

Abstract
Cholinergic stimulation of the hippocampal formation results in excitation and/or seizure. We report here, using whole-cell patch-clamp techniques in the hippocampal slice (34–35°C), a cholinergic-dependent slow afterdepolarization (sADP) and long-lasting plateau potential (PP). In the presence of 20 μm carbachol, action potential firing evoked by weak intracellular current injection elicited an sADP that lasted several seconds. Increased spike firing evoked by stronger depolarizing stimuli resulted in long-duration PPs maintained close to −20 mV. Removal of either Na+ or Ca2+ from the external media, intracellular Ca2+([Ca2+]i) chelation with 10 mmbis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid, or the addition of 100 μmCd2+ to the perfusate abolished both the sADP and PP. The sADP was depressed and the PP was abolished by either 10 μm nimodipine or 1 μmω-conotoxin, whereas 1.2 μm tetrodotoxin was ineffective. The involvement of a Na+/Ca2+ exchanger was minimal because both the sADP and PP persisted after equimolar substitution of 50 mm Li+for Na+ in the external media or reduction of the bath temperature to 25°C. Finally, in the absence of carbachol the sADP and PP could not be evoked when K+ channels were suppressed, suggesting that depression of K+conductances alone was not sufficient to unmask the conductance. Based on these data, we propose that a Ca2+-activated nonselective cation conductance was directly enhanced by muscarinic stimulation. The sADP, therefore, represents activation of this conductance by residual [Ca2+]i, whereas the PP represents a novel regenerative event involving the interplay between high-voltage-activated Ca2+ channels and the Ca2+-activated nonselective cation conductance. This latter mechanism may contribute significantly to ictal depolarizations observed during cholinergic-induced seizures.
Keywords: muscarinic stimulation, Ca2+-activated nonselective cation conductance, HVA Ca2+ currents, ictal depolarization, epilepsy, slice-patch technique
The hippocampal formation receives cholinergic afferents from the medial septum/diagonal band that terminate on dendritic segments located in both the cornu Ammonis and dentate gyrus (Wainer et al., 1993). This septohippocampal pathway is critical in the generation of rhythmical slow activity (Bland, 1986; Vanderwolf, 1988) and is implicated in some forms of learning and memory (Shen et al., 1994). Cholinergic stimulation is also effective in generating limbic seizures and, thus, injection of cholinergic agents is used as a model for temporal lobe epilepsy (Lothman et al., 1991). Indeed, hippocampal sclerosis after infusion of cholinergic agonists accurately mimics numerous pathological indices of human status epilepticus (Wasterlain et al., 1993).
The electrophysiological actions of cholinergic agonists on neurons have been studied extensively in vitro and, for the most part, are considered excitatory (Halliwell, 1990; Krnjević, 1993;McCormick, 1993). For example, cholinergic stimulation causes a sustained depolarization associated with an increased input resistance (RIN) and depression of spike frequency adaptation. These effects are attributed to a cholinergic-induced depression of four potassium conductances, including the M-current (IM) (Halliwell and Adams, 1982; Madison et al., 1987), a fast inactivating current (IA) (Nakajima et al., 1986), a slow Ca2+-activated current (IAHP) (Madison et al., 1987; Bernardo and Prince, 1982), and a background leak current (ILEAK) (Madison et al., 1987). Voltage-dependent Ca2+ channels are also modulated by cholinergic stimulation; high-voltage-activated (HVA) channels are depressed (IHVA) (Misgeld et al., 1986; Toselli and Lux, 1989), whereas low voltage-activated channels are enhanced (ILVA) (Toselli and Lux, 1989; Fraser and MacVicar, 1991). More recently, a nonselective cation conductance has been demonstrated to contribute to the initial cholinergic-induced depolarization (Benson et al., 1988; Shen and North, 1992; Colino and Halliwell, 1993). A slow afterdepolarization (sADP) has also been observed in the presence of cholinergic agents (Bernardo and Prince, 1982; Gähwiler, 1984; McCormick and Prince, 1986; Müller et al., 1988; Hasuo and Gallagher, 1990; Andrade, 1991; Müller and Connor, 1991), however, multiple ionic mechanisms may underlie this phenomenon. For instance, a Na+/Ca2+ exchanger (Friedman et al., 1992), a prolonged Ca2+conductance (Blitzer et al., 1991), deactivation of a K+ conductance by maintained Ca2+ influx (Constanti and Bagetta, 1991;Constanti et al., 1993), and a Ca2+-activated nonselective cation conductance (ICAN) (Schwindt et al., 1988; Hasuo et al., 1990; Caeser et al., 1993) are all plausible mechanisms.
Although there are multiple reports of sADPs after muscarinic stimulation, cholinergic-dependent plateau potentials (PPs) have not been reported. This PP may represent a novel regenerative event involving interactions between IHVA andICAN. It is conceivable that interplay between these channels could contribute significantly to ictal depolarizations observed during cholinergic-induced seizures (Lothman et al., 1991). The aim of the present study, therefore, was to identify the ionic mechanisms underlying the cholinergic-induced PP recorded in hippocampal CA1 pyramidal neurons with whole-cell patch-clamp techniques.
These results have been presented in abstract form (Fraser and MacVicar, 1995).
MATERIALS AND METHODS
Hippocampal slice preparation. Sprague–Dawley rats, postnatal 15–23 d, were decapitated and the brains were immersed in chilled artificial CSF (aCSF) containing (in mm): 126 NaCl, 2.5 KCl, 2 MgCl2, 2 CaCl2, 1.25 NaH2PO4, 26 NaHCO−3, and 10 d-glucose, pH 7.3. The hippocampi were isolated and attached to the stage of a vibrating micro-slicer with cyanoacrylate glue. Tissue orientation and stability were ensured by propping the hippocampi against an immobilized block of 2% agar dissolved in physiological saline followed by a light application of low-melt gelatin around the tissue base. Transverse slices (150–400 μm) were prepared by sectioning the hippocampus perpendicular to its septotemporal axis. The slices were then transferred to a glass vial filled with oxygenated aCSF and incubated at room temperature.
Whole-cell patch-clamp recording. Whole-cell current-clamp recordings from neurons within hippocampal slices were obtained using either the “blind-patch” technique (Blanton et al., 1989) or visual guidance (Edwards et al., 1989). Slices were individually transferred to a recording chamber located on an upright microscope (Standard 14; Zeiss, Thornwood, NY) and submerged in rapidly flowing (1 ml/min) oxygenated aCSF. Bath temperature was maintained at 34–35°C with a Peltier unit and Cambion bipolar controller. Individual cells were visualized through a 40×, 0.75 numerical aperture, water-immersion objective (Zeiss). The objective was first modified by filing away the metal shoulder around the lens (Brown and Flaming, 1992). This improved the working space beside the lens, thereby increasing the angle of electrode approach. The objective was then coated with SYLGARD for temperature and electrical isolation. Patch electrodes (5–7 MΩ) were pulled from 1.5 outer diameter thin-walled glass (150F-4, World Precision Instruments) in two stages on a Narishige puller (PP-83; Tokyo, Japan) and filled with intracellular solution (in mm): 140 K-gluconate, 1.1 EGTA, 0.1 CaCl2, 10 HEPES, 2 Mg-ATP, and 0.3 Na-GTP, pH 7.2. Intracellular Ca2+ concentration was calculated to be 16 nm. In some experiments, Cs-gluconate or KCl was substituted for equimolar K-gluconate (see Table 1 for description of intracellular solutions). Voltage recordings were obtained in bridge mode (Axoclamp-2A, Axon Instruments, Foster City, CA) and were low-pass filtered (4-pole Bessel) at 10 kHz (−3 dB). Capacitance neutralization was fully maximized, and the voltage drop across the electrode patch was subtracted by bridge circuit potentiometer. Data were digitized and analyzed using computer software (pClamp or Axotape) via a Tl-1 A/D interface (Axon Instruments). All data are presented as mean ± SE.
Table 1.
Membrane properties of CA1 pyramidal neurons under whole-cell current clamp
| rp(mV) | RIN(MΩ) | APAMP(mV) | APTH(mV) | APDUR (msec) | τ (msec) | n | |
|---|---|---|---|---|---|---|---|
| K-gluconate1_a | −65 ± 0.1 | 149 ± 3 | 92 ± 1 | −47 ± 0.3 | 1.6 ± 0.01 | 20.7 ± 0.4 | 188 |
| Cs-gluconate1_b | −59 ± 0.7 | 183 ± 12 | – | – | – | 23.6 ± 1.8 | 11 |
| KClc | −65 ± 0.4 | 173 ± 24 | 92 ± 5 | −48 ± 1.3 | 1.8 ± 0.06 | 22.8 ± 3.0 | 8 |
| BAPTAd | −67 ± 1.9 | 164 ± 47 | 88 ± 2 | −45 ± 1.4 | 2.4 ± 0.15 | 21.6 ± 1.1 | 4 |
Abbreviations: rp, resting potential;RIN, input resistance;APAMP, action potential amplitude;APTH, action potential threshold;APDUR, action potential duration; τ, tau.
Intracellular solutions (in mm):
140 K-gluconate; 1.1 EGTA; 0.1 CaCl2; 10 HEPES; 2 Mg-ATP; 0.3 Na-GTP.
40 Cs-gluconate; 100 K-gluconate; 1.1 EGTA; 0.1 CaCl2; 10 HEPES; 2 Mg-ATP; 0.3 Na-GTP.
c70 KCl; 70 K-gluconate; 1.1 EGTA; 0.1 CaCl2; 10 HEPES; 2 Mg-ATP; 0.3 Na-GTP.
d100 K-gluconate; 10 K-BAPTA; 40 HEPES; 2 Mg-ATP; 0.3 Na-GTP.
Measurement of electrophysiological properties. Membrane characteristics were determined after patch rupture and sampled throughout the experiment. Series resistance was determined via a bridge potentiometer by balancing the voltage drop across the patch in response to a negative current pulse (−30 pA; 10 msec).RIN was calculated from a steady-state potential response, free of membrane rectification, to a hyperpolarizing current step. The membrane time constant (τ) was calculated as the time necessary to reach 1 − e−1 (63%) of the maximum voltage deflection. Action potential threshold (APTH) was measured as the membrane potential at the base of the action potential, whereas, action potential amplitude (APAMP) was measured as the voltage difference between the threshold and peak amplitude. Action potential duration (APDUR) was measured at the threshold potential.
RESULTS
The results in this paper were obtained from 211 CA1 pyramidal neurons in the hippocampal slice preparation, recorded with whole-cell patch-clamp techniques. The average series resistance was 14.0 ± 0.2 MΩ (range 8–20; n = 211); recordings with series resistance >20 MΩ were discarded. The resting membrane potential, input resistance, membrane time constant, and action potential characteristics were similar to those reported previously (see Table 1) (Stabel et al., 1992; Williams et al., 1994).
Cholinergic stimulation of CA1 pyramidal neurons
Hippocampal CA1 pyramidal neurons were stimulated by bath application of carbachol, a nonhydrolyzable cholinergic agonist. Consistent with previous reports, 20 μmcarbachol depolarized the membrane potential by 3–11 mV (n = 188) and increased RINby 16 ± 1% (range 5–30; 50 neurons sampled). Spike frequency adaptation was reduced, and the action potential afterhyperpolarization was abolished (Fig. 1A). In thicker slices (300–400 μm), an increase in synaptic noise was commonly observed with carbachol application that could be differentiated into discrete excitatory and inhibitory postsynaptic potentials (data not shown) (MacVicar and Tse, 1989; Pitler and Alger, 1992; Behrends and Bruggencate, 1993). These effects were antagonized by coapplication of atropine (n = 7/7), indicating that carbachol acted specifically at muscarinic receptors, as shown previously (Halliwell, 1990). At a concentration of 20 μm, however, carbachol failed to induce rhythmic membrane potential oscillations or theta activity, reported previously for higher concentrations (>50 μm) (Konopacki et al., 1987; MacVicar and Tse, 1989).
Fig. 1.

In the presence of carbachol, a nonhydrolyzable cholinergic agonist, depolarizing current injection resulted in either an sADP or a PP. A, Typical responses of a hippocampal CA1 pyramidal neuron to hyperpolarizing and depolarizing current injection. Neither an sADP nor a PP was observed in the absence of cholinergic agonists. The resting membrane potential of this neuron was −66 mV. After application of 20 μmcarbachol, the membrane potential depolarized to −60 mV, spike frequency adaptation was reduced, and the action potential afterhyperpolarization was abolished. In addition, an sADP(arrow) was now evident after cessation of the current stimulus. After a 20 min wash of carbachol, the membrane potential repolarized to −64 mV, spike frequency adaptation was again observed, and the sADP was absent. At least one component of action potential afterhyperpolarization, however, failed to return after wash of carbachol. B, In a different CA1 pyramidal neuron, 0.8 sec depolarizing current injection resulted in a typical pattern of repetitive action potentials. The membrane potential returned to the baseline level after cessation of the stimuli. In the presence of 20 μm carbachol, however, spike firing evoked by identical stimuli resulted in not only a slow afterdepolarization (sADP; arrow), but also a long-lasting plateau potential (PP; arrow). Notice the low-amplitude oscillations superimposed on the latter portion of the PP. Both the sADP and PP were reversed after carbachol had been washed from the slice for 20 min. Action potentials were truncated by the digitization rate.
Cholinergic-dependent sADP and PP
In the absence of a cholinergic agonist, positive current injection (≥0.1 nA) resulted in repetitive action potential firing in all CA1 pyramidal neurons tested (Fig. 1A,B;n = 196). After cessation of current injection, the membrane potential rapidly returned to resting levels. In contrast, after a 5 min application of 20 μm carbachol, weak current injection (∼0.1 nA; 0.8 sec) resulted in repetitive firing at higher frequency, which was commonly superimposed on a depolarizing ramp. After cessation of evoked action potentials, a depolarizing afterpotential was observed that declined slowly over several seconds (Fig. 1A,B; n = 188/188). This potential was termed an sADP. The average duration and maximal amplitude of the sADP was 6.1 ± 0.2 sec (range 2.8–8.5;n = 50) and 9 ± 1 mV (range 3–20; n = 50), respectively. Injection of stronger current (≥0.2 nA) for the same duration resulted in higher frequency action potential firing superimposed on a steep depolarization that developed into a regenerative PP (Fig. 1B; n = 179/188). The average duration of the PP evoked under the above conditions was 9.9 ± 0.6 sec (range 2.2–21.5; n = 50), whereas the average membrane potential of the PP was −19 ± 1 mV (range −25 to −11;n = 50; measured at 0.2 sec after cessation of the stimuli). Oscillations in the membrane potential were commonly observed superimposed on the PP (Fig. 1B), probably consisting of Na+ and/or Ca2+oscillations well described by others (MacVicar, 1985; Alonso and Llinas, 1989; Leung and Yim, 1991; García-Muñoz et al., 1993). Both the sADP and PP were antagonized by coapplication of 1 μm atropine, again suggesting the specific involvement of muscarinic receptors (Fig. 2;n = 7/7).
Fig. 2.
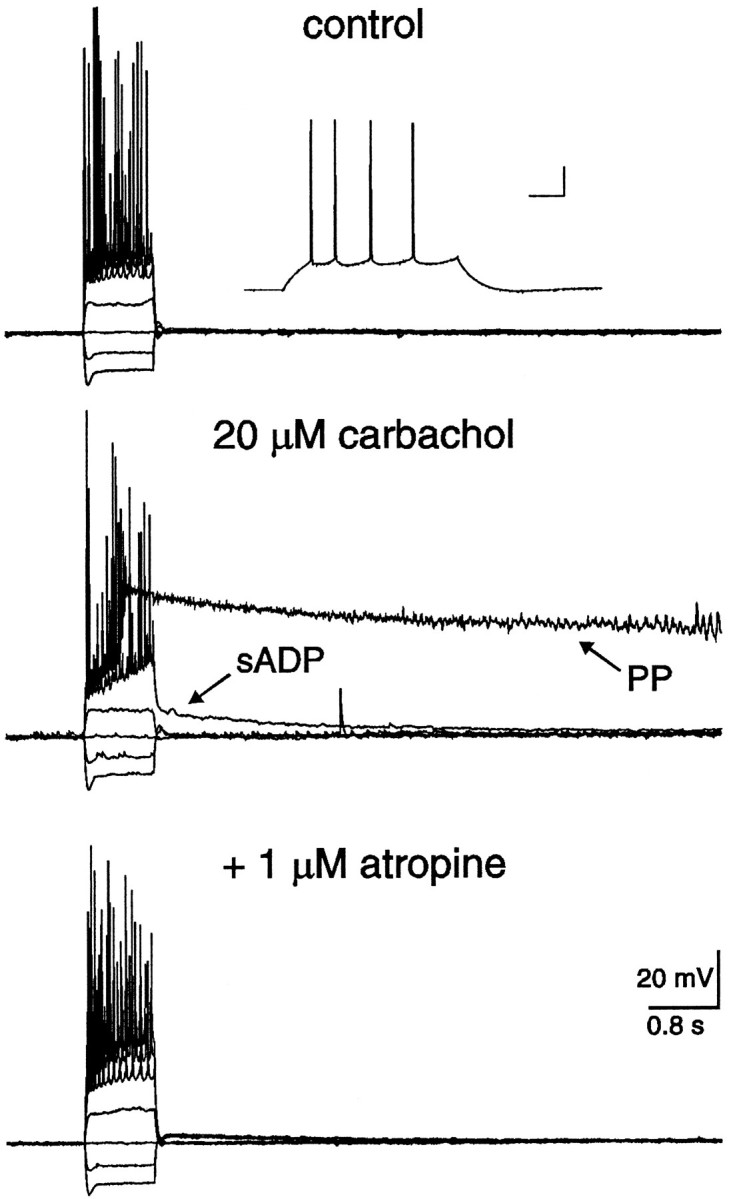
Both the sADP and PP were depressed by coapplication of the muscarinic receptor antagonist atropine. In control, the resting membrane potential of this neuron was −65 mV, and neither the sADP nor the PP was observed after action potential firing. The inset illustrates the typical firing pattern of this CA1 pyramidal neuron. The calibration bars in the insetrepresent 40 msec and 20 mV. In the presence of 20 μm carbachol, the membrane potential depolarized to −61 mV, and both the sADP and PP were observed after the spike firing. Coapplication of 1 μmatropine depressed both the sADP and PP, implicating the involvement of muscarinic receptors.
Each PP consisted of at least four distinct phases (Fig.3A, arrows). The first phase consisted of a steep depolarizing shift that accompanied action potential firing during intracellular current injection. The second phase consisted of the prolonged PP. The third phase, lasting up to several seconds, was a repolarization that commonly hyperpolarized beyond the resting membrane potential. The final phase consisted of this hyperpolarization, which, over a prolonged period of time, returned to the baseline. The data in Figure 3A also demonstrate the reproducibility of the PP waveform in any one cell. In this neuron, five PPs were evoked, separated by 10 min between stimuli.
Fig. 3.

The cholinergic-dependent PP consisted of at least four distinct phases, and the likelihood of generating a PP increased with the number of action potentials elicited. A, In the absence of cholinergic stimulation, the resting membrane potential of the neuron illustrated was −67 mV. Application of 20 μm carbachol depolarized this neuron to −62 mV. In addition, action potential firing evoked by depolarizing current injection (0.1 nA; 0.8 sec) elicited a long-lasting plateau potential in the CA1 pyramidal neuron illustrated. Notice that the spikes are superimposed on a depolarizing ramp that jumped suddenly to a depolarized membrane potential of −20 mV (phase 1;arrow). The PP remained at positive membrane potentials for a sustained period, long after cessation of the current injection (phase 2; arrow). After a prolonged period, the PP repolarized, surpassing the resting potential (phase 3; arrow). Finally, the membrane potential remained at a hyperpolarized potential for a period of ∼19 sec before a gradual return to the resting potential (phase 4; arrow). The PP was evoked five times and digitized on line at five different rates to illustrate not only the distinct phases of the PP, but also the reproducibility of the waveform. Action potentials were truncated by the digitization rate.B, In the presence of 20 μmcarbachol, the likelihood of generating a PP increased with the magnitude of depolarizing current injection. The responses to three amplitudes of current injection (0.1, 0.2, 0.3 nA; 0.8 sec) are illustrated. The resting membrane potential of the neuron before cholinergic stimulation was −68 mV, whereas in carbachol the neuron depolarized to −59 mV. C, The probability of generating a PP in carbachol also increased with the duration of depolarizing current injection. Depolarizing current injection for three durations (0.2, 0.4, 0.6 sec; 0.3 nA) is illustrated. The resting membrane potential of the neuron before cholinergic stimulation was −72 mV, whereas in carbachol the neuron depolarized to −65 mV.
Increasing the number of evoked action potentials enhanced the sADP amplitude until a critical threshold for PP genesis was achieved. For example, gradually increasing the intensity of the injected current resulted in increased spike firing (Fig. 3B). As a result, the amplitude of the sADP increased accordingly until the PP was elicited. Conversely, for a current pulse of a given amplitude, increasing the duration of the stimuli also increased the number of action potentials, thereby enhancing the sADP amplitude (Fig.3C). A critical threshold for PP genesis was also obtained. Even though a clear transition from sADP to PP genesis was observed, the two phenomena may be related because both required cholinergic stimulation and followed a burst of action potentials.
The amplitude of the sADP and the duration of the PP were dependent on the holding potential (Fig. 4). As before, in the absence of a cholinergic agonist, the membrane potential returned to resting levels after a burst of evoked action potentials (Fig.4A). In the presence of carbachol, however, both the sADP and PP were observed after identical stimuli (Fig. 4B). The membrane potential in this case was maintained at −60 mV with positive DC current injection. The sADP and PP were differentially affected when evoked from more negative membrane potentials. For example, the sADP was hardly detectable below −75 mV, and the duration of the PP was reduced with more negative membrane potentials (Fig. 4C–E;n = 6/6). The graph depicts the PP duration versus membrane potential (Fig. 4F; n = 3). The duration was measured from the end of the current pulse to the point at which the PP crossed or reached the prestimuli membrane potential. Notice the nonlinear DC current injection, also depicted in Figure4F (n = 3), required to maintain the membrane at negative potentials. This indicates the activation of a steady-state conductance at these negative membrane potentials; the most likely candidate in this cell type is the hyperpolarizing-activated cation conductance (Halliwell and Adams, 1982; Stabel et al., 1992).
Fig. 4.

The amplitude of the sADP and the duration of the PP decreased with negative membrane potentials. A, Typical response of a hippocampal pyramidal neuron to hyperpolarizing and depolarizing current injection. Neither an sADP nor a PP was observed in the absence of cholinergic stimulation. The resting membrane potential of this pyramidal neuron was −66 mV. B, In the presence of 20 μm carbachol, the neuron depolarized to −63 mV. After depolarizing current injection, both an sADP and a PP were evoked from a membrane potential of −60 mV.C–E, Decreasing the membrane potential with negative DC current injection decreased the amplitude of the sADP. In contrast, the PP was still generated, however, the duration decreased with lowered membrane potentials. F, A plot illustrating the PP duration and negative DC current injection versus membrane potential. The data were tabulated from three neurons.
Ionic dependency of the sADP and PP
The ion(s) mediating the sADP and PP were investigated either by substitution experiments or by including specific channel blockers in the perfusate. In the first experiment, a role for Na+ was investigated by replacing extracellular Na+ with equimolar choline. When extracellular Na+ was reduced from 152 to 26 mm, both the sADP and PP were reversibly depressed (Fig. 5A; n = 6/6). A small amplitude sADP was still observed in low extracellular Na+ (6 ± 1 mV; range 3–10; n = 6), however, the amplitude of the depolarizing current injected was twofold greater. In a similar experiment, both the sADP and PP were reversibly abolished when extracellular Ca2+ was reduced from 2 to 0.1 mm (Fig. 5B;n = 4/4). In this experiment, extracellular Mg2+ was simultaneously increased from 2 to 10 mm to maintain the divalent cation surface charge (Hille, 1984). Finally, a role for Cl− was also investigated by equimolar substitution of 70 mmKCl for K-gluconate in the pipette solution (Table 1; n = 8). Even though the estimated Cl− reversal potential was shifted to −17 mV, characteristic sADPs and PPs were still elicited in carbachol after depolarizing current injection and action potential firing (data not shown; n = 8/8). Hence, Na+ and Ca2+ influx were both critical for genesis of the sADP and PP, whereas the involvement of Cl− was negligible.
Fig. 5.

Both the sADP and PP were dependent on Na+ influx, independent of TTX-sensitive channels and Ca2+ influx via HVA channels. A, In 20 μm carbachol, depolarizing current injection evoked both an sADP and a PP. Lowering external Na+ from 152 to 26 mm reversibly depressed the sADP and abolished the PP, suggesting that Na+ influx was required for these afterpotentials. The resting potentials before and after cholinergic stimulation were −68 and −59 mV, respectively. The calibration bars in the inset represent 40 msec and 20 mV. B, In a different pyramidal neuron, both the sADP and PP were evoked by intracellular current injection. Lowering external Ca2+ from 2 to 0.1 mmreversibly abolished both the sADP and PP. The concentration of Mg2+ was simultaneously increased from 2 to 10 mm to maintain divalent cation charge screening. This finding suggests that Ca2+ influx was also necessary for these potentials. The resting potentials of this neuron before and after cholinergic stimulation were −67 and −60 mV, respectively. The calibration bars in the inset represent 40 msec and 20 mV. C, In 20 μmcarbachol, an sADP and a PP were observed after evoked action potentials. Coapplication of 1.2 μm TTX, a concentration sufficient to block action potentials, failed to depress either the sADP or the PP. Notice that the PP followed a slow regenerative potential, presumably Ca2+-dependent, whereas the sADP did not. Activation of voltage-dependent Na+ channels, therefore, was not required for either of these afterpotentials. The resting membrane potentials before and after carbachol were −65 and −62mV, respectively. D, In carbachol, an sADP and a PP were elicited by intracellular current injection. Coapplication of 100 μm Cd2+ abolished both the sADP and PP. Activation of HVA Ca2+ channels, therefore, was necessary to evoke these potentials. The resting membrane potential in control was −69 mV, whereas 20 μmcarbachol depolarized the membrane potential to −62 mV.
To determine whether voltage-activated Na+channels were involved in sADP and PP genesis, 1.2 μm tetrodotoxin (TTX) was included in the perfusate (Fig. 5C; n = 5). In all neurons, both the sADP and PP were still observed after application of TTX. In the presence of TTX, not only were fast action potentials blocked, but the low-amplitude oscillations observed superimposed on the PP were also abolished (MacVicar, 1985; Alonso and Llinas, 1989; Leung and Yim, 1991). In some neurons, a small amplitude sADP was observed after current injection in the absence of regenerative potentials (Fig.5C; n = 2/5). The average duration and maximal amplitude were 4.8 sec and 4 mV, respectively. The PP, however, was only observed after the depolarizing stimuli activated slow, regenerative potentials that were presumably Ca2+-dependent (Fig. 5C;n = 5). These data clearly eliminate the involvement of TTX-sensitive, voltage-activated Na+ channels in genesis of the sADP and PP. To determine whether HVA Ca2+ channels were required for genesis of the sADP and PP, 100 μmCd2+ was included in the perfusate (Fig.5D). In all neurons (n = 4), both the sADP and PP were blocked by coapplication of Cd2+, suggesting a critical role for HVA Ca2+ channels in genesis of these afterpotentials. In fact, the sADP and PP could not be evoked, even if the amplitude and duration of current injection was increased.
To determine the HVA channel subtypes involved in sADP and PP genesis, blockers of either L- or N-type channels were included in the perfusate (Fig. 6). In the presence of 20 μm carbachol, both a sADP and PP were evoked after intracellular current injection and spike firing. The addition of 10 μm nimodipine, an L-type channel blocker, to the perfusate depressed the sADP and abolished the PP (Fig.6A; n = 4/4). Although an sADP of 11 ± 4 mV (range 6–22; n = 4) was still observed, the amplitude of the depolarizing current had been increased twofold. Similarly, coapplication of 1 μm ω-conotoxin-GVIA, an N-type channel blocker, also depressed the sADP and abolished the PP (Fig. 6B; n = 4/4). As above, an sADP of 10 ± 2 mV (range 6–17; n = 4) was still observed, however, these stimuli were also increased twofold. The N-type channel blocker ω-conotoxin-GVIA was coapplied with 0.1 mg/ml cytochrome C to inhibit nonspecific binding. Cytochrome C, however, had no measurable effect on either the sADP or PP (Fig. 6B; n = 4). Interestingly, neither nimodipine nor ω-conotoxin-GVIA abolished the sADP, suggesting that both L- and N-type channels participate in elevating intracellular Ca2+([Ca2+]i) to a critical level for PP genesis. These data, however, cannot rule out the minor involvement of other HVA Ca2+ channels in sADP and PP genesis.
Fig. 6.

The sADP and PP required Ca2+ influx through L- and N-type Ca2+ channels. A, In a CA1 pyramidal neuron, intracellular current injection revealed an sADP and a PP in the presence of 20 μm carbachol. The resting membrane potentials before and after cholinergic stimulation were −64 and −60 mV, respectively. The sADP was reduced and the PP could not be evoked after coapplication of the L-type channel blocker nimodipine.B, In another pyramidal neuron, both the cholinergic-dependent sADP and PP were also evoked by intracellular current injection. The resting membrane potentials before and after cholinergic stimulation were −66 and −61 mV, respectively. Both the sADP and PP were reversibly depressed by coapplication of the N-type channel blocker ω-conotoxin-GVIA.
Taken together, these data demonstrated that both Na+ and Ca2+ influx was critical for sADP and PP genesis. Moreover, Ca2+influx occurred via HVA Ca2+ channels, predominantly of the L and N subtypes, whereas Na+ influx occurred independently of TTX-sensitive channels.
Ionic mechanisms underlying the sADP and PP
To elucidate whether Ca2+ influx functioned primarily as a charge carrier or as an intracellular messenger, [Ca2+]i was chelated by inclusion of 10 mmbis(2-aminophenoxy)ethane-N,N,N′,N′-tetra-acetic acid (BAPTA) in the pipette solution (Table 1). Application of 20 μm carbachol not only failed to depolarize these BAPTA-containing neurons, but neither the sADP nor the PP were observed after action potential firing evoked by depolarizing current injection (Fig. 7B; n = 4/4). In the same slices, however, both the cholinergic-dependent sADP and PP were evoked in pyramidal neurons dialyzed with control intracellular solution (Fig. 7A; n = 4/4). These data demonstrate that elevations in [Ca2+]i were critical for genesis of the sADP and PP. Hence, the primary role of Ca2+ influx was to serve as an intracellular messenger, not as a charge carrier.
Fig. 7.

Both the sADP and PP were blocked by intracellular BAPTA. A, Typical responses of a hippocampal pyramidal neuron to hyperpolarizing and depolarizing current injection. Neither an sADP nor a PP was observed in the absence of cholinergic stimulation (A1). The calibration bars in the inset represent 40 msec and 20 mV. In the presence of carbachol, however, depolarizing current injection evoked a long-lasting PP (A2). This pyramidal neuron was loaded with pipette solution containing 1.1 mm EGTA and 0.1 mmCa2+ and, thus, intracellular Ca2+ was bufferred to ∼16 nm. B, A different pyramidal neuron, recorded from the same slice as the neuron illustrated in A, loaded with 10 mm BAPTA. In control aCSF, neither an sADP nor a PP was observed (B1). The calibration bars in the inset represent 40 msec and 20 mV. In the presence of carbachol, neither an sADP nor a PP could be evoked, suggesting that elevated [Ca2+]i was necessary for these potentials (B2).
The goal of the next set of experiments was to determine whether the sADP and PP could be evoked by maximizing Ca2+influx via K+ channel suppression. Two methods were used independently to suppress K+ channels. First, external K+ channel blockers [10 mm tetraethylammonium (TEA), 5 mm 4-aminopyridine (4-AP), 100 μm Ba2+; Fig.8A; n = 9] were included in the perfusate, and when applied to pyramidal neurons, depolarized the membrane potential by 9 ± 1 mV (range 5–10; n = 9). Second, 40 mm Cs+ was included in the pipette solution ([Cs+]i; Fig.8B; Table 1; n = 11). Both conditions would independently increase Ca2+ influx, and yet neither the sADP nor the PP could be elicited from a membrane potential maintained at −60 mV with negative DC current injection. Even though K+ channels were suppressed, application of 20 μm carbachol still depolarized the membrane potential by an additional 4 ± 1 mV (range 2–7; n = 17) (Benson et al., 1988; Colino and Halliwell, 1993). In the presence of carbachol, both the sADP and PP were now easily evoked from a membrane potential of −60 mV. These afterpotentials followed regenerative Ca2+ spikes and, as before, were reversibly depressed by reduction of extracellular Na+ from 152 to 26 mm (Fig.8A,B; n = 17/17). A small sADP, however, was still observed in low extracellular Na+ with a maximal amplitude of 5 ± 1 mV (range 3–13; n = 17); however, the amplitude of the depolarizing current injection was twofold greater. Interestingly, the membrane potential of the PP measured at 0.2 sec after cessation of the stimuli was 10 mV more positive when K+ channels were suppressed (−9 ± 2 mV; range −21 to 3; n = 17; vs control −19 ± 1 mV). These experiments demonstrate that Ca2+influx alone was not sufficient for sADP and PP genesis. Moreover, Na+ was the main charge carrier underlying the sADP and PP, whereas deactivation of a K+conductance by maintained Ca2+ influx was excluded as a contributing mechanism (Constanti and Bagetta, 1991;Constanti et al., 1993). Finally, the membrane potential of the PP in the presence of K+ channel blockers (−9 mV) was consistent with activation of a Ca2+-activated nonselective cation conductance with little contribution of concurrent outward conductances.
Fig. 8.

Blockers of K+ channels failed to unmask either the sADP or the PP in the absence of cholinergic stimulation. A, The sADP and PP were not observed after application of 1.2 μm TTX and K+ channel blockers (10 mmTEA, 5 mm 4-AP, 100 μmBa2+). Cholinergic stimulation, however, revealed a PP that was reversibly depressed by lowering Na+ from 152 to 26 mm in the external media (A1). Notice that robust Ca2+ spikes remained in low Na+, whereas the PP was reversibly depressed (A2). B, In addition, neither the sADP nor the PP was observed in pyramidal neurons loaded with 40 mm intracellular Cs+ and bathed in 1.2 μm TTX. As above, cholinergic stimulation revealed a PP that was reversibly depressed by lowering Na+ in the external media (B1). In this example, robust Ca2+ spikes also remained in low Na+, whereas the PP was reversibly depressed (B2).
In the following experiments, the involvement of a Na+/Ca2+ exchanger was tested in two ways (Crépel et al., 1994). First, 50 mm LiCl was substituted for equimolar NaCl in the extracellular solution (Fig. 9A;n = 4). Immediately after ion substitution, the membrane potential depolarized by 9 ± 1 mV (range 6–11 mV;n = 4) corresponding to a Li+-induced depression of the Na+/K+ ATPase (Glynn, 1993). Negative DC current was applied via the recording pipette (−60 ± 9 pA; range −80 to −40 pA; n = 4), however, to maintain the membrane potential at the same presubstitution level. Depolarizing current was then injected and, as described previously, both the sADP and PP were evoked after spike firing in all neurons tested (n = 4/4). Because the Na+/Ca2+ exchanger is also depressed by Li+ (Reuter and Porzig, 1995), this experiment implies that the Na+/Ca2+ exchanger played little, if any, role in genesis of the sADP and PP. An alternative explanation favored by this experiment is the Ca2+-activated nonselective cation channel that is permeable to Li+ (Yellen, 1982). In the second experiment, spike firing elicited by depolarizing current injection evoked both the cholinergic-dependent sADP and PP. The bath temperature was then lowered from 35 to 25°C, and action potential firing was elicited as described previously (Fig. 9B; n = 3). In all cases, a decrease of 10°C did not depress either the sADP or the PP. Because the Na+/Ca2+ exchanger is extremely temperature-sensitive (Kimura and Reeves, 1979; seeCrépel et al., 1994), this second set of experiments also implies that the Na+/Ca2+ exchanger played a negligible role in genesis of the sADP and PP. Interestingly, if the bath temperature was lowered to 21–23°C, the PP could not be elicited (data not shown; n = 3).
Fig. 9.

The sADP and PP were independent of a Na+/Ca2+ exchanger.A, Intracellular current injection revealed an sADP and a PP in the presence of 20 μm carbachol. The resting membrane potentials before and after cholinergic stimulation were −70 and −62 mV, respectively. Both the sADP and PP were still observed after substitution of 50 mmLi+ for equimolar Na+ in the external media. After Li+ substitution, the membrane depolarized an additional 9 mV; however, negative DC current was injected to maintain the membrane potential at the presubstitution value. B, In a different neuron, intracellular current injection also revealed an sADP and a PP in the presence of 20 μm carbachol. The resting membrane potentials before and after cholinergic stimulation were −66 and −61 mV, respectively. Both the sADP and PP were still observed after a reduction in bath temperature from 35 to 25°C. The neuron depolarized after temperature reduction, however, negative DC current was injected to maintain the membrane potential at the same value as at 35°C.
Taken together, these experiments indicate that both the sADP and PP require three events: cholinergic stimulation, an elevation in [Ca2+]i via HVA Ca2+ channels (L- and N-type), and activation of the Ca2+-activated nonselective cation conductance.
Effects of individual K+ channel blockers on the sADP and PP
Although the sADP and PP could not be unmasked by K+ channel suppression, once elicited, these waveforms are likely to be modulated by concurrent outward K+ conductances. The effects of individual K+ channel blockers, therefore, were tested on the sADP and PP to determine whether individual K+ conductances influenced the shape and/or duration of the afterpotential waveforms. In the absence of carbachol, the addition of 10 mm TEA to the perfusate resulted in an initial 5 ± 2 mV (range 2–7 mV; n = 3) depolarization of the membrane potential. When the membrane potential was stable, spike firing elicited by intracellular current injection revealed a brief PP, probably mediated by HVA Ca2+ channels activated by action potential firing (Fig. 10A; n = 3/3) (Bourque et al., 1986). After cessation of the stimuli, neither an sADP nor a PP was observed. In a different pyramidal neuron, both the cholinergic-dependent sADP and PP were easily evoked after action potential firing. Coapplication of 10 mm TEA, however, reduced the amplitude of the sADP and the duration of the PP (Fig. 10A; n = 4/4). The duration of the PP was reduced from 11.6 ± 4.1 sec (range 10–21.4; n = 4) in carbachol to 6.9 ± 2 sec (range 1.3–10.4; n = 4) in the presence of TEA. Furthermore, the membrane potential of the PP measured at 0.2 sec after cessation of the stimuli was increased from −15 ± 3 mV (range −20 to −9; n = 4) in carbachol to −8 ± 3 mV (range −4 to 0) in the presence of TEA. Because TEA effectively increased Ca2+ influx via K+ channel depression, it seems reasonable to conclude that TEA inhibited the Ca2+-activated nonselective cation channel directly (see Crépel et al., 1994).
Fig. 10.

Individual K+ channel blockers modulate the PP waveform. A, Action potential firing in the presence of 10 mm TEA resulted in a brief plateau potential that was presumably Ca2+-dependent (A1). Neither an sADP nor a PP was observed after cessation of the depolarizing stimulus. The calibration bars in the inset represent 40 msec and 20 mV. In a different neuron, both the sADP and PP were observed in the presence of 20 μm carbachol (A2). Coapplication of 10 mm TEA abolished the sADP and decreased the duration of the PP. B, In the presence of 5 mm 4-AP, individual action potentials activated brief afterdepolarizations that presumably were Ca2+-dependent (B1). After cessation of the depolarizing current stimuli, a brief afterpotential was commonly observed. Neither the sADP nor the PP was observed. The calibration bars in the inset represent 40 msec and 20 mV. In another neuron, a PP was evoked in the presence of 20 μm carbachol (B2). Coapplication of 5 mm 4-AP slightly prolonged the PP.C, Application of 100 μmBa2+ to the perfusate resulted in a brief afterpotential and subsequent spike broadening (C1). Brief afterpotentials were also observed after depolarizing current stimuli, however, neither the sADP nor the PP was evoked. In the presence of 20 μm carbachol, a different neuron exhibited an sADP and a PP after cessation of stimuli (C2). Coapplication of 100 μm Ba2+ to the perfusate increased the PP duration.
In separate experiments, the addition of 5 mm4-AP to the perfusate, in the absence of carbachol, resulted in a 3 ± 1 mV (range 1–4 mV; n = 3) depolarization of the membrane potential. This K+ channel blocker also increased Ca2+ influx, but instead of a single PP, which was observed after TEA perfusion, a transient potential followed each action potential (Fig. 10B; n = 3). After a burst of evoked action potentials, only a brief afterpotential was observed in 4-AP. In a different pyramidal neuron, a PP was evoked in the presence of 20 μmcarbachol that was slightly prolonged by coapplication of 4-AP (Fig.10B; n = 3/3). The duration of the PP was 11.3 ± 0.9 sec (range 9.7–12.8; n = 3) in carbachol versus 14.3 ± 1.1 sec (range 11.4–15; n = 3) in 4-AP. In addition, the membrane potential of the PP measured at 0.2 sec after cessation of the stimuli was increased from −20 ± 2 mV (range −22 to −17; n = 3) in carbachol to −14 ± 1 mV (range −17 to −13) in the presence of 4-AP. The small increase in duration may reflect increased Ca2+ influx stimulated by K+ channel suppression.
In the absence of carbachol, the addition of 100 μm Ba2+ to the perfusate resulted in a 4 ± 1 mV (range 2–5; n = 3) depolarization of the membrane potential. This K+channel blocker increased Ca2+ influx as evidenced by increased spike broadening (Fig. 10C;n = 3) (Bourque et al., 1986). At this concentration, however, it is unlikely that Ba2+ permeated the HVA Ca2+ channels, because Ba2+ has a much lower binding affinity within the pore and therefore is repelled out of the channel by Ca2+ (Hess and Tsien, 1984). Even with increased Ca2+ influx, neither an sADP nor a PP was evoked after current injection. In another pyramidal neuron, an sADP and a PP were observed after current injection in the presence of 20 μm carbachol. Coapplication of 100 μm Ba2+ greatly prolonged the PP duration (Fig. 10C; n = 3/3). In carbachol, the average duration of the PP was 8.8 ± 2.1 sec (range 5.9–13.7; n = 3), whereas in Ba2+ the duration increased to 17.2 ± 3.1 sec (range 13.6–22.9; n = 3). In addition, the membrane potential of the PP measured at 0.2 sec after cessation of the stimuli was increased from −17 ± 1 mV (range −20 to −16; n = 3) in carbachol to −12 ± 1 mV (range −14 to −10) in the presence of Ba2+. These data imply that a Ba2+-sensitive conductance contributed to the shape of the PP waveform. A likely candidate expressed in hippocampal pyramidal neurons that opposes prolonged depolarizations and is Ba2+-sensitive is IM(Halliwell and Adams, 1982). Additional K+channels are, however, also suppressed by this divalent cation.
In the absence of carbachol, the simultaneous addition of Ca2+-activated K+ channel blockers [100 nm iberiotoxin (ITX), 100 nm apamin] to the perfusate depolarized the neurons by 3 ± 1 (range 0–4; n = 4) and completely blocked the afterhyperpolarization after each action potential. In the presence of these blockers, however, neither the sADP nor the PP could be evoked (data not shown; n = 4/4). In different pyramidal neurons, both ITX and apamin failed to affect either the sADP or PP observed after carbachol application (n = 3/3). These experiments indicate that Ca2+-activated K+ conductances did not shape either the sADP or PP waveforms.
Taken together, these experiments again illustrate that although Ca2+ influx was increased via K+ channel depression, the sADP and PP required cholinergic stimulation. In addition, TEA may have an inhibitory affect on the Ca2+-activated nonselective cation channel, whereas a Ba2+-sensitive current (e.g.,IM) was important for determining the duration of the PP waveform.
Conductance changes accompanying the PP
In the final experiments, changes inRIN were examined during the PP by injection of low-amplitude current pulses (−50 pA; 0.2 sec; 5 Hz; Fig.11). In all neurons examined, theRIN decreased during the PP by 81 ± 2% (range 71–87; n = 16), measured within 2 sec after cessation of the depolarizing current stimuli (Fig. 11A). In the presence of TTX and K+ channel blockers, theRIN measured during the PP also decreased by 80 ± 3% (range 75–84; n = 3; Fig.11B). Because similar changes inRIN were obtained in the absence and presence of both TTX and K+ channel blockers, the contributions of slow Na+ oscillations and K+ channel activation were minimal in analysis of conductance changes immediately after cessation of stimuli. In all cases, a reproducible pattern was observed, in which theRIN observed immediately after cessation of the depolarizing stimuli gradually diminished during the PP. Even though the RIN increased significantly during the PP, only a minor change in membrane potential was observed (Fig. 11C). Finally, injection of large-amplitude current pulses of long duration (−0.4 nA; 1 sec) failed to terminate the PP (Fig. 11D; n = 4/4).
Fig. 11.

The PP was associated with a largeRIN decrease and could not be terminated by hyperpolarizing pulses. A, Cholinergic-dependent PPs illustrated on two time scales. In the latter, brief current injection (−50 pA; 0.2 sec; 0.5 Hz) demonstrated a large decreasedRIN. B, PPs in the presence of carbachol, TTX, and K+ channel blockers (10 mm TEA, 5 mm 4-AP, 100 μm Ba2+). Under these conditions, brief current injection confirmed anRIN decrease in the absence of slow Na+ oscillations and suppression of K+ channels. C, A graph depicting the decreased RIN relative to control obtained at the resting potential, versus duration of the PP. In the same graph, membrane potential was also plotted versus PP duration. BothRIN and membrane potential values were obtained from the neuron illustrated in B (*). Notice that the change in membrane potential was minimal, whereas the increase inRIN was significant. D, Two superimposed PPs evoked from a CA1 pyramidal neuron. In the broken trace, hyperpolarizing current injection (−0.4 nA; 0.8 sec) failed to terminate the PP.
DISCUSSION
In this study, we report a cholinergic-induced sADP and PP recorded from hippocampal CA1 pyramidal neurons with the whole-cell patch-clamp technique. We propose that the PP represents a novel regenerative event involving the interplay between HVA Ca2+ channels (L- and N-type) and the Ca2+-activated nonselective cation conductance. These ionic mechanisms may contribute significantly to ictal depolarizations observed during temporal-lobe seizures of the partial-complex subtype (Lothman et al., 1991).
Both the sADP and PP were depressed by removal of either Na+ or Ca2+ from the external media, and blocked by either extracellular Cd2+ or chelation of [Ca2+]i by inclusion of BAPTA in the intracellular solution. Moreover, the sADP was depressed and the PP was abolished by either the L-type channel blocker, nimodipine, or the N-type channel blocker ω-conotoxin-GVIA. Superfusion of TTX at a concentration sufficient to block fast action potentials, however, did not suppress either the sADP or PP. These data imply that both the sADP and PP rely on elevations in [Ca2+]i via HVA Ca2+ channels (L- and N-type) and Na+ influx independent of TTX-sensitive channels.
One possible mechanism contributing to the sADP and PP described here is the Na+/Ca2+ exchanger (Friedman et al., 1992). Our experiments, however, do not support a role for this mechanism. First, equimolar substitution of extracellular Na+ with 50 mmLi+ did not affect either the sADP or the PP. The Na+/Ca2+ exchanger does not use Li+ as a charge carrier (Kimura et al., 1987;Reuter and Porzig, 1995); however, the Ca2+-activated nonselective cation channel is permeable to Li+ (Yellen, 1982). Second, lowering the temperature from 35 to 25°C also did not affect either the sADP or the PP (see Crépel et al., 1994). Because the Na+/Ca2+ exchanger is highly temperature-sensitive (Kimura and Reeves, 1979), this latter experiment also suggests that this mechanism plays a negligible role in sADP and PP genesis. Another possible mechanism that may have contributed to the sADP and PP is deactivation of a K+ conductance by maintained Ca2+ influx (Constanti and Bagetta, 1991;Constanti et al., 1993). This mechanism was also ruled out by our experiments because both the sADP and the PP were observed in the presence of either extracellular K+ channel blockers or [Cs+]i. Furthermore, an increased conductance was observed during both the sADP and PP, which is not consistent with this mechanism (Constanti and Bagetta, 1991). Based on our experiments, a Ca2+-activated nonselective cation conductance is the prime candidate for sADP genesis (Schwindt et al., 1988; Hasuo et al., 1990; Caeser et al., 1993). The PP, however, relied not only on the Ca2+-activated nonselective cation conductance, but also on prolonged activation of HVA Ca2+ channels (Blitzer et al., 1991). After Ca2+ was elevated to a critical level by spike firing, the Ca2+-activated nonselective cation channel was sufficiently activated to depolarize the membrane potential positive enough to sustain Ca2+ influx, and the regenerative potential was initiated. Hence, the sADP represents a weak depolarization mediated by residual [Ca2+]i, whereas the PP represents a novel regenerative event relying on continual activation of both HVA Ca2+ channels and the Ca2+-activated nonselective cation conductance.
A major question is whether the Ca2+-activated nonselective cation conductance was directly enhanced by carbachol or simply unmasked by cholinergic-induced suppression of K+ conductances (Caesar et al., 1993). Our data indicate that the Ca2+-activated nonselective cation conductance was directly enhanced by cholinergic stimulation. In the presence of either K+ channel blockers or [Cs+]i, neither the sADP nor the PP was observed. The sADP and PP were only elicited after carbachol was added to the perfusate. Because HVA Ca2+ channels are depressed by cholinergic stimulation (Toselli and Lux, 1989), the Ca2+-activated nonselective cation channel must have been augmented by carbachol. Cholinergic-induced depression of K+ channels would, however, facilitate genesis of the sADP and PP by increasing the net inward current. Moreover, because neither the sADP nor the PP was observed with K+channel depression, the Ca2+-activated nonselective cation channel must have a low open-channel probability in the absence of cholinergic stimulation.
Previous studies have shown that PPs can be generated by either voltage-activated Ca2+ or Na+ channels. PPs generated by Ca2+ channels alone, however, are observed only when K+ channel blockers are present, intracellular Ca2+ is chelated, and/or Ba2+ ions are used as the charge carriers (Llinas and Yarom, 1981; Bourque et al., 1986). PPs generated solely by voltage-activated Ca2+ channels are depressed by muscarinic agonists (Misgeld et al., 1986). This effect is consistent with voltage-clamp studies demonstrating cholinergic-induced depression of HVA Ca2+ channels (Toselli and Lux, 1989). Alternatively, fast-inactivating Na+ channels may gate in a bimodal manner, whereby bursts of channel openings are prolonged (Taylor, 1993). In hippocampal pyramidal neurons, the role of persistent Na+ channel openings in generating PPs is unclear, because these events have been observed only in 0 Ca2+ external media (Leung and Yim, 1991;García-Muñoz et al., 1993; Segal, 1994). PPs generated by these Na+ channels are identified by their sensitivity to the specific channel blocker TTX. In contrast to the above mechanisms, the sADP and PP described here were evoked in physiological solutions, requiring only cholinergic stimulation. The expression of PPs in intact tissue, therefore, could occur as a result of burst firing and activation of the cholinergic system.
The Ca2+-activated nonselective cation conductance implicated in this study is present in a broad range of cell types (Partridge and Swandulla, 1988) and is distinct from voltage-gated cation channels. For example, voltage-gated cation channels have been described in a variety of neuronal cell types (Hoehn et al., 1993; Alzheimer, 1994) and participate in afterpotential genesis after spike repolarization (Penington and Kelly, 1993). These depolarizing afterpotentials are not sensitive to [Ca2+]i, are only milliseconds in duration, and mediate burst firing at rapid frequencies (Deisz, 1996). Hence, the depolarizing afterpotentials mediated by voltage-activated cation channels are clearly distinguishable from the sADP and PP described in this paper.
Based on our findings, the sADP represents an afterpotential due to the Ca2+-activated nonselective cation conductance activated by Ca2+ influx via HVA channels. The PP, however, represents a novel regenerative event involving the interplay between HVA Ca2+ channels and the Ca2+-activated nonselective cation conductance. This interplay between channels may occur at a distal dendritic site because both a Ca2+-sensitive inward current (Benson et al., 1988; Colino and Halliwell, 1993) and HVA Ca2+ channels (L-type, Westenbroek et al., 1990; Magee and Johnston, 1995; N-type, Jones et al., 1989; Mills et al., 1994) have been localized to dendritic processes. Dendritically located currents may be one reason why the PP has not been reported previously; for example, the PP may have been masked by a large somatic current shunt produced by microelectrode impalement (Spruston and Johnston, 1992; Staley et al., 1992). In addition, many studies using whole-cell patch-clamp techniques have either chelated [Ca2+]i or worked at room temperature, where the PP was not observed. Under our experimental conditions, the ability to evoke the sADP and PP was both reliable and reproducible.
The sADP and PP described here involved intrinsic mechanisms directly modulated by cholinergic stimulation. These included enhancement of the Ca2+-activated nonselective cation conductance and depression of several K+ conductances. It is unlikely that other neurotransmitters (e.g., glutamate) or synaptic interactions played a significant role in this study because both the sADP and PP were observed in TTX. Moreover, thin slices (150 μm) were commonly used where synaptic connections and recurrent collaterals were minimal. Although other neurotransmitters and synaptic interactions were presumably not involved in sADP and PP genesis in this study, they likely play a significant physiological role. For example, previous work in cortical neurons has demonstrated a cholinergic-sensitive slow inward conductance potentiated by low-frequency synaptic stimulation of white matter (Andrade, 1991). It is therefore conceivable that a PP could also be evoked during synaptic stimulation. For instance, cholinergic stimulation elevates intradendritic Ca2+ accumulation in hippocampal pyramidal neurons during repetitive firing (Müller and Connor, 1991) and enhances Ca2+ influx via postsynaptic NMDA receptors (Markram and Segal, 1992; Segal, 1992). This elevation in dendritic [Ca2+]i could activate the Ca2+-activated nonselective cation conductance. Depolarization of the membrane would then activate HVA Ca2+ channels, and a regenerative PP could be initiated. Similar mechanisms may underlie long-duration rhythmic bursts observed after either cholinergic or metabotropic stimulation of hippocampal slices (Bianchi and Wong, 1994, 1995). In our hands, stimulation of metabotropic glutamate receptors withtrans-(I)-1-amino-1,3-cyclopentanedicarboxylic acid also resulted in sADP and PP generation (our unpublished observations). Interestingly, both cholinergic and metabotropic glutamate receptors are linked to phospholipase C via G-proteins, suggesting a common final pathway.
Functional significance
Cholinergic agents are effective in generating hippocampal seizures both in vivo and in vitro (Lothman et al., 1991; Wasterlain et al., 1993). In the hippocampal slice preparation, these same analogs generate prolonged depolarizations (Bianchi and Wong, 1994) and exacerbate ictal depolarizations observed during high K+-induced seizures (Yaari and Jensen, 1989). In this paper, we have identified a novel regenerative PP involving the interplay between HVA Ca2+channels and the Ca2+-activated nonselective cation conductance, and have also presented evidence that this latter conductance was directly enhanced by cholinergic stimulation. Because this PP has properties reminiscent of ictal depolarizations observed during cholinergic-induced seizures, it seems reasonable to conclude that these mechanisms could play a central role in hippocampal epileptogenesis.
Footnotes
This work was supported by a grant from the Medical Research Council of Canada (MRC). D.D.F. is a recipient of studentships from the MRC, Alberta Heritage Foundation for Medical Research (AHFMR), and Savoy Foundation for Epilepsy Research. B.A.M. is an AHFMR scientist and the Ciba Geigy chair for schizophrenia research. We are grateful to Drs. S. Barnes and G. Spencer for critical comments on this manuscript and Drs. A. R. McQuiston and W. F. Colmers for slice-patch advice.
Correspondence should be addressed to Douglas D. Fraser, Department of Neuroscience, 3330 Hospital Drive N.W., Health Sciences Building, University of Calgary, Alberta, Canada, T2N 4N1.
REFERENCES
- 1.Alonso A, Llinas RR. Subthreshold Na+-dependent theta-like rhythmicity in stellate cells of entorhinal cortex layer II. Nature. 1989;342:175–177. doi: 10.1038/342175a0. [DOI] [PubMed] [Google Scholar]
- 2.Alzheimer C. A novel voltage-dependent cation current in rat neocortical neurones. J Physiol (Lond) 1994;479:199–205. doi: 10.1113/jphysiol.1994.sp020288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Andrade R. Cell excitation enhances muscarinic cholinergic responses in rat association cortex. Brain Res. 1991;548:81–93. doi: 10.1016/0006-8993(91)91109-e. [DOI] [PubMed] [Google Scholar]
- 4.Behrends JC, Bruggencate G. Cholinergic modulation of synaptic inhibition in the guinea pig hippocampus in vitro: excitation of GABAergic interneurons and inhibition of GABA-release. J Neurophysiol. 1993;69:626–629. doi: 10.1152/jn.1993.69.2.626. [DOI] [PubMed] [Google Scholar]
- 5.Benson DM, Blitzer RD, Landau EM. An analysis of the depolarization produced in guinea-pig hippocampus by cholinergic receptor stimulation. J Physiol (Lond) 1988;404:479–496. doi: 10.1113/jphysiol.1988.sp017301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bernardo LS, Prince DA. Cholinergic excitation of mammalian hippocampal pyramidal cells. Brain Res. 1982;249:315–331. doi: 10.1016/0006-8993(82)90066-x. [DOI] [PubMed] [Google Scholar]
- 7.Bianchi R, Wong RKS. Carbachol-induced synchronized rhythmic bursts in CA3 neurons of guinea pig hippocampus in vitro. J Neurophysiol. 1994;72:131–138. doi: 10.1152/jn.1994.72.1.131. [DOI] [PubMed] [Google Scholar]
- 8.Bianchi R, Wong RKS. Excitatory synaptic potentials dependent on metabotropic glutamate receptor activation in guinea pig hippocampal pyramidal neurons. J Physiol (Lond) 1995;487:663–676. doi: 10.1113/jphysiol.1995.sp020908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bland BH. The physiology and pharmacology of hippocampal formation theta rhythms. Prog Neurobiol. 1986;26:1–54. doi: 10.1016/0301-0082(86)90019-5. [DOI] [PubMed] [Google Scholar]
- 10.Blanton MG, Lo Turco JJ, Kriegstein AR. Whole cell recording from neurons in slices of reptilian and mammalian cerebral cortex. J Neurosci Methods. 1989;30:203–210. doi: 10.1016/0165-0270(89)90131-3. [DOI] [PubMed] [Google Scholar]
- 11.Blitzer RD, Gil O, Omri G, Landau E. Nifedipine blocks calcium-dependent cholinergic depolarization in the guinea pig hippocampus. Brain Res. 1991;542:293–299. doi: 10.1016/0006-8993(91)91581-k. [DOI] [PubMed] [Google Scholar]
- 12.Bourque CW, Brown DA, Renaud LP. Barium ions induce prolonged plateau depolarizations in neurosecretory neurones of the adult rat supraoptic nucleus. J Physiol (Lond) 1986;375:573–586. doi: 10.1113/jphysiol.1986.sp016134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Brown KT, Flaming DG. Wiley; New York: 1992. Advanced micropipette techniques for cell physiology, pp 176–203. . [Google Scholar]
- 14.Caeser M, Brown DA, Gähwiler BH, Knöpfel T. Characterization of a calcium-dependent current generating a slow afterdepolarization of CA3 pyramidal cells in rat hippocampal slice cultures. Eur J Neurosci. 1993;5:560–569. doi: 10.1111/j.1460-9568.1993.tb00521.x. [DOI] [PubMed] [Google Scholar]
- 15.Colino A, Halliwell JV. Carbachol potentiates Q current and activates a calcium-dependent non-specific conductance in rat hippocampus in vitro. Eur J Neurosci. 1993;5:1198–1209. doi: 10.1111/j.1460-9568.1993.tb00974.x. [DOI] [PubMed] [Google Scholar]
- 16.Constanti A, Bagetta G. Muscarinic receptor activation induces a prolonged post-stimulus afterdepolarization with a conductance decrease in guinea-pig olfactory cortex neurones in vitro. Neurosci Lett. 1991;131:27–32. doi: 10.1016/0304-3940(91)90329-r. [DOI] [PubMed] [Google Scholar]
- 17.Constanti A, Bagetta G, Libri V. Persistent muscarinic excitation in guinea pig olfactory cortex neurons: involvement of a slow post-stimulus afterdepolarizing current. Neuroscience. 1993;56:887–904. doi: 10.1016/0306-4522(93)90135-3. [DOI] [PubMed] [Google Scholar]
- 18.Crépel V, Aniksztejn L, Ben-Ari Y, Hammond C. Glutamate metabotropic receptors increase a Ca2+-activated nonspecific cationic current in CA1 hippocampal neurons. J Neurophysiol. 1994;72:1561–1569. doi: 10.1152/jn.1994.72.4.1561. [DOI] [PubMed] [Google Scholar]
- 19.Deisz RA. A tetrodotoxin-insensitive sodium current initiates burst firing of neocortical neurons. Neuroscience. 1996;70:341–351. doi: 10.1016/0306-4522(95)00362-2. [DOI] [PubMed] [Google Scholar]
- 20.Edwards FA, Konnerth A, Sakmann B, Takahashi T. A thin slice preparation for patch clamp recordings from neurons of the mammalian central nervous system. Pflügers Arch. 1989;414:600–612. doi: 10.1007/BF00580998. [DOI] [PubMed] [Google Scholar]
- 21.Fraser DD, MacVicar BA. Low-threshold transient Ca2+ current in lacunosum-moleculare interneurons: kinetics and modulation by neurotransmitters. J Neurosci. 1991;11:2812–2820. doi: 10.1523/JNEUROSCI.11-09-02812.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fraser DD, MacVicar BA. Ionic mechanisms underlying the cholinergic-dependent plateau potentials in hippocampal CA1 pyramidal neurons. Soc Neurosci Abstr. 1995;25:32. doi: 10.1523/JNEUROSCI.16-13-04113.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Friedman A, Arens J, Heinemann U, Gutnick MJ. Slow depolarizing afterpotentials in neocortical neurons are sodium and calcium dependent. Neurosci Lett. 1992;135:13–17. doi: 10.1016/0304-3940(92)90125-q. [DOI] [PubMed] [Google Scholar]
- 24.Gähwiler BH. Facilitation by acetylcholine of tetrodotoxin-resistant spikes in rat hippocampal pyramidal cells. Neuroscience. 1984;11:381–388. doi: 10.1016/0306-4522(84)90030-7. [DOI] [PubMed] [Google Scholar]
- 25.García-Muñoz A, Barrio LC, Buno W. Membrane potential oscillations in CA1 hippocampal pyramidal neurons in vitro: intrinsic rhythms and fluctuations entrained by sinusoidal injected current. Exp Brain Res. 1993;97:325–333. doi: 10.1007/BF00228702. [DOI] [PubMed] [Google Scholar]
- 26.Glynn IM. All hands to sodium pump. J Physiol (Lond) 1993;462:1–30. doi: 10.1113/jphysiol.1993.sp019540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Halliwell JV. Physiological mechanisms of cholinergic action in the hippocampus. Prog Brain Res. 1990;84:255–272. doi: 10.1016/s0079-6123(08)60910-3. [DOI] [PubMed] [Google Scholar]
- 28.Halliwell JV, Adams PR. Voltage-clamp analysis of muscarinic excitation in hippocampal neurons. Brain Res. 1982;250:71–92. doi: 10.1016/0006-8993(82)90954-4. [DOI] [PubMed] [Google Scholar]
- 29.Hasuo H, Gallagher JP. Facilitatory action of muscarine on the slow afterdepolarization of rat dorsolateral septal nucleus neurons in vitro. Neurosci Lett. 1990;112:234–238. doi: 10.1016/0304-3940(90)90209-r. [DOI] [PubMed] [Google Scholar]
- 30.Hasuo H, Phelan KD, Twery MJ, Gallagher JP. A calcium-dependent afterdepolarization recorded in rat dorsolateral septal nucleus neurons in vitro. J Neurophysiol. 1990;64:1838–1846. doi: 10.1152/jn.1990.64.6.1838. [DOI] [PubMed] [Google Scholar]
- 31.Hess P, Tsien RW. Mechanism of ion permeation through calcium channels. Nature. 1984;309:453–456. doi: 10.1038/309453a0. [DOI] [PubMed] [Google Scholar]
- 32.Hille BH. Sinauer; Sunderland, MA: 1984. Ionic channels of excitable membranes. [Google Scholar]
- 33.Hoehn K, Watson TWJ, MacVicar BA. A novel tetrodotoxin-insensitive, slow sodium current in striatal and hippocampal neurons. Neuron. 1993;10:543–552. doi: 10.1016/0896-6273(93)90341-n. [DOI] [PubMed] [Google Scholar]
- 34.Jones OT, Kunze DL, Angelides KJ. Localization and mobility of ω-conotoxin-sensitive Ca2+ channels in hippocampal CA1 neurons. Science. 1989;244:1189–1193. doi: 10.1126/science.2543080. [DOI] [PubMed] [Google Scholar]
- 35.Kimura JE, Reeves H. The effect of temperature on the asymmetrical charge movement in squid giant axons. J Physiol (Lond) 1979;289:479–500. doi: 10.1113/jphysiol.1979.sp012748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kimura J, Miyamae S, Noma A. Identification of a sodium-calcium exchange current in single ventricular cells of guinea-pig. J Physiol (Lond) 1987;384:199–222. doi: 10.1113/jphysiol.1987.sp016450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Konopacki J, Bland BH, MacIver MB, Roth SH. Cholinergic theta rhythm in transected hippocampal slices: independent CA1 and dentate generators. Brain Res. 1987;417:399–407. doi: 10.1016/0006-8993(87)91664-7. [DOI] [PubMed] [Google Scholar]
- 38.Krnjević K. Central cholinergic mechanisms and function. Prog Brain Res. 1993;98:285–292. doi: 10.1016/s0079-6123(08)62410-3. [DOI] [PubMed] [Google Scholar]
- 39.Leung LS, Yim CYC. Intrinsic membrane potential oscillations in hippocampal neurons in vitro. Brain Res. 1991;553:261–274. doi: 10.1016/0006-8993(91)90834-i. [DOI] [PubMed] [Google Scholar]
- 40.Llinas R, Yarom Y. Properties and distribution of ionic conductances generating electroresponsiveness of mammalian inferior olivary neurones in vitro. J Physiol (Lond) 1981;315:569–584. doi: 10.1113/jphysiol.1981.sp013764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lothman EW, Bertram EH, Stringer JL. Functional anatomy of hippocampal seizures. Prog Neurobiol. 1991;37:1–82. doi: 10.1016/0301-0082(91)90011-o. [DOI] [PubMed] [Google Scholar]
- 42.MacVicar BA. Depolarizing prepotentials are Na+ dependent in CA1 pyramidal neurons. Brain Res. 1985;333:378–381. doi: 10.1016/0006-8993(85)91597-5. [DOI] [PubMed] [Google Scholar]
- 43.MacVicar BA, Tse FWY. Local neuronal circuitry underlying cholinergic rhythmical slow activity in CA3 area of rat hippocampal slices. J Physiol (Lond) 1989;417:197–212. doi: 10.1113/jphysiol.1989.sp017797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Madison DV, Malenka RC, Nicoll RA. Voltage clamp analysis of cholinergic action in the hippocampus. J Neurosci. 1987;7:733–741. doi: 10.1523/JNEUROSCI.07-03-00733.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Magee JC, Johnston D. Synaptic activation of voltage-gated channels in the dendrites of hippocampal pyramidal neurons. Science. 1995;268:301–304. doi: 10.1126/science.7716525. [DOI] [PubMed] [Google Scholar]
- 46.Markram H, Segal M. The inositol 1,4,5-triphosphate pathway mediates cholinergic potentiation of rat hippocampal neuronal responses to NMDA. J Physiol (Lond) 1992;447:513–533. doi: 10.1113/jphysiol.1992.sp019015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCormick DA. Actions of acetylcholine in the cerebral cortex and thalamus and implications for function. Prog Brain Res. 1993;98:303–308. doi: 10.1016/s0079-6123(08)62412-7. [DOI] [PubMed] [Google Scholar]
- 48.McCormick DA, Prince DA. Mechanisms of action of acetylcholine in the guinea-pig cerebral cortex in vitro. J Physiol (Lond) 1986;375:169–194. doi: 10.1113/jphysiol.1986.sp016112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Mills LR, Niesen CE, So AP, Carlen PL, Spigelman I, Jones OT. N-type Ca2+ channels are located on somata, dendrites, and a subpopulation of dendritic spines on live hippocampal pyramidal neurons. J Neurosci. 1994;14:6815–6824. doi: 10.1523/JNEUROSCI.14-11-06815.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Misgeld U, Calabresi P, Dodt HU. Muscarinic modulation of calcium dependent plateau potentials in rat neostriatal neurons. Pflügers Arch. 1986;407:482–487. doi: 10.1007/BF00657504. [DOI] [PubMed] [Google Scholar]
- 51.Müller W, Connor JA. Cholinergic input uncouples Ca2+ changes from K+conductance activation and amplifies intradendritic Ca2+ changes in hippocampal neurons. Neuron. 1991;6:901–905. doi: 10.1016/0896-6273(91)90230-w. [DOI] [PubMed] [Google Scholar]
- 52.Müller W, Misgeld U, Heinemann U. Carbachol effects on hippocampal neurons in vitro: dependence on the rate of rise of carbachol tissue concentration. Exp Brain Res. 1988;72:287–298. doi: 10.1007/BF00250251. [DOI] [PubMed] [Google Scholar]
- 53.Nakajima Y, Nakajima S, Leonard RJ, Yamaguchi K. Acetylcholine raises excitability by inhibiting the fast transient potassium current in cultured hippocampal neurons. Proc Natl Acad Sci USA. 1986;83:3022–3026. doi: 10.1073/pnas.83.9.3022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Partridge LD, Swandulla D. Ca2+-activated non-specific cation channels. Trends Neurosci. 1988;11:69–72. doi: 10.1016/0166-2236(88)90167-1. [DOI] [PubMed] [Google Scholar]
- 55.Penington NJ, Kelly JS. Ionic dependence of a slow inward tail current in rat dorsal raphe neurones. J Physiol (Lond) 1993;464:33–48. doi: 10.1113/jphysiol.1993.sp019623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pitler TA, Alger BE. Cholinergic excitation of GABAergic interneurons in the rat hippocampal slices. J Physiol (Lond) 1992;450:127–142. doi: 10.1113/jphysiol.1992.sp019119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Reuter H, Porzig H. Localization and functional significance of the Na+/Ca2+ exchanger in presynaptic boutons of hippocampal cells in culture. Neuron. 1995;15:1077–1084. doi: 10.1016/0896-6273(95)90096-9. [DOI] [PubMed] [Google Scholar]
- 58.Schwindt PC, Spain WJ, Foehring RC, Chubb MC, Crill WE. Slow conductances in neurons from cat sensorimotor cortex in vitro and their role in slow excitability changes. J Neurophysiol. 1988;59:450–467. doi: 10.1152/jn.1988.59.2.450. [DOI] [PubMed] [Google Scholar]
- 59.Segal M. Acetylcholine enhances NMDA-evoked calcium rise in hippocampal neurons. Brain Res. 1992;587:83–87. doi: 10.1016/0006-8993(92)91430-m. [DOI] [PubMed] [Google Scholar]
- 60.Segal MM. Endogenous bursts underlie seizurelike activity in solitary excitatory hippocampal neurons in microcultures. J Neurophysiol. 1994;72:1874–1884. doi: 10.1152/jn.1994.72.4.1874. [DOI] [PubMed] [Google Scholar]
- 61.Shen KZ, North RA. Muscarine increases cation conductance and decreases potassium conductance in rat locus coeruleus neurones. J Physiol (Lond) 1992;455:471–485. doi: 10.1113/jphysiol.1992.sp019312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shen Y, Specht SM, De Saint Ghislain I, Li R. The hippocampus: a biological model for studying learning and memory. Prog Neurobiol. 1994;44:485–496. doi: 10.1016/0301-0082(94)90008-6. [DOI] [PubMed] [Google Scholar]
- 63.Spruston N, Johnston D. Perforated patch-clamp analysis of the passive membrane properties of three classes of hippocampal neurons. J Neurophysiol. 1992;67:508–529. doi: 10.1152/jn.1992.67.3.508. [DOI] [PubMed] [Google Scholar]
- 64.Stabel J, Ficker E, Heinemann U. Young CA1 pyramidal cells of rats, but not dentate gyrus granule cells, express a delayed inward rectifying current with properties of IQ. Neurosci Lett. 1992;135:231–234. doi: 10.1016/0304-3940(92)90443-b. [DOI] [PubMed] [Google Scholar]
- 65.Staley KJ, Otis TS, Mody I. Membrane properties of dentate gyrus granule cells: comparison of sharp microelectrodes and whole-cell recordings. J Neurophysiol. 1992;67:1346–1358. doi: 10.1152/jn.1992.67.5.1346. [DOI] [PubMed] [Google Scholar]
- 66.Taylor CP. Na+ currents that fail to inactivate. Trends Neurosci. 1993;16:455–460. doi: 10.1016/0166-2236(93)90077-y. [DOI] [PubMed] [Google Scholar]
- 67.Toselli M, Lux HD. Opposing effects of acetylcholine on the two classes of voltage-dependent calcium channels in hippocampal neurons. In: Frotscher M, Misgeld U, editors. Central cholinergic synaptic transmission. Birkhäuser; Basel: 1989. pp. 97–103. [DOI] [PubMed] [Google Scholar]
- 68.Vanderwolf CH. Cerebral activity and behavior: control by central cholinergic and serotonergic systems. Int Rev Neurobiol. 1988;30:225–340. doi: 10.1016/s0074-7742(08)60050-1. [DOI] [PubMed] [Google Scholar]
- 69.Wainer BH, Steininger TL, Roback JD, Burke-Watson MA, Mufson EJ, Kordower J. Ascending cholinergic pathways: functional organization and implications for disease models. Prog Brain Res. 1993;98:9–30. doi: 10.1016/s0079-6123(08)62378-x. [DOI] [PubMed] [Google Scholar]
- 70.Wasterlain CG, Fujikawa DG, Penix L, Sankar R. Pathophysiological mechanisms of brain damage from status epilepticus. Epilepsia. 1993;34:S37–S53. doi: 10.1111/j.1528-1157.1993.tb05905.x. [DOI] [PubMed] [Google Scholar]
- 71.Westenbroek RE, Ahlijanian MK, Catterall WA. Clustering of L-type Ca2+ channels at the base of major dendrites in hippocampal pyramidal neurons. Nature. 1990;347:281–284. doi: 10.1038/347281a0. [DOI] [PubMed] [Google Scholar]
- 72.Williams S, Samulack DD, Beaulieu C, Lacaille JC. Membrane properties and synaptic responses of interneurons located near the stratum lacunosum-moleculare/radiatum border of area CA1 in whole-cell recordings from rat hippocampal slices. J Neurophysiol. 1994;71:2217–2235. doi: 10.1152/jn.1994.71.6.2217. [DOI] [PubMed] [Google Scholar]
- 73.Yaari Y, Jensen MS. Cholinergic modulation of hippocampal epileptic activity in vitro. In: Frotscher M, Misgeld U, editors. Central cholinergic synaptic transmission. Birkhäuser; Basel: 1989. pp. 150–158. [DOI] [PubMed] [Google Scholar]
- 74.Yellen G. Single Ca2+-activated non-selective cation channels in neuroblastoma. Nature. 1982;296:357–359. doi: 10.1038/296357a0. [DOI] [PubMed] [Google Scholar]