

Abstract
Copper is one of the most abundant and less toxic transition metals. Nature takes advantage of the bioavailability and rich redox chemistry of Cu to carry out oxygenase and oxidase organic transformations using O2 (or H2O2) as oxidant. Inspired by the reactivity of these Cu-dependent metalloenzymes, chemists have developed synthetic protocols to functionalize organic molecules under enviormentally benign conditions. Copper also promotes other transformations usually catalyzed by 4d and 5d transition metals (Pd, Pt, Rh, etc.) such as nitrene insertions or C–C and C–heteroatom coupling reactions. In this review, we summarized the most relevant research in which copper promotes or catalyzes the functionalization of organic molecules, including biological catalysis, bioinspired model systems, and organometallic reactivity. The reaction mechanisms by which these processes take place are discussed in detail.
Graphical Abstract

1. INTRODUCTION AND SCOPE
Copper is one of the most abundant transition metals in earth’s crust, seawater, and human plasma.1,2 In 2017, 19.7 million tons of copper were produced with an average cost of $6.2 per kg.3 Of the late transition 3d metals (i.e., Fe, Co, Ni, Cu, and Zn), only Fe is produced in higher quantities and with lower costs. Copper can reach four oxidation states (Cu0, CuI, CuII, and CuIII) in a one-electron (e.g., CuI/CuII) or two-electron fashion (e.g., CuI/CuIII), which leads to plentiful and complex redox reactivity. In general, copper has lower toxicity than most 4d and 5d transition metals and similar toxicity to other late transition 3d metals (only iron is believed to have lower toxicity).4 Nature, synthetic chemists in academia, and chemical industry take advantage of the properties of copper (i.e., availability, low cost, relatively low toxicity, and rich redox chemistry) to carry out many chemical transformations.
Copper plays a key role in many natural processes.5 The ability of Cu to switch between two oxidation states (CuI and CuII) is used by nature to transfer electrons (blue copper proteins).6,7 Copper-dependent enzymes catalyze the decomposition of damaging reactive oxygen species (ROS) such as superoxide (Cu superoxide dismutase).8 Current research has shown that copper is involved in several diseases like Alzeheimer’s, heart failure, cancer, DNA damage, and antibiotic resistance.9–11 It has also been reported that Cu plays a key role in the oxidative modification of lipoprotein that causes atherosclerosis.12 In medicinal chemistry, copper complexes are currently used as anticancer drugs,13,14 small molecule sensors (e.g., detection of H2O2, NO),15–17 contrast reagents,18 and as catalysts to decompose toxic peroxynitrite (OONO−).19 Among all biological functions of copper, a wide variety of Cu-dependent metalloproteins are dedicated to O2 reduction, usually coupling this process with the oxidation of organic substrates.6,20–22 The 4H+/4e– reduction of O2 to water (cellular respiration in mitochondria) takes place in cytochrome C oxidase (CcO), which contains a binuclear active site consisting of a heme center and copper ion bound by three histidines and an unusual histidine-tyrosine residue.23 It has been also found that CcO also regulates the intracellular concentration of O2 using NO and NO2− as electron donor and acceptor, respectively.24,25 In other copper-dependent oxidases such as catechol oxidase or galactose oxidase, O2 acts as a trap for the protons and electrons formed during the catalytic oxidation of organic substrates.26 Cu-dependent monooxygenases also use O2 as an H+/e− sink, but they also are able to incorporate an oxygen atom into the substrate.27–29
Copper is utilized in important chemical processes including ATRP polymerization,30,31 phenol polymerization,32 CO reduction,33 Wacker oxidation,34 the selective oxidation of methane to methanol,35 alkyne–azide cycloaddition (“Click” chemistry)36 and Glaser-Hay alkyne coupling.37 Over the past few years, research has shown that copper carries out organometallic transformations such as C–C and C–heteroatom coupling reactions that are traditionally catalyzed by heavy metals such as palladium, opening new avenues to replace these expensive metals by copper in industrial applications.38–41
Recent review articles have focused on the detailed description of the enzymology of copper-dependent metalloproteins,6 the generation, characterization, and reactivity of biologically relevant synthetic Cu–O2 species,20,21,42 and the copper-catalyzed oxidation chemistry using oxygen as the oxidant,43 and the recent advances in the organometallic reactivity of copper.39 In this review, we discuss in detail the most recent and relevant research in which copper promotes (or catalyzes) the functionalization of organic molecules in the presence or absence of O2, emphasizing the reaction mechanisms by which these processes are proposed to take place. This compilation ranges from synthetic model complexes (including Cu/O2 systems and organocopper species) to examples of Cu-promoted functionalizations useful in organic synthesis. The reactivity is divided in two sections (see Figure 1), the first one focuses on bioinspired oxidations and oxygenations (e.g., hydroxylation of C–H bonds, dehydrogenation of alcohols, etc.) and the second one describes reactions that expand beyond enzymatic mimicry such as Cu-nitrene reactivity and organometallic-like transformations (e.g., Ullman-like coupling reactions and C–H activation). Research articles in which Cu complexes are used as catalysts for the O2 reduction to H2O (using external electron and proton sources) and as catalysts for water oxidation were not included in this compendium, but detailed mechanistic explanations and references can be found in recent publications.44,45 The numerous and outstanding research in which copper is used to reduce organic substrates (e.g., Buchwald CuH chemistry)46–48 was also outside of the scope of this review.
Figure 1.

Scope of this review article.
2. BIOINSPIRED Cu-PROMOTED OXIDATION OF SUBSTRATES USING O2 (OR ITS REDUCED FORMS) AS OXIDANTS
2.1. Overview of the Natural and Synthetic Cu–O2 Intermediates Formed Upon O2 Reduction
Aerobic life emerged 2.5 billion years ago with the propagation of organisms able to use solar energy to reduce CO2 to form C–C bonds with concomitant formation of O2 (i.e., photosynthesis).49 The increase in O2 concentrations led to the spread of eukaryotic systems able to consume the glucose produced in photosynthesis to generate CO2, water, and energy. These organisms also developed strategies to carry out the synthesis of complex metabolites, which usually entailed the oxidation of organic molecules using O2 as oxidant (oxidase chemistry) and O atom source (oxygenase chemistry). In order to bypass the inertness of O2 (S = 1 in the triplet ground state) toward organic substrates (most S = 0 in the ground state), nature designed catalysts able to reduce O2 to more reactive species (e.g., superoxide or peroxide).2,50 Most of these natural catalysts usually contain bioavailable metal cofactors in the active center (Fe, Mn, Cu, Ni, and Mo) that provide the electrons necessary to reduce O2 and bind these reduced species to generate metastable M-O2 species, which permits control of the reactivity of these “hot” natural oxidants and bypasses free-radical ROS damage (see formation of superoxide and hydroxyl radical in Figure 2A).
Figure 2.

(A) Energetics of the stepwise 4H+/4e− reduction of dioxygen to water. (B) Mononuclear, (C) dinuclear, (D) trinuclear and tetranuclear Cu/O2 species formed in the reduction of O2 (and its reduced forms).
Copper is used by nature to “activate” small molecules such as O2 in a wide array of biological processes including O2 transport, O2 reduction to water, oxidation of O–H bonds, oxygenation of C–H bonds (e.g., oxidation of methane to methanol in particulate methane monooxygenase, pMMO), among others. In these Cu-dependent metalloproteins, copper occupies mononuclear active/PHM, respectively, heterobimetallic active sites (e.g., heme-copper in CcO), or even trinuclear and tetranuclear copper sites (e.g., laccase and N2O reductase).6 Inspired by the natural systems, synthetic inorganic chemists have explored the use of low-weight Cu complexes that can mimic some of the features of the active center of Cu-dependent metalloenzymes (structure, spectroscopy, and reactivity).21,42,51,52 Cryogenic temperatures, organic solvents with various polarity, different copper sources (i.e., CuI or CuII salts with different counteranions), and the use of ligand donors beyond the ones found in nature (e.g., pyridines vs amino acids) have allowed for generating, stabilizing, and characterizing a wide array of Cu/O2 species (Figure 2). These can be systematically classified according to (see Figure 2B, top right): (i) number of copper ions that form the Cu/O2 species with their corresponding oxidation state; (ii) identity of the reduced O2 species (e.g., superoxide, (hydro)peroxide, oxyl, oxide, hydroxide, etc.), and (iii) binding mode of these reduced species to the copper center(s) (end-on and side-on).51,52 Pioneering this research field, Prof. K. D. Karlin reported in 1988 the first crystal structure of an L2CuII2(O22−) system bearing a tetradentate ligand in which the peroxide was coordinated in an end-on fashion (EPII,II in Figure 2B).53 Shortly after that, Kitajima published the crystal structure of a side-on L2CuII2(O22−) complex (SPII,II) that enabled the prediction for the structure of the Cu2O2 intermediate in tyrosinase (published 20 years after Kitajima’s article).54,55 Tolman research lab reported the first example of an L2Cu2O2 complex in which the O–O bond was cleaved reversibly, an L2CuIII2(O2−)2 species (O,OIII,III) that was in equilibrium with a side-on L2CuII2 (O22−) complex.56 Similarly, Stack reported a model system of tyrosinase in which an SPII,II species isomerized to an O,OIII,III complex upon addition of phenolate and prior to the oxidation of the substrate.57
The discovery of monooxygenase metalloezymes containing mononuclear Cu sites in the active center (i.e., lytic polysaccharide monooxygenase, LPMO) has triggered the development of model systems able to generate monocopper-O2 species.58 However, it has been challenging due to the tendency of mononuclear LCuO2 systems to form more stable L2Cu2O2 moieties. By rational ligand design, Schindler, Karlin, and Itoh have used N4 and N3 ligands, respectively, to stabilize mononuclear LCuII(O2•−) species that mimic the structure (end-on CuII-superoxo, ESII) and reactivity (oxidation of weak C–H bonds) of peptidylglycine α-hydroxylating monooxygenase (PHM).59–63 The recent discovery of Cu metalloenzymes able to oxidize strong C–H bonds (i.e., pMMO) has inspired the development of mononuclear and dinuclear Cu/O2 species able to carry out these transformations.64 For example, Tolman has reported the stoichiometric oxidation of C–H bonds using a mononuclear LCuIII–OH complex (MOHIII) that is generated via 1e− oxidation of a stable LCuII(OH) complex (MOHIII).65 Other mechanistic proposals for pMMO or PHM suggest that the oxidation of C–H bonds could only be achieved via formation of mononuclear copper(II)-hydroperoxo species (EHPII), which could react directly with the C–H substrate or could undergo O–O cleavage to generate copper(II) or copper(III)-oxyl species (O·II or O·III) before C–H oxidation.66 It has been proposed that these reactive intermediates (O·II and O·III) can be formed via reductive protonation of ESII species67 or by using ROOH oxidants (hydrogen peroxide or alkyl- and acyl-hydroperoxides) to generate LCuII(OOR) complexes (EHPII, EAPII) before O–O cleavage.68
Methodical design of low-weight model complexes has also allowed for generating Cu/O2 species that are not observed (but could be formed) in biological systems (Figure 2). These include: (i) mononuclear side-on copper(II)-superoxide species (SSII) and side-on copper(III)-peroxide species (SPIII)69,70 and(ii) reduced, protonated, and oxidized forms of other Cu/O2 species such as mononuclear copper(III)-alkylperoxide complexes (EAPIII),71 end-on superoxide dicopper species (ESI,II and ESII,II),72,73 dicopper(II) bis-hydroxide (OH,OHII,II), dicopper-(II) oxide-hydroxide (O,OHII,II), dicopper(II) oxide (OII,II) and dicopper(II) hydroxide (OHII,II) complexes,44,74 dicopper-(II) η1-hydroperoxide (η1HPII,II), and dicopper(II) η1-alkylper-oxide species (η1APII,II).75–77 Metalloenzymes and synthetic models also form trinuclear and tetranuclear Cu–O2 species in which dioxygen is reduced to the peroxide or oxide/hydroxide form (see Figure 2D).78–80
2.2. Oxidase-Like Reactivity
Dehydrogenative oxidations are very common in organic chemistry. These transformations include the oxidation of alcohols to aldehydes and ketones,81 aromatizations,82 oxidation of amines to imines83 and nitriles,83 coupling of phenols,84 among others. Large-scale industrial dehydrogenations are usually carried out using stoichiometric oxidants such as NaOCl, MnO2, Br2, or NaBrO3 that generate undesired waste.85 Metalloenzymes usually couple the dehydrogenation of organic substrates with the reduction of O2 (to H2O2 or water).86,87 Many research endeavors are focused on developing green synthetic protocols that can oxidize organic molecules using catalytic amounts of first row metal complexes and O2. In this section, we compiled the most relevant examples of Cu-promoted aerobic dehydrogenations, including galactose oxidase-like oxidations of alcohols (and other substrates) and dehydrogenative coupling of phenols.
2.2.1. Alcohol Oxidation to Aldehydes and Ketones. Galactose-Oxidase Model Systems.
Galactose oxidase (GAO) is a fungal metalloenzyme that performs the catalytic 2e− oxidation of alcohols to aldehydes using O2 and generates H2O2 (Figure 3). In its active center, a copper(II) ion is bound to two histidines, a tyrosine and an unusual tyrosine-cysteine (Tyr-Cys) cross-linked residue (Figure 3, i). In the active state of GAO (species A in Figure 3, iii) the Tyr-Cys residue is oxidized, which generates a CuII-phenoxyl radical species (EPR silent due to antiferromagnetic coupling).88,89 The catalytic reaction is separated in two half-reactions, oxidation of alcohol to aldehyde and reduction of O2 to H2O2 via a ping-pong mechanism (Figure 3, iii). It is proposed that initial deprotonation of the substrate by the tryosinate residue (species A) leads to coordination of the substrate alkoxide (species B), which would be oxidized in the rate-determining step of the reaction (kinetic isotope effect, KIE = 4–6) via hydrogen atom abstraction by the Tyr-Cys phenoxyl ligand (species C). Release of the aldehyde product would generate a CuI complex (species D), which would be oxidized by O2 to produce a mononuclear CuII-superoxide intermediate (ESII, species E). The Tyr-Cys phenoxyl moiety is then regenerated via formation of a mononuclear CuII-hydroperoxide complex (EHPII, species F) that would reform the active intermediate (species A) via deprotonation of the tyrosinate and release of H2O2.
Figure 3.

Galactose oxidase (GAO): active center (i), reaction catalyzed (ii), and proposed catalytic cycle (iii).
Stack developed one of the first copper systems able to mimic some of the reactivity features of galactose oxidase (Figure 4).90,91 In one of these contributions, a family of copper(II) complexes bearing diimino-diphenolate ligands (N2O2 coordination) catalyzed the oxidation of benzyl alcohol using tris(4-bromophenyl)ammonium hexachloroantimoniate as oxidant and in the presence of a strong base (Figure 4, iii).90 The authors observed that addition of the 1e− oxidant to the copper(II) complex under anaerobic conditions quenched the CuII EPR signal to generate a putative CuII-phenoxyl radical species that, after coordination of the substrate, could accept 2e− to produce benzaldehyde and the corresponding CuI complex (Figure 4, v). In a second landmark article, it was found that the oxidation of the same copper(II) complex (Figure 4, i) in the presence of an alkoxide substrate formed a pentacoordinate CuII-phenoxyl-substrate intermediate (species B in Figure 4, v), which was characterized by EXAFS spectroscopy.91 Interestingly, the study included the catalytic conversion of a series of alcohols (Figure 4, iv) with low catalyst loadings (0.01–0.06%) in the presence of small amounts of base (0.3–1.2%) using O2 as oxidant (Figure 4, iii). The aerobic reaction conditions (Figure 4, ii) achieved remarkable turnover number (TN up to 1300), but modest yields were obtained (up to 13%). A kinetic isotope effect (KIE) of 5.3 was found in the kinetic analysis of the oxidation of PhCH2O− versus PhCD2O−, which suggested that cleavage of the benzylic C–H bond occurred in the rate-determining step of the reaction (r.d.s.). The authors proposed that in the presence of O2 and deprotonated substrate, the CuII complex (species A in Figure 4) was oxidized to the corresponding CuII-phenoxyl-substrate intermediate (speciesB), which would carry out the 2e− oxidation of the substrate (via hydrogen atom abstraction during the r.d.s.) to generate the aldehyde product and a CuI complex (species C in Figure 4, v). The CuII-phenoxyl-substrate species could be regenerated by reaction of the CuI complex with O2 to produce a mononuclear CuII-hydroperoxide intermediate (EHPII, species D) that then releases H2O2 upon substrate coordination.
Figure 4.

Chaudhuri and Wieghardt reported a dinuclear CuII-phenoxyl species that catalyzed the oxidation of alcohols to aldehydes and 1,2-glycol derivatives with concomitant reduction of O2 to H2O2 (Figure 5A).92 Exposing mixtures of 2,2′-thiobis(2,4′-di-tert-butylphenol), CuCl, and NEt3 to O2 led to the assembly of a diamagnetic dicopper(II)-diphenoxyl complex (Figure 5A, i), which oxidized various alcohols to the corresponding 1e− coupling products and 2e− aldehyde products. Small amounts of the dinuclear complex (0.1%) were used to reach significant yields of the corresponding oxidation products (up to 68%) under mild conditions (Figure 5A, ii). It was proposed that the dicopper(II)-diphenoxyl complex promoted the 2e− oxidation of the substrate or the 1e− oxidation of two substrate equivalents depending on the alcohol identity (Figure 5A, iv). The authors emphasized that the oxidation equivalents were stored on two phenoxyl radicals of the ligand scaffolds (species A in Figure 5A). The facile oxidation of the dicopper(II)-diphenol complex (species D in Figure 5A) by O2 to generate H2O2 could explain the high reactivity of this system.
Figure 5.

(A) Dinuclear and (B) mononuclear galactose oxidase model systems.92,93
In a related system, the same authors used the N,N-bis(2-hydroxy-3,5-di-tert-butylphenol) ligand to synthesize a mononuclear CuII-phenoxyl complex by oxidation of a mixture of the deprotonated ligand, CuI and NEt3 with O2 (Figure 5B, i).93 The resulting copper(II) complex was characterized by X-ray diffraction analysis and EPR spectroscopy (EPR silent). This mononuclear system catalyzed the selective oxidation of alcohols to the corresponding aldehyde products (no C–C coupling product) utlizing low catalyst loadings (0.02%) at room temperature and using O2 as oxidant (Figure 5B, ii). A catalytic cycle analogous to the one proposed for galactose oxidase was suggested (Figure 5B, iv): ligand-promoted deprotonation of the substrate leads to coordination of alkoxide (species B in Figure 5B) with subsequent H atom abstraction (species C) and CuI/aldehyde formation (species D). Reaction of the reduced copper(I) complex with O2 could then produce a CuII-superoxide intermediate (species E in Figure 5B) that at room temperature releases H2O2 via intramolecular oxidation of the ligand framework. Interestingly, the authors were able to generate and isolate the putative CuII-superoxide species at low temperature, which was characterized by resonance Raman spectroscopy (ν(O–O): 964 cm−1; Δ18O2 = 55 cm−1).
Chaudhuri and Wieghardt also reported the GAO-like reactivity of copper and zinc complexes bearing a tetradentate redox-active ligand (Figure 6).94 It was found that both metal complexes catalyzed the oxidation of ethanol to acetaldehyde, with concomitant reduction of O2 to H2O2. Interestingly, a higher activity was observed for the Cu analogue (50% vs 2% yield). A detailed mechanistic analysis was provided, which suggested that the CuII-diimine-phenoxylphenolate form of the catalyst (species A in Figure 6) could deprotonate and coordinate the alcohol substrate (species B) and promote its oxidation via H atom abstraction (species C) followed by fast e− transfer (species D). The CuII-diimine-phenoxylphenolate state could then be regenerated via 2e−/2H+ reduction of O2 to H2O2.The authors showed that the CuII-semiquinonediiminato-diphenolate complex was oxidized to the corresponding CuII-diimine-phenoxylphenolate form using O2 in the presence of 2 equiv of acetic acid, which generated stoichiometric amounts of H2O2. The stoichiometric oxidation of substrates under anaerobic conditions was also analyzed. These studies included the use of 2-chloroethanol as substrate (see Figure 6, iii). It was proposed that the CuII-diimine-phenoxylphenolate complex was reduced by the substrate to generate the CuII-diimine-diphenolate species (1e− reduction) and a ketyl radical anion ClCH2 C•HO−. It was suggested that this radical undergoes fast rearrangement to Cl− and C•H2CHO, which dimerized to form succinic dialdehyde. The lack of chloroacetaldehyde product suggested that the oxidation occurs via H atom abstraction (1e− pathway) rather than hydride transfer (2e− pathway).
Figure 6.

Copper (and zinc) galactose oxidase model systems.94
Markó developed one of the first synthetically relevant Cu-based protocols for the oxidation of alcohols (Figure 7).95 The authors combined catalytic amounts of CuCl, phenanthroline (phen), and di-tert-butylazodicarboxylic acid (DBADH2) to oxidize a wide array of primary and secondary alcohols to the corresponding aldehydes and ketones in the presence of stoichiometric amounts of base (2 equiv) at moderate temperatures (70–90 °C) and short reactions times (1–2 h). It was proposed that a mononuclear CuI–OH/phen/DBAD complex (species A in Figure 7) performed the initial deprotonation and coordination of the alcohol substrate (species B), which could be oxidized via hydride transfer by the coordinated DBAD moieity. The resulting CuI/phen/DBADH−/product adduct (species C) could react with dioxygen to generate a dicopper(II)-peroxide intermediate (speceis D), which could promote the intramolecular oxidation of the two DBADH− ligands to regenerate the active CuI catalyst.
Figure 7.

Cu-catalyzed oxidation of alcohols.95
In a follow-up article, the same research group slightly varied the reaction conditions (i.e., use of C6H5F instead of toluene and replacing the 2 equiv of K2CO3 with 5% of tBuOK) to oxidize a wider range of primary and secondary alcohols.96
Inspired by pioneering research by Semmelhack, Markó, and others,95,97–100 Stahl reported one of the breakthrough findings on modern copper catalysis in which a practical, mild, and efficient method for the oxidation of a widespread range of alcohols was described (Figure 8).101 Primary alcohols were oxidized using air as the O2 source by combining catalytic amounts of a CuI source, bpy (4,4′-bipyridine), TEMPO, and NMI (1-methylimidazole). The reaction mechanism was extensively studied, including kinetic analysis, spectroscopic characterization of reaction intermediates, and computational analysis.102,103 In the mechanistic proposal, a CuI/bpy/NMI complex (species A in Figure 8A) reacts with O2 to generate sequentially a mononuclear CuII-superoxide complex (speciesB) and a dicopper(II)-peroxide intermediate (species C), which abstracts a H atom from TEMPO-H to form a mononuclear CuII-hydroperoxide complex (species D in Figure 8A, iv). This putative EHPII,II intermediate could be protonated by water to release H2O2 (that could be further reduced to water via disproportionation by the Cu/bpy system) to produce a CuII–OH complex (species E in Figure 8A, v) that then deprotonates and binds the alcohol substrate (species F). Ligand exchange (TEMPO by NMI) could generate a CuII-alkoxy-TEMPO species (species G in Figure 8A), which could then promote the intramolecular oxidation of the alkoxy substrate via H atom abstraction and e− transfer to form a CuI-TEMPO-H adduct (species H) and the corresponding aldehyde product.
Figure 8.

Cu-catalyzed oxidation of (A and B) primary and (B) secondary alcohols.101–105
In a related system, Stahl reported that replacing TEMPO for the less sterically hindered N-oxide ABNO and the use of a slightly more electron-donating ligand, MeObpy, permitted the oxidation of secondary alcohols (Figure 8B).104 Interestingly, this method allows for oxidizing α-chiral alcohols without erosion of enantiomeric excess (Figure 8B, iii). The reaction was proposed to occur via a mechanism analogous to the one proposed for the Cu/TEMPO/bpy system.
Lancaster and Stahl have recently studied the electronic structure of the putative reactive intermediates in the CuII-TEMPO/ABNO alcohol oxidations described above (species G in Figure 8), which was found to be better described as CuI-TEMPO+ species.105
Lumb and Arndtsen reported a method for the catalytic oxidation of alcohols with copper(I), amine, and pyridine ligands using dioxygen as oxidant (Figure 9).106 The catalytic system oxidized a wide array of primary and secondary alcohols with good yields in short reaction times at room temperature. A mechanistic study suggested that the secondary amine DBED was oxidized to the corresponding N-oxide product to generate reaction intermediates similar to the ones formed in the Cu/TEMPO/bpy system, including the key CuII/N-oxide/alkoxy adduct (species D in Figure 9).107 These findings might trigger the development of oxidation protocols based on the in situ generation of these oxidants (i.e., N-oxides). Given the ubiquity of amines in catalysis, the implication of N-oxides in oxidative functionalizations might not be unusual.108
Figure 9.

2.2.2. Other Cu-Catalyzed Oxidase-Like Reactions.
The copper-nitroxyl catalytic systems developed for the oxidation of alcohols (see sections above) have also been used for other oxidase-like transformations (Figure 10). For example, Oisaki and Kanai described the oxidation of amines to imines catalyzed by CuI in a combination of ketoABNO, 2tBubpy, DMAP, and O2 (Figure 10A).109 This catalytic system was used for the oxidation of secondary amines to imines and for the oxidative homocoupling of primary amines to imines via in situ condensation of the starting amines and the generated imines (see Figure 10A, ii). It was proposed that the oxidation occurred via formation of CuII-nitroxyl intermediates similar to the ones proposed in the oxidation of alcohols in the Cu/TEMPO system (see Figure 8 above).
Figure 10.

Catalytic copper-nitroxyl systems beyond oxidation of alcohols: oxidation of amines to (A) imines109 and to (B) nitriles;110 (C) oxidative coupling of alcohols and amines111 and (D) lactonization of diols.112
Stahl studied the catalytic performance of the CuI/ABNO/4tBubpy/DMAP in the oxidation of primary amines to nitriles (Figure 10B).110 It was found that this catalytic system was able to avoid the formation of the homocoupling imine products generated in the CuI/ketoABNO/2tBubpy/DMAP described above. Preliminary mechanistic studies suggested that an active CuII/nitroxyl intermediate species promoted two consecutive 2H+/2e− oxidations of amines to imines and imines to nitriles.
The same research group also reported the use of the CuI/ABNO/4MeObpy/NMI catalytic system for the synthesis of amides via oxidative coupling of alcohols and amines (Figure 10C).111 The catalytic system was selective toward the formation of amides, avoiding the generation of products derived from the oxidation of alcohol (aldehydes) and amines (nitriles or imines). This methodology was applied for a wide range of substrates with excellent yields (Figure 10C, iii). It was suggested that the oxidation of primary alcohols formed the corresponding aldehydes, which reacted with amines to generate hemiaminal intermediates that could be further oxidized by the CuII/nitroxyl system to yield the final amide products.
Stahl also reported that the catalytic CuI/nitroxyl/bpy/NMI could be used for the lactonization of diols (Figure 10D).112 The system was very efficient in the lactonization of a wide array of diols using air as a dioxygen source in short reaction times and at room temperature. The reaction occurred via oxidation of one of the alcohol moieties followed by fast cyclization to produce hemiacetal intermediates (avoiding formation of dialdehyde products), which could be oxidized by a CuII/nitroxyl species to the final lactone products.
Lumb and Arndtsen also developed a synthetic protocol based on copper and O2 to oxidize amines to the corresponding homocoupling imine products and nitriles (Figure 11).113 Surprisingly, it was found that copper(I) sources, O2, and molecular sieves promoted these oxidations without using N-oxides (e.g., TEMPO, ABNO) or additional ligands. They also reported that changing the Cu source had a deep impact on the selectivity of the reaction; [CuI(CH3CN)4](PF6) catalyzed the oxidation of amines to nitriles and copper(I) iodide catalyzed the oxidation of amines to the coupling imine products (Figure 11, ii). It was proposed that the amines had a dual role in the catalytic system, serving as substrates and as ligands for the copper ion (Figure 11, iv). It was suggested that a CuI-amine complex (species A in Figure 11) reacted with O2 to form a putative Cu2O2 intermediate (SPII,II or O,OIII,III, species B) that could oxidize one of the amine ligands in an intramolecular fashion to produce a CuI-imino-amine complex (species C). This intermediate could react with another equivalent of amine substrate to generate the homocoupling imine product (species D in Figure 11) when [CuI(CH3CN)4](PF6) is used as Cu source. In the case of copper(I) iodide, the CuI-imino-amine complex could react with O2 to form another Cu2O2 adduct (SPII,II or O,OIII,III, species E) that could oxidize the imine ligand to the nitrile product.
Figure 11.

Copper-catalyzed oxidation of amines to imines and nitriles.113
2.2.3. 1H+/1e− Oxidation of Phenols: Stoichiometric and Catalytic Reactions.
The 1H+/1e− oxidation of phenols catalyzed by copper/O2 species is a common reaction in natural metabolic processes. For example, plant laccases couple the 4e−/4H+ reduction of O2 to water with the oxidation of phenols to form phenoxyl radicals, which polymerize to form phenolic oligomers.114 Another example of Cu-mediated oxidation of phenols is the cofactor biogenesis in copper amine oxidases in which the protein-derived organic cofactors 2,4,5-trihydroxyphenylalanine-quinone (TPQ) and lysine tyrosyl-quinone (LTQ) are synthesized via 1H+/1e− oxidation of a tyrosine residue and subsequent coupling with O2 and lysine, respectively (Figure 12).115
Figure 12.

Biosynthesis of 2,4,5-trihydroxyphenylalanine-quinone (TPQ) and lysine tyrosyl-quinone (LTQ).115
The mechanism by which phenols are oxidized by Cu/O2 systems has been studied by several research groups (Figure 13). These reports usually included generation of metastable Cu/O2 species at low temperature and kinetic analysis of their reactivity toward phenol substrates with varying stereoelectronic properties (note: the reaction of phenolates with Cu2O2 cores usually leads to the ortho-hydroxylation of the substrate, see section 2.3.2). Comparison of the reaction rates with the thermodynamic properties of the O–H bond that will undergo oxidation, including pKa, redox potential (E0ox.), and bond dissociation free energy (BDFE), is commonly used to determine the reaction pathway by which Cu/O2 species carry out the oxidation of phenols (Marcus plot in Figure 13A).116 These pathways may consist of (i) proton-transfer followed by electron transfer (PT-ET); (ii) initial electron transfer followed by proton transfer (ET-PT); or (iii) coupled-proton electron transfer or hydrogen atom transfer (CPET or HAT).
Figure 13.

1H+/1e− oxidation of phenols promoted by Cu/O2 species: (A) mechanistic tools to identify possible reaction pathways and research reports in which this methodology has been used to understand the reactivity of Cu/O2 species [(B) mononuclear and (C) dinuclear] toward phenols.
Karlin provided a detailed analysis of the reactivity of a monononuclear CuII-superoxide complex (ESII) bearing a tetradentate electron-donating TMPA derivative ligand toward 2,6-di-tert-butyl-4-substituted phenols (Figure 13B).117 It was found that the rate-determining step of the reaction was the oxidation of the phenol by the LCuII(O2•−), which generated a phenoxyl radical that was trapped by remaining ESII species to form a peroxy CuII–O2–X–PhO intermediate (species C in Figure 13B). With dependence on the identity of the 4-X-substitutent of the phenol, the peroxy intermediate could evolve to form H2O2 (4-MeO) or to eliminate isobutylene (4-tBu) with concomitant formation of the benzoquinone product. Kinetic analysis of the reaction using 4-substituted phenols with different E0ox. led to a Marcus plot with a slope value between −0.5 and 0 (slope = −0.29). Comparison of the reaction rates obtained in the oxidation of phenols with the rates found for the deuterated analogues led to relatively high kinetic isotope effects (KIE = 4.2–11). The mechanistic evidence suggested that during the r.d.s. an H atom was transferred from the PhOH to the Cu-superoxide species via HAT.
Dinuclear Cu2O2 species can also promote the stoichiometric oxidation of phenols to the corresponding coupling products (Figure 13C). Itoh and Fukuzumi reported that side-on dicopper(II)-peroxo (SPII,II) and dicopper(III) bis-μ-oxo (O,OIII,III) complexes reacted with phenols via endergonic electron transfer (from the phenol to the Cu2O2 core) coupled with the proton transfer (slope between −0.5 and −1.0, KIE = 1.2–1.6).118 Interestingly, the O,OIII,III species was found to oxidize phenols with reaction rates 20–50 times faster than the SPII,II complex.
Costas and Ribas also studied the reactivity of an end-on dicopper(II)-peroxo complex (EPII,II) toward phenols.119 It was found that the Cu2O2 core reacted with phenols in a two-step fashion via fast substrate association followed by slow H atom abstraction in the r.d.s. The slope obtained in the Marcus plot (−0.5) and the KIE (1.5–2.0) were in accordance with a CPET mechanism during the r.d.s. It was emphasized that the unique structure of the EPII,II complex, in which one of the copper(II) centers was coordinatively unsaturated (three nitrogens and one oxygen), allowed for binding and substrate oxidation in a Cu2O2 core that is typically considered nucleophilic.120
Karlin, Solomon, and Garcia-Bosch have also recently reported the reactivity toward phenols of a family of O,OIII,III complexes bound by Lewis acids (LA: Sc3+, H+, B(C6F5)3) in the secondary coordination sphere (Figure 13C).121 The authors proposed that the reaction occurred via slow electron-transfer (r.d.s.) followed by fast proton transfer (ET-PT), which was supported by a Marcus plot slope of −1.0 and KIE of 1.0. The authors also showed that the reaction rates were dependent on the Lewis acid identity, with the L2CuIII2(O2−)2-2H+ and L2CuIII2(O2−)2-Sc3+ cores more reactive than the corresponding L2CuIII2(O2−)2-2B(C6F5)3 analogue (see Figure 13 bottom left and Figure 30).
Figure 30.

Generation of O,OIII,III cores bound by Lewis-acids and their reactivity toward weak C–H bonds.121
The oxidative coupling of phenols was popularized in the 1960s when Hay reported the polymerization of 2,6-dimethylphenol using Cu and O2.32 Despite this important finding, the controlled coupling of phenols for synthetic purposes has been challenging, due to the inherent reactivity of the substrate-based radical intermediates which can generate multiple C–C and C–O coupling products.
Lumb reported that slight changes in the reaction conditions (ligand and copper source used, presence of water or anhydrous conditions) allows for obtaining oxygenation and oxidation products, selectively (Figure 14).122 The authors showed that combining [CuI(CH3CN)4](PF6), triethylamine, molecular sieves (4 Å), and phenol substrates in the presence of dioxygen led to the formation of the oxygenation ortho-quinone product. However, if molecular sieves were not added and the CuICl was used as a Cu source, the phenol substrate was converted to the 1H+/1e− C–C coupling product. It was also found that if NEt3 was replaced with DBED, the C–C coupling product could be further oxidized to the benzoxepine product. This methodology was applied to a wide array of phenol substrates with excellent yields and selectivity (some of these results are included in Figure 14, iii). It was suggested that phenols (species A in Figure 14) in the presence of [CuI(CH3CN)4](PF6), NEt3, and O2 are deprotonated to form phenolates that could be oxidized by Cu2O2 species (e.g., side-on dicopper(II)-peroxo or dicopper-(III) bis-μ-oxo) via a tyrosinase-like mechanism (see sections below). When phenols are exposed to CuI/base/O2 and no desiccant, the reaction could occur via 1H+/1e− oxidation promoted by a putative Cu2O2 species to generate a phenoxyl radical (species D in Figure 14). After dimerization, the C–C coupling product could be further oxidized to benzoxepine by a putative Cu2O2 species in the presence of DBED. Ortho-silyl phenol was used to provide further evidence for the proposed mechanism since the 1H+/1e− oxidation of this substrate leads to the formation of the byproduct derived from intramolecular C–Si fragmentation and Si–O bond formation (Brook rearrangement), which was observed under the Cu/O2 oxidation conditions.
Figure 14.
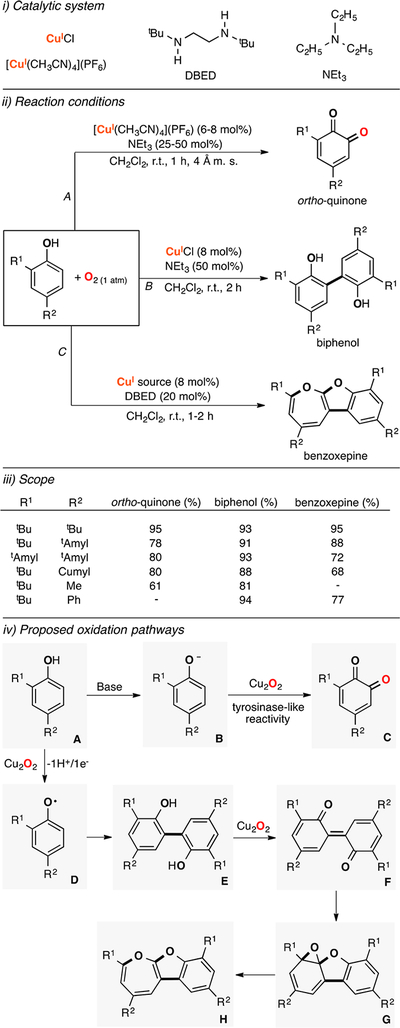
Cu-catalyzed aerobic oxidation of phenols.122
In 2001, Kozlowski reported a Cu-catalyzed method for the stereoselective C–C coupling of napthols (Figure 15).123 A copper(II)-hydroxide-iodide complex bearing a bidentate ligand (1,5-diaza-cis-decalin) was utilized to catalyze the oxidative coupling of a series of naphthols to the corresponding BINOL derivatives with high yields and excellent ee’s using O2 as oxidant (Figure 15, ii). In collaboration with Stahl, a mechanistic study was reported that included a kinetic analysis of the oxidant consumption under different conditions (varying the concentration of substrate, O2, and catalyst).124 It was proposed that the oxidation of the naphthol substrate could occur via two different routes (Figure 15, iv). The first oxidation pathway involved initial deprotonation and coordination of the substrate to LCuII(OH) (species A in Figure 15) to form a LCuII(NapH) species (species B) that upon intramolecular electron transfer could generate an LCuI(NapH•) intermediate (species C). This ligand-centered radical species could react with another equivalent of LCuI(NapH•) to produce the cuprous complex (species D in Figure 15) and the C–C coupling product (Nap-Nap). Oxidation of LCuI with molecular oxygen could regenerate the active LCuII(OH) catalyst (species A) or coordinate the substrate (species E), which could be oxygenated to form the cofactor NapOX (two structures were proposed). The LCuI(NapOX) complex (species F in Figure 15) could oxidize the naphthol substrate via coordination (species G) and subsequent intramolecular electron transfer and dimerization.
Figure 15.

Lumb reported a Cu-based catalytic system for the oxidative C–O coupling of phenols and quinones (Figure 16).125 The authors found that catalytic amounts of CuI sources and a ligand (DBED or 4-MeO-pyridine) promoted the C–O coupling of phenols and quinones using O2 as oxidant (Figure 16, ii). A wide array of ether cross-coupling products were synthesized with excellent yields and selectivity (Figure 16, iii). It was suggested that the oxidation occurred via coordination of the quinone substrate to the CuI ion to generate a CuII-semiquinone complex (species A in Figure 16, iv) that could react with O2 to produce a CuII-quinone-superoxide intermediate (species B in Figure 16). This complex could couple the phenol substrate to generate a putative copper(II)-semiquinone-phenol hydroperoxide intermediate (species C in Figure 16) that could release H2O2 to produce a CuII-semiquinonephenol species (species D in Figure 16). Substitution of the quinone-phenol product by the starting quinone substrate could regenerate the initial CuII-semiquinone species.
Figure 16.

Cu-catalyzed oxidative C–O coupling of phenols and quinones.125
Very recently, the Baran research lab has used stroichiometric amounts of CuI and TMEDA (tetramethylethylenediamine) to synthesize arylomycin derivatives via intramolecular oxidative macrocyclization of phenols (Figure 17).126 On the basis of literature precedent, it was suggested that a putative O,OIII,III core mediated the 1H+/1e− oxidation of the phenol moieties.
Figure 17.

Cu-promoted synthesis of arylomycin cores.126
2.3. Monooxygenase-Like Reactivity
The oxygenation of organic substrates is commonly found in many natural metabolic pathways, including the degradation of pharmaceuticals,127 biosynthesis of hormones,128 and cellular energy production (e.g., oxidation of methane to methanol in methanotropic bacteria129). Many of these reactions are catalyzed by metalloenzymes, in which O2 serves as an oxygen donor and as a proton and electron sink (to generate H2O).130,131 Despite the ubiquity of these processes in nature, many of the oxygenations in organic synthesis rely on the use of stoichiometric oxidants such as NaOCl, KMnO4, MnO2, etc.132,133 Inspired by the reactivity of Fe-dependent metalloenzymes, several research groups have developed iron catalysts able to catalyze C–H hydroxylations, epoxidations, and cis-dihydroxylations using H2O2 as oxidant.134–136 On the other hand, the use of Cu to carry out oxygenations is less explored.137 In this section, we describe the reactivity of Cu systems that perform bioinspired C–H hydroxylations (sp3 and sp2 C–H bonds) and other oxygenations.
2.3.1. sp3 C–H Hydroxylations.
Peptidylglycine α-hydroxylating monooxygenase (PHM) catalyzes the hydroxylation of a C-terminal glycine residue of a prohormone (Figure 18). This copper-containing monooxygenase enzyme is part of a group of natural systems that have similar structure and function that also includes dopamine β-monooxygenase (DβM) and tyramine β-monooxygenase (TβM).27 These enzymes contain a noncoupled dinuclear active center where the two copper ions are separated by ~11 Å. The CuH site is coordinated by three histidine residues, and it is proposed to provide electrons during catalysis. The CuM is coordinated by one methionine and two histidine residues, and it is believed to be the site responsible for O2 binding, reduction, and substrate oxidation. The mechanism by which these enzymes carry out the hydroxylation of C–H bonds has been studied in detail, but there is still disagreement on some reaction key steps (Figure 18, iii). The main divergence stands on which of the putative CuO2 species abstracts an H atom from the substrate and in which step CuH transfers the electron. All the mechanistic proposals agree on the first reaction step, in which substrate coordination triggers dioxygen binding to form a mononuclear CuII-(O2•−) intermediate (species B Figure 18). Klinman and Solomon proposed that this ESII species, which could be crystallographically characterized,138 oxidizes the C–H bond via hydrogen atom transfer.139–141
Figure 18.

Peptidylglycine α-hydroxylating monooxygenase (PHM): (i) active center, (ii) reaction catalyzed, and (iii) proposed catalyztic cycle. Note: For C2, it was proposed that a radical cation, located over the three ligands bound to the Cu, was formed.
Other proposals suggest that before substrate oxidation, e− transfer from the CuH center with concomitant protonation produces a mononuclear LCuII(OOH) complex (species C in Figure 18) which can undergo O–O cleavage to generate high-valent Cu-oxyl intermediates (species C2 and D in Figure 18) that could be the active oxidants.66
Particulate methane monoooxygenase (pMMO): Little is known about how these Cu-dependent metalloenzymes oxidize methane to methanol. In the active center, two histidine and an unusual histidine-amine residue acting as bidentate ligand (histidine brace) are found (Figure 19A).64 The number of copper ions involved in the active center is under debate: initial structural reports by Rosenzweig indicated that a dicopper center might be responsible for substrate oxidation, but recent investigations suggest that the active center might bind only one copper ion.142,143 On the basis of reactivity studies, a dicopper(II) side-on peroxide (SPII,II) species has been proposed as a possible competent reactive intermediate.144 Other species such as CuIII2(O2−)2 and CuIII(O2−)2 CuII havess also been invoked (note: several authors currently suggest that a mononuclear Cu species is involved in the oxidation of methane in pMMO).64,145,146
Figure 19.

(A) Particulate methane monoooxygenase (pMMO) and (B) lytic polysaccharide monooxygenase (LPMO): active centers, reactions catalyzed, (C) histidine brace binding motif, and (D) putative Cu/O2 species responsible of C–H oxidation.
Lytic polysaccharide monooxygenase (LPMO): Recently discovered, these copper metalloenzymes promote the degradation of polysaccharides (e.g., cellulose, chitin, and starch) by oxidative cleavage of the glycosidic bond (Figure 19B).147–149 In the mononuclear active site, the Cu ion is bound by a histidine amino acid and a histidine-amine residue (similar to the histidine-brace in pMMO).150,151 Mechanistic studies have suggested the involvement of mononuclear species derived from the reaction of CuI with O2 [e.g., CuII(O2•−), CuII(OOH), CuII(O•), or CuIII(OH)].152 Recent studies by Eijsink and Walton proposed that the active oxidant in LPMO could be H2O2, which, in combination with CuI, could generate hydroxyl radicals that could be responsible for C–H bond oxidation or could generate an active O·II species via H atom abstraction of an OHII species.153,154
2.3.1.1. Stoichiometric sp3 C–H Hydroxylations with Model Systems.
Itoh reported the first example of metastable CuII-superoxide bearing a tridentate ligand (species B in Figure 20).62,155 The complex reacted in an intramolecular fashion to oxidize the benzylic position of the ligand. Mechanistic studies suggested that the ESII species abstracted an H atom from the ligand to generate a fleeting EHPII complex (species C in Figure 20), which upon rebound with the C-centered radical could produce a CuII-hydroxyl radical intermediate (species E in Figure 20). This species could be trapped by unreacted CuI complex to form the final Cu-alkoxide product (species F in Figure 20). The yields obtained (35%) were consistent with the proposed mechanism (50% threshold).
Figure 20.

Intramolecular C–H hydroxylation promoted by an ESII species.62,155
Karlin reported the first mononuclear CuII-superoxide species capable of performing intermolecular C–H oxidations (Figure 21).60 The authors made use of a TMPA derivative to stabilize the ESII adduct via H-bonding interactions.156 It was suggested that the LCuII(O2•−) complex reacted with substrates containing weak C–H bonds (e.g., BNAH, 1-benzyl-1,4-dihydronicotinamide) via HAT. The substrates used (hydride donors) were proposed to produce a mixture of the unstable mononuclear LCuIIOOH and LCuII(O2 2−) species, which could evolve to form the final EPII,II product (quantified by UV–vis). A similar reactivity was reported recently by Castillo, in which a tetradentate N3S ligand system was able to stabilize a side-on LCuII-superoxide species that reacted with 9,10-dihydroanthracene to form 0.5 equiv of the corresponding dehydrogenation product (anthracene).157
Figure 21.

Intermolecular C–H hydroxylation promoted by an ESII species.60
Karlin has also studied the reactivity of a mononuclear copper(II) hydroperoxide species (Figure 22).158 An EHPII intermediate was generated by addition of H2O2 and a base (triethylamine) to the corresponding LCuII(ClO4)2 complex. The LCuII(OOH) complex decayed at low temperatures to oxidize the ligand scaffold (i.e., intramolecular N-dealkylation). Mechanistic studies suggested that the CuII-hydroperoxide underwent homolytic Cu–O cleavage to produce a hydroperoxyl radical and CuI (species B, Figure 22). The reaction was carried out under excess amounts of H2O2 that would permit the oxidation of the resulting CuI complex to the LCuII-OH and hydroxyl radical (species C), which could abstract one of the benzylic H atoms of the ligand scaffold (species D). Rebound of the radical with the CuII-OH center could form the C–O bond (species E in Figure 22), which, upon N-dealkylation, forms benzaldehyde, the cleaved ligand scaffold, and CuI (species F). This mechanistic proposal was supported by the KIE observed (KIE = 1.0). It was also found that the copper complexes bearing analogues of the ligand (4-substitution of the internal benzylic substrate) decayed with similar rates to the ones found for the nonsubstituted systems.
Figure 22.

Intramolecular N-dealkylaiton oxidation promoted by an EHPII species.158
In collaboration with Baran, Garcia-Bosch has also studied the reactivity of Cu systems toward intramolecular hydroxylations.159 This contribution was inspired by the initial findings of Schönecker and co-workers (based on earlier work by Réglier 160), in which they reported the hydroxylation of sp3 γ–C-H bonds of cyclic ketones using imino-pyridine systems as directing groups, Cu and O2 (Figure 23A).161 On the basis of the yields obtained (below 50%), the involvement of dinuclear L2Cu2O2 species (e.g., SPII,II, O,OIII,III) as active oxidants was suggested (Figure 23A, right). Baran was able to overcome the 50% threshold by the addition of excess amounts of reductant, which was proposed to reduce the nonhydroxylated LCuII system to LCuI in order to regenerate the putative L2Cu2O2 cores (Figure 23A).162 The copper complexes bearing the substrate–ligand scaffolds were isolated and characterized (e.g., S1-CuI and S3-Me-CuI in Figure 23B). Interestingly, the Cu ion and the C atom that undergoes C–H oxidation were found by X-ray diffraction analysis to be in close proximity (Cu–Coxid.: 3.1–3.3 Å). With the use of the isolated complexes, the oxidation with O2 was carried out, and it was observed that the hydroxylation yields overcame the 50% theoretical maximum imposed by the L2Cu2O2 intermediacy. The new mechanistic scenario (Figure 23C) proposed the formation of a mononuclear LCuII(OOH) species prior to C–H bond oxidation. The extensive experimental evidence suggested that the copper(I) complexes reacted with O2 to generate a putative LCuII(O2•−) species (species B in Figure 23C), which could release free superoxide and the LCuII complex (species C). The free superoxide could oxidize the solvent (products derived from acetone oxidation were quantified) to generate H2O2, which reacts with the LCuII complex to form an LCuII(OOH) intermediate (species D in Figure 23). This LCuII(OOH) species, along with related LCuII(OOR) analogues, could be independently generated by the addition of ROOH oxidants (R:H, tBu, Cum) to LCuI or LCuII in the presence of a base. During the reaction, the putative LCuII(OOH) could undergo homolytic O–O bond cleavage to generate hydroxyl radical that abstracts an H atom from the ligand scaffold in an intramolecular fashion (species E in Figure 23). The ligand C-centered radical generated could rebound with the O·II core to form the C–O bond (radical trap experiments confirmed the formation of ligand C-centered radicals). On the basis of the proposed mechanism, the oxidation protocol was redesigned to use stoichiometric amounts of inexpensive CuII sources (e.g., CuII(NO3)2) and H2O2 as oxidant (Figure 23A), which improved the previous methods in terms of practicability (no need for O2 gas, and reactions could be perfomed at room temperature for shorter periods of time), cost (no addition of external reductant, cheaper copper source), and yields.
Figure 23.

Intramolecular C–H hydroxylations (promoted by putative EHPII species) for synthetic purposes.159,161,162
Karlin reported the reactivity of an isolable CuII-superoxide species using TMG3Tren as ligand (previously crystallized by Schindler’s lab).67 It was found that an ESII complex (species A in Figure 24) reacted at low temperatures with external substrates containing weak O–H bonds (e.g., TEMPO-H) to generate a fleeting LCuII(OOH) intermediate (species B in Figure 24). With the use of UV–vis and EPR, the authors proposed that the EHPII species underwent homolytic O–O bond cleavage to form an O·II intermediate (species C in Figure 24) that could oxidize the ligand scaffold in an intramolecular fashion (species D in Figure 24). The EHPII species (LCuII + H2O2 + base) and the fleeting O·II species (LCuI + PhIO) were independently generated, and in both cases ligand oxidation was observed.
Figure 24.

Cu-promoted intermolecular oxidations and intramolecular hydroxylation involving ESII, EHPII, and O·II intermediates.67
Tolman has reported a series of research articles on the reactivity of a new type of mononuclear copper–oxygen core (Figure 25). In the first article, the synthesis of an LCuIII(OH) intermediate via 1e− oxidation of a LCuII(OH) complex bearing a tridentate dianionic ligand was described.65 This OHIII species was characterized using UV–vis, EPR, EXAFS, and DFT calculations. It was found that these high-valent cores oxidize various C–H bonds (BDFE: 75 to 99 kcal/mol) via H atom transfer (slower reaction rates were measured in the reaction with substrates containing strong C–H bonds). The use of ligand scaffolds with different stereoelectronic properties allowed for tuning the redox potential and the basicity of the OHIII cores.163 It was reported that the reaction rates were dependent on the reduction potential of LCuIII(OH), with the species with higher E0 having the faster rate. The authors described that the OHIII core carried out the intermolecular oxidation of C–H bonds via HAT (KIE > 20) to generate the corresponding C-centered radical and the LCuII(OH2) complex(i.e., the LCuIII(OH) core acts as a 1H+/1e− oxidant).
Figure 25.

Intermolecular 1H+/1e− oxidation of C−H bonds by high-valent OHIII complexes.65,163
In 2017, Meyer reported the synthesis, characterization, and reactivity of a CuII(O2•−)CuII core (species A in Figure 26).72 This species was generated via 1e− oxidation of a CuII(O22−)CuII core (species B in Figure 26). Interestingly, the report also described the synthesis and characterization of the corresponding CuII(OOH)CuII intermediate (species C in Figure 26), which is the reaction product of the reductive protonation (1e−/1H+) of the ESII,II complex. The Bordwell equation was used (E0 and pKa measured in CH3CN) to calculate the thermodynamic driving force of the ESII,II core to perform 1H+/1e− oxidations. The BDFE value obtained (72 kcal/mol) fitted the reactivity observed, in which the CuII(O2•−)CuII species oxidized TEMPO-H (BDFE: 67 kcal/mol) but not DHA (BDFE: 78 kcal/mol).
Figure 26.

Thermodynamic analysis of the 1H+/1e− reactivity of an ESII,II core.72
Suzuki studied the reactivity of a SPII,II species toward external substrates (Figure 27).164 The Cu2O2 core was reactive toward substrates containing weak and relatively strong C–H bonds (75–92 kcal/mol). It was proposed that the dicopper(II) peroxide (species A in Figure 27) abstracted an H atom from C–H bonds via HAT in the rate-determining step (KIE = 9–34). The resulting putative CuII,CuIII-oxo-hydroxo complex (species B in Figure 27) could react with the C-centered radical to form the hydroxylation product (species C in Figure 27). The formation of C–C coupling products was also observed (species D in Figure 27) that could be derived from a second HAT carried out by the mixed-valence oxo-hydroxo core. The C–C coupling products derived from the coupling between the susbtrate and the ligand scaffold were also characterized and quantified. It was proposed that the LCuIICuIII(O2−)(OH) core (species B) could abstract an H atom from the ligand scaffold (species E), which could be coupled with the substrate C-centered radical produced in the first reaction step (species F). It is worth mentioning that the intermolecular oxidations performed by this SPII,II species led to the oxygenation of the substrate (i.e., formation of C–O bonds). This differs from the oxidations performed by Tolman’s mononuclear OHIII complex and by Meyer’s CuII(O2•−)CuII core (see above), in which the substrate was not oxygenated.
Figure 27.

Inter- and intramolecular C–H oxidations performed by an SPII,II species.164
Itoh pioneered the study of the reactivity of O,OIII,III cores.165 A remarkable research article described that small changes in the structure of the ligand led to intramolecular or intermolecular reactivity (Figure 28).166 For example, the use of a bidentate ligand in which the benzylic positions are protected (L:HPy1Et‑Bz‑d2) led to intermolecular C–H oxidation of external substrates with weak C–H bonds (e.g., 1,4-cyclohexadiene). Interestingly, second-order kinetics were observed, which suggested disproportionation of the O,OIII,III complex (species A in Figure 28) to form a dicopper(III)-oxo-oxyl complex (species B) and a mixed-valent CuII(O2−)2CuIII core (species C) that could be responsible for C–H oxidation. When the ligand was slightly modified (L: HPy1Et,Phe), the authors observed generation of a O,OIII,III species that decayed to form the ligand hydroxylation product (species E in Figure 28).167
Figure 28.

Inter- and intramolecular C–H oxidations performed by O,OIII,III cores.165–167
Stack has used a series of diamine ligands that combined with CuI and O2 to generate O,OIII,III cores at very low temperatures (Figure 29).22,168 The authors studied the reactivity of the different L2CuIII(O2−)2 intermediates toward C–H bonds. It was found that the rates of H atom abstraction (KIE: 14–31) and stability of the formation of the Cu2O2 species were dependent on the ligand sterics, with the most accessible structures being more reactive than the sterically hindered systems (e.g., RNH2 vs RNMe2). It was emphasized that the ligands used resemble the ones found in the active site of pMMO (histidine-brace), which could suggest that an O,OIII,III core might be formed during enzymatic catalytic turnover.
Figure 29.

Intermolecular oxidation of weak C–H bonds by O,OIII,III cores bearing biologically relevant ligands.168
Lewis acids have been recently used to tune the reactivity of metal-O2 and metal-oxo(hydroxo) species.169–172 Karlin, Solomon, and Garcia-Bosch have reported the reactivity of a series of O,OIII,III cores bound by Lewis acids (Figure 30).121 It was described that an L2CuII2(O22−) species isomerized to the L2CuIII2(O2−)2 core upon addition of stoichiometric amounts of Lewis acids (2 equiv of DMF·CF3SO3H, 1 equiv of Sc-(CF3SO3)3 or 2 equiv of B(C6F5)3). Detailed spectroscopic (UV–vis, rRaman) and computational analysis (TD-DFT calculations) suggested that isomerization occurred via coordi-nation of the Lewis acid to the ligand scaffold (species B in Figure 30). It was found that the starting SPII,II species was unreactive toward substrates with weak C–H bonds but that the O,OIII,III-LA adducts were able to perform C–H oxidations (note: the substrates underwent a 1H+/2e− oxidation without formation of the C–O bond). Interestingly, the H atom abstraction rates were dependent on the Lewis acid identity. The KIE observed, along with the faster reaction rates observed for substrates with weaker C–H bonds, indicated HAT during the rate-determining step.
In 2017, Kieber-Emmons reported the reactivity of a LCuII-(μ-O2−)-CuIIL core toward H-abstraction of substrates with weak C–H and O–H bonds (i.e., 1,2-cyclohexadiene, TEMPO-H).74 An OII,II species was generated via deprotonation of the isolated OHII,II complex, which allowed for calculation of the pKa of this conversion. The reduction potential of OHII,II complex (species B in Figure 31) was also obtained, which, in combination with the pKa, enabled the calculation of the thermodynamic driving force of the OII,II core (species A in Figure 31) to perform 1H+/1e− oxidations (BDFEO–H = 77 kcal/mol, note: pKa and reduction potentials were obtained in CH3CN). It was observed that the dicopper(II) μ-oxo intermediate was able to abstract an H atom from 1,4-cyclohexadiene (BDFEC–H = 73 kcal/mol) to generate benzene (75% yield) and to stoichiometrically oxidize TEMPO-H (BDFEO–H = 67 kcal/mol) to TEMPO radical.
Figure 31.

Thermodynamic analysis of the 1H+/1e− reactivity of an OII,II core.74
2.3.1.2. Catalytic Hydroxylation of sp3 C–H Bonds.
Sir D.H. R. Barton reported one of the first examples of Cu-catalyzed hydroxylation of alkanes that combined Cu powder, pyridine, acetic acid, and H2O2.173 Alkane oxidation occurred via formation of O-centered and C-centered radical species, although low yields were obtained (<5%).174 Using a similar system, Sawyer found that the CuI(bpy)2/pyridine/H2O2 system (bpy: 2,2′-bipyridine) reached moderate efficiency in terms of oxidant used (up to 30%), but the yields obtained were too low for synthetic purposes (<5%).175 Kitagawa also reported the catalytic hydroxylation of cyclohexane with H2O2 using Cu complexes bearing N3S tetradentate ligands.176 The authors observed modest yields, which suggested catalyst decomposition during catalytic turnover via intramolecular sulfoxidation of ligand scaffold.
Pombeiro reported one of the first examples of Cu-catalyzed oxidation of C–H bonds that reached substantial yields (Figure 32).177,178 A tetranuclear CuII-oxo cluster (OII,II,II,II) catalyzed oxidation of cyclohexane to cyclohexanol and cyclohexanone with yields up to 38%. Surprisingly, methane was oxidized to methanol with modest yields (TON: 44, 2.2% yield). It was suggested that the active oxidants were O-centered radicals (i.e., hydroxyl and hydroperoxyl) that formed a cyclohexylhydroperoxide product, which decayed to cyclohexanol and cyclohexanone under the reaction conditions, via 1e− oxidation pathways.
Figure 32.

Cu-catalyzed hydroxylation of cyclohexane using H2O2 as oxidant.177,178
Garcia-Bosch has also explored the use of mononuclear Cu complexes to catalyze the oxidation of strong C–H bonds with H2O2 (Figure 33).179 An oxidation method was developed by which a wide array of ligand scaffolds could be tested, which consisted of mixing catalytic equimolar amounts of the ligand and the Cu source (1 mol %), cyclohexane (1 equiv), and hydrogen peroxide (10 equiv) in CH3CN. The reaction was stirred for 6 h at room temperature, and the organic products derived from the oxidation of the substrate were analyzed by gas chromatography (Figure 33, top left). The GC analysis was carried out by (i) direct injection of the crude reaction in the presence of an internal standard and (ii) injection of the crude reaction previously treated with PPh3 to convert the cyclohexyl hydroperoxide product to cyclohexanol. This analysis protocol, developed by Shul’pin,180 permitted for quantification of the alkyl hydroperoxide along with the alcohol and ketone products. It was observed that copper complexes bearing tetradentate ligands such TMPA (tris(2-pyridilmethyl)amine) and (R,R)-BPBP ((2R,2’R)-1,1′-bis(2-pyridilmethyl)-2,2′-pipyrrolidine) were able to catalyze the oxidation of cyclohexane with H2O2 in unprecedented yields (up to 56%). While tetradentate and tridentate ligands bearing alkylic amines (e.g., Me6Tren, MeAN) were unable to catalyze this transformation, other copper complexes with simple ligands such as bpy were able to promote this catalytic oxidation with remarkable yields. The Cu/TMPA and Cu/(R,R)-BPBP systems catalyzed the oxidation of a series of cyclic alkanes (C5–C8) reaching notable yields (36–56%).
Figure 33.

Cu-catalyzed peroxidation of strong C–H bonds using H2O2 as oxidant.179
In all the oxidations with Cu and H2O2, the alkyl hydroperoxide was obtained as major product, which suggested the formation of 1e− oxidants during catalysis (•OH, •OOH). The formation of O-centered radicals is representative of Fenton-like chemistry (i.e., Fe + H2O2). In the oxidation of cyclohexane when [FeII((R,R)-BPBP)]2+ was used as catalyst in the presence of AcOH as cocatalyst, formation of cyclohexyl hydroperoxide was not observed (Figure 33, iii).181,182 It has been proposed that in the LFeII/H2O2/AcOH system, a high-valent iron(V)-oxo intermediate acts as a 2e− oxidant that will oxidize C–H substrates via H atom abstraction followed by Fe(IV)−OH/C• rebound which avoids the formation of O-centered radicals.183 The possible involvement of O-centered radicals in the LCuI/H2O2 oxidations was also reflected in the results found in the oxidation of n-hexane, which formed a mixture of C1, C2, and C3 oxidation products. White observed that in the LFeII/H2O2/AcOH system, selective methylenic oxidation was accomplished, suggesting the involvement of a selective oxidant (i.e., Fe(V)-oxo).182 In the oxidations with the LCu/H2O2 in the presence of CCl3Br (radical trap), the authors observed the formation of cyclohexyl bromide and cyclohexyl chloride as main products (C-X/C–O ratio: 12–14), which suggested the generation of C-centered radicals (cyclohexyl radical) during the catalytic turnover. The substrate cis-1,2-dimethylcyclohexane (cis-1,2-DMCH) was also used as a mechanistic tool to determine the formation of C-centered radicals. Oxidation of this substrate by 1e− oxidants (e.g., O-centered radicals) leads to epimerization of the C–C bond to form a mixture of cis- and trans-hydroxylation products. Otherwise, if the oxidation follows a hydrogen-abstraction/radical-rebound mechanism, the stereochemistry is retained.
The results observed were indicative of formation of 1e− oxidants under the LCu/H2O2 oxidation conditions (cis:trans product mixtures) and 2e− oxidants in the LFe/H2O2/AcOH oxidations (stereoretention).
In the kinetic analysis of the oxidation of cyclohexane with LCuI and H2O2, first-order dependence on the concentration of metal and oxidant and zero-order dependence on the concentration of substrate was observed. With all the mechanistic information in hand, it was proposed that in the first reaction step the LCuI complexes (species A in Figure 33) were oxidized to the corresponding LCuII (species B in Figure 33), which is the resting state of the catalysts (observed spectroscopically). It was suggested that during the rate-determining step, LCuII reacted with H2O2 to generate hydroxyl radicals and a LCuIII–OH complex (species C in Figure 33). The resting state of the catalysts could be regenerated via reduction of the OHIII complex with H2O2 that produced hydroperoxyl radicals. Both O-centered radicals (•OH, •OOH) could react with the substrate after the rate-determining step to generate the oxidation products.
Pérez established that mononuclear Cu complexes could also catalyze the oxidation of strong C–H bonds using H2O2 as oxidant (Figure 34).184 Interestingly, the oxidation of cyclohexane generated products derived from the dehydrogenation of alkanes (i.e., cyclohexene) along with cyclohexanol and cyclohexanone (yields up to 25%). The authors proposed a mechanism in which the LCuI complex (species A in Figure 34) was oxidized by H2O2 to form an O·II intermediate that could abstract an H atom from the substrate to produce a LCuII–OH complex (species C in Figure 34) and a C-centered radical. A second H atom abstraction from the C-centered radical could regenerate the CuI complex and form the alkene product (species D in Figure 34). Alternatively, the OHII complex obtained in the first H atom abstraction could react with the C-centered radical via hydroxyl rebound to form the 2e− oxygenation product (species E in Figure 34). The mechanism proposed was supported by DFT calculations and experimental data by varying the oxidants used (i.e., the O-transfer reagent oxone generated the same oxidation products with similar ratios).
Figure 34.

Cu-catalyzed oxidation of alkanes using H2O2 as oxidant.184
Simaan has recently reported the LPMO-like reactivity of mononuclear Cu(II) complexes using H2O2 as oxidant (Figure 35).185 These copper complexes catalyzed the oxidative cleavage of 4-nitrophenyl-β-D-glucopyronaside via C–H oxidation of the 1-position of the glucose derivative to produce glucolactone and 4-NO2-phenolate (observed and quantified by UV–vis). It was suggested that the LCuII reacted with H2O2 under aqueous conditions to form a mononuclear LCuII(OOH) species that could be characterized by UV–vis (λ: 305 nm) and EPR spectroscopy.
Figure 35.

LPMO-like oxidation of C–H bonds with H2O2.185
Itoh has also shown that H2O2 can be used to oxidize alkane using copper complexes bearing β-diketiminato ligands (Figure 36).186 These copper(II) complexes catalyzed the oxidation of strong C–H bonds such as cyclohexane with low yields but good efficiency in terms of H2O2 used (up to 26 TN). It was found that the initial copper(II) complexes generated O,OIII,III species in their reaction with H2O2. It was also reported that the same species could be formed in the reaction of the corresponding LCuI complexes with O2. The experimental data suggested that the L2CuIII2(O2−)2 intermediate (species A in Figure 36) carried out the H atom abstraction of the C–H substrate in the rate-determining step (KIE: 2.4). The resulting C-centered radical could rebound with the putative CuIIICuII-hydroxo-oxyl (species B in Figure 36) to form a product-bound dicopper(II) complex (species C), which can regenerate the active O,OIII,III core by reaction with H2O2 with concomitant release of water and the oxidized alkane.
Figure 36.

Cu-catalyzed hydroxylation of C–H bonds with H2O2. Note: these oxidations were carried out under excess amounts of substrate (i.e., H2O2 was the limiting reagent), which led to very low product yields.186
Only a handful of copper complexes are able to carry out the oxidation of strong C–H bonds using O2 as oxidant.187 One of the landmark examples was reported by Schindler, where isolable end-on LCuII(O22−)CuIIL species (L: TMPA and Me6Tren and using BPh4− as counteranion) were utilized as catalysts for the oxidation of toluene (Figure 37). It was found that the EPII,II complexes were able to convert toluene to benzaldehyde using O2 as oxidant with yields up to 20%.
Figure 37.

Cu-catalyzed sp3 C–H oxidation of toluene using O2 as oxidant.187
Chan research group has used trinuclear copper complexes to catalyze the oxidation of substrates containing strong C–H bonds (Figure 38).188 Oxidation of cyclohexane to cyclohexanol and cyclohexanone with H2O2 reached low yields (up to 10%) but moderate efficiency in the use of oxidant.189 The authors proposed a reaction mechanism in which the tricopper(I) complex (species A in Figure 38) is oxidized with two equivalents of H2O2 to form sequentially a CuICuIICuII (O2−)2 intermediate (species B) and a CuIICuIICuIII(O2−)2 core (species C), which could oxidize the C–H bond. It was proposed that the tricopper(I) complex was regenerated by H2O2 which could also act as sacrificial reductant. In a second seminal contribution, they reported that the trinuclear complex could also catalyze the oxidation of methane to methanol.190 A mixture of O2 and H2O2 was used to carry out the oxidation, in which it was proposed that O2 was responsible for the formation of the active O,OII,II,III intermediate and that H2O2 could act as a 2e− electron source (species C in Figure 38, iii).
Figure 38.

Cu-catalyzed hydroxylation of strong C–H bonds using O2 and H2O2 as oxidants.188–190
2.3.2. sp2 C–H Hydroxylations.
Tyrosinase is a binuclear Cu enzyme responsible for the aromatic hydroxylation of tyrosine to form catechols and subsequently quinones in the first step of melanin biosynthesis (Figure 39).55,191 Related metalloproteins such as hemocyanin (oxygen carrier for arthropods and molluscs) and catechol oxidase (enzyme that catalyzes the oxidation of catechols to quinones) share the same active site, but only tyrosinase is able to both bind O2 and carry out the 4e−/4H+ oxidation of the natural substrates.192,193 Analysis of the crystal structure of the three enzymes indicates that substrate access to the active center is critical for oxygenase and oxidase reactivity (i.e., the active center of hemocyanin is buried in the protein). Mechanistic studies suggest that the reduced form of tyrosinase (species A in Figure 39) reacts with O2 to form a stable SPII,II species (species B) that binds the deprotonated form of tyrosine before aromatic electrophilic hydroxylation (species C).
Figure 39.

Tyrosinase: active center (i), reaction catalyzed (ii) and proposed catalytic cycle (iii).
2.3.2.1. Tyrosinase-Like Oxidations: Stoichiometric and Catalytic Reactions.
Over the last decades, many research groups have studied the reactivity of Cu2O2 species with phenolates in order to understand the reaction mechanism of tyrosinase (Figure 40). In 2000, Casella reported one of the first model systems able to perform the stoichiometry ortho-hydroxylation of phenolates (species B in Figure 40).194,195 A bridging dinucleating ligand with two symmetric N3 sites was used to stabilize a side-on CuII2(O22−) intermediate that upon addition of 4-substituted phenolates formed a putative CuII2 (O22−)-phenolate adduct prior to electrophilic aromatic oxidation (Hammett constant ρ = −1.84). Shortly after, Itoh reported the reactivity of another SPII,II bearing tridentate ligands capable of performing the hydroxylation of phenolates via electrophilic aromatic attack (species C in Figure 40).196 Interestingly, the same Cu2O2 core reacted with phenols to form the C–C coupling product derived from the 1H+/1e− oxidation, suggesting that in tyrosinase the substrate is deprotonated before binding the Cu2O2 center and being oxidized.118 In a seminal report, Stack reported a SPII,II complex (species F in Figure 40) that, upon addition of phenolate, isomerized to a metastable O,OIII,III-phenolate adduct (characterized by UV–vis, rRaman, EXAFS) before electrophilic hydroxylation, hypothesizing that a similar high-valent core could be formed in the natural system.57 Costas and Ribas described a CuIII2(O2−)2 core (species E in Figure 40) able to bind and oxidize phenolates in a analogous fashion (i.e., via formation of a O,OIII,III-phenolate adduct).197 The same research lab reported the tyrosinase-like reactivity of an end-on dicopper(II)-peroxide intermediate stabilized by a N3–N4 unsymmetric dinucleating ligand (species A in Figure 40).198 This example of EPII,II species able to perform electrophilic oxidation of substrates. On the basis of DFT calculations, Solomon suggested that the EPII,II core isomerized to an O,OIII,III-phenolate intermediate before oxygenation of the substrate.199 Stack has also shown that small changes in the stereoelectronic features of the ligands binding O,OIII,III cores (species G in Figure 40) have a deep impact on the reactivity toward phenols and phenolates (i.e., ortho-hydroxylation vs 1H+/1e− C–C coupling vs no reaction).200
Figure 40.

Cu2O2 model systems able to perform tyrosinase-like stoichiometric ortho-hydroxylation of phenolates: types of cores, (i) mechanistic evidence, and (ii) proposed hydroxylation pathways.
In a groundbreaking article, Stack has recently reported the generation of metastable SPII,II cores bearing monodentate imidazole ligands (species H and I in Figure 40).201 Interestingly, these tyrosinase-like cores are able to perform the hydroxylation of phenolates at very low temperatures (−135 °C). A recent study by the same research group reported that these CuII2(O22−) species also isomerize to the corresponding CuIII2(O2−)2-phenolate adduct before substrate hydroxylation.202
Costas reported that O,OIII,III cores are able to perform the electrophilic ortho-hydroxylation of 2-F-substituted phenolates (species E in Figure 40).203 Karlin, Solomon, and Garcia-Bosch have showed that an unreactive SPII,II core (species D in Figure 40) performed this oxidation (ortho-hydroxylation of 2,6-F2-phenolate) via formation of a O,OIII,III-phenolate core (characterized by UV–vis and rRaman spectroscopy).121 It is worth mentioning that the ortho-hydroxylation of these substrates is not an oxidative transformation (the C–F bond is replaced by a C–O bond) but that the above-mentioned Cu2O2 cores are proposed to perform this C–F functionalization via reaction pathways similar to the ones found in the C–H ortho-hydroxylation of phenolates.
The above-mentioned reports were focused on the stoichiometric reactivity of Cu/O2 species toward phenols or phenolates. On the basis of precedents by Réglier204 and Casella,205 Tuczek described that a mononuclear copper(I) complex (Figure 41, i) bearing a bidentate ligand performed the catalytic oxidation of 2,4-tBu-phenol in the presence of 2 equiv of triethylamine.206 The reaction was carried out under very dilute conditions, the substrate was not fully converted, and a mixture of reaction products was obtained (catechol: 3%, quinone: 52%), which precluded its utilization for synthetic purposes. Kinetic studies of the reaction suggested that the CuI complex (species A in Figure 41) reacted with PhOH and base to form a CuI-phenolate intermediate (species B). Oxygenation of the cuprous-phenolate species led to the formation of a putative SPII,II-phenolate adduct (species C) before generation of the quinone product.
Figure 41.

Catalytic tyrosinase-like reactivity.206
Stack and Herres-Pawlis reported a Cu2O2 system (Figure 42, i) able to perform the hydroxylation of a series of phenolic substrates.207 The catalytic oxidation of phenols was accomplished at room temperature under O2 (1 atm) and in the presence of NEt3 (2 equiv), which allowed for achievement of remarkable yields (up to 60%) but modest turnover numbers (4 to 15 TN). It was proposed that the reaction of the CuI complex (species A in Figure 42) with O2 formed a SPII,II core (species B) that could bind the substrate in the phenolate form (species C) before product generation.
Figure 42.

Catalytic tyrosinase-like reactivity.207
In a series of articles, Lumb reported a catalytic method that utilizes Cu and dioxygen to oxygenate and functionalize phenols. It was described that combining CuI with amines (DBED or NEt3) promoted the ortho-hydroxylation of phenols and subsequent coupling with nucleophiles (Figure 43).208 For example, oxidation of 4-methoxyphenol produced the corresponding ortho-quinone with another unit of 4-MeO-PhOH oxidatively coupled in the fifth position (Figure 43, iii). The oxygenation/oxidative-coupling of a series of substituted phenols was accomplished, reaching excellent yields for most of the substrates. In a follow up article, the reaction mechanism was studied using 4-tBu-PhOH as substrate.209 It was proposed that the CuI complex reacted with O2 to form a SPII,II intermediate, which was characterized at low temperature by UV–vis spectroscopy (species B in Figure 43). It was suggested that the side-on dicopper(II)peroxo complex reacted with phenols to produce the corresponding catechol (species C) that upon protonation released CuI and a product-bound Cu(II)-semiquinone complex (species D). It was proposed that the cupric-semiquinone species reacted with phenol to form the oxidation C–O coupling product (species E in Figure 43). In fact, oxidation of 3,5-substituted phenols led to the selective formation of the uncoupled ortho-quinone product (see Figure 14).122
Figure 43.

Cu-catalyzed oxygenation and functionalization of phenols for synthetic purposes.208,209
The Lumb research group has followed a similar approach(i.e., CuI/DBED/O2) to synthesize ortho-azophenols (via dehydrogenative coupling and phenols with hydrazines or hydrazides) and to synthesize dehydronornuciferine.210,211
2.3.2.2. sp2 Hydroxylations of Nonphenolic Systems: Stoichiometric and Catalytic Reactions.
Karlin reported the generation of a mononuclear cupric hydroperoxide species (species B in Figure 44) via addition of H2O2 and a base (NEt3) to the corresponding copper(II) complex.212 The EHPI complex was stable at low temperatures (−80 °C) but decayed fast at higher temperatures to form a copper(II) complex in which the ligand scaffold has undergone intramolecular hydroxylation of the aryl substituent (species C in Figure 44). Although the mechanism by which the LCuII(OOH) oxidized the arene ring was not described, the authors suggested that dinuclear Cu2O2 species were not involved in the intramolecular oxidation (generation of the O,OIII,III intermediate bearing the same ligand scaffold did not lead to sp2 hydroxylation of the arene system).
Figure 44.

Intramolecular sp2 C–H hydroxylation involving an EHPII species.212
Itoh research lab has also studied in detail the reactivity of mononuclear CuII-hydroperoxide and CuII-alkylperoxide species (Figure 45). Addition of stoichiometric amounts of H2O2 and NEt3 into an acetone solution of a copper(II) complex bearing a tridentate ligand led to the formation of an unusual copper(II)-alkylperoxo species that was characterized by UV–vis, EPR, and rRaman spectroscopy.213 The LCuII(OOR) species decayed at low temperatures (−70 °C) to oxidize the ligand in an intramolecular fashion. Computational studies suggested that the sp2 hydroxylation occurred via formation of a metastable carbocation intermediate (species C in Figure 45) before generation of the final hydroxylation product (speciesD).214 Interestingly, the analogous mononuclear LCuII-cumylperoxo intermediate (species E in Figure 45) decayed via homolytic O–O bond cleavage to generate a putative LCuII-oxyl species (O·II) that was not able to oxidize the ligand scaffold but reacted with external substrates containing weak C–H bonds (e.g., 1,4-cyclohexadiene).68
Figure 45.

Intramolecular sp2 C–H hydroxylation and intermolecular oxidations involving EAPII species.213,214
In 2007, Tolman reported that the reaction of copper(I)-α-ketocarboxylate complexes with O2 led to intramolecular sp2 hydroxylation (Figure 46).215 It was suggested that oxygenation of the cuprous complex generated a CuII-superoxide adduct (species B in Figure 46), which released CO2 (decarboxylation) to form a CuI-peracid complex (species C) before ligand hydroxylation. Computational studies proposed that the CuI-peracid adduct could oxidize the arene ring via a concerted mechanism (species D1 in Figure 46) or via heterolytic O–O cleavage to generate a CuII-oxyl species before ligand oxidation (species D2).
Figure 46.

Intramolecular hydroxylation of sp2 C–H bonds in a mononuclear copper complex using O2 as oxidant.215
In 1984, Karlin reported one of the first copper systems able to hydroxylate sp2 C–H bonds (Figure 47).216,217 Dinuclear copper(I) complexes were found to react with O2 and oxidize intramolecularly the bridging arene ring with high yields (Figure 47, (i).218 Oxygenation of the dicopper(I) complex at low temperatures generated a metastable side-on peroxodicopper-(II) intermediate (species B in Figure 47) that was spectroscopically characterized. Computational studies suggested that the SPII,II adduct attacked the phenyl ring with concomitant O–O bond cleavage in the transition state (species C) to produce a dicopper(II)-phenoxyl–oxyl intermediate (species D) that upon radical recombination and deprotonation could sequentially form a dicopper(II) μ-oxo complex and a dicopper(II) μ-hydroxo species bearing the hydroxylated ligand scaffold (species E and F in Figure 47).219
Figure 47.

Intramolecular hydroxylation of sp2 C–H bonds in a dinuclear copper complex using O2 as oxidant.216–219
The Tolman research group also studied the ability of Cu2O2 species to promote the intramolecular hydroxylation of sp2 C–H bonds (Figure 48).220 A series of metastable O,OIII,III cores bearing bidentate aminopyridine ligands were generated at low temperatures (−70 °C). These Cu2O2 cores decayed (slow decomposition at −70 °C or fast decay upon warming up) to generate equimolar mixtures of the starting ligand scaffolds (LH in Figure 48) and the intramolecular hydroxylation products (LOH in Figure 48). Two competitive pathways for the degradation of the CuIII2(O2−)2 species were suggested, one consisting of electrophilic attack to oxidize the arene ring (favored for the electron-rich systems R1: MeO, H) and an unknown self-decay pathway that did not generate the hydroxylation product (favored for electron-poor systems R1:NO2).
Figure 48.

Intramolecular hydroxylation of sp2 C–H bonds promoted by O,OIII,III cores.220
A similar ligand scaffold was used by Schindler to analyze the reactivity of Cu2O2 cores toward sp2 C–H bonds (Figure 49).221 It was proposed that the bidentate imino-amine system reacted with a CuI source and O2 at low temperatures to form an O,OIII,III core (characterized by UV–vis spectroscopy and favored by DFT), which could attack the phenyl ring to produce a dicopper(II) complex in which one of the ligands is hydroxylated (species C in Figure 49). GC/MS confirmed that only 50% of the ligand scaffold was oxidized. The hydroxylation imine product was hydrolyzed to isolate salicylaldehyde with modest yields (8%).
Figure 49.

Intramolecular hydroxylation of sp2 C–H bonds promoted by O,OIII,III cores.221
Casella described a unique dicopper system able to undergo two consecutive intramolecular sp2 hydroxylations of an m-xylene linker (Figure 50).222 It was proposed that addition of excess amounts of H2O2 to the dicopper(II) complex (species A in Figure 50) generates a dicopper(II) μ-hydroperoxo complex (species B), which could attack the meta position of the arene ring to form the monohydroxylation product (species D). A similar reaction pathway was proposed for the hydroxylation of the ortho position of the phenyl ring (species E and F in Figure 50). Spectroscopic evidence (UV–vis, EPR) for the formation of the first dicopper(II) μ-hydroperoxo complex was provided (species B in Figure 50).
Figure 50.

Intramolecular double hydroxylation of sp2 C–H bonds in a dinuclear copper complex using H2O2 as oxidant.222
Pérez and Diaz-Requejo discovered that trispyrazolylborate cuprous systems catalyzed the hydroxylation of benzene using H2O2 as oxidant (Figure 51).223,224 The authors used small catalysts loadings (1 mol %) and excess amounts of oxidant (10 equiv) to convert benzene to phenol (24%) and 1,4-benzoquinone (4%) using CH3CN at 80 °C. Toluene could also be converted to a myriad of oxidation products (benzaldehyde, phenols, and quinone) with remarkable yields (up to 35%). DFT calculations were carried out in order to understand the reaction mechanism, and it was proposed that the copper(I) complex reacted with H2O2 to generate a LCuII-oxyl intermediate (species B in Figure 51) that could oxidize benzene via H atom abstraction followed by hydroxyl rebound (species C and D) or via electrophilic substitution (species E and F). Fenton species were not invoked in the mechanism of substrate oxidation since the addition of radical traps (halogenated solvents) did not affect the catalytic performance of the system (similar conversion and reaction yields). The small KIE values observed (1.1) also supported their mechanistic proposal (a KIE value of 1.7 was previoulsy reported for Fenton-type sp2 hydroxylations).225
Figure 51.

Cu-catalyzed hydroxylation of sp2 C–H bonds using H2O2 as oxidant.223,224
Karlin and Fukuzumi also reported the use of a well-defined Cu complex for the catalytic conversion benzene using H2O2 (Figure 52).226 The copper(II)-TMPA complex oxidized benzene to phenol under mild conditions with modest yields (2%) but high turnover numbers when the catalyst was immobilized in mesoporous silica–alumina (NOTE: use of higher amounts of catalyst and excess of H2O2 reached 17% yield of phenol, with 100% selectivity). A detailed mechanistic study, including kinetic analysis and the use of radical traps, suggested that the copper(II) complex reacted with H2O2 to generate a LCuIII(OH) core and hydroxyl radical (species B in Figure 52), which could further react with hydrogen peroxide to produce hydroperoxyl radicals (observed by EPR using DMPO as radical trap). A radical chain reaction initiated by the hydroperoxyl radicals would lead to the formation of phenol (see Figure 52, iii).
Figure 52.

Cu-catalyzed hydroxylation of benzene using H2O2 as oxidant.226
2.3.3. Other Cu-Promoted Oxygenase-Like Reactions.
In a milestone contribution, Karlin reported a comparison in reactivity of dicopper(II)-peroxo species.120 It was found that EPII,II complexes were basic and nucleophilic, reacting with Brönsted acids to release hydrogen peroxide and reacting with CO2 to generate a percarbonate complex. The authors also found that SPII,II cores were better described as nonbasic/electrophilic species, reacting with 2,4-tBu2-PhOH to form the C–C coupling product and with PPh3 to form O=PPh3 but not reacting with acids. Since then, many reports have shown that these initial findings could be applied in a universal fashion to describe the reactivity of Cu2O2 cores with only a few selected exceptions in which end-on CuII2(O22−) species could act as electrophilic oxidants and side-on CuII2(O22−) complexes could behave as basic cores.198,227
McDonald has studied the reactivity of a mononuclear CuII-superoxide complex, which was previously described by Tolman (Figure 53).228 It was found that the ESII species had nucleophilic character and was able to oxidize acyl chloride substrates (benzoyl chloride to benzoic acid) and carry out aldehyde deformylation reactions (cyclohexanecarboxaldehyde to cyclohexanone). The authors proposed that the reaction with acyl chloride substrates occurred via nucleophilic attack of the Cu–O2 moiety to produce a CuIII–peracetate complex intermediate (species C in Figure 53), which upon O–O cleavage generates the oxygenation product. On the other hand, reaction with alkylic aldehydes was proposed to occur via Criegee rearrangement prior to O–O scission (species D) to produce the ketone products.
Figure 53.

Nucleophilic reactivity of an ESII complex.228
Cho has recently reported the nucleophilic character of mononuclear copper systems. Mononuclear copper(II)-alkylperoxide complexes were generated by addition of alkylhydroperoxides in the presence of triethylamine (Figure 54).229 These EAPII species were characterized by low-temperature UV–vis, CSI-MS, rRaman, and EPR. It was also found that these complexes reacted with benzoyl chloride and 2-phenylpropionaldehyde to yield the corresponding oxidation products (benzoic acid and acetophenone). A reaction mechanism was proposed in which Cu–O bond cleavage could promote formation of a nonmetallic peroxyhemiacetal-like intermediate before formation of the oxidation products and release of the initial LCuII complex. Interestingly, it was found that the copper(II) complex, under excess of alkyl-hydroperoxide oxidant, carried out the catalytic dehydrogenation of 1,4-cyclohexadiene to benzene (up to 10 turnover numbers).
Figure 54.

Nucleophilic reactivity of EAPII complexes.229
In 2017, Betley reported the reactivity of a mononuclear Cu–O2 species bearing a dipyrromethene bidentate ligand (Figure 55).230 The Cu/O2 adduct was characterized by X-ray diffraction analysis, which confirmed the side-on binding mode (the Cu–O2 core could be described as CuIII-peroxide or CuII-superoxide). The mononuclear Cu-dioxygen complex performed electrophilic oxidations including oxygen-transfer reactions (PPh3 to O=PPh3) and 2,4,6-tBu3-phenol oxygenation (formation of quinone) and 2,4-tBu2-phenol 1e− oxidations (C–C coupling product). Moreover, it was found that the LCuO2 species reacted with phenol to release H2O2 in an acid–base fashion. This is an unique example of mononuclear Cu–O2 species depicting chameleonic reactivity and is similar to the ambiguous character found for some unsymmteric Cu2O2 cores.198,231
Figure 55.

Electrophilic and acid–base reactivity of a mononuclear Cu/O2 species.230
In an unique example, Masuda reported that a mononuclear copper(II) complex bearing a tridentate N3 ligand catalyzed the sulfoxidation of dimethyl sulfide and thioanisole under low catalyst loadings (0.2 mol %), mild conditions (CH3CN at 0 °C), and short reaction times (Figure 56).232 The reaction of the copper(II) complex with H2O2/NEt3 mixtures generated a mononuclear LCuII(OOH) species (characterized by UV–vis, EPR, and ESI-MS), which was proposed to react as oxygen transfer reagent.
Figure 56.

Cu-catalyzed sulfoxidations using H2O2 as oxidant.232
3. Cu-PROMOTED FUNCTIONALIZATION OF ORGANIC MOLECULES BEYOND ENZYMATIC MIMICRY
After millions of years of evolution, nature has mastered the direct hydroxylation of C–H bonds utilizing oxygenase enzymes.6 On the other hand, enzymatic processes in which C–H bonds can be converted directly to C–C, C–N, and C–halogen bonds are scarce.233–235 Outstanding research is in progress to “teach” metalloenzymes to promote these organic transformations under in vitro conditions.236–238 Inspired by the features of metal-dependent oxidase and oxygenase reactions(i.e., use of bioavailable metals with rich redox chemistry in combination with rationally designed ligand scaffolds and mild reaction conditions), chemists have utilized metals such as copper to promote oxidative transformations beyond enzymatic activity. In this section, we summarize some of these processes, including Cu-nitrene reactivity (that can lead to C–H amination and C=C aziridination),239 organometallic transformations such as C–C and C–heteroatom coupling reactions (classically catalyzed by heavy metals such as Pd), and a “hybrid” class of reactions in which organometallic Cu–O2 species (Cu/O2 intermediates similar to the ones formed in Cu-dependent metalloenzymes) carry out diverse oxidative transformations.
3.1. Cu-Nitrene Species in C–N Bond Formation
The predominance of nitrogen atoms found in natural products, synthetic drugs, and new petroleum-derived materials has caused the emergence of synthetic methods for the formation of C–N bonds.240 The classic approach to introduce the N functionality is based on multistep nucleophilic displacement routes that generate stoichiometric amounts of leaving groups. The discovery of the Pd-catalyzed C–N cross-coupling reaction has been a major breakthrough in this field, leading to the efficient synthesis of a wide-range of amines.241 Apart from the high cost of Pd, they require the use of preactivated substrates such as aryl halides (Ar–X) that generate stoichiometric amounts of halide salts. Also, their functional group tolerance is limited. To address these shortcomings, direct amination of C–H bonds catalyzed by transition metals via metal-nitrenoid/imido species has emerged as a one-step, economical, and environmentally friendly alternative.242–244
Metal–nitrene species are considered reactive intermediates in some metal-catalyzed C–N bond formation reactions, including C=C aziridinations and C–H aminations.245 The study of their structure, spectroscopy, and reactivity can also give some insight into the chemical properties of biologically relevant fleeting intermediates: copper-oxyl species (i.e., CuII-oxyl) are proposed to be formed in copper-catalyzed C–H hydroxylations but have not yet been been isolated; however, some of the isoelectronic copper-imido radical intermediates have been characterized (Figure 57). In this section, we describe the most relevant examples of copper-nitrene species, including their generation, isolation, and characterization, and the organic transformations that they perform.
Figure 57.

Putative intermediates formed in the reaction between Cu and oxene and nitrene sources.
Copper-catalyzed nitrene transfer reactions were discovered in the early 90s. In two back-to-back research articles, Jacobsen and Evans reported the use of copper catalysts to promote the enantioselective aziridination of alkenes (Figure 58).246,247 Jacobsen used a copper(I) source (10 mol %), chiral diimine ligands (10 mol %), and PhI = NTs as nitrene source (1.5 equiv) to convert the alkene substrates to the corresponding aziridines with good yields and remarkable ee’s when the reaction was carried out at low temperatures. It was suggested that the active oxidant, a putative Cu-nitrene species, was bound by a single chiral ligand. Using a similar approach (i.e., CuI, chiral ligand and PhI = NTs), Evans found that high enantioselectivites (up to 97%) could be obtained for cinnamate esters. Interestingly, similar enantioselectivities were observed when a CuII source was employed, which led to the speculation that the copper was functioning as a catalyst in the CuII oxidation state. In a follow-up article, Evans expanded the substrate scope of the aziridination reaction and studied in detail the reaction mechanism.248 The use of copper sources in different oxidation states, analysis of the epimerization of cis and trans olefins and the oxidation of substrates containing a cyclopropyl-based radical trap suggested that the oxidation occurred via a concerted mechanism involving a CuII species.
Figure 58.

Later on, the Che research group reported that enantioselective aziridination reactions could be accomplished in a one-pot fashion using catalytic amounts of CuI and a chiral bisoxazoline ligand (previously used by Evans), sulfonamides, and PhI(OAc)2 as oxidant (Figure 58C).249 This advantageous procedure avoided the independent synthesis of the nitrene source, which was proposed to be generated “in situ” in the reaction of the sulfonamide and the iodine(III) oxidant. The authors reported the synthesis of aziridine products derived from styrene substrates and aromatic sulfonamides with good yields (up to 95%) and moderate enantioselectivity (30–70%).
Since Jacobsen’s and Evans’ initial findings, several reports have described the use of copper catalysts for the aziridination of olefins using PhI=NTs as a nitrene source.250,251 Pérez reported that mononuclear CuI complexes bearing tris-(pyrazolylborate) ligands catalyzed the aziridination of styrene and alkylic olefins (e.g., 1-hexene) using PhI=NTs and chloramine-T as nitrene sources (Figure 59).252 In a follow-up article, the mechanism of aziridination was studied, including competition experiments between different olefins (Hammett’s analysis for the aziridination of 4-substituted styrene substrates), use of radical inhibitors, oxidation of substrates containing radical clock probes, and stereoretention experiments (e.g., aziridination of cis and trans olefins). On the basis of the DFT calculations and the experimental findings, the authors proposed a reaction mechanism that involves the formation of a Cu-nitrene species in the triplet state ([CuII-(N•)-Ts]), which could react with alkenes in a stepwise pathway maintaining the stereoretention of the substrate (Figure 59, iii).
Figure 59.

In two key contributions, the same research group reported that cuprous tris(pyrazolylborate) complexes also catalyzed the amination of strong C–H bonds using the nitrene donors PhI=NTs and chloramine T (Figure 59, iv).253,254 Oxidation of cyclohexane and benzene formed the corresponding amination products with remarkable yields (40–65%) using the organic substrates as solvents (note: the yields are based on equivalents of oxidant, the limiting reagent of the reaction). Interestingly, the oxidation of toluene selectively produced the amination product derived from sp3 C–H oxidation (95% yield). Oxidation of cumene formed a mixture of products derived from the oxidation of the tertiary sp3 C–H bond (30%) and the primary sp3 C–H bond (20%). It was also found that the regioselectivity of the amination reactions was dependent on the ligand scaffold. All these studies seem to indicate that these oxidations are metal-centered (i.e., C–H abstraction and C–N bond formation occur in the vicinity of the copper center).
The same cuprous tris(pyrazolylborate) complexes have been recently used to synthesize sulfinamides and isothiazoles via nitrene transfer to alkynes (Figure 60).255 In the presence of catalytic amounts of copper, terminal alkynes (e.g., 1-phenyl-1-propyne) reacted with PhI=NTs to form mixtures of sulfinamides and isothiazoles (Figure 60, ii). For terminal alkynes (e.g., phenyl acetylene), selective formation of sulfinamides was observed. The reaction mechanism was studied computationally, and it was suggested that a putative copper-nitrene species reacted with the alkynes to form an intermediate in a triplet state with 2 unpaired electrons on the benzylic carbon of the substrate (species C in Figure 60). This species could produce a copper–carbene intermediate (species D) or isomerize to a second imine complex (species E), with either species evolving to generate a cyclization product (species F). It was suggested that this cyclization complex could undergo S–O bond cleavage to form the sulfinamide product. The second imine intermediate (species E in Figure 60) could also undergo intramolecular C–C bond formation (species H) to form the isothiazole product. It was proposed that both reaction products could be formed from terminal alkynes, but the reversibility of the last reaction step in the generation of the isothiazole product leads to the selective formation of the sulfinamide product since it was computed to be a thermodynamic sink.
Figure 60.
Cu-catalyzed synthesis of sulfinamides and isothiazoles.255
Over the past decade, the Warren research lab has contributed enormously to the understanding of Cu-catalyzed C–N bond formation reactions. A dicopper nitrene species was generated in the reaction between 1-adamantylazide (N3-Ad) and 2 equiv of a CuI complex bearing a bidentate β-diketiminato ligand (Figure 61, i).256 This reaction intermediate was characterized using X-ray diffraction analysis in which a short Cu–Cu distance (2.9 Å) was observed (note: Warren previously reported the synthesis of a similar dicopper nitrene species using aryl azides257). Interestingly, the use of excess amounts of alkyl azide formed a dimeric product in which the ligand scaffold underwent intramolecular insertion of the adamantylnitrene species (Figure 61, i). It was found that the use of chemically inert substituents in the ortho-positions of the β-diketiminate ligands precluded intramolecular C–H amination and enabled exogenous C–H amination reactions. The isolated dicopper nitrene complex stoichiometrically oxidized strong C–H bonds reaching excellent reaction yields. Interestingly, alkanes could also be converted to the corresponding amination products using catalytic amounts of the CuI complex (Figure 61, ii). A reaction mechanism was suggested in which the CuI complex reacted with the alkylic azide to generate a dicopper-nitrene complex (isolated), which is in equilibrium with a terminal Cu-nitrene complex (species C in Figure 61). This terminal nitrene could then react with C–H bonds via H abstraction followed by NH-R rebound. This mechanistic proposal was supported with kinetic experiments, in which the rates of amination were found to be dependent on the strength of the C–H bond oxidized (i.e., stronger C–H bonds, slower reaction rates). The KIE values observed (5.3–6.6) were also in agreement with the proposed mechanism, which suggests that H atom abstraction is the rate-determining step. DFT calculations predicted that the electronic structure of the terminal monocopper-nitrene species is ambiguous (triplet and singlet states were close in energy), with the singlet species being probably responsible for C–H amination due to the bent coordination of the nitrene to the copper ion, which would allow access of the substrate to the Cu–N core. In a related research article, the reactivity of a dicopper alkylnitrene complex was studied in detail and it was implied that the active intermediate in the C–H amination reactions was the terminal mononuclear Cu-nitrene species.258 It was noted that the dicopper nitrene species derived from 1° and 2° alkyl azides were unstable and tautomerized to the corresponding CuI imine complexes.
Figure 61.

In 2010, the Warren research group reported the synthesis of the analogous LCuII-amide complex using a dicopper(II)-dichloride complex and LiNHAd (Figure 62).259 This cupric complex reacted with benzylic C–H bonds to generate 0.5 equiv of the amination product and 0.5 equiv of the alkylic amine (Ad-NH2). On the basis of the stoichiometry of the reaction, it was suggested that the CuII-amide complex reacted with the C–H substrates to form a LCuI-amine complex and a C-centered radical, which could be trapped with a second equivalent of CuII-amide to yield the C–N reaction product. It was also found that catalytic amounts of the LCuI complex could perform the amination of C–H bonds using tBuOOtBu as oxidant and alkylic amines as N sources (Figure 62, ii).260 The authors proposed that the LCuI complex reacted with tBuOOtBu to produce a LCuII-OtBu species (independently synthesized and characterized with X-ray diffraction analysis) and tBuO•, which abstracts H atoms from the C–H substrates (note: it was previously shown that in the absence of tBuOOtBu, the Cu-amide complex was the active H abstractor259). The resulting carbon-centered radicals could be trapped by a LCuII-amide species (species C in Figure 62), generated in an acid–base reaction between the LCuII-OtBu and the corresponding amine, to form the amination organic product and regenerating the LCuI catalysts. In a follow-up article, Warren reported that aromatic amines could also be used as nitrogen sources under similar C–H amination reaction conditions (catalytic amounts of CuI and tBuOOtBu as oxidant).261 A further detailed mechanistic study on the reaction mechanism pointed toward the mechanistic hypothesis proposed for the amination with alkylic amines (i.e., H atom abstraction by tBuO• followed by C–N bond formation via reaction between the C-centered radical and a CuII-anilide species).260
Figure 62.

Cu-catalyzed amination of C–H bonds using tBuOOtBu as sacrificial oxidant and amines as N-sources.259–261
In a recent attempt to isolate terminal Cu-nitrene species, Warren reported the characterization of a dicopper(II) ketimide complex derived from the dimerization of a LCu-nitrene species and a k2-diazametallocyclobutene complex derived from the addition of CH3CN to a terminal LCu-nitrene intermediate (Figure 63).262 It was reported that this species can act as “masked” terminal Cu-nitrene complex, releasing the mononuclear copper-nitrene intermediate that can then perform the 2e− amination of benzylic C–H bonds (H atom abstraction followed by C–N bond formation).
Figure 63.

Generation and reactivity of “masked” copper-nitrene species.262
In a landmark research article, Ray reported that a Lewis acid could be used to stabilize and characterize a terminal copper-nitrene complex (Figure 64).263 The reaction of a LCuI complex bearing a tridentate ligand (previously used by Karlin research group to stabilize an O,OIII,III core264) with a soluble nitrene source SPhI=NTs at room temperature formed a CuII-amide complex (characterized by X-ray diffraction analysis, EPR, and UV–vis). It was suggested that this species was formed via generation of a metastable Cu-nitrene complex, which could then abstract an H atom from the solvent. Reaction between the CuI complex and the nitrene source in the presence of Sc(CF3SO3)3 at −90 °C produced a purple species that was formulated as a Cu-nitrene-Sc adduct (Figure 64, i). The authors were unable to characterize this species by X-ray diffraction analysis, but detailed spectroscopic evidence was provided, including UV–vis, resonance Raman, and X-ray absorption spectroscopy (XAS and XANES). DFT calculations suggested that the Cu-nitrene-Sc adduct was a terminal Cu-nitrene species in a singlet state with the Sc3+ ion bound to the N atom of the nitrene moiety. Analysis of the reactivity of the Cu-nitrene-Sc core toward a series of hydrocarbons was also carried out (Figure 64, ii). The reaction rates were found to be linearly dependent on the BDFEC–H of the substrate (i.e., higher BDFEC–H, slower reaction rates), and a KIE of 5.1 was observed, which suggested that the Cu-nitrene-Sc species oxidized C–H bonds via H atom abstraction followed by NH-R rebound (i.e., Cu-nitrene-Sc species acted as a 2e− oxidant). In a second contribution, Ray characterized a second Cu-nitrene-Sc core derived from the oxidation of the same CuI complex with an organic azide in the presence of the Lewis acid (Figure 64, i).265 DFT calculations suggested that in this case, the copper ion was only bound by two N atoms of the ligand scaffold and the N atom of the nitrene, which was also coordinated by Sc3+. Reactivity studies found that this second Cu-nitrene-Sc species was a better nitrene transfer reagent (faster reaction rates with PPh3 to form RN=PPh3) but a worse H atom abstractor (slower rates in the reaction with C–H bonds) than their previous example.
Figure 64.

Generation and reactivity of copper-nitrene species stabilized by Lewis acids.263
Ribas, Ray, and Company have recently reported the characterization and reactivity of a mononuclear Cu-nitrene species that is generated using an azide triazamacrocycle which serves as ligand and nitrene source (Figure 65).266 The authors were unable to obtain X-ray diffraction data but UV–vis, rRaman, CSI-MS (cold spray ionization mass spectrometry), and X-ray absorption spectroscopy (XANES and EXAFS) confirmed the formation of a mononuclear Cu-nitrene complex. DFT calculations suggested that the Cu-nitrene intermediate was a triplet system in the ground state (S = 1), which matched the effective magnetic moment determined using the Evans method. The mononuclear CuII-(L-N•) species decayed slowly at room temperature to produce a dimeric complex in which the ligand scaffold had undergone N–N bond formation (Figure 65). It was also reported that the CuII-nitrene species reacted with PPh3 to form the corresponding L–N=PPh3 product with moderate yields. It was also found that the Cu-nitrene core reacted with weak C–H bonds such as 1,4-cyclohexadiene (note: the organic products derived from those reactions were not characterized nor quantified).
Figure 65.

Generation and reactivity of a mononuclear copper-nitrene species.266
Bertrand reported the first crystal structure of a monocopper-dinitrene complex (Figure 66).267 A phosphonitrene ligand reacted with 2 equiv of CuI(CF3SO3) to form a dicopper-nitrene complex (colorless crystal), a species similar to the dicopper-nitrene species reported by Warren (see above).256 On the other hand, the use of 0.5 equiv or 1 equiv of CuI produced the corresponding Cu-dinitrene and Cu-mononitrene complexes, respectively. X-ray diffraction analysis of the dicopper-nitrene and copper-dinitrene species was carried out. Attempts to isolate the copper-mononitrene complex (characterized by 31P NMR) led to the formation of a mixture of the dicopper-nitrene and the copper-dinitrene complexes, which suggested that the terminal Cu-nitrene complex underwent dismutation.
Figure 66.

Generation of mononuclear and dinuclear copper- (di)nitrene species.267
The Ottenwaelder research group has recently reported the generation and characterization of a dicopper(II)-arylnitroso complex (species A in Figure 67).268 The Cu2II-arylnitroso species was characterized by X-ray diffraction analysis, in which the Ar-NO is bound in a side-on fashion with one of the trifluoromethanesulfonate anions bridging between the copper centers. Interestingly, this structure is reminiscent of the SPII,II complexes derived from the oxygenation of similar LCuI complexes.52 This unusual intermediate (stable at room temperature) was able to react with 2,4-tBu2-PhONa to perform a tyrosinase-like nitrene insertion in the ortho-position of the substrate (species B in Figure 67). After nitrene insertion, the resulting iminoquinone-CuII complex could be reduced with Na2S2O4 to isolate the corresponding 2-aminophenol product with good yields.
Figure 67.

Generation and tyrosinase-like reactivity of a dicopper(II)- arylnitroso complex.268
3.2. Cu-Promoted Organometallic Functionalization Reactions
C–C and C–heteroatom coupling reactions are widely applied in organic chemistry and have replaced, in some instances, other classic routes of molecule synthesis. A more appealing approach is the direct functionalization of C–H bonds, but it entails overcoming some synthetic challenges such as the inherent inertness of C–H bonds, group tolerance (i.e., other functionalities are usually more prone to react), and their ubiquity, which usually leads to nonselective product formation. For economical and environmental reasons, enormous research efforts are currently devoted to the development of synthetic protocols to carry out organic transformations based on first row transitions metals (Ti, Mn, Fe, Co, Ni, and Cu) that are classically catalyzed by expensive metals such as Pd, Pt, Ru, or Rh (e.g., C–H functionalizations, coupling reactions, etc.).269–272 The ability of copper to promote organometallic transformations has been know since the beginning of the 20th century, but recent research developments have shown that this metal could replace (or complement) Pd in selected applications such as C–C and C–heteroatom coupling reactions.273,274 In this section, we summarized the most relevant examples of Cu-promoted and Cu-catalyzed organometallic transformations, focusing on specific examples of high mechanistic relevance. Nucleophilic organocopper(I) reactions are not discussed in detail in this review article, but they have been meticulously described by Nakamura in a recent publication.275 Research in which copper promotes reductive transformations (i.e., CuH catalysis) is outside of the scope of this review article.276
3.2.1. Two-Electron Reactivity (CuI/CuIII Catalytic Cycles).
3.2.1.1. Ullman-Like Coupling Reactions.
Copper-promoted Ullman-Goldberg coupling reactions were discovered more than a century ago.277 Stoichiometric amounts of copper(II) sources were used to perform the C-heteroatom bond formation (C–O, C–N, and C–S) of aryl halide substrates with nucleophiles under harsh conditions (extremely high temperatures and use of stoichiometric amounts of base).278 A significant advance was made when Hartwig, Taillefer, and Buchwald reported that addition of ancillary ligands allowed for milder reaction conditions and catalytic amounts of copper.279,280 Taillefer studied the catalytic performance of different neutral bidentate ligands in the oxidative coupling of aryl iodide and phenols (Figure 68).281 It was described that simple imino-pyridine and bipyridine ligands, in combination with CuI, could catalyze these coupling reactions. Interestingly, they found that changes in the stereoelectronic properties of the ligands had an impact on the overall reaction yields. For example, addition of electron-donating groups in the aromatic substituent of the imine diminished the reaction yields. On the other hand, an increase in the reaction yields was observed when electron-rich bipyridine scaffolds (e.g., 4-MeO-bpy) were used. It was rationalized that this behavior was based on the proposed mechanism, in which the LCuI catalyst reacts with aryl iodide to form a putative copper(III)-aryl-iodide intermediate (oxidative addition) that could undergo nucleophilic substitution of the iodide ligand to produce an organocopper(III)-aryl-phenolate complex (species C in Figure 68). Reductive elimination could form the C–O coupling product and regenerate the active cuprous catalyst. It was suggested that electron-donating groups in the imine-pyridine systems would disfavor the reaction steps in which a more electrophilic copper center is required (i.e., nucleophilic substitution and/or reductive elimination), and that use of electron-rich systems in the bipyridine ligand would accelerate the oxidative addition step (i.e., favor the formation of the copper(III)-aryl-iodide complex).
Figure 68.

Cu-catalyzed C–O coupling reactions between aryl halides and phenols.281
The Hartwig research lab studied in detail the reaction mechanism of Ullman amination reactions (oxidative coupling of aryl iodides and amines using catalytic CuI and ligand auxiliaries).282,283 The authors synthesized a series of CuI-amido complexes that were competent intermediates in the Ullman reaction (Figure 69). Reactions of the isolated CuI-amido compounds with aryl iodide in the presence of dative ligands (phen) enhanced the reactivity of the CuI diamido complexes by replacing the amide groups (Figure 69, ii). Kinetic experiments showed that the reaction was first-order on iodoarene concentration. Selectivity experiments (competition between amination of ortho- and para-iodotoluene) suggested that a CuI-phen-amide was formed before addition of the aryl iodide. The formation of free aryl radicals was excluded by using ortho-(allyloxy)-iodobenzene as a substrate (i.e., formation of aryl radical would lead to the cyclization product, see Figure 69, ii). On the basis of the extensive experimental data and DFT calculations, the authors proposed a reaction mechanism that could be initiated by the phen-CuI-iodide complex, which could be converted to an alkali-metal cuprate in the presence of amine (species B in Figure 69). This complex could generate the active CuI-phen-amide intermediate (species C) that could then react with aryl iodide via oxidative addition to produce a copper(III) species (species D) before reductive elimination of the amine aryl product. In a similar research article, Hartwig and co-workers studied the reaction mechanism of etherification of aryl halides catalyzed by CuI-iodide and phenanthroline.284 The experimental findings suggested that in those coupling reactions, a copper(III)-aryl-phenoxide complex was formed before reductive elimination of the ether product.
Figure 69.

Cu-catalyzed C–N coupling reactions between aryl iodides and amines.282,283
Buchwald have also studied the mechanism of Cu-catalyzed coupling reactions. Kinetic analysis of the reaction between amides and aryl iodide was catalyzed by CuI and diamine ligands.285 Like in Hartwig’s proposal, a CuI-amide-diamine complex was identified as the active intermediate, which reacts with the aryl iodide substrate before C–N bond formation (note: both CuI/CuII and CuI/CuIII aryl iodide activation pathways were proposed). Suresh and co-workers reported a noteworthy application of the Ullman reaction in a tandem copper-catalyzed synthesis of 1,4-benzodiazepines and imida-zobenzodiazepines (Figure 70).286 The reaction of 1,2-diamines with ortho-haloaryl carbonyl compounds in the presence of catalytic amounts of copper(I) iodide and DMEDA (N,N′-dimethylethylenediamine) under basic conditions produced 1,4-benzodiazepines in high yields. Interestingly, a one-pot strategy to synthesize imidazobenzodiazepines was developed; these compounds were generated in the reaction of the 1,4-benzodiazepines with TosMIC (toluenesulfonylmethyl isocyanide). This tandem reaction was proposed to be initiated by the coordination of the diamine to the CuI source (species A in Figure 70) followed by the formation of the imine synthon (species B), which triggers oxidative addition of the C-halogen bond to generate an organocopper(III) intermediate (species C). Deprotonation of the organocopper(III) (species D) followed by the reductive elimination step could then generate the 1,4-benzodiazepine product (species E in Figure 70) and regenerate the CuI catalyst. In the presence of base, the TosMIC could add to the imine product to form an anionic intermediate (species F in Figure 70) that could then undergo cyclization (species G) and elimination to produce imidazobenzodiazepine (species H).
Figure 70.

Tandem copper-catalyzed synthesis of 1,4-benzodiazepines and imidazobenzodiazepines.286
Hartwig has also studied the reaction mechanism of the Cu-catalyzed Ullman C–O coupling of phenols and aryl halides (Figure 71).287 In that research article, the authors focused on understanding the reactions catalyzed by CuI and anionic ligand donors. Three-coordinate heteroleptic complexes were characterized, which were proposed to be formed before the oxidative addition of the aryl iodide substrate (Figure 71, i). Kinetic analysis of the reaction using 13C-labeled aryl iodide revealed a KIE that was in agreement with an irreversible oxidative addition of aryl halide to the CuI anionic species to form the organocopper(III) intermediate (species B in Figure 71). DFT calculations suggested that after oxidative addition, dissociation of the halide substituent occurred (species C) before the reductive elimination step. Very recently, Warren proposed the formation of a similar organocopper(III)-phenolate intermediate in the stoichiometric reaction between a β-diketiminato arylcopper(II) complex with phenolates which afforded C–O coupling products.288
Figure 71.

Mechanistic study on Cu-catalyzed C–O coupling reactions with auxiliary anionic ligands.287
In 2012, the Hartwig research lab reported the mechanism of copper-catalyzed Hurtley-type reactions (e.g., C–C coupling of diethyl malonate with phenyl iodide).289 Bis-phenanthroline copper(I)-enolate complexes were synthesized by addition of 2 equiv of phenanthroline to an equimolar mixture of CuIOtBu and enolate sources (Figure 72, i). It was found that the reactivity of the [(phen)2CuI](CH(CO2Et)2) complex with PhI to produce PhCH(CO2Et)2 was accelerated when copper iodide was added, which suggested that a monophen CuI-enolate species was the active catalytic intermediate (species A in Figure 72). As in Hartwig’s Cu-catalyzed amination reactions,282,283 the oxidation of ortho-(allyloxy)iodobenzene did not form the cyclization product (see Figure 72), which suggested nonradical oxidation pathways. With all the mechanistic information in hand, it was proposed that the reaction occurred via oxidative addition (formation of an arylcopper(III) iodide enolate complex) followed by reductive addition to generate the C–C coupling product (Figure 72, iii).
Figure 72.

Mechanistic study on the Cu-catalyzed Hurtley-type reactions.289
Ribas and Stahl have extensively explored the use of model systems to understand the Ullman-Goldberg reaction mechanism.39 Macrocylic ligands with an aryl-halide internal substrate allowed for observation, isolation, and characterization of organocopper intermediate species. In one of their first joined efforts, they reported that C–N bond formation could be achieved using catalytic amounts of copper(I) and pyridone as a nucleophile (Figure 73).290 In a follow-up article, Ribas reported that other nucleophiles also promoted C-nucleophile bond formation (C–S, C–Se, and C–P).291 On the basis of their evidence, it was proposed that copper(I) coordination to the macrocyclic ligand would trigger oxidative addition to produce an organocopper(III)-bromide intermediate (species B in Figure 73) that reacts with nucleophiles to generate the C-nucleophile product via reductive elimination. Kinetic analysis of the reaction was performed (by low-temperature UV–vis and NMR) in which the organocopper(III) intermediate formed and decayed with concomitant generation of the coupling product (note: see section below for further details on organocopper-(III) synthesis and reactivity).
Figure 73.

Cu-catalyzed coupling reactions in macrocylic ligands.290,291
In a milestone research article, the Ribas research lab used the same macrocylic system to study nucleophilic aryl fluorinations and aryl halide exchange reactions (Figure 74).292 Catalytic amounts of copper(I) and excess amounts of halide sources (NBu4X, NaX, LiX for X: Cl, Br, I) promoted halide exchange in the aryl-halide substrate–ligand system (Figure 74, i). Exchange of the aryl-I substrate was rapidly achieved when exposed to CuI and NBu4Cl or NBu4Br due to the higher reactivity of the aryliodide systems. Interestingly, the aryl-iodide product could also be obtained from the less reactive aryl-chloride and aryl-bromide taking advantage of the poor solubility of NaCl and NaBr in CH3CN: addition of a CuI source and excess NaI (soluble in CH3CN) triggered halide-exchange with concomitant precipitation of the NaCl or NaBr byproducts. In order to generate the aryl-fluoride coupling product, AgF was used as fluoride source (yields up to 76%). It was proposed that the oxidative coupling occurred via oxidative addition to form an organocopper(III)-halide intermediate (species B in Figure 74) that could exchange the halide ligand (species C) before reductive elimination.
Figure 74.

Cu-catalyzed aryl halide exchange reactions in macrocylic ligands.292
In 2012, Hartwig reported that copper could be utilized to mediate the fluorination of aryl iodides.293 The use of a cationic copper reagent [CuI(tBuCN)2](CF3SO3) (3 equiv) and AgF (2 equiv) under relatively harsh conditions (DMF, 140 °C, 22h) converted a wide array of aryl iodides to the corresponding aryl fluorides (yields: 40–96%). Similar to the mechanism proposed by Ribas, it was suggested that the copper(I) complex could undergo oxidative addition to form an organocopper(III)-iodide intermediate that upon halide exchange forms an organocopper-(III)-fluoride complex before reductive elimination of the aryl-fluoride product. Using the same copper(I) complex, the Hartwig research group reported that arylboronate esters could also be fluororinated.294 In this case, [CuI(tBuCN)2](CF3SO3) (2 equiv), excess amounts of the F+ source [Me3pyF](PF6) (3 equiv), and AgF (2 equiv) were used to fluorinate Ar-BPin substrates under milder conditions (THF, 50 °C, 18h). It was proposed that an arylboronate copper(III)-fluoride complex underwent transmetalation to generate an aryl copper(III) fluoride intermediate before reductive C–F bond formation.
In 2013, the Sanford research lab reported a mild fluorination method of aryl stannanes and aryl trifluoroborates using [CuI(tBuCN)2](CF3SO3) and N-fluoro-2,4,6-trimethylpyridinium triflate (NFTPT), which acts both as an oxidant and fluorine source.295 Surprisingly, the fluorination of aryl stannanes could be accomplished at room temperature using stoichiometric amounts of copper (1 equiv) and 2 equiv of the F+ source in EtOAc for 5 min. A CuI/CuIII reaction mechanism with the formation of an aryl copper(III) fluoride complex was proposed as a key intermediate.
The Chan-Evans-Lam coupling reaction uses boronic acids (or arylboronic esters) as aryl donors, which, in combination with nucleophiles such as amines, yields C–X coupling products.296 Stahl studied the mechanism of the copper-catalyzed aerobic oxidative C–O coupling of arylboronic esters and methanol (Figure 75).297 Kinetic studies, EPR characterization of the reaction intermediates, and oxidation of the substrate under anaerobic and aerobic conditions all supported a reaction mechanism that could be initiated by coordination of the arylboronic ester (species A in Figure 75) before transmetalation to generate a copper(II)-aryl intermediate in the rate-determining step (species B). It was proposed that oxidation of this organocopper(II) complex via disproportionation would produce aryl-copper(III) intermediates (species C and D) before C–O bond formation. In a follow-up article, Stahl studied the copper(II) species that led to the formation of the key copper(II)-aryl intermediate.298 Warren has recently reported the reactivity of a CuII-aryl species, which reacts with phenolates to produce the C–O coupling product via formation of a CuIII-aryl-phenolate intermediate (analogous to species D in Figure 75).288
Figure 75.

Mechanistic study of the Cu-catalyzed Chan–Evans–Lam C–O coupling reaction.297,298
Watson have recently studied in detail the reaction between boronic acids and amines in the presence of CuII (Figure 76, i).299 The C–N coupling products and the byproducts derived from the oxidation/protodeboronation side reaction were characterized and quantified. The inhibitory effect of species present in solution such as AcOH, AcOK, and pinacol was also analyzed. With all the mechanistic information in hand, the authors proposed a complex reaction mechanism that relies on the formation of a mononuclear copper(II)-amine complex (species A in Figure 76) that is in equilibrium with other dinuclear and tetranuclear copper species. The mononuclear CuII complex could react with aryl boronic acid to generate a copper(II)-hydroxo/boronic acid adduct (species B) that could undergo transmetalation to produce an organocopper(II) aryl intermediate (species C). Oxidation of this copper(II) complex via disproportionation could generate an organocopper(III) intermediate (species D) that upon reductive elimination forms the C–N coupling product. On the basis of the proposed mechanism, it was found that addition of B(OH)3 and an increase in the amine concentration improved the reaction yields (Figure 76, iii). These reaction conditions were applied for the synthesis of a wide variety of C–N coupling products.
Figure 76.

Mechanistic study and synthetic applications of the Cu-catalyzed Chan–Evans–Lam C–N coupling reaction.299
In a recent contribution, Taylor described the selective copper-mediated O-arylation of carbohydrate derivatives.300 Arylboronic acids and excess amounts of CuII(OAc)2 were used to synthesize sugar-derived aryl esters with remarkable yields and selectivity. It was proposed that reaction of the boronic acid and the carbohydrate formed a substrate-derived boronic ester that could direct the oxidative coupling via formation of an arylcopper(III)/substrate-boronic ester intermediate before reductive C–O elimination.
The Zhang research group has recently studied the reactivity of copper trifluoromethyl complexes in C–C coupling reactions (Figure 77).301 The authors isolated an ion-pair CuI/CuIIItrifluoromethyl species and a monocopper(III) tris-trifluoromethyl complex and used them as stoichiometric trifluoromethylation reagents of arylboronic acids. It was found that the ion-pair CuI/CuIII complex was unable to generate the C–C oxidation product. On the other hand, the [(bpy)CuIII(CF3)3] transferred the CF3 group with high yields in the presence of KF. A reaction mechanism was proposed in which the mononuclear copper(III) complex reacted with the arylboronic acid in the presence of a fluoride source to generate a copper(III)-aryl intermediate (species B in Figure 77), which would undergo reductive elimination to form the aryl-CF3 coupling product. Oxidation of the resulting [(bpy)CuI(CF3)] complex in the presence of O2 and water could produce a LCuIII(OH)2(CF3) intermediate (species D) that could activate another molecule of arylboronic acid to form a second equivalent of C–C coupling product. In a follow-up article, the synthesis of ion-pair CuI/CuIII trifluoromethyl complexes bearing bisphosphine ligand donors was described.302 In this case, it was found that these complexes resulted in C–C bond formation when reacted with arylboronic acids. It was proposed that the ion-pair complex was in equilibrium with a monocopper(III) species, which is the active CF3 transfer reagent.
Figure 77.

Stochiometric Cu-promoted trifluoromethylation of arylboronic acids.301
Grushin research lab studied the reaction mechanism by which Cu complexes carry out oxidative trifluororomethylation reactions in the presence of O2 (Figure 78).303 A well-defined copper(II) fluoride complex reacted with TMSCF3 to form a mixture of CuIII and CuI species. Of those, only one of the CuI complexes reacted with O2 to produce a key intermediate [CuIII(bpy)(CF3)2(OSiMe3)] (species C), which, upon reaction with aryl boronic acid, would generate a CuIII–aryl complex before C–C reductive elimination. It was proposed that oxidation of the TMS ligand occurred via generation of dinuclear Cu2O2 species, which were not spectroscopically observed.
Figure 78.

Mechanism of Cu-catalyzed oxidative aerobic trifluoromethylation.303
The Grushin research lab also developed a synthetic protocol for the pentafluoroethylation of acid chlorides using well-defined CuIC2F5 complexes (Figure 79).304 The reaction of stoichiometric amounts of [CuI(PPh3)(phen)(C2F5)] with acid chlorides in THF at 65 °C generated the corresponding RCOC2F5 products with good-to-excellent yields. The reaction mechanism was not studied in detail, but based on the chemoselectivity, the authors proposed that C2F5• radicals were not formed and that the oxidative coupling probably occurred via oxidative addition of the C–Cl to CuI followed by C–C2F5 reductive elimination of the resulting CuIII intermediate.
Figure 79.

Cu-promoted pentafluoroethylation of acid chlorides.304
Desage-El Murr and Fensterbank have studied the reactivity of copper-CF3 species bearing redox-active ligands (Figure 80). In their first report, they described the synthesis of a copper(II)-trifluoromethyl complex by reaction of the bis-iminosemiquinone copper(II) species with a CF3+ source, which triggered the 1e− oxidation of the two ligand scaffolds to form the copper(II)-CF3 bond (Figure 80, i).305 It was observed that the CuII–CF3 species decayed slowly at room temperature to generate a C–CF3 ligand oxidation product (CF3 umpolung transfer product). Heating up the copper(II)-CF3 complex formed a second C–CF3 coupling product, which could be also obtained by heating up the C–CF3 oxidation product formed at room temperature. Interestingly, the L2CuII (CF3) complex catalyzed the CF3 transfer to aminophenol substrates with similar structure to the ligand scaffold (Figure 80, i). In a second contribution, the authors reported that the bis-iminobenzoquinone CuII–CF3 complex reacted with arylboronic acids to generate the C–N coupling product derived from the oxidation of the ligand scaffold (Figure 80, ii).306 It was proposed that intramolecular CF3− transfer produced a copper(III) complex bearing the oxidized ligand scaffold and an iminosemiquinone radical species (species C in Figure 80). It was also implied that addition of PhB(OH)2 could generate a copper(II) species with the phenyl bound to the copper in a trans position relative to the CF3 group (species D in Figure 80). Reductive C–N bond formation with concomitant ligand dissociation could achieve the ligand C–N coupling product.
Figure 80.

Mechanistic studies of the Cu-promoted intramolecular C– C and C–N coupling reactions using copper complexes bearing redox-active ligands.305,306
Zhang and Xi described the synthesis, characterization, and reactivity of organocopper(III) spiro complexes (Figure 81).307 The biphenyl dilithio reagent was reacted with CuIBr·SMe2 to form an organocuprate(I) complex that upon reaction with a stoichiometric oxidant (e.g., p-benzoquinone) produced the spiro organocopper(III) in good yields. Remarkably, both copper(I) and copper(III) complexes could be characterized by X-ray diffraction analysis. The reactivity of the organocopper-(III) complex toward a series of electrophiles was also described (Figure 81, ii). Addition of HCl promoted the formation of the o-quaterphenyl product. Excess amounts of I2 produced a diiodo-o-quaterphenyl product. Addition of 1 equiv of [Me3S]-(BF4) afforded a novel arylcopper(I) species that could react further with other reagents such as MeI, I2, or PhCOCl to generate the corresponding coupling products.
Figure 81.

Synthesis, characterization, and reactivity of organocopper- (III) spiro species.307
3.2.1.2. C–H Activation Reactions Involving CuIII Species.
Ribas et al. reported one of the first examples of copper-promoted aryl C–H activation to generate stable organocopper-(III) complexes (Figure 82).308 It was observed that addition of 1 equiv of a copper(II) source to a triazamacrocylic ligand triggered a disproportionation reaction that produced equimolar amounts of an arylcopper(III) complex and the copper(I) complex. Later on, Ribas studied the C–H activation mechanism, which involved coordination of the copper(II) ion to the ligand scaffold to form an unstable three-center three-electron C–H⋯CuII species before disproportionation, which was proposed to occur via CPET with simultaneous C–H bond cleavage and copper(II) oxidation (note: kinetic analysis of the reaction suggested the formation of an L2CuII complex that acted as an electron and proton acceptor during C–H activation to generate LH+, L, and CuI).309
Figure 82.

Synthesis, characterization, and reactivity of organocopper- (III) species.308–314
With a similar ligand scaffold, Stahl and Ribas reported that addition of CuII halide sources promoted the formation of the corresponding arylcopper(III)-halide complexes via disproportionation (Figure 82, i).290 In a remarkable contribution, the same authors showed that these copper(III)-halide complexes could be generated stoichiometrically using acetone as a solvent and O2 as a sacrificial oxidant.310 Interestingly, it was described that the functionalization of the triazamacrocylcic ligand could be accomplished using catalytic amounts of copper(II) and O2 (Figure 82, ii). Ribas and Stahl have studied extensively the stoichiometric reactivity of the isolated copper(III) species with nucleophiles that leads to the formation of the C-nucleophile coupling products.311–314 Under catalytic amounts of copper and using MeOH as nucleophile, they observed the formation and decay of the arylcopper(III)-halide complex with concomitant formation of the aryl-OMe coupling product. It was proposed that the reaction of the triazamacrocylcic ligand with CuII produced the corresponding copper(II) complex (species B in Figure 82) that could react with the CuII present in solution to generate the arylcopper(III)-halide intermediate via disproportionation (species C). This organocopper(III) complex could react with nucleophiles to generate the Ar-Nuc products via reductive elimination (species D). It was suggested that O2 could be reduced to H2O by capturing electrons from CuI (regenerating CuII) and protons from the substrate and the nucleophile.
Wang have also studied the ability of copper to activate C–H bonds. In one of their first contributions, they described the reaction between a tetrazacalix[1]arene[3]pyridine ligand with CuII in a CHCl3:MeOH mixture (1:1) that under aerobic conditions formed an arylcopper(III) complex, which was characterized using X-ray diffraction analysis (Figure 83).315 Like in the Ribas organocopper system (described above),308 it was proposed that the copper(III) complex was formed via a disproportionation reaction. In this article, the authors also included the reactivity toward nucleophile sources such as halide sources (e.g., NEt4Cl) or carboxylate salts (e.g., PhCO2Na), which reacted at room temperature with the arylcopper(III) complex to generate the aryl-nucleophile coupling products (Figure 83, ii). In a series of research articles, the same research group also described the reactivity of the organocopper(III) species toward alcohols (e.g., EtOH in the presence of a strong base led to the formation of the aryl-OEt product) and alkynyllithium reagents (e.g., phenylethynyl lithium generated the C–C coupling product).316,317
Figure 83.

Synthesis, characterization, and reactivity of organocopper- (III) species.315–317
In a remarkable research article, the Wang research lab described the synthesis of a series of organometallic arylcopper-(II) and arylcopper(III) complexes and the reaction pathways by which these species are formed (Figure 84).318 The authors used the tetrazacalix[1]arene[3]pyridine system and derivatives in which the 4-position of the arene system had been substituted with electron-donating (R: MeO, Me) and electron-withdrawing groups (R: F, Cl, CO2Me, CN, and NO2). It was discovered that the arylcopper(II) and arylcopper(III) complexes could be isolated selectively with minor variations of the reaction conditions. If the reaction was carried out using 1.5 equiv of the CuII under their standard conditions (i.e., CH2Cl2:MeOH (1:1), air at room temperature), the organocopper(III) complex was obtained with excellent yields (99%). On the other hand, combining 1 equiv of the CuII source with 5 equiv of base (e.g., 2,4,6-trimethylpyridine or triethylamine) led to the isolation of the organocopper(II) complexes, which were characterized by different means, including X-ray diffraction analysis and EPR. It was also found that the isolated arylcopper(II) complexes could be quantitatively transformed to the corresponding arylcopper(III) analogues by addition of an equivalent of CuII (i.e., disproportionation to form CuI and the CuIII complex). The authors studied the electrochemistry of the arylcopper(II) complexes, which could be oxidized reversibly at redox potentials that depended on the aryl substituent: electron-donating groups such as MeO led to a lower E1/2 than the systems with electron-withdrawing groups (e.g., E1/2NO2 – E1/2MeO = 200 mV). Kinetic analysis of the formation of the arylcopper(II) complex bearing the different ligand scaffolds was also performed using stopped-flow technology. It was found that the generation of the arylcopper(II) complex was faster for the electron-donating ligand systems (e.g., 4-MeO-arene), and Hammett analysis supported the formation of a positive charge during the course of the reaction (ρ = −1.56). The reaction rates of the tetrazacalix[1]arene[3]pyridine system with the corresponding deuterium-substituted analogue were compared. The KIE observed (1.13) suggested that C–H bond cleavage was not involved in the rate-determining step of the arylcopper(II) formation. With all the mechanistic evidence in hand, it was proposed that the reaction between CuII and the ligand scaffold produced a tridentate copper(II) complex (species A in Figure 84) that could form a Wheland intermediate during the rate-determining step of the reaction (species B) before deprotonation and formation of the arylcopper(II) complex (species C).
Figure 84.

Synthesis, characterization, and reactivity of organocopper- (II) and organocopper(III) species.318
Wang have also reported that the tetrazacalix[1]arene[3]-pyridine system could be oxidized to the corresponding C–C coupling product using catalytic amounts of CuII, boronic acids, and oxygen as oxidant (Figure 85).319 Interestingly, the coupling products were obtained in remarkable yields using aryl, alky, and alkenyl boronic acids. In a follow-up article, the authors studied the mechanism of the C–C coupling reaction in detail (Figure 85, ii).320 Surprisingly, it was found that addition of arylboronic acids to the isolated arylcopper(III) complexes did not generate the coupling products. On the other hand, under the same reaction conditions the isolated arylcopper(II) complexes stoichiometrically formed the aryl–aryl coupling products. It was suggested that reaction of the arylcopper(II) complex with aryl boronic acid could generate an aryl-CuII-aryl′ intermediate (species B in Figure 85), which could then react with the initial arylcopper(II) complex to produce an aryl-CuIII-aryl′ complex (species C). In the last reaction step, reductive elimination could form the aryl–aryl′ coupling product.
Figure 85.

Mechanistic studies of the Cu-catalyzed functionalization of sp2 C–H bonds using a macrocylic ligand as substrate.319,320
In 2013, the Stahl research group reported a study of the reaction pathways by which copper(II) can promote aerobic C–H activation (Figure 86).321 A model substrate (N-(8-quinolinyl)benzamide) was used, which, under different reaction conditions, underwent various C–H oxidation processes. For example, addition of 2 equiv of CuII(OAc)2 under basic and aerobic conditions in MeOH led to the C–O functionalization of the ligand (directed C–H methoxylation). If CuCl2 was used as copper source and Na2CO3 as base, ligand chlorination was observed (directed C–H chlorination). On the other hand, if the functionalization reactions were carried out under acidic conditions and catalytic amounts of CuICl, the authors observed the selective chlorination of the quinoline directing group (nondirected C–H chlorination). The mechanism of both reactions (directed and nondirected C–H functionalizations) was studied in detail, including kinetic analysis (substrates with different substituents and KIE) and DFT calculations. The authors suggested that the CuII(OAc)2 promoted the methoxylation of the substrate–ligand via coordination of the copper source (species A in Figure 86) followed by deprotonation to form an organocopper(II) acetic acid complex (species B) that reacts with methanol to generate an arylcopper(II) methoxide intermediate (species C). Disproportionation could promote the formation of an organocopper(III) complex (species D) that, upon reductive elimination (triggered by methoxide coordination), could generate the aryl-OMe coupling product (species E). DFT calculations predicted that the r.d.s. of the reaction was the formation of the arylcopper(II) complex, which was in agreement with the KIE observed experimentally (KIE(exp.), 5.7; KIE(DFT), 4.9). The nondirected chlorination reaction was proposed to occur via a single-electron transfer mechanism (see SET reactivity in the section 3.2.2 below), which was initiated by coordination of CuCl2 (species F in Figure 86). Electron transfer from the quinoline ring to the copper center and disproportionation would produce a CuII complex bound by a radical-cation amidoquinoline ligand (species G). Chlorination of the radicalcation system was proposed to occur via chlorine-atom transfer from CuCl2 with the involvement of acetic acid (species H). Deprotonation of the Wheland intermediate (species I) would afford the chlorination product. The proposed SET mechanism was consistent with the kinetic isotope effect observed (KIE:1.0).
Figure 86.

Mechanistic study of the Cu-promoted functionalization of sp2 C–H bonds using N-(8-quinolinyl)benzamide as model substrate.321
Yu have also used amides as directing groups to functionalize sp2 C–H bonds (Figure 87).322 The ortho-trifluoromethylation of arenes was achieved using the Ruppert-Prakash reagent (TMS-CF3), stoichiometric amounts of CuII(OAc)2, and various additives (1.5 equiv of Ag2CO3, 4 equiv of KF, and 2 equiv of N-methylmorpholine-N-oxide). These oxidation conditions were used to functionalize various arene substrates, including heteroarene systems, with good yields and excellent selectivity (Figure 87, i). A primary KIE (3.2–3.5) in the intra- and intermolecular C–H/C–D competition experiments was observed. It was also observed that addition of TEMPO (radical scavenger) did not affect the final reaction yield, which suggested that the oxidation did not involve the formation of radical species. It was proposed that the reaction was initiated by a copper-mediated C–H activation step to form an organocopper(II) complex (species A in Figure 87) that upon disproportionation would generate an organocopper(III) intermediate (species B). Transmetalation would produce a CuIII–CF3 complex (species C in Figure 87) before reductive C–C bond formation.
Figure 87.

Cu-promoted trifluoromethylation of sp2 C–H bonds using directing groups.322
In a series of research articles, the same research group have used similar approaches (i.e., utilization of amide as directing groups) to functionalize sp2 C–H bonds, including hydroxylations (using stoichiometric amounts of copper and O2 as oxygen source),323 aminations,324 alkynylations,325 and arylations326 (using catalytic amounts of CuII and arylboron reagents). In some of these reports, the authors suggested a reaction mechanism similar to the one proposed for the Cu-mediated trifluoromethylation reaction, where the formation of organocopper(II) and organocopper(III) intermediates occur before reductive elimination (see Figure 87, ii).
3.2.1.3. Other Relevant Organometallic Reactions Involving CuIII Species.
In 2014, the Hartwig research group explored the use of copper to catalyze the amidation and imidation of unactivated alkanes using amides and imides as nitrogen sources and tBuOOtBu as oxidant (Figure 88).327 Catalytic amounts of CuII (2 mol %), (MeO)2phen (2.5 mol %), and tBuOOtBu (2 equiv) converted cyclohexane to the corresponding C–N coupling products using benzene as solvent (100 °C, 24 h). These reaction conditions were applied to functionalize other alkanes such as cis-1,4-dimethylcyclohexane, adamantane, 3-ethylpentane, etc. Strikingly, the amidation of cis-1,4-dimethylcyclohexane using benzamide led to the functionalization of the primary and secondary C–H bonds (i.e., no oxidation of the tertiary C–H bonds), which is unusual since the products derived from the cleavage of the tertiary C–H bonds (weaker C–H bond) are usually the major products in most of the oxidations involving 1e− oxidants (e.g., O-centered radicals) or 2e− oxidants (i.e., metal-oxo or metal-nitrene species). The copper(I) and copper(II) complexes containing imide and amide as ligands were isolated and characterized by X-ray diffraction analysis. With the use of these copper complexes, it was found that C–N functionalization was only accomplished in the presence of tBuOOtBu (i.e., the Cu complexes were not able to oxidize strong C–H bonds). The authors proposed a reaction mechanism initiated by the reaction between the LCuI-imidate complexes (species A in Figure 88) and tBuOOtBu that generates tert-butoxy radical (tBuO•) and the LCuII(NR2)2 complex (species B). The tBuO• could then abstract a H atom from the alkane to form an alkyl radical that could be trapped by the LCuII(NR2)2 complex to form an organocopper(III) intermediate (species C) that upon reductive elimination would afford the C–N coupling product.
Figure 88.

Cu-catalyzed amidation and imidation of unactivated alkanes using amides and imides as N-sources and tBuOOtBu as oxidant.327
Stahl research lab also used CuI and tBuOOtBu to functionalize sp3 C–H bonds (Figure 89).328 Arylboronic esters and benzylic substrates (e.g., toluene) were converted to the corresponding C(sp3)-aryl coupling products in the presence of catalytic amounts of a CuI source (3 mol %) and phenantholine (15 mol %) using toluene as substrate and solvent (note: for other substrates such as ethylbenzene, PhCl was used as solvent). It was observed that the selectivity of the reaction was similar to the one found in the Cu-catalyzed amidations reported by Hartwig (see above),327 in which the secondary and primary C–H bonds were more reactive than tertiary C–H bonds. A reaction mechanism based on the one proposed for the Kharasch-Sosnovsky C–O coupling reactions was proposed, in which the LCuI complex triggers the homolytic O–O bond cleavage of tBuOOtBu to produce tBuO• and LCuII–OtBu. Hydrogen atom abstraction from the alkane by the tert-butoxy radical could then produce a C-centered radical that then reacts with the LCuII–OtBu and the arylboronic ester to generate an aryl-alkyl-copper(III) intermediate (species C in Figure 89) before C–C bond formation.
Figure 89.

Cu-catalyzed C–C coupling reaction of benzylic C–H bonds and arylboronic esters using tBuOOtBu as oxidant.328
Stahl and Liu have recently reported the enantioselective cyanation of benzylic C–H bonds catalyzed by copper complexes (Figure 90).329 It was envisioned that hydrogen atom abstraction of these substrates could generate achiral benzylic radicals that could then be trapped by a chiral copper catalyst to form asymmetric Csp3-CN bonds (radical relay). It was found that using catalytic amounts of CuI(OAc) (10 mol %), bis(oxazoline) ligands (12 mol %) in the presence of excess amounts of a cyanide source (3 equiv of TMSCN) and N-fluorobenzenesulfonylimide (NSFI) as oxidant (1.5 equiv) allowed for the synthesis of a wide variety of benzylic nitriles with very good yields and enantioselectivities. It was proposed that the reaction between the CuI complex and the oxidant generated a reactive imidyl radical that could be the H atom abstractor in the reactions. The benzylic radical could then be trapped by the LCuII(CN)2 complex (formed in the reaction between LCuII–F and 2 equiv of TMSCN) to produce a benzyl-copper(III) intermediate (species D in Figure 90) before the enantioselective reductive elimination of the C–C coupling product.
Figure 90.

Cu-catalyzed enantioselective cyanation of benzylic substrates.329
Nicholas reported that simple CuI sources [e.g., [CuI(CH3CN)4](PF6)] can catalyze allylic aminations (Figure 91).330 Under excess amounts of olefin, mixing catalytic amounts of CuI (10 mol %) and phenyl-hydroxylamine (1 equiv) in 1,4-dioxane produced N-aryl-N-allylamines in good yields. It was found that the key intermediate [CuI(R-PhNO)3](PF6) could be isolated, which was characterized by X-ray diffraction analysis and various spectroscopic techniques (FT-IR, NMR, etc.). Kinetic analysis of the reaction between the isolated [CuI(PhNO)3]+ species and α-metylstyrene as substrate was carried out, and it was found that the rate of the reaction was first order in the concentration of CuI and substrate. Computational studies were conducted to gather information on the reaction mechanism. It was proposed that CuI reacted with the PhNHOH to generate PhNH2 and CuII. The resulting CuII could be reduced to CuI with concomitant oxidation of PhNHOH to PhNO. Reaction of 3 equiv of PhNO with CuI could form a stable [CuI(PhNO)3]+ complex (species A in Figure 91) that could coordinate the alkene substrate to produce a [(η2-alkene)Cu(PhNO)3]+ intermediate (species B). This putative complex would then be converted to an allyl-CuI species via allyl-H transfer followed by Cu-haptotropic shift (species C). Reductive elimination (species D) would generate an allyl hydroxylamine product that upon reduction with CuI would form the allyl amine product.
Figure 91.

Cu-catalyzed allylic aminations.330
In a recent report, Mankad described the synthesis of allylic alcohols via hydrocarbonylative coupling of alkynes with alkyl halides using CuI catalysts (Figure 92).331 Catalytic amounts of a copper(I) carbene complex (10 mol %) in the presence of CO (6 atm), a base (3 equiv of KOMe), and a silane reducing reagent (PHMS: polymetyhlhydrosiloxane, 6 equiv) allowed for the conversion of alkynes and alkyl bromide reagents to the corresponding allylic alcohols with remarkable yields and selectivity. Mechanistic studies indicated that the alkyl halide reacted via radical activation (no product formation was observed when TEMPO was added to the reaction mixture). It was also observed that stoichiometric coupling of the alkenyl copper intermediate with an acyl halide formed the α,β-unsaturated ketone product. It was also found that this α,β-unsaturated ketone could generate the allylic alcohol product, consistent with the ketone being an intermediate product. With all the information in hand, the authors proposed a reaction mechanism that is initiated by formation of an alkyl radical (from the reaction between alkyl bromide and a silyl radical) that could undergo carbonylation with CO to generate an acyl radical. Reaction of this acyl radical with alkyl halide could form an acyl halide product that could then react with the alkenyl copper complex via oxidative addition to generate a copper(III)-alkenyl-acyl complex (species B in Figure 92). Reductive elimination could produce the α,β-unsaturated ketone that upon Cu-catalyzed 1,2-reduction (species C and D) provides the allylic alcohol product.
Figure 92.

Cu-catalyzed synthesis of allylic alcohols via hydrocabonylative coupling of alkynes with alkyl halides.331
3.2.2. One-Electron Reactivity (Cun/Cun+1).
3.2.2.1. Ullman-Like Reactivity via Single Electron-Transfer (SET).
During the last years, Peters and Fu have revolutionized the field of copper-catalyzed cross-coupling reactions by developing synthetic methods that use light to trigger reactivity (i.e., photoredox catalysis). In one of their first reports, the authors studied the stoichiometric and catalytic cross-coupling C–N bond formation reaction using a CuI catalyst and light (Figure 93).332 The copper(I) complex utilized was bound by two phosphine ligands and a carbazolide moiety that, upon irradiation with a fluorescent light bulb and in the presence of aryl halides (Ph-I, Ph-Br, and Ph-Cl), produced the C–N coupling product with good yields (Figure 93, i). It was also observed that catalytic amounts of the copper-carbazolide complex could be used in the coupling reaction between Ph-I and lithium carbazolide. It was proposed that the reaction is initiated by the irradiation of the CuI complex to promote it to the excited state. This could trigger an electron transfer (SET) from the copper(I) complex to the aryl halide substrate to generate a radical ion pair (aryl-halide radical species/copper-(II) radical species). C-halide bond cleavage (species C in Figure 93) and radical rebound could then form the Ar-carbazolyl product. Alternatively, it was also proposed that SET could generate a CuII-halide complex (species B′ in Figure 93), which could react with the aryl radical to form the C–N bond.
Figure 93.

Cu-catalyzed C–N cross-coupling reaction with light.332
In one of their most prominent research articles, Peters and Fu reported the enantioselective C–N bond formation between racemic tertiary alkyl chlorides and amines (Figure 94).333 It was found that irradiation of a solution of the reactants (α-halocarbonyl compounds and carbazoles) with a blue light at low temperatures in the presence of a Brönsted base (1.5 equiv), catalytic amounts of CuCl (1 mol %), and a chiral phosphine ligand (1.2 mol %) provided the desired C–N coupling products with high yields and high enantiomeric excess (Figure 94, i). The authors proposed a reaction mechanism that is initiated by the irradiation of the CuI-nucleophile complex (species A in Figure 94), which is promoted to the excited state (species B) and reacts with the alkyl halide via single electron-transfer to generate an alkyl radical and a copper(II)-nucleophile adduct (species C). Inner-sphere C–N bond formation (explains the high enantiomeric excesses observed) forms the coupling product with concomitant regeneration of the CuI catalyst. A CuI complex bearing two phosphine ligands and carbazole was isolated and characterized. It was found that the isolated complex was stoichiometrically converted to the C–N product only under light, reaching ee’s similar to the ones observed in the catalytic reaction. It was hypothesized that this complex could be involved in the catalytic cycle (species A in Figure 94) or could be the precursor of a more active copper(I) complex bound by only one phosphine ligand and carbazole.
Figure 94.

Cu-catalyzed enantioselective C–N cross-coupling reaction with light.333
The same research groups have also used copper and light to catalyze Ullman-like cross-coupling reactions using a variety of nucleophiles and electrophiles.334–337 A photoredox copper(I) complex able to catalyze the synthesis of carbamate-protected amines using carbamate nucleophiles and unactivated secondary alkyl bromides was designed (Figure 95).338 The experimental findings suggested that the reaction proceeded via photo-activation of the copper(I) catalyst to generate an excited state (species B in Figure 95) that could trigger the formation of carbon-centered radicals by transferring an electron to the alkyl bromide with concomitant C–Br bond cleavage. The presence of another copper catalyst could allow nucleophile activation via formation of a CuI–Nuc complex (species D) that upon reaction with the copper(II) complex resulting from electrophile activation (species C) could form a copper(II)-nucleophile intermediate (species E), which then can trap alkyl radicals to afford the C–N coupling product.
Figure 95.

Cu-catalyzed synthesis of carbamate-protected amines using carbamate nucleophiles, unactivated secondary alkyl bromides and light.338
Hwang have also used light, in combination with copper, to synthesize ketones via oxidative coupling of phenols and terminal alkynes (Figure 96).339 Acetonitrile solutions of the organic substrates were combined with CuCl (5 mol %) and O2 and exposed to a blue light LED to afford the corresponding C–C oxygenation coupling products (coupling of the phenol in the para position) with remarkable yields and excellent regioselectivity. It was suggested that the reaction proceeded via activation of the alkyne substrate by the Cu complex to form a CuI-acetylide intermediate (species A in Figure 96) that upon visible light irradiation could generate the excited state of the CuI complex. This species could then react with O2 via a SET mechanism to generate superoxide and the corresponding CuII-acetylide adduct (species B in Figure 96). Simultaneously, the phenol substrate would be transformed to the corresponding 1,4-quinone in the presence of O2, CuCl, and light. A Paterno-Buchi type 2 + 2 reaction between the CuII-acetylide species and the 1,4-quinone product could generate a copper(II)-oxetane ring (species C) that upon rearrangement would produce a copper(II)-quinone/methide intermediate (species D). Homolytic CuII–C cleavage could generate an acyl radical intermediate that could react with molecular oxygen and protons to produce an unstable peracid product (species E in Figure 96), which then could undergo radical cleavage and aromatization to form the aryl ketone product. With the use of 1,4-quinone as a starting point, incorporation of labeled 18O was observed (experiments under an 18O2 atmosphere). It was also found that the use of the radical trap TEMPO precluded the formation of the aryl ketone product. EPR experiments supported the formation of a CuII-acetylide intermediate and superoxide.
Figure 96.

Cu-catalyzed oxidative coupling of phenols and terminal alkynes to synthesize ketones using light.339
In 2016, Nishikata reported that CuBr2 and CsF promoted the formation of C–F bonds from reaction with α-bromoamide substrates (Figure 97).340 It was found that these transformations could be carried out for a remarkable array of amides under catalytic amounts of copper(II) using N-containing ligands such as PMEDTA and excess amounts of the fluoride source. A series of control experiments revealed that (i) the C–F bond formation reaction was quenched in the presence of the radical trap BHT (2,6-Di-tert-butyl-4-methyl-phenol) and the formation of the C–O product derived from the C–O coupling between the amide substrate and the phenoxyl radical was observed; and (ii) substrates with tertiary amides were not converted to the corresponding C–F product, which suggested the amide was acting as directing group; and (iii) other less ionic fluoride sources such as KF or NaF did not lead to C–F bond formation. With this mechanistic information, it was proposed that in the first reaction step the copper(I) catalyst promoted C–Br cleavage to generate a copper(II)-Br complex and a carbon-centered radical (species A in Figure 97), which could be trapped by CuF2 (generated in situ from CuBr2 and CsF) via coordination to the amide group (species B). Subsequent intramolecular C–F bond formation yields the C–F product and the active CuI catalyst.
Figure 97.

Cu-promoted intramolecular fluorinations of α-bromoamide substrates.340
3.2.2.2. Direct C–H Activation Reactions Involving CuI/CuII or CuII/CuIII Cycles.
In a landmark research article, Yu described that catalytic amounts of CuII(OAc)2 or CuIICl2 could be used to carry out the ortho-functionalization of aryl C–H bonds using pyridyl as directing group (Figure 98).341 It was also described that using CuII(OAc)2 and anion sources allowed for the synthesis of the C-anion coupling products with good yields (Figure 98, ii). It was proposed that a single-electron transfer mechanism (SET) was initiated by the coordination of the CuII source (species A in Figure 98), which triggered electron transfer from the arene group to the CuII ion to form a CuI/radical-cation intermediate (species B) before anion transfer. The KIE observed (1.0) supported the proposed reaction mechanism, although other mechanisms involving the formation of an organocopper(III) intermediate could not be discarded.273 In a follow-up article, it was found that the 2-phenylpyridine systems were oxidatively dimerized at the ortho-position of the phenyl group in the presence of stoichiometric amounts of CuII(OAc)2 and I2.342 In this article, it was stated that Cu-mediated oxidation of 2-phenylpyridine could occur via SET or via electrophilic metalation followed by Ullman coupling.
Figure 98.

Cu-promoted ortho-functionalization of aryl C–H bonds using pyridyl as directing group.341
Baran has reported the Ritter-type C–H amination of sp3 C–H bonds catalyzed by copper and selectflour reagent (F-TEDA-PF6) using CH3CN as N source in the presence of a Lewis acid (Figure 99).343 It was found that substrates containing directing groups such as alcohols and ketones were aminated in the γ-position with remarkable yields and selectivity (Figure 99, i). More importantly, under similar reaction conditions, unactivated sp3 substrates (e.g., cyclohexane) were also converted to the C–N coupling products (Figure 99, ii). A reaction mechanism initiated by the oxidation of the copper(II) salt by F-TEDA+ via a SET mechanism was proposed that, in the presence of the substrate, could generate a copper(III) intermediate and HF. The resulting carbon-centered radical could then react with the copper(III) complex to regenerate the copper(II) catalyst and a carbocation center, which would be trapped by CH3CN to form a Ritter nitrilium intermediate (species B in Figure 99, iii). The authors observed a primary KIE in the competition experiments between cyclohexane and cyclohexane-D12, which suggests that the C–H cleavage step was rate-limiting. It was also found that addition of TEMPO (radical trap) inhibited the formation of the C–N coupling product, in agreement with the formation of radical species during the amination reaction. A recent research article by Zare studied the mechanism of this amination reaction in detail using UV–vis spectroscopy and online ESI-MS spectrometry, which allowed the characterizion and analysis of the evolution of the reaction intermediates over time.344 The results obtained were consistent with the mechanism proposed previously by Baran in which the copper(II) catalyst reacts with the oxidant to generate a radical carbocation intermediate (species A in Figure 99, iii) that could act as H atom abstractor.
Figure 99.

Cu-catalyzed Ritter-type C–H amination of sp3 C–H bonds.343
Yu reported the copper-catalyzed bromination of sp3 C–H bonds distal to functional groups (Figure 100).345 This transformation required catalytic amounts of copper(II) (10 mol %), phenanthroline (10 mol %), NBS (3.0 equiv), and TMSN3 (3.0 equiv), which enabled the bromination of various amides (γ and δ-functionalization) and amines (δ and ε-functionalization) with good yields and good selectivities. It was proposed that the reaction between NBS and TMSN3 produces BrN3, which undergoes homolytic cleavage to generate azidyl radical, which can then abstract an H atom from the N–H bond of the substrate (amine or amide) to form HN3 and the N-centered radical product (species B in Figure 100). The Br• radical generated during BrN3 cleavage could then be trapped by the CuII catalyst to form a CuIII–Br species that reacts with the substrate C-centered radical (produced via intramolecular H atom abstraction by the N-centered radical species) to form the C–Br oxidation product.
Figure 100.

Cu-catalyzed bromination of sp3 C–H bonds distal to functional groups.345
Su used catalytic amounts of copper(II) acetate, bipyridine, and stoichiometric amounts of TEMPO radical to promote the β-functionalization of saturated ketones by sequential dehydrogenation and subsequent conjugate addition with nucleophiles (Figure 101).346 This protocol was used to functionalize a wide range of ketones using amines, alcohols, and 1,3-dicarboyl compounds as nucleophiles with good yields and excellent selectivity. It was found that under the reaction conditions (CuII, TEMPO) and without the presence of nucleophile, the dehydrogenation product was formed. It was also observed that addition of CBr4 (radical trap) formed the α-bromosubstituted product, which supported that the dehydrogenation step involved the formation of a C-centered radical. Kinetic analysis of the reaction showed that the rates were first-order dependent on the concentration of catalyst and substrate and zero-order dependent on the concentration of TEMPO radical. It was also observed that deuteration of the α position of the substrate led to a primary kinetic isotope effect (KIE: 5.3), but when the β position of the unsaturated ketone was deuterated, similar reaction rates were observed for the undeuterated substrate (KIE: 1.02). With all that information in hand, it was proposed that the copper(II) catalyst reacted with the substrate to form a metal-enolate complex (species A in Figure 101) that upon homolysis would generate a copper(I) and a ketone-radical (species B). This radical species would be trapped by TEMPO to produce an α-TEMPO-substituted ketone intermediate (species C in Figure 101). This intermediate was independently synthesized and characterized, and it could be converted to the saturated ketone under the reaction conditions. The α-TEMPO-substituted ketone was also detected during the oxidation of the saturated substrate. Fast elimination of TEMPO-H, most likely promoted by another TEMPO molecule, could produce the enone synthon that is able to react with the nucleophiles present in solution to form the coupling product. Regeneration of the copper(II) catalyst could proceed via reduction of TEMPO-H (generated during the oxidation of the substrate) to form 2,2,6,6-tetramethylpiperidine and water.
Figure 101.

Cu-catalyzed β-functionalization of saturated ketones.346
3.2.2.3. Cu-Catalyzed C–O Coupling Reactions Involving the Generation of O-Centered Radicals.
In 2012, the Warren research lab reported that copper(I) complexes were able to catalyze etherification of strong C–H bond using dialkylper-oxides as oxidants (Figure 102). It was described that cyclohexane (substrate and solvent) was converted to the Cy-OtBu product using catalytic amounts of the diketiminato CuI catalysts at room temperature (up to 60% yields).347 A detailed mechanistic analysis of the reaction (including kinetic analysis, isolation of Cu intermediates, analysis of the organic products derived from the decomposition of the ROOR species and DFT calculations) suggested that the copper(I) complex triggered the homolytic O–O bond cleavage of the tBuOOtBu oxidant to generate an O-centered radical (tBuO•) and the copper(II)-alkoxide complex (isolated and characterized by X-ray diffraction analysis). The tBuO• species would abstract an H atom from the substrate to produce a C-centered radical that could be rapidly trapped by the CuII–OtBu to form a product-bound CuI complex. In a recent article, the same research group reported that the Cu-catalyzed etherification of sp3 C–H bonds could also be accomplished using acyl protected phenols as O-sources.348 Catalytic amounts of a diketiminato CuI complex (5 mol %) and 1.2 equiv of tBuOOtBu were used to transform the C–H substrates (10 equiv, neat) and the acyl phenol to the corresponding C–O coupling products. The oxidation of isobutylbenzene formed a mixture of products, in agreement with the poor selectivity of the proposed active H atom abstractor (tBuO•). Like in their previous report, it was proposed that the LCuI complex reacted with tBuOOtBu to form tBuO• and LCuII(OtBu). A transesterification reaction could replace the alkoxide ligand to produce the LCuII-phenolate adducts (synthesized and characterized independently), which could then trap the C-centered radicals (generated in the reaction between tBuO• and the C–H substrates) and result in the C-OAr oxidation products.
Figure 102.

Cu-catalyzed etherification of alkanes using tBuOOtBu as oxidant.347
In 2014, Hartwig used copper(I) sources to catalyze the dehydrogenative carboxylation of unactivated alkanes to form allylic esters (Figure 103).349 It was observed that warming up benzene solutions containing catalytic amounts of CuI (5 mol %), carboxylic acids (1 equiv), C–H substrates (20 equiv), and tBuOOtBu (3 equiv) generated the corresponding allylic esters with good yields. A well-defined CuI complex bearing the BPI anionic ligand was used to study the reaction mechanism (Figure 103, ii). The [(BPI)CuII(O2CPh)] complex was synthesized (an analogue was also characterized by X-ray diffraction analysis), and it was used as a stoichiometric reagent to functionalize C–H bonds. It was found that this copper(II)-benzoate complex was only able to oxidize C–H bonds in the presence of tBuOOtBu and that when used as a catalyst in the oxidation of cyclohexane and tBuOOtBu without carboxylic acid, cyclohexene was obtained as an oxidation product. The authors proposed a reaction mechanism that is initiated by the reaction of the CuI complex (species A in Figure 103) and tBuOOtBu in the presence of the carboxylic acid to give the CuII-benzoate complex (species B) and tBuO•, which could react with the C–H substrates to generate C-centered radicals. The alkyl radicals would be trapped by the CuII-benzoate complex via SET and deprotonation (species C) to generate the dehydrogenation product. This alkene could then undergo a second H atom abstraction to produce an allylic radical that could react with the CuII-benzoate to form the C–O coupling product. A KIE of 2.8 (C6H12 vs C6D12) was observed, which indicated that C–H bond cleavage occurred during the turnover limiting step.
Figure 103.

Cu-catalyzed dehydrogenative carboxylation of unactivated alkanes to form allylic esters using tBuOOtBu as oxidant.349
In a series of research articles, Li reported the functionalization of tertiary amines using copper(I), nucleophiles, and tBuOOH as oxidant (Figure 104).350–352 Klussmann, Thiel, and Bietti studied the reaction mechanism of these copper-catalyzed oxidative coupling transformations using N-aryl tetrahydroisoquinolines as substrates and without adding nucleophiles.353 It was observed that under these reaction conditions, the substrates were converted to the peroxide product derived from the C–O coupling between the tetrahydroisoquinolines and tBuOOH. Kinetic analysis of the reaction using N-aryl amines with different substituents in the fourth position of the aryl ring supported a HAT mechanism (the Hammett plot was compared with the one obtained when CumO• was used as oxidant). A primary KIE suggested that the C–H cleavage occurred during the rate-determining step. A reaction mechanism was proposed in which CuIBr would undergo oxidation to the corresponding CuIIBr(OH) intermediate with concomitant formation of tBuO•. The CuIBr catalyst would be regenerated via reduction of the tBuOOH by the CuIIBr(OH) that could then produce tBuOO• and water. The generated O-centered radicals (i.e., tBuO• and tBuOO•) could abstract an H atom from the substrate to produce a C-centered radical intermediate (species B in Figure 104) that would then be trapped by tBuOO• to yield the C–O coupling product (species C). This peroxy product was proposed to be in equilibrium with the iminium ion (species D), which, in the presence of nucleophiles, could form the C-Nuc coupling product (species E).
Figure 104.

Mechanistic studies of the Cu-catalyzed functionalization of N-aryl tetrahydroisoquinolines.350–353
The Ball research lab have reported the remote sp3 C–H chlorination of alkyl hydroperoxides catalyzed by CuICl (10 mol %), PMDTA ligand (N,N,N′,N″,N″-pentamethyldiethylenetri-amine, 12 mol %), acetic acid (4 equiv), and NH4Cl as chlorine source, which gave chlorinated alcohols with good yields and notable selectivity (Figure 105).354 The authors proposed a reaction mechanism initiated by the CuICl catalyst (species B in Figure 105) which triggers the homolytic cleavage of the O–O bond to generate a CuII(OH)(Cl) complex and an alkoxy radical (species C) that abstracts an H atom intramolecularly (1,5-hydrogen atom abstraction). This radical would be trapped by the CuII(OH)(Cl) complex to form the chlorination product.
Figure 105.

Cu-catalyzed remote sp3 C–H chlorination of alkyl hydroperoxides.354
In 2015, Zhu reported the synthesis of functionalized epoxides via Cu-promoted alkylative epoxidation of allylic alcohols with alkyl nitriles (Figure 106).355 Substoichiometric amounts of CuII(OAc)2 (0.5 equiv), phenanthroline (0.5 equiv), and excess amounts of tBuOOtBu (4 equiv) were used to transform allylic alcohols (tertiary and secondary) to the corresponding epoxide using CH3CN as a C and N source. It was proposed that the reaction was initiated via Cu-promoted homolytic O–O bond cleavage of the peroxide that generates LCuII(OAc) (or LCuII(OtBu)), which then reacts with acetonitrile to form an organocopper(II) complex (species D in Figure 106). Homolytic Cu–C cleavage would produce a cyanomethyl radical (species E) that could add across the double bond to generate a radical intermediate (species G), which could then be trapped by the CuII complex to produce an organocopper(III) intermediate before reductive C–O bond formation. Alternative reaction pathways were also suggested, which involved the generation of a second organocopper(II) complex (species F) and a carbocation intermediate (species H) before C–O bond formation.
Figure 106.

Cu-promoted synthesis of functionalized epoxides.355
3.2.2.4. Other Organometallic Reactions Involving CuI/CuII or CuII/CuIII Cycles.
During the last few years, Q. Wang has used copper to functionalize alkenes using hypervalent iodine or hydroxylamines as oxidants.356–360 In a prominent research article, the intramolecular aminoazidination of alkenes using catalytic amounts of copper(I) acetate and excess amounts of azidoiodinane (1.5 equiv) was described (Figure 107, i).359 Mechanistic studies suggested the formation of alkyl radical species (trapped using TEMPO radical). On the basis of their results, it was proposed that the oxidation of the substrate could occur via two plausible pathways. The first one involved an intramolecular aminocupration of the substrate via activation of the alkene by CuII (species B in Figure 107) that could form two organocopper(II) intermediates (species C), which could either form the exo (5-membered ring) and endo (6-membered ring) oxidation products. It was also suggested that the reaction could occur via formation of copper(III)-azido intermediate (species A′) that would coordinate the substrate to produce two organocopper(III) species before reductive C–N bond formation. In both proposals, it was proposed that the organocopper species were in equilibrium with copper-alkyl radical intermediates, which could be trapped by the radical scavenger TEMPO.D.
Figure 107.

Cu-catalyzed intramolecluar functionalization of alkenes.359
H. Wang has recently reported the ortho-amination of phenols catalyzed by CuII(OAc)2 using R1R2NCH2BF3 as nitrogen sources (Figure 108).361 The reaction was found to be very selective for a wide array of phenolic substrates and reached remarkable yields (up to 98%). It was proposed that the reaction between phenol and CuII produced a CuII-phenolate species that could react with the N source to form an aminophenol-CuII adduct (species B in Figure 108). Oxidation of the trifluoroborate by another copper(II) center could generate a boron radical species that, upon homolytic B–C cleavage, would form a C-centered radical (species D), which could then intramolecularly attack the phenol substrate via a six-membered cyclic transition state. The resulting CuI complex (species E) could release the ketone synthon, which, upon tautomerization, would form the final C–C coupling product. The copper(II) catalyst could then be regenerated by the reaction of the resulting CuI complex and O2.
Figure 108.

Cu-catalyzed ortho-amination of phenols.361
3.2.3. Reactivity without Change in the Oxidation State of Copper (Cun/Cun).
Organometallic oxidative transformations in which the copper ion is proposed to maintain the same oxidation state during the catalytic cycle are rare. Instead, for other Cu-catalyzed organic reactions such as Cu–H hydrogenations and Cu-catalyzed cycloadditions (e.g., click chemistry), the copper ion is believed to stay in one oxidation state.36,276 Bertrand has described the dehydrogenative borylation of terminal alkynes using copper(I) complexes bearing cyclic alkylamino carbene ligands (Figure 109).362 It was observed that using benzene as solvent, NEt3 as base (5 mol %), and catalytic amounts of the copper complex (2.5 mol %) resulted in the corresponding C–B coupling products with excellent yields and remarkable selectivity. On the basis of previous research findings,363 kinetic studies, and labeling experiments, the authors proposed a mechanism that was initiated by the reaction between the copper catalyst and the substrate to produce a dicopper(I) intermediate (species B in Figure 109). This dicopper(I) species was proposed to react with pinacolborane to generate the C–B product in one step via σ-bond metathesis in the transition state (species C). The resulting copper(I)-hydride complex (species D) could then react with the protons generated in the first reaction step to form H2.
Figure 109.

Cu-catalyzed dehydrogenative borylation of terminal alkynes.362
In 2015, Lei described the cross-coupling reaction between N-methoxyarylamides and allylic substrates using catalytic amounts of copper(II) triflate and tBuOOtBu as oxidant (Figure 110).364 EPR spectroscopy, DFT calculations, and high-resolution ESI-MS analysis were carried out in order to gather some insight into the reaction mechanism. It was proposed that the copper(II) source coordinates the N-methoxyarylamide substrate (species B in Figure 110) before H atom abstraction of the bound substrate by the tBuO• radical (generated by homolysis of tBuOOtBu) to produce the copper(II) complex bound by the oxidized substrate (species C). The C-centered radical, generated in the reaction between tBuO• and the allylic C–H substrates, could react with the CuII-radical complex (species C) to form the product-bound cupric adduct (species D) which releases the C–N coupling product and the copper(II) ion before initiating the catalytic cycle again.
Figure 110.

Cu-catalyzed C–N coupling reactions using tBuOOtBu as oxidant.364
3.3. Organic Transformations Involving Organocopper-O2 Species
During the past decade, several research reports have proposed the formation of organometallic copper/O2 during the catalytic oxidative functionalization of organic molecules. However, it is important to note that most of the proposed intermediates throughout this section have yet to be trapped and characterized. Chiba research lab has pioneered this field, carrying out oxygenase-like and oxidase-like syntheses of useful organic molecules. One of their first contributions described the Cu-catalyzed C–C cleavage of α-azido carbonyl compounds to generate the corresponding nitriles and carboxylic acids (Figure 111).365 This methodology was applied in the synthesis of a wide variety of alkyl and aryl nitriles with remarkable yields (Figure 111, ii). A reaction mechanism was suggested, in which the copper(II) salt reacted with the α-azido substrate to generate an iminyl copper(II) intermediate (species B in Figure 111), which could react with dioxgen to form a putative peroxycopper-(III) species (species C). This peroxy complex then releases the nitrile product via homolytic C–C cleavage to produce an acylperoxocopper(II) complex (species D) that, upon protonation, regenerates the CuII catalyst and forms the carboxylic acid product via an unknown O–O cleavage event.
Figure 111.

Cu-catalyzed sysnthesis of nitriles via C–C cleavage of α- azido carbonyl compounds.365
Chiba used a similar approach to synthesize azaspirocyclo-hexadienones by oxidation of α-azido-N-arylamides with copper and O2 (Figure 112).366 Copper(II) acetate catalyzed this transformation for a varied range of substrates in the presence of stoichiometric amounts of a base (Figure 112, ii). It was suggested that the copper(II) catalyst reacted with the α-azido-N-arylamides susbtrates to generate a copper(II)-iminyl complex (species B in Figure 112) that reacts with O2 to produce a copper(III)-peroxo intermediate (species C). It was proposed that an intramolecuar imino-cupration would form an ipso C–N and a para Cu–C bond (species D) that, upon isomerization, could generate a copper(II)-peroxydiene intermediate (species E), which then eliminates the oxygenation products.
Figure 112.

Cu-catalyzed synthesis of azaspirocyclohexadienones.366
In a similar fashion, Chiba’s research group reported the Cu-catalyzed spirocyclization of biaryl-N-H-imines (Figure 113).367 Biaryl-2-carbonitrile were used as starting materials that, upon reaction with Grignard reagants, generated biaryl N–H imine intermediates which reacted with Cu and O2 to form the oxidation products via intramolecular 1,4-aminooxygenation. It was proposed that an intermediate iminyl-Cu-superoxo was formed (similar to species C in Figure 112) before sp2 oxygenation. It was observed that addition of nitrogen ligands (phen) and water led to remarkable reaction yields for a wide array of biaryl-2-carbonitrile substrates and various Grignard reagents (Figure 113, ii). The scope of this reaction was expanded (i.e., use of N–H imines to direct Cu oxygenations) to synthesize azaheterocycles and to oxidize benzylic C–H bonds.368,369
Figure 113.

Cu-catalyzed spirocyclization of biaryl-N-H-imines.367
Chiba has also explored the use of N-alkylamidines to direct C–H oxygenation and cyclization for the synthesis dihydrooxazoles (Figure 114).370 It was found that a mixture of CuBr· SMe2 (20 mol %) and 2,2′-bipyridine (20 mol %) in DMSO-PhCF3 (5:1) under O2 (1 atm) triggered the oxygenation/cyclization reaction for a wide range of N-alkylamidines with good yields. It was proposed that the amidine substrates reacted with CuII (generated via oxidation of the CuI source) to form a CuII-substrate adduct (species B in Figure 114) that undergoes 1e− oxidation to produce CuI and a 1,3-diazaallyl radical (species C). 1,5-H radical shift could form a tertiary carbon radical (species D), which could then be trapped by O2 to generate a C-superoxo intermediate (species E). This radical species was proposed to react with CuI to form a CuII-alkoxide intermediate (species F) in a Fenton-like fragmentation. Intramolecular nucleophilic attack followed by elimination of ammonia (species G in Figure 114) produces the dihydrooxazole product. The authors also showed that the dihydrooxazole products could be transformed into vicinal amino alcohols via reduction (LiAlH4, AlCl3) or by acid-mediated hydrolysis (Figure 114, iv).
Figure 114.

Cu-catalyzed synthesis of dihydrooxazoles.370
The Jiao research group has also contributed significantly to the field of copper-catalyzed aerobic organic transformations.371 One of their first reports described the synthesis of α-ketoamides from amines, terminal alkynes, and dioxygen using catalytic amounts of Cu (Figure 115, i).372 An array of terminal alkynes (mainly phenylic) and aromatic amines were coupled with good reaction yields (Figure 115, ii). It was proposed that copper(II) reacted with the amine to generate an amino-CuII species (species A in Figure 115) that could couple with the terminal alkyne to form an organocopper(II) intermediate (species B). Homolytic cleavage of the Cu–C bond could produce CuI, which could then react with O2 to regenerate the CuII catalyst and form a C-centered radical (species C). This radical species could then be trapped by O2 to generate a superoxide radical species (species D in Figure 115). Intramolecular cycloaddition would form an aminyl radical intermediate (species E), which could undergo hydrogen atom abstraction by reacting with TEMPO to produce a cyclic peroxide intermediate (species F) which, upon fragmentation, would yield the final dioxygenation α-ketoamide product.
Figure 115.

Cu-catalyzed synthesis of α-ketoamides.372
The same research group reported another protocol for the synthesis of α-ketoamides that improved the previous methodology (see above) in terms of substrate scope and efficiency. In this new approach, copper catalyzed the aerobic oxidative coupling of aryl acetaldehydes with anilines (Figure 116, i).373 The optimized oxidative conditions were applied to produce an array of α-ketoamides with different substituents reaching remarkable yields (Figure 116, ii). A reaction mechanism was proposed that involved initial formation of an imine substrate (species A in Figure 116), which, in the presence of pyridine and O2, could generate an organic superoxide intermediate (species B) that, upon reacting with CuII, would produce a putative copper(III)-alkylperoxide complex (species C). Intramolecular cyclization (species D in Figure 116) could be followed by hydrogen atom abstraction to form peroxide intermediate (species E) that could undergo fragmentation to achieve the reaction product. Oxidation reactions with 18O2 generated the labeled dioxygenation product with 80% incorporation of 18O. In a follow-up article, a third method for the synthesis of α-ketoamides was descibed based on oxidative cross-coupling of amines and α-carbonylaldehydes using copper(I) bromide as catalyst and oxygen as terminal oxidant.374
Figure 116.

Cu-catalyzed synthesis of α-ketoamides via oxidative coupling of aryl acetaldehydes and anilines.373
Using a similar approach, Kumar reported the synthesis of primary α-ketoamides by copper-mediated aerobic oxidation of benzylimidates (Figure 117).375 The substrates were converted to the corresponding oxygenation products using stoichiometric amounts of Cu(OAc)2 (2 equiv) and O2 in DMF (80 °C, 6 h). For most of the substrates, yields higher than 50% were observed (Figure 117, ii). The authors envisioned a mechanistic scenario in which the benzylimidate substrate was coordinated by copper(II) to generate an aminyl-copper(II) complex (species A in Figure 117). This CuII-aminyl species could react with O2 to produce a substrate-centered superoxide radical intermediate (species B) or could sequentially form a copper(III)-superoxide (species C) and copper(II)-peroxide complex (species D) before formation of a 6-membered transition state (species E). Intramolecular addition to the imine group (species F in Figure 117) followed by H atom abstraction assisted by copper or O2 (species G) would lead to peroxide fragmentation to form the final dioxygenation product. Addition of radical scavengers such as TEMPO led to a substantial decrease of the reaction yields, which suggested that the reaction involved the formation of radical species.
Figure 117.

Cu-mediated the synthesis of primary α-ketoamides.375
In 2012, Jiao also described the synthesis of oxazoles via aerobic oxidative dehydrogenative annulation and oxygenation of aldehydes and amines using O2 as oxidant and stoichiometric amounts of copper (Figure 118).376 An array of oxazoline synthons with different substituents were synthesized reaching moderate-to-good yields (Figure 118, (ii). The reaction was proposed to proceed via initial formation of an imine (species A in Figure 118) that, in the presence of a base and copper, would be oxidized to form an alkylsuperoxide intermediate (species B). Copper could trigger the 1,5-H atom abstraction and subsequent O–O cleavage to produce a diradical intermediate (species C in Figure 118) that could then undergo intramolecular radical coupling to form a 4,5′-dihydrooxazole ring (species D). This ring could be easily oxidized under the reaction conditions to form the final aromatic oxazole product (species E). Similar reaction conditions have been employed by Jiao to synthesize esters from ketones via C–C(O) cleavage followed by coupling with alcohols and to produce α-ketoesters by esterification of 1,3-diones.377,378
Figure 118.

Cu-promoted synthesis of oxazoles.376
Li reported the Cu-catalyzed intramolecular 6-exo-trig cyclization of 1,6-enynes to synthesize 1,4-napthoquinones (Figure 119).379 CuCl2, Ce(SO4)2, O2, and H2O were combined with the substrates to obtain the oxygenation products with moderate-to-good yields (Figure 119, ii). It was suggested that substrate coordination to CuII (species A in Figure 119) could trigger a Wacker oxidation of the C–C double bond with H2O as oxygen source (species B). Subsequent cyclization could produce an organocopper(II) intermediate (species C) that could then react with O2 to generate an organocopper(III)-superoxide complex (species D). Reductive elimination (species E) followed by alcohol oxidation would deliver the oxidation product (species F) and regenerate the active copper(II) catalyst. Oxidation experiments in the presence of H218O confirmed that one of the oxygen atoms in the napthoquinone product originated from water and the other from dioxygen.
Figure 119.

Cu-catalyzed synthesis of 1,4-napthoquinones.379
Li research lab has also used Cu to catalyze the C–C cleavage of α-aminocarbonyl compounds to formylamides and carboxylic acids (Figure 120).380 A reusable heterogeneous copper on iron catalyst was utilized, which, in combination with TEMPO and O2, generated the C–C cleavage products with moderate-to-good yields (Figure 120, i). The authors suggested a reaction mechanism that was initiated by the 1H+/1e− oxidation of the α-aminocarbonyl substrate (involving Cu, O2, and TEMPO) to generate an imine intermediate (species B in Figure 120) that could react with a putative Cu2O2 core to form an unusual dicopper complex (species C). C–C cleavage (species D) followed by reductive O–O cleavage generates the oxidation products.
Figure 120.

Cu-catalyzed C–C cleavage of α-aminocarbonyl substrates.380
Zhang and Zhu discovered that copper(II) catalyzed the formation of formyl-substituted aromatic N-heterocycles via intramolecular dehydrogenative aminooxygenation (Figure 121).381 The reaction conditions were optimized (use of 20 mol% of CuII(hfacac)2, hfacac: hexafluoroacteylacetonate; O2: 1 atm; in DMF at 105 °C) to synthesize substituted imidazo[1,2-a]pyridine-3-carbaldehydes with good yields (Figure 121, ii). A reaction mechanism was proposed in which the copper(II) source coordinated the pyridine substrate to form an imino-copper(II) complex (species A in Figure 121) that could react with O2 to generate a putative CuIII-superoxide intermediate (species B). This CuIII(O2•−) species could undergo insertion into the C–C double bond to form a putative organocopper-(III)-superoxide complex (species C). Isomerization of the copper-superoxide complex could produce a copper(II)-alkylperoxide intermediate (species D) that upon elimination could generate CuII–OH and an aldehyde product (species E), which could then be spontaneously aromatized to the final oxidation product. The proposed mechanism was supported by labeling experiments (use of 18O2 led to the formation of the oxygenation product with 95% 18O incorporation) and by using the radical scavenger TEMPO (no formation of C-TEMPO products was observed, which suggested that C-centered radicals were not formed during the reaction).
Figure 121.

Cu-catalyzed synthesis of formyl-substituted aromatic N-heterocycles.381
The synthesis of 4-carbonyl-quinolines by oxidative cyclization of enynes using CuII as catalyst and O2 as oxidant was reported by Liang (Figure 122).382 Intramolecular cyclization of a wide range of substrates was accomplished using CuCl2 (10 mol %), phenanthroline as ligand (20 mol %), and 2 equiv of DABCO under 1 atm of O2 (DMF,100 °C). A reaction mechanism was proposed in which the substrate reacted with a putative CuIII-superoxide species to form a CuIII-superoxosubstrate adduct (species A in Figure 122), which could be in equilibrium with an organocopper(III)-superoxide intermediate (species B). Carbocupration could produce a vinyl copper(III)-superoxo intermediate (species C) that could then isomerize to a copper(II)-alkylperoxo complex (species D) before under-going deprotonative O–O bond cleavage to yield the oxidation product. The mechanistic proposal was supported by labeling experiments (incorporation of 18O into the products when 18O2 was used as oxidant) and by ESI-MS (a mass matching that of species C in Figure 122 was detected). Interestingly, the reaction conditions were optimized to carry out this transformation in an intermolecular fashion by reacting substituted 2-(phenylethynyl)anilines and but-2-eyenedioate substrates with CuICl (15 mol %) and O2 (1 atm) in DMA at 100 °C.
Figure 122.

Cu-catalyzed synthesis of 4-carbonyl-quinolines.382
Lu and Wang reported the synthesis of 3-functionalized indoles in a three-component reaction that combined indoles, sulfonyl azides, and alkynes, using copper(I) bromide as a catalyst and O2 as an oxidant (Figure 123).383 Interestingly, this transformation was accomplished under mild conditions (room temperature and 1 atm of O2) for an extensive variety of substrates (17 indole derivatives were synthesized and characterized). A cascade mechanism with two Cu-catalyzed independent cycles was proposed. In the first cycle, a click reaction between the azide, the alkyne, and the CuI ion generates a triazole-copper complex (species A in Figure 123), which upon Dimorth rearrangement and concomitant N2 release could form a ketenimine intermediate (species B). Nucleophilic attack of this species on the electron-rich sp2 carbon in the indole substrate closes the first catalytic cycle and generates the C–C coupling product (species C). In the presence of a base, this coupling product could react with CuII-OH (generated in situ under the reaction conditions) to produce an amidocopper(II) complex (species D in Figure 123) that in the presence of O2 could generate a putative copper(III)-superoxide adduct (species E). Intramolecular electron transfer and subsequent isomerization could form a putative organocopper(III)-super-oxo intermediate (species F) that could isomerize to a copper(II)-alkylperoxide complex (species G) before elimination of CuII-OH and formation of the oxygenation product. As predicted by the proposed reaction mechanism, when the reaction was carried out under anaerobic conditions (i.e., Ar atmosphere, no O2), the indole-imine coupling products were obtained (species C in Figure 123).
Figure 123.

Cu-catalyzed synthesis of 3-functionalized indoles.383
In 2016, the Sun research lab reported the synthesis of benzoimidazo[1,2-a]imidazolone in a one-pot three-component coupling cascade reaction using 2-aminobenzimidazoles, aldehydes, and terminal alkynes in the presence of catalytic amounts of copper and employing O2 as an oxidant (Figure 124).384 A reaction mechanism was proposed which was initiated by the “in situ” formation of the imine from the 2-aminobenzylimidazole (amine) and the aldehyde substrate (species A in Figure 124). In the presence of CuI and base, this imine could react with the alkyne to afford a propargylic amine CuI adduct (species B). Copper would trigger a 5-exo dig cyclization to produce an organocopper(I) intermediate (species C in Figure 124), which could react with O2 to generate sequentially an organocopper(II)-superoxide complex (species D) and a copper(I)-alkylperoxide intermediate (species E). In the presence of a base, this species could undergo homolytic O–O bond cleavage to achieve the oxygenation product and CuI hydroxide.
Figure 124.

Cu-catalyzed synthesis of benzoimidazo[1,2-a]-imidazolone.384
In a follow-up article, the same authors prepared and isolated the benzimidazole Schiff base before exposing it to the alkyne substrate, copper, and O2, which allowed preparation of benzimidazole-linked pyrroles via intramolecular 6-endo-dig cyclization of the propargylamine intermediate.385
Very recently, Yin and Newhouse reported the vinylogous aerobic oxidation of unsaturated compounds (Figure 125).386 It was found that catalytic amounts of copper(II) trifate (5 mol %), stoichiometric amounts of a strong base (1.2 equiv of TMG: tetramethylguanidine), and a reductant (PPh3, 1.5 equiv) led to the hydroxylation of wide variety of γ-β and α-β-unsaturated esters using air as oxidant under mild conditions (THF, r.t., 10–24 h). Interestingly, the report also included the hydroxylation of unsaturated aldehydes, ketones, nitriles, amides, and sulfones under similar reaction conditions (Figure 125, ii). On the basis of labeling experiments and other experimental observations, it was proposed that the reaction proceeded via coordination of the TMG base to the copper(II) ion to generate a copper(II) complex (species A in Figure 125), which could trap the substrate to form a copper(II) dienolate intermediate (species B′). This complex would be in equilibrium with an organocopper(II) intermediate (species B) that could be oxidized by O2 to produce a copper(II)-peroxo complex (species D), which, upon reduction by PPh3, could form the hydroxylation product.
Figure 125.

Cu-catalyzed vinylogous aerobic oxidation of unsaturated compounds.386
A novel methodology for the functionalization of C–H bonds has been recently introduced by Hirano and Uchimaya, in which the hydroxylation and amination of sp2 C–H bonds was accomplished using amide directing groups, stoichiometric amounts of a cuprate base, and excess amounts of oxidant (Figure 126).387 The ortho-hydroxylation and ortho-amination products of aromatic N,N-diisopropyl amides were synthesized with very good yields. DFT calculations were carried out to understand the reaction mechanism, which was proposed to be initiated by the reaction between the cuprate base and the substrate (via deprotonation) to form an arylcopper(I) complex (species A in Figure 126). Oxidant coordination and displacement of the amide ligand (species B in Figure 126) could trigger the oxidative addition of the oxidant to generate an organocopper(III) intermediate (species C in Figure 126) before reductive C–O (or C–N) bond formation.
Figure 126.

Cu-promoted hydroxylation and amination of sp2 C–H bonds using directing groups.387
4. SUMMARY AND FUTURE PERSPECTIVES
In this review, we illustrated the high versatility of copper to carry out transformations useful in organic synthesis. Inspired by metalloenzymes that use bioavailable nontoxic metals such as copper and green oxidants (i.e., O2 and H2O2), current research efforts aim to develop selective, cheap, and environmentally benign oxidation and oxygenation synthetic protocols. For example, White’s research lab has shown that nonheme Fe complexes can catalyze the regioselective hydroxylation of C–H bonds with H2O2.181,182 Despite the established nature of this approach, most of the oxidations in academia and industry still rely on the use of stoichiometric oxidants such as NaOCl, OsO4, or KMnO4.85 So why are these green protocols not widely employed in organic synthesis? The first reason could be the cost of implementing these technologies, such as the optimization of big-scale conditions. Most research articles report these functionalization reactions at a μmol/mmol scale, and they employ expensive ligand scaffolds. The use of 5–10 mol % catalysts bearing ligand systems that are synthesized in a multistep fashion increases the overall cost, environmental impact and practicality of the method. A second factor to consider is the fact that researchers have been devoting most of their efforts on expanding the substrate scope of these reactions. Rather than focusing on the functionalization of similar substrates with comparable C–H bonds, we should dedicate our resources to target specific substrates and amplify their product scope (e.g., functionalization of the same C–H bond with different organic substitutents), which is more appealing for late stage derivatization for pharmaceutical applications.388,389 Fortunately, academics are actively collaborating with chemical companies to develop synthetic protocols that utilize first row metals to synthesize useful organic molecules.162,390 With the numerous examples of dehydrogenations and hydroxylations promoted by bioinspired metal-O2 species,183,391 the promising findings on the use of redox-active ligands in 3d metal catalysis,392,393 which can provide reactivity features characteristic of 4d and 5d metals, and the new approaches to perform selective functionalization via formation and control of C-centered radicals394 (i.e., metal-catalyzed radical relay), we foresee that the implementation of these synthetic routes will increase in the coming years.
With new examples appearing on a daily basis in the literature, most of the mechanisms by which Cu-promoted tranformations take place are still unknown. Spectroscopic characterization of relevant reaction intermediates such as the Cu/O2 species responsible for the oxidation of strong C–H bonds like methane, kinetic analysis, oxidation of substrates designed to serve as mechanistic probes, the effect of the addition of radical traps, and computational analysis are part of the box of tools that will be increasingly employed to investigate the reaction pathways of these Cu-promoted processes. A deeper knowledge of these mechanisms must allow for designing cheaper, greener, safer, and more practical synthetic methods based on Cu catalysis with higher efficiency and divergent selectivity. With all of these considerations in mind, it is certain that copper will be in the avante garde of metal catalysis in the 21st century.
ACKNOWLEDGMENTS
We thank the Robert A. Welch Foundation (Grant N-1900) and the National Institutes of Health (NIH Award R15GM128078) for financial support.
Biography
Biographies
Rachel Trammell studied Biology at Tyler Junior College and transferred to the University of Texas at Tyler where she graduated with her B.S. in Chemistry in 2016 and her A.S. in Biology in 2015. She joined the Garcia-Bosch Lab at Southern Methodist University in 2016 as a Ph.D. student to work on sp2 and sp3 C–H bond functionalizations with copper.
Khashayar Rajabimoghadam was born in 1989 in Arak, Iran. He received his B.S. and M.S. at Shiraz University under the supervision of Prof. Nasser Iranpoor and Prof. Habib Firouzabadi. He is currently a Ph.D. student at Southern Methodist University in the Garcia-Bosch Lab. His research in the group is focused on green catalysis using first row metal complexes bearing redox-active ligands containing H-bonding donors.
Isaac Garcia-Bosch was born in Catalonia in 1984. After obtaining his B.Sc. in Chemistry in 2006 at the University of Girona, he continued on to graduate studies under the supervision of Prof. Miquel Costas and Prof. Xavi Ribas at the University of Girona (Ph.D. granted in 2011). In 2012, he was awarded a Marie Curie IOF Fellowship to pursue his career as a postdoc under the supervision of Prof. Kenneth D. Karlin at The Johns Hopkins University (Baltimore). In 2015, he joined the faculty at Southern Methodist University (Dallas) where he is currently an assistant professor (Harold A. Jeskey Endowed Chair in Inorganic Chemistry). The Garcia-Bosch lab research focuses on the use of bioinspired metal catalysts (Fe, Mn, Co, Ni, and Cu) for synthetic organic transformations.
Footnotes
Notes
The authors declare no competing financial interest.
REFERENCES
- (1).Egorova KS; Ananikov VP Which Metals are Green for Catalysis? Comparison of the Toxicities of Ni, Cu, Fe, Pd, Pt, Rh, and Au Salts. Angew. Chem., Int. Ed 2016, 55, 12150–12162. [DOI] [PubMed] [Google Scholar]
- (2).Bertini I; Gray HB; Lippard SJ; Valentine JS Bioinorganic Chemistry; University Science Books, 1994. [Google Scholar]
- (3).U. S. Geological Survey. https://minerals.usgs.gov/minerals/ (accessed May 2018).
- (4).Egorova KS; Ananikov VP Toxicity of Metal Compounds: Knowledge and Myths. Organometallics 2017, 36, 4071–4090. [Google Scholar]
- (5).Festa RA; Thiele DJ Copper: An essential metal in biology. Curr. Biol 2011, 21, R877–R883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Solomon EI; Heppner DE; Johnston EM; Ginsbach JW; Cirera J; Qayyum M; Kieber-Emmons MT; Kjaergaard CH; Hadt RG; Tian L Copper Active Sites in Biology. Chem. Rev 2014, 114, 3659–3853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Solomon EI; Hadt RG Recent advances in understanding blue copper proteins. Coord. Chem. Rev 2011, 255, 774–789. [Google Scholar]
- (8).Peterson RL; Galaleldeen A; Villarreal J; Taylor AB; Cabelli DE; Hart PJ; Culotta VC The Phylogeny and Active Site Design of Eukaryotic Copper-only Superoxide Dismutases. J. Biol. Chem 2016, 291, 20911–20923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Brady DC; Crowe MS; Turski ML; Hobbs GA; Yao X; Chaikuad A; Knapp S; Xiao K; Campbell SL; Thiele DJ; Counter CM Copper is required for oncogenic BRAF signalling and tumorigenesis. Nature 2014, 509, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Turski ML; Thiele DJ New Roles for Copper Metabolism in Cell Proliferation, Signaling, and Disease. J. Biol. Chem 2009, 284, 717–721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Hao Z; Lou H; Zhu R; Zhu J; Zhang D; Zhao BS; Zeng S; Chen X; Chan J; He C; et al. The multiple antibiotic resistance regulator MarR is a copper sensor in Escherichia coli. Nat. Chem. Biol 2014, 10, 21–28. [DOI] [PubMed] [Google Scholar]
- (12).Burkitt MJA Critical Overview of the Chemistry of Copper-Dependent Low Density Lipoprotein Oxidation: Roles of Lipid Hydroperoxides, α-Tocopherol, Thiols, and Ceruloplasmin. Arch. Biochem. Biophys 2001, 394, 117–135. [DOI] [PubMed] [Google Scholar]
- (13).Paterson BM; Donnelly PS Copper complexes of bis(thiosemicarbazones): from chemotherapeutics to diagnostic and therapeutic radiopharmaceuticals. Chem. Soc. Rev 2011, 40, 3005–3018. [DOI] [PubMed] [Google Scholar]
- (14).Santini C; Pellei M; Gandin V; Porchia M; Tisato F; Marzano C Advances in Copper Complexes as Anticancer Agents. Chem. Rev 2014, 114, 815–862. [DOI] [PubMed] [Google Scholar]
- (15).Hyman LM; Franz KJ A cell-permeable fluorescent prochelator responds to hydrogen peroxide and metal ions by decreasing fluorescence. Inorg. Chim. Acta 2012, 380, 125–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Hyman LM; Franz KJ Probing oxidative stress: Small molecule fluorescent sensors of metal ions, reactive oxygen species, and thiols. Coord. Chem. Rev 2012, 256, 2333–2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Rosenthal J; Lippard SJ Direct Detection of Nitroxyl in Aqueous Solution Using a Tripodal Copper(II) BODIPY Complex. J. Am. Chem. Soc 2010, 132, 5536–5537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Xie D; King TL; Banerjee A; Kohli V; Que EL Exploiting Copper Redox for 19F Magnetic Resonance-Based Detection of Cellular Hypoxia. J. Am. Chem. Soc 2016, 138, 2937–2940. [DOI] [PubMed] [Google Scholar]
- (19).Soon CPW; Donnelly PS; Turner BJ; Hung LW; Crouch PJ; Sherratt NA; Tan J-L; Lim NK-H; Lam L; Bica L; et al. Diacetylbis(N(4)-methylthiosemicarbazonato) Copper(II) (CuII(atsm)) Protects against Peroxynitrite-induced Nitrosative Damage and Prolongs Survival in Amyotrophic Lateral Sclerosis Mouse Model. J. Biol. Chem 2011, 286, 44035–44044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Liu JJ; Diaz DE; Quist DA; Karlin KD Copper(I)-Dioxygen Adducts and Copper Enzyme Mechanisms. Isr. J. Chem 2016, 56, 738–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Elwell CE; Gagnon NL; Neisen BD; Dhar D; Spaeth AD; Yee GM; Tolman WB Copper–Oxygen Complexes Revisited: Structures, Spectroscopy, and Reactivity. Chem. Rev 2017, 117, 2059–2107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Citek C; Herres-Pawlis S; Stack TDP Low Temperature Syntheses and Reactivity of Cu2O2 Active-Site Models. Acc. Chem. Res 2015, 48, 2424–2433. [DOI] [PubMed] [Google Scholar]
- (23).Yoshikawa S; Shimada A Reaction Mechanism of Cytochrome c Oxidase. Chem. Rev 2015, 115, 1936–1989. [DOI] [PubMed] [Google Scholar]
- (24).Castello PR; David PS; McClure T; Crook Z; Poyton RO Mitochondrial cytochrome oxidase produces nitric oxide under hypoxic conditions: Implications for oxygen sensing and hypoxic signaling in eukaryotes. Cell Metab 2006, 3, 277–287. [DOI] [PubMed] [Google Scholar]
- (25).Hematian S; Garcia-Bosch I; Karlin KD Synthetic Heme/Copper Assemblies: Toward an Understanding of Cytochrome c Oxidase Interactions with Dioxygen and Nitrogen Oxides. Acc. Chem. Res 2015, 48, 2462–2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Siegbahn PEM The catalytic cycle of catechol oxidase. JBIC, J. Biol. Inorg. Chem 2004, 9, 577–590. [DOI] [PubMed] [Google Scholar]
- (27).Klinman JP The Copper-Enzyme Family of Dopamine beta-Monooxygenase and Peptidylglycine alpha-Hydroxylating Monoox-ygenase: Resolving the Chemical Pathway for Substrate Hydroxylation.J. Biol. Chem 2006, 281, 3013–3016. [DOI] [PubMed] [Google Scholar]
- (28).Phillips CM; Beeson WT; Cate JH; Marletta MA Cellobiose Dehydrogenase and a Copper-Dependent Polysaccharide Monooxygenase Potentiate Cellulose Degradation by Neurospora crassa. ACS Chem. Biol 2011, 6, 1399–1406. [DOI] [PubMed] [Google Scholar]
- (29).Balasubramanian R; Smith SM; Rawat S; Yatsunyk LA; Stemmler TL; Rosenzweig AC Oxidation of methane by a biological dicopper centre. Nature 2010, 465, 115–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Tsarevsky NV; Matyjaszewski K Green” Atom Transfer Radical Polymerization: From Process Design to Preparation of Well-Defined Environmentally Friendly Polymeric Materials. Chem. Rev 2007, 107, 2270–2299. [DOI] [PubMed] [Google Scholar]
- (31).Matyjaszewski K; Tsarevsky NV Macromolecular Engineering by Atom Transfer Radical Polymerization. J. Am. Chem. Soc 2014, 136, 6513–6533. [DOI] [PubMed] [Google Scholar]
- (32).Hay AS; Blanchard HS; Endres GF; Eustance JW Polymerization by oxidative coupling. J. Am. Chem. Soc 1959, 81, 6335–6336. [Google Scholar]
- (33).Feng X; Jiang K; Fan S; Kanan MW A Direct Grain-Boundary-Activity Correlation for CO Electroreduction on Cu Nanoparticles. ACS Cent. Sci 2016, 2, 169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Keith JA; Nielsen RJ; Oxgaard J; Goddard WA Unraveling the Wacker Oxidation Mechanisms. J. Am. Chem. Soc 2007, 129, 12342–12343. [DOI] [PubMed] [Google Scholar]
- (35).Snyder BER; Bols ML; Schoonheydt RA; Sels BF; Solomon EI Iron and Copper Active Sites in Zeolites and Their Correlation to Metalloenzymes. Chem. Rev 2018, 118, 2718–2768. [DOI] [PubMed] [Google Scholar]
- (36).Worrell BT; Malik JA; Fokin VV Direct Evidence of a Dinuclear Copper Intermediate in Cu(I)-Catalyzed Azide-Alkyne Cycloadditions. Science 2013, 340, 457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Hay AS Oxidative Coupling of Acetylenes. II1. J. Org. Chem 1962, 27, 3320–3321. [Google Scholar]
- (38).McCann SD; Stahl SS Copper-Catalyzed Aerobic Oxidations of Organic Molecules: Pathways for Two-Electron Oxidation with a Four-Electron Oxidant and a One-Electron Redox-Active Catalyst. Acc. Chem. Res 2015, 48, 1756–1766. [DOI] [PubMed] [Google Scholar]
- (39).Casitas A; Ribas X The role of organometallic copper(iii) complexes in homogeneous catalysis. Chem. Sci 2013, 4, 2301–2318. [Google Scholar]
- (40).Evans RW; Zbieg JR; Zhu S; Li W; MacMillan DWC Simple Catalytic Mechanism for the Direct Coupling of α-Carbonyls with Functionalized Amines: A One-Step Synthesis of Plavix. J. Am. Chem. Soc 2013, 135, 16074–16077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhu S; MacMillan DWC Enantioselective Copper-Catalyzed Construction of Aryl Pyrroloindolines via an Arylation–Cyclization Cascade. J. Am. Chem. Soc 2012, 134, 10815–10818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Quist DA; Diaz DE; Liu JJ; Karlin KD Activation of dioxygen by copper metalloproteins and insights from model complexes. JBIC, J. Biol. Inorg. Chem 2017, 22, 253–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Allen SE; Walvoord RR; Padilla-Salinas R; Kozlowski MC Aerobic Copper-Catalyzed Organic Reactions. Chem. Rev 2013, 113, 6234–6458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Koepke SJ; Light KM; VanNatta PE; Wiley KM; Kieber-Emmons MT Electrocatalytic Water Oxidation by a Homogeneous Copper Catalyst Disfavors Single-Site Mechanisms. J. Am. Chem. Soc 2017, 139, 8586–8600. [DOI] [PubMed] [Google Scholar]
- (45).Das D; Lee Y-M; Ohkubo K; Nam W; Karlin KD; Fukuzumi S Acid-Induced Mechanism Change and Overpotential Decrease in Dioxygen Reduction Catalysis with a Dinuclear Copper Complex. J. Am. Chem. Soc 2013, 135, 4018–4026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Shi S-L; Wong ZL; Buchwald SL Copper-catalysed enantioselective stereodivergent synthesis of amino alcohols. Nature 2016, 532, 353–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Yang Y; Perry IB; Lu G; Liu P; Buchwald SL Copper-catalyzed asymmetric addition of olefin-derived nucleophiles to ketones. Science 2016, 353, 144–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Yang Y; Shi S-L; Niu D; Liu P; Buchwald SL Catalytic asymmetric hydroamination of unactivated internal olefins to aliphatic amines. Science 2015, 349, 62–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Semenza GL Life with Oxygen. Science 2007, 318, 62–64. [DOI] [PubMed] [Google Scholar]
- (50).Biological Inorganic Chemistry: Structure and Reactivity; Bertini I., Gray HB, Stiefel EI, Valentine JS, Eds.; University Science Books: Sausalito, CA, 2007. [Google Scholar]
- (51).Lewis EA; Tolman WB Reactivity of Dioxygen-Copper Systems. Chem. Rev 2004, 104, 1047–1076. [DOI] [PubMed] [Google Scholar]
- (52).Mirica LM; Ottenwaelder X; Stack TDP Structure and Spectroscopy of Copper-Dioxygen Complexes. Chem. Rev 2004, 104, 1013–1045. [DOI] [PubMed] [Google Scholar]
- (53).Jacobson RR; Tyeklár Z; Karlin KD; Liu S; Zubieta J; Farooq A A Cu2-O2 Complex. Crystal Structure and Characterization of a Reversible Dioxygen Binding System. J. Am. Chem. Soc 1988, 110, 3690–3692. [Google Scholar]
- (54).Kitajima N; Fujisawa K; Moro-oka Y; Toriumi K μ-η2:η2-Peroxo Binuclear Copper Complex, [Cu(HB(3,5-iPr2Pz)3)]2(O2). J. Am. Chem. Soc 1989, 111, 8975–8976. [Google Scholar]
- (55).Matoba Y; Kumagai T; Yamamoto A; Yoshitsu H; Sugiyama M Crystallographic Evidence That the Dinuclear Copper Center of Tyrosinase Is Flexible during Catalysis. J. Biol. Chem 2006, 281, 8981–8990. [DOI] [PubMed] [Google Scholar]
- (56).Halfen JA; Mahapatra S; Wilkinson EC; Kaderli S; Young VG Jr.; Que L Jr.; Zuberbühler AD; Tolman WB Reversible Cleavage and Formation of the Dioxygen O-O Bond Within a Dicopper Complex. Science 1996, 271, 1397–1400. [DOI] [PubMed] [Google Scholar]
- (57).Mirica LM; Vance M; Rudd DJ; Hedman B; Hodgson KO; Solomon EI; Stack TDP Tyrosinase Reactivity in a Model Complex: An Alternative Hydroxylation Mechanism. Science 2005, 308, 1890–1892. [DOI] [PubMed] [Google Scholar]
- (58).Schatz M; Raab V; Foxon SP; Brehm G; Schneider S; Reiher M; Holthausen MC; Sundermeyer J; Schindler S Combined spectroscopic and theoretical evidence for a persistent end-on copper superoxo complex. Angew. Chem., Int. Ed 2004, 43, 4360–4363. [DOI] [PubMed] [Google Scholar]
- (59).Maiti D; Fry HC; Woertink JS; Vance MA; Solomon EI; Karlin KDA 1:1 Copper-Dioxygen Adduct is an End-on Bound Superoxo Copper(II) Complex which Undergoes Oxygenation Reactions with Phenols. J. Am. Chem. Soc 2007, 129, 264–265. [DOI] [PubMed] [Google Scholar]
- (60).Peterson RL; Himes RA; Kotani H; Suenobu T; Tian L; Siegler MA; Solomon EI; Fukuzumi S; Karlin KD Cupric Superoxo-Mediated Intermolecular C–H Activation Chemistry. J. Am. Chem. Soc 2011, 133, 1702–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Lee JY; Peterson RL; Ohkubo K; Garcia-Bosch I; Himes RA; Woertink J; Moore CD; Solomon EI; Fukuzumi S; Karlin KD Mechanistic Insights into the Oxidation of Substituted Phenols via Hydrogen Atom Abstraction by a Cupric–Superoxo Complex. J. Am. Chem. Soc 2014, 136, 9925–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Kunishita A; Kubo M; Sugimoto H; Ogura T; Sato K; Takui T; Itoh S Mononuclear Copper(II)–Superoxo Complexes that Mimic the Structure and Reactivity of the Active Centers of PHM and DβM. J. Am. Chem. Soc 2009, 131, 2788–2789. [DOI] [PubMed] [Google Scholar]
- (63).Würtele C; Gaoutchenova E; Harms K; Holthausen MC; Sundermeyer J; Schindler S Crystallographic Characterization of a Synthetic 1:1 End-On Copper Dioxygen Adduct Complex. Angew. Chem., Int. Ed 2006, 45, 3867–3869. [DOI] [PubMed] [Google Scholar]
- (64).Ross MO; Rosenzweig AC A tale of two methane monooxygenases. JBIC, J. Biol. Inorg. Chem 2017, 22, 307–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Donoghue PJ; Tehranchi J; Cramer CJ; Sarangi R; Solomon EI; Tolman WB Rapid C–H Bond Activation by a Monocopper(III)–Hydroxide Complex. J. Am. Chem. Soc 2011, 133, 17602–17605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Crespo A; Marti MA; Roitberg AE; Amzel LM; Estrin DA The Catalytic Mechanism of Peptidylglycine alpha-Hydroxylating Monooxygenase Investigated by Computer Simulation. J. Am. Chem. Soc 2006, 128, 12817–12828. [DOI] [PubMed] [Google Scholar]
- (67).Maiti D; Lee D-H; Gaoutchenova K; Würtele C; Holthausen MC; Narducci Sarjeant AA; Sundermeyer J; Schindler S; Karlin KD Reactions of a Copper(II) Superoxo Complex Lead to C–H and O–H Substrate Oxygenation: Modeling Copper-Monooxygenase C–H Hydroxylation. Angew. Chem., Int. Ed 2008, 47, 82–85. [DOI] [PubMed] [Google Scholar]
- (68).Kunishita A; Ishimaru H; Nakashima S; Ogura T; Itoh S Reactivity of Mononuclear Alkylperoxo Copper(II) Complex. O-O Bond Cleavage and C-H Bond Activation. J. Am. Chem. Soc 2008, 130, 4244–4245. [DOI] [PubMed] [Google Scholar]
- (69).Fujisawa K; Tanaka M; Moro-oka Y; Kitajima N A Monomeric Side-On Superoxocopper(II) Complex: Cu(O2)(HB-(3-tBu-5-iPrpz)3). J. Am. Chem. Soc 1994, 116, 12079–12080. [Google Scholar]
- (70).Aboelella NW; Lewis EA; Reynolds AM; Brennessel WW; Cramer CJ; Tolman WB Snapshots of Dioxygen Activation by Copper: The Structure of a 1:1 Cu/O2 Adduct and Its Use in Syntheses of Asymmetric Bis(μ-oxo) Complexes. J. Am. Chem. Soc 2002, 124, 10660–10661. [DOI] [PubMed] [Google Scholar]
- (71).Neisen BD; Gagnon NL; Dhar D; Spaeth AD; Tolman WB Formally Copper(III)–Alkylperoxo Complexes as Models of Possible Intermediates in Monooxygenase Enzymes. J. Am. Chem. Soc 2017, 139, 10220–10223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Kindermann N; Günes C-J; Dechert S; Meyer F Hydrogen Atom Abstraction Thermodynamics of a μ−1,2-Superoxo Dicopper(II) Complex. J. Am. Chem. Soc 2017, 139, 9831–9834. [DOI] [PubMed] [Google Scholar]
- (73).Saracini C; Ohkubo K; Suenobu T; Meyer GJ; Karlin KD; Fukuzumi S Laser-Induced Dynamics of Peroxodicopper(II) Complexes Vary with the Ligand Architecture. One-Photon Two-Electron O2 Ejection and Formation of Mixed-Valent CuICuII–Superoxide Intermediates. J. Am. Chem. Soc 2015, 137, 15865–15874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Ali G; VanNatta PE; Ramirez DA; Light KM; Kieber-Emmons MT Thermodynamics of a μ-oxo Dicopper(II) Complex for Hydrogen Atom Abstraction. J. Am. Chem. Soc 2017, 139, 18448–18451. [DOI] [PubMed] [Google Scholar]
- (75).Cao R; Saracini C; Ginsbach JW; Kieber-Emmons MT; Siegler MA; Solomon EI; Fukuzumi S; Karlin KD Peroxo and Superoxo Moieties Bound to Copper Ion: Electron-Transfer Equilibrium with a Small Reorganization Energy. J. Am. Chem. Soc 2016, 138, 7055–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Mahroof-Tahir M; Murthy NN; Karlin KD; Blackburn NJ; Shaikh SN; Zubieta J New Thermally Stable Hydroperoxo-and Peroxo-Copper Complexes. Inorg. Chem 1992, 31, 3001–3003. [Google Scholar]
- (77).Karlin KD; Ghosh P; Cruse RW; Farooq A; Gultneh Y; Jacobson RR; Blackburn NJ; Strange RW; Zubieta J Dioxygen-Copper Reactivity: Generation, Characterization and Reactivity of a Hydroperoxo-Dicopper(II) Complex. J. Am. Chem. Soc 1988, 110, 6769–6780. [Google Scholar]
- (78).Tolman WB Binding and Activation of N2O at Transition-Metal Centers: Recent Mechanistic Insights. Angew. Chem., Int. Ed 2010, 49, 1018–1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Reim J; Werner R; Haase W; Krebs B From Tetranuclear μ4-Oxo to μ4-Peroxocopper(II) Complexes. Chem. - Eur. J 1998, 4, 289–298. [Google Scholar]
- (80).Cole AP; Root DE; Mukherjee P; Solomon EI; Stack TDP A Trinuclear Intermediate in the Copper-Mediated Reduction of O2: Four Electrons from Three Coppers. Science 1996, 273, 1848–1850. [DOI] [PubMed] [Google Scholar]
- (81).Semmelhack MF; Chou CS; Cortes DA Nitroxyl-mediated electrooxidation of alcohols to aldehydes and ketones. J. Am. Chem. Soc 1983, 105, 4492–4494. [Google Scholar]
- (82).Zhang X; Xu Z; Si W; Oniwa K; Bao M; Yamamoto Y; Jin T Synthesis of extended polycyclic aromatic hydrocarbons by oxidative tandem spirocyclization and 1,2-aryl migration. Nat. Commun 2017, 8, 15073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Jaiswal G; Landge VG; Jagadeesan D; Balaraman E Iron-based nanocatalyst for the acceptorless dehydrogenation reactions. Nat. Commun 2017, 8, 2147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Sarhan AAO; Bolm C Iron(III) chloride in oxidative C–C coupling reactions. Chem. Soc. Rev 2009, 38, 2730–2744. [DOI] [PubMed] [Google Scholar]
- (85).Caron S; Dugger RW; Ruggeri SG; Ragan JA; Ripin DHB Large-Scale Oxidations in the Pharmaceutical Industry. Chem. Rev 2006, 106, 2943–2989. [DOI] [PubMed] [Google Scholar]
- (86).Quintanar L; Stoj C; Taylor A; Hart P; Kosman D; Solomon E Shall We Dance? How A Multicopper Oxidase Chooses Its Electron Transfer Partner. Acc. Chem. Res 2007, 40, 445–452. [DOI] [PubMed] [Google Scholar]
- (87).Turner NJ Enantioselective Oxidation of C–O and C–N Bonds Using Oxidases. Chem. Rev 2011, 111, 4073–4087. [DOI] [PubMed] [Google Scholar]
- (88).Whittaker JW Free radical catalysis by galactose oxidase. Chem. Rev 2003, 103, 2347–2363. [DOI] [PubMed] [Google Scholar]
- (89).Lee Y-K; Whittaker MM; Whittaker JW The Electronic Structure of the Cys-Tyr· Free Radical in Galactose Oxidase Determined by EPR Spectroscopy. Biochemistry 2008, 47, 6637–6649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Wang Y; Stack TDP Galactose Oxidase Model Complexes: Catalytic Reactivities. J. Am. Chem. Soc 1996, 118, 13097–13098. [Google Scholar]
- (91).Wang Y; DuBois JL; Hedman B; Hodgson KO; Stack TDP Catalytic Galactose Oxidase Models: Biomimetic Cu(II)-Phenoxyl-Radical Reactivity. Science 1998, 279, 537–540. [DOI] [PubMed] [Google Scholar]
- (92).Chaudhuri P; Hess M; Flörke U; Wieghardt K From structural models of galactose oxidase to homogeneous catalysis: efficient aerobic oxidation of alcohols. Angew. Chem., Int. Ed 1998, 37, 2217–2220. [DOI] [PubMed] [Google Scholar]
- (93).Chaudhuri P; Hess H; Weyhermüller T; Wieghardt K Aerobic Oxidation of Primary Alcohols by a New Mononuclear CuII-Radical Catalyst. Angew. Chem., Int. Ed 1999, 38, 1095–1098. [DOI] [PubMed] [Google Scholar]
- (94).Chaudhuri P; Hess M; Mueller J; Hildenbrand K; Bill E; Weyhermueller T; Wieghardt K Aerobic Oxidation of Primary Alcohols (Including Methanol) by Copper(II)-and Zinc(II)-Phenoxyl Radical Catalysts. J. Am. Chem. Soc 1999, 121, 9599–9610. [Google Scholar]
- (95).Markó IE; Giles PR; Tsukazaki M; Brown SM; Urch CJ Copper-Catalyzed Oxidation of Alcohols to Aldehydes and Ketones: An Efficient, Aerobic Alternative. Science 1996, 274, 2044–2046. [DOI] [PubMed] [Google Scholar]
- (96).Markó IE; Tsukazaki M; Giles PR; Brown SM; Urch CJ Anaerobic Copper-Catalyzed Oxidation of Alcohols to Aldehydes and Ketones. Angew. Chem., Int. Ed. Engl 1997, 36, 2208–2210. [Google Scholar]
- (97).Semmelhack MF; Schmid CR; Cortes DA; Chou CS Oxidation of alcohols to aldehydes with oxygen and cupric ion, mediated by nitrosonium ion. J. Am. Chem. Soc 1984, 106, 3374–3376. [Google Scholar]
- (98).Ragagnin G; Betzemeier B; Quici S; Knochel P Copper-catalysed aerobic oxidation of alcohols using fluorous biphasic catalysis. Tetrahedron 2002, 58, 3985–3991. [Google Scholar]
- (99).Jiang N; Ragauskas AJ Cu(II)-Catalyzed Selective Aerobic Oxidation of Alcohols under Mild Conditions. J. Org. Chem 2006, 71, 7087–7090. [DOI] [PubMed] [Google Scholar]
- (100).Markó IE; Gautier A; Dumeunier R; Doda K; Philippart F; Brown SM; Urch CJ Efficient, Copper-Catalyzed, Aerobic Oxidation of Primary Alcohols. Angew. Chem., Int. Ed 2004, 43, 1588–1591. [DOI] [PubMed] [Google Scholar]
- (101).Hoover JM; Stahl SS Highly Practical Copper(I)/TEMPO Catalyst System for Chemoselective Aerobic Oxidation of Primary Alcohols. J. Am. Chem. Soc 2011, 133, 16901–16910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Hoover JM; Ryland BL; Stahl SS Mechanism of Copper(I)/TEMPO-Catalyzed Aerobic Alcohol Oxidation. J. Am. Chem. Soc 2013, 135, 2357–2367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Ryland BL; McCann SD; Brunold TC; Stahl SS Mechanism of Alcohol Oxidation Mediated by Copper(II) and Nitroxyl Radicals. J. Am. Chem. Soc 2014, 136, 12166–12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Steves JE; Stahl SS Copper(I)/ABNO-Catalyzed Aerobic Alcohol Oxidation: Alleviating Steric and Electronic Constraints of Cu/TEMPO Catalyst Systems. J. Am. Chem. Soc 2013, 135, 15742–15745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Walroth RC; Miles KC; Lukens JT; MacMillan SN; Stahl SS; Lancaster KM Electronic Structural Analysis of Copper(II)–TEMPO/ABNO Complexes Provides Evidence for Copper(I)–Oxoammonium Character. J. Am. Chem. Soc 2017, 139, 13507–13517. [DOI] [PubMed] [Google Scholar]
- (106).Xu B; Lumb J-P; Arndtsen BA A TEMPO-Free Copper-Catalyzed Aerobic Oxidation of Alcohols. Angew. Chem., Int. Ed 2015, 54, 4208–4211. [DOI] [PubMed] [Google Scholar]
- (107).McCann SD; Lumb J-P; Arndtsen BA; Stahl SS Second-Order Biomimicry: In Situ Oxidative Self-Processing Converts Copper(I)/Diamine Precursor into a Highly Active Aerobic Oxidation Catalyst. ACS Cent. Sci 2017, 3, 314–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Rousselet G; Chassagnard C; Capdevielle P; Maumy M Copper-catalyzed olefin epoxidation by dioxygen or amine N-oxide. Tetrahedron Lett 1996, 37, 8497–8500. [Google Scholar]
- (109).Sonobe T; Oisaki K; Kanai M Catalytic aerobic production of imines en route to mild, green, and concise derivatizations of amines. Chem. Sci 2012, 3, 3249–3255. [Google Scholar]
- (110).Kim J; Stahl SS Cu/Nitroxyl-Catalyzed Aerobic Oxidation of Primary Amines into Nitriles at Room Temperature. ACS Catal 2013, 3, 1652–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Zultanski SL; Zhao J; Stahl SS Practical Synthesis of Amides via Copper/ABNO-Catalyzed Aerobic Oxidative Coupling of Alcohols and Amines. J. Am. Chem. Soc 2016, 138, 6416–6419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Xie X; Stahl SS Efficient and Selective Cu/Nitroxyl-Catalyzed Methods for Aerobic Oxidative Lactonization of Diols. J. Am. Chem. Soc 2015, 137, 3767–3770. [DOI] [PubMed] [Google Scholar]
- (113).Xu B; Hartigan EM; Feula G; Huang Z; Lumb J-P; Arndtsen BA Simple Copper Catalysts for the Aerobic Oxidation of Amines: Selectivity Control by the Counterion. Angew. Chem., Int. Ed 2016, 55, 15802–15806. [DOI] [PubMed] [Google Scholar]
- (114).Jeon J-R; Baldrian P; Murugesan K; Chang Y-S Laccase-catalysed oxidations of naturally occurring phenols: from in vivo biosynthetic pathways to green synthetic applications. Microb. Biotechnol 2012, 5, 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Klinman JP; Bonnot F Intrigues and Intricacies of the Biosynthetic Pathways for the Enzymatic Quinocofactors: PQQ, TTQ, CTQ, TPQ, and LTQ. Chem. Rev 2014, 114, 4343–4365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Marcus RA; Sutin N Electron transfers in chemistry and biology. Biochim. Biophys. Acta, Rev. Bioenerg 1985, 811, 265–322. [Google Scholar]
- (117).Lee JY; Peterson RL; Ohkubo K; Garcia-Bosch I; Himes RA; Woertink J; Moore CD; Solomon EI; Fukuzumi S; Karlin KD Mechanistic Insights into the Oxidation of Substituted Phenols via Hydrogen Atom Abstraction by a Cupric–Superoxo Complex. J. Am. Chem. Soc 2014, 136, 9925–9937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (118).Osako T; Ohkubo K; Taki M; Tachi Y; Fukuzumi S; Itoh S Oxidation mechanism of phenols by dicopper-dioxygen (Cu2O2) complexes. J. Am. Chem. Soc 2003, 125, 11027–11033. [DOI] [PubMed] [Google Scholar]
- (119).Garcia-Bosch I; Ribas X; Costas M Electrophilic Arene Hydroxylation and Phenol O–H Oxidations Performed by an Unsymmetric μ-η1:η1-O2-Peroxo Dicopper(II) Complex. Chem. - Eur. J 2012, 18, 2113–2122. [DOI] [PubMed] [Google Scholar]
- (120).Paul PP; Tyeklár Z; Jacobson RR; Karlin KD Reactivity Patterns and Comparisons in Three Classes of Synthetic Copper-Dioxygen {Cu2-O2} Complexes: Implication for Structure and Biological Relevance. J. Am. Chem. Soc 1991, 113, 5322–5332. [Google Scholar]
- (121).Garcia-Bosch I; Cowley RE; Díaz DE; Peterson RL; Solomon EI; Karlin KD Substrate and Lewis Acid Coordination Promote O–O Bond Cleavage of an Unreactive L2CuII2(O22–) Species to Form L2CuIII2 (O)2 Cores with Enhanced Oxidative Reactivity. J. Am. Chem. Soc 2017, 139, 3186–3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Esguerra KVN; Fall Y; Petitjean L; Lumb J-P Controlling the Catalytic Aerobic Oxidation of Phenols. J. Am. Chem. Soc 2014, 136, 7662–7668. [DOI] [PubMed] [Google Scholar]
- (123).Li X; Yang J; Kozlowski MC Enantioselective Oxidative Biaryl Coupling Reactions Catalyzed by 1,5-Diazadecalin Metal Complexes. Org. Lett 2001, 3, 1137–1140. [DOI] [PubMed] [Google Scholar]
- (124).Hewgley JB; Stahl SS; Kozlowski MC Mechanistic Study of Asymmetric Oxidative Biaryl Coupling: Evidence for Self-Processing of the Copper Catalyst to Achieve Control of Oxidase vs Oxygenase Activity. J. Am. Chem. Soc 2008, 130, 12232–12233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (125).Huang Z; Lumb J-P A Catalyst-Controlled Aerobic Coupling of ortho-Quinones and Phenols Applied to the Synthesis of Aryl Ethers. Angew. Chem., Int. Ed 2016, 55, 11543–11547. [DOI] [PubMed] [Google Scholar]
- (126).Peters DS; Romesberg FE; Baran PS Scalable Access to Arylomycins via C–H Functionalization Logic. J. Am. Chem. Soc 2018, 140, 2072–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Ortiz de Montellano PR Hydrocarbon Hydroxylation by Cytochrome P450 Enzymes. Chem. Rev 2010, 110, 932–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Klinman JP Mechanisms Whereby Mononuclear Copper Proteins Functionalize Organic Substrates. Chem. Rev 1996, 96, 2541–2561. [DOI] [PubMed] [Google Scholar]
- (129).Hanson RS; Hanson TE Methanotrophic bacteria. Microbiol. Rev 1996, 60, 439–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (130).Costas M Selective C–H oxidation catalyzed by metalloporphyrins. Coord. Chem. Rev 2011, 255, 2912–2932. [Google Scholar]
- (131).Costas M; Mehn MP; Jensen MP; Que L Dioxygen activation at mononuclear nonheme iron active sites: Enzymes, models, and intermediates. Chem. Rev 2004, 104, 939–986. [DOI] [PubMed] [Google Scholar]
- (132).McGarrigle EM; Gilheany DG Chromium– and Manganese–salen Promoted Epoxidation of Alkenes. Chem. Rev 2005, 105, 1563–1602. [DOI] [PubMed] [Google Scholar]
- (133).Sundermeier U; Döbler C; Beller M: Modern Oxidation Methods In Modern Oxidation Methods; Backvall J-E, Ed.; Wiley-VCH: Weinheim, 2004. [Google Scholar]
- (134).White MC Adding Aliphatic C–H Bond Oxidations to Synthesis. Science 2012, 335, 807–809. [DOI] [PubMed] [Google Scholar]
- (135).Gómez L; Garcia-Bosch I; Company A; Benet-Buchholz J; Polo A; Sala X; Ribas X; Costas M Stereospecific C–H Oxidation with H2O2 Catalyzed by a Chemically Robust Site-Isolated Iron Catalyst. Angew. Chem., Int. Ed 2009, 48, 5720–5723. [DOI] [PubMed] [Google Scholar]
- (136).Cussó O; Cianfanelli M; Ribas X; Klein Gebbink RJM; Costas M Iron Catalyzed Highly Enantioselective Epoxidation of Cyclic Aliphatic Enones with Aqueous H2O2. J. Am. Chem. Soc 2016, 138, 2732–2738. [DOI] [PubMed] [Google Scholar]
- (137).Garcia-Bosch I Copper-Catalyzed Oxidation of Alkanes under Mild Conditions. Synlett 2017, 28, 1237–1243. [Google Scholar]
- (138).Prigge ST; Eipper B; Mains R; Amzel LM Dioxygen Binds End-On to Mononuclear Copper in a Precatalytic Enzyme Complex. Science 2004, 304, 864–867. [DOI] [PubMed] [Google Scholar]
- (139).Evans JP; Ahn K; Klinman JP Evidence that dioxygen and substrate activation are tightly coupled in dopamine β-monooxygenase - Implications for the reactive oxygen species. J. Biol. Chem 2003, 278, 49691–49698. [DOI] [PubMed] [Google Scholar]
- (140).Chen P; Solomon EI Oxygen Activation by the Non-Coupled Binuclear Copper Site in Peptidylglycine a-Hydroxylating Monooxygenase. Reaction Mechanism and Role of Non-Coupled Nature of the Active Sites. J. Am. Chem. Soc 2004, 126, 4991–5000. [DOI] [PubMed] [Google Scholar]
- (141).Cowley RE; Tian L; Solomon EI Mechanism of O2 activation and substrate hydroxylation in noncoupled binuclear copper monooxygenases. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 12035–12040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Lieberman RL; Rosenzweig AC Crystal structure of a membrane-bound metalloenzyme that catalyses the biological oxidation of methane. Nature 2005, 434, 177–182. [DOI] [PubMed] [Google Scholar]
- (143).Cao L; Caldararu O; Rosenzweig AC; Ryde U Quantum Refinement Does Not Support Dinuclear Copper Sites in Crystal Structures of Particulate Methane Monooxygenase. Angew. Chem., Int. Ed 2018, 57, 162–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (144).Culpepper MA; Cutsail GE; Hoffman BM; Rosenzweig AC Evidence for Oxygen Binding at the Active Site of Particulate Methane Monooxygenase. J. Am. Chem. Soc 2012, 134, 7640–7643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Yoshizawa K; Shiota Y Conversion of Methane to Methanol at the Mononuclear and Dinuclear Copper Sites of Particulate Methane Monooxygenase (pMMO): A DFT and QM/MM Study. J. Am. Chem. Soc 2006, 128, 9873–9881. [DOI] [PubMed] [Google Scholar]
- (146).Ciano L; Davies GJ; Tolman WB; Walton PH Bracing copper for the catalytic oxidation of C–H bonds. Nat. Catal 2018, 1, 571–577. [Google Scholar]
- (147).Meier KK; Jones SM; Kaper T; Hansson H; Koetsier MJ; Karkehabadi S; Solomon EI; Sandgren M; Kelemen B Oxygen Activation by Cu LPMOs in Recalcitrant Carbohydrate Polysaccharide Conversion to Monomer Sugars. Chem. Rev 2018, 118, 2593–2635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Quinlan RJ; Sweeney MD; Lo Leggio L; Otten H; Poulsen J-CN; Johansen KS; Krogh KBRM; Jörgensen CI; Tovborg M; Anthonsen A; et al. Insights into the oxidative degradation of cellulose by a copper metalloenzyme that exploits biomass components. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 15079–15084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Vaaje-Kolstad G; Westereng B; Horn SJ; Liu Z; Zhai H; Sorlie M; Eijsink VGH An Oxidative Enzyme Boosting the Enzymatic Conversion of Recalcitrant Polysaccharides. Science 2010, 330, 219–222. [DOI] [PubMed] [Google Scholar]
- (150).Frandsen KEH; Simmons TJ; Dupree P; Poulsen J-CN; Hemsworth GR; Ciano L; Johnston EM; Tovborg M; Johansen KS; von Freiesleben P; et al. The molecular basis of polysaccharide cleavage by lytic polysaccharide monooxygenases. Nat. Chem. Biol 2016, 12, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Hemsworth GR; Henrissat B; Davies GJ; Walton PH Discovery and characterization of a new family of lytic polysaccharide monooxygenases. Nat. Chem. Biol 2014, 10, 122–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Span EA; Suess DLM; Deller MC; Britt RD; Marletta MA The Role of the Secondary Coordination Sphere in a Fungal Polysaccharide Monooxygenase. ACS Chem. Biol 2017, 12, 1095–1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (153).Wang B; Johnston EM; Li P; Shaik S; Davies GJ; Walton PH; Rovira C QM/MM Studies into the H2O2-Dependent Activity of Lytic Polysaccharide Monooxygenases: Evidence for the Formation of a Caged Hydroxyl Radical Intermediate. ACS Catal 2018, 8, 1346–1351. [Google Scholar]
- (154).Bissaro B; Røhr ÅK; Müller G; Chylenski P; Skaugen M; Forsberg Z; Horn SJ; Vaaje-Kolstad G; Eijsink VGH Oxidative cleavage of polysaccharides by monocopper enzymes depends on H2O2. Nat. Chem. Biol 2017, 13, 1123–1128. [DOI] [PubMed] [Google Scholar]
- (155).Kunishita A; Ertem MZ; Okubo Y; Tano T; Sugimoto H; Ohkubo K; Fujieda N; Fukuzumi S; Cramer CJ; Itoh S Active Site Models for the CuA Site of Peptidylglycine α-Hydroxylating Monooxygenase and Dopamine β-Monooxygenase. Inorg. Chem 2012, 51, 9465–9480. [DOI] [PubMed] [Google Scholar]
- (156).Wada A; Harata M; Hasegawa K; Jitsukawa K; Masuda H; Mukai M; Kitagawa T; Einaga H Structural and Spectroscopic Characterization of a Mononuclear Hydroperoxo-Copper(II) Complex with Tripodal Pyridylamine Ligands. Angew. Chem., Int. Ed 1998, 37, 798–799. [DOI] [PubMed] [Google Scholar]
- (157).Sanchez-Eguia BN; Flores-Alamo M; Orio M; Castillo I Side-on cupric-superoxo triplet complexes as competent agents for H-abstraction relevant to the active site of PHM. Chem. Commun 2015, 51, 11134–11137. [DOI] [PubMed] [Google Scholar]
- (158).Kim S; Ginsbach JW; Lee JY; Peterson RL; Liu JJ; Siegler MA; Sarjeant AA; Solomon EI; Karlin KD Amine Oxidative N-Dealkylation via Cupric Hydroperoxide Cu-OOH Homolytic Cleavage Followed by Site-Specific Fenton Chemistry. J. Am. Chem. Soc 2015, 137, 2867–2874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (159).Trammell R; See YY; Herrmann AT; Xie N; Díaz DE; Siegler MA; Baran PS; Garcia-Bosch I Decoding the Mechanism of Intramolecular Cu-Directed Hydroxylation of sp3 C–H Bonds. J. Org. Chem 2017, 82, 7887–7904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Blain I; Giorgi M; De Riggi I; Réglier M Substrate-Binding Ligand Approach in Chemical Modeling of Copper-Containing Monooxygenases, 1 Intramolecular Stereoselective Oxygen Atom Insertion into a Non-Activated C-H Bond. Eur. J. Inorg. Chem 2000, 2000, 393–398. [Google Scholar]
- (161).Schonecker B; Zheldakova T; Liu Y; Kotteritzsch M; Gunther W; Gorls H Biomimetic hydroxylation of nonactivated CH2 groups with copper complexes and molecular oxygen. Angew. Chem., Int. Ed 2003, 42, 3240–3244. [DOI] [PubMed] [Google Scholar]
- (162).See YY; Herrmann AT; Aihara Y; Baran PS Scalable C–H Oxidation with Copper: Synthesis of Polyoxypregnanes. J. Am. Chem. Soc 2015, 137, 13776–13779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Dhar D; Yee GM; Spaeth AD; Boyce DW; Zhang H; Dereli B; Cramer CJ; Tolman WB Perturbing the Copper(III)– Hydroxide Unit through Ligand Structural Variation. J. Am. Chem. Soc 2016, 138, 356–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Matsumoto T; Ohkubo K; Honda K; Yazawa A; Furutachi H; Fujinami S; Fukuzumi S; Suzuki M Aliphatic C–H Bond Activation Initiated by a (μ-η2:η2-Peroxo)dicopper(II) Complex in Comparison with Cumylperoxyl Radical. J. Am. Chem. Soc 2009, 131, 9258–9267. [DOI] [PubMed] [Google Scholar]
- (165).Itoh S; Nakao H; Berreau LM; Kondo T; Komatsu M; Fukuzumi S Mechanistic Studies of Aliphatic Ligand Hydroxylation of a Copper Complex by Dioxygen: A Model Reaction for Copper Monooxygenases. J. Am. Chem. Soc 1998, 120, 2890–2899. [Google Scholar]
- (166).Taki M; Itoh S; Fukuzumi S C-H. Bond Activation of External Substrates with a Bis(μ-oxo)dicopper(III) Complex. J. Am. Chem. Soc 2001, 123, 6203–6204. [DOI] [PubMed] [Google Scholar]
- (167).Itoh S; Taki M; Nakao H; Holland PL; Tolman WB; Que L Jr.; Fukuzumi S Aliphatic Hydroxylation by a Bis(μ-oxo)dicopper(III) Complex. Angew. Chem., Int. Ed 2000, 39, 398–400. [DOI] [PubMed] [Google Scholar]
- (168).Citek C; Lin B-L; Phelps TE; Wasinger EC; Stack TDP Primary Amine Stabilization of a Dicopper(III) Bis(μ-oxo) Species: Modeling the Ligation in pMMO. J. Am. Chem. Soc 2014, 136, 14405–14408. [DOI] [PubMed] [Google Scholar]
- (169).Baglia RA; Zaragoza JPT; Goldberg DP Biomimetic Reactivity of Oxygen-Derived Manganese and Iron Porphyrinoid Complexes. Chem. Rev 2017, 117, 13320–13352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (170).Li F; Van Heuvelen KM; Meier KK; Münck E; Que L Sc3+-Triggered Oxoiron(IV) Formation from O2 and its Non-Heme Iron(II) Precursor via a Sc3+–Peroxo–Fe3+ Intermediate. J. Am. Chem. Soc 2013, 135, 10198–10201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Bang S; Lee Y-M; Hong S; Cho K-B; Nishida Y; Seo MS; Sarangi R; Fukuzumi S; Nam W Redox-inactive metal ions modulate the reactivity and oxygen release of mononuclear non-haem iron(III)–peroxo complexes. Nat. Chem 2014, 6, 934–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Baglia RA; Dürr M; Ivanović-Burmazović I; Goldberg DP Activation of a High-Valent Manganese-Oxo Complex by a Non-metallic Lewis Acid. Inorg. Chem 2014, 53, 5893–5895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (173).Barton DHR; Beviere SD; Chavasiri W; Csuhai E; Doller D The Functionalization of Saturated Hydrocarbons. Part XXI +. The Fe(III)-Catalyzed and the Cu(II)-Catalyzed Oxidation of Saturated Hydrocarbons by Hydrogen Peroxide: A Comparative Study. Tetrahedron 1992, 48, 2895–2910. [Google Scholar]
- (174).Barton DHR; Doller D The Selective Functionalization of Saturated Hydrocarbons: Gif Chemistry. Acc. Chem. Res 1992, 25, 504–512. [Google Scholar]
- (175).Sobkowiak A; Qui A; Liu X; Llobet A; Sawyer DT Copper(I)/(tert-BuOOH)-induced activation of dioxygen for the ketonization of methylenic carbons. J. Am. Chem. Soc 1993, 115, 609–614. [Google Scholar]
- (176).Ohta T; Tachiyama T; Yoshizawa K; Yamabe T; Uchida T; Kitagawa T Synthesis, Structure and H2O2-Dependent Catalytic Functions of Disulfide-Bridged Dicopper(I) and Related Thioether-Copper(I) and Thioether-Copper(II) Complexes. Inorg. Chem 2000, 39, 4358–4369. [DOI] [PubMed] [Google Scholar]
- (177).Kirillov AM; Kopylovich MN; Kirillova MV; Haukka M; da Silva MFCG; Pombeiro AJL Multinuclear Copper Triethanolamine Complexes as Selective Catalysts for the Peroxidative Oxidation of Alkanes under Mild Conditions. Angew. Chem., Int. Ed 2005, 44, 4345–4349. [DOI] [PubMed] [Google Scholar]
- (178).Kirillov AM; Kopylovich MN; Kirillova MV; Karabach EY; Haukka M; da Silva MFCG; Pombeiro AJL Mild Peroxidative Oxidation of Cyclohexane Catalyzed by Mono-, Di-, Tri-, Tetra- and Polynuclear Copper Triethanolamine Complexes. Adv. Synth. Catal 2006, 348, 159–174. [Google Scholar]
- (179).Garcia-Bosch I; Siegler MA Copper-Catalyzed Oxidation of Alkanes with H2O2 under a Fenton-like Regime. Angew. Chem., Int. Ed 2016, 55, 12873–12876. [DOI] [PubMed] [Google Scholar]
- (180).Shul’pin GB Metal-catalyzed hydrocarbon oxygenations in solutions: the dramatic role of additives: a review. J. Mol. Catal. A: Chem 2002, 189, 39–66. [Google Scholar]
- (181).Chen MS; White MC A Predictably Selective Aliphatic C-H Oxidation Reaction for Complex Molecule Synthesis. Science 2007, 318, 783–787. [DOI] [PubMed] [Google Scholar]
- (182).Chen MS; White MC Combined Effects on Selectivity in Fe-Catalyzed Methylene Oxidation. Science 2010, 327, 566–571. [DOI] [PubMed] [Google Scholar]
- (183).Oloo WN; Que L Bioinspired Nonheme Iron Catalysts for C–H and C = C Bond Oxidation: Insights into the Nature of the Metal-Based Oxidants. Acc. Chem. Res 2015, 48, 2612–2621. [DOI] [PubMed] [Google Scholar]
- (184).Conde A; Vilella L; Balcells D; Díaz-Requejo MM; Lledós A; Pérez PJ Introducing Copper as Catalyst for Oxidative Alkane Dehydrogenation. J. Am. Chem. Soc 2013, 135, 3887–3896. [DOI] [PubMed] [Google Scholar]
- (185).Concia AL; Beccia MR; Orio M; Ferre FT; Scarpellini M; Biaso F; Guigliarelli B; Réglier M; Simaan AJ Copper Complexes as Bioinspired Models for Lytic Polysaccharide Mono-oxygenases. Inorg. Chem 2017, 56, 1023–1026. [DOI] [PubMed] [Google Scholar]
- (186).Shimokawa C; Teraoka J; Tachi Y; Itoh S A functional model for pMMO (particulate methane monooxygenase). Hydroxylation of alkanes with H2O2 catalyzed by [beta]-diketiminatocopper-(II) complexes. J. Inorg. Biochem 2006, 100, 1118–1127. [DOI] [PubMed] [Google Scholar]
- (187).Würtele C; Sander O; Lutz V; Waitz T; Tuczek F; Schindler S Aliphatic C–H Bond Oxidation of Toluene Using Copper Peroxo Complexes That Are Stable at Room Temperature. J. Am. Chem. Soc 2009, 131, 7544–7545. [DOI] [PubMed] [Google Scholar]
- (188).Chen PP-Y; Yang RB-G; Lee JC-M; Chan SI Facile O-atom insertion into C-H bonds by a trinuclear copper complex designed to harness a singlet oxene. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 14570–14575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Nagababu P; Maji S; Kumar MP; Chen PPY; Yu SSF; Chan SI Efficient Room-Temperature Oxidation of Hydrocarbons Mediated by Tricopper Cluster Complexes with Different Ligands. Adv. Synth. Catal 2012, 354, 3275–3282. [Google Scholar]
- (190).Chan SI; Lu Y-J; Nagababu P; Maji S; Hung M-C; Lee MM; Hsu IJ; Minh PD; Lai JCH; Ng KY; Ramalingam S; Yu SSF; Chan MK Efficient Oxidation of Methane to Methanol by Dioxygen Mediated by Tricopper Clusters. Angew. Chem., Int. Ed 2013, 52, 3731–3735. [DOI] [PubMed] [Google Scholar]
- (191).Decker H; Schweikardt T; Tuczek F The First Crystal Structure of Tyrosinase: All Questions Answered? Angew. Chem., Int. Ed 2006, 45, 4546–4550. [DOI] [PubMed] [Google Scholar]
- (192).Solem E; Tuczek F; Decker H Tyrosinase versus Catechol Oxidase: One Asparagine Makes the Difference. Angew. Chem., Int. Ed 2016, 55, 2884–2888. [DOI] [PubMed] [Google Scholar]
- (193).Rolff M; Schottenheim J; Decker H; Tuczek F Copper-O2 reactivity of tyrosinase models towards external monophenolic substrates: molecular mechanism and comparison with the enzyme. Chem. Soc. Rev 2011, 40, 4077–4098. [DOI] [PubMed] [Google Scholar]
- (194).Santagostini L; Gullotti M; Monzani E; Casella L; Dillinger R; Tuczek F Reversible Dioxygen Binding and Phenol Oxygenation in a Tyrosinase Model System. Chem. - Eur. J 2000, 6, 519–522. [DOI] [PubMed] [Google Scholar]
- (195).Palavicini S; Granata A; Monzani E; Casella L Hydroxylation of phenolic compounds by a peroxodicopper(II) complex: Further insight into the mechanism of tyrosinase. J. Am. Chem. Soc 2005, 127, 18031–18036. [DOI] [PubMed] [Google Scholar]
- (196).Itoh S; Kumei H; Taki M; Nagatomo S; Kitagawa T; Fukuzumi S Oxygenation of phenols to catechols by a (μ-η2:η2-peroxo)dicopper(II) complex: mechanistic insight into the phenolase activity of tyrosinase. J. Am. Chem. Soc 2001, 123, 6708–6709. [DOI] [PubMed] [Google Scholar]
- (197).Company A; Palavicini S; Garcia-Bosch I; Mas-Balleste R; Que L; Rybak-Akimova EV; Casella L; Ribas X; Costas M Tyrosinase-like reactivity in a Cu2(III)(μ-O)2 species. Chem. - Eur. J 2008, 14, 3535–3538. [DOI] [PubMed] [Google Scholar]
- (198).Garcia-Bosch I; Company A; Frisch JR; Torrent-Sucarrat M; Cardellach M; Gamba I; Güell M; Casella L; Que L Jr.; Ribas X; Luis JM; Costas M O2 Activation and Selective Phenolate ortho Hydroxylation by an Unsymmetric Dicopper μ-η1:η1-Peroxido Complex. Angew. Chem., Int. Ed 2010, 49, 2406–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (199).Kieber-Emmons MT; Ginsbach JW; Wick PK; Lucas HR; Helton ME; Lucchese B; Suzuki M; Zuberbühler AD; Karlin KD; Solomon EI Observation of a CuII2(μ−1,2-peroxo)/CuIII2(μ-oxo)2 Equilibrium and its Implications for Copper–Dioxygen Reactivity. Angew. Chem 2014, 126, 5035–5039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Herres-Pawlis S; Verma P; Haase R; Kang P; Lyons CT; Wasinger EC; Flörke U; Henkel G; Stack TDP Phenolate Hydroxylation in a Bis(μ-oxo)dicopper(III) Complex: Lessons from the Guanidine/Amine Series. J. Am. Chem. Soc 2009, 131, 1154–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Citek C; Lyons CT; Wasinger EC; Stack TDP Self-assembly of the oxy-tyrosinase core and the fundamental components of phenolic hydroxylation. Nat. Chem 2012, 4, 317–322. [DOI] [PubMed] [Google Scholar]
- (202).Chiang L; Keown W; Citek C; Wasinger EC; Stack TDP Simplest Monodentate Imidazole Stabilization of the oxy-Tyrosinase Cu2O2 Core: Phenolate Hydroxylation through a CuIII Intermediate. Angew. Chem., Int. Ed 2016, 55, 10453–10457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (203).Serrano-Plana J; Garcia-Bosch I; Miyake R; Costas M; Company A Selective ortho-Hydroxylation–Defluorination of 2-Fluorophenolates with a Bis(μ-oxo)dicopper(III) Species. Angew. Chem., Int. Ed 2014, 53, 9608–9612. [DOI] [PubMed] [Google Scholar]
- (204).Réglier M; Jorand C; Waegell B Catalytic Phenol oxidation mediated by a binuclear copper complex. J. Chem. Soc., Chem. Commun 1990, 1752–1755. [Google Scholar]
- (205).Casella L; Gullotti M; Radaelli R; Di Gennaro P A Tyrosinase Model System. Phenol ortho-Hydroxylation of a Binuclear Three-coordinated Copper(I) Complex and Dioxygen. J. Chem. Soc., Chem. Commun 1991, 1611–1612. [Google Scholar]
- (206).Rolff M; Schottenheim J; Peters G; Tuczek F The First Catalytic Tyrosinase Model System Based on a Mononuclear Copper(I) Complex: Kinetics and Mechanism. Angew. Chem., Int. Ed 2010, 49, 6438–6442. [DOI] [PubMed] [Google Scholar]
- (207).Hoffmann A; Citek C; Binder S; Goos A; Rübhausen M; Troeppner O; Ivanović-Burmazović I; Wasinger EC; Stack TDP; Herres-Pawlis S Catalytic Phenol Hydroxylation with Dioxygen: Extension of the Tyrosinase Mechanism beyond the Protein Matrix. Angew. Chem., Int. Ed 2013, 52, 5398–5401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Esguerra KVN; Fall Y; Lumb J-P A Biomimetic Catalytic Aerobic Functionalization of Phenols. Angew. Chem., Int. Ed 2014, 53, 5877–5881. [DOI] [PubMed] [Google Scholar]
- (209).Askari MS; Esguerra KVN; Lumb J-P; Ottenwaelder X A Biomimetic Mechanism for the Copper-Catalyzed Aerobic Oxygenation of 4-tert-Butylphenol. Inorg. Chem 2015, 54, 8665–8672. [DOI] [PubMed] [Google Scholar]
- (210).Esguerra KVN; Lumb J-P Synthesis of ortho-Azophenols by Formal Dehydrogenative Coupling of Phenols and Hydrazines or Hydrazides. Chem. - Eur. J 2017, 23, 8596–8600. [DOI] [PubMed] [Google Scholar]
- (211).Esguerra KVN; Lumb J-P Selectivity in the Aerobic Dearomatization of Phenols: Total Synthesis of Dehydronornuciferine by Chemo- and Regioselective Oxidation. Angew. Chem., Int. Ed 2018, 57, 1514–1518. [DOI] [PubMed] [Google Scholar]
- (212).Maiti D; Lucas HR; Sarjeant AAN; Karlin KD Aryl Hydroxylation from a Mononuclear Copper-Hydroperoxo Species. J. Am. Chem. Soc 2007, 129, 6998–6999. [DOI] [PubMed] [Google Scholar]
- (213).Kunishita A; Teraoka J; Scanlon JD; Matsumoto T; Suzuki M; Cramer CJ; Itoh S Aromatic Hydroxylation Reactivity of a Mononuclear Cu(II)-Alkylperoxo Complex. J. Am. Chem. Soc 2007, 129, 7248–7249. [DOI] [PubMed] [Google Scholar]
- (214).Kunishita A; Scanlon JD; Ishimaru H; Honda K; Ogura T; Suzuki M; Cramer CJ; Itoh S Reactions of copper(II)-H2O2 adducts supported by tridentate bis(2-pyridylmethyl)amine ligands: Sensitivity to solvent and variations in ligand substitution. Inorg. Chem 2008, 47, 8222–8232. [DOI] [PubMed] [Google Scholar]
- (215).Hong S; Huber SM; Gagliardi L; Cramer CC; Tolman WB Copper(I)-alpha-Ketocarboxylate Complexes: Characterization and O2 Reactions That Yield Copper-Oxygen Intermediates Capable of Hydroxylating Arenes. J. Am. Chem. Soc 2007, 129, 14190–14192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (216).Karlin KD; Cruse RW; Gultneh Y; Hayes JC; Zubieta J Peroxide Coordination to a Dicopper(II) Center. Dioxygen Binding to a Structurally Characterized Phenoxide Bridged Binuclear Copper(I) Complex. J. Am. Chem. Soc 1984, 106, 3372–3374. [Google Scholar]
- (217).Karlin KD; Gultneh Y; Hayes JC; Cruse RW; McKown JW; Hutchinson JP; Zubieta J Copper Mediated Hydroxylation of an Arene: Model System for the Action of Copper Monooxygenases and the Structures of a Binuclear Cu(I) Complex and its Oxygenated Product. J. Am. Chem. Soc 1984, 106, 2121–2128. [Google Scholar]
- (218).Karlin KD; Nasir MS; Cohen BI; Cruse RW; Kaderli S; Zuberbühler AD Reversible Dioxygen Binding and Aromatic Hydroxylation in O2-Reactions with Substituted Xylyl Dinuclear Copper(I) Complexes: Syntheses and Low-Temperature Kinetic/Thermodynamic and Spectroscopic Investigations of a Copper Monooxygenase Model System. J. Am. Chem. Soc 1994, 116, 1324–1336. [Google Scholar]
- (219).Qayyum MF; Sarangi R; Fujisawa K; Stack TDP; Karlin KD; Hodgson KO; Hedman B; Solomon EI L-Edge X-ray Absorption Spectroscopy and DFT Calculations on Cu2O2 Species: Direct Electrophilic Aromatic Attack by Side-on Peroxo Bridged Dicopper(II) Complexes. J. Am. Chem. Soc 2013, 135, 17417–17431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Holland PL; Rodgers KR; Tolman WB Is the Bis(μ-oxo)dicopper Core Capable of Hydroxylating an Arene. Angew. Chem., Int. Ed 1999, 38, 1139–1142. [DOI] [PubMed] [Google Scholar]
- (221).Becker J; Gupta P; Angersbach F; Tuczek F; Näther C; Holthausen MC; Schindler S Selective Aromatic Hydroxylation with Dioxygen and Simple Copper Imine Complexes. Chem. - Eur. J 2015, 21, 11735–11744. [DOI] [PubMed] [Google Scholar]
- (222).Battaini G; Monzani E; Perotti A; Para C; Casella L; Santagostini L; Gullotti M; Dillinger R; Nather C; Tuczek F A double arene hydroxylation mediated by dicopper(II)-hydroperoxide species. J. Am. Chem. Soc 2003, 125, 4185–4198. [DOI] [PubMed] [Google Scholar]
- (223).Conde A; Mar Díaz-Requejo M; Pérez PJ Direct, copper-catalyzed oxidation of aromatic C-H bonds with hydrogen peroxide under acid-free conditions. Chem. Commun 2011, 47, 8154–8156. [DOI] [PubMed] [Google Scholar]
- (224).Vilella L; Conde A; Balcells D; Díaz-Requejo MM; Lledos A; Pérez PJ A competing, dual mechanism for catalytic direct benzene hydroxylation from combined experimental-DFT studies. Chem. Sci 2017, 8, 8373–8383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Augusti R; Dias AO; Rocha LL; Lago RM Kinetics and Mechanism of Benzene Derivative Degradation with Fenton’s Reagent in Aqueous Medium Studied by MIMS. J. Phys. Chem. A 1998, 102, 10723–10727. [Google Scholar]
- (226).Yamada M; Karlin KD; Fukuzumi S One-step selective hydroxylation of benzene to phenol with hydrogen peroxide catalysed by copper complexes incorporated into mesoporous silica-alumina. Chem. Sci 2016, 7, 2856–2863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Garcia-Bosch I; Cowley RE; Díaz DE; Siegler MA; Nam W; Solomon EI; Karlin KD Dioxygen Activation by a Macrocyclic Copper Complex Leads to a Cu2O2 Core with Unexpected Structure and Reactivity. Chem. - Eur. J 2016, 22, 5133–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Pirovano P; Magherusan AM; McGlynn C; Ure A; Lynes A; McDonald AR Nucleophilic Reactivity of a Copper(II)– Superoxide Complex. Angew. Chem., Int. Ed 2014, 53, 5946–5950. [DOI] [PubMed] [Google Scholar]
- (229).Kim B; Jeong D; Cho J Nucleophilic reactivity of copper(II)-alkylperoxo complexes. Chem. Commun 2017, 53, 9328–9331. [DOI] [PubMed] [Google Scholar]
- (230).Iovan DA; Wrobel AT; McClelland AA; Scharf AB; Edouard GA; Betley TA Reactivity of a stable copper-dioxygen complex. Chem. Commun 2017, 53, 10306–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Tachi Y; Aita K; Teramae S; Tani F; Naruta Y; Fukuzumi S; Itoh S Dicopper–Dioxygen Complex Supported by Asymmetric Pentapyridine Dinucleating Ligand. Inorg. Chem 2004, 43, 4558–4560. [DOI] [PubMed] [Google Scholar]
- (232).Fujii T; Naito A; Yamaguchi S; Wada A; Funahashi Y; Jitsukawa K; Nagatomo S; Kitagawa T; Masuda H Construction of a square-planar hydroperoxo-copper(II) complex inducing a higher catalytic reactivity. Chem. Commun 2003, 2700–2701. [DOI] [PubMed] [Google Scholar]
- (233).Barry SM; Kers JA; Johnson EG; Song L; Aston PR; Patel B; Krasnoff SB; Crane BR; Gibson DM; Loria R; Challis GL Cytochrome P450–catalyzed L-tryptophan nitration in thaxtomin phytotoxin biosynthesis. Nat. Chem. Biol 2012, 8, 814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Matthews ML; Chang W-C; Layne AP; Miles LA; Krebs C; Bollinger JM, Jr Direct nitration and azidation of aliphatic carbons by an iron-dependent halogenase. Nat. Chem. Biol 2014, 10, 209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Srnec M; Wong SD; Matthews ML; Krebs C; Bollinger JM; Solomon EI Electronic Structure of the Ferryl Intermediate in the α-Ketoglutarate Dependent Non-Heme Iron Halogenase SyrB2: Contributions to H Atom Abstraction Reactivity. J. Am. Chem. Soc 2016, 138, 5110–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Kan SBJ; Lewis RD; Chen K; Arnold FH Directed evolution of cytochrome c for carbon–silicon bond formation: Bringing silicon to life. Science 2016, 354, 1048–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Hyster TK; Ward TR Genetic Optimization of Metalloenzymes: Enhancing Enzymes for Non-Natural Reactions. Angew. Chem., Int. Ed 2016, 55, 7344–7357. [DOI] [PubMed] [Google Scholar]
- (238).Key HM; Dydio P; Clark DS; Hartwig JF Abiological catalysis by artificial haem proteins containing noble metals in place of iron. Nature 2016, 534, 534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Gephart RT; Warren TH Copper-Catalyzed sp3 C–H Amination. Organometallics 2012, 31, 7728–7752. [Google Scholar]
- (240).Hili R; Yudin AK Making carbon-nitrogen bonds in biological and chemical synthesis. Nat. Chem. Biol 2006, 2, 284–287. [DOI] [PubMed] [Google Scholar]
- (241).Hartwig JF Carbon-heteroatom bond formation catalysed by organometallic complexes. Nature 2008, 455, 314–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Shin K; Kim H; Chang S Transition-Metal-Catalyzed C-N Bond Forming Reactions Using Organic Azides as the Nitrogen Source: A Journey for the Mild and Versatile C-H Amination. Acc. Chem. Res 2015, 48, 1040–1052. [DOI] [PubMed] [Google Scholar]
- (243).Collet F; Dodd RH; Dauban P Catalytic C-H amination: recent progress and future directions. Chem. Commun 2009, 5061–5074. [DOI] [PubMed] [Google Scholar]
- (244).Varela-Álvarez A; Yang T; Jennings H; Kornecki KP; Macmillan SN; Lancaster KM; Mack JBC; Du Bois J; Berry JF; Musaev DG Rh2(II,III) Catalysts with Chelating Carboxylate and Carboxamidate Supports: Electronic Structure and Nitrene Transfer Reactivity. J. Am. Chem. Soc 2016, 138, 2327–2341. [DOI] [PubMed] [Google Scholar]
- (245).Hennessy ET; Liu RY; Iovan DA; Duncan RA; Betley TA Iron-mediated intermolecular N-group transfer chemistry with olefinic substrates. Chem. Sci 2014, 5, 1526–1532. [Google Scholar]
- (246).Li Z; Conser KR; Jacobsen EN Asymmetric alkene aziridination with readily available chiral diimine-based catalysts. J. Am. Chem. Soc 1993, 115, 5326–5327. [Google Scholar]
- (247).Evans DA; Faul MM; Bilodeau MT; Anderson BA; Barnes DM Bis(oxazoline)-copper complexes as chiral catalysts for the enantioselective aziridination of olefins. J. Am. Chem. Soc 1993, 115, 5328–5329. [Google Scholar]
- (248).Evans DA; Bilodeau MT; Faul MM Development of the Copper-Catalyzed Olefin Aziridination Reaction. J. Am. Chem. Soc 1994, 116, 2742–2753. [Google Scholar]
- (249).Kwong H-L; Liu D; Chan K-Y; Lee C-S; Huang K-H; Che C-M Copper(I)-catalyzed asymmetric alkene aziridination mediated by PhI(OAc)2: a facile one-pot procedure. Tetrahedron Lett 2004, 45, 3965–3968. [Google Scholar]
- (250).Halfen JA; Hallman JK; Schultz JA; Emerson JP Remarkably Efficient Olefin Aziridination Mediated by a New Copper(II) Complex. Organometallics 1999, 18, 5435–5437. [Google Scholar]
- (251).Vedernikov AN; Caulton KG Angular Ligand Constraint Yields an Improved Olefin Aziridination Catalyst. Org. Lett 2003, 5, 2591–2594. [DOI] [PubMed] [Google Scholar]
- (252).Mairena MA; Díaz-Requejo MM; Belderraín TR; Nicasio MC; Trofimenko S; Pérez PJ Copper-Homoscorpionate Complexes as Very Active Catalysts for the Olefin Aziridination Reaction. Organometallics 2004, 23, 253–256. [Google Scholar]
- (253).Díaz-Requejo MM; Belderraín TR; Nicasio MC; Trofimenko S; Pérez PJ Cyclohexane and Benzene Amination by Catalytic Nitrene Insertion into C–H Bonds with the Copper- Homoscorpionate Catalyst TpBr3Cu(NCMe). J. Am. Chem. Soc 2003, 125, 12078–12079. [DOI] [PubMed] [Google Scholar]
- (254).Fructos MR; Trofimenko S; Díaz-Requejo MM; Pérez PJ Facile Amine Formation by Intermolecular Catalytic Amidation of Carbon–Hydrogen Bonds. J. Am. Chem. Soc 2006, 128, 11784–11791. [DOI] [PubMed] [Google Scholar]
- (255).Rodríguez MR; Beltran A; Mudarra AL;Álvarez E; Maseras F; Díaz-Requejo MM; Pérez PJ Catalytic Nitrene Transfer To Alkynes: A Novel and Versatile Route for the Synthesis of Sulfinamides and Isothiazoles. Angew. Chem., Int. Ed 2017, 56, 12842–12847. [DOI] [PubMed] [Google Scholar]
- (256).Badiei YM; Dinescu A; Dai X; Palomino RM; Heinemann FW; Cundari TR; Warren TH Copper-Nitrene Complexes in Catalytic C–H Amination. Angew. Chem., Int. Ed 2008, 47, 9961–9964. [DOI] [PubMed] [Google Scholar]
- (257).Badiei YM; Krishnaswamy A; Melzer MM; Warren TH Transient Terminal Cu–Nitrene Intermediates from Discrete Dicopper Nitrenes. J. Am. Chem. Soc 2006, 128, 15056–15057. [DOI] [PubMed] [Google Scholar]
- (258).Aguila MJB; Badiei YM; Warren TH Mechanistic Insights into C-H Amination via Dicopper Nitrenes. J. Am. Chem. Soc 2013, 135, 9399–9406. [DOI] [PubMed] [Google Scholar]
- (259).Wiese S; Badiei YM; Gephart RT; Mossin S; Varonka MS; Melzer MM; Meyer K; Cundari TR; Warren TH Catalytic C–H Amination with Unactivated Amines through Copper(II) Amides. Angew. Chem., Int. Ed 2010, 49, 8850–8855. [DOI] [PubMed] [Google Scholar]
- (260).Jang ES; McMullin CL; Käβ M; Meyer K; Cundari TR; Warren TH Copper(II) Anilides in sp3 C-H Amination. J. Am. Chem. Soc 2014, 136, 10930–10940. [DOI] [PubMed] [Google Scholar]
- (261).Gephart RT; Huang DL; Aguila MJB; Schmidt G; Shahu A; Warren TH Catalytic C–H Amination with Aromatic Amines. Angew. Chem., Int. Ed 2012, 51, 6488–6492. [DOI] [PubMed] [Google Scholar]
- (262).Bakhoda A; Jiang Q; Bertke JA; Cundari TR; Warren TH Elusive Terminal Copper Arylnitrene Intermediates. Angew. Chem., Int. Ed 2017, 56, 6426–6430. [DOI] [PubMed] [Google Scholar]
- (263).Kundu S; Miceli E; Farquhar E; Pfaff FF; Kuhlmann U; Hildebrandt P; Braun B; Greco C; Ray K Lewis Acid Trapping of an Elusive Copper–Tosylnitrene Intermediate Using Scandium Triflate. J. Am. Chem. Soc 2012, 134, 14710–14713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Liang H-C; Zhang CX; Henson MJ; Sommer RD; Hatwell KR; Kaderli S; Zuberbuehler AD; Rheingold AL; Solomon EI; Karlin KD Contrasting Copper-Dioxygen Chemistry Arising from Alike Tridentate Alkyltriamine Copper(I) Complexes. J. Am. Chem. Soc 2002, 124, 4170–4171. [DOI] [PubMed] [Google Scholar]
- (265).Abram S-L; Monte-Perez I; Pfaff FF; Farquhar ER; Ray K Evidence of two-state reactivity in alkane hydroxylation by Lewis-acid bound copper-nitrene complexes. Chem. Commun 2014, 50, 9852–9854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Corona T; Ribas L; Rovira M; Farquhar ER; Ribas X; Ray K; Company A Characterization and Reactivity Studies of a Terminal Copper–Nitrene Species. Angew. Chem., Int. Ed 2016, 55, 14005–14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Dielmann F; Andrada DM; Frenking G; Bertrand G Isolation of Bridging and Terminal Coinage Metal–Nitrene Complexes. J. Am. Chem. Soc 2014, 136, 3800–3802. [DOI] [PubMed] [Google Scholar]
- (268).Askari MS; Orio M; Ottenwaelder X Controlled nitrene transfer from a tyrosinase-like arylnitroso-copper complex. Chem. Commun 2015, 51, 11206–11209. [DOI] [PubMed] [Google Scholar]
- (269).Lyons TW; Sanford MS Palladium-Catalyzed Ligand-Directed C–H Functionalization Reactions. Chem. Rev 2010, 110, 1147–1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Camasso NM; Sanford MS Design, synthesis, and carbonheteroatom coupling reactions of organometallic nickel(IV) complexes. Science 2015, 347, 1218–1220. [DOI] [PubMed] [Google Scholar]
- (271).Shang R; Ilies L; Nakamura E Iron-Catalyzed C–H Bond Activation. Chem. Rev 2017, 117, 9086–9139. [DOI] [PubMed] [Google Scholar]
- (272).Bauer I; Knölker H-J Iron Catalysis in Organic Synthesis. Chem. Rev 2015, 115, 3170–3387. [DOI] [PubMed] [Google Scholar]
- (273).Hickman AJ; Sanford MS High-valent organometallic copper and palladium in catalysis. Nature 2012, 484, 177–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).van Koten G Organocopper Compounds: From Elusive to Isolable Species, from Early Supramolecular Chemistry with RCuI Building Blocks to Mononuclear R2–nCuII and R3–mCuIII Compounds. A Personal View. Organometallics 2012, 31, 7634–7646. [Google Scholar]
- (275).Yoshikai N; Nakamura E Mechanisms of Nucleophilic Organocopper(I) Reactions. Chem. Rev 2012, 112, 2339–2372. [DOI] [PubMed] [Google Scholar]
- (276).Deutsch C; Krause N; Lipshutz BH CuH-Catalyzed Reactions. Chem. Rev 2008, 108, 2916–2927. [DOI] [PubMed] [Google Scholar]
- (277).Evano G; Blanchard N; Toumi M Copper-Mediated Coupling Reactions and Their Applications in Natural Products and Designed Biomolecules Synthesis. Chem. Rev 2008, 108, 3054–3131. [DOI] [PubMed] [Google Scholar]
- (278).Sambiagio C; Marsden SP; Blacker AJ; McGowan PC Copper catalysed Ullmann type chemistry: from mechanistic aspects to modern development. Chem. Soc. Rev 2014, 43, 3525–3550. [DOI] [PubMed] [Google Scholar]
- (279).Monnier F; Taillefer M Catalytic C-C, C-N, and C-O Ullmann-Type Coupling Reactions. Angew. Chem., Int. Ed 2009, 48, 6954–6971. [DOI] [PubMed] [Google Scholar]
- (280).Monnier F; Taillefer M Catalytic C–C, C–N, and C–O Ullmann-Type Coupling Reactions: Copper Makes a Difference. Angew. Chem., Int. Ed 2008, 47, 3096–3099. [DOI] [PubMed] [Google Scholar]
- (281).Ouali A; Spindler JF; Jutand A; Taillefer M Nitrogen Ligands in Copper-Catalyzed Arylation of Phenols: Structure/Activity Relationships and Applications. Adv. Synth. Catal 2007, 349, 1906–1916. [Google Scholar]
- (282).Giri R; Hartwig JF Cu(I)–Amido Complexes in the Ullmann Reaction: Reactions of Cu(I)–Amido Complexes with Iodoarenes with and without Autocatalysis by CuI. J. Am. Chem. Soc 2010, 132, 15860–15863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Tye JW; Weng Z; Johns AM; Incarvito CD; Hartwig JF Copper Complexes of Anionic Nitrogen Ligands in the Amidation and Imidation of Aryl Halides. J. Am. Chem. Soc 2008, 130, 9971–9983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Tye JW; Weng Z; Giri R; Hartwig JF Copper(I) Phenoxide Complexes in the Etherification of Aryl Halides. Angew. Chem., Int. Ed 2010, 49, 2185–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Strieter ER; Bhayana B; Buchwald SL Mechanistic Studies on the Copper-Catalyzed N-Arylation of Amides. J. Am. Chem. Soc 2009, 131, 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Murugesh V; Harish B; Adiseshu M; Babu Nanubolu J; Suresh S Tandem Copper-Catalyzed N-Arylation–Condensation and van Leusen Reaction: Synthesis of 1,4-Benzodiazepines and Imidazobenzodiazepines (ImBDs). Adv. Synth. Catal 2016, 358, 1309–1321. [Google Scholar]
- (287).Giri R; Brusoe A; Troshin K; Wang JY; Font M; Hartwig JF Mechanism of the Ullmann Biaryl Ether Synthesis Catalyzed by Complexes of Anionic Ligands: Evidence for the Reaction of Iodoarenes with Ligated Anionic CuI Intermediates. J. Am. Chem. Soc 2018, 140, 793–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Kundu S; Greene C; Williams KD; Salvador TK; Bertke JA; Cundari TR; Warren TH Three-Coordinate Copper(II) Aryls: Key Intermediates in C–O Bond Formation. J. Am. Chem. Soc 2017, 139, 9112–9115. [DOI] [PubMed] [Google Scholar]
- (289).Huang Z; Hartwig JF Copper(I) Enolate Complexes in α-Arylation Reactions: Synthesis, Reactivity, and Mechanism. Angew. Chem., Int. Ed 2012, 51, 1028–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Casitas A; King AE; Parella T; Costas M; Stahl SS; Ribas X Direct observation of CuI/CuIII redox steps relevant to Ullmann-type coupling reactions. Chem. Sci 2010, 1, 326–330. [Google Scholar]
- (291).Font M; Parella T; Costas M; Ribas X Catalytic C–S, C– Se, and C–P Cross-Coupling Reactions Mediated by a CuI/CuIII Redox Cycle. Organometallics 2012, 31, 7976–7982. [Google Scholar]
- (292).Casitas A; Canta M; Solà M; Costas M; Ribas X Nucleophilic Aryl Fluorination and Aryl Halide Exchange Mediated by a CuI/CuIII Catalytic Cycle. J. Am. Chem. Soc 2011, 133, 19386–19392. [DOI] [PubMed] [Google Scholar]
- (293).Fier PS; Hartwig JF Copper-Mediated Fluorination of Aryl Iodides. J. Am. Chem. Soc 2012, 134, 10795–10798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Fier PS; Luo J; Hartwig JF Copper-Mediated Fluorination of Arylboronate Esters. Identification of a Copper(III) Fluoride Complex. J. Am. Chem. Soc 2013, 135, 2552–2559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Ye Y; Sanford MS Mild Copper-Mediated Fluorination of Aryl Stannanes and Aryl Trifluoroborates. J. Am. Chem. Soc 2013, 135, 4648–4651. [DOI] [PubMed] [Google Scholar]
- (296).Ley SV; Thomas AW Modern Synthetic Methods for Copper-Mediated C(aryl)-O, C(aryl)-N, and C(aryl)-S Bond For-mation. Angew. Chem., Int. Ed 2003, 42, 5400–5449. [DOI] [PubMed] [Google Scholar]
- (297).King AE; Brunold TC; Stahl SS Mechanistic Study of Copper-Catalyzed Aerobic Oxidative Coupling of Arylboronic Esters and Methanol: Insights into an Organometallic Oxidase Reaction. J. Am. Chem. Soc 2009, 131, 5044–5045. [DOI] [PubMed] [Google Scholar]
- (298).King AE; Ryland BL; Brunold TC; Stahl SS Kinetic and Spectroscopic Studies of Aerobic Copper(II)-Catalyzed Methoxylation of Arylboronic Esters and Insights into Aryl Transmetalation to Copper(II). Organometallics 2012, 31, 7948–7957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Vantourout JC; Miras HN; Isidro-Llobet A; Sproules S; Watson AJB Spectroscopic Studies of the Chan–Lam Amination: A Mechanism-Inspired Solution to Boronic Ester Reactivity. J. Am. Chem. Soc 2017, 139, 4769–4779. [DOI] [PubMed] [Google Scholar]
- (300).Dimakos V; Garrett GE; Taylor MS Site-Selective, Copper-Mediated O-Arylation of Carbohydrate Derivatives. J. Am. Chem. Soc 2017, 139, 15515–15521. [DOI] [PubMed] [Google Scholar]
- (301).Zhang S-L; Bie W-F Isolation and characterization of copper(iii) trifluoromethyl complexes and reactivity studies of aerobic trifluoromethylation of arylboronic acids. RSC Adv 2016, 6, 70902–70906. [Google Scholar]
- (302).Zhang S-L; Bie W-F Ligand-dependent formation of ion-pair CuI/CuIII trifluoromethyl complexes containing bisphosphines. Dalton Trans 2016, 45, 17588–17592. [DOI] [PubMed] [Google Scholar]
- (303).Nebra N; Grushin VV Distinct Mechanism of Oxidative Trifluoromethylation with a Well-Defined Cu(II) Fluoride Promoter: Hidden Catalysis. J. Am. Chem. Soc 2014, 136, 16998–17001. [DOI] [PubMed] [Google Scholar]
- (304).Panferova LI; Miloserdov FM; Lishchynskyi A; Martínez Belmonte M; Benet-Buchholz J; Grushin VV Well-Defined CuC2F5 Complexes and Pentafluoroethylation of Acid Chlorides. Angew. Chem., Int. Ed 2015, 54, 5218–5222. [DOI] [PubMed] [Google Scholar]
- (305).Jacquet J; Salanouve E; Orio M; Vezin H; Blanchard S; Derat E; Desage-El Murr M; Fensterbank L Iminosemiquinone radical ligands enable access to a well-defined redox-active CuII-CF3 complex. Chem. Commun 2014, 50, 10394–10397. [DOI] [PubMed] [Google Scholar]
- (306).Jacquet J; Chaumont P; Gontard G; Orio M; Vezin H; Blanchard S; Desage-El Murr M; Fensterbank L C–N Bond Formation from a Masked High-Valent Copper Complex Stabilized by Redox Non-Innocent Ligands. Angew. Chem., Int. Ed 2016, 55, 10712–10716. [DOI] [PubMed] [Google Scholar]
- (307).Liu L; Zhu M; Yu H-T; Zhang W-X; Xi Z Organocopper(III) Spiro Complexes: Synthesis, Structural Characterization, and Redox Transformation. J. Am. Chem. Soc 2017, 139, 13688–13691. [DOI] [PubMed] [Google Scholar]
- (308).Ribas X; Jackson DA; Donnadieu B; Mahia J; Parella T; Xifra R; Hedman B; Hodgson KO; Llobet A; Stack TDP Aryl C-H activation by CuII to form an organometallic aryl-CuIII species: A novel twist on copper disproportionation. Angew. Chem., Int. Ed 2002, 41, 2991–2994. [DOI] [PubMed] [Google Scholar]
- (309).Ribas X; Calle C; Poater A; Casitas A; Gómez L; Xifra R; Parella T; Benet-Buchholz J; Schweiger A; Mitrikas G; Solà M; Llobet A; Stack TDP Facile C–H Bond Cleavage via a Proton- Coupled Electron Transfer Involving a C–H···CuII Interaction. J. Am. Chem. Soc 2010, 132, 12299–12306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (310).King AE; Huffman LM; Casitas A; Costas M; Ribas X; Stahl SS Copper-Catalyzed Aerobic Oxidative Functionalization of an Arene C–H Bond: Evidence for an Aryl-Copper(III) Intermediate.J. Am. Chem. Soc 2010, 132, 12068–12073. [DOI] [PubMed] [Google Scholar]
- (311).Huffman LM; Stahl SS Carbon–Nitrogen Bond Formation Involving Well-Defined Aryl–Copper(III) Complexes. J. Am. Chem. Soc 2008, 130, 9196–9197. [DOI] [PubMed] [Google Scholar]
- (312).Huffman LM; Casitas A; Font M; Canta M; Costas M; Ribas X; Stahl SS Observation and Mechanistic Study of Facile C–O Bond Formation between a Well-Defined Aryl–Copper(III) Complex and Oxygen Nucleophiles. Chem. - Eur. J 2011, 17, 10643–10650. [DOI] [PubMed] [Google Scholar]
- (313).Huffman LM; Stahl SS Mechanistic analysis of trans C-N reductive elimination from a square-planar macrocyclic aryl-copper(iii) complex. Dalton Trans 2011, 40, 8959–8963. [DOI] [PubMed] [Google Scholar]
- (314).Casitas A; Ioannidis N; Mitrikas G; Costas M; Ribas X Aryl-O reductive elimination from reaction of well-defined aryl-Cuiii species with phenolates: the importance of ligand reactivity. Dalton Trans 2011, 40, 8796–8799. [DOI] [PubMed] [Google Scholar]
- (315).Yao B; Wang D-X; Huang Z-T; Wang M-X Room-temperature aerobic formation of a stable aryl-Cu(iii) complex and its reactions with nucleophiles: highly efficient and diverse arene C-H functionalizations of azacalix[1]arene[3]pyridine. Chem. Commun 2009, 2899–2901. [DOI] [PubMed] [Google Scholar]
- (316).Wang Z-L; Zhao L; Wang M-X Regiospecific Functionalization of Azacalixaromatics through Copper-Mediated Aryl C–H Activation and C–O Bond Formation. Org. Lett 2011, 13, 6560–6563. [DOI] [PubMed] [Google Scholar]
- (317).Wang Z-L; Zhao L; Wang M-X Construction of Caryl– Calkynyl Bond from Copper-Mediated Arene–Alkyne and Aryl Iodide–Alkyne Cross-Coupling Reactions: A Common Aryl-CuIII Intermediate in Arene C–H Activation and Castro–Stephens Reaction. Org. Lett 2012, 14, 1472–1475. [DOI] [PubMed] [Google Scholar]
- (318).Zhang H; Yao B; Zhao L; Wang D-X; Xu B-Q; Wang M-X Direct Synthesis of High-Valent Aryl–Cu(II) and Aryl–Cu(III) Compounds: Mechanistic Insight into Arene C–H Bond Metalation. J. Am. Chem. Soc 2014, 136, 6326–6332. [DOI] [PubMed] [Google Scholar]
- (319).Liu Y; Long C; Zhao L; Wang M-X Functionalization of Azacalixaromatics by Cu(II)-Catalyzed Oxidative Cross-Coupling Reaction between the Arene C–H Bond and Boronic Acids. Org. Lett 2016, 18, 5078–5081. [DOI] [PubMed] [Google Scholar]
- (320).Zhang Q; Liu Y; Wang T; Zhang X; Long C; Wu Y-D; Wang M-X Mechanistic Study on Cu(II)-Catalyzed Oxidative Cross- Coupling Reaction between Arenes and Boronic Acids under Aerobic Conditions. J. Am. Chem. Soc 2018, 140, 5579–5587. [DOI] [PubMed] [Google Scholar]
- (321).Suess AM; Ertem MZ; Cramer CJ; Stahl SS Divergence between Organometallic and Single-Electron-Transfer Mechanisms in Copper(II)-Mediated Aerobic C–H Oxidation. J. Am. Chem. Soc 2013, 135, 9797–9804. [DOI] [PubMed] [Google Scholar]
- (322).Shang M; Sun SZ; Wang HL; Laforteza BN; Dai HX; Yu JQ Exceedingly Fast Copper(II)-Promoted ortho C-H Trifluoromethylation of Arenes using TMSCF3. Angew. Chem., Int. Ed 2014, 53, 10439–10442. [DOI] [PubMed] [Google Scholar]
- (323).Sun S-Z; Shang M; Wang H-L; Lin H-X; Dai H-X; Yu J-Q Cu(II)-Mediated C(sp2)–H Hydroxylation. J. Org. Chem 2015, 80, 8843–8848. [DOI] [PubMed] [Google Scholar]
- (324).Shang M; Shao Q; Sun S-Z; Chen Y-Q; Xu H; Dai H-X; Yu J-Q Identification of monodentate oxazoline as a ligand for copperpromoted ortho-C-H hydroxylation and amination. Chem. Sci 2017, 8, 1469–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Shang M; Wang H-L; Sun S-Z; Dai H-X; Yu J-Q Cu(II)-Mediated ortho C–H Alkynylation of (Hetero)Arenes with Terminal Alkynes. J. Am. Chem. Soc 2014, 136, 11590–11593. [DOI] [PubMed] [Google Scholar]
- (326).Shang M; Sun S-Z; Dai H-X; Yu J-Q Cu(OAc)2-Catalyzed Coupling of Aromatic C–H Bonds with Arylboron Reagents. Org. Lett 2014, 16, 5666–5669. [DOI] [PubMed] [Google Scholar]
- (327).Tran BL; Li B; Driess M; Hartwig JF Copper-Catalyzed Intermolecular Amidation and Imidation of Unactivated Alkanes. J. Am. Chem. Soc 2014, 136, 2555–2563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (328).Vasilopoulos A; Zultanski SL; Stahl SS Feedstocks to Pharmacophores: Cu-Catalyzed Oxidative Arylation of Inexpensive Alkylarenes Enabling Direct Access to Diarylalkanes. J. Am. Chem. Soc 2017, 139, 7705–7708. [DOI] [PubMed] [Google Scholar]
- (329).Zhang W; Wang F; McCann SD; Wang D; Chen P; Stahl SS; Liu G Enantioselective cyanation of benzylic C–H bonds via copper-catalyzed radical relay. Science 2016, 353, 1014–1018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (330).Srivastava RS; Tarver NR; Nicholas KM Mechanistic Studies of Copper(I)-Catalyzed Allylic Amination. J. Am. Chem. Soc 2007, 129, 15250–15258. [DOI] [PubMed] [Google Scholar]
- (331).Cheng L-J; Islam SM; Mankad NP Synthesis of Allylic Alcohols via Cu-Catalyzed Hydrocarbonylative Coupling of Alkynes with Alkyl Halides. J. Am. Chem. Soc 2018, 140, 1159–1164. [DOI] [PubMed] [Google Scholar]
- (332).Creutz SE; Lotito KJ; Fu GC; Peters JC Photoinduced Ullmann C–N Coupling: Demonstrating the Viability of a Radical Pathway. Science 2012, 338, 647–651. [DOI] [PubMed] [Google Scholar]
- (333).Kainz QM; Matier CD; Bartoszewicz A; Zultanski SL; Peters JC; Fu GC Asymmetric copper-catalyzed C-N crosscouplings induced by visible light. Science 2016, 351, 681–684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (334).Matier CD; Schwaben J; Peters JC; Fu GC Copper- Catalyzed Alkylation of Aliphatic Amines Induced by Visible Light. J. Am. Chem. Soc 2017, 139, 17707–17710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (335).Do H-Q; Bachman S; Bissember AC; Peters JC; Fu GC Photoinduced, Copper-Catalyzed Alkylation of Amides with Unactivated Secondary Alkyl Halides at Room Temperature. J. Am. Chem. Soc 2014, 136, 2162–2167. [DOI] [PubMed] [Google Scholar]
- (336).Ziegler DT; Choi J; Muñoz-Molina JM; Bissember AC; Peters JC; Fu GC A Versatile Approach to Ullmann C–N Couplings at Room Temperature: New Families of Nucleophiles and Electrophiles for Photoinduced, Copper-Catalyzed Processes. J. Am. Chem. Soc 2013, 135, 13107–13112. [DOI] [PubMed] [Google Scholar]
- (337).Uyeda C; Tan Y; Fu GC; Peters JC A New Family of Nucleophiles for Photoinduced, Copper-Catalyzed Cross-Couplings via Single-Electron Transfer: Reactions of Thiols with Aryl Halides Under Mild Conditions (O °C). J. Am. Chem. Soc 2013, 135, 9548–9552. [DOI] [PubMed] [Google Scholar]
- (338).Ahn JM; Peters JC; Fu GC Design of a Photoredox Catalyst that Enables the Direct Synthesis of Carbamate-Protected Primary Amines via Photoinduced, Copper-Catalyzed N-Alkylation Reactions of Unactivated Secondary Halides. J. Am. Chem. Soc 2017, 139, 18101–18106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (339).Sagadevan A; Charpe VP; Ragupathi A; Hwang KC Visible Light Copper Photoredox-Catalyzed Aerobic Oxidative Coupling of Phenols and Terminal Alkynes: Regioselective Synthesis of Functionalized Ketones via C≡C Triple Bond Cleavage. J. Am. Chem. Soc 2017, 139, 2896–2899. [DOI] [PubMed] [Google Scholar]
- (340).Nishikata T; Ishida S; Fujimoto R Site-Selective Tertiary Alkyl–Fluorine Bond Formation from α-Bromoamides Using a Copper/CsF Catalyst System. Angew. Chem., Int. Ed 2016, 55, 10008–10012. [DOI] [PubMed] [Google Scholar]
- (341).Chen X; Hao X-S; Goodhue CE; Yu J-Q Cu(II)- Catalyzed Functionalizations of Aryl C–H Bonds Using O2 as an Oxidant. J. Am. Chem. Soc 2006, 128, 6790–6791. [DOI] [PubMed] [Google Scholar]
- (342).Chen X; Dobereiner G; Hao X-S; Giri R; Maugel N; Yu J-Q Cu(II)-mediated oxidative dimerization of 2-phenylpyridine derivatives. Tetrahedron 2009, 65, 3085–3089. [Google Scholar]
- (343).Michaudel Q; Thevenet D; Baran PS Intermolecular Ritter- Type C–H Amination of Unactivated sp3 Carbons. J. Am. Chem. Soc 2012, 134, 2547–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (344).Sathyamoorthi S; Lai Y-H; Bain RM; Zare RN Mechanistic Analysis of the C–H Amination Reaction of Menthol by CuBr2 and Selectfluor. J. Org. Chem 2018, 83, 5681. [DOI] [PubMed] [Google Scholar]
- (345).Liu T; Myers MC; Yu JQ Copper-Catalyzed Bromination of C(sp3)–H Bonds Distal to Functional Groups. Angew. Chem., Int. Ed 2017, 56, 306–309. [DOI] [PubMed] [Google Scholar]
- (346).Jie X; Shang Y; Zhang X; Su W Cu-Catalyzed Sequential Dehydrogenation–Conjugate Addition for β-Functionalization of Saturated Ketones: Scope and Mechanism. J. Am. Chem. Soc 2016, 138, 5623–5633. [DOI] [PubMed] [Google Scholar]
- (347).Gephart RT; McMullin CL; Sapiezynski NG; Jang ES; Aguila MJB; Cundari TR; Warren TH Reaction of CuI with Dialkyl Peroxides: CuII-Alkoxides, Alkoxy Radicals, and Catalytic C–H Etherification. J. Am. Chem. Soc 2012, 134, 17350–17353. [DOI] [PubMed] [Google Scholar]
- (348).Salvador TK; Arnett CH; Kundu S; Sapiezynski NG; Bertke JA; Raghibi Boroujeni M; Warren TH Copper Catalyzed sp3 C–H Etherification with Acyl Protected Phenols. J. Am. Chem. Soc 2016, 138, 16580–16583. [DOI] [PubMed] [Google Scholar]
- (349).Tran BL; Driess M; Hartwig JF Copper-Catalyzed Oxidative Dehydrogenative Carboxylation of Unactivated Alkanes to Allylic Esters via Alkenes. J. Am. Chem. Soc 2014, 136, 17292–17301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (350).Li Z; Li C-J CuBr-Catalyzed Efficient Alkynylation of sp3 C– H Bonds Adjacent to a Nitrogen Atom. J. Am. Chem. Soc 2004, 126, 11810–11811. [DOI] [PubMed] [Google Scholar]
- (351).Li Z; Li C-J Highly Efficient Copper-Catalyzed Nitro- Mannich Type Reaction: Cross-Dehydrogenative-Coupling between sp3 C–H Bond and sp3 C–H Bond. J. Am. Chem. Soc 2005, 127, 3672–3673. [DOI] [PubMed] [Google Scholar]
- (352).Li Z; Li C-J CuBr-Catalyzed Direct Indolation of Tetrahydroisoquinolines via Cross-Dehydrogenative Coupling between sp3 C–H and sp2 C–H Bonds. J. Am. Chem. Soc 2005, 127, 6968–6969. [DOI] [PubMed] [Google Scholar]
- (353).Boess E; Wolf LM; Malakar S; Salamone M; Bietti M; Thiel W; Klussmann M Competitive Hydrogen Atom Transfer to Oxyl- and Peroxyl Radicals in the Cu-Catalyzed Oxidative Coupling of N-Aryl Tetrahydroisoquinolines Using tert-Butyl Hydroperoxide. ACS Catal 2016, 6, 3253–3261. [Google Scholar]
- (354).Kundu R; Ball ZT Copper-Catalyzed Remote sp3 C–H Chlorination of Alkyl Hydroperoxides. Org. Lett 2010, 12, 2460–2463. [DOI] [PubMed] [Google Scholar]
- (355).Bunescu A; Wang Q; Zhu J Synthesis of Functionalized Epoxides by Copper-Catalyzed Alkylative Epoxidation of Allylic Alcohols with Alkyl Nitriles. Org. Lett 2015, 17, 1890–1893. [DOI] [PubMed] [Google Scholar]
- (356).Shen K; Wang Q Copper-catalyzed diamination of unactivated alkenes with hydroxylamines. Chem. Sci 2015, 6, 4279–4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (357).Shen K; Wang Q Copper-catalyzed aminotrifluoromethylation of alkenes: a facile synthesis of CF3-containing lactams. Org. Chem. Front 2016, 3, 222–226. [Google Scholar]
- (358).Hemric BN; Shen K; Wang Q Copper-Catalyzed Amino Lactonization and Amino Oxygenation of Alkenes Using O-Benzoylhydroxylamines. J. Am. Chem. Soc 2016, 138, 5813–5816. [DOI] [PubMed] [Google Scholar]
- (359).Shen K; Wang Q Copper-Catalyzed Alkene Aminoazidation as a Rapid Entry to 1,2-Diamines and Installation of an Azide Reporter onto Azahetereocycles. J. Am. Chem. Soc 2017, 139, 13110–13116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (360).Shen K; Wang Q Copper-catalyzed aminoalkynylation of alkenes with hypervalent iodine reagents. Chem. Sci 2017, 8, 8265–8270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Dai J-L; Shao N-Q; Zhang J; Jia R-P; Wang D-H Cu(II)- Catalyzed ortho-Selective Aminomethylation of Phenols. J. Am. Chem. Soc 2017, 139, 12390–12393. [DOI] [PubMed] [Google Scholar]
- (362).Romero EA; Jazzar R; Bertrand G Copper-catalyzed dehydrogenative borylation of terminal alkynes with pinacolborane. Chem. Sci 2017, 8, 165–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Jin L; Romero EA; Melaimi M; Bertrand G The Janus Face of the X Ligand in the Copper-Catalyzed Azide–Alkyne Cycloaddition.J. Am. Chem. Soc 2015, 137, 15696–15698. [DOI] [PubMed] [Google Scholar]
- (364).Zhou L; Yi H; Zhu L; Qi X; Jiang H; Liu C; Feng Y; Lan Y; Lei A Tuning the Reactivity of Radical through a Triplet Diradical Cu(II) Intermediate in Radical Oxidative Cross-Coupling. Sci. Rep 2015, 5, 15934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).Chiba S; Zhang L; Ang GY; Hui BW-Q Generation of Iminyl Copper Species from α-Azido Carbonyl Compounds and Their Catalytic C–C Bond Cleavage under an Oxygen Atmosphere. Org. Lett 2010, 12, 2052–2055. [DOI] [PubMed] [Google Scholar]
- (366).Chiba S; Zhang L; Lee J-Y Copper-Catalyzed Synthesis of Azaspirocyclohexadienones from α-Azido-N-arylamides under an Oxygen Atmosphere. J. Am. Chem. Soc 2010, 132, 7266–7267. [DOI] [PubMed] [Google Scholar]
- (367).Tnay YL; Chen C; Chua YY; Zhang L; Chiba S Copper- Catalyzed Aerobic Spirocyclization of Biaryl-N-H-imines via 1,4- Aminooxygenation of Benzene Rings. Org. Lett 2012, 14, 3550–3553. [DOI] [PubMed] [Google Scholar]
- (368).Toh KK; Sanjaya S; Sahnoun S; Chong SY; Chiba S Copper-Catalyzed Aerobic Intramolecular Carbo- and Amino-Oxygenation of Alkynes for Synthesis of Azaheterocycles. Org. Lett 2012, 14, 2290–2292. [DOI] [PubMed] [Google Scholar]
- (369).Zhang L; Ang GY; Chiba S Copper-Catalyzed Benzylic C– H Oxygenation under an Oxygen Atmosphere via N-H Imines as an Intramolecular Directing Group. Org. Lett 2011, 13, 1622–1625. [DOI] [PubMed] [Google Scholar]
- (370).Wang Y-F; Chen H; Zhu X; Chiba S Copper-Catalyzed Aerobic Aliphatic C–H Oxygenation Directed by an Amidine Moiety.J. Am. Chem. Soc 2012, 134, 11980–11983. [DOI] [PubMed] [Google Scholar]
- (371).Liang Y-F; Jiao N Oxygenation via C–H/C–C Bond Activation with Molecular Oxygen. Acc. Chem. Res 2017, 50, 1640–1653. [DOI] [PubMed] [Google Scholar]
- (372).Zhang C; Jiao N Dioxygen Activation under Ambient Conditions: Cu-Catalyzed Oxidative Amidation–Diketonization of Terminal Alkynes Leading to α-Ketoamides. J. Am. Chem. Soc 2010, 132, 28–29. [DOI] [PubMed] [Google Scholar]
- (373).Zhang C; Xu Z; Zhang L; Jiao N Copper-Catalyzed Aerobic Oxidative Coupling of Aryl Acetaldehydes with Anilines Leading to α-Ketoamides. Angew. Chem., Int. Ed 2011, 50, 11088–11092. [DOI] [PubMed] [Google Scholar]
- (374).Zhang C; Zong X; Zhang L; Jiao N Copper-Catalyzed Aerobic Oxidative Cross-Dehydrogenative Coupling of Amine and α-Carbonyl Aldehyde: A Practical and Efficient Approach to α-Ketoamides with Wide Substrate Scope. Org. Lett 2012, 14, 3280–3283. [DOI] [PubMed] [Google Scholar]
- (375).Kumar Y; Shaw M; Thakur R; Kumar A Copper(II)- Mediated Aerobic Oxidation of Benzylimidates: Synthesis of Primary α-Ketoamides. J. Org. Chem 2016, 81, 6617–6625. [DOI] [PubMed] [Google Scholar]
- (376).Xu Z; Zhang C; Jiao N Synthesis of Oxazoles through Copper-Mediated Aerobic Oxidative Dehydrogenative Annulation and Oxygenation of Aldehydes and Amines. Angew. Chem., Int. Ed 2012, 51, 11367–11370. [DOI] [PubMed] [Google Scholar]
- (377).Zhang C; Feng P; Jiao N Cu-Catalyzed Esterification Reaction via Aerobic Oxygenation and C–C Bond Cleavage: An Approach to α-Ketoesters. J. Am. Chem. Soc 2013, 135, 15257–15262. [DOI] [PubMed] [Google Scholar]
- (378).Huang X; Li X; Zou M; Song S; Tang C; Yuan Y; Jiao N From Ketones to Esters by a Cu-Catalyzed Highly Selective C(CO)– C(alkyl) Bond Cleavage: Aerobic Oxidation and Oxygenation with Air.J. Am. Chem. Soc 2014, 136, 14858–14865. [DOI] [PubMed] [Google Scholar]
- (379).Wang Z-Q; Zhang W-W; Gong L-B; Tang R-Y; Yang X-H; Liu Y; Li J-H Copper-Catalyzed Intramolecular Oxidative 6-exotrig Cyclization of 1,6-Enynes with H2O and O2. Angew. Chem., Int. Ed 2011, 50, 8968–8973. [DOI] [PubMed] [Google Scholar]
- (380).Song R-J; Liu Y; Hu R-X; Liu Y-Y; Wu J-C; Yang X-H; Li J-H Oxidative Cleavage of the Carbon-Carbon σ-Bond Using Reusable Copper on Iron. Adv. Synth. Catal 2011, 353, 1467–1473. [Google Scholar]
- (381).Wang H; Wang Y; Liang D; Liu L; Zhang J; Zhu Q Copper-Catalyzed Intramolecular Dehydrogenative Aminooxygenation: Direct Access to Formyl-Substituted Aromatic N-Heterocycles. Angew. Chem., Int. Ed 2011, 50, 5678–5681. [DOI] [PubMed] [Google Scholar]
- (382).Xia X-F; Zhang L-L; Song X-R; Liu X-Y; Liang Y-M Copper-Catalyzed Oxidative Cyclization of Enynes for the Synthesis of 4-Carbonyl-quinolines with O2. Org. Lett 2012, 14, 2480–2483. [DOI] [PubMed] [Google Scholar]
- (383).Wang J; Wang J; Zhu Y; Lu P; Wang Y Copper-cascade catalysis: synthesis of 3-functionalized indoles. Chem. Commun 2011, 47, 3275–3277. [DOI] [PubMed] [Google Scholar]
- (384).Selvaraju M; Ye T-Y; Li C-H; Ho P-H; Sun C-M Copper catalyzed aerobic oxidative cyclization and ketonization: one pot synthesis of benzoimidazo[1,2-a]imidazolones. Chem. Commun 2016, 52, 6621–6624. [DOI] [PubMed] [Google Scholar]
- (385).Ye T-Y; Selvaraju M; Sun C-M Cascade Synthesis of Benzimidazole-Linked Pyrroles via Copper Catalyzed Oxidative Cyclization and Ketonization. Org. Lett 2017, 19, 3103–3106. [DOI] [PubMed] [Google Scholar]
- (386).Zhang H-J; Schuppe AW; Pan S-T; Chen J-X; Wang B-R; Newhouse TR; Yin L Copper-Catalyzed Vinylogous Aerobic Oxidation of Unsaturated Compounds with Air. J. Am. Chem. Soc 2018, 140, 5300–5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (387).Tezuka N; Shimojo K; Hirano K; Komagawa S; Yoshida K; Wang C; Miyamoto K; Saito T; Takita R; Uchiyama M Direct Hydroxylation and Amination of Arenes via Deprotonative Cupration.J. Am. Chem. Soc 2016, 138, 9166–9171. [DOI] [PubMed] [Google Scholar]
- (388).Blakemore DC; Castro L; Churcher I; Rees DC; Thomas AW; Wilson DM; Wood A Organic synthesis provides opportunities to transform drug discovery. Nat. Chem 2018, 10, 383–394. [DOI] [PubMed] [Google Scholar]
- (389).Cernak T; Dykstra KD; Tyagarajan S; Vachal P; Krska SW The medicinal chemist’s toolbox for late stage functionalization of drug-like molecules. Chem. Soc. Rev 2016, 45, 546–576. [DOI] [PubMed] [Google Scholar]
- (390).Friedfeld MR; Zhong H; Ruck RT; Shevlin M; Chirik PJ Cobalt-catalyzed asymmetric hydrogenation of enamides enabled by single-electron reduction. Science 2018, 360, 888–893. [DOI] [PubMed] [Google Scholar]
- (391).Nam W; Lee Y-M; Fukuzumi S Hydrogen Atom Transfer Reactions of Mononuclear Nonheme Metal–Oxygen Intermediates. Acc. Chem. Res 2018, 51, 2014–2022. [DOI] [PubMed] [Google Scholar]
- (392).Chirik PJ; Wieghardt K Radical Ligands Confer Nobility on Base-Metal Catalysts. Science 2010, 327, 794–795. [DOI] [PubMed] [Google Scholar]
- (393).Broere DLJ; Plessius R; van der Vlugt JI New avenues for ligand-mediated processes – expanding metal reactivity by the use of redox-active catechol, o-aminophenol and o-phenylenediamine ligands. Chem. Soc. Rev 2015, 44, 6886–6915. [DOI] [PubMed] [Google Scholar]
- (394).Wang F; Chen P; Liu G Copper-Catalyzed Radical Relay for Asymmetric Radical Transformations. Acc. Chem. Res 2018, 51, 2036–2046. [DOI] [PubMed] [Google Scholar]