

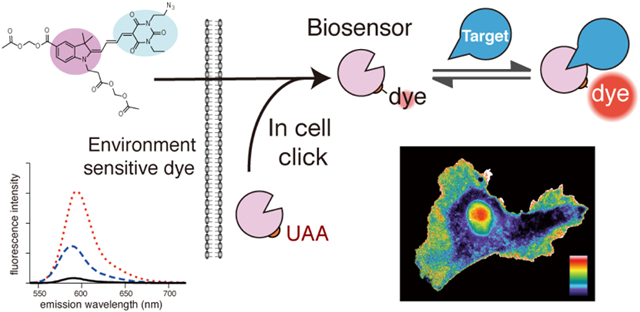
Abstract
Dyes with environment-sensitive fluorescence have proven useful to study the spatio-temporal dynamics of protein activity in living cells. When attached to proteins, their fluorescence can reflect protein conformational changes, posttranslational modifications or protein interactions. However, the utility of such dye-protein conjugates has been limited because it is difficult to load them into cells. They usually must be introduced using techniques that perturb cell physiology, limit throughput, or generate fluorescent vesicles (e.g. electroporation, microinjection, or membrane transduction peptides). Here we circumvent these problems by modifying a proven, environment-sensitive biosensor fluorophore so that it can pass through cell membranes without staining intracellular compartments, and can be attached to proteins within living cells using unnatural amino acid (UAA) mutagenesis. Reactive groups were incorporated for attachment to UAAs or small molecules (mero166, azide; mero167, alkyne; mero76, carboxylic acid). These dyes are bright and fluoresce at long wavelengths (reaching ε = 100,000 M−1cm−1, φ = 0.24, with excitation 565 nm and emission 594 nm). The utility of mero166 was demonstrated by in-cell labeling of an UAA to generate a biosensor for the small GTPase Cdc42. In addition, conjugation of mero166 to a small molecule produced a membrane-permeable probe that reported the localization of the DNA methyltransferase G9a in cells. This approach provides a strategy to access biosensors for many targets, and to more practically harness the varied environmental sensitivities of synthetic dyes.
Graphical Abstract
INTRODUCTION
The same protein can be activated at different subcellular locations or with different kinetics to produce very different cell behaviors. To understand how signaling networks are regulated, it is often essential to quantify the spatio-temporal dynamics of protein conformational changes in living cells. This is frequently accomplished using biosensors based on fluorescence resonance energy transfer (FRET) between fluorescent proteins; many such biosensors are fully genetically encoded so can be readily expressed in cells.1-8 Another class of biosensor is based on covalently attaching environment-sensitive fluorescent dyes to proteins at positions where their fluorescence responds to conformational changes or protein-protein interactions. This approach can be more sensitive than FRET because the fluorophore is excited directly (rather than indirectly by the FRET donor) and because one can use dyes that are brighter than fluorescent proteins. The dye is either attached directly to the protein of interest,9-10 or to a protein fragment that binds selectively to one conformation of the targeted protein. In the latter case, the protein-dye “affinity reagent” produces a unique fluorescence signature when it binds to the activated target.11-16 The ability to interrogate endogenous, unmodified proteins is an important advantage of dye-based biosensors. Despite their advantages, dye-based biosensors are used much less frequently than genetically encoded biosensors because they are difficult to load into living cells. Delivery has been accomplished using import-transducing peptides that rely on endocytic uptake followed by rupture of internal vesicles,17-19 but this can produce fluorescent vesicles that interfere with imaging. Techniques that pass labeled proteins through holes in the cell membrane (e.g. microinjection, electroporation, scrape loading, bead loading, syringe loading)20-22 are difficult for many cell types to tolerate.
Here we develop a membrane-permeant variant of an environment sensitive fluorophore that has proven utility for biosensor imaging. Attachment of appropriate side chains enabled it to pass effectively through the membrane, and importantly, not stain intracellular compartments. Using unnatural amino acid (UAA) mutagenesis, the dye was site-specifically attached to an expressed protein, effectively assembling a biosensor within cells. Alternately, the dye was attached not to a protein, but to a small molecule with specific binding affinity for the targeted protein. This generated a biosensor that in its entirety could pass through the cell membrane.
Intracellular labeling of proteins using unnatural amino acids has been accomplished with bright dyes suitable for intracellular imaging, but they are not environment-sensitive, so report protein localization but not conformation.23-29 To function as part of a biosensor that reports protein conformation, the dye must undergo fluorescence changes that can be detected even at low intracellular concentrations where the biosensor does not perturb cell behavior. The ability to detect fluorescence changes is a function of both the dye’s brightness and the magnitude of the changes. Some small environment-sensitive dyes pass through membranes and have strong environment-sensitivity, but they are too dim for most biosensor applications.30 We based our dye development on extensively characterized merocyanine fluorophores that have a useful combination of brightness and environment sensitivity and with extended conjugation for excitation above 550 nm (to avoid cellular autofluorescence).6, 11,31-37 We sought to confer membrane permeability on these proven dyes, to enable biosensor production within cells, and for generating membrane-permeable “small molecule biosensors.”
RESULTS AND DISCUSSION
We began with our merocyanine fluorophores that have been optimized for use in biosensors and have a proven track record of reporting protein function in live cells.11-12, 15, 31-33 Merocyanine dyes contain electron donor and acceptor components that are linked through a system of conjugation, usually double bonds. This configuration results in a ground state that may be represented as a resonance hybrid of charged and uncharged forms, depending on the relative strength of donor and acceptor groups.38-39 The potential for charge redistribution across the polyene system renders these dyes especially sensitive to polarity and hydrogen bonding interactions from the solvent environment, as shown in previous studies from our lab and others.31, 34, 36-40 Thus, changes in the local environment produced by protein interactions can result in significant changes in fluorescence intensity as well as shifts in excitation and/or emission maxima.41
Because they bear charged groups for water solubility and have extended conjugation to emit brightly at > 500 nm, merocyanine dyes are poorly membrane permeant. When side chains are manipulated to reduce water solubility and thereby enhance membrane permeability, the dyes stain intracellular membranes including the Golgi apparatus, mitochondria, and endoplasmic reticulum. We attempted to reduce these interactions and achieve uniform intracellular distribution of the dyes by attaching acetoxymethyl (AM) esters to several of the previously developed merocyanine fluorophores (data not shown). These studies revealed that both staining of intracellular membranes and membrane permeability were strongly impacted by the number and position of the AM ester side chains. Further, the effects of the AM ester depended on the specific merocyanine fluorophore tested, likely because of the differences in overall charge distribution among the dyes.34-35, 37, 41-45 Based on these preliminary observations, the indolenine-barbituric acid fluorophore (I-BA, an indolenine ring I linked to a barbituric acid BA by four carbons) was chosen for further optimization (compound 1, Figure 1a). This fluorophore showed both reduced membrane staining and a suitable trade-off between brightness, photostability, and solvent sensitivity. We examined the membrane permeability and subcellular distribution of the underivatized parent merocyanine 1, two versions of the dye with single AM esters (2 and 3), and two versions of the dye containing two AM esters (4 and 5).
Figure 1.
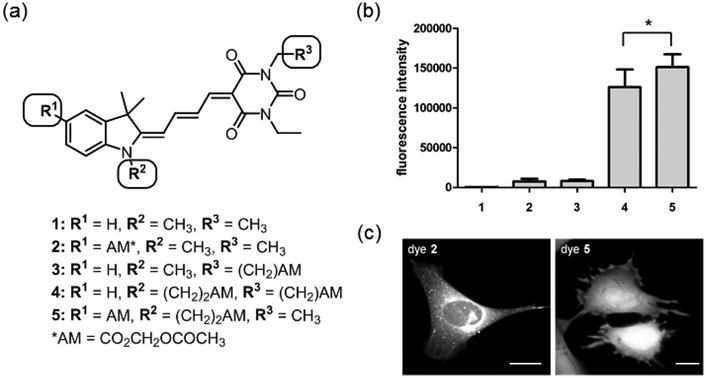
Design and screening of merocyanine derivatives for cell entry. (a) Overview of structures. (b) Fluorescence intensity of each dye in the initial screening set. Fluorescence intensity (FI) values were recorded at the peak of the emission curve for each dye when excited at the excitation maximum (p < 0.5, n = 7 samples). (c) Distribution of dyes in MEF cells. These images were scaled individually to show subcellular distribution, so do not indicate the relative brightness of each dye. The intensity difference between the nucleus and the cell edge is due to variation in cell thickness, as shown by normalization with volume indicators. (Scale bar 20 μm, excitation filter 545/50, emission filter 630/45).
To screen the dyes for potential cellular uptake, mouse embryonic fibroblast cells (MEFs) were incubated in medium containing a test compound (10 μM) for 30 minutes, followed by aspiration of the dye solution and washing of the cells with compound-free medium. The cells were suspended and fluorescence intensity (FI) was determined for each dye, with values normalized for cell number based on the relative intensity of the nuclear stain Hoechst 33342, which was included with the dye in each test solution. As expected, the unmodified merocyanine dye 1 showed negligible import into cells (Figure 1b). Merocyanines containing a single AM ester showed only small improvements in fluorescent dye uptake relative to 1, but the addition of two AM esters produced a substantial increase in dye retention. Compound 5, which bore two AM esters on the indolenine ring, produced significantly more fluorescence than did compound 4, which had one AM ester on the indolenine ring (I) and the other on the barbituric acid ring (BA). Direct visualization of adherent cells showed that dyes 2 and 3 had entered cells to some extent, but were primarily located in intracellular membranes and vesicles (Figure 1c). This may have been due to selective membrane staining or enhanced vesicular uptake. Dye 5 showed the same uniform intracellular distribution as homogenously localized “volume indicators” loaded in the same cell, with no accumulation in subcellular membranes or organelles (YFP or carboxyfluorescein diacetate acetoxymethyl ester, Figure S1). Cells loaded with dye 5 showed no signs of toxicity during 90 minutes of imaging. Uniform distribution of dye 5 was maintained until the extracellular dye concentration was reduced to 2.5 μM, presumably when dye fluorescence became comparable to cellular autofluorescence. The intensity of dye 5 in cells decreased rapidly following initial treatment (Figure S2), an advantage for biosensors, as unattached dye would not contribute to background fluorescence. We were concerned that this indicated lack of AM ester cleavage, but Cdc42 biosensor made with the dye behaved normally (see below). Based on these results, merocyanine 5 was chosen as the lead structure for further development into conjugatable versions.
Synthesis of mero76, a conjugatable version of compound 5 bearing a carboxylic acid side chain, began from the barbituric acid derivative 6 (Scheme 1), obtained in 3 steps from β-alanine methyl ester hydrochloride. Compound 6 was first converted to the activated diene 7 and the free carboxylic acid was then coupled with the dimethoxy nitrobenzyl (DMNB)46-47 photolabile protecting group to give compound 8. The di-carboxy indolenine salt 9 was reacted with 8 under mildly basic conditions to give a merocyanine product that after installation of the two acetoxymethyl esters and photodeprotection of DMNB provided mero76. Azide- and alkyne-containing merocyanines, mero166 and mero167 respectively, were prepared through an analogous route involving reaction of compound 9 with an azide or alkyne containing barbituric acid, but without the need to utilize protecting group chemistry (see Supporting Information). All dyes maintained good brightness and solvent sensitivity relative to the non-derivatized parent structure (Table 1, Figure 2, Figure S3).33 Merol66 was sensitive to viscosity (Figure S4), indicating that rigidification upon protein interaction could contribute to fluorescence changes. The dye was not toxic to HeLa cells when incubated for up to 60 minutes at 10 μM concentration (Figure S5).
Scheme 1.

Synthesis of the conjugatable dye mero76.
Table 1.
Photophysical properties of conjugatable merocyanines bearing acetoxymethyl ester groups.
 |
|||||||
|---|---|---|---|---|---|---|---|
| mero # | R | Solventa | ex λmax (nm) |
em λmax (nm) |
ε (M−1cm−1) | Φ | brightness (ε × φ) |
| 76 | CO2H | DMSO | 564 | 594 | 70000 | 0.12 | 8400 |
| MeOH | 555 | 588 | 56000 | 0.08 | 4480 | ||
| H2O | 569 | 590 | 28000 | 0.02 | 560 | ||
| 166 | N3 | DMSO | 565 | 594 | 100000 | 0.24 | 24000 |
| MeOH | 559 | 588 | 91000 | 0.11 | 10010 | ||
| H2O | 570 | 591 | 30000 | 0.02 | 600 | ||
| 167 | CCH | DMSO | 566 | 594 | 116000 | 0.21 | 24360 |
| MeOH | 558 | 588 | 85000 | 0.09 | 7650 | ||
| H2O | 569 | 591 | 22000 | 0.06 | 1320 | ||
H2O contained 0.1% DMSO.
Figure 2.

Emission fluorescence spectra of conjugatable merocyanine dyes.
We next examined whether the dye mero166 could be used to build a fluorescent small molecule that served as a reporter/sensor for a specific protein in living cells. For this we targeted the DNA methyltransferase G9a. A selective ligand for G9a was functionalized with an alkynyl side chain and conjugated to mero166 via click chemistry to give compound 10 (Scheme 2).48 MEF cells were incubated with either the dye alone (mero166) or the reporter compound 10 for 20 minutes, washed, and imaged. Cells treated with mero166 showed uniform fluorescence intensity throughout the cell, indicating that the reactive side chain did not appreciably affect cellular uptake or uniform distribution (Figure 3). In contrast, the inhibitor conjugate 10 (G9a sensor) was localized primarily in the nucleus, consistent with the function of G9a, a well-characterized histone methyltransferase.49 Some localization in cytoplasmic vesicles was also observed. In the future, this approach can be applied to small molecules with specific affinity for the target but without target inhibition. Application of this G9a probe in biological studies would require titration to levels where cell physiology was not perturbed.
Scheme 2.

Compound mero166 with azide side chain and conjugation to G9a ligand.
Figure 3.

MEF treated with either mero166 alone, or mero166 conjugated to an inhibitor of histone methyl transferase G9a. Only the latter shows nuclear localization, consistent with the function of G9a. (Scale bar 20 μm, excitation filter 545/50, emission filter 630/45).
We also used mero166 to generate a biosensor by covalently labeling a protein within living cells. Previous dye-based biosensors required protein expression and isolation, followed by dye labeling, re-purification, and mechanical introduction into cells. Use of such biosensors could be greatly enhanced if this cumbersome process were replaced by transfection followed by in-cell labeling. This was made possible by recent methods to label proteins bearing unnatural amino acids (UAA) in mammalian cells.28, 50 We tested feasibility by using a mammalian expression vector containing a nuclear localization signal (NLS), followed by a short sequence containing the amber stop codon (TAG) and EGFP. For cells transfected with this construct, EGFP fluorescence depended on successful incorporation of the UAA into the expressed protein. EGFP expression should also only be present in cells co-transfected with the pyrrolysyl-tRNA synthetase (PylRS)/tRNACUA pair. We chose to use a variant of the M. barkeri (Mb) synthetase with mutations Y271M, L274G, and C321A that confer specificity for the unnatural amino acid bicyclononyne lysine (BCN-lysine).51-52 The click reaction between BCN and azides has been shown to proceed under copper-free conditions with highly favorable reaction kinetics.53-54 HeLa cells were transfected with NLS-TAG-EGFP either with or without co-transfection with the PylRS/tRNACUA pair, and with or without addition of BCN-lysine (0.5 mM) to the cell culture media. Only among cells treated with all components was EGFP fluorescence observable (Figure 4). Due to the NLS, the fluorescence was concentrated in the nucleus, brighter in puncta morphologically consistent with nucleoli. Although cells in all conditions were treated with mero166 for 20 minutes, washed, and allowed to recover for 2 h, clear co-localization of mero166 and EGFP fluorescence was seen only in cells co-transfected with the PylRS/tRNACUA pair and in the presence of the BCN-lysine UAA (Figure S6). Cells that had not been treated with UAA showed virtually no background fluorescence from mero166. However, some background mero166 fluorescence was observed in cells that had been UAA treated, regardless of whether or not co-transfection with the PylRS/tRNACUA pair had taken place. This background mero166 fluorescence may have resulted from in-cell reaction between residual BCN-lysine and mero166, which could create a conjugate fluorescent product that is exported from the cell less efficiently than mero166 itself. The amount of time between UAA wash-out and subsequent dye labeling could be optimized for each protein target to reduce this background, depending on the stability of the expressed protein. Inhibition of organic anion transport caused retention of the dye in the cell, indicating that the AM esters are cleaved during the incubation times used, and that the anionic form of the dye must be actively exported (Figure S7).
Figure 4.

HeLa cells transfected with test construct NLS-TAG-EGFP with or without cotransfection of the plasmid containing the tRNA synthetase/tRNA pair. Only in cells containing both plasmids and in the presence of UAA was co-localization of nuclear EGFP fluorescence and mero166 fluorescence observed. NLS, nuclear localization signal. (Scale bar 20 μm, dye excitation filter 545/50, dye emission filter 630/45, EGFP excitation filter 470/40, EGFP emission filter 630/45).
Having established a method for in-cell labeling using mero166, we sought to generate a functional protein-based biosensor by reconstructing our meroCBD biosensor that reports the conformation of the small GTPase Cdc42.11 Cdc42 is a member of the Rho family of GTPases, proteins that act as molecular signaling switches as they cycle between inactive, GDP-bound and active, GTP-bound conformations.55 The Cdc42 binding domain (CBD) derived from the Wiskott-Aldrich syndrome protein (WASP) binds only to the activated, GTP-bound conformation of Cdc42.11, 56 In our published biosensor, this domain was mutated to include a single cysteine at position 271, a residue known to be near a hydrophobic pocket generated when CBD binds to activated Cdc42.57 Labeling with an environment-sensing fluorophore at this position resulted in a biosensor whose fluorescence intensity increased when it bound to activated Cdc42 in cells.11 We sought to generate this biosensor by introducing BCN-lysine at residue 271 of CBD, followed by reaction in the cell with mero166.
Mammalian expression vectors for both wild type CBD and a mutant with reduced Cdc42 binding (mCBD; H246D, H249D)11, 59 were created, each with the amber stop codon (TAG) introduced at F271 and fused to the amino terminus of EGFP. As with the NLS-TAG-EGFP constructs, due to the placement of the TAG codon, EGFP fluorescence depended on UAA incorporation into the expressed protein. Expression of both CBD-TAG-EGFP and mCBD-TAG-EGFP was carried out in HeLa cells via co-transfection with an optimized mammalian expression vector containing the PylRS/tRNACUA pair (see Supporting Information)50 and in the presence of the BCN-lysine UAA. Subsequent labeling of CBD-EGFP with mero166 showed clear colocalization of dye and EGFP fluorescence; no such co-localization was seen when cells were treated with dye 5, which does not contain a reactive functional group but is otherwise structurally analogous to mero166 (Figure S8). Flow cytometry analysis showed a clear correlation between the level of EGFP expression and the extent of dye labeling (Figure S9). Transfecting constitutively active Cdc42 (Cdc42Q61L) affected the dye/GFP fluorescence ratio of cells expressing CBD much more than cells expressing mCBD (Figure 5a). The Cdc42 inhibitor ML 141 significantly reduced the dye/GFP ratio of the wt biosensor. Finally, CBD-TAG-EGFP expressed in HeLa cells was labeled with mero166 and imaged over time to observe Cdc42 activation during cell migration. Consistent with earlier studies, this Cdc42 biosensor showed Cdc42 activity localized at active regions of the cell edges and in protrusions (Figure 5b).6, 11, 15 Uniform background fluorescence from unattached dye could be greatly reduced by washing the cells in fresh media. Use of the mCBD-TAG-EGFP control construct reduced the level of observed Cdc42 activation and failed to show the localization observed with CBD. Background fluorescence was present to some degree in these studies, in the nucleus and juxtanuclear area. Perhaps mutation of the WASP fragment enhanced affinity for non-Cdc42 cell components and/or off target localizations were more predominant when the biosensor could not bind Cdc42. Although this background was not particularly confounding for studies of protrusion and retraction, which occur away from the nucleus, it merits further study to enable other applications.
Figure 5.

Generating and using the Cdc42 biosensor in living cells. (a) HeLa cells transfected with either CBD or mutant CBD (mCBD) constructs and co-transfected with Cdc42Q61L or Cdc42T17N (n ≥ 7 cells per condition). The Cdc42 inhibitor ML 14158 was added to cells for 1 hour (10 μM) prior to fluorescence measurements. (b) Migrating HeLa cells expressing CBD-TAG-EGFP and labeled in-cell with mero166 show Cdc42 activity at motile portions of the cell edge and in cell protrusions, consistent with previous observations (left panel). Cells containing the mutant biosensor with reduced affinity for Cdc42 (mCBD-TAG-EGFP, right panel) showed no such localized activity. White arrows indicate protrusions that led to cell translocation. (Scale bar 20 μm, dye excitation filter 545/50, dye emission filter 630/45, EGFP excitation filter 470/40, EGFP emission filter 630/45).
CONCLUSIONS
In summary, we have developed environment-sensing dyes that are optimized to both report protein activity and to pass through membranes and label proteins in living cells. They have a suitable combination of brightness and solvent dependent fluorescence changes, long excitation and emission wavelengths, and are water soluble. They show efficient cell loading and uniform distribution within the cell and are derivatized for conjugation to proteins or small molecules. One such dye entered the cell when conjugated to a small molecule and showed the localization of this molecule’s protein target. Another variant was used for in-cell generation of a functional biosensor via reaction between the dye and an unnatural amino acid side chain on a protein affinity reagent. Through precise positioning of dyes on proteins, this strategy can produce biosensors for new protein targets, and provides a practical route to harness the many capabilities of environment-sensing dyes in living cells.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to Alexander Deiters (University of Pittsburgh) and Eric Brustad (University of North Carolina at Chapel Hill) for providing reagents and helpful advice. We gratefully acknowledge the American Cancer Society (C.J.M, 119169-PF-10-183-01-TBE), and the National Institute of General Medical Sciences of the National Institutes of Health (K.M.H., GM- R35GM122596; N.K.P., F32GM120958) for financial support of this research.
Footnotes
Supporting Information
Supporting figures, molecular biology, imaging and image analysis methods, experimental procedures and spectroscopic data for new compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
REFERENCES
- 1.Hahn K; Toutchkine A Curr. Live-cell fluorescent biosensors for activated signaling proteins. Opin. Cell. Biol 2002, 14, 167–172. [DOI] [PubMed] [Google Scholar]
- 2.Aoki K; Komatsu N; Hirata E; Kamioka Y; Matsuda M Stable expression of FRET biosensors: a new light in cancer research. Cancer Sci. 2012, 103, 614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zadran S; Standley S; Wong K; Otiniano E; Amighi A; Baudry M Fluorescence resonance energy transfer (FRET)-based biosensors: visualizing cellular dynamics and bioenergetics. Appl. Microbiol. Biot 2012, 96, 895–902. [DOI] [PubMed] [Google Scholar]
- 4.Sample V; Mehta S; Zhang J Genetically encoded molecular probes to visualize and perturb signaling dynamics in living biological systems. J. Cell Sci. 2014, 127, 1151–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pertz O; Hodgson L; Klemke RL; Hahn KM Spatiotemporal dynamics of RhoA activity in migrating cells. Nature 2006, 440, 1069–1072. [DOI] [PubMed] [Google Scholar]
- 6.Machacek M; Hodgson L; Welch C; Elliott H; Pertz O; Nalbant P; Abell A; Johnson GL; Hahn KM; Danuser G Coordination of Rho GTPase activities during cell protrusion. Nature 2009, 461, 99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.O'Shaughnessy EC; Yi JJ; Hahn KM In Optical Probes in Biology, Zhang J, Schultz C, Eds.; Taylor & Francis Books, Inc.: Boca Raton, FL, 2015. [Google Scholar]
- 8.Welch CM; Elliott H; Danuser G; Hahn KM Imaging the coordination of multiple signalling activities in living cells. Nat. Rev. Mol. Cell Bio 2011, 12, 749–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hahn K; Debiasio R; Taylor DL Patterns of Elevated Free Calcium and Calmodulin Activation in Living Cells. Nature 1992, 359, 736–738. [DOI] [PubMed] [Google Scholar]
- 10.Garrett SC; Hodgson L; Rybin A; Toutchkine A; Hahn KM; Lawrence DS; Bresnick AR A biosensor of S100A4 metastasis factor activation: Inhibitor screening and cellular activation dynamics. Biochemistry-US 2008, 47, 986–996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Nalbant P; Hodgson L; Kraynov V; Toutchkine A; Hahn KM Activation of endogenous Cdc42 visualized in living cells. Science 2004, 305, 1615–1619. [DOI] [PubMed] [Google Scholar]
- 12.Gulyani A; Vitriol E; Allen R; Wu J; Gremyachinskiy D; Lewis S; Dewar B; Graves LM; Kay BK; Kuhlman B; Elston T; Hahn KM A biosensor generated via high-throughput screening quantifies cell edge Src dynamics. Nat. Chem. Biol 2011, 7, 437–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Szent-Gyorgyi C; Schmidt BF; Creeger Y; Fisher GW; Zakel KL; Adler S; Fitzpatrick JA; Woolford CA; Yan Q; Vasilev KV; Berget PB; Bruchez MP; Jarvik JW; Waggoner A Fluorogen-activating single-chain antibodies for imaging cell surface proteins. Nat. Biotechnol 2008, 26, 235–240. [DOI] [PubMed] [Google Scholar]
- 14.Kummer L; Hsu CW; Dagliyan O; MacNevin C; Kaufholz M; Zimmermann B; Dokholyan NV; Hahn KM; Pluckthun A Knowledge-based design of a biosensor to quantify localized ERK activation in living cells. Chem. Biol 2013, 20, 847–856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.MacNevin CJ; Toutchkine A; Marston DJ; Hsu CW; Tsygankov D; Li L; Liu B; Qi T; Nguyen DV; Hahn KM Ratiometric Imaging Using a Single Dye Enables Simultaneous Visualization of Rac1 and Cdc42 Activation. J. Am. Chem. Soc 2016, 138, 2571–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Verveer PJ; Wouters FS; Reynolds AR; Bastiaens PI Quantitative imaging of lateral ErbB1 receptor signal propagation in the plasma membrane. Science 2000, 290, 1567–1570. [DOI] [PubMed] [Google Scholar]
- 17.Erazo-Oliveras A; Najjar K; Dayani L; Wang TY; Johnson GA; Pellois JP Protein delivery into live cells by incubation with an endosomolytic agent. Nat. Methods 2014, 11, 861–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bolhassani A; Jafarzade BS; Mardani G In vitro and in vivo delivery of therapeutic proteins using cell penetrating peptides. Peptides 2017, 87, 50–63. [DOI] [PubMed] [Google Scholar]
- 19.Kristensen M; Birch D; Morck Nielsen H Applications and Challenges for Use of Cell-Penetrating Peptides as Delivery Vectors for Peptide and Protein Cargos. Int. J. Mol. Sci 2016, 17, 185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McNeil PL Direct introduction of molecules into cells. Curr. Protoc. Cell Biol 2001, Chapter 20, Unit 20 1. [DOI] [PubMed] [Google Scholar]
- 21.Weaver JC Electroporation: a general phenomenon for manipulating cells and tissues. J. Cell. Biochem 1993, 51, 426–435. [DOI] [PubMed] [Google Scholar]
- 22.Zhang Y; Yu LC Microinjection as a tool of mechanical delivery. Curr. Opin. Biotech 2008, 19, 506–510. [DOI] [PubMed] [Google Scholar]
- 23.Lang K; Chin JW Cellular incorporation of unnatural amino acids and bioorthogonal labeling of proteins. Chem. Rev 2014, 114, 4764–4806. [DOI] [PubMed] [Google Scholar]
- 24.Lang K; Chin JW Bioorthogonal reactions for labeling proteins. ACS Chem. Biol 2014, 9, 16–20. [DOI] [PubMed] [Google Scholar]
- 25.Yan Q; Bruchez MP Advances in chemical labeling of proteins in living cells. Cell Tissue Res. 2015, 360, 179–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jing C; Cornish VW Chemical tags for labeling proteins inside living cells. Accounts Chem. Res 2011, 44, 784–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xue L; Karpenko IA; Hiblot J; Johnsson K Imaging and manipulating proteins in live cells through covalent labeling. Nat. Chem. Biol 2015, 11, 917–923. [DOI] [PubMed] [Google Scholar]
- 28.Chin JW Expanding and reprogramming the genetic code. Nature 2017, 550, 53–60. [DOI] [PubMed] [Google Scholar]
- 29.Lukinavicius G; Umezawa K; Olivier N; Honigmann A; Yang G; Plass T; Mueller V; Reymond L; Correa IR Jr.; Luo ZG; Schultz C; Lemke EA; Heppenstall P; Eggeling C; Manley S; Johnsson K A near-infrared fluorophore for live-cell super-resolution microscopy of cellular proteins. Nat. Chem 2013, 5, 132–139. [DOI] [PubMed] [Google Scholar]
- 30.Summerer D; Chen S; Wu N; Deiters A; Chin JW; Schultz PG A genetically encoded fluorescent amino acid. P. Natl. Acad. Sci. USA 2006, 103, 9785–9789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Toutchkine A; Kraynov V; Hahn K Solvent-sensitive dyes to report protein conformational changes in living cells. J. Am. Chem. Soc 2003, 125, 4132–4145. [DOI] [PubMed] [Google Scholar]
- 32.Toutchkine A; Nguyen DV; Hahn KM Merocyanine dyes with improved photostability. Org. Lett 2007, 9, 2775–2777. [DOI] [PubMed] [Google Scholar]
- 33.MacNevin CJ; Gremyachinskiy D; Hsu CW; Li L; Rougie M; Davis TT; Hahn KM Environment-sensing merocyanine dyes for live cell imaging applications. Bioconjugate Chem. 2013, 24, 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Toutchkine A; Han WG; Ullmann M; Liu TQ; Bashford D; Noodleman L; Hahn KM Experimental and DFT studies: novel structural modifications greatly enhance the solvent sensitivity of live cell imaging dyes. J. Phys. Chem. A 2007, 111, 10849–10860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Liu TQ; Han WG; Himo F; Ullmann GM; Bashford D; Toutchkine A; Hahn KM; Noodleman L Density Functional Vertical Self-Consistent Reaction Field Theory for Solvatochromism Studies of Solvent-Sensitive Dyes. J. Phys. Chem. A 2004, 108, 3545–3555. [Google Scholar]
- 36.Tan X, ; Nalbant P; Toutchkine A; Hu D; Vorpagel ER; Hahn KM; Lu HP Single-Molecule Study of Protein-Protein Interaction Dynamics in a Cell Signaling System. J. Phys. Chem. B 2004, 108, 737–744. [Google Scholar]
- 37.Han WG; Liu T; Himo F; Toutchkine A; Bashford D; Hahn KM; Noodleman L A theoretical study of the UV/visible absorption and emission solvatochromic properties of solvent-sensitive dyes. ChemPhysChem 2003, 4, 1084–1094. [DOI] [PubMed] [Google Scholar]
- 38.Shirinian VZ; Shimkin AA Merocyanines: Synthesis and Application. Top. Heterocycl. Chem 2008, 14, 75–105. [Google Scholar]
- 39.Kulinich AV; Ishchenko AA Merocyanine dyes: synthesis, structure, properties and applications. Russ. Chem. Rev+ 2009, 78, 141–164. [Google Scholar]
- 40.Marder SR; Gorman CB; Meyers F; Perry JW; Bourhill G; Bredas JL; Pierce BM A Unified Description of Linearand Nonlinear Polarizationin Organic Polymethine Dyes. Science 1994, 265, 632–635. [DOI] [PubMed] [Google Scholar]
- 41.Buncel E; Rajagopal S Solvatochromism and Solvent Polarity Scales. Accounts Chem. Res 1990, 23, 226–231. [Google Scholar]
- 42.Brooker LGS; Craig AC; Heseltine DW; Jenkins PW; Lincoln LL Color and Constitution. XIII. Merocyanines as Solvent Property Indicators. J. Am. Chem. Soc 1965, 87, 2443–2450. [Google Scholar]
- 43.Brooker LGS; Keyes GH; Sprague RH; VanDyke RH; VanLare E; VanZandt G; White FL; Cressman HWJ; Dent SG Color and Constitution. X. Absorption of the Merocyanines. J. Am. Chem. Soc 1951, 73, 5332–5350. [Google Scholar]
- 44.Liptay W Electrochromism and Solvatochromism. Angew. Chem. Int. Edit 1969, 8, 177–188. [Google Scholar]
- 45.Reichardt C, Solvents and Solvent Effects in Organic Chemistry, 2nd ed. VCH Verlagsgesellschaft: Weinheim, 1988. [Google Scholar]
- 46.Bochet CG Photolabile protecting groups and linkers. J. Chem. Soc. Perk. T 1 2002, 125–142. [Google Scholar]
- 47.Blanc A; Bochet CG Wavelength-controlled orthogonal photolysis of protecting groups. J. Org. Chem 2002, 67, 5567–5577. [DOI] [PubMed] [Google Scholar]
- 48.Vedadi M; Barsyte-Lovejoy D; Liu F; Rival-Gervier S; Allali-Hassani A; Labrie V; Wigle TJ; DiMaggio PA; Wasney GA; Siarheyeva A; Dong AP; Tempel W; Wang SC; Chen X; Chau I; Mangano TJ; Huang XP; Simpson CD; Pattenden SG; Norris JL; Kireev DB; Tripathy A; Edwards A; Roth BL; Janzen WP; Garcia BA; Petronis A; Ellis J; Brown PJ; Frye SV; Arrowsmith CH; Jin J A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells. Nat. Chem. Biol 2011, 7, 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shinkai Y; Tachibana M H3K9 methyltransferase G9a and the related molecule GLP. Gene. Dev 2011, 25, 781–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schmied WH; Elsasser SJ; Uttamapinant C; Chin JW Efficient multisite unnatural amino acid incorporation in mammalian cells via optimized pyrrolysyl tRNA synthetase/tRNA expression and engineered eRF1. J. Am. Chem. Soc 2014, 136, 15577–15583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lang K; Davis L; Wallace S; Mahesh M; Cox DJ; Blackman ML; Fox JM; Chin JW Genetic Encoding of bicyclononynes and trans-cyclooctenes for site-specific protein labeling in vitro and in live mammalian cells via rapid fluorogenic Diels-Alder reactions. J. Am. Chem. Soc 2012, 134, 10317–10320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Virdee S; Kapadnis PB; Elliott T; Lang K; Madrzak J; Nguyen DP; Riechmann L; Chin JW Traceless and site-specific ubiquitination of recombinant proteins. J. Am. Chem. Soc 2011, 133, 10708–10711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dommerholt J; Schmidt S; Temming R; Hendriks LJ; Rutjes FP; van Hest JC; Lefeber DJ; Friedl P; van Delft FL Readily accessible bicyclononynes for bioorthogonal labeling and three-dimensional imaging of living cells. Angew. Chem. Int. Edit 2010, 49, 9422–9425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Borrmann A; Milles S; Plass T; Dommerholt J; Verkade JM; Wiessler M; Schultz C; van Hest JC; van Delft FL; Lemke EA Genetic encoding of a bicyclo[6.1.0]nonyne-charged amino acid enables fast cellular protein imaging by metal-free ligation. ChemBioChem 2012, 13, 2094–2099. [DOI] [PubMed] [Google Scholar]
- 55.Jaffe AB; Hall A Rho GTPases: biochemistry and biology. Annu. Rev. Cell Dev. Bi 2005, 21, 247–269. [DOI] [PubMed] [Google Scholar]
- 56.Symons M; Derry JMJ; Karlak B; Jiang S; Lemahieu V; McCormick F; Francke U; Abo A Wiskott-Aldrich syndrome protein, a novel effector for the GTPase CDC42Hs, is implicated in actin polymerization. Cell 1996, 84, 723–734. [DOI] [PubMed] [Google Scholar]
- 57.Abdul-Manan N; Aghazadeh B; Liu GA; Majumdar A; Ouerfelli O; Siminovitch KA; Rosen MK Structure of Cdc42 in complex with the GTPase-binding domain of the 'Wiskott-Aldrich syndrome' protein. Nature 1999, 399, 379–383. [DOI] [PubMed] [Google Scholar]
- 58.Hong L; Kenney SR; Phillips GK; Simpson D; Schroeder CE; Noth J; Romero E; Swanson S; Waller A; Strouse JJ; Carter M; Chigaev A; Ursu O; Oprea T; Hjelle B; Golden JE; Aube J; Hudson LG; Buranda T; Sklar LA; Wandinger-Ness A Characterization of a Cdc42 protein inhibitor and its use as a molecular probe. J. Biol. Chem 2013, 288, 8531–8543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Miki H; Sasaki T; Takai Y; Takenawa T Induction of filopodium formation by a WASP-related actin-depolymerizing protein N-WASP. Nature 1998, 391, 93–96. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.