

Abstract
Mononuclear Mo and W enzymes require a unique ligand known as molybdopterin (MPT). This ligand binds the metal through a dithiolene chelate, and the dithiolene bridges a reduced pyranopterin group. Pyran scission and formation has been proposed as a reaction of the MPT ligand that may occur within the enzymes to adjust reactivity at the Mo atom. We address this issue by investigating oxo-Mo(IV) model complexes containing dithiolenes substituted by pterin or quinoxaline, and a hydroxyalkyl poised to form a pyran ring. hile the pterin-dithiolene model complex exhibits a low energy, eversible pyran cyclization, here we report that pyran cyclization does not spontaneously occur in the quinoxalyldithiolene model. However, protonating the quinoxalyl-dithiolene model induces pyran cyclization forming an unstable, pyrano-quinoxalyl-dithiolene complex which subsequently dehydrates and rearranges to a pyrrolo-quinoxlyl-dithiolene complex that was previously characterized. The protonated pyrano-quinoxalyl-dithiolene complex was characterized by absorption spectroscopy and cyclic voltammetry, and these results suggest pyran cyclization leads to a significant change in the Mo electronic structure exhibited as a strong ILCT transition and 370 mV positive shift of the Mo(V/IV) reduction potential. The influence of protonation on quinoxaline reactivity supports the hypothesis that the local protein environment in the second coordination sphere of Moco could control pyran cyclization. The results also demonstrate that the remarkable chemical reactivity of the pterin dithiolene ligand is subtlely distinct and not reproduced by the simpler quinoxaline analog that is often used to replace pterin in synthetic Moco models.
SYNOPSIS TOC.
An investigation of oxo-Mo(IV) model complexes for the molybdenum cofactor that possess dithiolene ligands substituted by pterin or quinoxaline reveals that pterin-dithiolene complexes exhibit low energy, reversible pyran cyclization, whereas pyran cyclization does not spontaneously occur in a quinoxalyl-dithiolene complex. However, protonation of the quinoxalyl-dithiolene model induces pyran cyclization t an unstable, pyranoquinoxalyl-dithiolene complex which dehydrates and rearranges to a pyrroloquinoxlyldithiolene complex.
Introduction.
Transition metal ions play remarkable roles in the chemistry of living systems. Though first row transition metals dominate bioinorganic chemistry, molybdenum (Mo) and tungsten (W) enzymes are distinctive as the only second and third row transition metals with well-defined functions in biology.1 Furthermore, Mo and W enzymes have been implicated as the earliest bioinorganic catalysts used by the last universal common ancestor (LUCA)2, foreshadowing the current knowledge that Mo or W enzymes are required by nearly all forms of life. In all Mo and W mononuclear enzymes, the metal ion is chelated by one or two dithiolenes substituted by a pterin and an alkylphosphate or dinucleotide (Fig. 1). The pterin-dithiolene ligand is most frequently observed in the pyranopterin form, as depicted in Fig. 1, and is commonly referred to as molybdopterin or MPT.
Figure 1.

The minimal coordination environment of the molybdenum cofactor (Moco) illustrating the pyranopterin dithiolene chelate. Remaining coordination sites are occupied by oxo, sulfido or selenido groups, and a variety of amino acids. P is a phosphate in eukaryotic proteins but can be one of several dinucleotides in certain prokaryotic proteins.
In molybdoenzymes, the catalytic site comprising Mo and MPT is called the molybdenum cofactor (Moco), and the composition of Moco is used to categorize these enzymes into three large families3,4 capable of catalyzing a broad variety of hydroxylation and oxygen atom transfer (OAT) reactions3,5 spanning a reduction potential range of approximately one volt.6 The ubiquity of Mo enzymes and the diversity of their substrates has been proposed to result from variation in the Mo inner coordination sphere, the number of MPT ligands, and bonding interactions between MPT and other parts of the enzyme.6
The synthesis and study of small molecule analogs for the catalytic site of metalloenzymes have been a frequent strategy employed to better understand how a metal site catalyzes chemical transformations. Efforts to model Moco using small molecule analogs have evolved over decades in accordance with increasingly detailed knowledge about the ligand environment for Mo in the catalytic site. Studies of small molecule analogs mimicking the first coordination spheres of the different molybdoenzyme families have demonstrated how variation in the Mo inner coordination sphere is partly responsible for adjusting reactivity of the oxidoreductases.7,8 The function of MPT, however, remains an enigma. Thus, the development of target model complexes possessing aspects of the second coordination sphere in Moco may reveal the function of the highly conserved pyranopterin dithiolene chelate.
A dissection of Moco into individual parts (Mo, dithiolene, and pterin) reveals surprising capability for redox activity. A total of eight electron equivalents are contained within the Mo-dithiolene-pterin motif (Fig. 2). Mo participates in two electron redox chemistry upon cycling between formal oxidation states (Mo(IV) and Mo(VI)), oxidation of the dithiolene to a dithione yields two electron equivalents, and oxidation of a fully reduced tetrahydropterin to an oxidized pterin provides four electron equivalents. However, the interplay between each individual component of the motif has yet to be fully explored.
Figure 2.

(Top) Three plausible oxidation states of the pterin moiety found in Moco. The most thermodynamically stable tautomer of the semi-reduced form is shown. (Bottom) The sequential two electron reduction of 1,2-dithione to ene-1,2-dithiolate.9
Protein crystal structures suggest possible functions of MPT in Moco such as serving as an electron transfer conduit to FeS clusters which are hydrogen-bound directly to the pterin and creating a hydrogenbonded anchor for the metal.10–14 In addition, it has been suggested that pterin protonation may have a role in catalysis,15,16 and that the pterin oxidation state12 may tune the redox capabilities of the metal center. We have also demonstrated the affinity for Mo to coordinate directly to pterin in several oxidation states.17,18 The likely non-innocence of MPT underscores the necessity of incorporating pterins or heteroaromatic substituents on dithiolenes in synthetic models.19 Most small molecule analogs for Moco have used simple aromatic systems to alleviate difficulties associated with pterin incorporation.7,8
We have developed a methodology to synthesize model compounds of the general formula Tp*Mo(≡X)(dithiolene-Het-R) where Tp* is the tridentate tris(3,5-dimethylpyrazolyl)hydroborate ligand, X is either an oxo or sulfido group, and dithiolene-Het-R designates a dithiolene chelate substituted by a N-heterocycle and an alkyl group. This methodology produced examples of model complexes containing pterin-dithiolenes and quinoxaline-dithiolenes.20 We and others have employed quinoxaline as a replacement for pterin in Moco models due to its synthetic accessibility and enhanced solubility versus pterin. The structural relationship and numbering schemes for quinoxaline and pterin are shown in Fig. 3.
Figure 3.

The structural relationship between pterin and quinoxaline, numerically labeled.
In 2012 we reported the first Moco model (Et4N)[Tp*Mo(IV)(O)(S2BMOPP)] (1) incorporating the full pyranopterin-dithiolene structural motif.21 This model exhibits reversible pyran cyclization where the predominant structure, 1o vs 1p, is determined by the solvent dielectric (Fig. 4, top).22 Subsequent studies of 1 revealed the significance of the pyran cyclization in modulating communication between an oxidized pterin and the dithiolene chelate which alters the electronic structure at Mo.23 These results prompted us to reinvestigate the previously reported quinoxaline-dithiolene complex (Et4N)[Tp*Mo(IV)(O)(S2BMOQO)] 224,25 to determine if reversible pyran cyclization occurs in 2, as was demonstrated for 1. Here we report that pyran cyclization does not spontaneously occur in 2, providing an important example of how quinoxaline fails to adequately model the pterin heterocycle in Moco. Under acidic conditions, however, 2 undergoes pyran cyclization to yield an unstable pyrano-quinoxalyl-dithiolene complex which subsequently dehydrates and rearranges to yield a pyrrole ring. As part of the work described herein, a new complex, (Et4N)[Tp*Mo(IV)(O)(S2BDMQO)] 4 (Fig. 4, bottom), was synthesized to serve as a control which precludes pyran cyclization.
Figure 4.

(Top) Pterin dithiolene model complex 1 which was previously reported to undergo reversible pyran cyclization. (Bottom) Quinoxaline dithiolene complexes studied in this work. Complex 2 was partially characterized in an earlier report, while 4 is a new complex synthesized for the current study.
Results and Discussion.
Synthesis.
Pterin and quinoxaline dithiolene model complexes are prepared by reacting a molybdenum polysulfide (Et4N)[Tp*Mo(IV)(S)(S4)] with an alkyne appended to the desired heterocycle.20 In the current work, a Sonogashira coupling reaction using 2-chloroquinoxaline and the appropriate alkyne yields the quinoxalylalkynes, BMOQO and BDMQO (Scheme 1, A). Subsequent reaction of the desired quinoxalylalkyne with (Et4N)[Tp*Mo(S)(S4)] produces (Et4N)[Tp*Mo(IV)(S)(quinoxaline-R-dithiolene)] (Scheme 1, B). Hydrolysis of the terminal sulfido ligand results in the target compounds (Scheme 1, C). This general reaction sequence was previously employed to prepare 2,24,25 however for the work presented here, small changes in the procedure led to more stable material than was previously obtained.
Scheme 1.

(A) Synthesis of the quinoxalylalkyne where R = -OH; -CH3; (B) reaction of the quinoxalylalkyne with (Et4N)[Tp*Mo(IV)(S)(S4)] to produce (Et4N)[Tp*Mo(IV)(S)(dithiolene-quinoxaline-R)]; (C) hydrolysis of the terminal sulfido ligand at Mo. (i) (PPh3)2PdCl2 CuI, Et3N, DMSO at 23 °C; (ii) CH3CN, 18–50 hrs at 23–45 °C; (iii) PBu3, 0.1% H2O in CH3CN, 1–2 hrs at 23 °C.
There are a few notable differences between the reaction conditions producing (Et4N)[Tp*Mo(IV)(O)(S2BMOQO)], 2 and (Et4N)[Tp*Mo(IV)(O)(S2BDMQO)], 4. A longer reaction time and an increase in equivalents of alkyne are required to prepare BDMQO (vs. BMOQO) due to the volatility of 3,3-dimethyl-1-butyne (Scheme 1, A), and both a longer reaction time and elevated temperature is required for synthesis of 5 (Scheme 1, B). The more forcing conditions required for 5 can be attributed to 1,3-allylic strain between the t-butyl and quinoxaline which succumbs to a favorable intramolecular hydrogen bond in 3. Subsequent hydrolysis of the terminal sulfido ligand in 3 and 5 is facilitated by the addition of tri-n-butylphosphine which has been suggested to labilize the terminal sulfur in the presence of adventitious water. Oxygen expedites the conversion albeit the reaction details are not well understood.
Compounds 2 and 4 were characterized in the solid state by X-ray crystallography and in solution by ESIMS, 1H NMR, HSQC, FTIR, absorption spectroscopy, and cyclic voltammetry.
X-ray Crystallography of 2 and 4.
The solid state structures of 2 and 4 were determined by single crystal X-ray diffraction studies (Table S1). Fig. 5 and 6 show the structures of the anionic complexes with line drawings to aid interpretation. Complexes 2 and 4 are structurally similar to other Tp*-Mo-dithiolene complexes21,24,26 in terms of the metrical parameters associated with the Tp* ligand and inner coordination sphere bond distances and angles. However, conformational differences between 2 and 4 are observed when comparing the dihedral angle between the quinoxaline and dithiolene planes. This angle is 53.98° in 2 and increases to 75.6° in 4. The smaller dihedral angle in 2 coincides with the formation of an intramolecular hydrogen bond between the hydroxyl group and the quinoxaline nitrogen atom, N8 (Fig. 5), consistent with the O2–N8 distance of 2.768 Å.
Figure 5.

Line drawing of the anionic complex 2 (left) illustrating the intramolecular H-bond between O2 and N8 in the molecular structure (right) with 50% probability ellipsoids. H-atoms are depicted with arbitrary radii. Countercation (Et4N) and disordered solvent molecules are omitted for clarity. Selected bond distances (Å) for 2: Mo1-O1 1.718(3), Mo1-S1 2.3655(9), Mo-S2 2.3622(10), S1- C16 1.794(3), S2-C17 1.776(3), C16-C17 1.344(5), C16-C21 1.480(4), N8-C21 1.319(4), C21-C22 1.426(5), N7-C22 1.318(5), N8-O2 2.768(5).
Figure 6.

Line drawing of the anionic complex, 4 (left), corresponding to the molecular diagram and labeling scheme (right) with 50% probability ellipsoids. Steric repulsion from the t-butyl in 4 forces the quinoxaline to rotate out of plane with respect to the dithiolene. H-atoms are depicted with arbitrary radii. Countercation (Et4N) and disordered solvent molecules are omitted for clarity. Selected bond distances (Å) for 4: Mo1-O1 1.688(2), Mo1-S1 2.3733(9), Mo-S2 2.3624(10), S1-C16 1.804(4), S2-C17 1.799(4), C16-C17 1.332(5), C16-C18 1.477(5), N7-C18 1.305(4), C18-C19 1.428(5), N8-C19 1.319(5).
A parameter of interest in metal dithiolene complexes is the dithiolene fold angle, which is defined as the angle between two planes consisting of atoms Mo-S1–S2 and S1–C16–C17–S2.27 The fold angle magnitude has been found to correlate with metal oxidation state and d electron count. X-ray crystal structures of metal dithiolene complexes reveal the fold angle increases from ~9° to ~46° for d2 and d0, respectively. In 2 and 4 the dithiolene fold angle is 13.3° and 9.1°, respectively, consistent with the formal Mo(IV) oxidation state.
NMR Studies.
1H NMR and Heteronuclear Single-Quantum Correlation (HSQC) spectra were employed to obtain information about the solution structures of 2 and 4. A comparison of 1H NMR spectra of 2 in CD3CN and CDCl3 is shown in Fig. 7 for the range of 10 to 5 ppm to highlight differences due to quinoxaline conformation. The 1H NMR spectrum of 2 in CD3CN (Fig. 7, bottom), exhibits one resonance at 9.37 ppm corresponding to the quinoxaline H3, four resonances at 8.03, 7.93, 7.77, and 7.71 ppm corresponding to quinoxaline protons H5–H8, three resonances at 5.98, 5.94, and 5.42 ppm corresponding to pyrazolyl H4 protons, and one resonance at 5.62 ppm corresponding to the hydroxyl group. In contrast, in CDCl3 the 1H NMR spectrum of 2 (Fig. 7, top), shows the hydroxyl resonance has shifted more than 2 ppm downfield to 8.04 ppm and the quinoxaline H3 shifted 0.5 ppm downfield. All other resonances of the quinoxaline and the pyrazoyl protons have minimal shifts of ~0.01 ppm. 1H-13C-HSQC experiments (Figs. S9–S12) confirm the assignments of the hydroxyl proton in CD3CN and CDCl3 since the hydroxyl resonance exhibits no crosspeak in either solvent, as expected for a proton on an oxygen atom. Boron is the only other heteroatom with a proton for which assignments have previously been made.20–22,25 Confirmation of the hydroxyl proton assignments in 2 is also obtained from comparison to 1H NMR spectra of 4 (Figs. S7, S8) which lacks a –OH group and exhibits no resonance at 5.62 ppm in CD3CN or at 8.04 ppm in CDCl3.
Figure 7.

1H NMR of 2 in (B) CDCl3 and in (C) CD3CN showing the solvent dependent –OH resonance (blue) due to hydrogen bonding of the hydroxyl group illustrated in (A). Resonances for Ha and Hb from Tp* are denoted accordingly, and the resonance from H3 of the quinoxaline is highlighted in red. H3 also shifts due to the changing local environment upon hydrogen bonding of the hydroxyl.
The large shift of the hydroxyl resonance in 2 between CD3CN and CDCl3 signals a significant change in the -OH environment consistent with a change in quinoxaline conformation. We speculate that in the nonpolar aprotic CDCl3, the –OH group forms an intramolecular hydrogen bond with quinoxaline N8, such as is observed in the solid state structure (Fig. 5), and this interpretation is consistent with the downfield 8.04 ppm resonance typical for a deshielded proton involved in H-bonding. Changing the solvent to CD3CN disrupts this H-bond, and the hydroxyl resonance shifts upfield to 5.62 ppm.
The most important information from the NMR studies is that which is absent: there is no set of resonances corresponding to a pyranoquinoxaline form of the dithiolene ligand in 2. Therefore, both in the solid state and in solution, the quinoxaline group in 2 does not behave in the same way as the pterin in the analogous pterin-dithiolene model complex (Et4N)[Tp*Mo(IV)(O)(S2BMOPP)] 1 (Fig. 4) where a pyranopterin structure readily forms in solution and is the structure observed in the solid state.21,22
Electronic Spectroscopy.
Absorption spectra of 2 and 4 in chloroform and acetonitrile have similar strong absorptions in the region 320–340 nm but have significant differences at lower energy. In CHCl3, 2 exhibits a broad strong band at 506 nm (4,960 M−1 cm−1) (Fig. 8) and this absorption in CH3CN exhibits a blue shift and a small but significant decrease in the extinction coefficient (490 nm, 3,484 M−1 cm−1) (Fig. S13). Complex 4 is nearly featureless in the lower energy region in both CHCl3 and CH3CN, except for an extremely broad and low intensity shoulder centered at 468 nm (2,139 M−1 cm−1) in CHCl3 that is largely unchanged in CH3CN (486 nm, 1,691 M−1 cm−1)
Figure 8.

Electronic Spectra of 2 (blue) and 4 (red) in CHCl3 at 3.00 × 10−5 M.
Electrochemistry.
Cyclic voltammetry was used to probe the environment of the molybdenum atom by measuring the Mo(V)/(IV) redox potential. The measurement was done in both chloroform and acetonitrile under identical conditions of a 100 mV/sec scan rate, a Ag/AgCl reference electrode and a Pt working electrode, with 0.1 M tetrabutylammonium perchlorate as the electrolyte, using the ferrocenium/ferrocene couple as an internal standard. The Mo(V/IV) couple is reversible for both 2 and 4 in both solvents and is shown in Fig. 9 for chloroform and in Fig. S14 for acetonitrile. A large difference in the Mo(V/IV) couple is observed in CHCl3 for 2 vs 4, where the Mo couple in 2 is 115 mV more positive than that for 4. In acetonitrile, the Mo(V/IV) couple in 2 is again more positive, albeit by a smaller 50 mV margin.
Figure 9.

Cyclic voltammograms of the MoV/MoIV couple of 2 (blue) and 4 (red). The voltammograms are plotted versus the potential of reference electrode Ag/AgCl in (n-Bu4N)(ClO4)/CHCl3 at a scan rate of 100 mV/s using a Pt working electrode. MoV/MoIV potentials: 2 −565 mV, 4 −681 mV, vs Fc/Fc+.
The spectroscopic and electrochemical results from 2 and 4 described above are quite similar to those obtained for two closely related pterin-dithiolene complexes (Fig. 10).23 In a detailed analysis of 1 and 6, it was determined that the strong low energy transition was a dithiolene to pterin intraligand charge transfer. On basis of the similarity of the spectra for 2 and 4 to 1 and 6, it is likely that an analogous dithiolene to quinoxaline intraligand charge transfer (ILCT) transition is the origin of the 506 nm absorption observed for 2 in CHCl3.23,25,28 The loss of absorption intensity near 500 nm when 2 is dissolved in CH3CN likely reflects the disruption of the intramolecular H-bond that pins the quinoxaline conformation more nearly co-planar with the dithiolene, thereby facilitating electronic delocalization between dithiolene and quinoxaline. As was reported for 6,23 the much attenuated 468 nm absorption of 4 correlates with the steric requirements of the tert-butyl group causing the quinoxaline to rotate further away from planarity with the dithiolene, effectively decoupling electronic delocalization from dithiolene to quinoxaline.
Figure 10.

The structural relationship of 2 and 4 to two pterin-dithiolene models 1 and 6.
The Mo(V)/(IV) reduction potential in 2 is significantly more positive (115 mV) than in 4. In a parallel manner, the Mo couple in 1 is more positive than that for 6, an observation that was attributed to electronic delocalization from the dithiolene sulfur atoms to the pterin ring that has a stabilizing effect for reduced Mo(IV). Similarly, we interpret the more positive Mo(V)/(IV) couple in 2 as due to delocalization of electron density from the dithiolene to the quinoxaline in response to the intramolecular H-bond formation, favoring the reduced Mo(IV). For 4 as for 6, the steric repulsion between the γ tert-butyl group and the N-heterocycle (quinoxaline or pterin) prevents planarity between the dithiolene and quinoxaline subunits and electronically decouples the dithiolene from the heterocycle.
Quinoxaline Protonation.
It has been established that protonation of N-heterocyclic substituents on dithiolenes causes significant changes in the electronic structure of the entire ligand and affects the properties of the dithiolene ligand as well as those of coordinated metal.28–31 On this basis, we explored the outcome of adding acid to complexes 2 and 4.
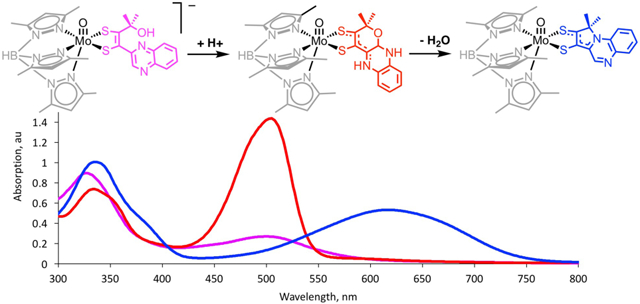
Upon addition of trifluoroacetic acid (TFAA) to 2 in acetonitrile, the solution color immediately changes from magenta to red as an absorption at 505 nm grows in having a five-fold increase in intensity (Fig. 11). Monitoring the reaction over 24 hours shows a decrease in the intensity of the 505 nm absorption in parallel with the appearance of a new band at 619 nm resulting in a color change of the solution from red to deep blue. This second reaction occurs cleanly as indicated by an isosbestic point at 539 nm. A similar reaction sequence was observed when TFAA was added to 2 in CHCl3 (Fig. S19).
Figure 11.

UV-Vis spectroscopic monitoring of reactions resulting from the protonation of 2 over 69 hours upon addition of 1 eq. of TFAA. Black line: initial spectrum of a 15 μM solution of 2. Red solid line: 505 nm absorption associated with the initial product and is assigned as the ILCT transition from formation of a short-lived pyrano-quinoxaline complex 7 (Fig. 12). Red dashed line: decay of 505 nm absorption of 7. Blue dashed line: growth of a 619 nm ILCT absorption of a previously reported pyrroloquinoxaline complex 8 (Fig. 12). Blue solid line: complete formation of 8 from 7.
In order to identify the red and blue species formed when 2 is protonated, the reaction progress was monitored by 1H NMR spectroscopy. The series of spectra in Fig. 12 were collected over 25 hours. Addition of 1 eq TFAA instantly results in loss of the quinoxaline H3 resonance at 9.37 ppm in CD3CN (Fig. 12, spectrum A) as the H3 signal shifts upfield to two new resonances at 6.26 and 6.28 ppm (Fig. 12, spectrum B), consistent with the formation of a pyrano-quinoxaline complex, 7 (Fig. 12, top). Simultaneously, the quinoxaline H5-H8 protons shift as a group from the 7.8–8.0 ppm region upfield to reside between 6.7–7.4 ppm. Subsequently, over 25 hours the 1H NMR spectral series indicates a decay of the quinoxaline H3 and H5-H8 resonances at 6.26 and 6.28 ppm in parallel with the growth of a new H3 resonance at 9.42 ppm and H5-H8 signals between 7.7–8.2 ppm (Fig. 12, spectra C–E).
Figure 12.

1H NMR spectra of a solution of 2 (spectrum A) in CD3CN over a 25 hr time course following addition of 0.99 eq of TFAA (spectra B-E). Spectrum A: 2 in absence of TFAA. Spectrum B after 0.99 equivalents of TFAA were added. Spectra C-E: after 4, 8, and 25 hours, respectively. Structural designations for the three species observed are shown (top) and correlated to prominent spectra by number designation (right).
Our interpretation of the 1H NMR data monitoring the protonation of 2 is shown in Fig. 12. The initial change in the quinoxaline H3 signal is consistent with its new environment in a pyranoquinoxaline dithiolene structure 7 where the appearance of two upfield H3 resonances are consistent with pyran cyclization and correspond to R- and S-isomers. This assignment is based on comparable changes in the corresponding proton (H7) during pyran cyclization in pterin models.22 The second transformation yields spectroscopic data that match the known 1H NMR spectra of a pyrroloquinoxaline complex 8, previously reported by us.25 Significant thione-thiolate character contributes to electronic ground state configurations for the dithiolene chelate of 8 based on observed differences in Mo-S and C-S bond lengths in the X-ray crystal structure of 8 which was consistent with a detailed electronic structure analysis (Fig. 13).24,25,32 Here, we speculate the protonated pyranoquinoxaline, 7, also has a significant amount of the thione, thiolate character (Fig. 13).
Figure 13.

Thione-thiolate resonance structures in 7 and 8.
Further probing of the protonation of 2 by cyclic voltammetry reveals a large (~370 mV) positive shift of the reversible MoV/ MoIV couple in 7 (Fig. 14). This positive shift indicates the Mo ion is now more electron deficient, and therefore, more easily reduced. This electron deficiency at Mo is consistent with the proposed thione, thiolate character of the chelating ligand, since this resonance structure effectively increasing the electron withdrawing capacity of the quinoxaline moiety. Once the pyran ring is cyclized, the quinoxaline is more co-planar with the dithiolene facilitating delocalization of electron density from the sulfur atoms to the N-heterocycle, as has been reported for the pyrano-pterin-dithiolene in 1.23 This redistribution of electron density away from the S atoms decreases their π-base donor character.33 A second, more negative reduction is also observed upon acid addition. This non-reversible redox event is initially being attributed to reduction of the quinoxaline based on previous work,34 but further analysis is currently underway.
Figure 14.

Cyclic voltammograms of a 1.0 mM solution of 2 (red) under a nitrogen atmosphere and after addition of 5 eq. of TFAA (blue). The voltammograms are plotted versus the potential of a Ag/AgCl reference electrode in (n-Bu4N)(ClO4)/CH3CN at a scan rate of 100 mV/s using a Pt working electrode. MoV/ MoIV potentials: Protonated 2 −135 mV, non-protonated 2 −501 mV, vs Fc/Fc+.
Protonation of 4 yields a smaller positive shift of the MoV/ MoIV which becomes non-Nerstian (Figs. S17 & S18). A second non-Nerstian wave is also observed due to a quinoxaline-based reduction. In contrast to the sequence of two intramolecular cyclization reactions induced by the protonation of 2, when TFAA is added to 4, 1H NMR spectra exhibit only broadening of all pyrazoyl and quinoxaline proton resonances. This broadening suggests either degradation of 4 or formation of paramagnetic Mo(V) species due to oxidation of the metal center. Initial observations by cyclic voltammetry favor the latter, and further investigation is underway.
Conclusions.
A reinvestigation of a molybdenum quinoxalyl-dithiolene model for Moco has shown that a quinoxalyl-dithiolene ligand exhibits its own unique chemistry distinct from that observed for a closely related pterin dithiolene model complex. The quinoxalyl-dithiolene ligand in this study does not undergo a spontaneous and reversible pyran cyclization as has been demonstrated for a comparable pterin dithiolene model complex.22 The difference in reactivity between pterin and quinoxaline in these two systems is consistent with the well-documented ability for pterins to undergo covalent addition of water, alcohols, and other small molecules across the 7,8-N=C bond.35 The larger number of nitrogen atoms in the pterin ring system, compared with only two N atoms in the quinoxaline ring system, significantly increases the electron deficient character of the pterin system and increases the electrophilic character of the ring carbon atoms. The electron deficient pterin system favors covalent addition of water, or in the case of model complex 1, favors the addition of the side chain hydroxyl group leading to formation of a pyran ring. Protonation of the quinoxaline at one or both N atoms likely induces pyran ring formation by increasing the electrophilicity of the adjacent C atom, thereby promoting addition of the hydroxyl group across the N=C bond. However, this pyranoquinoxaline species is unstable towards loss of the hydroxyl group in a dehydration reaction. Dehydration results in a second intramolecular cyclization yielding a pyrroloquinoxaline ligand on a Mo complex whose characterization was previously reported.24,25 This study has underscored that the remarkable chemical reactivity of the pterin dithiolene ligand is subtly distinct and not reproduced by the simpler quinoxaline analog that is often used to replace pterin in synthetic Moco models.
The work described here yields the first example of a small molecule analog of Moco where quinoxaline protonation induces pyran cyclization with an associated significant change in the Mo electronic structure exhibited as a strong ILCT transition. As such, these results support the hypothesis that the local protein environment in the second coordination sphere of Moco could control pyran cyclization.6,36,37 Our results suggest a critical role for amino acid protonation and how that may control pyran formation. Further degradation of 2 to form a pyrrolo-quinoxaline system points to the unique aspects of a Mo-dithiolenepterin moiety to be critical to cofactor stability.
Materials and Methods.
The synthesis of (Et4N)[Tp*Mo(S)(S4)] was performed using a previously published procedure.25 All other reagents, chemicals, and deuterated solvents were purchased from Sigma-Aldrich and used as received unless otherwise noted. All solvents for syntheses were purchased from Pharmco-AAPER and were deaerated with N2 gas over activated neutral alumina before use. ESI-MS analyses were performed using a Waters Micromass-ZQ mass spectrometer via infusion of samples as acetonitrile solutions. All NMR experiments were performed on a Bruker 400 MHz FT-NMR. Infrared spectra were obtained using a PerkinElmer Frontier FT-IR on samples prepared as KBr pellets. Electronic absorption spectra were obtained using an Agilent 8453 spectrophotometer on samples in deaerated, anhydrous solvents. Electrochemical analyses were performed using a BASi Epsilon-EC potentiostat using 0.1 M tetrabutylammonium perchlorate (TBAP) as the electrolyte in anhydrous solvents, platinum working and auxiliary electrodes, and a Ag/AgCl reference electrode. All potentials were measured in reference to an internal ferrocene potential at +0.40 V vs. the Ag/AgCl electrode.
Synthesis of 2-(3-Butynyl-2-methyl-2-ol)-quinoxaline (BMOQO).
Synthesized by a modification of previously described procedures.25 Bis(triphenylphosphine)palladium (II) dichloride (0.1023 g, 0.1457 mmol), copper (I) iodide (0.0342 g, 0.180 mmol), and 2-chloroquinoxaline (1.8129 g, 11.015 mmol) were combined in a magnetically stirred triethylamine (72.6 g, 717 mmol) and dimethylsulfoxide (DMSO) (44 g, 560 mmol) mixture previously dried over activated 3 Å molecular sieves. The mixture was stirred for 10 minutes before addition of 2-methyl-3-butyn-2-ol (1.1 g, 13 mmol) via syringe under N2 gas using standard Schlenk techniques. The resulting orange mixture was stirred for 6 hours at room temperature and subsequently chilled (4 °C) for 90 minutes to freeze the DMSO layer. The triethylamine layer was decanted and distilled water (100 mL) was added to the thawed DMSO layer. The organic layer was extracted with methylene chloride (30 mL) and washed with distilled water (3 × 30 mL). All aqueous layers were combined and back-extracted with methylene chloride (3 × 30 mL). All organic layers were combined and washed with saturated sodium chloride (2 × 20 mL) and dried over magnesium sulfate. The resulting solution was rotary evaporated to produce a mocha solid. Recrystallization of the residual solid from acetonitrile and diethyl ether yields BMOQO (1.9842 g, 84.9 %) as sandy tan crystals. 1H NMR (CDCl3) δ: 8.71 (1H, s, pyrazine H), 7.92–7.89 (2H, m, ArH), 7.58–7.26 (2H, m, ArH), 4.99 (1H, s, OH), 1.61 (6H, s, -C(CH3)2). FT-IR (KBr pellet, cm−1): ν(O–H) 3377, ν(C≡C) 2232, ν(C=N) 1542. Melting point range: 88.9–89.2.
Synthesis of (Et4N)[Tp*Mo(IV)(S)(S2BMOQO)] (3).
Synthesized by a modification of the procedure described by Matz et al.25 (Et4N)[Tp*Mo(IV)(S)(S4)] (0.2267 g, 0.3316 mmol) and BMOQO (0.0912 g, 0.430 mmol) were magnetically stirred for 18 hours in acetonitrile (40 mL) at room temperature to produce a brown solution under N2 gas. Vacuum concentration followed by addition of diethyl ether precipitated a dark brown solid that was subsequently filtered and dried under vacuum to yield 3 (0.2250 g, 81.6 %). FT-IR (KBr): ν(Mo≡S) 483. ESI+MS: m/z: [M+2TEA]: 963; ESI−MS: m/z:[M]: 703.
Synthesis of (Et4N)[Tp*Mo(IV)(O)(S2BMOQO)] (2).
Synthesized by a modification of the procedure described by Matz et al.25 Under N2 gas, a solution of (Et4N)[Tp*M(IV)(S)(S2BMOQO)] (0.2486 g, 0.2989 mmol) in wet acetonitrile (20 mL, 0.1 % H2O v/v) was treated with tributylphosphine (0.64 g, 3.2 mmol) drop-wise. The brown solution turned dark pink upon injection of 120 mL of air over the course of 30 minutes and proceeded to react for one hour post air addition. The sample was then concentrated under vacuum and precipitated with the addition of diethyl ether resulting in a carmine solid that was filtered and dried under vacuum to yield 2 (0.2043 g, 83.6 %). 1H NMR (CD3CN) δ: 9.37 (1H, s, pyrazine H), 8.03–8.01 (1H, dd, ArH), 7.93–7.91 (1H, dd, ArH), 7.77–7.73 (1H, td, ArH), 7.71–7.67 (1H, td, ArH), 5.98 (1H, s, Tp* C-H), 5.94 (1H, s, Tp* C-H), 5.62 (1H, s, -OH), 5.42 (1H, s, Tp* C-H), 2.72 (3H, s, -CH3), 2.62 (3H, s, CH3), 2.48 (3H, s, -CH3), 2.46 (3H, s, -CH3), 2.19 (3H, s, -CH3), 2.14 (6H, s, -C(CH3)2), 1.96 (3H, s, -CH3). FT-IR (KBr): ν(Mo≡O) 920. ESI+MS: m/z: [M+2TEA]: 947; ESI−MS: m/z: [M]: 687. UV/vis (CH3CN), nm (ε, M−1 cm−1): 325 (13,303), 490 (3,484). HRESI−MS M = C28H34O2N8B1Mo1S2: m/z: [M]: 687.14146. Calcd for M: m/z: 687.13878.
Synthesis of 2-(3-Butynyl-2,2-dimethyl)-quinoxaline (BDMQO).
Bis(triphenylphosphine)palladium (II) dichloride (0.1018 g, 0.1450 mmol), copper (I) iodide (0.0400 g, 0.210 mmol), and 2-chloroquinoxaline (1.8002 g, 10.937 mmol) were combined in a magnetically stirred triethylamine (72.6 g, 717 mmol) and dimethylsulfoxide (DMSO) (44 g, 560 mmol) mixture previously dried over activated 3 Å molecular sieves. The mixture was stirred for 10 minutes before addition of 3,3dimethyl-1-butyne (1.6 g, 19 mmol) via syringe under N2 gas using standard Schlenk techniques. The resulting orange mixture was stirred for 24 hours at room temperature and subsequently chilled (4 °C) for 90 minutes to freeze the DMSO layer. The triethylamine layer was decanted and distilled water (100 mL) was added to the thawed DMSO layer. The organic layer was extracted with methylene chloride (30 mL) and washed with distilled water (3 × 30 mL). All aqueous layers were combined and back-extracted with methylene chloride (3 × 30 mL). All organic layers were combined and washed with saturated sodium chloride (2 × 20 mL) and dried over magnesium sulfate. The resulting solution was rotary evaporated to yield BDMQO (2.0336 g, 88.4 %) as a burnt umber oil.23 1H NMR (CDCl3) δ: 8.77 (1H, s, pyrazine H), 8.02–7.99 (2H, m, ArH), 7.72–7.65 (2H, m, ArH), 1.36 (9H, s, -C(CH3)3). FT-IR (liquid, cm−1): ν(C≡C) 2232, ν(C=N) 1541.
Synthesis of (Et4N)[Tp*Mo(IV)(S)(S2BDMQO)] (5).
(Et4N)[Tp*Mo(IV)(S)(S4)] (0.4201 g, 0.6145 mmol) and BDMQO (0.35 g, 1.7 mmol) were magnetically stirred in acetonitrile (40 mL) to produce a brown solution that was heated for 50 hours at 45 °C under N2 gas. Vacuum concentration followed by addition of diethyl ether precipitated a dark brown solid that was subsequently filtered and dried under vacuum to yield 5 (0.4937 g, 96.6 %). FT-IR (KBr): ν(Mo≡S) 475. ESI+MS: m/z: [M+2TEA]: 961; ESI−MS: m/z: [M]: 701.
Synthesis of (Et4N)[Tp*Mo(IV)(O)(S2BDMQO)] (4).
Under N2 gas, a solution of (Et4N)[Tp*Mo(IV)(S)(S2BDMQO)] (0.3600 g, 0.4328 mmol) in wet acetonitrile (25 mL, 0.1 % H2O v/v) was treated with tributylphosphine (0.81 g, 4.0 mmol) drop-wise. The brown solution was stirred for two hours at room temperature, concentrated under vacuum, and precipitated by the addition of diethyl ether resulting in a seal brown solid that was filtered and dried under vacuum to yield 4 (0.3185 g, 90.2 %). 1H NMR (CD3CN) δ: 8.91 (1H, s, pyrazine H), 8.04–8.02 (1H, dd, ArH), 7.98–7.95 (1H, dd, ArH), 7.76–7.74 (1H, td, ArH), 7.72–7.69 (1H, td, ArH), 5.98 (1H, s, Tp* C-H), 5.90 (1H, s, Tp* C-H), 5.43 (1H, s, Tp* C-H), 2.74 (3H, s, -CH3), 2.58 (3H, s, CH3), 2.48 (3H, s, -CH3), 2.45 (3H, s, -CH3), 2.26 (3H, s, -CH3), 2.19 (3H, s, -CH), 1.26 (9H, s, -C(CH)3)3). FT-IR (KBr): ν(Mo≡O) 917. ESI+MS: m/z: [M+2TEA]: 945; ESI− MS: m/z: [M]: 685. UV/vis (CH3CN), nm (ε, M−1 cm−1): 323 (9,315), 460 (1,871). HRESI+MS: M=C29H37O1N8B1Mo1S2: m/z: [M+H as Mo5+]: 686.16810. Calcd for M: m/z: 686.16734.
Timecourse study of trifluoracetic acid protonation of 2 monitored by 1H NMR
The initial 1H NMR spectrum was obtained from a sample of 2 (0.0054 g, 0.0066 mmol) in CD3CN (0.400 mL). To this sample was added 0.050 mL of TFAA in CD3CN (0.131 M, 0.00655 mmol TFAA, 0.99 eq). Spectra were obtained at intervals of 1, 2, 4, 6, 8, 11, 13 and 25 h after the initial addition, a period over which the sample color changed from strong red to bright blue.
X-ray Crystallography.
X-ray structural analysis for 2 and 4: Pink, rectangular crystals of 2 were grown by vapor diffusion of diethyl ether into a solution of 2 in 1,2-dichloroethane. Brown rectangular crystals of 4 were grown by the slow evaporation of a solution of 4 in tetrahydrofuran-d8.
Crystals were mounted using viscous oil onto a plastic mesh and cooled to the data collection temperature. Data were collected on a Bruker-AXS APEX II DUO CCD diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å). Unit cell parameters were obtained from 36 data frames, 0.5° ω, from three different sections of the Ewald sphere.38 The systematic absences in the diffraction data are consistent, uniquely, with Pbcn. The data were treated with multi-scan absorption corrections.37 The structures were solved using intrinsic phasing methods39 and refined with full-matrix, least-squares procedures on F2.39
Initial solutions for 2 suggested a disordered di-1,2-chloroethane of solvation. The data sets were treated with Squeeze to model the solvent in 2 and other residual solvent accessible voids as diffused electron density contributions.41
Non-hydrogen atoms were refined with anisotropic displacement parameters. H-atoms were treated as idealized contributions with geometrically calculated positions and with Uiso equal to 1.2 Ueq (1.5 Ueq for methyl) of the attached atom. Atomic scattering factors are contained in the SHELXTL program library.40
The structures have been deposited at the Cambridge Structural Database under the following depositary numbers: CCDC 1891122–1891123.
Supplementary Material
Acknowledgements.
The authors acknowledge the National Institutes of Health (GM-081848 to S.J.N.B.) for continued financial support of our work on molybdoenzymes. S.J.N.B. also acknowledges the National Science Foundation (CHE-0958996). D.R.G., A.L.N., and S.J.N.B. thank Ben Rupert and Papa Nii Asare-Okai at the Mass Spectrometry Facility of the University of Delaware for HRESI analyses.
Footnotes
Supporting Information.
Supporting Information is available free of charge on the ACS Publications website at DOI:xx.xxxx/acs.inorgchem.xxxxxxx.
ESI-MS, 1H, and HSQC NMR data for 2 and 4; UV-vis spectra and CV data for 2 and 4 in acetonitrile and chloroform; X-ray crystal structure data for 2 and 4 (PDF).
Notes
The authors declare no competing financial interest.
References
- 1.Pushie MJ; George GN Spectroscopic studies of molybdenum and tungsten enzymes. Coor. Chem. Rev 2011, 255, 1055–1084. [Google Scholar]
- 2.Weiss MC; Sousa FL; Mrnjavac N; Neukirchen S; Roettger M; Nelson-Sathi S; Martin WF The physiology and habitat of the last universal common ancestor. Nat. Microbiol 2016, 1 (9). [DOI] [PubMed] [Google Scholar]
- 3.Schwarz G; Mendel RR; Ribbe MW Molybdenum cofactors, enzymes and pathways. Nature 2009, 460, 13, 839–847. [DOI] [PubMed] [Google Scholar]
- 4.Hille R; Hall J; Basu P The mononuclear molybdenum enzymes. Chem. Rev 2014, 114, 3963–4038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Maia LB; Moura I; Moura JJG Molybdenum and Tungsten Containing Enzymes: An Overview 2017, 5, 1–80. [Google Scholar]
- 6.Rothery RA; Weiner JH Shifting the metallocentric molybdoenzyme paradigm: the importance of pyranopterin coordination. J. Biol. Inorg. Chem 2015, 20, 349–372. [DOI] [PubMed] [Google Scholar]
- 7.Holm RH; Solomon EI; Majumdar A; Tenderholt A Comparative molecular chemistry of molybdenum and tungsten and its relation to hydroxylase and oxotransferase enzymes. Coord. Chem. Rev 2011, 255, 993–1015. [Google Scholar]
- 8.Young CG Scorpionate Complexes as Models for Molybdenum Enzymes. Eur. J. Inorg. Chem 2016, 2357–2376. [Google Scholar]
- 9.Basu P; Burgmayer SJN Pterin chemistry and its relationship to the molybdenum cofactor. Coord. Chem. Rev 2011, 255, 1016–1038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Burgmayer SJN Dithiolene Chemistry: Synthesis, Properties, and Applications 2004, 52, 491–538. [Google Scholar]
- 11.Enemark JH; Garner CD The coordination chemistry and function of the molybdenum centres of the oxomolybdoenzymes. J. Biol. Inorg. Chem 1997, 2, 817–822. [Google Scholar]
- 12.Rothery RA; Stein B; Solomonson M; Kirk ML; Weiner JH Pyranopterin conformation defines the function of molybdenum and tungsten enzymes. Proc. Natl. Acad. Sci 2012, 109, 37, 14773–14778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rajagopalan KV The molybdenum cofactors – perspective from crystal structure. J. Biol. Inorg. Chem 1997, 2, 786–789. [Google Scholar]
- 14.Burgmayer SJN; Pearsall DL; Blaney SM; Moore EM; Sauk-Schubert C Redox reactions of the pyranopterin system of the molybdenum cofactor. J. Biol. Inorg. Chem 2004, 9, 59–66. [DOI] [PubMed] [Google Scholar]
- 15.Duval S; Santini JM; Lemaire D; Chaspoul F; Russell MJ; Grimaldi S; Nitschke W; Schoepp-Cothenet B The H-bond network surrounding the pyranopterins modulates redox cooperativity in the molybdenum-bisPGD cofactor in arsenite oxidase. Biochim. Biophys. Acta 2016, 1857, 1353–1362. [DOI] [PubMed] [Google Scholar]
- 16.Adamson H; Simonov AN; Kierzek M; Rothery RA; Weiner JH; Bond AM; Parkin A Electrochemical evidence that pyranopterin redox chemistry controls the catalysis of YedY, a mononuclear Mo enzyme. Proc. Natl. Acad. Sci 2015, 112, 47, 14506–14511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Burgmayer SJN; Stiefel EI Synthesis and structure of the first molybdenum-pterin complex. J. Am. Chem. Soc 1986, 108, 8310–8311. [Google Scholar]
- 18.Burgmayer SJN; Everett K; Bostick L Molybdenum Enzymes, Cofactors, and Model Systems 1993, 114–129. [Google Scholar]
- 19.Mtei RP; Perera E; Mogesa B; Stein B; Basu P; Kirk ML A Valence Bond Description of Dizwitterionic Dithiolene Character in an Oxomolybdenum-Bis(ditihone) Complex. Eur. J. Inorg. Chem 2011, 5467–5470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Burgmayer SJN; Kim M; Petit R; Rothkopf A; Kim A; BelHamdounia S; Hou Y; Somogyi A; Habel-Rodriguez D; Williams A; Kirk ML Synthesis, characterization, and spectroscopy of model molybdopterin complexes. J. Inorg. Biochem, 2007, 101, 1601–1616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Williams BR; Fu Y; Yap GPA; Burgmayer SJN Structure and Reversible Pyran Formation in Molybdenum Pyranopterin Dithiolene Models of the Molybdenum Cofactor. J. Am. Chem. Soc 2012, 134, 19584–19587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Williams BR; Gisewhite D; Kalinski A; Esmail A; Burgmayer SJN Solvent-Dependent Pyranopterin Cyclization in Molybdenum Cofactor Model Complexes. Inorg. Chem 2015, 54, 8214–8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gisewhite DR; Yang J; Williams BR; Esmail A; Stein B; Kirk ML; Burgmayer SJN Implications of Pyran Cyclization and Pterin Conformation on Oxidized Forms of the Molybdenum Cofactor. J. Am. Chem. Soc 2018, 140, 12808–12818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Matz KG; Mtei RP; Leung B; Burgmayer SJN; Kirk ML Noninnocent Dithiolene Ligands: A New Oxomolybdenum Complex Possessing a Donor-Acceptor Dithiolene Ligand. J. Am. Chem. Soc 2010, 132, 7830–7831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Matz KG; Mtei RP; Rothstein R; Kirk ML; Burgmayer SJN Study of Molybdenum(4+) Quinoxalyldithiolenes as Models for the Noninnocent Pyranopterin in the Molybdenum Cofactor. Inorg. Chem 2011, 50, 9804–9815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Inscore FE; Knottenbelt SZ; Rubie ND; Joshi HK; Kirk ML; Enemark JH Understanding the Origin of Metal-Sulfur Vibrations in an Oxo-Molybdenum Dithiolene Complex: Relevance to Sulfite Oxidase. Inorg. Chem 2006, 45, 967–976. [DOI] [PubMed] [Google Scholar]
- 27.Joshi HK; Cooney JJA; Inscore FE; Gruhn NE; Lichtenberger DL; Enemark JH Investigation of metal-dithiolate fold angle effects: Implications for molybdenum and tungsten enzymes. Proc. Natl. Acad. Sci. U.S.A. 2003, 100, 7, 3719–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Hsu JK; Bonangelino CJ; Kaiwar SP; Boggs CM; Fettinger JC; Pilato RS Direct Conversion of ɑ-Substituted Ketones to Metallo-1,2-enedithiolates. Inorg. Chem 1996, 35, 4743–4751. [Google Scholar]
- 29.Dicks JP; Zubair M; Davies ES; Garner CD; Schulzke C; Wilson C; McMaster J Synthesis, Structure and Redox Properties of Asymmetric (Cyclopentadienyl)(ene-1,2- dithiolate)cobalt(III) Complexes Containing Phenyl, Pyridyl and Pyrazinyl Units. Eur. J. Inorg. Chem 2015, 3550–3561. [Google Scholar]
- 30.Porcher J; Fogeron T; Gomez-Mignot M; Derat E; Chamoreau L; Li Y; Fontecave M A Bioinspired Molybdenum Complex as a Catalyst for the Photo- and Electroreduction of Protons. Angew. Chem. Int. Ed 2015, 127, 14090–14093. [DOI] [PubMed] [Google Scholar]
- 31.Whalley AL; Blake AJ; Collison D; Davies ES; Disley HJ; Helliwell M; Mabbs FE; McMaster J; Wilson C; Garner CD Synthesis, structure and redox properties of bis(cyclopentadienyl)dithiolene complexes of molybdenum and tungsten. Dalton Trans. 2011, 40, 10457–10472. [DOI] [PubMed] [Google Scholar]
- 32.Pilato RS; Eriksen K; Greaney MA; Gea Y; Taylor EC; Goswami S; Kilptrick L; Spiro TG; Rheingold AL; Stiefel EI Molybdenum Enzymes, Cofactors, and Model Systems, 1993, 83–97. [Google Scholar]
- 33.Enemark JH; Cooney JJA; Wang J; Holm RH Synthetic Analogues and Reaction Systems Relevant to the Molybdenum and Tungsten Oxotransferases. Chem. Rev 2004, 104, 1175–1200. [DOI] [PubMed] [Google Scholar]
- 34.Raghavan R; Dryhurst G Redox chemistry of reduced pterin species. J. Electroanal. Chem 1981, 129, 189–212. [Google Scholar]
- 35.Albert A Advances in Heterocyclic Chemistry, 1977. 117–143. [DOI] [PubMed] [Google Scholar]
- 36.Rendon J; Biaso F; Ceccaldi P; Toci R; Seduk F; Magalon A; Guigliarelli B; Grimaldi S Elucidation the Structures of the Low- and High-pH Mo(V) Species in Respiratory Nitrate Reductase: A Combined EPR, 14,15N HYSCORE, and DFT Study. Inorg. Chem 2017, 56, 8, 4422–4434. [DOI] [PubMed] [Google Scholar]
- 37.Wu S-Y; Rothery RA; Weiner JH Pyranopterin Coordination Controls Molybdenum Electrochemistry in Escherichia coli Nitrate Reductase. J. Biol. Chem 2015, 290, 41, 25164–25173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Apex3; Bruker AXS Inc.: Madison, WI, 2015. [Google Scholar]
- 39.Sheldrick GM SHELXT – Integrated space-group and crystal-structure determination. Acta Cryst. 2015, A71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sheldrick GM Crystal structure refinement with SHELXL. Acta Cryst. 2015, C71, 3–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Spek AL PLATON SQUEEZE: a tool for the calculation of the disordered solvent contribution to the calculated structure factors. Acta Cryst. 2015, C71, 9–18. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.