

Abstract
Transcriptome analysis revealed a strong positive correlation between human SLFN11 expression and the sensitivity of tumor cells to DNA damaging agents (DDAs). We show here that SLFN11 preferentially inhibits translation of ATR or ATM upon DDAs treatment based on distinct codon usage without disrupting early DNA damage response signaling. Type II tRNAs, which include all serine and leucine tRNAs, are cleaved in a SLFN11-dependent manner in response to DDAs. mRNAs encoded by genes with high TTA (Leu) codon usage such as ATR display utmost susceptibility to translational suppression by SLFN11. Specific attenuation of tRNA-Leu-TAA sufficed to ablate ATR protein expression and restore DDA sensitivity of SLFN11-deficient cells. Our study uncovered a novel mechanism of codon-specific translational inhibition via SLFN11-dependent tRNA cleavage in the DNA damage response, and supports the notion that SLFN11-deficient tumor cells can be resensitized to DDAs by targeting ATR or tRNA-Leu-TAA.
Introduction
DNA-damaging agents (DDAs) represent the largest group of cancer drugs, but primary or secondary resistance severely limits their effectiveness. Two large-scale transcriptome analyses in cancer cells revealed that human Schlafen 11 (SLFN11) – a protein we previously found to inhibit translation of HIV proteins due to atypical codon-usage in the viral RNA1 – sensitizes cancer cells to DDAs2,3. SLFN11 belongs to the gene family of Schlafen (SLFN), which are exclusively found in mammals with the notable exception of orthopoxviruses4. SLFNs share no significant homology with other proteins beyond an N-terminal divergent AAA (ATPases associated with various cellular activities) domain, and in the case of the longer family members such as SLFN11, a putative C-terminal DNA-RNA helicase domain 5. Based on our knowledge gained from the study of SLFN11 in HIV proteins synthesis, we hypothesized that SLFN11 may sensitize cells to DNA damage by inhibiting the synthesis of proteins vital to survival after DNA damage if the corresponding genes also harbor deviant codon-usage. To this end, we calculated the Codon Adaptation Index (CAI) of genes involved in DNA damage response signaling and multiple DNA damage repair mechanisms including Homology Directed Repair (HDR), Nonhomologous End-Joining (NHEJ), Mismatch Mediated Repair (MMR), Nucleotide Excision Repair (NER) and Base Excision Repair (BER), using 80 highly-expressed ribosomal proteins as a reference gene set6–9. Contrasting the very high average CAI (0.79) of the most abundantly expressed cellular proteins10, the DNA damage response signaling related genes displayed an average CAI of as low as 0.66, comparable to the average CAI (0.60) of HIV-1 genes (Supplementary Table 1). Importantly, the two components central to theDNA damage response, ATR and ATM11,12, present CAIs as low as 0.65 for ATR and 0.64 for ATM, starkly contrasting the highly expressed GAPDH with a CAI of 0.81. Considering the additional impact of the long coding sequences of ATR (2644 a.a.) and ATM (3056 a.a.), it appeared that the translation of both ATR and ATM could indeed be a likely target for SLFN11. Interestingly, we also noted that genes involved in HDR, NHEJ and MMR also display lower average CAIs (0.67, 0.69 and 0.69 respectively) than genes affiliated with NER and BER (0.73 and 0.74) (Supplementary Table 2 & 3).
Results
To investigate this potential posttranscriptional control of ATR and ATM expression by SLFN11, we generated stable polyclonal derivatives from the pancreatic cancer cell line COLO 357 FG (hereafter referred to as FG cells)13 and HEK293 cells (hereafter referred to as 293 cells) using two independent lentiviral-based shRNA constructs against SLFN11 to permanently silence SLFN11 expression. Crucially, silencing of SLFN11 expression conferred significant resistance upon both FG and 293 cells to the Topoisomerase I inhibitor Camptothecin (CPT) (Fig. 1a, d), as well as other DDAs including the Topoisomerase II inhibitor Mitoxantrone, the nucleoside analog Gemcitabine and the DNA-alkylating and -cross-linking agent Chlorambucil (Fig. 1g-i). Further, microscopy imaging of live cell cultures confirmed that CPT treatment induced cell death in SLFN11-expressing cells but not in SLFN11-deficient cells (Fig. 1j).
Figure 1: SLFN11 selectively inhibits ATR/ATM protein expression and sensitizes cells to death upon DNA damaging agents treatment.
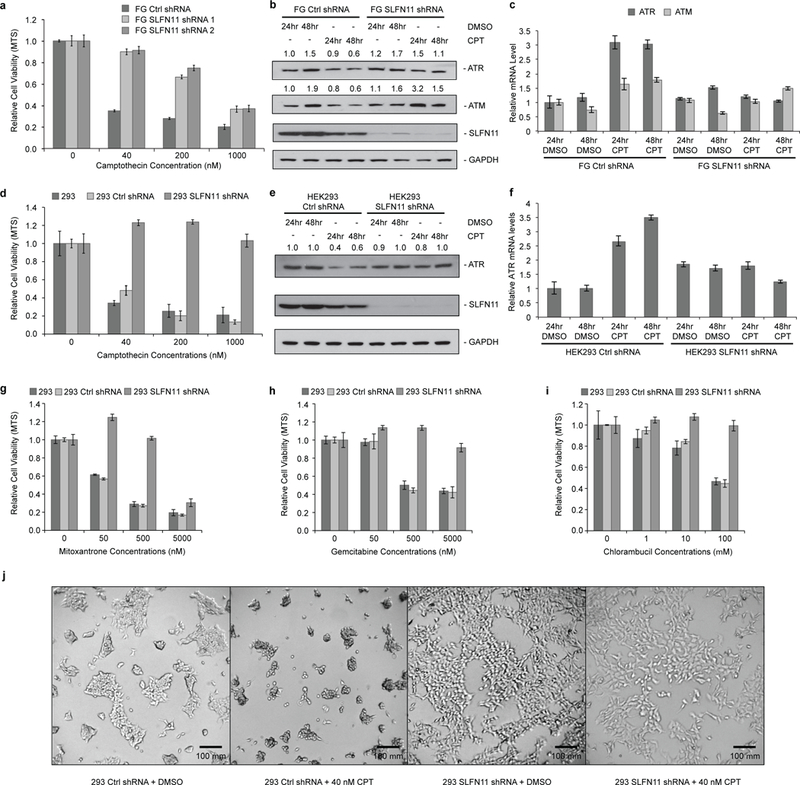
a, Relative viability of FG cells expressing control or SLFN11 shRNA was measured by MTS assay after 48 hours of CPT or DMSO treatment (biological replicates, mean ± s.d., n = 3). b, Immunoblot analysis of ATR and ATM protein levels after 40 nM CPT or DMSO treatment in FG cells expressing control or SLFN11 shRNA. c, Relative ATR and ATM mRNA levels as determined by qPCR in FG cells expressing control or SLFN11 shRNA after 40 nM CPT or DMSO treatment (mean ± s.d., n = 3). d, e, f, As in a, b, c, except with HEK293 cells. g, h, i, As in d, with additional DDAs as specified. j, Microscopic images of HEK293 cell cultures after 24 hours of CPT. Uncropped images are shown in Supplementary Data Set 1.
To determine whether SLFN11 affects the translation of ATR and ATM in response to DNA damage, we first analyzed the expression levels of both ATR and ATM after CPT treatment. Indeed, expression of both proteins was significantly down-regulated after 24 or 48 hours of CPT exposure in FG and 293 cells, but were barely affected in their SLFN11-lacking matched counterparts (Fig. 1b, e). In contrast, both ATR and ATM mRNA levels stayed constant or even got up-regulated upon CPT treatment (Fig. 1c, f). To determine whether the elevated expression of ATR or ATM proteins in SLFN11-deficient cells upon CPT treatment is essential for their resistance to DDAs, we abolished the expression of ATR or ATM with corresponding siRNAs in both parental and SLFN11-deficient FG and 293 cells (Fig. 2a, c). As shown, attenuation of ATR expression completely restored the sensitivity of both SLFN11-deficient cell lines to CPT treatment, while silencing of ATM expression failed to do so (Fig. 2b, d). The inherently SLFN11-deficient pancreatic tumor cell line, MIA Paca-2, was also sensitized to CPT treatment by siRNA-mediated ATR expression suppression, further corroborating the results (Fig. 2e-g). Inhibition of ATR kinase activity by the small molecule ATR inhibitor VE-822 also sensitized the SLFN11-deficient cells to CPT treatment in a dosage-dependent manner (Fig. 2h-k). Our data also suggested that the expression of SLFN11 did not affect the early DNA damage response signaling as represented by the phosphorylation of CHK1 at S317 by ATR (Fig. 2i, lanes 1, 2, 6, 7).
Figure 2: Selective inhibition of ATR sensitizes SLFN11-deficient cells to CPT treatment.
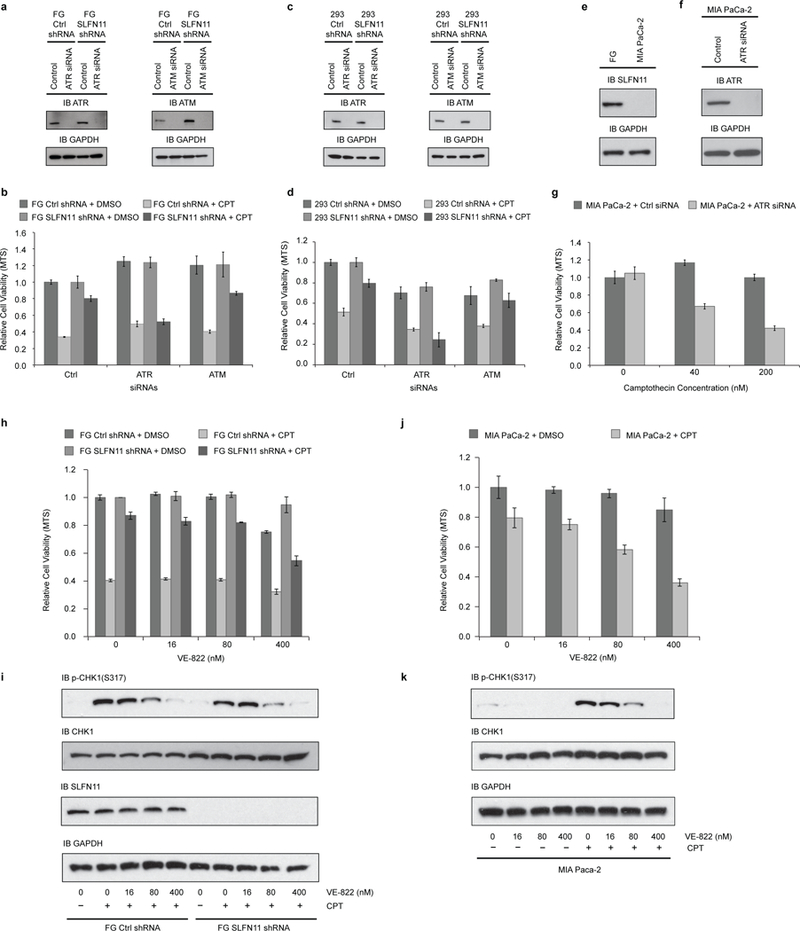
a, Efficiency of ATR/ATM knockdown by siRNAs in FG cells was monitored by immunoblotting 5 days after transfection. b, 72 hours after siRNAs transfection, FG cells expressing control or SLFN11 shRNA were treated by 200 nM CPT or DMSO for another 48 hours. Relative cell viabilities were determined by MTS assay. c, d, As in a, b, except with HEK293 cells. e, SLFN11 expression deficiency in MIA PaCa-2 cells was confirmed by immunoblotting. f, g, As in a, b, except with MIA PaCa-2 cells. h, Relative viabilities of FG cells expressing control or SLFN11 shRNA after 48 hours of ATR inhibitor VE-822 and 40 nM CPT treatment were measured by MTS assay. i, Phosphorylation of CHK1 was determined by immunoblotting after 6 hours of 40 nM CPT and VE-822 treatment. j, As in h, except with MIA PaCa-2 cells. k, As in i, with except with MIA Paca-2 cells. Samples were collected after 3 hours treatment. (b, d, g, h, i, biological replicates, mean ± s.d., n = 3). Uncropped images are shown in Supplementary Data Set 1.
We further performed 35S-methionine and 35S-cysteine labeling and pulse-chase experiments to determine the synthesis rate and stability of ATR protein upon CPT treatment. As anticipated, in the presence of SLFN11, ATR protein synthesis was nearly abolished 24 hours after CPT treatment started in both 293 and FG cells (Fig. 3a, lane 4) as compared to their SLFN11-deficient counterparts (Fig. 3a, lane 10). However, the stability of ATR protein did not seem to be significantly affected by CPT or SLFN11 as evidenced by the relatively stable levels of newly synthesized 35S-labeled ATR protein in the chase-part of the experiment (Fig. 3a, lanes 2, 3, 5, 6, 8, 9, 11, 12). Indeed, the inhibition of ATR protein synthesis, but not of GAPDH, was observed as early as 3 hours after CPT administration solely in SLFN11-expressing cells (Fig. 3b). After 6 hours of CPT treatment, slight inhibition of GAPDH translation became notable, while the inhibition of ATR protein synthesis consistently escalated (Figure 3b). Further, polysome profiling experiments also showed that 6 hours after CPT treatment started, the ATR polysome profile changed much more significantly in SLFN11-expressing FG cells than in SLFN11-deficient FG cells upon CPT treatment. Importantly, only minor changes were noticed in the GAPDH polysome profile upon CPT treatment, regardless of the presence of SLFN11 (Fig. 3c). Altogether, these results demonstrate that upon CPT treatment, a prominent inhibition of ATR protein synthesis occurs quickly following DNA damaging.
Figure 3: SLFN11 selectively inhibits ATR protein synthesis upon CPT treatment.

a. Pulse-chase analysis of ATR protein translation and stability via 35S-methionine and 35S-cysteine labeling of HEK293 and FG cells expressing control or SLFN11 shRNA. Cells were treated with 40 nM CPT or DMSO for 24 hours prior to 35S-labeling. b, 35S-methionine and 35S-cysteine protein labeling of FG cells expressing control or SLFN11 shRNA after 3 or 6 hours of 40 nM CPT or DMSO treatments. Numbers indicate quantified band intensity relative to DMSO treated cells expressing control shRNA. c, Polysome profiles of ATR and GAPDH in FG cells expressing control or SLFN11 shRNA. Cells were collected and analyzed 6 hours after DMSO or 40 nM CPT treatment (technical replicates, mean ± s.d., n = 3). Uncropped images are shown in Supplementary Data Set 1.
To facilitate our further investigations into the molecular mechanism of SLFN11 function, we established SLFN11-deficient FG and 293 cell lines using CRISPR-Cas9 technology. Complete abrogation of SLFN11 expression via CRISPR-Cas9 yielded an even more profound phenotype of DDA resistance in both FG and 293 cells (Fig. 4a-d) without significant effect on cell proliferation in the absence of DDAs (Fig. 4e). Significant increase of Caspase-3 and −7 activity in the presence of SLFN11 upon CPT treatment (Fig. 4f), as well as shrinkage of cells and generation of apoptotic bodies (Fig. 4g), suggest SLFN11 expression sensitizes cells to apoptosis upon CPT treatment.
Figure 4: CRISPR/Cas9 mediated SLFN11 gene knockout confers significant resistance to CPT-induced apoptosis on cells without affecting cell proliferation.

a, Relative viability of FG cells and FG SLFN11 knockout cells was measured by MTS assay after 48 hours of CPT or DMSO treatment (biological replicates, mean ± s.d., n = 3). b, Knockout of SLFN11 expression in FG cells by CRISPR/Cas9 technique and its effect on ATR/ATM protein expression upon 40 nM CPT or DMSO treatment as determined by immunoblotting. c, As in a, except with HEK293 cells. d, As in b, except with HEK293 cells. Cells were analyzed after 24 hours of 40 nM CPT or DMSO treatment. e, Proliferation of FG and FG SLFN11 knockout cells. f, Relative Caspase-3/7 activity after 24 hours CPT treatment. g, Microscopic images of cell cultures after 24 hours CPT treatment. Uncropped images are shown in Supplementary Data Set 1.
In our previous report on the codon-usage dependent, selective translational inhibition of HIV-1 proteins we provided the initial evidence that SLFN11 affects cellular transfer RNA (tRNA) levels, particularly in infected cells1. We further investigated whether SLFN11 would potentially alter tRNA levels during DNA damage response. The 70 to 87 nucleotides long tRNAs can be divided into two groups based on the structure and size of their variable loops: tRNAs with a short variable loop of 4 or 5 nucleotides are classified as type I, whereas those harboring a long variable stem, totaling 13–19 bases, are referred to as type II tRNAs. In human, the longer type II tRNAs are comprised of all leucine tRNAs including Leu-AAG, Leu-CAA, Leu-CAG, Leu-TAA and Leu-TAG, and all serine tRNAs including Ser-AGA, Ser-CGA, Ser-GCT and Ser-TGA14,15.
Unpredictably, the analysis of total tRNAs abundance revealed that as early as 3 hours after the CPT treatment started, the levels of type II tRNAs in FG cells already began to decline in a SLFN11-dependent manner (Fig. 5a, b). After 12 hours of CPT treatment, only ~50% of type II tRNAs were still present in SLFN11-expressing FG and 293 cells, while no such change was observed in their SLFN11-deficient counterparts. Strikingly, the expression levels of type I tRNAs appeared unaffected in response to the DDA regardless of SLFN11 expression (Fig. 5a-d). Similar down-regulation of type II tRNAs was also observed upon Mitoxantrone, Gemcitabine or Chlorambucil treatment in SLFN11-expressing FG cells (Fig. 5e, f). We further performed protein domain function analysis by expressing either the full length, the truncated N-terminal half (a.a. 1–579) or the C-terminal half (523–901) of SLFN11 in HEK293T cells, which is different from its parental cell line HEK293 by inherently lacking all endogenous SLFNs expression1. The result clearly indicated that the N-terminal half of SLFN11 contains the effector domain to suppress type II tRNA levels (Fig. 5g-i).
Figure 5: SLFN11 mediates the down regulation of type II tRNAs upon DDAs treatment.
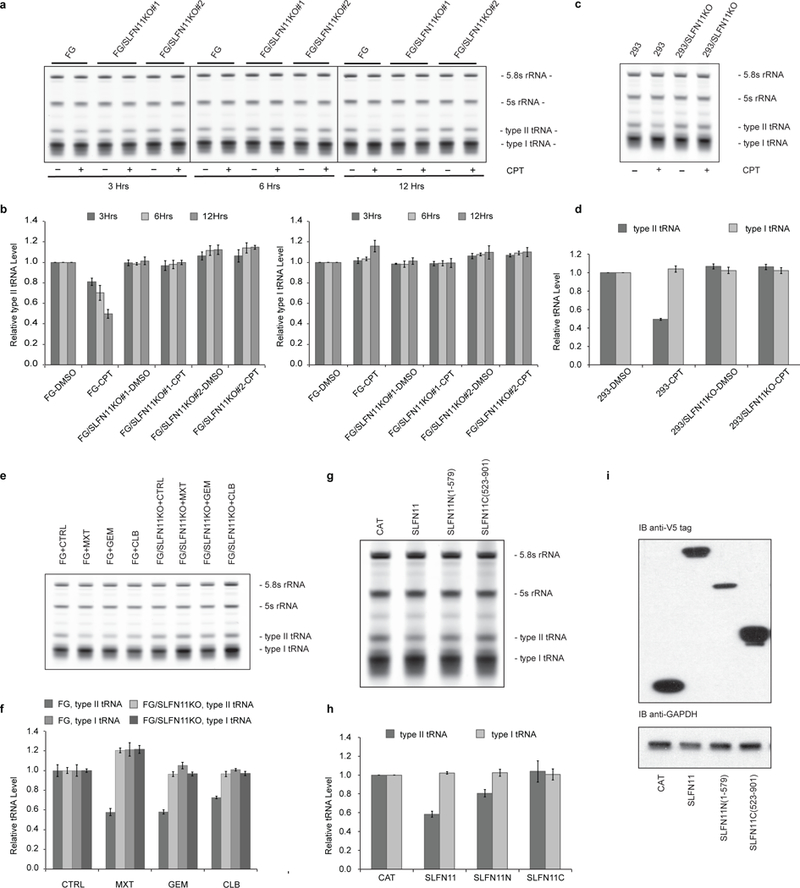
a, Total RNAs from FG cells and FG SLFN11 knockout cells treated with 200 nM CPT or DMSO were resolved by 10% denaturing Urea PAGE, visualized by SYBR Gold staining and quantified. b, Quantified results from a (biological replicates, mean ± s.d., n = 3; 5.8s rRNA served as the endogenous control for normalization; all data are presented relative to DMSO treated FG cell samples). c, As in a, except with HEK293 cells. Cells were analyzed after 12 hours of 200 nM CPT or DMSO treatment. d, As in b, quantified results from c. e, As in a, FG cells and FG SLFN11 knockout cells were treated with Mitoxantrone (MXT, 1μM), Gemcitabine (GEM, 1μM), Chlorambucil (CLB, 100μM), or DMSO for 12 hours. f, As in b, quantified results from e. g, Total RNAs from HEK293T cells exogenously expressing V5-tagged CAT (Chloramphenicol Acetyl Transferase), SLFN11, SLFN11N (a.a. 1–579) or SLFN11C (a.a. 523–901) were collected 48 hours after transfection and analyzed as in a. h, As in b, quantified results from g. i, Expression levels of the corresponding SLFN11 proteins was confirmed by immunoblotting against the V5 tag. Uncropped images are shown in Supplementary Data Set 1.
To identify the individual tRNAs down-regulated upon CPT treatment in the presence of SLFN11, we performed RT-qPCR-based microarray analysis on all nuclear-encoded tRNAs. The tRNAs were first demethylated using E. coli Alpha-ketoglutarate-dependent dioxygenase AlkB to allow efficient reverse transcription of tRNAs, following a protocol that we optimized based on two recent publications16,17. The qPCR-based microarray analysis clearly indicated that all leucine tRNAs as well as Ser-AGA, Ser-TGA and Ser-GGA tRNAs were significantly down-regulated after 12 hours of CPT treatment in SLFN11-expressing FG cells (Fig. 6a). Intriguingly, the only type I tRNA also affected was the initiator methionine tRNA Ini-CAT, whose complementary AUG codon represents the classical translation initiation site for most mRNAs18,19. Most important for our investigation, however, was the finding that not a single tRNA, regardless of its type, was subdued in response to CPT in the absence of SLFN11 (Fig. 6b).
Figure 6: SLFN11-mediated type II tRNAs cleavage inhibits translation mRNAs of genes with high frequency of codon TTA (Leu) usage.

a, Volcano plot of fold change of all cellular tRNAs after 12 hours of 200 nM CPT or DMSO treatment in FG cells as determined by qPCR. b, as in a, except in FG SLFN11 knockout cells. (log2(mean of fold change) vs. p-Value; n = 3; p-Value was calculated by performing a two-tailed two-sample equal variance (homoscedastic) Student’s t-test.) c, tRNA Northern blot analysis of total RNA isolated from FG cells and FG SLFN11 knockout cells treated with 200 nM CPT or DMSO. d, tRNA Northern blot analysis of total RNA from HEK293T cells exogenously expressing control protein (CAT) or SLFN11. Samples were collected 48 hours after transfection. (Numbers indicate quantified band intensity relative to DMSO treated FG cell samples in c or relative to CAT-expressing HEK293T cell controls in d.; 5.8s rRNA served as the endogenous control for normalization) e, Prolonged exposure of tRNA Northern blots of total RNA from FG cells and FG SLFN11 knockout cells treated with 200 nM CPT or DMSO for 12 hours, revealing the cleaved tRNA fragments. f, Protein expression of EGFP encoded by constructs using only the indicated codon for all leucine or serine residues in HEK293T cells in the absence or presence of exogenous SLFN11 expression 48 hours after transfection as determined by anti-GFP immunoblotting. g, As in f, relative mRNA levels derived from indicated EGFP constructs were determined by qPCR (mean ± s.d.; n = 3). Uncropped images are shown in Supplementary Data Set 1.
As the qPCR-based microarray evaluation is a very novel, as of yet unpublished method of tRNA quantification, we decided to further confirm our findings by Northern blot. There, we observed again that the down-regulation of tRNAs Leu-TAA, Leu-CAA, Leu-CAG, Ser-AGA and Ini-CAT started as early as 3 hours after CPT administration, followed by diminution of Leu-TAG, Leu-AAG, Ser-CGA, Ser-GCT and Ser-TGA tRNAs 3 hours later (Fig. 6c). Once more, the attenuation of these tRNAs was only evident in cells expressing SLFN11 (Fig. 6c).
To further evaluate the specific type II tRNAs suppression during DNA damage response as the direct result of SLFN11 activation, we transiently expressed SLFN11 in inherently SLFN11-deficient HEK293T cells. As shown in Fig. 6d, the exogenous expression of SLFN11 alone was sufficient to reduce the levels of all type II serine and leucine tRNAs as well as tRNA Ini-CAT, whereas the type I tRNAs Thr-TGT and Val-TAC remained unchanged. Extended exposures of tRNA Northern blots visualized a fragment of corresponding type II tRNAs in CPT treated, SLFN11-expressing FG cells, suggesting the SLFN11-mediated reduction of type II tRNAs upon CPT treatment is more likely to be the result of direct cleavage of tRNAs (Fig. 6e). The direct cleavage model is also supported by the quick decline of type II tRNAs right after CPT treatment started.
Our data thus far clearly illustrated that all leucine and serine tRNAs are targets of SLFN11. A logical follow-up investigation encompassed therefore a likely selective impact of SLFN11 on the protein translation of genes adopting corresponding codons. To address this point, we designed a series of EGFP-encoding vectors such that in each individual construct all leucine or serine residues were encoded by a single codon, with the original EGFP construct as the control. Each construct was then tested by transfection into the endogenous SLFN11-deficient HEK293T cells, either with or without co-transfection of SLFN11-encoding vector. As we had demonstrated previously1, expression of the original EGFP protein was essentially refractory to suppression by SLFN11 (Fig. 6f, lanes 1 and 2, both upper and lower panels). Remarkably, SLFN11 completely abolished the expression of EGFP_Leu(TTA), and to a much less extent of EGFP_Leu(CTT). EGFP protein derived from all other constructs exhibited marginal to no alteration on account of the presence of SLFN11 (Fig. 6f). Most importantly, the observed inhibition of specific EGFP protein expression definitely occurred at the translational level, as SLFN11 did not significantly affect the level of EGFP mRNA regardless of the deriving construct (Fig. 6g).
We showed that moderate down-regulation of tRNA-Leu-TAA by SLFN11 could significantly inhibit the protein expression of genes with high frequency of codon TTA (Leu) usage such as ATR. Since knockdown of ATR expression with siRNAs sensitized the SLFN11-deficient cells to DDA, we wondered whether knockdown of tRNA-Leu-TAA could down-regulate ATR protein expression thus sensitize SLFN11-deficient cells to DDA as well. Because of the heavy modification and extensive secondary structures of tRNAs, knocking-down specific tRNAs efficiently with either siRNAs or shRNAs has been proven an extremely challenging job. Based on a novel antisense oligonucleotide technique called Gapmer20,21, we designed specific Gapmers targeting all four tRNA-Leu-TAA isodecoders. As we expected, anti-tRNA-Leu-TAA Gapmer down-regulated the level of tRNA-Leu-TAA in both FG and FG-SLFN11KO cells, which led to the profound inhibition of ATR protein expression (Fig. 7a). Interestingly, although anti-tRNA-Leu-TAA Gapmer transfection alone triggered strong Caspase-3 and −7 activation (Fig. 7b), much more significant cell death was only observed when the anti-tRNA-Leu-TAA Gapmer transfected FG-SLFN11KO cells were treated by CPT sequentially, as evidenced by the MTS assay and the microscopy imaging of live cell cultures (Fig. 7c, d). The knockdown of tRNA-Leu-TAA by the corresponding Gapmer was quite specific since it only had a minor effect on tRNA-Ser-CGA, which was partially targeted by the anti-tRNA-Leu-TAA Gapmer (11 nt out of total 16 nt of Gapmer Leu-TAA align with most Ser-CGA tRNA isodecoders). Similar results were obtained in intrinsically SLFN11-deficient MIA Paca-2 cells (Fig. 8a-d).
Figure 7: Ablation of tRNA-Leu-TAA via Gapmer antisense oligonucleotides resensitizes SLFN11-deficient FG cells to CPT-induced apoptosis.

12 hours after transfection with control Gapmer (N.C.) or a Gapmer specific for tRNA-Leu-TAA (50 nM), FG cells and FG SLFN11 knockout cells were treated with DMSO or 200 nM CPT. a, Total RNA was collected 12 hours after CPT treatment and subject to Northern blot analysis. Protein for immunoblotting was collected 24 hours after CPT treatment. Numbers represent quantified band intensity relative to DMSO treated, Gapmer N.C.-transfected cell samples. 5.8s rRNA served as control for Northern blot. GAPDH served as control for immunoblotting. b, Relative Caspase-3/7 activity after 12 hours CPT treatment. c, relative cell viabilities after 60 hours CPT exposure were measured by MTS assay. (b, c, biological replicates, mean ± s.d., n = 3. Both are relative to DMSO treated, Gapmer N.C.-transfected cell samples.) d, Microscopic images of cell cultures after 24 hours CPT treatment. Uncropped images are shown in Supplementary Data Set 1.
Figure 8: Gapmers antisense oligonucleotides directed at tRNA-Leu-TAA sensitize intrinsically SLFN11-deficient MIA Paca-2 cells to CPT-induced apoptosis.
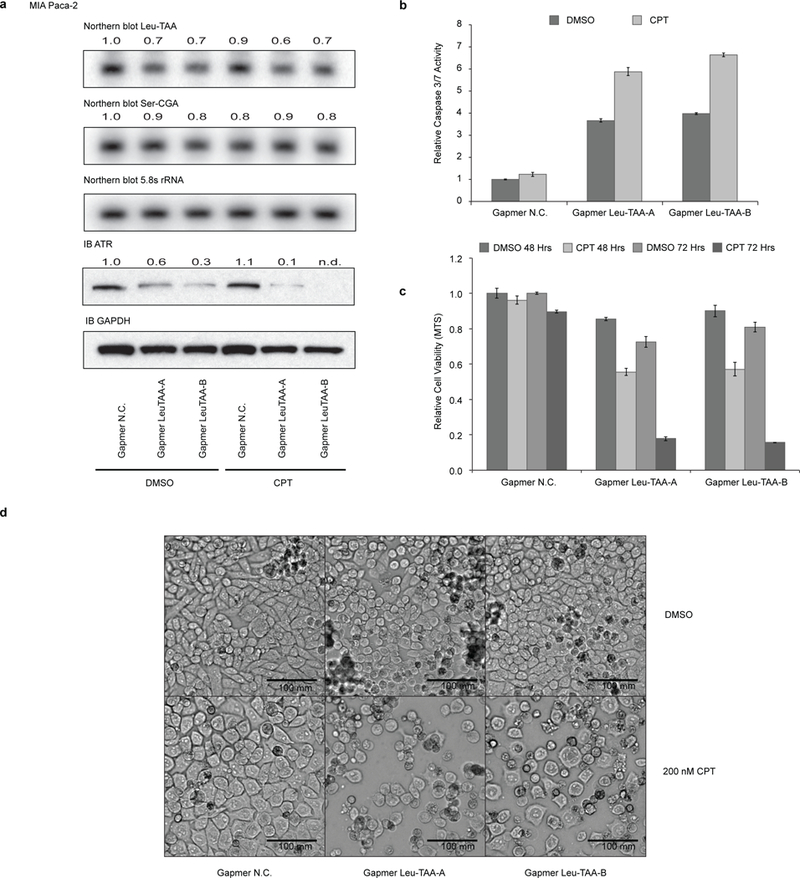
12 hours after transfection with control Gapmer (N.C.) or Gapmers against tRNA-Leu-TAA (12.5 nM), MIA Paca-2 cells were treated with DMSO or 200 nM CPT. a, Total RNA for Northern blots was collected after 12 hours CPT treatment. Protein samples for immunoblotting were collected after 36 hours CPT treatment. Numbers indicate quantified band intensity relative to DMSO treated, Gapmer N.C.-transfected control cell samples. 5.8s rRNA served as control for Northern blots. GAPDH served as control for immunoblotting. b, Relative Caspase-3/7 activity was measured after 24 hours CPT treatment. c, 48 or 72 hours after CPT exposure, relative cell viabilities were determined by MTS assay. (b, c, biological replicates, mean ± s.d., n = 3. Both relative to DMSO treated, Gapmer N.C. transfected control cell samples.) d, Microscopic images of cell cultures after 24 hours CPT treatment. Uncropped images are shown in Supplementary Data Set 1.
Discussion
DDAs are the earliest and most widely used therapeutics for cancer treatment, accounting for almost one third of all chemotherapeutic drugs. However, many tumors are resistant to therapies based on DNA damaging approaches. Even tumors initially responsive to the regimen routinely acquire resistance over the course of the treatment (reviewed in27). Two large-scale transcriptome profiling approaches revealed an indisputable requirement of SLFN11 in cancer cells for DDAs to trigger cell death2,3. Most recently, a study showed that in recurring small cell lung cancer, the silencing of SLFN11 expression mediated by histone modification H3K27me3 at the SLFN11 gene locus was responsible for the tumor’s acquired chemo-resistance. Inhibition of Histone-Lysine N-Methyltransferase EZH2 restored SLFN11 expression and re-sensitized the tumor cells to chemotherapy28. We found early DNA damage response signaling such as the phosphorylation of CHK1 not to be affected by the expression of SLFN11, concurring with other reports of SLFN11 not changing early DNA damage responses but inhibiting checkpoint maintenance and homologous recombination repair at significantly later stages. It was suggested that SLFN11 was promptly recruited to the sites of DNA damage via RPA124, which could be a step in the as of yet unknown activation mechanism of SLFN11. While this manuscript was being reviewed, another report agreed that upon CPT treatment, SLFN11 was promptly recruited to chromatin at stressed replication foci via RPA1 but did not affect the early DNA damage response signaling like the phosphorylation of CHK1 or the loading of CDC45 and PCNA26.
We describe here the novel molecular mechanism by which SLFN11 sensitizes cells to apoptosis upon DNA damage. The SLFN11-dependent down-regulation of type II tRNAs, most importantly tRNA-Leu-TAA, predisposes genes essential for DNA damage response and repair such as ATR or ATM to translational inhibition as they abundantly utilize the corresponding codon TTA (Leu). At the onset of this study we noted that the synthesis of proteins encoded by genes with a low CAI resembling that of HIV were drastically more repressed by SLFN11 than translation of genes with high CAI. The overall TTA (Leu) codon usage frequency is only about 8% for the entire human genome coding sequences, and 2% for the top 24 most highly expressed human cellular proteins (Supplementary Table 4). In striking contrast, genes involved in HDR, NHEJ and MMR displayed much higher average TTA (Leu) codon usage (18%, 15% and 13% respectively), even when compared to genes affiliated with NER and BER (9% and 6%) (Supplementary Table 5, 6). Indeed, out of the 352 leucines in ATR, 73 use the codon TTA (21%), and of 389 leucine residues in ATM, 91 are encoded by TTA (23%). In striking contrast, only a single TTA codon can be found among the 19 leucine residues of GAPDH (5%). Our previous work demonstrated that it is the distinct codon bias that facilitates the preferential translational inhibition of HIV proteins by SLFN11. We now find that select mammalian genes such as those associated with DNA damage response signaling (e.g. ATR or ATM) harbor a similar codon usage distinction as HIV. Our more detailed analysis revealed that specifically the frequency of TTA (Leu) codon usage is the apparent common denominator that subjects the so encoded proteins to translational suppression by SLFN11 (Supplementary Table 4).
An obvious challenge is the question why only the SLFN11-mediated degradation of tRNA-Leu-TAA significantly affected the translation efficiency of mRNAs harboring the corresponding codon, while the cleavage of other leucine or serine tRNAs appeared to be of lesser consequence. One possible explanation can be found in the apparently lower abundance of tRNA-Leu-TAA (for codon TTA) that we have noted in our qPCR and Northern blot analysis. Although neither technique permits a highly accurate quantitative assessment, they do support a reasonable estimation of relative tRNA abundance. For instance, the qPCR Ct value for tRNA-Leu-CAG (for codon CTG) was about 4 cycles lower than the Ct value of tRNA-Leu-TAA (for codon TTA), suggesting the abundance of tRNA-Leu-CAG is roughly 16-fold of tRNA-Leu-TAA. A similar observation was also made with regard to the relative signal intensity in our Northern Blot analysis. Thus, for genes requiring tRNA-Leu-TAA to support their translation, the availability of this tRNA might readily become the rate-limiting factor, and consequently a significant down-regulation of tRNA-Leu-TAA may cause the ribosome to stall and/or detach at the corresponding TTA codons. This effect are expected to be compounded with increasing appearance of the TTA codon and with the length of the mRNA, with the end result that the longer the mRNA and the higher the frequency of TTA codon usage for leucine residues is, the more likely a a premature translation termination will occur. In contrast, for proteins whose translation relies on more abundant synonymous tRNAs, translation initiation rates rather than the reduced yet still sufficient supply of the required tRNAs may remain the limiting factor.
Our data showed that the N-terminal half (residues 1–579) of human SLFN11 contains the functional domain responsible for the degradation of type II tRNAs. It was reported that a leporine SLFN14 N-terminal fragment in rabbit reticulocyte lysate harbors endoribonuclease activity22. Furthermore, while this manuscript was under final review, the crystallographic structure of the murine SLFN13 N-terminal domain (residues 14–353) was solved and demonstrated to possess intrinsic tRNA and rRNA-targeting endoribonuclease activity in vitro23. In concert, these findings establish SLFN family members as a novel class of tRNA specific endoribonucleases with the enzymatic activity residing in the N-terminal region of the protein and with an apparent specificity for type II tRNAs.
Mu et al reported that a SLFN11 deletion mutant missing the most C-terminal sequence (residues 741–901) was found unable to bind to RPA1 and thus failed to be recruited to the sites of DNA damage. This SLFN11 C-terminus deletion mutant also failed to re-sensitize SLFN11 deficient cells to CPT. In contrast, we found that an ever further deletion retaining only the N-terminal half of SLFN11 (residues 1–579) is sufficient for the degradation of type II tRNAs, and as we had previously shown, for attenuating the translation of HIV proteins. One possible explanation for these findings is that the C-terminal half of SLFN11 acts as a regulatory domain with auto-inhibitory properties that need to be relieved via post-translational modification, which might occur at the sites of DNA damage. A short truncation as in Mu et al. could possibly remove the site required for activation, but still retain enough of the C-terminal domain to inhibit such truncated SLFN11. Yet, a more extensive deletion as we employed might fully remove any inhibitory elements, and thus result in an active, albeit unregulated, SLFN11 fragment harboring the newly discovered tRNAse activity. Besides the activation mechanism(s) of SLFN11, the subcellular location where it exerts its tRNA-degrading effects also requires further investigation. Although mature tRNAs generally function outside the nucleus, they are not only transcribed in the nucleus, but move between the cytoplasm and the nucleus dynamically25. Therefore the tRNAs could be susceptible to cleavage by SLFN11 regardless of their subcellular localization.
While SLFN11’s cleavage of type II tRNAs and consequent ATR suppression are undoubtedly crucial components of DDA-induced cell death, it does not preclude that other aspects of the DNA damage response are not also affected by SLFN11. We cannot rule out the possibility that the induction of chromatin opening across replication initiation sites and blocking of fork progression by SLFN11 upon CPT treatment may be a second mechanism of SLFN11 function. However, as we show in this study, the tRNA-Leu-TAA degradation with specifically targeted Gapmers alone is capable to restore DNA damage-induced apoptosis via CPT in SLFN11-deficient cells, suggesting that this step encompasses indeed the crucial element of SLFN11 function.
In summary, our findings not only produce significant new insights into the molecular mechanism underlying SLFN11 function, but also provide one of the first examples that modulation of distinct tRNAs facilitates the specific targeting of proteins relying on those tRNAs for their translation. The extreme technical difficulty of manipulating specific tRNA expression has been a substantial hindrance to tRNA research. As we show here for the first time, Gapmer technology can be used efficiently to specifically attenuate individual tRNA levels. The direct targeting of tRNA-Leu-TAA also offers a new strategy to overcome tumor cell resistance to DDAs, which could hold significant clinical potential.
Materials and Methods
Cell lines, plasmids, antibodies and chemicals
All cell lines were maintained at 37 °C, 5% CO2 in high glucose DMEM supplemented with 10% heat-inactivated fetal bovine serum, 100 U/ml Penicillin, 100 μg/ml Streptomycin, 2 mM L-Glutamine, 1× MEM Non-essential Amino Acid, 1 mM Sodium Pyruvate, and 50 μM 2-Mercaptoethanol. The HEK293 (CRL-1573) and HEK293T (CRL-3216) cell lines were acquired from ATCC. Both COLO 357 FG and MIA Paca-2 cell lines were acquired from Dr. Tannishtha Reya at Univ. of California, San Diego. HEK293 and COLO 357 FG cell lines with stable expression of shRNAs were generated using lentiviral-based vectors as previously described1. To obtain HEK293 and COLO 357 FG derivative cell lines in which SLFN11 expression was obliterated using CRISPR-Cas9 technique, cells were transfected with pSpCas9 BB-2A-Puro (PX459) All-in-One CRISPR-Cas9 construct and selected based on puromycin resistance (for HEK293 cells: SLFN11 CRISPR-Cas9 guide RNA 4 - GCAGCCTGACAACCGAGAAA; for FG cells: SLFN11 CRISPR-Cas9 guide RNA 1 - GGCTTGACAGAGCGATCTTC; both obtained from Genscript). Surviving cells were cloned by limiting dilution and screened for SLFN11 expression by immunoblotting. The construction of pcDNA6-SLFN11-V5-His and pcDNA6.2-EGFP-Myc expression vectors were previously reported1. Anti-ATR antibody (N-19) (SC-1887), anti-CHK1 Antibody (G-4) (SC-8408), anti-SLFN11 antibody (E-4) (SC-374339), and anti-V5-Probe (G-14)-R (SC-374339) were obtained from Santa Cruz Biotechnology. Antibodies against ATR (#2790S), ATM (D2E2) (#2873S), Phospho-CHK1 (Ser317) (D12H3) (#12302S), GFP (D5.1) (#2956S) and GAPDH (14C10) (#2118S) were purchased from Cell Signaling Technology. Antibody for GAPDH immunoprecipitation was acquired from Abcam (ab110305). Camptothecin was procured from EMD Millipore, Mitoxantrone and Gemcitabine from ENZO Life Sciences, Chlorambucil from MP Biomedicals, and the ATR inhibitor VE-822 from Selleckchem.
siRNA transfection
Reverse transfection of cells was performed using ON-TARGETplus SMARTpool siRNAs (GE Dharmacon) and RNAiMax reagent (Life Technologies) in 96-well plates. 72 hours post transfection cells were treated with the indicated drugs for 48 hours prior to harvesting and immunoblotting analysis. Survival rates of siRNA transfected cells were determined by means of MTS based cell viability assays.
Design and transfection of Gapmers
Gapmers antisense oligonucleotides designed to target tRNA sequence regions with less secondary structures were synthesized by EXIQON (Qiagen). “*” represents phosphorothioate bond, and “+A”, “+T”, “+G”, “+C” represent corresponding LNA residues, respectively. Cells were transfected with Gapmers using RNAiMax (Thermo Fisher Scientific) at the indicated concentrations 24 hours after the cells were plated, followed by additional 12 hours treatment. Unless specified otherwise, Gapmer tRNA-Leu-TAA-B was used for tRNA-Leu-TAA knockdown due to its lesser cytotoxicity and more potent effect.
Gapmer tRNA-Leu-TAA-A:
+T*+C*+T*+T*A*A*G*T*C*C*A*A*+C*+G*+C*+C
Gapmer tRNA-Leu-TAA-B:
+C*+C*+A*+T*T*G*G*A*T*C*T*T*+A*+A*+G*+T
Gapmer Negative Control:
+T*+A*+C*+G*C*G*T*C*T*A*T*A*+C*+G*+C*+A
MTS cell viability assay
Cells were seeded in 96-well clear tissue culture plates 24 hours prior to the indicated regimen in complete phenol red-free DMEM medium. MTS was added to cell culture at the end of the intended treatments following the manufacturer’s protocol (Promega, G3581). Absorbance was measured at 490 nm after incubation at 37°C for 2 hours.
Caspase-3 and −7 activity assay
Caspase-3 and −7 activity was measured with Caspase-Glo® 3/7 Assay Systems (Promega, G8091) following the manufacturer’s protocol. Basically, cells were seeded in 96-well tissue culture plates with white wall and clear bottom in complete phenol red-free DMEM medium, and treated as indicated specifically. Equal volume of Caspase-Glo® 3/7 Reagent was added into the cell culture and gently mixed using a plate shaker at 600rpm for 30 seconds. The plate was incubated at room temperature for 30 minutes, and the generated luminescence signal was measured by a NOVOstar (BMG LABTECH) multi-function plate reader.
Whole cell lysis and immunoblotting
Cells were lysed in 1× Cell Lysis Buffer (Cell Signaling Technology, #9803S) supplemented with Phosphatase Inhibitor Cocktail Set I, Phosphatase Inhibitor Cocktail Set II, Protease Inhibitor Cocktail Set III (all EMD Millipore), and 1 mM PMSF. Samples were resolved by 4–12% SDS-PAGE (Invitrogen) and transferred onto PVDF membranes. After incubation with target-specific primary antibody and HRP-conjugated secondary antibody, signals were detected using Western Lightning ECL Pro (PerkinElmer) and film exposure. When needed, the film was scanned and quantified with ImageJ64.
Total RNA preparation and mRNAs qPCR
Total cellular RNA was isolated with TRIzol (Invitrogen) and cleaned with the TURBO DNA-free Kit (Ambion). The reverse transcription was performed using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems). The qPCR reactions were carried out on an Applied Biosystems StepOne Plus Real-Time PCR System using iTaq Universal SYBR Green Supermix (Bio-Rad) following the manufacturer’s protocols. Relative levels of mRNAs of interest were calculated based on ΔCt values and subsequent normalization to GAPDH mRNA levels. The following qPCR primers were used in these assays:
ATR forward 5′-CGCTGAACTGTACGTGGAAA-3′,
ATR reverse 5′-caattagtgcctggtgaacatc-3′;
ATM forward 5′-tttcttacagtaattggagcattttg-3′,
ATM reverse 5′-ggcaatttactagggccattc-3′;
GAPDH forward 5′-TCCACTGGCGTCTTCACC-3′,
GAPDH reverse 5′-GGCAGAGATGATGACCCTTTT-3′;
EGFP-tag forward 5′-CGCCGACCCAGCTTTCTTGTA-3′,
EGFP-tag reverse 5′-TGATCAGCTTCTGCTCGCCG-3′.
tRNA demethylation and qPCR-based tRNA microarray assay
Total cellular RNA was prepared with TRIzol (Invitrogen), treated with TURBO DNA-free Kit (Ambion) and re-purified with TRIzol. The tRNA demethylation was carried out following a manufacturer-provided protocol (Arraystar) modified and optimized in our lab: 2.5 μg total RNA was incubated with 2.5 μl AlkB demethylase, 1 μl RNasin® Plus RNase Inhibitor (Promega) in a total of 100 μl freshly prepared tRNA demethylation buffer (50 mM MES pH 6.0, 100 mM KCl, 2 mM L-ascorbate, 1 mM α-ketoglutarate, 50 μg/ml UltraPure BSA, 300 μM Fe(NH4)2(SO4)2.6H2O) at 37 °C for 2 hours. Reaction was terminated by sequential addition of EDTA and MgCl2 (final concentration 1 mM each). Total RNA was further purified by phenol-hloroform extraction and isopropanol precipitation. Reverse transcription was performed using rtStar™ tRNA-optimized First-Strand cDNA Synthesis Kit (Arraystar), and qPCR for all cellular tRNAs was performed employing nrStar™ Human tRNA Repertoire PCR Array (Arraystar) and Arraystar SYBR® Green qPCR Master Mix on a QuantStudio 7 Flex Real-Time PCR System. The data were analyzed with the Analysis tool for nrStar™ Human tRNA repertoire PCR Array - v1.01 (Arraystar) with the cut-off Ct set at 30. The p-values were calculated by two-tailed two-sample equal variance (homoscedastic) Student’s t-test.
Polysome profiling by sucrose gradient
Cells were seeded 24 hours prior to 6-hour treatment with DMSO or 40nM CPT, subsequently incubated with 100 μg/mL of cycloheximide (CHX) for 3 minutes at 37 °C and then washed with PBS containing 100 μg/mL CHX. Cells were lysed in polysome extraction buffer (0.5% Triton, 10 mM Tris pH7.4, 15 mM MgCl2, 150 mM NaCl, 1 unit/ μl RNase inhibitor, 100 μg/mL CHX), and resulting lysates were layered onto previously prepared linear sucrose density gradients (10–50%). Ultracentrifugation was performed at 35,000 rpm for 2.5 hours. Fractions were collected using ISCO Gradient Former (Model 160). Total RNA from each fraction was extracted using TRIzol LS (with 2 ng/ml in vitro transcribed EGFP mRNA added to monitor RNA extraction efficiency) and reverse transcribed into cDNA. qPCR was performed using primers specific for ATR, GAPDH and EGFP.
35S- protein labeling and immunoprecipitation
Cells were treated with DMSO or 40nM CPT as indicated, cultured in methionine and cysteine-free DMEM for 30 min at 37 °C prior to incubation with 250 μCi of 35S-methionine and cysteine (PerkinElmer EasyTag EXPRESS [35S]-protein labeling mix, 11 mCi/ml) for an additional 30 minutes. For pulse chase studies, labeled cells were returned into normal complete DMEM medium without 35S-methionine and cysteine, and cultured for the indicated time spans. At the end of the labeling or pulse chase experiment the cells were washed with PBS, harvested using trypsin and lysed as outlined above. The 35S-labeled cell lysates were incubated with anti-ATR (Santa Cruz Biotechnology, (N-19), SC-1887) or anti-GAPDH antibody (Abcam ab110305) for 2 hours at 4 °C. Antibody-antigen complexes were captured on Dynabeads Protein G (Life Technologies), resolved by 4–12% SDS-PAGE and transferred to PVDF membranes. The membrane was dried and analyzed using the Typhoon storage phosphorimager.
Northern blot analysis of tRNAs
TRIzol purified total RNA was resolved on 10% TBE-Urea gels, and subsequently transferred for 14 hours onto Zeta-Probe nylon membranes (Bio-Rad) in 0.5× TBE at 25 V and 4 °C. After transfer, membranes were cross-linked in an UV Stratalinker 2400 (Stratagene), prehybridized with 10 ml ULTRAhyb-oligo hybridization buffer (Ambion) for 1 hour at 42 °C and then subject to hybridization with 10 pmol 32P-labeled DNA oligo probes (Integrated DNA technologies) at 42 °C for at least 14 hours. Membranes were then rinsed and washed 2 times with wash-buffer (2× SSC with 0.5% SDS) at 42 °C for 1 hour, dried and analyzed using the Typhoon Phosphoimager. The following probe sequences were chosen based on predictions from GtRNAdb (tRNAscan-SE analysis of complete genomes, gtrnadb.ucsc.edu)14,15:
tRNA Leu-AAG: AGCCTTAATCCAGCGCCTTAGACCGCTCGGCCACGCTACC;
tRNA Leu-CAA: GGAGACCAGAACTTGAGTCTGGCGCCTTAGACCA
tRNA Leu-CAG: CACGCCTCCAGGGGAGACTGCGACCTGAACGCAGCGCCTT
tRNA Leu-TAA: CCATTGGATCTTAAGTCCAACGCCTTAACCACTC
tRNA Leu-TAG: GACTGGAGCCTAAATCCAGCGCCTTAGACC
tRNA Ser-TGA: TGGATTTCAAGTCCATCGCCTTAACCACTCGGCCACGACTAC
tRNA Ser-AGA: GATTTCTAGTCCATCGCCTTAACCACTCGGCCACGACTAC
tRNA Ser-CGA: CCCCATTGGATTTCGAGTCCAACGCCTTAACCACTCGGCCA
tRNA Ser-GCT: GGATTAGCAGTCCATCGCCTTAACCACTCGGCCACCTCGTC
tRNA Thr-TGT: AGGCCCCAGCGAGGATCGAACTCGCGACCCCTGG
tRNA Val-TAC: TGGTTCCACTGGGGCTCGAACCCAGGACCTTCTGCG
tRNA Ini-CAT: CCGCTGCGCCACTCTGCT
5.8s rRNA: TCCTGCAATTCACATTAATTCTCGCAGCTAGC
EGFP Expression constructs with synonymous leucine or serine codons
The parental Myc-tagged EGFP pcDNA6.2/gw/d-Topo vector has been previously described1. EGFP coding sequences in which all leucine or serine residues are encoded by one distinct codon were synthesized by Genscript and cloned into pcDNA6.2/gw/d-Topo using ApaI and NotI. Expression of EGFP protein was visualized by anti-GFP immunoblotting, and the corresponding EGFP mRNA levels were determined by qPCR using a primer set targeting the common C-terminal region of all EGFP constructs.
Microscopic Imaging
Microscopic imaging of cell cultures was done with a Nikon Eclipse E800 microscope system with Nuance FX Multispectral Imaging System. The scale bar was generated with ImageJ64 calibration data. Adobe Photoshop was used to minimally adjust the brightness and contrast of entire images. No further digital processing was applied.
Statistical analyses
For all statistical analyses, two-sample equal variance (homoscedastic) Student’s t-tests (two-tailed) were performed using Microsoft Excel. All biological replicates and technical replicates are specified accordingly. Experimental sample sizes were chosen according to commonly accepted ranges for in vitro studies in this field and to achieve statistical significance. For all experiments without statistical analyses, one representative result out of at least three independent experiments is shown.
Supplementary Material
Acknowledgements
The authors wish to thank R. Lardelli and M. Arribas-Layton for assistance with Northern blot and polysome profile analysis. This work was supported by grants R01-GM101982 and R21-AI124199 to M.D.
Footnotes
Author Contributions: M.L., J-Y.W. and M.D. conceived the experiments; E.K. and X.G. performed the cell viability studies, ATR experiments and polysome analysis; M.L., X.G. and D.M. are responsible for all tRNA data and codon usage studies; M.L. and X.G. designed and performed all Gapmer related studies; M.L., E.K. and D.M. generated the figures; and M.L. and M.D. wrote the manuscript.
Reprints and permissions information is available at www.nature.com/reprints
Competing financial interests
The authors declare no financial conflicts.
Data availability
All data generated or analyzed during this study are included in this published article (and its supplementary information files).
Supplementary Information is available online
References
- 1.Li M et al. Codon-usage-based inhibition of HIV protein synthesis by human schlafen 11. Nature 491, 125–128, doi: 10.1038/nature11433 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zoppoli G et al. Putative DNA/RNA helicase Schlafen-11 (SLFN11) sensitizes cancer cells to DNA-damaging agents. Proc Natl Acad Sci U S A 109, 15030–15035, doi: 10.1073/pnas.1205943109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Barretina J et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607, doi: 10.1038/nature11003 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schwarz DA, Katayama CD & Hedrick SM Schlafen, a New Family of Growth Regulatory Genes that Affect Thymocyte Development. Immunity 9, 657–668 (1998). [DOI] [PubMed] [Google Scholar]
- 5.Bustos O et al. Evolution of the Schlafen genes, a gene family associated with embryonic lethality, meiotic drive, immune processes and orthopoxvirus virulence. Gene 447, 1–11, 10.1016/j.gene.2009.07.006(2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sharp PM & Li WH The codon Adaptation Index--a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Research 15, 1281–1295 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puigbo P, Bravo IG & Garcia-Vallve S CAIcal: a combined set of tools to assess codon usage adaptation. Biology direct 3, 38, doi: 10.1186/1745-6150-3-38 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jalal S, Earley JN & Turchi JJ DNA Repair: From Genome Maintenance to Biomarker and Therapeutic Target. Clinical Cancer Research 17, 6973–6984, doi: 10.1158/1078-0432.ccr-11-0761 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brown JS, O’Carrigan B, Jackson SP & Yap TA Targeting DNA Repair in Cancer: Beyond PARP Inhibitors. Cancer Discovery 7, 20–37, doi: 10.1158/2159-8290.cd-16-0860 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Beck M et al. The quantitative proteome of a human cell line. Molecular Systems Biology 7, 549–549, doi: 10.1038/msb.2011.82 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blackford AN & Jackson SP ATM, ATR, and DNA-PK: The Trinity at the Heart of the DNA Damage Response. Molecular Cell 66, 801–817, doi: 10.1016/j.molcel.2017.05.015 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Cimprich KA & Cortez D ATR: an essential regulator of genome integrity. Nature Reviews Molecular Cell Biology 9, 616, doi: 10.1038/nrm2450 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Morgan RT et al. Human cell line (COLO 357) of metastatic pancreatic adenocarcinoma. Int J Cancer 25, 591–598 (1980). [DOI] [PubMed] [Google Scholar]
- 14.Chan PP & Lowe TM GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Research 37, D93–D97, doi: 10.1093/nar/gkn787 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chan PP & Lowe TM GtRNAdb 2.0: an expanded database of transfer RNA genes identified in complete and draft genomes. Nucleic Acids Res 44, D184–189, doi: 10.1093/nar/gkv1309 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zheng G et al. Efficient and quantitative high-throughput tRNA sequencing. Nat Meth 12, 835–837, doi:10.1038/nmeth.347810.1038/nmeth.3478http://www.nature.com/nmeth/journal/v12/n9/abs/nmeth.3478.html-supplementary-informationhttp://www.nature.com/nmeth/journal/v12/n9/abs/nmeth.3478.html-supplementary-information (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cozen AE et al. ARM-seq: AlkB-facilitated RNA methylation sequencing reveals a complex landscape of modified tRNA fragments. Nat Meth 12, 879–884, doi:10.1038/nmeth.350810.1038/nmeth.3508http://www.nature.com/nmeth/journal/v12/n9/abs/nmeth.3508.html-supplementary-informationhttp://www.nature.com/nmeth/journal/v12/n9/abs/nmeth.3508.html-supplementary-information (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hinnebusch AG Molecular Mechanism of Scanning and Start Codon Selection in Eukaryotes. Microbiology and Molecular Biology Reviews : MMBR 75, 434–467, doi: 10.1128/MMBR.00008-11 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kolitz SE & Lorsch JR Eukaryotic Initiator tRNA: Finely Tuned and Ready for Action. FEBS letters 584, 396–404, doi: 10.1016/j.febslet.2009.11.047 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Grünweller A et al. Comparison of different antisense strategies in mammalian cells using locked nucleic acids, 2′-O-methyl RNA, phosphorothioates and small interfering RNA. Nucleic Acids Research 31, 3185–3193 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kurreck J, Wyszko E, Gillen C & Erdmann VA Design of antisense oligonucleotides stabilized by locked nucleic acids. Nucleic Acids Research 30, 1911–1918 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pisareva VP, Muslimov IA, Tcherepanov A & Pisarev AV Characterization of Novel Ribosome-Associated Endoribonuclease SLFN14 from Rabbit Reticulocytes. Biochemistry 54, 3286–3301, doi: 10.1021/acs.biochem.5b00302 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang J-Y et al. Structure of Schlafen13 reveals a new class of tRNA/rRNA- targeting RNase engaged in translational control. Nature Communications 9, 1165, doi: 10.1038/s41467-018-03544-x (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mu Y et al. SLFN11 inhibits checkpoint maintenance and homologous recombination repair. EMBO Rep 17, 94–109, doi: 10.15252/embr.201540964 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hopper AK & Huang H-Y Quality Control Pathways for Nucleus-Encoded Eukaryotic tRNA Biosynthesis and Subcellular Trafficking. Molecular and Cellular Biology 35, 2052–2058, doi: 10.1128/mcb.00131-15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Murai J et al. SLFN11 Blocks Stressed Replication Forks Independently of ATR. Molecular Cell 69, 371–384.e376, doi: 10.1016/j.molcel.2018.01.012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cheung-Ong K, Giaever G & Nislow C DNA-Damaging Agents in Cancer Chemotherapy: Serendipity and Chemical Biology. Chemistry & Biology 20, 648–659, doi: 10.1016/j.chembiol.2013.04.007 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Gardner EE et al. Chemosensitive Relapse in Small Cell Lung Cancer Proceeds through an EZH2-SLFN11 Axis. Cancer Cell 31, 286–299, doi: 10.1016/j.ccell.2017.01.006 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.