

Abstract
Human small-cell lung carcinoma (SCLC) cells express neuronal-like voltage-operated calcium channels (VOCCs) and release mitogenic hormones such as serotonin (5-HT). Opioid peptides, on the other hand, have been shown to reduce SCLC cell proliferation by an effective autocrine pathway.
Here we show that in GLC8 SCLC cells, only δ-opioid receptor subtype mRNA is expressed. Consistently, the selective δ-opioid agonist [d-Pen2-Pen5]-enkephalin (DPDPE), but not μ and κ agonists, potently and dose-dependently inhibits high-threshold (HVA) VOCCs in these cells. As in peripheral neurons, this modulation is largely voltage-dependent, mediated by pertussis toxin (PTX)-sensitive G-proteins, cAMP-independent, and mainly affecting N-type VOCCs.
With the same potency and selectivity, DPDPE also antagonizes the Ca2+-dependent release of [3H]serotonin ([3H]5-HT) from GLC8 cells. However, DPDPE inhibits not only the depolarization-induced release, but also the Ca2+-dependent secretion induced by thapsigargin or ionomycin. This suggests that besides inhibiting HVA VOCCs, opioids also exert a direct depressive action on the secretory apparatus in GLC8 cells. This latter effect also is mediated by a PTX-sensitive G-protein but, contrary to VOCC inhibition, it can be reversed by elevations of cAMP levels.
These results show for the first time that opioids effectively depress both Ca2+ influx and Ca2+-dependent hormone release in SCLC cells by using multiple modulatory pathways. It can be speculated that the two mechanisms may contribute to the opioid antimitogenic action on lung neuroendocrine carcinoma cells.
Keywords: small-cell lung carcinoma, human, voltage-operated calcium channels, hormone secretion, opioid modulation, G-proteins
Modulation of neuronal excitability by opioids generally is linked to changes in ion channel properties (North, 1993). Activation of μ-, κ-, and δ-opioid receptors can mediate the inhibition of neurotransmitter release from nerve terminals (MacDonald and Nelson, 1978; Konishi et al., 1979; Mudge et al., 1979; Illes, 1989) as well as catecholamine release from adrenal chromaffin cells (Kumakura et al., 1980). However, the ability of opioids to inhibit neurotransmitter and hormone secretion cannot be accounted for fully by the modulation of ion channels alone. Direct effects of opioids (Capogna et al., 1993; Rekling, 1993; Lupica, 1995) and other inhibitory hormones (Jones et al., 1987; Ullrich and Wollheim, 1988;Man-Son-Hing et al., 1989; Dale and Kandel, 1990; Luini and De Matteis, 1990; Ullrich et al., 1990; Scholz and Miller, 1992) on the secretory machinery also have been described.
Human small-cell lung carcinoma (SCLC) cells contain secretory granules and release both amines and peptide hormones (Weynants et al., 1990;Woll, 1991). In particular, SCLC cells synthesize opioid peptides and express opioid receptors (Roth and Barchas, 1985; Maneckjee and Minna, 1990). SCLC cells also express heterogeneous voltage-operated calcium channels (VOCC) subtypes, some of which are typically “neuronal” (DeAizpurua et al., 1988; Sher et al., 1990), like the ω-conotoxin (ω-CTx)-sensitive GVIA VOCC (N-type), that are ubiquitous in all SCLC cell lines tested (Sher et al., 1990). Recently, mRNA coding for class D (L-type), class B (N-type), and class A (P/Q-type) α1 VOCC subunits has been detected in the GLC8 SCLC cell line (Codignola et al., 1993). Agents selective for the three VOCC subtypes [dihydropyridines (DHPs)] for the L-type, ω-CTx for the N-type, and ω-agatoxin (ω-Aga) IVA for the P/Q-type block separate components of the HVA Ca2+ currents and hormone release from these cells (Codignola et al., 1993). Thus, GLC8 cells represent an ideal preparation in which to test the relative contribution of Ca2+ channel modulation versus distal effects on the secretory apparatus in the overall control of secretion in neuroendocrine cells.
Here we report that DPDPE potently inhibits HVA Ca2+ channels in SCLC cells, an effect mediated by δ receptors, the only opioid receptor subtype detectable in these cells. The modulation is exerted primarily on N-type channels, with no effects on the L-type and small residual effects on non-L-, non-N-types. Ca2+ channel modulation typically is voltage-dependent and mediated by PTX-sensitive G-proteins. In parallel experiments, DPDPE was found to potently depress the release of [3H]5-HT induced by depolarizing concentrations of KCl. However, DPDPE also inhibits the thapsigargin- and ionomycin-stimulated release of [3H]5-HT with the same potency, suggesting that the opioid can act directly on the secretory apparatus downstream to Ca2+ influx. These opioid effects on the secretory machinery also are mediated by PTX-sensitive G-proteins but, in contrast to VOCC inhibition, are reversed by increasing cellular cAMP levels, suggesting that multiple opioid inhibitory pathways coexist in SCLC cells.
MATERIALS AND METHODS
Cell lines. The human SCLC cell line GLC8 was kindly provided by Dr. G. Gaudino (Department of Biomedical Science and Oncology, Turin, Italy). The cells were grown in suspension in RPMI 1640 medium supplemented with 10% fetal calf serum, 100 IU/ml penicillin, 100 μg/ml streptomycin, in 10-cm-diameter Corning tissue culture dishes (New York, NY). The cells were incubated at 37°C in a humidified atmosphere of 5% CO2 in air.
RT-PCR analysis. First-strand cDNA was synthesized starting from 200 ng of poly-A+ RNA extracted from GLC8 cells and from rat brains, using the first strand synthesis kit (Stratagene, La Jolla, CA) and the supplied random examer primers. Five percent of the cDNA synthesized was amplified using Taq polymerase (Perkin-Elmer, Emeryville, CA). The PCR reactions (94°C for 30 sec; 62°C for 1 min; 72°C for 45 sec, 45 cycles) were performed in the presence of primers drawn on the sequences of human δ-, μ-, and κ-opioid receptors (Knapp et al., 1994; Mansson et al., 1994; Wang et al., 1994). The primers were chosen to match on the corresponding rat sequences. The primers used were respectively S1op and AS2op for δ receptors, S3op and AS4op for μ receptors, S5op and AS6op for κ receptors. PCR products were analyzed by agarose gel electrophoresis, and their identity was confirmed by restriction analysis.
Primers were as follows: S1op: CTCTTCGCCAACGCCTCGGAC; AS2op: GTCACCGTGTCCCAGTACCAG; S3op: AACCCGAAAAGT-CTCGGTGCTC; AS4op:TTCAGCAGGTTTTCCCAGTACC;S5op: CTGGAGCCCGCGCACATCTC; AS6op: TTCATGAAGAGGTCC-CACCAG.
Electrophysiological recordings. Membrane currents were measured with the patch-clamp technique either in the whole-cell (Hamill et al., 1981) or in the perforated-patch configuration (Horn and Marty, 1988). Stimulation, acquisition, filtering, and data analysis were carried out as described previously (Codignola et al., 1993; Pollo et al., 1993). Step depolarizations (100–200 msec) of various amplitudes were applied every 10 sec to cells clamped at −60 to −90 mV holding potential (Vh). Capacitive transients and leakage currents were cancelled by subtracting from on-line compensated currents the Cd2+-insensitive component, recorded after bath addition of 200 μm Cd2+. The external solution composition was (in mm): NaCl 125, BaCl2 10, MgCl21, 10 HEPES, and 0.3 TTX. Solutions of higher Ba2+ concentration (50 mm) were prepared by reducing the NaCl concentration to maintain constant osmolarity (300 mOsm). The pipette solution contained (in mm): CsCl2 110, TEA 30, EGTA 10, MgCl2 2, HEPES 10, glucose 8. Nifedipine, Bay K 8644, and ω-CTx were handled as described elsewhere (Codignola et al., 1993; Pollo et al., 1993). ω-Aga was dissolved in distilled water and then diluted in the extracellular solution at the indicated concentrations (Magnelli et al., 1995). Data are expressed as mean ± SEM for n = number of cells.
[3H]serotonin release. GLC8 cells were recovered from the dishes and passed through a Pasteur pipette to dissociate cell clusters. After centri-fugation (5 min, 800 rpm), the cells were resuspended in a Krebs–Ringer–Hepes (KRH) solution containing (in mm): NaCl 150, KCl 5, MgSO4 and KH2PO4 1.2, CaCl2 2, glucose 6, and Hepes–NaOH 25, pH 7.4. The medium also contained equal amounts (0.1 mg/ml) of ascorbic acid and the monoamine oxydase inhibitor pargyline, plus [3H] 5-HT (final concentration 500 nm). After 30 min at 37°C, the cells were centrifuged (5 min, 800 rpm), resuspended in KRH containing the 5-HT reuptake inhibitor Cl-imipramine (1 μm), and aliquoted in Eppendorf tubes containing the desired drug concentrations. To assay the effects of opioid agents, the cells were preincubated with the drugs 15 min before and during the stimulation period at 37°C. To test PTX effects, the cells were incubated with the toxin (100 ng/ml) overnight. At the end of the stimulation at 37°C (for 5 min or as indicated), the cells were washed three times with ice-cold KRH. Finally, the pellets were dissolved in 300 μl of N NaOH 1, resuspended in 5 ml of Ultima Gold scintillation fluid, and counted in a beta counter (Packard Tri Carb 2100TR, Groningen, The Netherlands) with an efficiency of 65% to quantify the radioactivity remaining associated with the cells. The amount of release is expressed as percent increase over basal, i.e., the release measured in parallel tubes incubated in buffer alone (see also Codignola et al., 1993).
Materials. Cell culture supplements were obtained from Seromed (Berlin, Germany) and Petri dishes from Corning. Pargyline, DPDPE, N6 2′-O-dibutyryladenosine 3′:5′- cyclic-monophosphate (Bt2cAMP,) PTX, ionomycin, thapsigargin, EGTA, GTPγS, and GDPβS were purchased from Sigma (St. Louis, MO). ω-CTx and ω-Aga were from Peptides International (Louisville, KY). Cl-imipramine, U50488 [trans-(1S,2S)-3,4-dichloro-N-methyl-N-[(1-pyrrolidinyl)cyclohexyl]-benzeneacetamide hydrochloride], DAMGO [d-Ala2, MePhe4, Gly-ol5]enkephalin), and naloxone hydrochloride [(−)isomer] were from RBI (Natick, MA). Bay K 8644 and nifedipine were kindly provided by Dr. Seuter (Bayer A.G., Wuppertal, Germany). [3H]5-HT-creatinine sulfate (specific activity 10.1 Ci/mmol) was from Amersham (Buckinghamshire, UK). All other salts were reagent grade and purchased from Merck (Darmstadt, Germany).
RESULTS
Expression of opioid receptor subtypes in GLC8 cells
The sequences of human μ-, κ-, and δ-opioid receptors have been determined recently (Knapp et al., 1994; Mansson et al., 1994;Wang et al., 1994). Using RT-PCR and primers specific for the three receptor subtypes, we checked for the expression of mRNA encoding these receptors in GLC8 cells. As shown in Figure1A, a product of the expected size (595 bp) corresponding to the δ-opioid receptor subtype was amplified from GLC8 cell mRNAs. No amplification products corresponding to μ-opioid or κ-opioid receptors were detectable. As a positive control, RT-PCR with the same primers was performed, following the same procedure, on rat brain mRNAs yielding specific amplification products for all three receptor subtypes (Fig. 1A). The specificity of the GLC8 δ-opioid receptor (Fig. 1B) and of the rat brain δ-, μ-, and κ-opioid receptor amplification products (data not shown) was confirmed by restriction analysis. As shown in Figure1B, digestion of the GLC8 δ-opioid receptor amplification product with the HincII restriction enzyme resulted in two restriction fragments of 390 bp and 205 bp, as expected from sequence analysis.
Fig. 1.

Only δ-opioid receptor mRNA is expressed in GLC8 cells. A, One tenth of each RT-PCR reaction specific for δ-, μ-, and κ-opioid receptors was analyzed by agarose gel electrophoresis. Only the amplification product (595 bp) corresponding to the δ-opioid receptor is visible by ethidium bromide staining in the lanes of GLC8 cells, whereas amplification products corresponding to δ-, μ-, and κ-opioid receptors are present in the lanes of rat brain (RB). Control reactions without reverse transcriptase were negative. Molecular weight markers (M) were (in bp): 2176, 1766, 1230, 1033, 653, 517, 453, 394, 298, 234, 220, and 154. B, Digestion of GLC8 δ receptor RT-PCR product withHincII restriction enzyme was performed to confirm its specificity. One fifth of RT-PCR product undigested (UC) andHincII digested (C) was analyzed on agarose gel electrophoresis. The band in lane UC (595 bp) is digested in lane C to give fragments of the expected size (390 and 205 bp). Molecular weight markers (M) are as in A.
Other RT-PCRs were performed with different primers specific for each receptor subtype, confirming the above results in both GLC8 and rat brain (data not shown). Furthermore, 45 supplementary cycles with nested primers were performed on the previous amplification products to both increase the sensitivity of the assay and confirm the identity of the PCR products. The results still confirmed the previous evidence.
Therefore, although different SCLC cell lines have been shown to coexpress multiple opioid receptor subtypes (Maneckjee and Minna, 1990), GLC8 cells represent a suitable model system in which to study δ-opioid receptor mechanisms in isolation.
Opioid inhibition of HVA Ca2+ channels in GLC8 cells
As reported previously (Codignola et al., 1993), GLC8 cells express only HVA VOCCs that can be studied easily under conditions in which Na+ and K+ channels are blocked effectively, and Ba2+ is used as charge carrier at high concentrations (10–50 mm). In 50 mmBa2+ and Vh −80 mV, inward HVA Ba2+ currents activated around 0 mV, reached maximal amplitude at approximately +30 mV, and reversed above +70 mV. The ability of opioid agonists to affect HVA Ba2+ currents was tested at +30 mV. Figure2A1 shows the typical inhibition of Ba2+ currents induced by saturating doses of the δ-opioid agonist DPDPE (300 nm). The current depression was accompanied by a marked slowdown of channel activation and fully reversed at washing. Except in a few cases, DPDPE caused reversible inhibitions that ranged between 48 and 87% measured at the time corresponding to the peak of the control current (69.4 ± 0.9, n = 87). Repetitive exposures to saturating concentrations of DPDPE over 10–12 min gave comparable HVA current depressions, suggesting that desensitization of DPDPE action was either absent or occurred slowly during our recording conditions (data not shown).
Fig. 2.

Selective and dose-dependent action of DPDPE on whole-cell Ba2+ currents in GLC8 cells.A, In A1, Ba2+ currents at +30 mV were recorded before (C), during (DPDPE), and after application of 0.3 μm DPDPE in 50 mm Ba2+. In A2and A3, the action of the μ-opioid-selective agonist DAMGO (1 μm) and the κ-opioid-selective agonist U50488 (1 μm) on Ba2+currents is compared with that of DPDPE (0.1 μm) on the same cell in 50 mm Ba2+. The sequential recordings were control (C), DPDPE, and the second agonist. In A4, the action of DPDPE (0.1 μm) was fully prevented by subsequent addition of the nonselective opioid antagonist naloxone (1 μm). Test depolarizations were to +30 mV from Vh −90 mV in all four panels. B, Effects of increasing doses of DPDPE (0.1, 0.3, 1, and 10 nm) on the Ba2+ currents of the same cell bathed in 10 mm Ba2+. Test depolarizations to +20 mV from Vh −90 mV.C, Percentage of Ba2+ current inhibition as a function of log [DPDPE] expressed in molar concentrations. Data points were collected from 20 GLC8 cells and plotted as mean ± SEM for n values as indicated. Thesolid line is a curve fit using equation:IBa inhibition (%) = 72/(1 + IC50/[DPDPE]), with IC50 = 0.64 nm.
DPDPE action was mediated by δ-opioid receptors. Both the μ-selective agonist DAMGO (1 μm, n = 7) and the κ-selective agonist U50488 (1 μm, n = 14) proved to be ineffective in GLC8 cells that normally responded to a subsequent application of 300 nm DPDPE (Figs. 2A2, 3). DPDPE action was fully prevented by the nonselective opioid antagonist naloxone (1 μm, n = 16), which had no effect on its own on the time course and size of the Ba2+ currents (Fig. 2A4). DPDPE action was dose-dependent when tested on the same cell (Fig. 2B) or on different cells (Fig. 2C). Significant inhibitions were evident with doses as low as 0.1–0.3 nm DPDPE, whereas saturating effects were reached at doses above 10 nm. The dose–response curve was best fitted by a one-to-one agonist–receptor stoichiometry with an IC50 of 0.64 nm and a maximal inhibition of 72% (n = 20) (solid linein Fig. 2C). On the basis of these data, we conclude that δ-opioid receptors are very efficient in inhibiting HVA VOCCs in GLC8 cells.
Voltage-dependent inhibition and facilitation of HVA VOCCs by DPDPE
Opioid inhibition of Ca2+ channels in GLC8 cells possesses all the features of the voltage-dependent VOCC inhibition by neurotransmitters reported in most neurons and neurosecretory cells (for review, see Carbone et al., 1996). At moderate potentials (+30 mV in 50 mmBa2+), the inhibition was stronger at the peak of the control current and weaker at the end of the pulse (73 vs 54% in Fig. 3A), suggesting a substantial time-dependent recovery from inhibition. At higher voltages, the inhibited currents activated more quickly and the percentage of removal of inhibition increased markedly. At the end of the +50 mV pulse, for instance, the percentage of inhibition was 34%, i.e., less than half the maximal inhibition at +30 mV. Voltage dependency of opioid inhibition also is evident from the I–Vrelationships obtained with ramp commands (inset in Fig.3A). Maximal block by DPDPE occurs at +20 mV (62%) and is reduced gradually with increasing voltages (30% at +60 mV and <15% above +70 mV).
Fig. 3.

Voltage dependency of DPDPE action on HVA Ba2+ currents. A, Ba2+ currents recorded with the perforated patch-clamp method in a GLC8 cell bathed in 50 mmBa2+. The two traces in each panel were recorded before (C) and during application of 0.3 μm DPDPE (open circles) at the potential indicated. Vh −90 mV.Inset, I–V curves in 50 mm Ba2+ recorded using a depolarizing ramp of 1.2 mV/msec from −90 mV holding potential before and during exposure to 1 μm DPDPE.B, Facilitation of DPDPE-inhibited Ba2+ currents induced by conditioning prepulses in whole-cell clamp recordings. The two overlapping traces in each panel were recorded on test depolarizations to +30 mV without (trace a) and with (trace b) a 50 msec conditioning prepulse to +90 mV. The double-pulse protocol was delivered before (top) and during (bottom) application of DPDPE with 6 sec interval between pulses. Vertical arrows indicate the time of peak control current at which DPDPE inhibition is estimated during test pulses. Horizontal arrows indicate the current amplitude reached at the test potential (+30 mV) soon after the prepulse.
The presence of a marked voltage dependency in the modulatory action of DPDPE also was demonstrated by the marked current facilitation induced by strong conditioning prepulses applied in the continuous presence of DPDPE (trace b in Fig. 3B). Prepulses of 50 msec to +90 mV were sufficient to recover >80% of the current amplitude that was depressed by DPDPE. This suggests that as in other cells (Tsunoo et al., 1986; Bean, 1989; Kasai, 1992; Rhim and Miller, 1994; Rusin and Moises, 1995), opioid modulation in GLC8 cells is largely V-dependent. V-independent modulation also was evident and accounted for 20–30% of the total modulation (Fig.3A,B) (also data not shown). We found, however, no indications that the two processes could be mediated by different receptors or G-proteins or directed against different VOCC subtypes (see below). Interestingly, in 20% of the cells not exposed to DPDPE, the conditioning prepulse induced a small but significant (10–50%) facilitation of Ba2+ currents. This suggests that as in sympathetic neurons (Ikeda, 1991) and chromaffin cells (Fenwick et al., 1982; Albillos et al., 1995), a “tonic inhibition” also is present in GLC8 cells, possibly attributable to a feedback inhibition caused by the hormones released by the cells themselves (Doupnik and Pun, 1994).
DPDPE inhibition is mediated by PTX-sensitive G-proteins and is independent of cAMP
In some neurons and neurosecretory cells, Ca2+ channel modulation by opioids is mediated by PTX-sensitive G-proteins (Hescheler et al., 1987; Kasai, 1992; Taussig et al., 1992; Albillos et al., 1995), very likely through direct membrane-delimited pathways (Shen and Suprenant, 1991). This also was the case for Ca2+ channel inhibition in GLC8 cells.
In cells showing slowly activating Ba2+ currents soon after the establishment of the whole-cell configuration (tonic inhibition) (Ikeda, 1991), intracellular application of GTPγS via the patch pipette (100 μm, n = 6) preserved the initial kinetic slowing (Fig. 4, top left). This current kinetics modification persisted after 3 min of perfusion and could be largely reversed by conventional prepulses (top middle). Application of DPDPE after 4–5 min caused a slightly increased prolongation of the activation kinetics (top right) that persisted after washout of the agonist (data not shown). On the contrary, in cells with Ba2+currents inhibited tonically at the start of the recordings, DPDPE action could be prevented completely by 1–2 min cell dialysis with GDPβS (500 μm, n = 18). In the example of Figure 4 middle, GDPβS could both remove the initial tonic inhibition (middle left) and prevent the voltage-dependent action of DPDPE after 3 min (middle right). Overnight cell incubation with PTX (100 ng/ml, 15 hr at 37°C) also was very effective in preventing both the tonic inhibition of Ba2+ currents and DPDPE action (Fig. 4bottom). In 9 of 11 cells, PTX treatment abolished completely the DPDPE-induced inhibition; Ba2+currents were fast-activating and showed no sign of initial tonic inhibition. In the two remaining cells, a small effect (∼20% of inhibition) still was present. All this confirms that Ca2+ channel modulation by opioid in GLC8 cells is mediated primarily by PTX-sensitive G-proteins.
Fig. 4.

Ba2+ current inhibition by opioids is mediated by PTX-sensitive G-proteins. Top, Intracellular GTPγS (100 μm) mimics and preserves the inhibitory action of DPDPE in a GLC8 cell already inhibited tonically from the start of the whole-cell recordings (t = 15 sec) (left). The recordings consisted of a double-pulse protocol similar to that in Figure 3B, which allows the determination sequentially of the degree of inhibition and facilitation (filled circles) either present at control (left and middle panels) or induced by the opioid (right panels). After 3 min of internal perfusion, GTPγS preserved both the tonic (middle) and the 0.5 μm DPDPE-induced (right) inhibition. The more pronounced kinetics slowing induced by DPDPE was preserved after washout of the agonist, suggesting that GTPγS makes irreversible the usually reversible effect of DPDPE. Middle, Intracellular GDPβS (500 μm) rapidly removed both tonic and DPDPE-induced inhibition. The GLC8 cell was inhibited tonically at the start of internal dialysis (t = 10 sec) (left). After 2 min of internal perfusion with GDPβS, the inhibition was abolished completely (middle), and subsequent application of DPDPE (0.5 μm) had no effects (right). Bottom, Cell preincubation with PTX (100 ng/ml, 15 hr) prevented fully the Ba2+ current inhibition by opioids. The PTX-treated cell exhibited no sign of tonic inhibition after 15 sec (left) and 1 min (middle) from the beginning of intracellular perfusion and was insensitive to 0.5 μm DPDPE (right). Note the total absence of voltage-dependent facilitation of Ba2+currents in this PTX pretreated cell. The slight decrease of current amplitude observed during exposure to DPDPE (right) was not reversible and was probably attributable to partial channel rundown.
PTX inactivates both Gi- and Go-proteins. Go-proteins are likely to couple the opioid receptor directly to the channels via a membrane-delimited pathway (Hille, 1994), whereas Gi-proteins may affect the channels indirectly by inhibiting the activity of adenylate cyclase. To test whether Ca2+ channel inhibition by opioid receptor activation derives from a decrease of cytoplasmic cAMP levels, we assayed the effects of DPDPE in GLC8 cells that were preincubated with the permeant analog Bt2cAMP (1 mm) and perfused intracellularly with cAMP (1 mm) in the whole-cell configuration, or preincubated with Bt2cAMP and studied in the perforated-patch configuration to preserve the cytoplasmic content. In both cases, Ba2+ currents were found similarly sensitive to DPDPE action. Their activation was delayed at potentials of maximal inhibition (+10 mV in 10 mmBa2+ and +30 mV in 50 mmBa2+) (Fig.5A1,B1), and the percentage of inhibition decreased with increasing voltages (mean depression 64.0 ± 3.5% at +30 mV and 43 ± 4.5% at +60 mV in 50 mm Ba2+, n = 5). This suggests that as in other cells (Schultz et al., 1990; Hille, 1994), Ca2+ channel inhibition by opioids is not mediated by reductions of cAMP levels (Hescheler et al., 1987; Taussig et al., 1992). On the contrary, the fast onset and offset of channel inhibition would rather suggest a direct, membrane-delimited interaction between G-proteins and Ca2+ channels, as proposed previously (Hescheler et al., 1987; Taussig et al., 1992;McEnery et al., 1994; Georgoussi et al., 1995; Wilding et al., 1995) (see also Hille, 1994). The persistence of opioid action in perforated patches (Fig. 5B) also excludes the possibility that cell dialysis may affect significantly the size and voltage dependency of opioid inhibition (Merinery et al., 1994).
Fig. 5.

DPDPE (0–1.0) μm inhibition of Ca2+ channels is not cAMP-dependent.A, Effects of DPDPE (filled circles) on Ba2+ currents (10 mm) recorded in the whole-cell patch-clamp configuration at the potential indicated. The cell was preincubated with 1 mmBt2cAMP for 4 hr and dialyzed internally with the standard Cs/EGTA solution containing 1 mm cAMP. Holding potential −90 mV. B, Effects of DPDPE (filled circles) on Ba2+ (50 mm) currents recorded with the perforated patch-clamp method at the potential indicated. The cell was preincubated for 4 hr with 1 mm cAMP to ensure the loading of the cAMP analog inside the cell. Vh −90 mV.
DPDPE does not affect L-type Ca2+ channels in GLC8 cells
GLC8 cells express several HVA Ca2+channels, namely the N-, the L-, and a “non-L-, non-N-type” sensitive to high concentrations of ω-Aga (P/Q) (Codignola et al., 1993). To identify the Ca2+ channels targeted by the δ-opioid-activated G-proteins, the action of DPDPE was tested in combination with agents selectively affecting each one of the channel subtypes.
We first assayed the sensitivity of L-type channels to DPDPE by using the DHP agonist Bay K 8644. Results from two representative cells are shown in Figure 6A and B. In one case, 1 μm Bay K 8644 doubled the currents at −10 mV, caused no changes at +20 mV, and prolonged the Ba2+ current deactivation on return to −50 mV (traces with filled circles in A1,A3). On the contrary, DPDPE had a mild action at −10 mV (A2), caused a marked inhibition at +20 mV (A4), and showed little or no effects on the slow component of the tail current at −50 mV associated to Bay K 8644-modified L-type channels (inset in Fig. 6A4). Most of the depressive action of DPDPE was indeed on the fast-tail current component associated to the non-L- type channels activating above 0 mV in 10 mm Ba2+ (Codignola et al., 1993; Pollo et al., 1993). The fast tails (τfast 0.96 msec) were depressed by 42% by the δ-opioid agonist. In the second case (Fig.6B), the cell showed a remarkably high sensitivity to Bay K 8644 at −20 mV, indicative of a prevalence of L-type channels at this potential and in this particular cell (left). DPDPE, however, had no clear effects on the Bay K 8644-modified currents both at −20 mV and on return to −50 mV (right). Little or no effects of DPDPE on L-type channels also were suggested by experiments with cells that were preincubated with 3 μmnifedipine to block L-type Ca2+ channels completely or with cells that exhibited little sensitivity to the DHP antagonist (Fig. 6C, left). In both cases, DPDPE still was able to induce a marked depression of the Ba2+ currents (71.6 ± 1.2% inhibition in 25 nifedipine-treated cells), which was largely removed by double-pulse protocols (Fig. 6C, right). This confirms that opioid modulation in GLC8 cells is selective for the recently described non-L-type Ca2+ channels of these cells (Sher et al., 1990; Codignola et al., 1993).
Fig. 6.

DPDPE acts primarily on DHP-resistant Ca2+ channels. A, Effects of DPDPE (0.3 μm) on Bay K 8644-modified Ba2+ currents in 10 mmBa2+. Control (C) and Bay K 8644-modified currents at −10 mV and +20 mV are shown in A1and A3. Notice the increased current amplitude at −10 mV, the prolonged time course of tails at −50 mV (filled circles), and the unchanged current size at +20 mV in the presence of 1 μm Bay K 8644 (Bay K). Contrary to the Ca2+ agonist, DPDPE had a marked inhibitory effect at +20 mV (A4) and nearly no effects on the amplitude and time course of the slow-tail component associated to L-type channels (open circles in A2,A4). Inset, Analysis of Bay K 8644-modified-tail currents on a more expanded time scale. The two tails, without (filled circles) and with (open circles) DPDPE were best fitted by double exponentials with the following time constants τfast and τslow and amplitude coefficientsIf and Is: 7.01 msec (−139 pA) and 0.97 msec (−1071 pA) (filled circles); 7.32 msec (−117 pA) and 0.94 msec (−488 pA) (open circles). B, DPDPE has nearly no effect on GLC8 cells containing almost exclusively L-type channels. Control currents in 10 mm Ba2+ (C) increase markedly at −20 mV and deactivate monoexponentially at −50 mV in the presence of 1 μm Bay K 8644 (BayK) (filled circles). Addition of DPDPE (0.3 μm) causes no sizable change to Bay K 8644-modified L-type currents (open circles). C, Nifedipine does not affect DPDPE action. Left panel, Control currents at +20 mV in 10 mmBa2+ (C) are affected little by exposure to 3 μm nifedipine (nife); DPDPE (0.3 μm) preserves its strong inhibitory effect on the nifedipine-resistant current (nife + DPDPE).Right panel, On the same cell, the time- and voltage-dependent inhibition of nifedipine-resistant Ba2+ current by DPDPE (noisy trace a) is facilitated effectively by a double pulse protocol (noisy trace b). Overlapped dashed traces (a, b) indicate the nearly absent facilitory action of preconditioning pulses in control conditions.
DPDPE action is primarily on N-type VOCCs in GLC8 cells
The N-type VOCC is the most abundant Ca2+channel of GLC8 cells (Sher et al., 1990; Codignola et al., 1993). It is expressed variably from cell to cell and accounts for 50–85% of the total current. Given its predominance, we expected that most of the DPDPE inhibition (70–75% of the total current) was exerted on this channel subtype. The contribution of the N-type channel to the DPDPE modulation thus was assayed by testing the opioid action before and after the application of saturating doses of ω-CTx (3 μm), which selectively block this channel subtype (Sher and Clementi, 1991). Nifedipine (3 μm) also was present in all the solutions.
As shown in Figure 7, short applications of ω-CTx (60 sec) in 2 mm Ca2+ were sufficient to maximally block the N-type current in GLC8 cells (Fig.7A). Subsequent additions of the toxin caused no additional current reductions (traces b, c in Fig.7A; traces e, h in Fig.8B). ω-CTx had marked blocking effects on the total Ba2+ current (69.5 ± 2.9% block in 17 cells) and caused a comparable reduction of the DPDPE inhibition (71.7 ± 5.6%; n = 10) that was calculated by comparing the DPDPE current inhibition before (ΔIb in Fig.7A1) and after toxin application (ΔIa in Fig. 7A2). Thus, as in peripheral neurons, δ-opioid inhibition of HVA currents in GLC8 cells is directed primarily on the N-type channel. In some cells, however, DPDPE also exerted significant modulatory effects on residual DHP- and ω-CTx-insensitive currents, suggesting also that non-L-, non-N-type channels can be modulated by the opioid agonist. This effect was quite variable. Figure 7 shows two extreme cases. In A, DPDPE preserved most of its efficacy after ω-CTx application (65% inhibition after toxin treatment vs 75% inhibition before application) (compare 7A1 and A2). In B, the δ-opioid inhibition decreased from 77% (trace b) to 20% (trace e). On the average (n = 10), DPDPE action decreased from 69.8 ± 2.3% before toxin treatment to 39.2 ± 4.9% after toxin application.
Fig. 7.

ω-CTx–GVIA removes a large proportion of DPDPE inhibition in nifedipine-treated cells. A, Comparison of 0.3 μm DPDPE action before (1) and after (2) two acute applications of 3 μmω-CTx in 2 mm Ca2+. On the left diagram, the dots represent peak Ba2+ currents recorded at +30 mV every 12 sec from Vh −90 mV. All bath solutions contained 3 μm nifedipine. Lower bars mark the time of toxin application (shadowed bars) and the interval in which DPDPE action was tested (filled bars). Upper bars indicate the Ba2+ and Ca2+concentrations of external solutions (in mm). On the right are the current traces recorded at the times (a–c) and intervals (1, 2) labeled on the left. ΔIb and ΔIa indicate the amount of DPDPE inhibition before and after the two toxin applications. B, Same as in A, but from a cell containing a higher density of N-type channels as proven by the potent block of Ba2+ currents by ω-CTx (tracesc, d). The strong inhibitory effect of DPDPE before toxin application (traces a–c) is partially preserved on residual ω-CTx-resistant currents.
Fig. 8.

Effects of ω-CTx on DPDPE action in ω-Aga and nifedipine-treated cells. The two cells of A andB were preincubated with ω-Aga (250 nm) for 15 min in Tyrode’s solution (2 mm Ca2+) and tested with bath solutions containing 3 μm nifedipine. InA, a short application of 3 μmω-CTx blocked almost all the currents resistant to ω-Aga and DHPs (traces d, e), suggesting that nearly all the inhibitory action of DPDPE (traces a–c) is on N-type channels. In B, ω-CTx blocks only 70% of the ω-Aga- and DHP-resistant currents (traces d,e, h), and DPDPE preserves a strong voltage-dependent action on the residual current that is not N-, L-, or P-type. Test depolarizations to +30 mV fromVh −90 mV. Bars andsymbols are as in Figure 7.
To further identify the Ca2+ channel subtypes responsible for the DPDPE inhibition of ω-CTx-resistant currents, we tested the effects of the δ-opioid agonist on cells pretreated with nifedipine to block L-type channels and high concentrations of ω-Aga to fully block P-type channels (Mintz et al., 1992). In cells pretreated with 250 nm ω-Aga for 17 min in 2 mm Ca2+, and in the presence of nifedipine, ω-CTx blocked a slightly larger component of current (79 ± 3.1%; n = 5) than in control cells. Currents resistant to nifedipine, ω-CTx, and ω-Aga, and thus deprived of L-, N-, and P-type channels, contributed to ∼15–20% of the total and were variably depressed by DPDPE (Fig.8A,B). In seven cells pretreated with ω-Aga, nifedipine, and ω-CTx, the δ-opioid depressed the residual current by 38 ± 7.5%, i.e., about half of the inhibition of N-type channels. In conclusion, DPDPE inhibition of HVA Ca2+ channels in GLC8 cells appears selective for non-L-type channels, with a predominant effect on N-type and a reduced action on residual “non-L-type, non-N-type, and non-P-type” channels.
DPDPE inhibits depolarization-induced [3H]5-HT release
DPDPE also was tested for its ability to inhibit the depolarization-induced release of [3H]5-HT from GLC8 cells. KCl-induced [3H]5-HT release from GLC8 cells recently was shown to be attributable to VOCC activation, being completely blocked by Cd2+, DHPs, ω-CTx, and ω-Aga (Codignola et al., 1993).
Figure 9 shows that DPDPE dose-dependently inhibits KCl (50 mm)-induced [3H]5-HT release, with an IC50 of 90 pm and a maximal inhibition of 72 ± 0.5%. Furthermore, the effects of DPDPE on KCl-induced [3H]5-HT release were antagonized by naloxone but not mimicked by μ and k agonists (Fig. 9, inset), confirming the selective involvement of δ receptors also in the modulation of release.
Fig. 9.

DPDPE selectively inhibits KCl-stimulated [3H]5-HT release from GLC8 cells. DPDPE inhibits KCl (50 mm)-induced [3H]5-HT release from preloaded GLC8 cells in a dose-dependent manner. The IC50 is 90 pm, and the maximal inhibition ∼72%.Inset, Neither μ-opioid (DAMGO) (b) nor κ-opioid (U50488) (c) agonists inhibit the KCl-stimulated release of [3H]5-HT. The opioid antagonist naloxone, which alone (d) does not have any effect, totally prevents DPDPE action on secretion (e). Cells were loaded and release-assayed as described in Materials and Methods. Each point in the curve, as well as each column in the inset, represents the mean ± SEM of five experiments, each performed in quintuplicate.
The potency of DPDPE in inhibiting [3H]5-HT release depended on the KCl concentration, being more effective at higher KCl (Fig. 10A). A very similar trend has been shown for morphine inhibition of KCl-stimulated acetylcholine release from Torpedo nerve terminals (Michaelson et al., 1984). One possibility is that stronger depolarizations recruit VOCC subtypes, which are more sensitive to opioid modulation. We have shown previously that the N- and P/Q-type VOCCs (opioid-sensitive) have a higher threshold of activation than the L-type VOCC (opioid-insensitive) (see above) in GLC8 cells (Codignola et al., 1993). In line with this hypothesis, we have found that DPDPE does not inhibit the selective component of Bay K 8644-induced release of [3H]5-HT, which instead is completely antagonized by nifedipine (Fig. 10B).
Fig. 10.
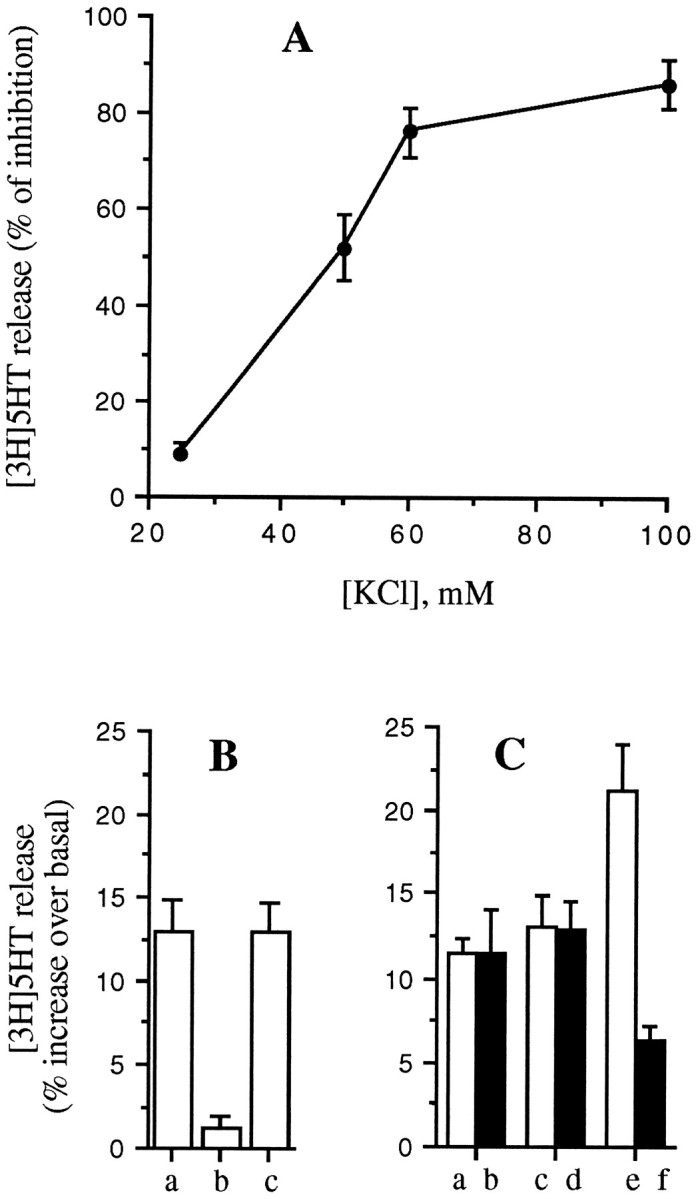
DPDPE inhibition is related to KCl concentrations. A, The ability of DPDPE (100 nm) to inhibit [3H]5-HT release from GLC8 cells increases with increasing KCl concentrations. B, DPDPE (100 nm) does not inhibit the release of [3H]5-HT, which is stimulated by 1 μm Bay K 8644 at basal KCl concentrations (c), whereas this was antagonized completely by 1 μm nifedipine (b).C, DPDPE (100 nm) does not inhibit the release stimulated by low KCl alone (25 mm) (a, b) or BayK8644 alone (1 μm) (c, d), but does inhibit the increased release obtained with the two secretagogues together (e, f). GLC8 cells were loaded and the release assayed as described in Materials and Methods. Each point in the curve, as well as each column, represents the mean ± SEM of four experiments, each performed in quintuplicate.
A second possibility, however, is that opioids inhibit (irrespective of VOCC modulation) (see below) one or more steps of exocytosis, such as granule translocation or docking, that need higher or more diffuse intracellular Ca2+ levels achieved only at the higher KCl concentrations. In line with this second hypothesis, we have found that when [3H]5-HT release is stimulated by the simultaneous addition of Bay K 8644 and a low (25 mm) concentration of KCl (neither alone inhibited by DPDPE), the release is not only higher, presumably attributable to an increase in Ca2+ influx, but it is now inhibited by DPDPE (Fig. 10C).
DPDPE inhibits thapsigargin- and ionomycin-stimulated [3H]5-HT release
We have shown previously that thapsigargin and ionomycin stimulate dose-dependently [3H]5-HT release from GLC8 cells, both in the presence and in the absence of extracellular Ca2+ (Codignola et al., 1993). This form of Ca2+-dependent secretion clearly bypasses VOCC activation, and when studied in Ca2+-free buffers, is attributable to Ca2+ release from intracellular stores.
Interestingly, DPDPE also potently blocked this form of Ca2+-dependent release. Figure 11shows the dose dependency of DPDPE inhibition of thapsigargin-induced release. Both the IC50 (20 pm) and the maximal inhibition (73 ± 2%) are very similar to those obtained in cells in which [3H]5-HT release was stimulated with KCl (see above).
Fig. 11.

DPDPE inhibits thapsigargin- and ionomycin-stimulated [3H]5-HT release. DPDPE dose-dependently inhibits the release of [3H]5-HT from GLC8 cells stimulated with 1 μm thapsigargin in a Ca2+-free medium. The IC50is 20 pm, and the maximal inhibition 73 ± 2%.Inset, ionomycin (1 μm) stimulates [3H]5-HT release in both the presence (a, b) and absence (c, d) of extracellular Ca2+. DPDPE (100 nm) blocks ionomycin-stimulated release under both conditions. Cell-loading and -release assays were performed as described in Materials and Methods. Each point in the curve and the values of the columns represent the mean ± SEM of four experiments, each performed in quintuplicate.
Ionomycin-stimulated release also was strongly inhibited by 100 nm DPDPE both in the presence (Fig.11B, inset, a, b) and in the absence (c, d) of external Ca2+.
These data suggest that opioid peptides still inhibit Ca2+-dependent hormone release even when intracellular Ca2+ is increased by very unspecific means.
DPDPE effects on secretion are G-protein-mediated and cAMP-modulated
Similarly to the inhibition of VOCCs, DPDPE inhibition of both KCl- and thapsigargin-induced release was mediated by a PTX-sensitive G-protein.
As shown in Figure 12A, PTX (100 ng/ml, 12 hr at 37°C) completely prevented DPDPE inhibition of both KCl-induced secretion and thapsigargin-induced secretion without affecting, by itself, either the spontaneous release of [3H]5-HT or that stimulated by the two secretagogues (data not shown).
Fig. 12.
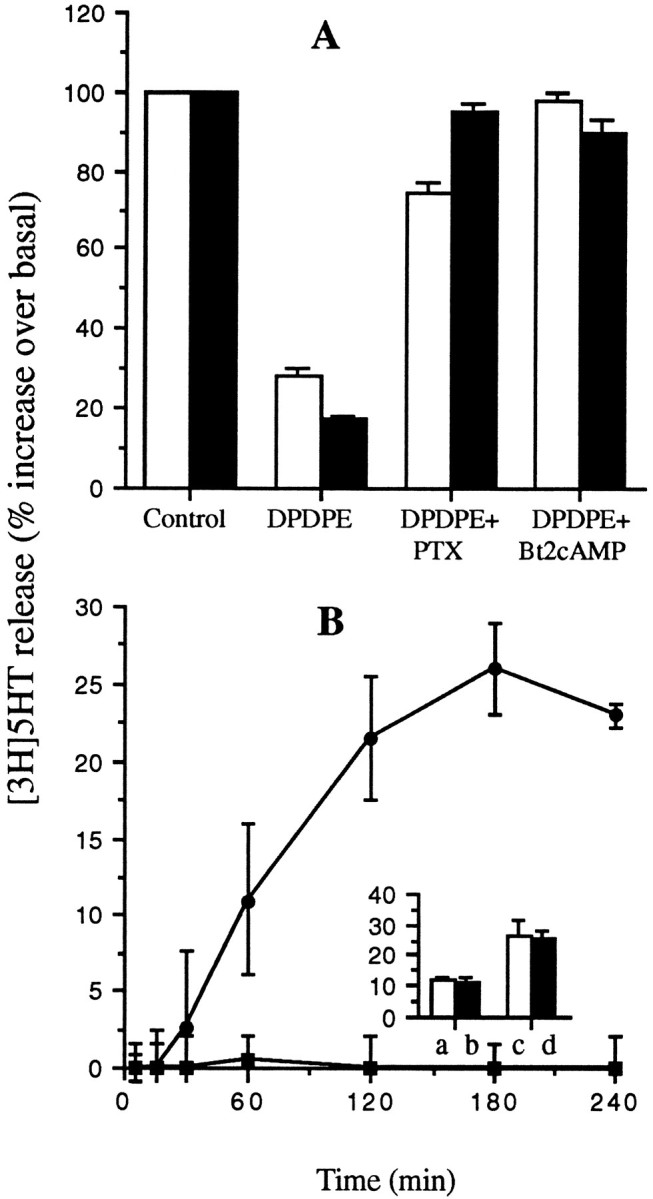
DPDPE effects on secretion are mediated by a PTX-sensitive G-protein and are counteracted by Bt2cAMP. A, The release of [3H]5-HT from GLC8 cells stimulated by either 50 mm KCl (open columns) or 1 μm thapsigargin (filled columns) is inhibited by 100 nm DPDPE in control cells as shown above (Figs. 9, 11). However, DPDPE does not inhibit the stimulated release of [3H]5-HT in cells pretreated with either PTX (100 ng/ml, overnight at 37°C) or with Bt2cAMP (1 mm, 15 min at 37°C). B, Two concentrations of Bt2cAMP [(1 mm(filled squares) and 10 mm(filled circles)] were tested for their ability to stimulate [3H]5-HT in the absence of other secretagogues. Whereas 1 mmBt2cAMP is ineffective, 10 mm Bt2cAMP stimulates a substantial release over several hours. Inset, 1 mm Bt2cAMP (filled columns) does not synergize with either 25 mm (a, b) or 50 mm (c, d) KCl in the stimulation of [3H]5-HT release. GLC8 cells were loaded and drug-treated, and the release assayed as described in Materials and Methods. Each point in the curves and each column represent the mean ± SEM of three experiments, each performed in quadruplicate.
At variance with VOCC modulation, however, DPDPE modulation of both KCl- and thapsigargin-stimulated release was counteracted completely by incubating the cells in 1 mmBt2cAMP (Fig. 12A). Bt2cAMP was equipotent when applied for 2–4 hr or for as few as 5 min.
Bt2cAMP (1 mm) did not stimulate [3H]5-HT release on its own (Fig.12B) nor did it synergize with KCl-induced secretion over the time scale of these experiments (Fig. 12B,inset). A time-dependent stimulatory effect of Bt2cAMP on [3H]5-HT release, however, was observed at a 10 mmBt2cAMP concentration (Fig.11B).
Regardless of the exact mechanism of Bt2cAMP action, its differential effects help in discriminating between two distinct δ-opioid inhibitory pathways: one on the VOCCs (which is cAMP-independent) and one on the secretory apparatus (which is cAMP-modulated).
DISCUSSION
Activation of adenosine (DeAizpurua et al., 1988) and muscarinic (Williams and Lennon, 1990) receptors has been reported previously to inhibit KCl-stimulated45Ca2+ influx in SCLC cells. We show here, for the first time, that opioids also are potent inhibitors of Ca2+ fluxes through HVA VOCCs in isolated SCLC cells and of hormone release from cell populations.
Opioid receptor subtypes in GLC8 cells
We found a selective expression of δ-opioid receptor mRNA in GLC8 cells. It has been shown previously that different SCLC cell lines can express various combinations of the different opioid receptor subtypes (Maneckjee and Minna, 1990). It is known, furthermore, that μ- and κ-opioid receptor subtypes also can inhibit Ca2+ channels and secretion in other neuronal preparations (North, 1993). On the other hand, our results suggest that GLC8 cells can provide a cellular system in which “pure” δ-opioid receptor effects can be evaluated.
Opioid inhibition of VOCCs
As shown previously in other cell types, the opioid modulation of HVA VOCCs in GLC8 cells is mainly voltage-dependent (Tsunoo et al., 1986; Kasai, 1992; Rhim and Miller, 1994) and G-protein-mediated (Hescheler et al., 1987; Kasai, 1992; Taussig et al., 1992). The G-protein involved is PTX-sensitive (Go–Gi family), but a reduction of cAMP levels is not the mechanism by which the modulation is exerted.
The modulation by opioids primarily affects N-type VOCCs (Figs. 6, 7), which are the most abundant in GLC8 cells (Codignola et al., 1993). On the other hand, opioid agonists had no effects on L-type VOCCs. We found also that the non-L-, non-N-type channels (P/Q) are modulated effectively by DPDPE. However, their small contribution to the total current (20–25%) in GLC8 cells and the subtle differences distinguishing P- from Q-type channels make it difficult to evaluate the relevance of these VOCC subtypes to the overall Ca2+ channel modulation by opioids. A voltage-dependent inhibitory action of opioids on N- and P/Q-type channels without affecting L-type channels has been reported in CNS (Rhim and Miller, 1994) and dorsal root ganglion (Rusin and Moises, 1995) neurons. This selectivity also is present in NG108 neuroblastoma cells (Kasai, 1992), but not in bovine chromaffin cells in which the marked voltage-dependent inhibition of non-L-type channels is accompanied by a significant voltage-independent inhibition of L-type channels (Albillos et al., 1995) .
Opioids effects on [3H]serotonin release
KCl-stimulated [3H]5-HT release from GLC8 cells was strongly inhibited by DPDPE. The easiest explanation of this expected result is that the inhibition of KCl-stimulated [3H]5-HT release is a direct consequence of VOCC modulation. However, DPDPE was found to inhibit also the “VOCC-independent”, thapsigargin-, and ionomycin-stimulated [3H]5-HT release in a PTX-sensitive manner.
Opioid action on [3H]5-HT release in GLC8 cells, therefore, is more complex than expected; it includes the inhibition of Ca2+ influx caused by Ca2+ channel modulation and the inhibition of additional, still undefined, distal steps of Ca2+-dependent secretion. Although novel for SCLC cells, the presence of multiple mechanisms by which hormones inhibit secretion has been reported previously in other preparations. Several hormones, such as adrenaline, FMRFamide, adenosine, serotonin, and somatostatin, inhibit secretion using multiple mechanisms that include ion channel modulation and distal effects on the secretory apparatus (Jones et al., 1987; Ullrich and Wollheim, 1988; Man-Son-Hing et al., 1989; Dale and Kandel, 1990; Luini and DeMatteis, 1990; Ullrich et al., 1990; Scholz and Miller, 1992). Opioids, in particular, have been shown to exert direct inhibitory effects on the secretory apparatus of GABA-releasing hippocampal interneurons (Capogna et al., 1993; Rekling, 1993; Lupica, 1995). The target(s) of this distal, G-protein-mediated inhibition still are unknown. An inhibition of vesicle translocation to release sites and/or a reduction in the Ca2+sensitivity of already releasable vesicles represent possible mechanisms. More is known about the classes of G-proteins involved in mediating the distal effects on secretion. Go-proteins are present in the membrane of secretory granules (Toutant et al., 1987), and Goantibodies are able to antagonize G-protein-mediated effects on secretion from permeabilized chromaffin cells (Ohara-Imaizumi et al., 1992; Vitale et al., 1993). Recent data on pancreatic β-cells show that both Gi and Go are involved in mediating the adrenaline inhibition of insulin release from permeabilized cells (Lang et al., 1995).
Bt2cAMP effects discriminate between the two opioid inhibitory pathways
Opioid modulation of Ca2+ channels in GLC8 cells is not affected by changes in cAMP levels. This is in agreement with most literature on hormone-induced VOCC modulation and that of opioid modulation, in particular (Schultz et al., 1990; Carbone et al., 1996).
On the other hand, the PTX-sensitive, G-protein-mediated, opioid inhibition of secretion in GLC8 cells is reversed completely in the presence of Bt2cAMP, suggesting that inhibition of [3H]5-HT secretion by δ-opioid receptors may result from a receptor-mediated reduction in intracellular cAMP levels.
The involvement of cAMP in mediating the inhibitory effects of opioids on secretion still is controversial. In most cells studied, the inhibitory effects of opioids are not counteracted by increasing cAMP levels (Schoffelmeer et al., 1986; Johnson, 1990; Heijna et al., 1992;Lupica, 1995), but in the enkephalin-releasing nerve terminals of the myenteric plexus, this clearly is the case (Xu et al., 1989; Gintzler and Xu, 1991; Wang and Gintzler, 1994). cAMP potentiates hormone release in neurons and endocrine cells, and also in GLC8 cells, it can stimulate [3H]5-HT release. cAMP is suggested to increase the Ca2+-sensitivity of the secretory apparatus in other endocrine cells such as pancreatic β-cells (Ammala et al., 1993). An opioid-induced reduction in cAMP levels could affect the same distal target in an inhibitory way.
A second attractive hypothesis is that the opioid inhibition of the secretory apparatus is not caused by an opioid-induced reduction in cAMP levels, but it occurs via an independent biochemical pathway. This being the case, the Bt2cAMP counteracting effects could be attributable to a cAMP-dependent phosphorylation of one element of this pathway with consequent “uncoupling” of the inhibitory mechanism. It has been shown, for example, that desensitization of δ-opioid receptors can be caused by receptor phosphorylation, although BARK-like kinases, and not PKA, seem to be implicated primarily (Pei et al., 1995). The different sensitivity to cAMP of the two inhibitory opioid pathways described here (VOCC inhibition vs secretory machinery inhibition) suggests that the two transducing pathways are distinct and, possibly, independent of each other.
Physiological relevance of VOCC modulation versus inhibition of the secretory apparatus
As discussed above, Bt2cAMP is able to counteract the opioid inhibition of both KCl- andthapsigargin-induced [3H]5-HT release. Because VOCC modulation by opioids is not counteracted by Bt2cAMP, these results suggest that also in the case of KCl-induced release, the inhibitory effects of DPDPE are primarily exerted directly on the secretory apparatus, and are not a consequence of VOCC modulation.
This raises the question of the physiological role of the HVA VOCC modulation here described: if opioids block secretion by acting downstream to Ca2+ influx, what is the need of VOCC modulation? We know that Ca2+ influx through HVA VOCCs is an important trigger of hormone release from GLC8 cells, as evidenced by the blockade of KCl-induced [3H]5-HT release by Cd2+or other Ca2+ channel antagonists (Codignola et al., 1993). However, the inhibitory effects of the opioids on HVA VOCCs are different from those of the Ca2+ antagonists, especially in terms of kinetics and voltage dependency. VOCC modulation by opioids could be relevant under conditions in which Ca2+ channels are activated transiently as, for example, during the spontaneous, Ca2+-dependent action potentials occurring in these cells (McCann et al., 1981). On the other hand, when SCLC cells are depolarized more intensively or repetitively, or with long-lasting exposures to KCl as in our conditions, the modulation of the channels might become less important, primarily because of the voltage- and time-dependent relief of hormone inhibition of HVA VOCCs (Bean, 1989; Pollo et al., 1992) (this paper). Under these conditions, however, SCLC cells still have an efficient, distal “checkpoint” at which opioids can act to inhibit secretion.
It should be mentioned that an increase in intracellular Ca2+ concentrations can be achieved in SCLC cells also by the activation of surface receptors that stimulate IP3 production and Ca2+release from internal stores, without the involvement of VOCCs. By acting on some distal step of the secretory process, opioids also can prevent this form of Ca2+-dependent release.
CONCLUSIONS
The work reported here is a first step in understanding, at the cellular and molecular levels, the complex mechanisms of opioid action in SCLC cells and suggests some pathways (a reduction of Ca2+ influx plus a reduction of released mitogens) by which δ-opioids could selectively inhibit GLC8 cells proliferation as well (Codignola et al., 1994).
Footnotes
This work was supported by Telethon-Italy (Grant 627 to E.C.). A.C. is a fellow of the Italian Association for Cancer Research. We thank Dr. Janet Richmond for helpful discussions.
Correspondence should be addressed to Emanuele Sher, CNR Center of Cellular and Molecular Pharmacology, Via Vanvitelli 32, 20129 Milan, Italy.
Dr. Cesare’s present address: Biomedical Science Division, King’s College London, London WC1N 1AX, UK.
REFERENCES
- 1.Albillos A, Carbone E, Gandia L, Garcia AG, Pollo A. Selective voltage-dependent and voltage-independent inhibition of calcium channel subtypes by opioids in bovine chromaffin cells. Soc Neurosci Abstr. 1995;21:1. [Google Scholar]
- 2.Ammala C, Ashcroft FM, Rorsman P. Calcium-independent potentiation of insulin release by cyclic AMP in single β-cells. Nature. 1993;363:356–358. doi: 10.1038/363356a0. [DOI] [PubMed] [Google Scholar]
- 3.Bean BP. Neurotransmitter inhibition of neuronal calcium currents by changes in channel voltage-dependence. Nature. 1989;340:153–156. doi: 10.1038/340153a0. [DOI] [PubMed] [Google Scholar]
- 4.Capogna M, Gahwiler BH, Thompson SM. Mechanism of μ-opioid receptor-mediated presynaptic inhibition in the rat hippocampus in vitro. J Physiol (Lond) 1993;470:539–558. doi: 10.1113/jphysiol.1993.sp019874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Carbone E, Magnelli V, Pollo A, Carabelli V, Albillos A, Zucker H. Biophysics of voltage-dependent Ca2+ channels: gating kinetics and their modulation. In: Soria B, Cena V, editors. Ion channel pharmacology. Oxford UP; Oxford: 1996. pp. 105–131. [Google Scholar]
- 6.Codignola A, Tarroni P, Clementi F, Pollo A, Lovallo M, Carbone E, Sher E. Calcium channel subtypes controlling serotonin release from human small-cell lung carcinoma cell lines. J Biol Chem. 1993;268:26240–26247. [PubMed] [Google Scholar]
- 7.Codignola A, Sher E, Clementi F, Missale C, Boroni F, Spano P, Pollo A, Carbone E. Opioid modulation of voltage-operated calcium channels, hormone release and cell proliferation in a small-cell lung carcinoma cell line. Eur J Neurosci [Suppl] 1994;7:28. [Google Scholar]
- 8.Dale N, Kandel ER. Facilitatory and inhibitory transmitters modulate spontaneous transmitter release at cultured Aplysia sensorimotor synapses. J Physiol (Lond) 1990;421:203–222. doi: 10.1113/jphysiol.1990.sp017941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.De Aizpurua HJ, Lambert EH, Griesmann GE, Olivera BM, Lennon VA. Antagonism of voltage-gated calcium channels in small-cell carcinoma of patients with and without Lambert–Eaton myasthenic syndrome by autoantibodies ω-conotoxin and adenosine. Proc Natl Acad Sci USA. 1988;48:4719–4724. [PubMed] [Google Scholar]
- 10.Doupnik CA, Pun RYK. G-protein activation mediates prepulse facilitation of Ca2+ currents in bovine chromaffin cells. J Membr Biol. 1994;140:47–56. doi: 10.1007/BF00234485. [DOI] [PubMed] [Google Scholar]
- 11.Fenwick EM, Marty A, Neher E. Sodium and calcium channels in bovine chromaffin cells. J Physiol (Lond) 1982;331:599–635. doi: 10.1113/jphysiol.1982.sp014394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Georgoussi Z, Milligan G, Zioudrou C. Immunoprecipitation of opioid receptor Go-protein complexes using specific GTP-binding-protein antisera. Biochem J. 1995;306:71–75. doi: 10.1042/bj3060071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Gintzler AR, Xu H. Different G-proteins mediate the opioid inhibition or enhancement of evoked [5-methionine]-enkephalin release. Proc Natl Acad Sci USA. 1991;88:4741–4745. doi: 10.1073/pnas.88.11.4741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high resolution current recording from cells and cell-free membrane patches. Pflügers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- 15.Heijna MH, Hogenboom F, Mulder AH, Schoffelmeer AN. Opioid receptor-mediated inhibition of [3H]-dopamine and [14C]-acetylcholine release from rat nucleus accumbens slices: a study on the possible involvement of K+ channels and adenylate cyclase. Naunyn Schmiedebergs Arch Pharmacol. 1992;345:627–632. doi: 10.1007/BF00164575. [DOI] [PubMed] [Google Scholar]
- 16.Hescheler J, Rosenthal W, Trautwein W, Schultz G. The GTP-binding protein, G0, regulates neuronal calcium channels. Nature. 1987;325:445–447. doi: 10.1038/325445a0. [DOI] [PubMed] [Google Scholar]
- 17.Hille B. Modulation of ion-channel function by G-protein-coupled receptors. Trends Neurosci. 1994;17:531–532. doi: 10.1016/0166-2236(94)90157-0. [DOI] [PubMed] [Google Scholar]
- 18.Horn R, Marty A. Muscarinic activation of ionic currents by a new whole-cell recording method. J Gen Physiol. 1988;92:145–159. doi: 10.1085/jgp.92.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ikeda SR. Double-pulse calcium current facilitation in adult rat sympathetic neurones. J Physiol (Lond) 1991;439:181–214. doi: 10.1113/jphysiol.1991.sp018663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Illes P. Modulation of transmitter and hormone release by multiple neuronal opioid receptors. Rev Physiol Biochem Pharmacol. 1989;112:139–233. doi: 10.1007/BFb0027497. [DOI] [PubMed] [Google Scholar]
- 21.Johnson SM. Opioid inhibition of cholinergic transmission in the guinea-pig ileum is dependent of intracellular cyclic AMP. Eur J Pharmacol. 1990;180:331–338. doi: 10.1016/0014-2999(90)90318-z. [DOI] [PubMed] [Google Scholar]
- 22.Jones PM, Fyles JM, Persaud SJ, Howell SL. Catecholamine inhibition of Ca2+-induced insulin secretion from electrically permeabilized islets of Langherans. FEBS Lett. 1987;219:134–144. doi: 10.1016/0014-5793(87)81206-1. [DOI] [PubMed] [Google Scholar]
- 23.Kasai H. Voltage- and time-dependent inhibition of neuronal calcium channels by a GTP-binding protein in a mammalian cell line. J Physiol (Lond) 1992;448:189–209. doi: 10.1113/jphysiol.1992.sp019036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Knapp RJ, Malatynska E, Fang L, Xiaoping L, Nguyen M, Santoro G, Varga EV, Hruby VJ, Roeske WR, Yamamura HI. Identification of a human delta receptor: cloning and expression. Life Sci. 1994;54:463–469. doi: 10.1016/0024-3205(94)90138-4. [DOI] [PubMed] [Google Scholar]
- 25.Konishi S, Tsunoo A, Otsuka M. Enkephalins presynaptically inhibit cholinergic transmission in sympathetic ganglia. Nature. 1979;282:515–516. doi: 10.1038/282515a0. [DOI] [PubMed] [Google Scholar]
- 26.Kumakura K, Karoum F, Guidotti A, Costa E. Modulation of nicotinic receptors by opiate receptor agonists in cultured adrenal chromaffin cells. Nature. 1980;283:489–492. doi: 10.1038/283489a0. [DOI] [PubMed] [Google Scholar]
- 27.Lang J, Nishimoto I, Okamoto T, Regazzi R, Kiraly K, Weller U, Wollheim CB. Direct control of exocytosis by receptor-mediated activation of the heterotrimeric GTPases Gi and Go or by the expression of their active Gα subunits. EMBO J. 1995;14:3635–3644. doi: 10.1002/j.1460-2075.1995.tb00033.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luini A, De Matteis MA. Evidence that receptor-linked G-protein inhibits exocytosis by a post-second-messenger mechanism in AtT-20 cells. J Neurochem. 1990;54:30–38. doi: 10.1111/j.1471-4159.1990.tb13279.x. [DOI] [PubMed] [Google Scholar]
- 29.Lupica CR. δ and μ Enkephalins inhibit spontaneous GABA-mediated IPSCs via a cyclic AMP-independent mechanism in the rat hippocampus. J Neurosci. 1995;15:737–749. doi: 10.1523/JNEUROSCI.15-01-00737.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.MacDonald RL, Nelson PG. Specific opiate-induced depression of transmitter release from dorsal root ganglion cells in culture. Science. 1978;199:1449–1451. doi: 10.1126/science.204015. [DOI] [PubMed] [Google Scholar]
- 31.Magnelli V, Pollo A, Sher E, Carbone E. Block of non-L, non-N-type Ca2+ channels in rat insulinoma RINm5F cells by ω-agatoxin IVA and ω-conotoxin MVIIC. Pflügers Arch. 1995;429:762–771. doi: 10.1007/BF00374799. [DOI] [PubMed] [Google Scholar]
- 32.Maneckjee R, Minna JD. Opioid and nicotine receptors affect growth regulation of human lung cancer cell lines. Proc Natl Acad Sci USA. 1990;87:3294–3298. doi: 10.1073/pnas.87.9.3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Man-Son-Hing H, Zoran MJ, Lukowiak K, Haydon PG. A neuromodulator of synaptic transmission acts on the secretory apparatus as well as on ion channels. Nature. 1989;341:237–239. doi: 10.1038/341237a0. [DOI] [PubMed] [Google Scholar]
- 34.Mansson E, Bare LA, Yang D. Isolation of a human κ-opioid receptor cDNA from placenta. Biochem Biophys Res Comm. 1994;202:1431–1437. doi: 10.1006/bbrc.1994.2091. [DOI] [PubMed] [Google Scholar]
- 35.McCann FV, Pettengill OS, Cole JJ, Russell JAG, Sorenson GD. Calcium spike electrogenesis and other electrical activity in continuously cultured small-cell carcinoma of the lung. Science. 1981;212:1156–1157. doi: 10.1126/science.6262914. [DOI] [PubMed] [Google Scholar]
- 36.McEnery MW, Snowmaan AM, Snyder SH. The association of endogenous Go-alpha with the purified ω-conotoxin GVIA receptor. J Biol Chem. 1994;269:5–8. [PubMed] [Google Scholar]
- 37.Merinery SD, Gray DB, Pilar GR. Somatostatin-induced induced inhibition of neuronal Ca2+ current modulated by cGMP-dependent protein kinase. Nature. 1994;369:336–339. doi: 10.1038/369336a0. [DOI] [PubMed] [Google Scholar]
- 38.Michaelson DM, McDowall G, Sarne Y. Opiates inhibit acetylcholine release from Torpedo nerve terminals by blocking Ca2+ influx. J Neurochem. 1984;43:614–618. doi: 10.1111/j.1471-4159.1984.tb12779.x. [DOI] [PubMed] [Google Scholar]
- 39.Mintz IM, Adams ME, Bean BP. P-type calcium channels in rat central and peripheral neurons. Neuron. 1992;9:85–95. doi: 10.1016/0896-6273(92)90223-z. [DOI] [PubMed] [Google Scholar]
- 40.Mudge AW, Leeman SE, Fischbach GD. Enkephalin inhibits release of Substance P from sensory neurons in culture and decrease action potential duration. Proc Natl Acad Sci USA. 1979;76:526–530. doi: 10.1073/pnas.76.1.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.North AR. Opioid actions on membrane ion channels. Handb Exp Pharmacol. 1993;104:773–796. [Google Scholar]
- 42.Ohara-Imaizumi M, Kameyama K, Kawae N, Takeda K, Muramatsu S, Kumakura K. Regulatory role of the GTP-binding protein, Go, in the mechanism of exocytosis in adrenal chromaffin cells. J Neurochem. 1992;58:2275–2284. doi: 10.1111/j.1471-4159.1992.tb10974.x. [DOI] [PubMed] [Google Scholar]
- 43.Pei G, Kieffer BL, Lefkowitz RJ, Freedman NJ. Agonist-dependent phosphorylation of the mouse δ-opioid receptor: involvement of G-protein-coupled receptor kinases but not protein kinase C. Mol Pharmacol. 1995;48:173–177. [PubMed] [Google Scholar]
- 44.Pollo A, Lovallo M, Sher E, Carbone E. Voltage-dependent noradrenergic modulation of ω-conotoxin-sensitive Ca2+ channels in human neuroblastoma IMR32 cells. Pflügers Arch. 1992;422:75–83. doi: 10.1007/BF00381516. [DOI] [PubMed] [Google Scholar]
- 45.Pollo A, Lovallo M, Biancardi E, Sher E, Socci C, Carbone E. Sensitivity to dihydropyridines, ω-conotoxin and noradrenaline reveals multiple high-voltage-activated Ca2+ channels in rat insulinoma and human pancreatic β-cells. Pflügers Arch. 1993;423:462–471. doi: 10.1007/BF00374942. [DOI] [PubMed] [Google Scholar]
- 46.Rekling JC. Effects of Met-enkephalin on GABAergic spontaneous miniature IPSPs in organotypic slice cultures of the rat hippocampus. J Neurosci. 1993;13:1954–1964. doi: 10.1523/JNEUROSCI.13-05-01954.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rhim H, Miller RJ. Opioid receptors modulate diverse types of calcium channels in the nucleus tractus solitarius of the rat. J Neurosci. 1994;14:7608–7615. doi: 10.1523/JNEUROSCI.14-12-07608.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roth KA, Barchas JD. Small-cell carcinoma cell lines contain opioid peptides and receptors. Cancer Res. 1985;57:769–773. doi: 10.1002/1097-0142(19860215)57:4<769::aid-cncr2820570415>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 49.Rusin IK, Moises HC. μ-Opioid receptor activation reduces multiple components of high-threshold calcium current in rat sensory neurons. J Neurosci. 1995;15:4315–4327. doi: 10.1523/JNEUROSCI.15-06-04315.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schoffelmeer ANM, Wierenga AA, Mulder AH. Role of adenylate cyclase in presynaptic α2-adrenoceptor- and μ-opioid receptor-mediated inhibition of [3H]noradrenaline release from rat brain cortex slices. J Neurochem. 1986;46:1711–1717. doi: 10.1111/j.1471-4159.1986.tb08488.x. [DOI] [PubMed] [Google Scholar]
- 51.Scholz KP, Miller RJ. Inhibition of quantal transmitter release in the absence of calcium influx by a G-protein-linked adenosine receptor at hippocampal synapses. Neuron. 1992;8:1139–1150. doi: 10.1016/0896-6273(92)90134-y. [DOI] [PubMed] [Google Scholar]
- 52.Schultz G, Rosenthal W, Hescheler J, Trautwein W. Role of G-proteins in calcium channel modulation. Annu Rev Physiol. 1990;52:275–292. doi: 10.1146/annurev.ph.52.030190.001423. [DOI] [PubMed] [Google Scholar]
- 53.Shen KZ, Suprenant A. Noradrenaline, somatostatin and opioids inhibit activity of single HVA/N-type calcium channels in excised neuronal membranes. Pflügers Arch. 1991;418:614–616. doi: 10.1007/BF00370580. [DOI] [PubMed] [Google Scholar]
- 54.Sher E, Clementi F. ω-Conotoxin-sensitive voltage-operated calcium channels in vertebrate cells. Neuroscience. 1991;42:301–307. doi: 10.1016/0306-4522(91)90376-y. [DOI] [PubMed] [Google Scholar]
- 55.Sher E, Pandiella A, Clementi F. Voltage-operated calcium channels in small-cell lung carcinoma cell lines: pharmacological, functional and immunological properties. Cancer Res. 1990;53:3892–3896. [PubMed] [Google Scholar]
- 56.Taussig R, Sanchez S, Rifo M, Gilman AG, Belardetti F. Inhibition of ω-conotoxin-sensitive calcium current by distinct G-proteins. Neuron. 1992;8:799–809. doi: 10.1016/0896-6273(92)90100-r. [DOI] [PubMed] [Google Scholar]
- 57.Toutant M, Aunis D, Bockaert J, Homburger V, Rouot B. Presence of three pertussis toxin substrates and Go alpha immunoreactivity in both plasma and granule membranes of chromaffin cells. FEBS Lett. 1987;215:339–344. doi: 10.1016/0014-5793(87)80174-6. [DOI] [PubMed] [Google Scholar]
- 58.Tsunoo A, Yoshi M, Narahashi T. Block of calcium channels by enkephalin and somatostatin in neuroblastoma-glioma hybrid NG108–15 cells. Proc Natl Acad Sci USA. 1986;83:9832–9836. doi: 10.1073/pnas.83.24.9832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ullrich S, Wolheim CB. GTP-dependent inhibition of insulin secretion by epinephrine in permeabilized RINm5F cells. J Biol Chem. 1988;263:8615–8620. [PubMed] [Google Scholar]
- 60.Ullrich S, Prentki M, Wolheim CB. Somatostatin inhibition of Ca2+-induced insulin secretion in permeabilized HIT-T15 cells. Biochem J. 1990;270:273–276. doi: 10.1042/bj2700273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Vitale N, Mukai H, Rouot B, Thierse’ D, Aunis D, Bader MF. Exocytosis in chromaffin cells. Possible involvement of the heterotrimeric GTP-binding protein Go. J Biol Chem. 1993;268:14715–14723. [PubMed] [Google Scholar]
- 62.Wang JB, Johnson PS, Persico AM, Hawkins AL, Griffin CA, Uhl GR. Human μ-opiate receptor, cDNA and genomic clones, pharmacologic characterization and chromosomal assignment. FEBS Lett. 1994;338:217–222. doi: 10.1016/0014-5793(94)80368-4. [DOI] [PubMed] [Google Scholar]
- 63.Wang L, Gintzler AR. Bimodal opioid regulation of cyclic AMP formation: implications for positive and negative coupling of opiate receptors to adenylate cyclase. J Neurochem. 1994;63:1726–1730. doi: 10.1046/j.1471-4159.1994.63051726.x. [DOI] [PubMed] [Google Scholar]
- 64.Weynants P, Humblet Y, Canon JL, Syman M. Biology of small-cell lung cancer: an overview. Eur Respir J. 1990;3:699–714. [PubMed] [Google Scholar]
- 65.Wilding TJ, Womack MD, McCleskey EW. Fast, local signal transduction between the μ-opioid receptor and Ca2+ channels. J Neurosci. 1995;15:4124–4132. doi: 10.1523/JNEUROSCI.15-05-04124.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Williams CL, Lennon VA. Activation of M3 muscarinic acetylcholine receptors inhibits voltage-dependent calcium influx in small-cell lung carcinoma. J Biol Chem. 1990;265:1443–1447. [PubMed] [Google Scholar]
- 67.Woll PJ. Neuropeptides growth factors and cancer. Br J Cancer. 1991;63:469–475. doi: 10.1038/bjc.1991.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Xu H, Smolens I, Gintzler AR. Opioids can enhance and inhibit the electrically evoked release of methionine-enkephalin. Brain Res. 1989;504:36–42. doi: 10.1016/0006-8993(89)91594-1. [DOI] [PubMed] [Google Scholar]