

Abstract
A deficit in glucose uptake and a deposition of amyloid β-peptide (Aβ) each occur in vulnerable brain regions in Alzheimer’s disease (AD). It is not known whether mechanistic links exist between Aβ deposition and impaired glucose transport. We now report that Aβ impairs glucose transport in cultured rat hippocampal and cortical neurons by a mechanism involving membrane lipid peroxidation. Aβ impaired 3H-deoxy-glucose transport in a concentration-dependent manner and with a time course preceding neurodegeneration. The decrease in glucose transport was followed by a decrease in cellular ATP levels. Impairment of glucose transport, ATP depletion, and cell death were each prevented in cultures pretreated with antioxidants. Exposure to FeSO4, an established inducer of lipid peroxidation, also impaired glucose transport. Immunoprecipitation and Western blot analyses showed that exposure of cultures to Aβ induced conjugation of 4-hydroxynonenal (HNE), an aldehydic product of lipid peroxidation, to the neuronal glucose transport protein GLUT3. HNE induced a concentration-dependent impairment of glucose transport and subsequent ATP depletion. Impaired glucose transport was not caused by a decreased energy demand in the neurons, because ouabain, which inhibits Na+/K+-ATPase activity and thereby reduces neuronal ATP hydrolysis rate, had little or no effect on glucose transport. Collectively, the data demonstrate that lipid peroxidation mediates Aβ-induced impairment of glucose transport in neurons and suggest that this action of Aβ may contribute to decreased glucose uptake and neuronal degeneration in AD.
Keywords: Alzheimer’s disease, apoptosis, excitotoxicity, GLUT3, hydroxynonenal, mitochondrial ATP
Alzheimer’s Disease (AD) is a progressive neurodegenerative disorder characterized by gradual impairment of memory function and accumulation of neurofibrillary tangles and neuritic plaques in brain regions subserving cognitive functions (for review, see Selkoe, 1993). A consistent feature of AD patients, detected by brain imaging methods, is impairment of glucose uptake in brain regions that exhibit neuritic plaques (Hoyer et al., 1988;Kalaria and Harik, 1989; Sims, 1990; Jagust et al., 1991). Studies of persons genetically at risk for AD suggest that reduced glucose uptake may occur early in the disease process before neuronal degeneration (Pettegrew et al., 1994; Kennedy et al., 1995; Reiman et al., 1996). Moreover, large decreases in the activities of two major mitochondrial enzyme systems, pyruvate dehydrogenase complex and ketoglutarate dehydrogenase complex, have been reported (for review, see Blass, 1993). When glucose uptake into neurons is compromised, mitocondrial production of ATP is suppressed, resulting in increased vulnerability to excitotoxic calcium overload (Novelli et al., 1988; Cheng and Mattson, 1992a,b), a mechanism of cell injury implicated in the pathogenesis of AD (for reviews, see Greenamyre and Young, 1989;Mattson et al., 1993a). Glucose uptake in the brain is mediated by specific transport proteins: GLUT1 in endothelial cells and GLUT3 in neurons (Simpson et al., 1994a). Recent data suggest that levels of these transporters are decreased in AD brain (Simpson et al., 1994b;Harr et al., 1995).
The major component of neuritic plaques is amyloid β-peptide (Aβ), a 40–42 amino acid proteolytic fragment of the β-amyloid precursor protein (βAPP) (Selkoe, 1993). Molecular genetic studies have causally linked βAPP mutations to some inherited forms of AD (for review, see Mullan and Crawford, 1993); the mutations may promote increased production of Aβ (Citron et al., 1992; Cai et al., 1993;Suzuki et al., 1994). Transgenic mice expressing a mutated form of human βAPP exhibit age-dependent and brain region-specific deposition of Aβ that is associated with neuronal degeneration (Games et al., 1995; Hsaio et al., 1996). Aβ, and an 11 amino acid fragment thereof (Aβ25-35), can be neurotoxic by a mechanism linked to peptide fibril formation (for review, see Yankner, 1996). The mechanism of Aβ toxicity may involve membrane lipid peroxidation (Behl et al., 1994;Butterfield et al., 1994), disruption of ion homeostasis (Mattson et al., 1992, 1993b), and apoptosis (Loo et al., 1993). Lipid peroxidation induced by Aβ has been linked to impairment of membrane transport and signaling systems, including ion-motive ATPases (Mark et al., 1995a), glutamate transporters (Harris et al., 1996; Keller et al., 1997), and the muscarinic cholinergic acetylcholine receptor–GTP-binding protein system (Kelly et al., 1996).
Oxidative stress is prevalent in AD brain: levels of protein (Smith et al., 1991) and lipid (Lovell et al., 1995) oxidation are increased in vulnerable regions and advanced glycation end products are associated with neuritic plaques and neurofibrillary tangles (for review, see Smith et al., 1995). 4-Hydroxynonenal (HNE), an aldehydic product of lipid peroxidation, is neurotoxic (Montine et al., 1996;Mark et al., 1997) and can impair ion-motive ATPase activities and disrupt calcium homeostasis in cultured hippocampal neurons (Mark et al., 1997). We now report that Aβ impairs glucose uptake and depresses ATP levels in cultured rat hippocampal and cortical neurons, and provide evidence that this action of Aβ is mediated by membrane lipid peroxidation and conjugation of HNE to GLUT3.
MATERIALS AND METHODS
Cell culture, experimental treatments, and quantification of neuron survival. Primary hippocampal and cortical cell cultures were established from embryonic rats (day 18 of gestation), as detailed elsewhere (Mattson et al., 1995). Cells were plated into polyethyleneimine-coated plastic culture dishes at a density of 70–120/mm2. The cultures were maintained in Eagle’s minimum essential medium supplemented with 10% (v/v) heat-inactivated fetal bovine serum (Life Technologies, Gaithersburg, MD), 20 mm KCl, and 1 mm pyruvate. The atmosphere consisted of 6% CO2/94% room air and was maintained near saturation with water. Experiments were performed in cultures that had been maintained for 6–10 d. With these culture conditions, ∼90% of the cells are neurons and the remaining cells are astrocytes, as judged by characteristic morphology and differential immunoreactivity with antibodies to neuron-specific (neurofilament, MAP2, and tau) and astrocyte-specific (glial fibrillary acidic protein and S-100β) proteins (Mattson et al., 1993b, 1995). In some experiments, cultures were prepared that contained only neurons or only astrocytes. Pure neuronal cultures were prepared by maintaining the embryonic cell cultures in serum-free medium (Neurobasal, Life Technologies). Pure astrocyte cultures were prepared by plating cells from postnatal rat cerebral cortex in uncoated plastic culture dishes and changing the serum-containing medium daily.
Immediately before experimental treatment, the culture maintenance medium was replaced with Locke’s solution containing (in mm): 154 NaCl, 5.6 KCl, 2.3 CaCl2, 1.0 MgCl2, 3.6 NaHCO3, 10 glucose, 5 HEPES buffer, pH 7.2. Aβ25-35 (lot ZM513) and Aβ1-40 (lot ZK600) were purchased from Bachem. Peptides were stored lyophilized, and 1 mm stocks (in water) were prepared 2–4 hr before use. HNE was purchased from Caymen Chemical (Ann Arbor, MI). Ouabain,n-propyl gallate, phloretin, and protein A–acrylic beads were purchased from Sigma (St. Louis, MO). Neuron survival was quantified by counting the number of viable neurons in premarked microscope fields before and at indicated time points after exposure to experimental treatments, as described previously (Mattson et al., 1992,1995).
Glucose transport assay. Uptake of [3H]-glucose was performed using a variation of the method of Horner et al. (1990). Before the addition of experimental treatments, the cultures were switched to Locke’s solution. After treatments and just before the uptake assay, cultures were switched to glucose-free Locke’s solution by first washing them three times with glucose-free Locke’s. The assay was started by the addition of 1.5 μCi of [3H]−2-deoxy-glucose (New England Nuclear), and cultures were maintained at 37°C. The assay was stopped 5 min later by aspiration of the supernatant and rapid washing with PBS (three rinses, 5–7 sec/rinse). Cells were lysed in 200 μl of a 0.5N NaOH/0.05% SDS solution; 10 μl was used for protein determination (Pierce BCA kit; Pierce, Rockford, IL), and the remainder was counted in a Packard 2500TR liquid scintillation counter. Data are expressed as cpm [3H]−2-deoxy-glucose per milligram protein per minute.
Immunoprecipitation and Western blot analysis.Immunoprecipitations were performed as described (Barger and Mattson, 1996). Briefly, after treatment cultures were lysed in RIPA buffer (50 mm Tris-HCl, 10% glycerol, 1% Triton X-100, 150 mm NaCl, 100 mm NaF, 5 mm EDTA, 2 mm phenylmethylsulfonyl fluoride, 1 mm sodium orthovanadate, and 1 μg/ml leupeptin, pH 7.5). GLUT3 protein was immunoprecipitated from 400 μg of total cellular protein using a polyclonal antibody directed against a C-terminus peptide of GLUT3 (Chemicon, Temecula, CA). The antibody-lysate solution was left overnight at 4°C on a rotary shaker, and the antibody–antigen complex was then pelleted using protein A linked to acrylic beads. The pellet was washed three times with ice-cold RIPA buffer, and the final pellet was suspended in 2× Laemmli sample buffer. Samples were boiled for 3 min and centrifuged at 3000 rpm for 30 sec, and the supernatant was loaded on a 7.5% SDS-PAGE gel. Protein was transferred to nitrocellulose, and the blot was incubated with a rabbit polyclonal antibody generated against HNE-protein conjugates (Uchida et al., 1993). The blot was processed further using HRP-conjugated secondary antibody and a chemiluminescence detection method (Amersham, Arlington Heights, IL).
Immunocytochemistry. Cells were fixed for 30 min in PBS containing 4% paraformaldehyde (4°C). Membranes were permeabilized by exposing the fixed cells to PBS containing 0.2% Triton X-100. Cells were then incubated sequentially in PBS solutions containing blocking serum (1% normal goat serum), primary anti-GLUT3 antibody (1:500; 4 hr), biotinylated anti-rabbit secondary antibody (Vector Labs, Burlingame, CA; 1 hr), avidin-peroxidase complex (Vector Labs; 30 min), and diaminobenzidine-tetrahydrochloride (Sigma; 5 min).
Quantification of cellular ATP levels. ATP levels were quantified using a luciferin/luciferase-based assay. Cells were exposed to experimental treatments in Locke’s solution. To begin the assay, cells were rinsed with PBS and lysed with 0.2 ml of ATP-releasing buffer (Sigma); 10 μl of the lysate was taken for protein determination. ATP concentrations in lysates were quantified using an ATP Bioluminescence Assay Kit CH II (Boehringer Mannheim, Mannheim, Germany) and a luminometer (Optocomp I, MGM Instruments) according to the manufacturers’ protocols. A standard curve was generated using solutions of known ATP concentrations; samples were diluted so that readings fell within the linear range. ATP levels were expressed as nanomole ATP per microgram protein.
RESULTS
Aβ impairs glucose transport in hippocampal and cortical neurons
Preliminary studies showed that, as expected from previous studies (Horner et al., 1990), >90% of the [3H]-glucose uptake in both hippocampal and cortical cells was blocked by phloretin, indicating mediation by a specific glucose transporter. Levels of [3H]-glucose uptake were (cpm · mg−1 · min−1): untreated control hippocampal cultures, 35,400 ± 580; hippocampal cultures exposed to 100 μm phloretin, 1908 ± 144; control cortical cultures, 16,500 ± 1300; cortical cultures exposed to 100 μm phloretin, 1372 ± 179; n = 6. The basal rate of glucose uptake in hippocampal cells was greater than twofold the rate of uptake in cortical cells (p < 0.001; paired t test; Fig.1A). Exposure of cultures to increasing concentrations of Aβ25-35 for 2 hr resulted in concentration-dependent decreases in the rate of uptake of [3H]-deoxy-glucose in both hippocampal and cortical cells (Fig. 1A). The minimum concentration of Aβ25-35 required to induce a significant decrease in glucose transport was 5 μm in hippocampal cells and 10 μm in cortical cells. The extent of inhibition of glucose transport in cultures exposed to 50 μm Aβ25-35 was ∼65% in hippocampal cells and 50% in cortical cells. The time courses of impairment of glucose transport in hippocampal and cortical cultures exposed to 10 μm Aβ25-35 were similar, with the first significant decrease occurring within 2 hr of exposure and a further decline to ∼50% inhibition by 3 hr (Fig. 1B). Between 4 and 10 hr of exposure to Aβ25-35, the rate of decrease in glucose transport slowed considerably. Impairment of glucose transport in cultures exposed to Aβ25-35 preceded cell degeneration; there was no significant cell loss during a 6 hr exposure to 10 μmAβ25-35, and ∼20% reduction in survival during a 12 hr exposure (Fig. 1C). Previous studies have shown that Aβ25-35 and Aβ1-40 exhibit similar neurotoxic profiles with similar, if not identical, mechanisms of action (Yankner et al., 1990; Mattson et al., 1992; Pike et al., 1993; Mark et al., 1995a). In the present study we found that, as expected, Aβ1-40 also significantly decreased glucose uptake in a concentration-dependent manner during a 6 hr exposure period (Fig. 1D). Exposure of hippocampal cultures to 50 μm of inactive (nontoxic) “overaged” Aβ25-35 (Mattson, 1995) for time periods of up to 10 hr had no significant effect on glucose transport (data not shown), further suggesting a link between impairment of glucose transport and neurotoxicity of the peptide.
Fig. 1.

Aβ induces a concentration-dependent decrease in glucose uptake in hippocampal and cortical cell cultures with a time course that precedes cell death. A, Neocortical and hippocampal cell cultures were exposed for 2 hr to vehicle or the indicated concentrations of Aβ25-35, and cellular uptake of [3H]-glucose uptake was quantified. Values are the mean and SD of determinations made in six separate cultures. *p < 0.01, **p < 0.001 for hippocampal cultures; #p < 0.01 for cortical cultures, compared to cultures exposed to vehicle (0 [Aβ]).B, Cortical and hippocampal cultures were exposed to 10 μm Aβ25-35 for the indicated time periods, and [3H]-glucose uptake was quantified. Values represent the mean and SD of determinations made in four to seven separate cultures. The decrease in glucose uptake was significant for both cortical and hippocampal cultures at the 2 hr (p < 0.05), and 4, 6, and 10 hr (p < 0.01) time points. ANOVA with Scheffe’s post hoc tests.C, Hippocampal cultures were exposed to vehicle or 10 μm Aβ25-35 for the indicated time periods, and neuronal survival was quantified (see Materials and Methods). Values represent the mean and SD of five separate cultures. *p < 0.005 (ANOVA with Scheffe’s post hoc test).D, Hippocampal cultures were exposed for 6 hr to Aβ1-40 at the indicated concentrations, and [3H]-glucose uptake was quantified. Values represent the mean and SD of determinations made in four separate cultures. *p < 0.05, **p < 0.001 compared to control (0 Aβ) value. ANOVA with Scheffe’s post hoc analysis.
Evidence that oxidative stress mediates Aβ-induced inhibition of glucose transport
Previous studies showed that Aβ can induce membrane lipid peroxidation in cultured neurons and that antioxidants can protect neurons against Aβ toxicity (Behl et al., 1994; Butterfield et al., 1994; Goodman and Mattson, 1994; Goodman et al., 1996) and Aβ-induced impairment of ion-motive ATPase activity (Mark et al., 1995a). When hippocampal and cortical cultures were exposed to 50 μmFeSO4 (2 hr), an agent that induces hydroxyl radical production and lipid peroxidation (Zhang et al., 1993; Goodman et al., 1996), a highly significant 50–60% decrease in glucose transport occurred (Fig. 2). Pretreatment of cultures with 10 μm of the antioxidant n-propyl gallate or 50 μg/ml vitamin E resulted in complete prevention of Aβ25-35-induced impairment of glucose transport (Fig. 2), indicating the involvement of free radicals in this action of Aβ25-35. Because Aβ can impair Na+/K+-ATPase activity (Mark et al., 1995a), the major utilizer of cellular ATP (Sweadner, 1989), it was conceivable that reduced glucose transport after exposure to Aβ resulted from a reduced demand of the cells for ATP. We therefore examined the effects of ouabain, a specific inhibitor of the Na+/K+-ATPase, on glucose transport rate. We exposed cultures to a concentration of ouabain (10 μm) that resulted, as we showed previously, in a 50% reduction in Na+/K+-ATPase activity in hippocampal cell cultures, a level of inhibition equivalent to that induced by 10 μm Aβ25-35 (Mark et al., 1995a). A 2 hr exposure to ouabain had no significant effect on glucose transport in either hippocampal or cortical cells (Fig. 2).
Fig. 2.

Evidence for the involvement of reactive oxygen species in Aβ-induced impairment of glucose uptake. Hippocampal and cortical cultures were pretreated for 16 hr with vehicle or 50 μg/ml vitamin E (Vit E), or for 2 hr with 10 μmn-propyl gallate (nPG). Cultures were then exposed for 2 hr to water (Control), 50 μm Aβ25-35, 10 μmouabain, or 50 μmFeSO4. The levels of [3H]-glucose uptake were quantified, and values (expressed as percentage of control) represent the mean and SD of determinations made in at least eight separate cultures. *p < 0.01 compared to control cultures; **p < 0.001 compared to Aβ-treated cultures. ANOVA with Scheffe’s post hoc analysis.
Aβ decreases cellular ATP levels
To determine whether the level of impairment of glucose transport induced by Aβ was sufficient to cause a decrease in intracellular ATP levels, we quantified ATP levels in cultured cortical cells at different time points after exposure to 10 μm Aβ25-35. Aβ25-35 induced a time-dependent decrease in ATP levels, with the first significant (20%) decrease occurring at the 4 hr time point (Fig. 3A). ATP levels thereafter declined at a very slow rate through 10 hr of exposure to Aβ25-35, with levels being reduced by 25–30% at that time point. Thus, impairment of glucose transport (Fig. 1B) precedes depletion of cellular ATP. If Aβ-induced impairment of glucose transport is mechanistically relevant to Aβ-induced neurotoxicity, then partial inhibition of glucose uptake by phloretin should cause toxicity. Phloretin induced a concentration-dependent neurotoxicity in hippocampal cell cultures (Fig. 3B).
Fig. 3.
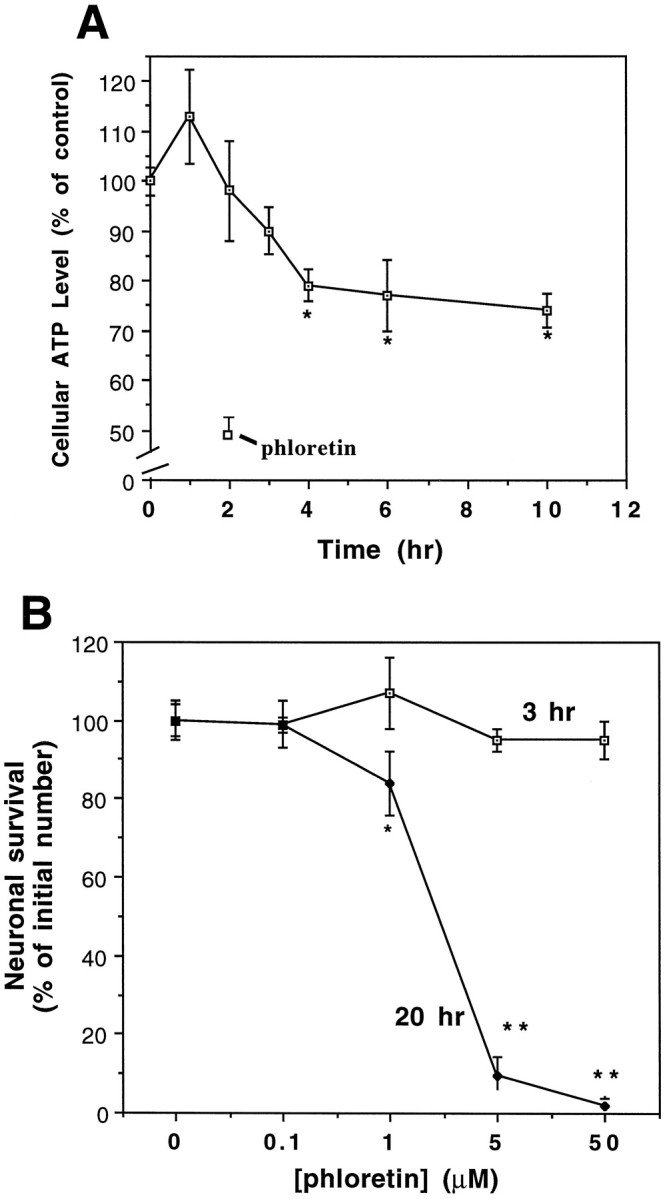
A, Aβ induces a decrease in cellular ATP levels with a time course that follows impairment of glucose uptake. Neocortical cultures were exposed to 10 μm Aβ25-35 for the indicated time periods and intracellular ATP levels were quantified (see Materials and Methods). Additional cultures were exposed to 5 μm phloretin for 2 hr. Values are expressed as percentage of the value in untreated control cultures and represent the mean and SD of determinations made in 5–15 separate cultures. *p < 0.05 compared to control cultures; ANOVA with Fischer’s post hocanalysis. The level of ATP in untreated control cultures was 29.2 ± 0.98 nmol ATP/mg protein. B, Phloretin induces a time- and concentration-dependent decrease in neuronal survival. Hippocampal cultures were exposed to the indicated concentrations of phloretin, and neuronal survival was determined 3 and 20 hr later. Values represent the mean and SD from five separate cultures. *p < 0.01, **p < 0.001 as compared to control cultures. ANOVA with Scheffe’s post hoc analysis.
Involvement of the lipid peroxidation product HNE in Aβ-induced impairment of glucose transport
We reported previously that Aβ induces the production of HNE, a cytotoxic product of lipid peroxidation (Esterbauer et al., 1991), in cultured rat hippocampal neurons (Mark et al., 1997). In the latter study we also showed that HNE impairs Na+/K+-ATPase activity and is neurotoxic. To determine whether HNE plays a role in impairment of glucose transport induced by Aβ, we determined whether HNE would impair glucose transport. Exposure of cultures to increasing concentrations of HNE for 3 hr resulted in a concentration-dependent inhibition of glucose transport (Fig. 4A). One micromolar HNE had no significant effect on glucose transport, whereas 10 and 100 μm HNE reduced glucose transport by 70% and 90%, respectively. Impairment of glucose transport occurred more rapidly in cells exposed to HNE than in cells exposed to Aβ such that a maximal decrease was observed within 3 hr of exposure to HNE, whereas it took 6–12 hr to observe maximal decreases in cells exposed to Aβ25-35 (Figs. 1B, 4A; and data not shown). The impairment of glucose transport activity was specific to HNE, because other aldehydic by-products of lipid peroxidation (pentanal, hexanal, heptanal, octanal, and nonanal) had little or no effect even at the very high concentration of 250 μm (Fig.4B). Trans-2-nonenal (250 μm) did cause a significant decrease in glucose transport, although 10 μm of this aldehyde did not impair glucose transport (data not shown).
Fig. 4.
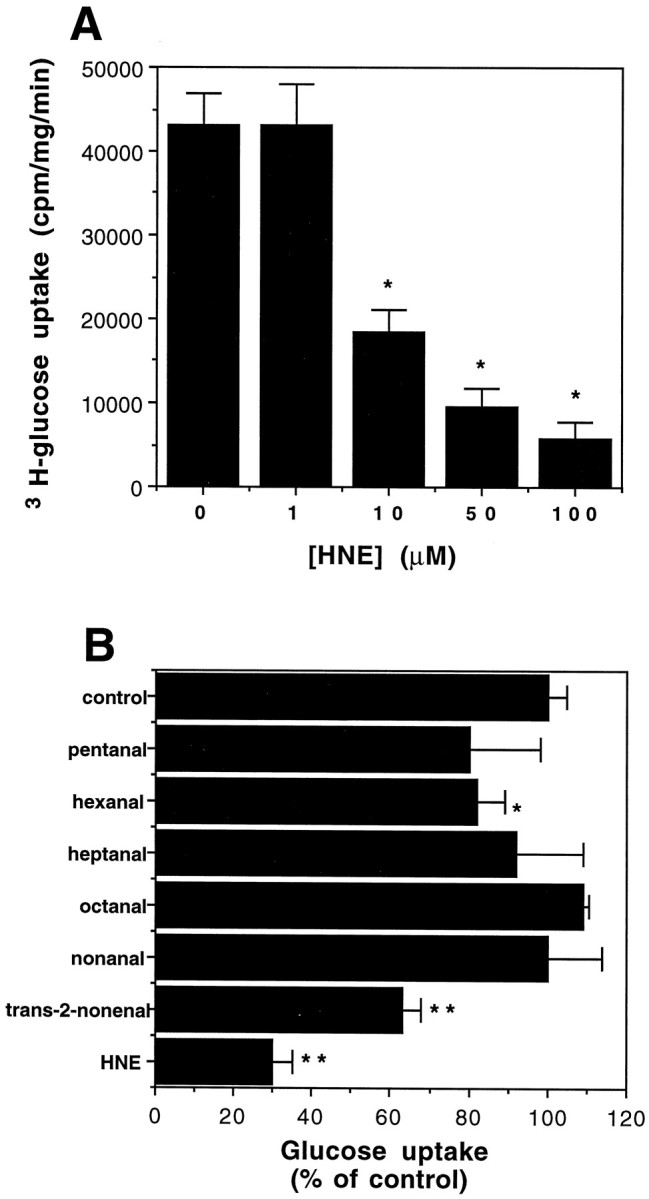
HNE impairs glucose uptake in hippocampal cell cultures. A, Cultures were exposed for 3 hr to the indicated concentrations of HNE, and [3H]-glucose uptake was quantified. Values represent the mean and SD of determinations made in five separate cultures. *p < 0.005.B, Other aldehydic products of lipid peroxidation have no effect on the rate of glucose uptake. Cortical cultures were treated with 10 μm HNE or 250 μm of the indicated aldehydes for 3 hr, and [3H]-glucose uptake was quantified. Values are the mean and SD of determinations made in six separate cultures. *p < 0.05, **p < 0.001 compared to control cultures. ANOVA with Scheffe’s post hoc analysis.
HNE is known to conjugate to lysine, histidine, and cysteine residues of proteins, and this interaction can impair the functions of those proteins (Uchida and Stadtman, 1992; Uchida et al., 1993; Siems et al., 1996; Mark et al., 1997). In preliminary studies we found that, consistent with previous in situ hybridization and immunocytochemical studies in adult rodent brain and cerebellar cell cultures (Maher and Simpson, 1994a,b; Simpson et al., 1994a; Maher et al., 1996), GLUT3 is expressed at high levels in cultured hippocampal neurons (Figs. 5, 6). GLUT1 immunoreactivity was not detectable in Western blot analysis of protein from our hippocampal or cortical cultures (data not shown). GLUT3 immunoreactivity was not present in astrocytes (Fig. 5), consistent with GLUT2 mediating glucose uptake in astrocytes (Simpson et al., 1994a). To determine whether HNE can conjugate directly to GLUT3, and whether Aβ can induce such conjugation, we used immunoprecipitation and Western blot analyses using antibodies to GLUT3 and HNE. Cultures were exposed for 4 hr to vehicle, 10 μm Aβ25-35, or 10 μm HNE. Cell proteins were then immunoprecipitated with a GLUT3 antibody and then Western-blotted using an anti-HNE antibody. A single HNE immunoreactive band at the molecular weight of GLUT3 (45 kDa) was observed in the cultures exposed to HNE and Aβ but not in control cultures (Fig. 6).
Fig. 5.
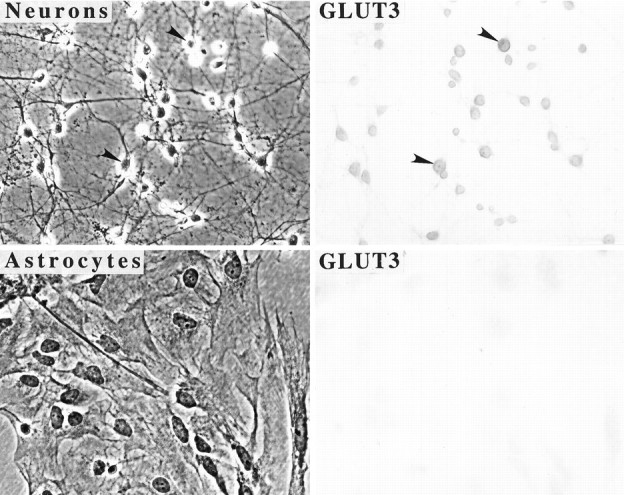
GLUT3 is expressed at high levels in cultured hippocampal neurons. Shown are phase-contrast (left) and bright-field (right) micrographs of cultured hippocampal neurons (top) and cortical astrocytes (bottom) immunostained with an antibody to GLUT3. Note that neurons exhibit considerable GLUT3 immunoreactivity, whereas astrocytes do not. Arrowheads point to neuron cell bodies (top panels).
Fig. 6.

Aβ induces HNE production and conjugation to the glucose transporter. Neocortical cultures were exposed for 4 hr to 10 μm Aβ25-35 (A), 10 μm HNE (H), or vehicle (V). Solubilized total cell protein was immunoprecipitated with an antibody against the glucose transporter, and the antibody-bound proteins were separated by SDS-PAGE, transferred to nitrocellulose, and immunoreacted with an HNE antibody (see Materials and Methods).
DISCUSSION
The present data demonstrate that Aβ can impair glucose uptake in cultured hippocampal and cortical neurons by a mechanism involving HNE, an aldehydic product of membrane lipid peroxidation. Aβ decreased the rate of glucose transport in a concentration-dependent manner; effective concentrations were in the range shown to be neurotoxic in the present study and previous studies (Yankner et al., 1990; Mattson et al., 1993b; Pike et al., 1993). The decrease in glucose transport after exposure to Aβ was relatively rapid, occurring within 1–2 hr of exposure, and preceded neurotoxicity, which did not become evident until after 6 hr of exposure. This time frame of impairment of glucose transport is consistent with a post-translational effect on function of the transporter, rather than an effect on expression of the transport protein. Our Western blot analysis of GLUT3 protein after Aβ treatment showed no change in GLUT3 protein levels, even after 6 hr of treatment (data not shown). FeSO4 and HNE also reduced glucose transport within 2 hr of exposure, and the antioxidants n-propyl gallate and vitamin E blocked impairment of glucose transport by Aβ, suggesting the involvement of oxyradical-mediated damage to the transporter. The observations that Aβ induced HNE production and conjugation to GLUT3, and HNE impaired glucose transport, further suggest a major role for lipid peroxidation in compromise of glucose transporter function. The concentrations of HNE produced in cultured hippocampal cells exposed to Aβ are in the range of 1–10 μm (Mark et al., 1997), concentrations that in the present study impaired glucose transport.
Aβ-induced impairment of glucose transport preceded the decrease in cellular ATP levels, suggesting the possibility that the reduced glucose uptake was causally linked to ATP depletion. With the same hippocampal culture system used in the present study, we showed previously that cellular ATP levels decrease by >30% within 1 hr after glucose withdrawal (Mattson et al., 1993c). In the present study we showed that phloretin, a specific inhibitor of glucose transport (Yokota et al., 1983), caused a decrease in cellular ATP levels. Thus, inhibition of glucose transport is sufficient to account for the suppressive effects of Aβ and HNE on cellular ATP levels. Previous studies have shown that Aβ impairs mitochondrial activity, as indicated by a decreased ability of neurons and synaptosomes exposed to Aβ to reduce the compound 3-(4:5-dimethylthiazol-2-yl)−2:5-diphenyltetrazolium bromide (MTT) (Shearman et al., 1994; Keller et al., 1997). Although not established in the present study, it is conceivable that reduced glucose availability to mitochondria may contribute to the observed decrease in MTT reduction. Metabolic impairment is known to increase neuronal vulnerability to excitotoxicity (Novelli et al., 1988; Bowling and Beal, 1995). Neuronal death induced by glucose deprivation involves activation of NMDA receptors and calcium overload (Cheng and Mattson, 1992a). Impairment of glucose transport may therefore provide an explanation for the fact that exposure of cultured neurons to Aβ increases their vulnerability to excitotoxicity (Koh et al., 1990;Mattson et al., 1992). Zhang et al. (1996) recently reported that Aβ1-40 induced a relatively slow decrease in ATP levels in cultured primary neurons that did not occur until after 6 hr of exposure; the rate of glucose utilization fell only slightly during the first 18 hr of exposure to Aβ1-40. 3H-glucose transport was not examined in the latter study. In the present study we found that the time course of impairment of glucose transport in neurons exposed to Aβ1-40 was slower than in neurons exposed to Aβ25-35, and in general we find that Aβ25-35 is more potent than Aβ1-40 in various cytotoxicity assays (e.g., Mark et al., 1995a). Differences in time course and concentration-dependence of the neurotoxic actions of Aβ, however, have previously been related to lot-to-lot variability in the aggregation kinetics of different batches of Aβ (May et al., 1992), which may explain the quantitative differences between our data and those of Zhang et al. (1996).
Taken together with previous findings (Behl et al., 1994; Butterfield et al., 1994; Mark et al., 1995a, 1997; Goodman et al., 1996; Harris et al., 1996), the present data suggest a scenario in which Aβ induces membrane lipid peroxidation and generation of HNE. HNE then binds to membrane proteins involved in transport of ions (Na+/K+-ATPase and Ca2+-ATPase) (Mark et al., 1995a, 1997), glutamate (Keller et al., 1997), and glucose and impairs their function. Impairment of the Na+/K+-ATPase results in membrane depolarization and promotes Ca2+ influx through NMDA receptors and voltage-dependent channels. Impairment of glutamate transport results in excessive accumulation of extracellular glutamate, with consequent overstimulation of glutamate receptors. Impairment of glucose transport results in ATP depletion, compromise of ion-motive ATPase function, and increased vulnerability to excitotoxic and oxidative insults. Previous studies have shown that Aβ (Koh et al., 1990; Mattson et al., 1992), HNE (Mark et al., 1997), and ouabain (Brines et al., 1995; Calabresi et al., 1995) increase neuronal vulnerability to excitotoxicity. Aβ (Loo et al., 1993), HNE (Kruman et al., 1996), and ouabain (Mark et al., 1995a) each also induce apoptosis in cultured neurons, and such cell death can be suppressed by agents that suppress lipid peroxidation (Behl et al., 1994;Goodman and Mattson, 1994; Goodman et al., 1996), detoxify HNE (Mark et al., 1997), or stabilize ion homeostasis (Mark et al., 1995a,b). These data suggest pivotal roles for lipid peroxidation, HNE production, and disruption of ion homeostasis in neuronal apoptosis induced by Aβ.
By inducing ATP depletion, Aβ might also alter protein phosphorylation reactions mediated by various kinases that could contribute to certain aspects of the neurodegenerative process. For example, both Aβ and metabolic/excitotoxic insults have been shown to alter the phosphorylation of various cytoskeletal proteins, including the microtubule-associated protein tau, a key component of neurofibrillary tangles in AD (Ko et al., 1990; Mattson, 1990; Cheng and Mattson, 1992b; Busciglio et al., 1995; Smith-Swintosky et al., 1996).
Although studies of neurons in dissociated cell cultures cannot provide conclusive evidence for mechanisms operative in AD patients, the present data suggest that lipid peroxidation induced by Aβ may underlie the well documented impairment of glucose transport in AD brain. Evidence linking Aβ to increased oxidative stress in AD brain is accumulating and includes data that show the following: increased levels of lipid peroxidation and protein oxidation in brain regions where fibrillar Aβ accumulates (Smith et al., 1991; Lovell et al., 1995); the presence of advanced glycation end-products, markers of oxidative stress, in neuritic plaques (Smith et al., 1994; Vitek et al., 1994); and genetic and biochemical data suggesting impaired mitochondrial function in AD brain cells (for reviews, see Luft, 1994;Benzi and Moretti, 1995). Associations between Aβ deposition and impaired glucose transport in AD have not been established; however, brain imaging data indicate that deficits in glucose uptake are greatest in brain regions such as the temporal (entorhinal cortex and hippocampus) and parietal cortex (Piert et al., 1996), where there is the greatest amyloid burden (Cummings and Cotman, 1995). Studies of the inter-relationships of Aβ deposition, oxidative stress, glucose transport, and neuron degeneration in transgenic mice expressing human βAPP mutations (Games et al., 1995; Hsaio et al., 1996) may prove useful in establishing sequences of events and cause–effect relationships in the neurodegenerative process in AD.
Footnotes
This work was supported by grants to M.P.M. from National Institutes of Health (NS30583 and AG10836), the Alzheimer’s Association (Zenith Award), and the Metropolitan Life Foundation; to J.W.G. from National Institutes of Health (AG05144); and a National Institute on Aging training grant fellowship to R.J.M. We thank S. Bose, W. Fu, R. Pelphrey, and J. G. Begley for technical assistance.
Correspondence should be addressed to Mark P. Mattson, 211 Sanders-Brown Building, University of Kentucky, 800 South Limestone, Lexington, KY 40536-0230.
REFERENCES
- 1.Barger SW, Mattson MP. Induction of neuroprotective κB-dependent transcription by secreted forms of the Alzheimer’s β-amyloid precursor. Mol Brain Res. 1996;40:116–126. doi: 10.1016/0169-328x(96)00036-8. [DOI] [PubMed] [Google Scholar]
- 2.Behl C, Davis J, Lesley R, Schubert D. Hydrogen peroxide mediates amyloid β protein toxicity. Cell. 1994;77:817–827. doi: 10.1016/0092-8674(94)90131-7. [DOI] [PubMed] [Google Scholar]
- 3.Benzi G, Moretti A. Age- and peroxidative stress-related modifications of the cerebral enzymatic activities linked to mitochondria and the glutathione system. Free Radic Biol Med. 1995;19:77–101. doi: 10.1016/0891-5849(94)00244-e. [DOI] [PubMed] [Google Scholar]
- 4.Blass JP. Metabolic alterations common to neural and non-neural cells in Alzheimer’s disease. Hippocampus. 1993;3:45–54. [PubMed] [Google Scholar]
- 5.Bowling AC, Beal MF. Bioenergetic and oxidative stress in neurodegenerative diseases. Life Sci. 1995;56:1151–1171. doi: 10.1016/0024-3205(95)00055-b. [DOI] [PubMed] [Google Scholar]
- 6.Brines ML, Dare AO, de Lanerolle NC. The cardiac glycoside ouabain potentiates excitotoxic injury of adult neurons in rat hippocampus. Neurosci Lett. 1995;191:145–148. doi: 10.1016/0304-3940(95)11577-j. [DOI] [PubMed] [Google Scholar]
- 7.Busciglio J, Lorenzo A, Yeh J, Yankner BA. β-amyloid fibrils induce tau phosphorylation and loss of microtubule binding. Neuron. 1995;14:879–888. doi: 10.1016/0896-6273(95)90232-5. [DOI] [PubMed] [Google Scholar]
- 8.Butterfield DA, Hensley K, Harris M, Mattson MP, Carney J. β-amyloid peptide free radical fragments initiate synaptosomal lipoperoxidation in a sequence-specific fashion: implications to Alzheimer’s disease. Biochem Biophys Res Commun. 1994;200:710–715. doi: 10.1006/bbrc.1994.1508. [DOI] [PubMed] [Google Scholar]
- 9.Cai X, Golde T, Younkin S. Release of excess amyloid β protein from a mutant amyloid β protein precursor. Science. 1993;259:514–516. doi: 10.1126/science.8424174. [DOI] [PubMed] [Google Scholar]
- 10.Calabresi P, De Murtas M, Pisani A, Stefani A, Sancesario G, Mercuri NB, Bernardi G. Vulnerability of medium spiny striatal neurons to glutamate: role of Na+/K+ ATPase. Eur J Neurosci. 1995;7:1674–1683. doi: 10.1111/j.1460-9568.1995.tb00689.x. [DOI] [PubMed] [Google Scholar]
- 11.Cheng B, Mattson MP. IGF-I and IGF-II protect cultured hippocampal and septal neurons against calcium-mediated hypoglycemic damage. J Neurosci. 1992a;12:1558–1566. doi: 10.1523/JNEUROSCI.12-04-01558.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheng B, Mattson MP. Glucose deprivation elicits neurofibrillary tangle-like antigenic changes in hippocampal neurons: prevention by NGF and bFGF. Exp Neurol. 1992b;117:114–123. doi: 10.1016/0014-4886(92)90120-f. [DOI] [PubMed] [Google Scholar]
- 13.Citron M, Oltersdorf T, Haass C, McConlogue L, Hung A, Seubert P, Vigo-Pelfrey C, Lieberburg I, Selkoe D. Mutation of the beta-amyloid precursor protein in familial Alzheimer’s disease increases beta-protein production. Nature. 1992;360:672–674. doi: 10.1038/360672a0. [DOI] [PubMed] [Google Scholar]
- 14.Cummings BJ, Cotman CW. Image analysis of β-amyloid load in Alzheimer’s disease and relation to dementia severity. Lancet. 1995;346:1524–1528. doi: 10.1016/s0140-6736(95)92053-6. [DOI] [PubMed] [Google Scholar]
- 15.Esterbauer H, Schaur RJ, Zollner H. Chemistry and biochemistry of 4-hydroxynonenal, malonaldehyde and related aldehydes. Free Radic Biol Med. 1991;11:81–128. doi: 10.1016/0891-5849(91)90192-6. [DOI] [PubMed] [Google Scholar]
- 16.Games D, Adams D, Alessandrini R, Barbour R, Berthelette P, Blackwell C, Carr T, Clemens J, Donaldson T, Gillespie F, Guido T, Hagoplan S, Johnson-Wood K, Khan K, Lee M, Lelbowitz E, McConlogue S, Montoya-Zavala M, Mucke L, Paganini L, Penniman E, Power M, Schenk D, Seubert P, Snyder B, Soriano F, Tan H, Vitale J, Wadsworth S, Wolozin B, Zhao J. Alzheimer-type neuropathology in transgenic mice overexpressing V717F β-amyloid precursor protein. Nature. 1995;373:523–527. doi: 10.1038/373523a0. [DOI] [PubMed] [Google Scholar]
- 17.Goodman Y, Mattson MP. Secreted forms of β-amyloid precursor protein protect hippocampal neurons against amyloid β-peptide-induced oxidative injury. Exp Neurol. 1994;128:1–12. doi: 10.1006/exnr.1994.1107. [DOI] [PubMed] [Google Scholar]
- 18.Goodman Y, Bruce AJ, Cheng B, Mattson MP. Estrogens attenuate and corticosterone exacerbates excitotoxicity, oxidative injury and amyloid β-peptide toxicity in hippocampal neurons. J Neurochem. 1996;66:1836–1844. doi: 10.1046/j.1471-4159.1996.66051836.x. [DOI] [PubMed] [Google Scholar]
- 19.Greenamyre JT, Young AB. Excitatory amino acids and Alzheimer’s disease. Neurobiol Aging. 1989;10:593–602. doi: 10.1016/0197-4580(89)90143-7. [DOI] [PubMed] [Google Scholar]
- 20.Harr SD, Simonian NA, Hyman BT. Functional alterations in Alzheimer’s disease: decreased glucose transporter 3 immunoreactivity in the perforant pathway terminal zone. J Neuropathol Exp Neurol. 1995;54:38–41. [PubMed] [Google Scholar]
- 21.Harris ME, Wang Y, Pedigo NW, Jr, Hensley K, Butterfield DA, Carney JM. Amyloid beta peptide (25–35) inhibits Na+-dependent glutamate uptake in rat hippocampal astrocyte cultures. J Neurochem. 1996;67:277–286. doi: 10.1046/j.1471-4159.1996.67010277.x. [DOI] [PubMed] [Google Scholar]
- 22.Horner HC, Packan DR, Sapolsky RM. Glucocorticoids inhibit glucose transport in cultured hippocampal neurons and glia. Neuroendocrinology. 1990;52:57–64. doi: 10.1159/000125539. [DOI] [PubMed] [Google Scholar]
- 23.Hoyer S, Oesterreich K, Wagner O. Glucose metabolism as the site of the primary abnormality in early-onset dementia of Alzheimer type? J Neurol. 1988;235:143–148. doi: 10.1007/BF00314304. [DOI] [PubMed] [Google Scholar]
- 24.Hsaio K, Chapman P, Nilsen S, Eckman C, Harigaya Y, Younkin S, Yang F, Cole G. Correlative memory deficits, Ab elevation, and amyloid plaques in transgenic mice. Science. 1996;274:99–103. doi: 10.1126/science.274.5284.99. [DOI] [PubMed] [Google Scholar]
- 25.Jagust WJ, Seab JP, Huesman RH, Valk PE, Mathis CA, Reed BR, Coxson PG, Budinger TF. Diminished glucose transport in Alzheimer’s disease: dynamic PET studies. J Cereb Blood Flow Metab. 1991;11:323–330. doi: 10.1038/jcbfm.1991.65. [DOI] [PubMed] [Google Scholar]
- 26.Kalaria RN, Harik SI. Reduced glucose transporter at the blood-brain barrier and in cerebral cortex in Alzheimer’s disease. J Neurochem. 1989;53:1083–1088. doi: 10.1111/j.1471-4159.1989.tb07399.x. [DOI] [PubMed] [Google Scholar]
- 27.Keller JN, Mark RJ, Bruce AJ, Blanc EM, Rothstein JD, Uchida K, Mattson MP (1997) 4-hydroxynonenal, an aldehydic product of membrane lipid peroxidation, impairs glutamate transport and mitochondrial function in synaptosomes. Neuroscience, in press. [DOI] [PubMed]
- 28.Kelly J, Furukawa K, Barger SW, Mark RJ, Rengen MR, Roth G, Mattson MP. Amyloid β-peptide disrupts carbachol-induced muscarinic cholinergic signal transduction in cortical neurons. Proc Natl Acad Sci USA. 1996;93:6753–6758. doi: 10.1073/pnas.93.13.6753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kennedy AM, Frackowiak RS, Newman SK, Bloomfield PM, Seaward J, Roques P, Lewington G, Cunningham VJ, Rossor MN. Deficits in cerebral glucose metabolism demonstrated by positron emission tomography in individuals at risk of familial Alzheimer’s disease. Neurosci Lett. 1995;186:17–20. doi: 10.1016/0304-3940(95)11270-7. [DOI] [PubMed] [Google Scholar]
- 30.Ko LW, Sheu KF, Young O, Thaler H, Blass JP. Expression in cultured human neuroblastoma cells of epitopes associated with affected neurons in Alzheimer’s disease. Am J Pathol. 1990;136:867–879. [PMC free article] [PubMed] [Google Scholar]
- 31.Koh J-Y, Yang LL, Cotman CW. β-amyloid protein increases the vulnerability of cultured cortical neurons to excitotoxic damage. Brain Res. 1990;533:315–320. doi: 10.1016/0006-8993(90)91355-k. [DOI] [PubMed] [Google Scholar]
- 32.Kruman I, Guo Q, Bruce AJ, Bredesen DE, Mattson MP. Hydroxynonenal may mediate apoptotic neuronal death induced by trophic factor withdrawal and oxidative insults. Soc Neurosci Abstr. 1996;22:1481. [Google Scholar]
- 33.Loo DT, Copani A, Pike CJ, Whittemore ER, Walencewicz AJ, Cotman CW. Apoptosis is induced by beta-amyloid in cultured central nervous system neurons. Proc Natl Acad Sci USA. 1993;90:7951–7955. doi: 10.1073/pnas.90.17.7951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lovell MA, Ehmann WD, Butler SM, Markesbery WR. Elevated thiobarbituric acid-reactive substances and antioxidant enzyme activity in the brain in Alzheimer’s disease. Neurology. 1995;45:1594–1601. doi: 10.1212/wnl.45.8.1594. [DOI] [PubMed] [Google Scholar]
- 35.Luft R. The development of mitochondrial medicine. Proc Natl Acad Sci USA. 1994;91:8731–8738. doi: 10.1073/pnas.91.19.8731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maher F, Simpson IA. The GLUT3 glucose transporter is the predominant isoform in primary cultured neurons: assessment by biosynthetic and photoaffinity labelling. Biochem J. 1994a;301:379–384. doi: 10.1042/bj3010379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Maher F, Simpson IA. Modulation of expression of glucose transporters GLUT3 and GLUT1 by potassium and N-methyl-d-aspartate in cultured cerebellar granule neurons. Mol Cell Neurosci. 1994b;5:369–375. doi: 10.1006/mcne.1994.1044. [DOI] [PubMed] [Google Scholar]
- 38.Maher F, Davies-Hill TM, Simpson IA. Substrate specificity and kinetic parameters of GLUT3 in rat cerebellar granule neurons. Biochem J. 1996;315:827–831. doi: 10.1042/bj3150827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mark RJ, Hensley K, Butterfield DA, Mattson MP. Amyloid β-peptide impairs ion-motive ATPase activities: evidence for a role in loss of neuronal Ca2+ homeostasis and cell death. J Neurosci. 1995a;15:6239–6249. doi: 10.1523/JNEUROSCI.15-09-06239.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Mark RJ, Ashford JW, Mattson MP. Anticonvulsants attenuate amyloid β-peptide neurotoxicity and promote maintenance of calcium homeostasis. Neurobiol Aging. 1995b;16:187–198. doi: 10.1016/0197-4580(94)00150-2. [DOI] [PubMed] [Google Scholar]
- 41.Mark RJ, Lovell MA, Markesbery WR, Uchida K, Mattson MP (1997) A role for 4-hydroxynonenal in disruption of ion homeostasis and neuronal death induced by amyloid β-peptide. J Neurochem, in press. [DOI] [PubMed]
- 42.Mattson MP. Antigenic changes similar to those seen in neurofibrillary tangles are elicited by glutamate and calcium influx in cultured hippocampal neurons. Neuron. 1990;4:105–117. doi: 10.1016/0896-6273(90)90447-n. [DOI] [PubMed] [Google Scholar]
- 43.Mattson MP. Untangling the pathophysiochemistry of β-amyloid. Nat Struct Biol. 1995;2:926–928. doi: 10.1038/nsb1195-926. [DOI] [PubMed] [Google Scholar]
- 44.Mattson MP, Cheng B, Davis D, Bryant K, Lieberburg I, Rydel RE. β-amyloid peptides destabilize calcium homeostasis and render human cortical neurons vulnerable to excitotoxicity. J Neurosci. 1992;12:376–389. doi: 10.1523/JNEUROSCI.12-02-00376.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mattson MP, Barger SW, Cheng B, Lieberburg I, Smith-Swintosky VL, Rydel RE. β-amyloid precursor protein metabolites and loss of neuronal calcium homeostasis in Alzheimer’s disease. Trends Neurosci. 1993a;16:409–415. doi: 10.1016/0166-2236(93)90009-b. [DOI] [PubMed] [Google Scholar]
- 46.Mattson MP, Tomaselli K, Rydel RE. Calcium-destabilizing and neurodegenerative effects of aggregated β-amyloid peptide are attenuated by basic FGF. Brain Res. 1993b;621:35–49. doi: 10.1016/0006-8993(93)90295-x. [DOI] [PubMed] [Google Scholar]
- 47.Mattson MP, Zhang Y, Bose S. Growth factors prevent mitochondrial dysfunction, loss of calcium homeostasis and cell injury, but not ATP depletion in hippocampal neurons deprived of glucose. Exp Neurol. 1993c;121:1–13. doi: 10.1006/exnr.1993.1066. [DOI] [PubMed] [Google Scholar]
- 48.Mattson MP, Barger SW, Begley JG, Mark RJ. Calcium, free radicals, and excitotoxic neuronal death in primary cell culture. Methods Cell Biol. 1995;46:187–216. doi: 10.1016/s0091-679x(08)61930-5. [DOI] [PubMed] [Google Scholar]
- 49.May PC, Gitter BD, Waters DC, Simmons LK, Becker GW, Small JS, Robison PM. β-Amyloid in vitro toxicity: lot-to-lot variability. Neurobiol Aging. 1992;13:605–607. doi: 10.1016/0197-4580(92)90064-5. [DOI] [PubMed] [Google Scholar]
- 50.Montine TJ, Amarnath V, Martin ME, Strittmatter WJ, Graham DG. E-4-hydroxy-2-nonenal is cytotoxic and cross-links cytoskeletal proteins in P19 neuroglial cultures. Am J Pathol. 1996;148:89–93. [PMC free article] [PubMed] [Google Scholar]
- 51.Mullan M, Crawford F. Genetic and molecular advances in Alzheimer’s disease. Trends Neurosci. 1993;16:398–403. doi: 10.1016/0166-2236(93)90007-9. [DOI] [PubMed] [Google Scholar]
- 52.Novelli A, Reilly JA, Lyska PC, Henneberry RC. Glutamate becomes neurotoxic via the N-methyl-d-aspartate receptor when intracellular energy levels are reduced. Brain Res. 1988;451:205–212. doi: 10.1016/0006-8993(88)90765-2. [DOI] [PubMed] [Google Scholar]
- 53.Pettegrew JW, Panchalingam K, Klunk WE, McClure RJ, Muenz LR. Alterations of cerebral metabolism in probable Alzheimer’s disease: a preliminary study. Neurobiol Aging. 1994;15:117–132. doi: 10.1016/0197-4580(94)90152-x. [DOI] [PubMed] [Google Scholar]
- 54.Piert M, Koeppe RA, Giordani B, Berent S, Kuhl DE. Diminished glucose transport and phosphorylation in Alzheimer’s disease determined by dynamic FDG-PET. J Nucl Med. 1996;37:201–208. [PubMed] [Google Scholar]
- 55.Pike C, Burdick D, Walencewicz A, Glabe C, Cotman C. Neurodegeneration induced by β-amyloid peptides in vitro: the role of peptide assembly state. J Neurosci. 1993;13:1676–1686. doi: 10.1523/JNEUROSCI.13-04-01676.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reiman EM, Caselli RJ, Yun LS, Chen K, Bandy D, Minoshima S, Thibodeau SN, Osborne D. Preclinical evidence of Alzheimer’s disease in persons homozygous for the epsilon 4 allele for apolipoprotein E. New Engl J Med. 1996;334:752–758. doi: 10.1056/NEJM199603213341202. [DOI] [PubMed] [Google Scholar]
- 57.Selkoe DJ. The molecular pathology of Alzheimer’s disease. Neuron. 1993;6:487–498. doi: 10.1016/0896-6273(91)90052-2. [DOI] [PubMed] [Google Scholar]
- 58.Shearman MS, Ragan CI, Iversen LL. Inhibition of PC12 cell redox activity is a specific, early indicator of the mechanism of β-amyloid-mediated cell death. Proc Natl Acad Sci USA. 1994;91:1470–1474. doi: 10.1073/pnas.91.4.1470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Siems WG, Hapner SJ, van Kuijk FJGM. 4-hydroxynonenal inhibits Na+-K+-ATPase. Free Radic Biol Med. 1996;20:215–223. doi: 10.1016/0891-5849(95)02041-1. [DOI] [PubMed] [Google Scholar]
- 60.Simpson IA, Vannucci SJ, Maher F. Glucose transporters in mammalian brain. Biochem Soc Trans. 1994a;22:671–675. doi: 10.1042/bst0220671. [DOI] [PubMed] [Google Scholar]
- 61.Simpson IA, Chundu KR, Davies-Hill T, Honer WG, Davies P. Decreased concentrations of GLUT1 and GLUT3 glucose transporters in the brains of patients with Alzheimer’s disease. Ann Neurol. 1994b;35:546–551. doi: 10.1002/ana.410350507. [DOI] [PubMed] [Google Scholar]
- 62.Sims NR. Altered glucose metabolism in Alzheimer’s disease. Ann Neurol. 1990;27:691–693. doi: 10.1002/ana.410270621. [DOI] [PubMed] [Google Scholar]
- 63.Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc Natl Acad Sci USA. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Smith MA, Taneda S, Richey PL, Miyata S, Yan SD, Stern D, Sayre LM, Monnier VM, Perry G. Advanced Maillard reaction end products are associated with Alzheimer disease pathology. Proc Natl Acad Sci USA. 1994;91:5710–5714. doi: 10.1073/pnas.91.12.5710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Smith MA, Sayre LM, Monnier VM, Perry G. Radical ageing in Alzheimer’s disease. Trends Neurosci. 1995;18:172–176. doi: 10.1016/0166-2236(95)93897-7. [DOI] [PubMed] [Google Scholar]
- 66.Smith-Swintosky VL, Pettigrew LC, Sapolsky RM, Phares C, Craddock SD, Brooke SM, Mattson MP. Metyrapone, an inhibitor of glucocorticoid production, reduces brain injury induced by focal and global ischemia and seizures. J Cereb Blood Flow Metab. 1996;16:585–598. doi: 10.1097/00004647-199607000-00008. [DOI] [PubMed] [Google Scholar]
- 67.Suzuki N, Cheung T, Cai X, Odaka A, Otovos L, Eckman L, Golde T, Younkin S. An increased percentage of long amyloid β protein secreted by familial amyloid β protein precursor (βAPP717) mutants. Science. 1994;264:1336–1340. doi: 10.1126/science.8191290. [DOI] [PubMed] [Google Scholar]
- 68.Uchida K, Stadtman ER. Modification of histidine residues in proteins by reaction with 4-hydroxynonenal. Proc Natl Acad Sci USA. 1992;89:4544–4548. doi: 10.1073/pnas.89.10.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Uchida K, Szweda LI, Chae HZ, Stadtman ER. Immunochemical detection of 4-hydroxynonenal protein adducts in oxidized hepatocytes. Proc Natl Acad Sci USA. 1993;90:8742–8746. doi: 10.1073/pnas.90.18.8742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vitek MP, Bhattacharya K, Glendening JM, Stopa E, Vlassara H, Bucala R, Manogue K, Cerami A. Advanced glycation end products contribute to amyloidosis in Alzheimer disease. Proc Natl Acad Sci USA. 1994;91:4766–4770. doi: 10.1073/pnas.91.11.4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yankner BA. Mechanisms of neuronal degeneration in Alzheimer’s disease. Neuron. 1996;16:921–932. doi: 10.1016/s0896-6273(00)80115-4. [DOI] [PubMed] [Google Scholar]
- 72.Yankner BA, Duffy LK, Kirschner DA. Neurotrophic and neurotoxic effects of amyloid beta protein: reversal by tachykinin neuropeptides. Science. 1990;250:279–282. doi: 10.1126/science.2218531. [DOI] [PubMed] [Google Scholar]
- 73.Yokota K, Nishi Y, Takesue Y. Effect of phloretin on Na+-dependent D-glucose uptake by intestinal brush border membrane vesicles. Biochem Pharmacol. 1983;32:3453–3457. doi: 10.1016/0006-2952(83)90376-3. [DOI] [PubMed] [Google Scholar]
- 74.Zhang Y, Tatsuno T, Carney J, Mattson MP. Basic FGF, NGF, and IGFs protect hippocampal neurons against iron-induced degeneration. J Cereb Blood Flow Metab. 1993;13:378–388. doi: 10.1038/jcbfm.1993.51. [DOI] [PubMed] [Google Scholar]
- 75.Zhang Z, Rydel RE, Drzewiecki GJ, Fuson K, Wright S, Wogulis M, Audia JE, May PC, Hyslop PA. Amyloid β-mediated oxidative and metabolic stress in rat cortical neurons: no direct evidence for a role for H2O2 generation. J Neurochem. 1996;67:1595–1606. doi: 10.1046/j.1471-4159.1996.67041595.x. [DOI] [PubMed] [Google Scholar]