5:219–225. doi: 10.1089/dna.1986.5.219. [DOI] [PubMed] [Google Scholar]
- 13.Fukushima T, Ishikawa S, Yamamoto H, Ogasawara N, Sekiguchi J. Transcriptional, functional and cytochemical analyses of the veg gene in Bacillus subtilis. J Biochem. 2003;133:475–483. doi: 10.1093/jb/mvg062. [DOI] [PubMed] [Google Scholar]
- 14.Bhavsar AP, Zhao X, Brown ED. Development and characterization of a xylose-dependent system for expression of cloned genes in Bacillus subtilis: conditional complementation of a teichoic acid mutant. Appl Environ Microbiol. 2001;67:403–410. doi: 10.1128/AEM.67.1.403-410.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hartl B, Wehrl W, Wiegert T, Homuth G, Schumann W. Development of a new integration site within the Bacillus subtilis chromosome and construction of compatible expression cassettes. J Bacteriol. 2001;183:4393. doi: 10.1128/JB.183.8.2696-2699.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Silbersack J, Jurgen B, Hecker M, Schneidinger B, Schmuck R, Schweder T. An acetoin-regulated expression system of Bacillus subtilis. Appl Microbiol Biotechnol. 2006;73:895–903. doi: 10.1007/s00253-006-0549-5. [DOI] [PubMed] [Google Scholar]
- 17.Phan TT, Schumann W. Development of a glycine-inducible expression system for Bacillus subtilis. J Biotechnol. 2007;128:486–499. doi: 10.1016/j.jbiotec.2006.12.007. [DOI] [PubMed] [Google Scholar]
- 18.Le Thuy AT, Schumann W. A novel cold-inducible expression system for Bacillus subtilis. Protein Expr Purif. 2007;53:264–269. doi: 10.1016/j.pep.2006.12.023. [DOI] [PubMed] [Google Scholar]
- 19.Yang MM, Zhang WW, Chen YL, Gong YS. Development of a Bacillus subtilis expression system using the improved Pglv promoter. Microb Cell Fact. 2010;9:55. doi: 10.1186/1475-2859-9-90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Promchai R, Promdonkoy B, Tanapongpipat S, Visessanguan W, Eurwilaichitr L, Luxananil P. A novel salt-inducible vector for efficient expression and secretion of heterologous proteins in Bacillus subtilis. J Biotechnol. 2016;222:86–93. doi: 10.1016/j.jbiotec.2016.02.019. [DOI] [PubMed] [Google Scholar]
- 21.Yue J, Fu G, Zhang DW, Wen JP. A new maltose-inducible high-performance heterologous expression system in Bacillus subtilis. Biotechnol Lett. 2017;39:1237–1244. doi: 10.1007/s10529-017-2357-7. [DOI] [PubMed] [Google Scholar]
- 22.Kerovuo J, von Weymarn N, Povelainen M, Auer S, Miasnikov A. A new efficient expression system for Bacillus and its application to production of recombinant phytase. Biotechnol Lett. 2000;22:1311–1317. doi: 10.1023/A:1005694731039. [DOI] [Google Scholar]
- 23.Bongers RS, Veening JW, Van Wieringen M, Kuipers OP, Kleerebezem M. Development and characterization of a subtilin-regulated expression system in Bacillus subtilis: strict control of gene expression by addition of subtilin. Appl Environ Microbiol. 2005;71:8818–8824. doi: 10.1128/AEM.71.12.8818-8824.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nijland R, Lindner C, van Hartskamp M, Hamoen LW, Kuipers OP. Heterologous production and secretion of Clostridium perfringens beta-toxoid in closely related Gram-positive hosts. J Biotechnol. 2007;127:361–372. doi: 10.1016/j.jbiotec.2006.07.014. [DOI] [PubMed] [Google Scholar]
- 25.Toymentseva AA, Schrecke K, Sharipova MR, Mascher T. The LIKE system, a novel protein expression toolbox for Bacillus subtilis based on the liaI promoter. Microb Cell Fact. 2012;11:143. doi: 10.1186/1475-2859-11-143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lee SJ, Pan JG, Park SH, Choi SK. Development of a stationary phase-specific autoinducible expression system in Bacillus subtilis. J Biotechnol. 2010;149:16–20. doi: 10.1016/j.jbiotec.2010.06.021. [DOI] [PubMed] [Google Scholar]
- 27.Guan C, Cui W, Cheng J, Zhou L, Guo J, Hu X, Xiao G, Zhou Z. Construction and development of an auto-regulatory gene expression system in Bacillus subtilis. Microb Cell Fact. 2015;14:150. doi: 10.1186/s12934-015-0341-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Guan C, Cui W, Cheng J, Zhou L, Liu Z, Zhou Z. Development of an efficient autoinducible expression system by promoter engineering in Bacillus subtilis. Microb Cell Fact. 2016;15:66. doi: 10.1186/s12934-016-0464-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yu X, Xu J, Liu X, Chu X, Wang P, Tian J, Wu N, Fan Y. Identification of a highly efficient stationary phase promoter in Bacillus subtilis. Sci Rep. 2015;5:18405. doi: 10.1038/srep18405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Helmann JD. Compilation and analysis of Bacillus subtilis sigma(a)-dependent promoter sequences-evidence for extended contact between RNA-polymerase and upstream promoter DNA. Nucleic Acids Res. 1995;23:2351–2360. doi: 10.1093/nar/23.13.2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Phan TT, Nguyen HD, Schumann W. Development of a strong intracellular expression system for Bacillus subtilis by optimizing promoter elements. J Biotechnol. 2012;157:167–172. doi: 10.1016/j.jbiotec.2011.10.006. [DOI] [PubMed] [Google Scholar]
- 32.Meijer WJ, Salas M. Relevance of UP elements for three strong Bacillus subtilis phage phi29 promoters. Nucleic Acids Res. 2004;32:1166–1176. doi: 10.1093/nar/gkh290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samarrai W, Liu DX, White AM, Studamire B, Edelstein J, Srivastava A, Widom RL, Rudner R. Differential responses of Bacillus subtilis rRNA promoters to nutritional stress. J Bacteriol. 2011;193:723–733. doi: 10.1128/JB.00708-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Estrem ST, Gaal T, Ross W, Gourse RL. Identification of an UP element consensus sequence for bacterial promoters. Proc Natl Acad Sci USA. 1998;95:9761–9766. doi: 10.1073/pnas.95.17.9761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Luedeking R, Piret EL. A kinetic study of the lactic acid fermentation Batch process at controlled pH. Biotechnol Bioeng. 2000;67:636–644. doi: 10.1002/(SICI)1097-0290(20000320)67:6<636::AID-BIT3>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 36.Guiziou S, Sauveplane V, Chang HJ, Clerté C, Declerck N, Jules M, Bonnet J. A part toolbox to tune genetic expression in Bacillus subtilis. Nucleic Acids Res. 2016;44:7495–7508. doi: 10.1093/nar/gkw624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Song Y, Fu G, Dong H, Li J, Du Y, Zhang D. High-efficiency secretion of beta-mannanase in Bacillus subtilis through protein synthesis and secretion optimization. J Agric Food Chem. 2017;65:2540–2548. doi: 10.1021/acs.jafc.6b05528. [DOI] [PubMed] [Google Scholar]
- 38.Zhang XZ, Cui ZL, Hong Q, Li SP. High-level expression and secretion of methyl parathion hydrolase in Bacillus subtilis WB800. Appl Environ Microbiol. 2005;71:4101–4103. doi: 10.1128/AEM.71.7.4101-4103.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Meng C, He Q, Huang JW, Cao Q, Yan X, Li SP, Jiang JD. Degradation of chlorothalonil through a hydrolytic dehalogenase secreted from Bacillus subtilis WB800. Int Biodeterior Biodegrad. 2015;104:97–104. doi: 10.1016/j.ibiod.2015.05.017. [DOI] [Google Scholar]
- 40.Tran DTM, Phan TT, Huynh TK, Dang NTK, Huynh PTK, Nguyen TM, Truong TTT, Tran TL, Schumann W, Nguyen HD. Development of inducer-free expression plasmids based on IPTG-inducible promoters for Bacillus subtilis. Microb Cell Fact. 2017;16:130. doi: 10.1186/s12934-017-0747-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Phan TT, Tran LT, Schumann W, Nguyen HD. Development of Pgrac100-based expression vectors allowing high protein production levels in Bacillus subtilis and relatively low basal expression in Escherichia coli. Microb Cell Fact. 2015;14:72. doi: 10.1186/s12934-015-0255-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Panahi R, Vasheghani-Farahani E, Shojaosadati SA, Bambai B. Induction of Bacillus subtilis expression system using environmental stresses and glucose starvation. Ann Microbiol. 2014;64:879–882. doi: 10.1007/s13213-013-0719-5. [DOI] [Google Scholar]
- 43.Wenzel M, Muller A, Siemann-Herzberg M, Altenbuchner J. Self-inducible Bacillus subtilis expression system for reliable and inexpensive protein production by high-cell-density fermentation. Appl Environ Microbiol. 2011;77:6419–6425. doi: 10.1128/AEM.05219-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Widner B, Thomas M, Sternberg D, Lammon D, Behr R, Sloma A. Development of marker-free strains of Bacillus subtilis capable of secreting high levels of industrial enzymes. J Ind Microbiol Biotechnol. 2000;25:204–212. doi: 10.1038/sj.jim.7000051. [DOI] [Google Scholar]
- 45.Jan J, Valle F, Bolivar F, Merino E. Construction of protein overproducer strains in Bacillus subtilis by an integrative approach. Appl Microbiol Biotechnol. 2001;55:69. doi: 10.1007/s002530000448. [DOI] [PubMed] [Google Scholar]
- 46.Hirooka K, Tamano A. Bacillus subtilis highly efficient protein expression systems that are chromosomally integrated and controllable by glucose and rhamnose. Biosci Biotechnol Biochem. 2018;82:1942–1954. doi: 10.1080/09168451.2018.1497945. [DOI] [PubMed] [Google Scholar]
- 47.Hirooka K, Kodoi Y, Satomura T, Fujita Y. Regulation of the rhaEWRBMA operon involved in l-rhamnose catabolism through two transcriptional factors, RhaR and CcpA, in Bacillus subtilis. J Bacteriol. 2015;198:830. doi: 10.1128/JB.00856-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Blazeck J, Alper HS. Promoter engineering: recent advances in controlling transcription at the most fundamental level. Biotechnol J. 2013;8:46–58. doi: 10.1002/biot.201200120. [DOI] [PubMed] [Google Scholar]
- 49.Liu X, Yang H, Zheng J, Ye Y, Pan L. Identification of strong promoters based on the transcriptome of Bacillus licheniformis. Biotechnol Lett. 2017;39:873–881. doi: 10.1007/s10529-017-2304-7. [DOI] [PubMed] [Google Scholar]
- 50.Liao Y, Huang L, Wang B, Zhou F, Pan L. The global transcriptional landscape of Bacillus amyloliquefaciens XH7 and high-throughput screening of strong promoters based on RNA-seq data. Gene. 2015;571:252–262. doi: 10.1016/j.gene.2015.06.066. [DOI] [PubMed] [Google Scholar]
- 51.Phan TT, Nguyen HD, Schumann W. Establishment of a simple and rapid method to screen for strong promoters in Bacillus subtilis. Protein Expr Purif. 2010;71:174–178. doi: 10.1016/j.pep.2009.11.010. [DOI] [PubMed] [Google Scholar]
- 52.Song Y, Nikoloff JM, Fu G, Chen J, Li Q, Xie N, Zheng P, Sun J, Zhang D. Promoter screening from Bacillus subtilis in various conditions hunting for synthetic biology and industrial applications. PLoS ONE. 2016;11:e0158447. doi: 10.1371/journal.pone.0158447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hammer K, Mijakovic I, Jensen PR. Synthetic promoter libraries—tuning of gene expression. Trends Biotechnol. 2006;24:53–55. doi: 10.1016/j.tibtech.2005.12.003. [DOI] [PubMed] [Google Scholar]
- 54.Siegl T, Tokovenko B, Myronovskyi M, Luzhetskyy A. Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab Eng. 2013;19:98–106. doi: 10.1016/j.ymben.2013.07.006. [DOI] [PubMed] [Google Scholar]
- 55.Portela RM, Vogl T, Kniely C, Fischer JE, Oliveira R, Glieder A. Synthetic core promoters as universal parts for fine-tuning expression in different yeast species. ACS Synth Biol. 2017;6:471–484. doi: 10.1021/acssynbio.6b00178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hartner FS, Claudia R, David L, Johnson SN, Petr H, Lin-Cereghino GP, Joan LC, Karin K, Cregg JM, Anton G. Promoter library designed for fine-tuned gene expression in Pichia pastoris. Nucleic Acids Res. 2008;36:e76. doi: 10.1093/nar/gkn369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu SM, Feng C, Zhong J, Huan LD. Enhanced production of recombinant nattokinase in Bacillus subtilis by promoter optimization. World J Microbiol Biotechnol. 2011;27:99–106. doi: 10.1007/s11274-010-0432-5. [DOI] [Google Scholar]
- 58.Voskuil MI, Chambliss GH. The TRTGn motif stabilizes the transcription initiation open complex. J Mol Biol. 2002;322:521–532. doi: 10.1016/S0022-2836(02)00802-1. [DOI] [PubMed] [Google Scholar]
- 59.Paul BJ, Ross W, Gaal T, Gourse RL. rRNA transcription in Escherichia coli. Annu Rev Genet. 2004;38:749–770. doi: 10.1146/annurev.genet.38.072902.091347. [DOI] [PubMed] [Google Scholar]
- 60.Rao L, Ross W, Appleman JA, Gaal T, Leirmo S, Schlax PJ, Record MT, Jr, Gourse RL. Factor independent activation of rrnB P1. An “extended” promoter with an upstream element that dramatically increases promoter strength. J Mol Biol. 1994;235:1421–1435. doi: 10.1006/jmbi.1994.1098. [DOI] [PubMed] [Google Scholar]
- 61.Agaisse H, Lereclus D. STAB-SD: a Shine–Dalgarno sequence in the 5′ untranslated region is a determinant of mRNA stability. Mol Microbiol. 2010;20:633–643. doi: 10.1046/j.1365-2958.1996.5401046.x. [DOI] [PubMed] [Google Scholar]
- 62.Salis HM, Mirsky EA, Voigt CA. Automated design of synthetic ribosome binding sites to control protein expression. Nat Biotechnol. 2009;27:946–950. doi: 10.1038/nbt.1568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Borkowski O, Goelzer A, Schaffer M, Calabre M, Mäder U, Aymerich S, Jules M, Fromion V. Translation elicits a growth rate-dependent, genome-wide, differential protein production in Bacillus subtilis. Mol Syst Biol. 2016;12:870. doi: 10.15252/msb.20156608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Phulara SC, Chaurasia D, Diwan B, Chaturvedi P, Gupta P. In-situ isopentenol production from Bacillus subtilis through genetic and culture condition modulation. Process Biochem. 2018;72:47–54. doi: 10.1016/j.procbio.2018.06.019. [DOI] [Google Scholar]
- 65.Liu Y, Zhang BB, Sun BY, Liu LL, Zou LH, Shi YG. Optimization of fermentation conditions for production of neutral protease by Bacillus subtilis 10075. Food Sci. 2014;35:166–170. [Google Scholar]
- 66.Anagnostopoulos C, Spizizen J. Requirements for transformation In Bacillus Subtilis. J Bacteriol. 1961;81:741–746. doi: 10.1128/jb.81.5.741-746.1961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wiegert T, Homuth G, Versteeg S, Schumann W. Alkaline shock induces the Bacillus subtilis sigma(W) regulon. Mol Microbiol. 2001;41:59–71. doi: 10.1046/j.1365-2958.2001.02489.x. [DOI] [PubMed] [Google Scholar]
- 68.Veening JW, Smits WK, Hamoen LW, Kuipers OP. Single cell analysis of gene expression patterns of competence development and initiation of sporulation in Bacillus subtilis grown on chemically defined media. J Appl Microbiol. 2006;101:531–541. doi: 10.1111/j.1365-2672.2006.02911.x. [DOI] [PubMed] [Google Scholar]
- 69.Cui ZL, Zhang XZ, Zhang ZH, Li SP. Construction and application of a promoter-trapping vector with methyl parathion hydrolase gene mpd as the reporter. Biotechnol Lett. 2004;26:1115–1118. doi: 10.1023/B:BILE.0000035481.03854.41. [DOI] [PubMed] [Google Scholar]
- 70.Wang G, Li R, Li S, Jiang J. A novel hydrolytic dehalogenase for the chlorinated aromatic compound chlorothalonil. J Bacteriol. 2010;192:2737–2745. doi: 10.1128/JB.01547-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ye B, Zhou C, Zhao L, Cheng S, Cheng D, Yan X. Unmarked genetic manipulation in Bacillus subtilis by natural co-transformation. J Biotechnol. 2018;284:57–62. doi: 10.1016/j.jbiotec.2018.08.001. [DOI] [PubMed] [Google Scholar]
- 72.Wu SC, Yeung JC, Duan Y, Ye R, Szarka SJ, Habibi HR, Wong SL. Functional production and characterization of a fibrin-specific single-chain antibody fragment from Bacillus subtilis: effects of molecular chaperones and a wall-bound protease on antibody fragment production. Appl Environ Microbiol. 2002;68:3261–3269. doi: 10.1128/AEM.68.7.3261-3269.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Fig. S1–S2 The expression pattern of BgaB in mutant strains with different promoters. Fig. S3–S4 The expression pattern of BgaB controlled by mutant Pylb. Fig. S5-S6 The expression pattern of BgaB controlled by mutant P3510. Fig. S7–S8 The expression pattern of sfGFP in mutant strains. Fig. S9–S10 The control of MPH (a) and Chd (b) expression in strain WB800. Fig. S11 Purification of BgaB and sfGFP. Fig. S12 Purification of MPH and Chd. Fig. S13 Verification of BgaB, sfGFP, MPH and Chd by western blot. Fig. S14-S16 The kinetics of bacterial growth. Fig. S17 The kinetic model for BgaB production in mutant strains. Table S1 Primers used in this study. Table S2 the parameters in the kinetics of bacterial growth. Table S3 the parameters in the kinetics of BgaB production.
Data Availability Statement
All data generated or analyzed during this study are included in this published article.