

Abstract
A brief exposure to high concentrations of glutamate kills cultured forebrain neurons by an excitotoxic process that is dependent on Ca2+ influx through the NMDA receptor. In this study, we have measured striking changes in mitochondrial function during and immediately after intense glutamate receptor activation. Using indo-1 microfluorometry and a specific inhibitor of the mitochondrial Na+/Ca2+ exchanger, CGP-37157, we have demonstrated that mitochondria accumulate large quantities of Ca2+ during a toxic glutamate stimulus and further that Ca2+ efflux from mitochondria contributes to the prolonged [Ca2+]i elevation after glutamate removal. We then used JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide), a ratiometric indicator of mitochondrial membrane potential (Δψ), to show that Ca2+ accumulation within the organelle dissipates Δψ. The abrupt loss of Δψ after glutamate stimulation did not occur in the presence of MK801 or in the absence of extracellular Ca2+. The mitochondrial depolarization was also cyclosporin A-sensitive, indicating a probable role for the permeability transition pore. Hence mitochondrial Ca2+accumulation and the subsequent permeability transition may be a critical early event specific to the NMDA receptor-mediated excitotoxic cascade.
Keywords: intracellular calcium, permeability transition pore, NMDA, excitotoxicity, forebrain neurons, CGP-37157
Pathological glutamate stimulation likely mediates the neuronal injury associated with acute trauma and ischemia (Siesjo and Bengtsson, 1989; Lynch and Dawson, 1994; Choi, 1995), and cultures of rat forebrain neurons have proven useful in studying the cellular consequences of excitotoxic glutamate exposure (Rothman et al., 1987). Previous studies have demonstrated that this “delayed neuronal death” is dependent on NMDA receptor activation and massive Ca2+ influx (Michaels and Rothman, 1990; Hartley et al., 1993). Interestingly, the excitotoxic cascade is initiated during the exposure period, but cells remain viable and can even be rescued for several hours after the insult (Hartley and Choi, 1989; Rothman et al., 1993). It is known that excitotoxic stimulation results in a delayed increase in [Ca2+]i (Randall and Thayer, 1992) and that this secondary loss of [Ca2+]ihomeostasis is associated with the loss of viability (Tymianski et al., 1993; Rajdev and Reynolds, 1994). Little is understood, however, about the Ca2+-mediated changes in cellular function that initiate an irreversible progression toward cell death. Given the impressive metabolic requirements of the brain and the large capacity that mitochondria have for buffering Ca2+ (Nicholls, 1985;Gunter et al., 1994), we hypothesized that these organelles might be a critical target in the excitotoxic cascade.
In preparations of isolated mitochondria, Ca2+ uptake via the mitochondrial Ca2+ uniporter effects a number of processes. The available evidence suggests that mitochondrial Ca2+ accumulation could conceivably increase and/or decrease mitochondrial membrane potential (Fig. 1). Limited Ca2+ uptake increases the availability of substrate nucleotides as well as the rate of electron transport (McCormack and Denton, 1993) (Fig. 1). It has been proposed that these Ca2+-dependent changes in mitochondrial respiration allow excitable cells to couple ATP production with cellular activity (McCormack and Denton, 1993), and there is some evidence that mitochondrial function plays a role in synaptic plasticity (Nguyen and Atwood, 1994).
Fig. 1.
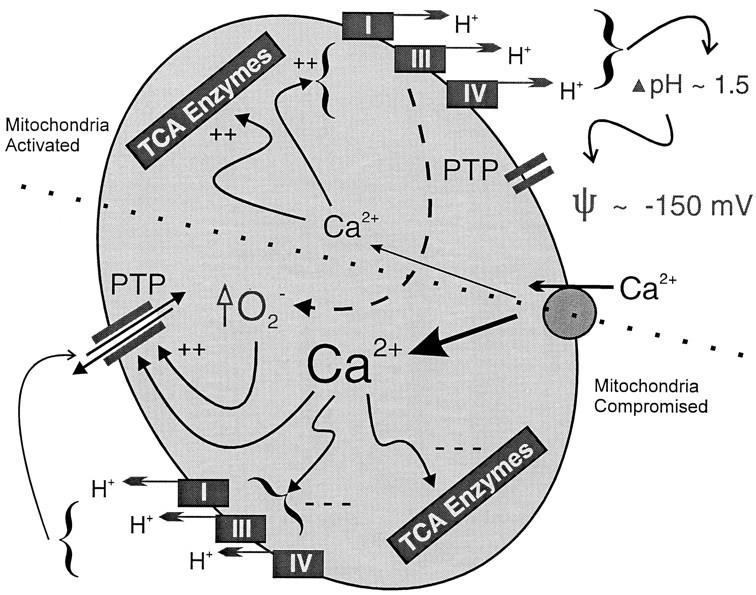
Changes in mitochondrial matrix [Ca2+] can cause hyperpolarization as well as depolarization. In this model of a neuronal mitochondrion, Ca2+ is transported from the cytoplasm into the mitochondria by the Ca2+ uniporter shown on theright. The top half of the picture indicates the positive effects that limited Ca2+uptake has on mitochondrial metabolism. Two rate-limiting steps in the tricarboxylic acid cycle (TCA Enzymes) are activated by matrix Ca2+ in the range of 300–800 nm. A parallel, but poorly understood, process increases the speed of electron transport through the respiratory chain. This latter Ca2+-induced activation probably involves expansion of the volume of the mitochondrion secondary to a compensatory movement of phosphate anions and water. Thus, an activated TCA cycle provides an increased supply of reducing equivalents to an accelerated set of respiratory complexes. At modest matrix Ca2+, the permeability transition pore (PTP) is not activated, and thus the net effect is an increase in the electrochemical potential across the mitochondrial membrane. In the bottom half, the more debilitating effects of massive Ca2+ accumulation are illustrated. Ultimately, unchecked Ca2+ accumulation will inhibit both the TCA cycle and the respiratory chain. In addition, oxygen free radicals produced by the respiratory chain (which was activated initially), as well as those produced in the cytoplasm, will sensitize the PTP to increasing levels of matrix Ca2+. Thus, the PTP is likely to open when it has been oxidized and then exposed to a severe Ca2+ load in the matrix. Opening of the PTP will result in mitochondrial dysfunction and swelling as the pore allows the free passage of many solutes <2 kDa in size. The electrochemical gradient is mostly eliminated under these conditions and is not readily established again until the pore closes.
In consideration of the large capacity of mitochondria for Ca2+ transport, Nicholls (1985) proposed a role for mitochondria in the protection of cells from Ca2+ overload a decade ago; however, excessive mitochondrial Ca2+ uptake has been implicated in the cascades leading to death in cardiac myocytes (Minezaki et al., 1994; Griffiths and Halestrap, 1995). Large increases in matrix [Ca2+]m interfere with mitochondrial ATP production by activating the permeability transition pore (PTP) (Gunter et al., 1994), a nuclear-encoded protein whose structure and identity remain elusive (Andreeva and Crompton, 1994). Activation of the pore results in mitochondrial depolarization (Fig. 1) and swelling as all solutes and proteins smaller than 2 kDa move freely through the mitochondrial inner membrane (Bernardi et al., 1994). The pore is regulated by various physiological signals, including adenosine nucleotides (particularly ADP), polyamines, Mg2+, and both matrix and cytosolic [Ca2+] (Novgorodov et al., 1994). Pharmacological modifiers of pore gating have also been identified. Cyclosporin A is the classic inhibitor of pore opening, whereas recent evidence suggests that several different oxidizing agents increase the likelihood of the permeability transition (Connern and Halestrap, 1994). Pore opening seems to be a critical event common to Ca2+-mediated damage in the heart (Griffiths and Halestrap, 1995) and free-radical injury in the liver (Nieminen et al., 1995).
Here, we present evidence in cultured forebrain neurons that mitochondria accumulate significant stores of Ca2+ during exposure to glutamate stimuli that are generally considered excitotoxic. Using a fluorescent probe of mitochondrial membrane potential, we demonstrate that the same intense NMDA receptor activation causes subsequent mitochondrial depolarization.
MATERIALS AND METHODS
Cell culture. Primary cultures of forebrain neurons on glass coverslips were obtained from embryonic day 17 rats, as described previously (White and Reynolds, 1995); measurements of [Ca2+]i were made between 14 and 18 din vitro. Because the experiments involving mitochondrial membrane potential were designed specifically to address the issue of excitotoxicity, we performed those experiments at 21 or 22 din vitro, a time at which the neurons are more sensitive to excitotoxic cell death. Each type of experiment described in this paper generally was performed on pyramidally shaped neurons from 5–10 different culture dates.
Indo-1 microfluorometry. Neurons were loaded for 40–50 min at 37°C in a solution of 5 μm indo-1/AM in HEPES buffered salt solution (HBSS) containing (in mm): 137 NaCl, 5 KCl, 0.9 MgSO4, 1.4 CaCl2, 10 NaHCO3, 0.6 Na2HPO4, 0.4 KH2PO4, 5.6 glucose, and 20 HEPES, pH adjusted to 7.4 with NaOH, supplemented with 5 mg/ml bovine serum albumin to enhance dye penetration into the cells. The dye was washed away, and the cells were reincubated at 37°C for an additional 20–30 min to allow for uniform cleavage of the acetyl-methoxyester moiety on the dye. The glass coverslips were fitted to the bottom of a 1.5 ml recording chamber, and buffer was perfused continuously over the cells at 20 ml/min, allowing for rapid solution exchange.
[Ca2+]i recordings were made at room temperature as described previously (White and Reynolds, 1995), except that the excitation path was fitted with a computer-controlled shutter. The custom data acquisition software was modified to drive a Uniblitz VA10 shutter (Vincent Associates, Princeton, NJ), which opened briefly to illuminate the neurons for 50 msec while the photomultiplier tubes were sampled. The shutter allowed us to increase the intensity of excitation while still reducing the light-associated changes in the dynamic range of the dye (Randall and Thayer, 1992; White and Reynolds, 1995). While the shutter was open, excitation light (peak transmittance, 340 ± 7.5 nm) from a 100 W mercury lamp was directed onto the sample; emitted fluorescence was reflected off a series of dichroic mirrors and filtered at 405 ± 17 nm and 490 ± 10 nm. Two photomultiplier tubes were used to measure the emitted light in parallel, and the raw fluorescence signals were background-subtracted before being converted to [Ca2+]i as first described (Grynkiewicz et al., 1985).
Time to recovery was measured from the time that glutamate was washed out of the chamber until the time at which the neurons first reached a [Ca2+]i value equal to twice the [Ca2+]i before glutamate stimulation.
Imaging of mitochondrial membrane potential. Mitochondrial membrane potential was assessed with 5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolocarbocyanine iodide (JC-1) on the basis of methods described elsewhere (Moudy et al., 1995). Neurons at 21 or 22 d in vitro were loaded for 20–30 min with 3 μm JC-1 in HBSS at 37°C; recordings were begun after the cells had incubated at room temperature for an additional 20 min. A 1 ml perfusion chamber was fitted to an Olympus microscope, and the cells were perfused at 8 ml/min while images were acquired by an ACAS-570 laser scanning confocal imaging system (Meridian Instruments, Okemos, MI). A 488 nm excitation line from an argon laser was used in conjunction with a 510 nm dichroic mirror. Emitted light was separated further by a 575 nm dichroic mirror before being filtered at 530 ± 10 nm and 590 ± 17 nm, the wavelengths corresponding to the peak fluorescences from the monomer and aggregate signals, respectively. A 400 μm pinhole and a 40× phase-contrast objective yielded an approximate optical slice thickness of 5.7 μm, whereas each pixel corresponded to a 1.6 μm2area. Phase-contrast images were used to draw polygons around the image data, and the software then summed the marked pixels within the two detectors to generate a fluorescence ratio for each time point. The polygons included the nucleus; however, the nucleus never contained any signal above background, and the ratio calculation does not include pixels below the background value. We have not attempted to calibrate the ratios to true values of Δψ. Because we were interested in the change of mitochondrial membrane potential in a single cell, we simply normalized the ratio at each time point to the ratio of the second scan. We chose the second scan because in most of the cells the laser caused a drop in the ratio that was notable only after the first scan (see Fig. 3, diamonds). Although filters were never applied to the data during analysis, the pictures shown in Figure 4 were “pixilated” in the absence of smoothing. We used Corel PhotoPaint v4.0 for aesthetic purposes only, to smooth, soften, and blend the panels that are shown.
Fig. 3.

Fluorescent measurements of changes in mitochondrial membrane potential using the probe JC-1. Ratios of aggregate fluorescence over monomer fluorescence were normalized to the value obtained at the second scan; multiple trials were averaged and plotted as a function of time (mean ± SEM). FCCP (750 nm) was applied for 1 min at the bar(squares, n = 7). Nigericin (1 μm) was applied at the bar(triangles, n = 12), and those same cells were subsequently treated with a combination of oligomycin (1 μm) and antimycin-A (1 μm) at thelarge arrow. Controls (diamonds;n = 101) remained under constant perfusion.
Fig. 4.
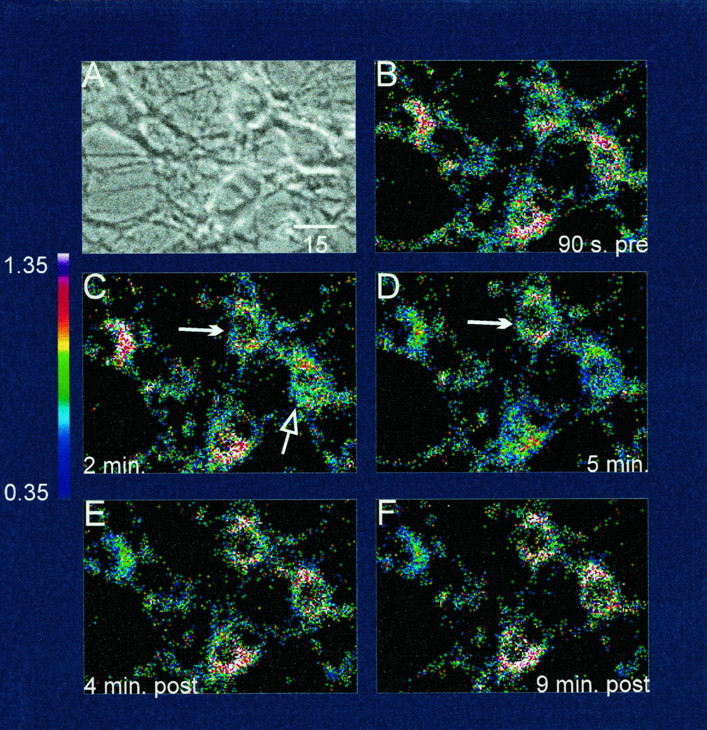
Phase and ratio scans of cultured neurons loaded with the fluorescent probe JC-1 and treated with glutamate.A, Phase-contrast photomicrograph of 21 d forebrain neurons in culture; B, ratiometric image (aggregate signal/monomer signal) of the mitochondrial membrane potential 90 sec before glutamate exposure; C, 2 min after 500 μm glutamate and 10 μm glycine were applied to the neurons; the filled arrow indicates a cell whose mitochondria do not depolarize during glutamate application, whereas the open arrow points to the cell whose mitochondria first depolarize. Of those neuronal mitochondria that would ultimately depolarize, individual cells did so at differing speeds, and this difference is seen more clearly in D. D, At the end of the 5 min exposure to glutamate, the difference between stable (filled arrow) and depolarized (open arrow) mitochondria is now obvious. The raw ratio tracing for the neuron that depolarized and then recovered in subsequent panels (open arrow) corresponded to thegreen line in Figure 5A. In contrast, the mitochondria in the neuron marked by the filled arrownever depolarized in this experiment, and thus its raw ratio tracing most closely resembled the blue line in Figure5A. E, Four minutes after glutamate was removed from the bath. Two of the three neurons with depolarized mitochondria have now recovered, whereas the mitochondria of one remain depolarized. F, Nine minutes after glutamate was removed, the leftmost neuron remained depolarized throughout the period after glutamate removal, and its raw ratio tracing corresponded most closely to the red line in Figure 5A. Thecolor scale shows a spectral representation of aggregate/monomer ratio values between 0.35 and 1.35. Scale bar (shown in A): 15 μm.
Analysis of the normalized JC-1 ratio data. The heterogeneity of responses precluded simple averaging of the JC-1 data; therefore, neurons were segregated in a blinded fashion using three sequential binary criteria. The mean response of 101 control experiments (see Fig. 3, diamonds) was used as the standard. We averaged the normalized ratios collected between 1 and 4 min after the onset of the glutamate stimulation, and an a priori choice of 2 SD was used as the limit of normal variation. Thus, any experimental cell whose normalized fluorescence ratio rose 2 SD greater than control during that period (1–4 min after stimulus onset) was marked as having an increased ratio (see Fig. 5, purple lines), and no controls (0/101) met this criterion. This first pass divided the cells into two groups (see Fig. 5, blue lines and purple lines); we subsequently looked for mitochondrial depolarization during and immediately after glutamate exposure. An a priori choice of 3 SD was used as the criterion for normal variation. This more stringent criterion was chosen because the experiments with drugs that directly modify Δψ (FCCP and nigericin) indicated that the mitochondria can make larger downward than upward deflections (see Fig.3; compare squares to triangles). Thus, any cell whose normalized fluorescence ratio was >3 SD below control at any time during or 3 min after glutamate exposure was said to have undergone a mitochondrial depolarization; only 2 of 101 controls met this criterion. For the experiments described in Figure5A–C, this second criterion divided the cells into three groups, because the increase or decrease was mutually exclusive in the vast majority of cells not treated with CGP-37157. Figure 5Dis more complicated because a substantial fraction of the cells met both the “hyperpolarization” and the “depolarization” criteria at different times during the experiment. Finally, for cells that met the “depolarization” criteria, we made one further binary division and separated the cells whose normalized fluorescence ratio was still <3 SD below control at the end of the experiment. This “recovery” criterion further subdivided the depolarization group into two subgroups, giving a total of four groups for Figure 5A–Cand six groups for Figure 5D. In Figure 5A, both the upper and lower limits of “normal” are illustrated by theopen black circles; in summary, any cell that deviated outside the graphed range at any time was flagged and categorized separately.
Fig. 5.
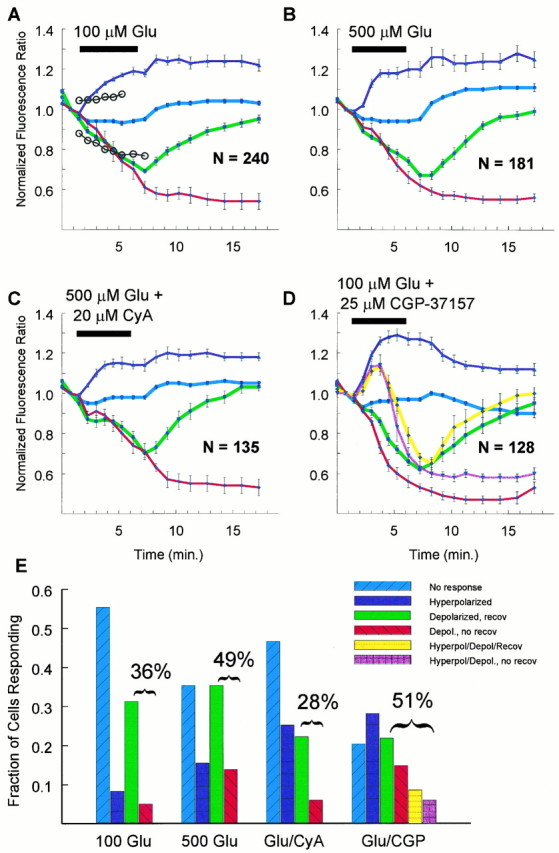
Distinct mitochondrial responses to excitotoxic glutamate stimulation. The data were collected and sorted as described in Materials and Methods. For each of the respective categories, data are plotted as mean and SEM. A, Two hundred forty neurons were stimulated with 100 μm glutamate (Glu) and 1 μm gly. This panel illustrates the “criteria” described in Materials and Methods as open circles. Any neuron that deviated above orbelow the circles would be separated out of the nonresponding (blue line) group and into one of the other three categories. B, One hundred eighty-one neurons were stimulated with 500 μm Glu and 10 μm gly and segregated. C, One hundred thirty-five neurons were stimulated with 500 μm Glu and 10 μm gly in the presence of 20 μmcyclosporin A and categorized. D, One hundred twenty-eight neurons were stimulated with 100 μm Glu and 1 μm gly in the presence of 25 μmCGP-37157, an inhibitor of mitochondrial Na+/Ca2+ exchange. This panel is particularly complex because the combined Glu + CGP-37157 treatment resulted in some cells demonstrating an increase that was followed by a decrease in mitochondrial membrane potential. The two extra linesand bars (in E) reflect this additional level of complexity. E, For each set of averaged responses, the corresponding set of bars illustrates the relative frequency of the particular response. For 100 μmGlu alone, 36% of the cells met the criteria for mitochondrial “depolarization” (sum of green and red bars), whereas only 10% met the criteria for an increase in mitochondrial membrane potential (purple bar); most of the cells with depolarized mitochondria recovered (green bar). With 500 μm Glu alone, 49% of the mitochondria of the cells underwent depolarization, and 19% increased their membrane potential (purple bar); here, 15% of the cells whose mitochondria depolarized never recovered (red bar). When neurons were treated with 500 μm Glu in the presence of cyclosporin A, only 28% of the mitochondria of the neurons depolarized, whereas 29% displayed an increased membrane potential (purple bar); as with 100 μm Glu alone, most of the neurons recovered (green bar). When CGP-37157 was co-applied with 100 μm Glu, fully 51% of the cells had depolarized mitochondria (sum of green, red, yellow, andmagenta bars), and 44% of the cells had increased mitochondrial membrane potentials (sum of purple, magenta, and yellow bars). Here, more than half of the neurons whose mitochondria depolarized never recovered (compare sum of green and yellow bar with sum ofred and magenta).
The graphs qualitatively illustrate the validity of the groupings, but we tested this quantitatively using a 4 × 4 contingency table and a γ2 test of independence. The relationship between treatment and response was highly significant (γ2 = 82.04; df = 9; total n = 684; p < 0.0001).
Materials. Nominally calcium-free solutions contained no added calcium and no chelating agents. Stock 750 μm FCCP solutions were made in methanol, giving a final solvent concentration of 0.1%. Nigericin was made as a 10 mm stock in ethanol. Indo-1/AM and 4Br-A23187 were obtained from Molecular Probes (Eugene, OR), aliquoted in anhydrous dimethylsulfoxide, and frozen. CGP-37157 was a generous gift from Ciba-Geigy Pharmaceuticals (Basel, Switzerland). It was stored as a dry powder at room temperature; 25 mm stocks were prepared in anhydrous DMSO and frozen (−20°C) for use within 1 month. The final solvent concentration was 0.1%. Cyclosporin A (Sandoz Pharmaceuticals, E. Hanover, NJ) was dissolved in methanol. The final methanol concentration was 0.1%. Cell culture supplies were purchased from Life Technologies (Gaithersburg, MD), and unless otherwise noted, all other reagents were of the highest grade available from Sigma (St. Louis, MO).
RESULTS
A glutamate exposure, which has been shown to be excitotoxic in cultured neurons (100 μm, 5 min) (Rajdev and Reynolds, 1994), caused an immediate rise in [Ca2+]ithat persisted long after the stimulus had been washed out, even in the absence of extracellular Ca2+ (Fig.2A). Mitochondria sequester glutamate-induced [Ca2+]i transients in forebrain neurons via a ruthenium red-sensitive, membrane potential-driven uniporter (Wang et al., 1994; White and Reynolds, 1995). In excitable cells, they extrude their Ca2+ store into the cytoplasm via a Na+-dependent exchanger that is specifically inhibited by CGP-37157 (Chiesi et al., 1988; Cox et al., 1993; Kiedrowski and Costa, 1995; Wang and Thayer, in press). Applied immediately after the glutamate stimulus, CGP-37157 significantly reduced the time for recovery to near basal [Ca2+]i (Fig. 2B) (100 μm glutamate followed by CGP-37157, 468 ± 306 sec,n = 7; glutamate followed by control, 2179 ± 893 sec, n = 13; F(0.464);p < 0.0005) Despite the absence of extracellular Ca2+ in the recovery buffer, [Ca2+]i rose again after CGP-37157 washout. This rise presumably reflected a resumption of the mitochondrial Na+/Ca2+ exchange and the subsequent unloading of the Ca2+, which had accumulated in the matrix during and after glutamate stimulation. Mitochondria thus sequester a large amount of Ca2+ during an excitotoxic stimulation only to make a substantial contribution to the prolonged elevation of [Ca2+]i after the stimulus is terminated. Interestingly, experiments with CGP-37157 did not reveal any significant mitochondrial Ca2+ accumulation after a more modest (3 μm, 15 sec) glutamate stimulus (Fig.2C), even though mitochondria clearly contribute to Ca2+ buffering in this paradigm (White and Reynolds, 1995). Accumulation of significant stores of Ca2+ by mitochondria is thus associated specifically with more intense glutamate receptor activation (R. J. White and I. J. Reynolds, unpublished observations).
Fig. 2.
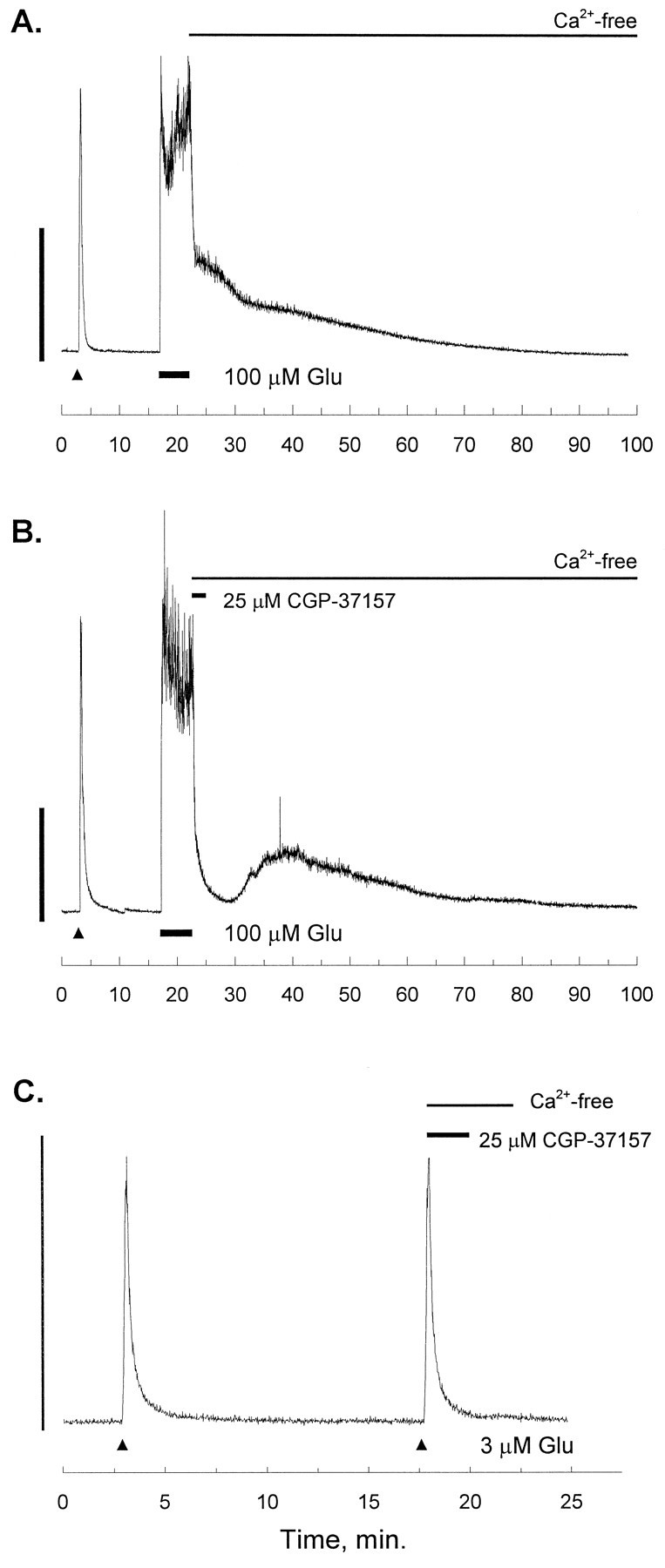
[Ca2+]i recordings after intense glutamate stimulation. In each trace, the small triangle indicates a 15 sec stimulus of 3 μmglutamate (Glu) and 1 μm glycine (gly) as an internal control (White and Reynolds, 1995). The experimental stimulus is applied after a 15 min recovery, and in each trace, the second Glu stimulus is washed out with a nominally Ca2+-free solution (no EGTA). A, After recovery from the internal control, 100 μm Glu and 1 μm gly were applied for 5 min (at theline), and the Glu was followed immediately by a nominally Ca2+-free solution (no EGTA). The trace is representative of 13 experiments. B, After recovery from the internal control, 100 μm Glu and 1 μmgly were applied for 5 min (at the line), and the Glu was followed immediately by a solution of the mitochondrial Na+/Ca2+-exchange inhibitor CGP-37157 (25 μm) in Ca2+-free buffer. After 2 min the CGP-37157 was washed out with Ca2+ -free buffer. The trace is representative of seven experiments.C, After recovery from the internal control, a second identical Glu pulse (15 sec, 3 μm Glu and 1 μm gly) was applied (at the small triangle), and the Glu was followed immediately by a Ca2+-free solution of CGP-37157 (25 μm). After 2 min, the CGP-37157 was washed out with Ca2+-free buffer for an additional 2 min before bath application of ordinary HBSS. The trace is representative of six experiments. In each panel, the scale bar indicates 1 μm[Ca2+]i.
Changes in mitochondrial and cellular function as a result of mitochondrial Ca2+ uptake have been implicated in the cascades leading to death in cardiac myocytes (Minezaki et al., 1994;Griffiths and Halestrap, 1995). Thus, we employed confocal laser microscopy and the ratiometric indicator JC-1 to probe for changes of mitochondrial membrane potential (Δψ) in cultured forebrain neurons. JC-1 monomers accumulate selectively in mitochondria and subsequently aggregate as a function of the membrane potential (Reers et al., 1991; Smiley et al., 1991). The ratio of the aggregate fluorescence to that of the monomer is thus a useful indicator of Δψ, which should be independent of dye loading, light scattering, and other optical difficulties encountered when making comparisons between cells loaded with single wavelength dyes. Furthermore, because the J-aggregate that is formed does not suffer from the fluorescence quenching associated with other lipophilic cations that have been used to study Δψ (e.g., rhodamine 123), JC-1 is suitable for a semiquantitative measurement of changes in Δψ (Reers et al., 1991).
We first sought to demonstrate the utility of JC-1 as a tool in cultured forebrain neurons by using chemicals with well described actions on isolated mitochondria (Wainio, 1970). The electron transport chain normally maintains an electrochemical potential of approximately −150 mV across the inner mitochondrial membrane by moving protons out of the matrix and into the cytosol (Gunter and Gunter, 1994). Acting as a protonophore, 750 nm carbonyl cyanidep-trifluoromethoxyphenylhydrazone (FCCP) caused the rapid collapse of Δψ (Fig. 3, squares), but the neurons were capable of recovering and subsequently increasing Δψ above baseline within minutes of FCCP washout. Nigericin equalizes the H+ gradient across biological membranes via the countertransport of K+. Smiley et al. (1991) showed that the membrane potential component of the proton motive force increases as a result of nigericin exchanging H+ for K+and thus dissipating the pH gradient (for details, see Discussion). As predicted, 1 μm nigericin treatment elevated Δψ (Fig.3, triangles). Finally, the combination of an F1Fo-ATPase inhibitor and a complex III blocker, oligomycin and antimycin-A, respectively, dissipated the membrane potential (Fig. 3, triangles, added at thearrow). More than 100 neurons subjected to constant bath perfusion showed almost no change (Fig. 3, diamonds). Thus, JC-1 is stable under control conditions and quite capable of reflecting downward as well as upward deflections in membrane potential.
Excitotoxic glutamate stimulation also caused mitochondrial depolarization (Figs. 4, 5), although the neurons did not respond uniformly. Glutamate stimulation caused rapid mitochondrial depolarization in many of the neurons studied (Fig.4C,D). Most of the neurons whose mitochondria depolarized recovered their Δψ by the end of the recording (Fig.4F), but some did not. A fraction of the neurons did not undergo mitochondrial depolarization (Fig. 4C,D,filled arrow), and mitochondria in some neurons depolarized before those in other neurons (compare 4C with 4D). Glutamate was applied only once, and we did not evaluate mitochondrial membrane potential longer than the 20 min period shown.
Detailed analysis of the JC-1 ratiometric data revealed four distinct patterns of changes in Δψ after pathological glutamate receptor activation (Fig. 5). Individual neurons were segregated as described in Materials and Methods into categories determined on the basis of their deviation (or lack thereof) from the pattern exhibited by control neurons (Fig. 3, diamonds) and then were grouped for statistical analysis and graphing. We did not attempt to assess individual mitochondria in a neuron but rather evaluated all of the mitochondria together, because the mitochondria of a single cell seemed to respond uniformly. One hundred micromolar glutamate did not alter Δψ in the majority of neurons tested (Fig. 5A,blue line), and some actually increased their mitochondrial membrane potential (Fig. 5A, purple line); however, one third of the neurons had lost their Δψ at the end of the 5 min stimulation (Fig. 5A, red andgreen lines). Of the cells with depolarized mitochondria, most would recover partially, whereas a few had mitochondria that remained depolarized. A higher concentration of glutamate was more efficacious at dissipating Δψ (Fig. 5B, redand green lines; 5E, red andgreen bars). Half of the neurons had depolarized mitochondria at the end of 5 min, and the higher glutamate concentration left 15% of the cells without energized mitochondria at the end of the experiment. Interestingly, the stronger stimulus was also more capable of eliciting the increase in Δψ (Fig.5B,E, purple).
Opening of the PTP seems to be a critical event in Ca2+-mediated damage to the heart (Griffiths and Halestrap, 1995) and free-radical injury in the liver (Nieminen et al., 1995). Furthermore, the fact that the two glutamate concentrations depolarized the mitochondria to the same extent (Fig. 5A,B) suggested an all-or-none response that could easily be ascribed to the permeability transition. Twenty micromolar cyclosporin A reduced by 50% the number of neurons undergoing mitochondrial depolarization (Fig.5C,E, red and green). For those neurons that did lose their mitochondrial membrane potential, the dissipation of Δψ seemed to be delayed when compared with those treated without cyclosporin (Fig. 5B). Once the permeability transition has occurred, cyclosporin A can facilitate pore closure (Novgorodov et al., 1994), and it is thus interesting to note that only a few cells remained without normal mitochondrial membrane potentials at the end of the experiment when cyclosporin was co-applied with glutamate (Fig. 5C,E, red). Cyclosporin A did not change the mitochondrial membrane potential in either direction in the absence of glutamate (data not shown; n = 10; mean normalized fluorescence ratio at end of 5 min = 0.91 ± 0.02).
CGP-37157, which promoted net mitochondrial Ca2+accumulation by inhibiting the mitochondrial Na+/Ca2+ exchange (Fig. 2B), also provoked changes in the mitochondrial membrane potential. In control experiments, a 5 min exposure to 25 μm CGP-37157 caused an increase in Δψ that was sustained throughout drug application and reversed after washout (Fig. 6, diamonds;n = 17). When applied together with 100 μm glutamate, CGP-37157 increased the fraction of cells responding with an elevated Δψ to 50% (Fig. 5D,purple, blue, and yellow lines). Interestingly, a much larger fraction of cells underwent mitochondrial depolarization when CGP-37157 was co-applied with glutamate, and more of those neurons were left with depolarized mitochondria at the close of the recording. Thus, CGP-37157 caused some cells to hyperpolarize first but then to depolarize abruptly, a pattern not seen in the absence of this intriguing compound. Neurons exposed to both glutamate and CGP-37157 also tended to lose their mitochondrial membrane potential faster than in the absence of CGP-37157 (Fig. 5D, red trace; compare with Fig. 5A).
Fig. 6.

NMDA receptor-dependent Ca2+ influx is required for mitochondrial depolarization. Ratios of JC-1 aggregate fluorescence over monomer fluorescence were normalized to the value obtained at the second scan; multiple trials were averaged and plotted as a function of time (mean ± SEM). When glutamate (Glu) (100 or 500 μm, data not different and thus combined) was applied in the presence of 5 μmMK801 (at the bar; squares,n = 31), none of the individual neurons met the criteria for depolarization as described in Materials and Methods. Glu (100 or 500 μm; data not different and thus combined) was also applied in the nominal absence of Ca2+ (at thebar; triangles,n = 35); none of the individual neurons met the criteria for depolarization, and as reflected in the average increase, 60% of the cells met the criteria for hyperpolarization. CGP-37157 (25 μm, an inhibitor of mitochondrial Na+/Ca2+ exchange) was applied for 5 min (at the bar) in a set of control experiments and caused a reversible hyperpolarization without exception (diamonds, n = 17).
The bar graph in Figure 5E summarizes this interesting set of data. Approximately one third of the neurons exposed to 100 μm glutamate underwent mitochondrial depolarization (sum of the red and green bars). An increased concentration of glutamate raised the total fraction of cells whose mitochondria depolarized to 50% and tripled the number of cells that did not recover (red bars). Applied together with the higher concentration of glutamate, cyclosporin A reduced the number of neurons whose mitochondria depolarized by half, diminished the number of cells that showed no recovery, and increased the number of cells whose mitochondria increased their membrane potential (purple bars). Co-application of CGP-37157 with 100 μm glutamate caused the cells to respond as though they had been treated with 500 μm glutamate. CGP-37157 quadrupled the number of cells that had hyperpolarized mitochondria (purple, magenta, and yellow bars), raised the number of neurons whose mitochondria depolarized (green, red, yellow, and magentabars), and dramatically increased the number of cells left without functioning mitochondria (red and magenta bars).
Glutamate may be neurotoxic by activating NMDA or non-NMDA receptors. We wanted to test the hypothesis that NMDA-mediated Ca2+entry was required for the rapid dissipation of the mitochondrial membrane potential. Co-application of the noncompetitive NMDA antagonist MK801 (5 μm) with either 500 μmor 100 μm glutamate prevented any cells from meeting the transition criterion (Fig. 6, squares; n = 31). Glutamate application at either concentration in a nominally Ca2+-free media caused an average increase in Δψ, and none of the cells met the depolarization criteria (Fig. 6,triangles; n = 35). Finally, the incidence of mitochondrial depolarization was reduced dramatically at glutamate concentrations that do not produce substantial toxicity in our cultures. Less than 10% of the neurons stimulated with 3 μm glutamate met the criteria for depolarization, and the average membrane potential was unchanged from control at the end of the 5 min stimulation (n = 43; mean normalized fluorescence ratio = 1.03 ± 0.02).
DISCUSSION
The combined use of CGP-37157 and JC-1 has provided a number of insights into the role that mitochondria play in neuronal cell biology. The data extend our understanding of neuronal Ca2+homeostasis by demonstrating net mitochondrial Ca2+accumulation after intense, but not more modest, glutamate receptor activation (compare Fig. 2, B and C). CGP-37157 seems to block mitochondrial Ca2+ efflux (R. J. White and I. J. Reynolds, unpublished observations). These data confirm the reports in isolated preparations of neuronal mitochondria, which indicated that the Na+-dependent Ca2+ efflux predominated over the Na+-independent form (Satrustegui and Richter, 1984; Gunter et al., 1994). The description of this pathway sheds new light on our understanding of cytoplasmic Na+ and its influence on the processes governing recovery from glutamate-induced Ca2+ transients (Kiedrowski et al., 1994;Koch and Barish, 1994; Kiedrowski and Costa, 1995; Stout et al., 1996; Wang and Thayer, in press). Increases in cytoplasmic Na+will reduce the gradient for Ca2+ extrusion via the plasma membrane Na+/Ca2+ exchanger (Kiedrowski et al., 1994), but Na+ will also force Ca2+ out of the mitochondrial matrix space and hence prolong the [Ca2+]i transient even in the absence of extracellular Ca2+ (Fig. 2A). The rapid recovery of [Ca2+]i shown in Figure 2Bsuggests that the latter effect may be more important than the former, because the plasma membrane Na+/Ca2+exchanger seems quite capable of clearing Ca2+ from the cytoplasm when mitochondrial Ca2+ efflux is blocked.
Ca2+ and metabolic control
The use of JC-1 to measure mitochondrial membrane potential in single neurons over time has substantiated a fundamental hypothesis concerning metabolic control previously untested in intact cells. Using purified enzymes and isolated mitochondria, several groups have proposed an important role for matrix Ca2+ in matching ATP production with cellular activity (Denton and McCormack, 1990;Hansford, 1994). Two key enzymes of the tricarboxylic acid cycle as well as the overall activity of the electron transport chain are upregulated by levels of matrix Ca2+ that are achieved in stimulated cardiac myocytes (Fig. 1). These investigators have thus advanced the hypothesis that elevated mitochondrial Ca2+will activate cellular energy production in anticipation of increased ATP demands. The increase in Δψ that we saw (Fig. 5, purple lines) provides potentially important in situcorroboration for their in vitro observations. The fact that CGP-37157 was able to promote an elevated Δψ by itself confirms speculation that the mitochondrial Na+/Ca2+exchange is tonically active and that inhibiting it will promote increases in matrix Ca2+ even in resting cells. The increase in Δψ could reflect Ca2+-dependent increases in metabolic activity, although other explanations are possible (see below). The Ca2+ dependence of the increased mitochondrial membrane potential is suggested by the fact that when co-applied with CGP-37157, 100 μm glutamate increased the membrane potential in a much larger proportion of the neurons (compare Fig.5E, purple bars). Cox and Matlib (1993) have proposed the use of drugs like CGP-37157 in cardiac pathologies associated with impaired mitochondrial function (e.g., congestive heart failure), and our findings suggest that a carefully titrated dose might indeed have a positive effect on cellular respiration. Mitochondrial Ca2+ accumulation sufficient to activate the PTP, however, can dissipate Δψ and potentially damage the cells even when mitochondrial respiration is stimulated initially by the Ca2+ uptake (Fig. 5D, magenta andyellow lines).
Another explanation for the hyperpolarization that we observed is possible. Mitochondria eject H+ from the matrix into the cytoplasm, thus forming the proton motive force. This electrochemical gradient has two components, a pH gradient (ΔpH) and the transmembrane potential Δψ (Nicholls, 1985; Gunter et al., 1994). The total proton motive force (ΔμH) is thought to be a relatively constant value such that an increase in one part of ΔμH is compensated by a corresponding decrease in the other, according to the equation ΔμH = Δψ−60ΔpH (Nicholls, 1985).
Reasonable estimates of these values from energized mammalian mitochondria are ΔpH of 0.5–1.0 and Δψ = 150–180 mV (matrix negative with respect to the cytosol) (Gunter and Gunter, 1994; Czyz et al., 1995). The expulsion of 1 nmol H+/mg mitochondrial protein, however, would establish a ΔpH of 0.05 and Δψ = 200 mV. It is clear then that an influx of other cations must be reducing some of the transmembrane potential to allow for a compensatory increase in the pH gradient. Even in the absence of a permeability transition event, mitochondrial Ca2+ transport will decrease Δψ and increase ΔpH to a maximum of ∼2 pH units for 10 nmol Ca2+ · min−1 · mg−1protein (Nicholls, 1985). This movement of cations obviously does not account for the observed hyperpolarization, because the mitochondria would have been retaining Ca2+ under both conditions in which we measured increases in Δψ (Fig. 5, purple lines; Fig. 6, diamonds). More recent work (Czyz et al., 1995) has identified K+ as a cation whose flux across the inner membrane may control the relationship between transmembrane potential and ΔpH. Czyz et al. (1995) used BCECF to monitor pH and a TPP+-sensitive electrode to measure Δψ in isolated mitochondria. In their system, an increase in the “cytosolic” [K+] caused a decrease in the Δψ, with a concomitant increase in the ΔpH. During the 5 min glutamate stimulations (Fig.5), it is possible that decreases in cytosolic [K+] might have decreased the ΔpH and thus caused a parallel increase in Δψ, with no net change in the total proton motive force.
Changes in K+ flux are less likely to account for the hyperpolarization that we observed in control experiments with CGP-37157 by itself (Fig. 6, diamonds). Although it is attractive to speculate that increases in matrix Ca2+ were activating mitochondrial metabolism, another explanation is available. Early research suggested that the mitochondrial Na+/Ca2+ exchange was distinct from that of the plasma membrane with respect to its stoichiometry in that the mitochondrial protein was not electrogenic (Gunter et al., 1994). More recent efforts, however, have provided reasonable proof that the mitochondrial exchange is in fact electrogenic and thus probably transports 2.5 or more Na+ for each Ca2+, similar to the plasma membrane protein (Jung et al., 1995). If we accept the latter study as correct, CGP-37157 would stop the tonic influx of positive charge and thus cause mitochondrial hyperpolarization (a more negative membrane potential). In favor of this explanation is the rapid change in membrane potential that we observed (Fig. 6, diamonds).
Mitochondrial dysfunction and lethal injury
Mitochondrial dysfunction has been suggested as a final common denominator in several chronic neurodegenerative disorders (Beal, 1992), and investigators have documented increased lactate levels in Huntington’s disease, which may correlate with impaired mitochondrial ATP generation (Jenkins et al., 1993). We now propose that mitochondria may be an early target of injury in the cascade of events after intense NMDA receptor stimulation. Our study has identified a critical intracellular target for Ca2+ that may play a central role in excitotoxic neuronal damage. One recent investigation complements our present findings and invites speculation with respect to the mechanism of cell death. Zhang et al. (1994) reported an NMDA receptor-mediated activation of the enzyme poly(ADP-ribose) synthase (PARS), and they hypothesized that elevated PARS activity would drain neuronal reserves of ATP and nicotinamide nucleotides. Thus, an early compromise in mitochondrial function (e.g., the induction of the permeability transition) may leave cells ill-prepared to deal with the later cellular energy drain associated with PARS activation.
Consistent with the suggestion that mitochondrial depolarization may be the cause of neuronal injury, cyclosporin A can inhibit neuronal death induced by glutamate under the same stimulation conditions used here (E. Aizenman, personal communication). Although neuroprotection with cyclosporin A supports a critical role for mitochondrial depolarization in excitotoxicity, however, it is difficult to exclude the possibility that the protective action of cyclosporin A is also attributable in part to alteration of the phosphorylation status of nitric oxide synthase (Zhang et al., 1994). The lack of selective PTP inhibitors makes it difficult to address this point unequivocally. We are also unable to provide a certain explanation for the underlying mechanisms that caused the cells to respond in categorically different ways. At least two possibilities seem reasonable. In this otherwise heterogenous preparation of both cortical and subcortical neurons, it is quite likely that some of the between-cell differences that we encountered were the result of making comparisons between several populations with stable but distinct responses. Particularly with respect to the distribution of NMDA receptors and their different subtypes, combining neurons from different brain regions is less than ideal for experiments exploring this level of subcellular physiology. A second, independent set of variables would be related to the intrinsic state of the neurons before glutamate stimulation. The present measurements of mitochondrial function may be reporting differences in intracellular redox state, substrate availability, and metabolic enzyme activity. The neurons whose mitochondria remained stable may already have been upregulated maximally and thus were unable to respond with hyperpolarized mitochondria. Similarly, those same neurons may have been better equipped with cellular reducing equivalents and thus less sensitive to oxidation and opening of the PTP; for that reason, they failed to depolarize, despite massive mitochondrial Ca2+ uptake. Additional experimentation to limit the variability that we encountered will help clarify these intriguing issues.
Given the metabolic demands of the brain, it is reasonable to speculate that mitochondria are a central target for several Ca2+-dependent effectors of excitotoxicity. The components of the electron transport chain are sensitive to oxidants in general and to nitric oxide in particular (Zhang et al., 1990; Schweizer and Richter, 1994), and the redox state of a key pair of vicinal thiols regulates the opening of the PTP (Petronilli et al., 1994). Mg2+ ions modulate mitochondrial Ca2+ transport (Gunter et al., 1994) as well as the gating of the PTP (Novgorodov et al., 1994), and indeed, Mg2+ may be requisite for PTP closure once it has opened. Thus, mitochondria are poised to integrate a diverse set of glutamate-induced signals: massive Ca2+influx, Mg2+ mobilization, nitric oxide generation, and free-radical production (Brocard et al., 1993; Dawson et al., 1993;Hartley et al., 1993; Reynolds and Hastings, 1995). It seems likely that future studies of mitochondrial physiology and dysfunction will provide significant insight into the mechanism of neuronal injury that underlies the damage in stroke and acute CNS trauma.
Footnotes
This work was supported by National Institutes of Health Grant NS34138 (I.J.R.). I.J.R. is an Established Investigator of the American Heart Association, and R.J.W. is supported by a National Institutes of Health Medical Scientist Training Program Grant (5T32GM08208). We gratefully acknowledge Kristi Rothermund for the preparation of cell cultures and Dr. Kari Hoyt for assistance with the Meridian equipment. We thank Drs. Elias Aizenman, Anne Murphy, and Amy Stout for critically reading this manuscript.
Correspondence should be addressed to Dr. Ian J. Reynolds, Center for Neuroscience and Department of Pharmacology, University of Pittsburgh School of Medicine, E1354 Biomedical Science Tower, Pittsburgh, PA 15261.
REFERENCES
- 1.Andreeva L, Crompton M. An ADP-sensitive cyclosporin-A-binding protein in rat liver mitochondria. Eur J Biochem. 1994;221:261–268. doi: 10.1111/j.1432-1033.1994.tb18737.x. [DOI] [PubMed] [Google Scholar]
- 2.Beal MF. Does impairment of energy metabolism result in excitotoxic neuronal death in neurodegenerative illnesses? Ann Neurol. 1992;31:119–130. doi: 10.1002/ana.410310202. [DOI] [PubMed] [Google Scholar]
- 3.Bernardi P, Broekemeier KM, Pfeiffer DR. Recent progress on regulation of the mitochondrial permeability transition pore: a cyclosporin-sensitive pore in the inner mitochondrial membrane. J Bioenerg Biomembr. 1994;26:509–517. doi: 10.1007/BF00762735. [DOI] [PubMed] [Google Scholar]
- 4.Brocard JB, Rajdev S, Reynolds IJ (1993) Glutamate-induced increases in intracellular free Mg2+ in cultured cortical neurons. Neuron 11: 751–757. [DOI] [PubMed]
- 5.Chiesi M, Schwaller R, Eichenberger K. Structural dependency of the inhibitory action of benzodiazepines and related compounds on the mitochondrial Na+-Ca2+exchanger. Biochem Pharmacol. 1988;37:4399–4403. doi: 10.1016/0006-2952(88)90623-5. [DOI] [PubMed] [Google Scholar]
- 6.Choi DW. Calcium: still center-stage in hypoxic-ischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 7.Connern CP, Halestrap AP. Recruitment of mitochondrial cyclophilin to the mitochondrial inner membrane under conditions of oxidative stress that enhance the opening of a calcium-sensitive non-specific channel. Biochem J. 1994;302:321–324. doi: 10.1042/bj3020321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox DA, Matlib MA. Modulation of intramitochondrial free Ca2+ concentration by antagonists of Na+-Ca2+ exchange. Trends Pharmacol Sci. 1993;14:408–413. doi: 10.1016/0165-6147(93)90063-P. [DOI] [PubMed] [Google Scholar]
- 9.Cox DA, Conforti L, Sperelakis N, Matlib MA. Selectivity of inhibition of Na+-Ca2+ exchange of heart mitochondria by benzothiazepine CGP-37157. J Cardiovasc Pharmacol. 1993;21:595–599. doi: 10.1097/00005344-199304000-00013. [DOI] [PubMed] [Google Scholar]
- 10.Czyz A, Szewczyk A, Nalecz MJ, Wojtczak L. The role of mitochondrial potassium fluxes in controlling the proton motive force in energized mitochondria. Biochem Biophys Res Commun. 1995;210:98–104. doi: 10.1006/bbrc.1995.1632. [DOI] [PubMed] [Google Scholar]
- 11.Dawson VL, Dawson TM, Bartley DA, Uhl GR, Snyder SH. Mechanisms of nitric oxide-mediated neurotoxicity in primary brain cultures. J Neurosci. 1993;13:2651–2661. doi: 10.1523/JNEUROSCI.13-06-02651.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Denton RM, McCormack JG (1990) Ca2+ as a second messenger within mitochondria of the heart and other tissues. Annu Rev Physiol 52: 451–466. [DOI] [PubMed]
- 13.Griffiths EJ, Halestrap AP. Mitochondrial non-specific pores remain closed during cardiac ischaemia, but open upon reperfusion. Biochem J. 1995;307:93–98. doi: 10.1042/bj3070093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grynkiewicz G, Poenie M, Tsien RY. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J Biol Chem. 1985;260:3440–3450. [PubMed] [Google Scholar]
- 15.Gunter KK, Gunter TE. Transport of calcium by mitochondria. J Bioenerg Biomembr. 1994;26:471–485. doi: 10.1007/BF00762732. [DOI] [PubMed] [Google Scholar]
- 16.Gunter TE, Gunter KK, Sheu SS, Gavin CE. Mitochondrial calcium transport: physiological and pathological relevance. Am J Physiol. 1994;267:C313–C339. doi: 10.1152/ajpcell.1994.267.2.C313. [DOI] [PubMed] [Google Scholar]
- 17.Hansford RG. Physiological role of mitochondrial Ca2+ transport. J Bioenerg Biomembr. 1994;26:495–508. doi: 10.1007/BF00762734. [DOI] [PubMed] [Google Scholar]
- 18.Hartley DM, Choi DW. Delayed rescue of N -methyl-d-aspartate receptor-mediated neuronal injury in cortical culture. J Pharmacol Exp Ther. 1989;250:752–758. [PubMed] [Google Scholar]
- 19.Hartley DM, Kurth MC, Bjerkness L, Weiss JH, Choi DW. Glutamate receptor-induced45Ca2+ accumulation in cortical cell culture correlates with subsequent neuronal degeneration. J Neurosci. 1993;13:1993–2000. doi: 10.1523/JNEUROSCI.13-05-01993.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jenkins BG, Koroshetz WJ, Beal MF, Rosen BR. Evidence for impairment of energy metabolism in vivo in Huntington’s disease using localized 1H NMR spectroscopy. Neurology. 1993;43:2689–2695. doi: 10.1212/wnl.43.12.2689. [DOI] [PubMed] [Google Scholar]
- 21.Jung DW, Baysal K, Brierley GP. The sodium-calcium antiport of heart mitochondria is not electroneutral. J Biol Chem. 1995;270:672–678. doi: 10.1074/jbc.270.2.672. [DOI] [PubMed] [Google Scholar]
- 22.Kiedrowski L, Costa E. Glutamate-induced destabilization of intracellular calcium concentration homeostasis in cultured cerebellar granule cells: role of mitochondria in calcium buffering. Mol Pharmacol. 1995;47:140–147. [PubMed] [Google Scholar]
- 23.Kiedrowski L, Brooker G, Costa E, Wroblewski JT. Glutamate impairs neuronal calcium extrusion while reducing sodium gradient. Neuron. 1994;12:295–300. doi: 10.1016/0896-6273(94)90272-0. [DOI] [PubMed] [Google Scholar]
- 24.Koch RA, Barish ME. Perturbation of intracellular calcium and hydrogen ion regulation in cultured mouse hippocampal neurons by reduction of the sodium ion concentration gradient. J Neurosci. 1994;14:2585–2593. doi: 10.1523/JNEUROSCI.14-05-02585.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lynch DR, Dawson TM. Secondary mechanisms in neuronal trauma. Curr Opin Neurol. 1994;7:510–516. doi: 10.1097/00019052-199412000-00007. [DOI] [PubMed] [Google Scholar]
- 26.McCormack JG, Denton RM. Mitochondrial Ca2+transport and the role of intramitochondrial Ca2+ in the regulation of energy metabolism. Dev Neurosci. 1993;15:165–173. doi: 10.1159/000111332. [DOI] [PubMed] [Google Scholar]
- 27.Michaels RL, Rothman SM. Glutamate neurotoxicity in vitro: antagonist pharmacology and intracellular calcium concentrations. J Neurosci. 1990;10:283–292. doi: 10.1523/JNEUROSCI.10-01-00283.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Minezaki KK, Suleiman MS, Chapman RA. Changes in mitochondrial function induced in isolated guinea-pig ventricular myocytes by calcium overload. J Physiol (Lond) 1994;476:459–471. doi: 10.1113/jphysiol.1994.sp020147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Moudy AM, Handran SD, Goldberg MP, Ruffin N, Karl I, Kranz-Eble P, DeVivo DC, Rothman SM. Abnormal calcium homeostasis and mitochondrial polarization in a human encephalomyopathy. Proc Natl Acad Sci USA. 1995;92:729–733. doi: 10.1073/pnas.92.3.729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen PV, Atwood HL. Altered impulse activity modifies synaptic physiology and mitochondria in crayfish phasic motor neurons. J Neurophysiol. 1994;72:2944–2955. doi: 10.1152/jn.1994.72.6.2944. [DOI] [PubMed] [Google Scholar]
- 31.Nicholls DG. A role for the mitochondrion in the protection of cells against calcium overload? Prog Brain Res. 1985;63:97–106. doi: 10.1016/S0079-6123(08)61978-0. [DOI] [PubMed] [Google Scholar]
- 32.Nieminen AL, Saylor AK, Tesfai SA, Herman B, Lemasters JJ. Contribution of the mitochondrial permeability transition to lethal injury after exposure of hepatocytes to t -butylhydroperoxide. Biochem J. 1995;307:99–106. doi: 10.1042/bj3070099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Novgorodov SA, Gudz TI, Brierley GP, Pfeiffer DR (1994) Magnesium ion modulates the sensitivity of the mitochondrial permeability transition pore to cyclosporin A and ADP. Arch Biochem Biophys 311: 219–228. [DOI] [PubMed]
- 34.Petronilli V, Costantini P, Scorrano L, Colonna R, Passamonti S, Bernardi P. The voltage sensor of the mitochondrial permeability transition pore is tuned by the oxidation-reduction state of vicinal thiols: increase of the gating potential by oxidants and its reversal by reducing agents. J Biol Chem. 1994;269:16638–16642. [PubMed] [Google Scholar]
- 35.Rajdev S, Reynolds IJ. Glutamate-induced intracellular calcium changes and neurotoxicity in cortical neurons in vitro: effect of chemical ischemia. Neuroscience. 1994;62:667–679. doi: 10.1016/0306-4522(94)90468-5. [DOI] [PubMed] [Google Scholar]
- 36.Randall RD, Thayer SA. Glutamate-induced calcium transient triggers delayed calcium overload and neurotoxicity in rat hippocampal neurons. J Neurosci. 1992;12:1882–1895. doi: 10.1523/JNEUROSCI.12-05-01882.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Reers M, Smith TW, Chen LB. J-aggregate formation of a carbocyanine as a quantitative fluorescent indicator of membrane potential. Biochemistry. 1991;30:4480–4486. doi: 10.1021/bi00232a015. [DOI] [PubMed] [Google Scholar]
- 38.Reynolds IJ, Hastings TG. Glutamate induces the production of reactive oxygen species in cultured forebrain neurons following NMDA receptor activation. J Neurosci. 1995;15:3318–3327. doi: 10.1523/JNEUROSCI.15-05-03318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rothman SM, Thurston JH, Hauhart RE. Delayed neurotoxicity of excitatory amino acids in vitro. Neuroscience. 1987;22:471–480. doi: 10.1016/0306-4522(87)90347-2. [DOI] [PubMed] [Google Scholar]
- 40.Rothman SM, Yamada KA, Lancaster N. Nordihydroguaiaretic acid attenuates NMDA neurotoxicity: action beyond the receptor. Neuropharmacology. 1993;32:1279–1288. doi: 10.1016/0028-3908(93)90022-u. [DOI] [PubMed] [Google Scholar]
- 41.Satrustegui J, Richter C. The role of hydroperoxides as calcium release agents in rat brain mitochondria. Arch Biochem Biophys. 1984;233:736–740. doi: 10.1016/0003-9861(84)90501-0. [DOI] [PubMed] [Google Scholar]
- 42.Schweizer M, Richter C. Nitric oxide potently and reversibly deenergizes mitochondria at low oxygen tension. Biochem Biophys Res Commun. 1994;204:169–175. doi: 10.1006/bbrc.1994.2441. [DOI] [PubMed] [Google Scholar]
- 43.Siesjo BK, Bengtsson F. Calcium fluxes, calcium antagonists, and calcium-related pathology in brain ischemia, hypoglycemia, and spreading depression: a unifying hypothesis. J Cereb Blood Flow Metab. 1989;9:127–140. doi: 10.1038/jcbfm.1989.20. [DOI] [PubMed] [Google Scholar]
- 44.Smiley ST, Reers M, Mottola-Hartshorn C, Lin M, Chen A, Smith TW, Steele GD, Jr, Chen LB. Intracellular heterogeneity in mitochondrial membrane potentials revealed by a J-aggregate-forming lipophilic cation JC-1. Proc Natl Acad Sci USA. 1991;88:3671–3675. doi: 10.1073/pnas.88.9.3671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stout AK, Li-Smerin Y, Johnson JW, Reynolds IJ. Mechanisms of glutamate-stimulated Mg2+influx and subsequent Mg2+ efflux in rat forebrain neurones in culture. J Physiol (Lond) 1996;492:641–657. doi: 10.1113/jphysiol.1996.sp021334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tymianski M, Charlton MP, Carlen PL, Tator CH. Secondary Ca2+ overload indicates early neuronal injury which precedes staining with viability indicators. Brain Res. 1993;607:319–323. doi: 10.1016/0006-8993(93)91523-u. [DOI] [PubMed] [Google Scholar]
- 47.Wainio WW. Academic; New York: 1970. The mammalian mitochondrial respiratory chain. . [Google Scholar]
- 48.Wang GJ, Thayer SA (1996) Sequestration of glutamate-induced Ca2+-loads by mitochondria in cultured rat hippocampal neurons. J Neurophysiol, in press. [DOI] [PubMed]
- 49.Wang GJ, Randall RD, Thayer SA. Glutamate-induced intracellular acidification of cultured hippocampal neurons demonstrates altered energy metabolism resulting from Ca2+loads. J Neurophysiol. 1994;72:2563–2569. doi: 10.1152/jn.1994.72.6.2563. [DOI] [PubMed] [Google Scholar]
- 50.White RJ, Reynolds IJ. Mitochondria and Na+/Ca2+ exchange buffer glutamate-induced calcium loads in cultured cortical neurons. J Neurosci. 1995;15:1318–1328. doi: 10.1523/JNEUROSCI.15-02-01318.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- 52.Zhang Y, Marcillat O, Giulivi C, Ernster L, Davies KJ. The oxidative inactivation of mitochondrial electron transport chain components and ATPase. J Biol Chem. 1990;265:16330–16336. [PubMed] [Google Scholar]