

Abstract
Astrocytes have been proposed to have multiple roles in the development and maintenance of the vertebrate CNS. To facilitate documentation of these roles, we designed a transgene to enable their ablation at selectable times. The transgene consists of the coding region for the herpes simplex virus-thymidine kinase (HSV-TK) under the control of the human glial fibrillary acidic protein gene promoter. The HSV-TK is innocuous but converts the antiherpetic agent ganciclovir (GCV) to a toxic product that interferes with DNA replication in proliferating cells. In a developmental study, transgenic mice were treated with GCV during the first postnatal week, with evaluation at P19. Treated mice displayed severe ataxia. Histological examination revealed disrupted astrocyte development, particularly in the cerebellum, with marked secondary effects on other cell types. Cerebellar defects included a loss in the numbers of astrocytes and an overall reduction in cerebellar size and disruption of the normally well defined cellular layers. Radial glia were disordered, Purkinje cells were ectopically distributed and displayed abnormal dendritic trees, and granule cells were markedly depleted. These effects were more severe in animals treated on postnatal day 1 versus treatment at day 5. A major factor causing granule cell death was excitotoxicity attributable to activation of NMDA receptors. These results suggest a critical role for astrocytes in cerebellar development.
Keywords: astrocyte, cerebellum, development, glial fibrillary acidic protein, herpes simplex virus-thymidine kinase, transgenic
Astrocytes are the most abundant cell type in the vertebrate CNS, yet their versatility and importance are only beginning to be appreciated. These cells display a range of receptors for neurotransmitters and express several ion channels and transporters (MacVicar et al., 1989; Oh and Waxman, 1994; Rothstein et al., 1994), suggesting that they respond to and participate in a variety of physiological processes. In addition, after injury to the CNS, astrocytes are activated to proliferate, hypertrophy, and secrete cytokines and growth factors. Whether this glial response promotes or hinders neural regeneration is not certain (Gage et al., 1988; Kawaja and Gage, 1991; Norenberg, 1994). During development, astrocytes are believed to provide radial scaffolding to guide cell migration (Rakic, 1971) and also to secrete factors that regulate the growth and differentiation of neurons and oligodendrocytes (Noble et al., 1988;Raff et al., 1988; Richardson et al., 1988; Bogler et al., 1990;McKinnon et al., 1990).
Surprisingly, although multiple critical roles now have been assigned to astrocytes, few CNS defects have been attributed specifically to faulty functioning of these cells. For instance, generation of GFAP-null mice by gene targeting revealed relatively normal development of the mutant mice (Gomi et al., 1995; Pekny et al., 1995; McCall et al., 1996) but subtle changes in both cerebellar long-term depression (Shibuki et al., 1996) and hippocampal long-term potentiation (McCall et al., 1996). In addition, astrocytes have been implicated in the pathogenesis of both hepatic encephalopathy (Butterworth, 1993) and Alexander’s disease (Borrett and Becker, 1985). A useful strategy to understand the role of cell–cell interactions is to ablate an individual cell type specifically. Such an approach has been used widely in Drosophila and Caenorhabditis elegans(Sentry et al., 1993; Hutter and Schnabel, 1995). Previous strategies for inactivating vertebrate glial cells in vivo have included radiation (Kalderon et al., 1990; Pippenger et al., 1990) or antimitotic agents (Politis and Houle, 1985) to kill dividing cells or toxic agents such as fluorocitrate or aminoadipic acid to inhibit metabolism (Khurgel et al., 1996; Largo et al., 1996). However, each of these approaches has its drawbacks. Antimitotic agents and radiation are also likely to kill proliferating oligodendrocytes, adult progenitor cells, and microglia, complicating interpretation of results. In addition, the radiation doses used may not harm radial glia (Pippenger et al., 1990). Aminoadipic acid may kill neurons at high doses (Olney et al., 1980).
An alternative approach to selective ablation of cells in vivo uses transgenic techniques to target expression of a toxic gene product. Expression of diphtheria toxin has been used previously in this manner to study cell lineage and cell–cell interactions in the pancreas, lens, pituitary, retina, cerebellum, and peripheral nerve (Breitman et al., 1987; Palmiter et al., 1987; Behringer et al., 1988;Lem et al., 1991; Messing et al., 1992; Smeyne et al., 1995). Although diphtheria toxin is a powerful killing agent, it has the disadvantage that cells are killed when they first express the transgene, which may produce undesirable lethal phenotypes. Several attempts have been made to devise conditional ablation strategies that permit cell killing at selected times during the life of the animal. For instance, photoablation of lacZ-expressing cells has been used conditionally to kill postmitotic neurons in transgenic mice but requires direct access to the tissue for illumination (Nirenberg and Cepko, 1993). In addition, expression of the human interleukin-2 receptor under the control of the dopamine β-hydroxylase promoter allowed selective ablation of some neuronal populations by immunotoxins (Kobayashi et al., 1995). Evans and colleagues (Borrelli et al., 1988,1989; Heyman et al., 1989) developed an alternative method for conditional ablation based on the cell-specific expression of the herpes thymidine kinase (TK) gene. Expression of the herpes TK in mammalian cells is innocuous unless an antiherpes drug is administered. For example, ganciclovir (GCV) is one of a family of drugs that are phosphorylated by the TK to toxic metabolites that kill DNA-synthesizing cells.
During the past few years we (Brenner et al., 1994) and others (Mucke et al., 1991) have identified regulatory regions in the human and mouse GFAP genes that direct expression of the Escherichia coli lacZ reporter gene to astrocytes in vivo. More recently, these GFAP regulatory regions have been used to modify functional properties of astrocytes to produce disease models (reviewed in Brenner and Messing, 1996) by directing expression of genes such as the gp120 of HIV-1 (Toggas et al., 1994), interleukin-6 (Campbell et al., 1993), human β-amyloid precursor (Mucke et al., 1994), and transforming growth factor-β (Galbreath et al., 1995; Wyss-Coray et al., 1995). To develop an improved system in which to explore the contribution of astrocytes to CNS development, we generated transgenic mice expressing the herpes TK gene under the control of the human GFAP promoter. We have focused our analysis of these mice to the cerebellum because of its simple cytoarchitecture relative to other regions of the CNS and because the majority of its cells mature postnatally, making it accessible for experimental perturbations. In addition, development of the cerebellum has been described in detail, including the birth dates and migration patterns of neuronal populations (Miale and Sidman, 1961;Fujita et al., 1966; Yuasa et al., 1991; Ryder and Cepko, 1994), and glial cells have been postulated to be significant contributors to its development (Rakic, 1971, 1972; Hatten and Liem, 1981; Edmondson and Hatten, 1987; Gao et al., 1991; Baptista et al., 1994).
We report here that treatment of GFAP-TK transgenic mice with ganciclovir during the early postnatal period results in ataxia, marked abnormalities in cerebellar granule cell survival, and fewer astrocytes. Excitotoxicity attributable to excess activation of NMDA receptors was a major factor in causing the granule cell death.
MATERIALS AND METHODS
Transgenic mouse production. Plasmid pGfa2-TK1 was constructed by excising the metallothionein promoter from pmK′ (Stuart et al., 1984) by cleavage withEcoRI and BglII and replacing it with the gfa2 promoter obtained by digestion of pGfa2-CAT (formerly called pGfaCAT-2;Besnard et al., 1991) with BglII and HindIII. Both fragments were blunt-ended by filling in before ligation. The injection fragment was excised from the plasmid by partial digestion with PvuII and by complete digestion with SspI. Plasmid pGfa2-TK2 was constructed by excising the lacZcoding region from pGfa2-lacZ (Brenner et al., 1994) by digestion with BamHI and replacing it with HSV-TK sequences consisting of the 1150 base pair (bp)BglII/BssHII fragment isolated from pmK′ joined to a synthetic 52 bp (including single-stranded ends) deoxyoligonucleotide extending from the BssHII site to 12 nucleotides downstream of the TK protein stop codon (Wagner et al., 1981). The antisense strand of the oligonucleotide had a GA added to its 5′ end to commence with a single-stranded GATC for BamHI ligation. The injection fragment was excised from the plasmid withBglII. For both plasmids, proper joining of the fragments was confirmed by sequencing.
Transgenic mice were produced according to standard techniques by microinjecting 2 pl of a solution of DNA into the male pronucleus of fertilized eggs obtained from the mating of FVB/N mice (Brinster et al., 1985). Founder mice and subsequent offspring within lines were identified by PCR analysis of DNA prepared from tail biopsies.
Nuclease protection assay. Transgene mRNA was detected by a probe protection assay. Both the riboprobes and the RNA standards were produced by in vitro transcription with SP6 RNA polymerase in the presence of 32P-UTP of PCR-generated templates. Probe protection experiments were performed by solution hybridization, followed by digestion with T1 nuclease and resolution of the products on a denaturing polyacrylamide gel.
Drug treatment. A stock solution of ganciclovir (GCV) (gift from Syntex, Palo Alto, CA) was prepared at 0.25 mg/ml in PBS. Pregnant females were checked daily, and the day of birth was considered as day 0. Neonatal mice received a single subcutaneous injection of 50 μl of stock solution/gm body weight (12.5 mg/kg) at postnatal day 1 (P1), P3, or P5. At P19, all animals were killed for collection of tissues.
In some experiments, treatment with GCV at P5 was followed immediately by MK-801 (NMDA receptor antagonist), CNQX (non-NMDA receptor antagonist), or PBS (vehicle). Stock solutions (1.2 mm) of MK-801 and CNQX (Research Biochemicals, Natick, MA) were prepared at 0.4 mg/ml in PBS and 0.28 mg/ml in dimethylsulfoxide, respectively. Neonatal mice received a single subcutaneous injection of 50 μl of MK-801 (20 mg/kg) or CNQX (14 mg/kg). Animals were killed 48 hr later (P7) for collection of tissues.
Histology and immunohistochemistry. For routine histology, the brain was removed rapidly and immersion-fixed in 10% neutral buffered formalin overnight and embedded in paraffin. Sections (6 μm) were stained with hematoxylin and eosin H&E. For immunohistochemistry, unstained paraffin sections were mounted on poly-l-lysine-coated slides (Cel-Tek, Glenviev, IL). Slides were deparaffinized in xylenes, dehydrated in graded ethanols, and washed in 0.009% hydrogen peroxide with 0.25% Triton X-100 in PBS followed by overnight incubation in 3% normal goat serum in PBS. The primary antibody was applied at a dilution of 1:500 for GFAP (Dako, Carpinteria, CA) or 1:5000 for calbindin (S Want, Bellinzona, Switzerland) for 1 hr at room temperature. The product was visualized via the Vector ABC system (Vector Laboratories, Burlingame, CA) and DAB (0.001% in PBS) as a chromagen, according to the manufacturers’ recommendations.
In situ hybridization. The DNA template for preparing a35S-UTP-labeled riboprobe was obtained by PCR amplification of a 625 bp fragment from the HSV-TK coding region spanning bp +480 to +1104 relative to the transcriptional start site. A T7 RNA polymerase promoter sequence was incorporated into the beginning of the antisense primer for use in generating the riboprobe. Animals, the tissues of which were to be used for in situ hybridization, were deeply anesthetized with Avertin and perfused transcardially through the left ventricle with 50 ml (adults) or 10 ml (newborns) of freshly prepared 4% paraformaldehyde in 0.1 m phosphate buffer. Tissues were removed, fixed overnight at 4°C, and embedded in paraffin. Unstained sections (6 μm) were mounted on poly-l-lysine-coated slides, hybridized, and washed as described previously (Lyons et al., 1991). Then slides were dipped in NTB-2 emulsion (Kodak, Rochester, NY) diluted 2:1 in 0.6 mammonium acetate and exposed in a light-tight box at 4°C for 2 weeks. The slides were developed with D-19 developer (Kodak), fixed, coverslipped, and viewed under light and dark-field microscopy.
Astrocyte cell counts. To identify astrocyte nuclei for cell counts, we crossed the GFAP-TK transgenic mice with a previously described transgenic line (TgN3Mes) carrying a nuclear form of thelacZ reporter gene under the control of the gfa2 promoter (Brenner et al., 1994). Offspring from such crosses were treated with GCV, as described above, and the tissues were processed forlacZ histochemistry. After killing, brains were rapidly removed and immersion-fixed in 10% ice-cold neutral buffered formalin for 2–3 hr and then stored in PBS until embedding. Brains were embedded in 2% Difco agar in PBS to facilitate cutting of vibratome sections (50 μm). Floating sagittal sections were washed three times for 15 min in a rinse buffer that consisted of 0.1 m PBS, 2.0 mm MgCl2, 0.24 mm Na deoxycholate, and 0.02% Nonidet P-40. The sections then were stained in the rinse buffer with the addition of 5 mm potassium ferricyanide, 5 mm potassium ferrocyanide, and 1 mg/ml X-gal for 3–4 hr at room temperature. The sections were washed three times for 15 min in the rinse buffer, mounted in glycerol/PBS (1:1), and viewed with light microscopy.
Cell counts were made in two anterior folia of the cerebellum, the central lobule and the culmen. These folia provided discrete and consistent anatomical locations in midline sagittal sections. Counts were restricted to the distal 500 μm length of each folium. The X-gal (blue)-stained nuclei in all cell layers within this length were counted. No correction was used for counting of partial nuclei in the sections. Triplicate counts were performed on each section and typically varied by <4% of the mean. A total of four lacZtransgenic (control) and four lacZ-TK double transgenic (experimental) mice were analyzed, derived from four separate litters. To allow calculation of astrocyte density in the cerebellum, we determined the area of each folia in which cell counting was performed by tracing the outline on a Zidas digitizing tablet. To account for possible interlitter variability in precise age, we performed a nested ANOVA.
Animal use. All animal protocols were approved by the Institutional Animal Care and Use Committee of the University of Wisconsin-Madison.
RESULTS
Generation of transgenic mice
We generated transgenic mice expressing the herpes TK gene under the control of a human GFAP promoter (Gfa2) that was shown previously to direct astrocyte-specific expression of a lacZreporter gene in vivo (Brenner et al., 1994). The Gfa2 promoter spans base pairs −2163 to +47 relative to the transcriptional start site of the GFAP gene. The HSV-TK was derived from a metallothionein-TK construct shown previously to express well in transgenic mice (Brinster et al., 1981). A diagram of the Gfa2-TK1 transgene is shown in Figure 1. In a separate series of experiments, we generated transgenic mice with mouse protamine-1 sequences on the 3′ end of the transgene (Gfa2-TK2), as used in the original Gfa2-lacZ mice (Brenner et al., 1994).
Fig. 1.

GFAP-HSV-TK transgene. The transgene consists of the human glial fibrillary acidic protein gfa2 promoter joined to the HSV-TK gene. The gfa2 fragment spans bp −2163 to +47 relative to the transcriptional start site, with the natural-initiating ATG at bp +15 converted to TTG by site-directed mutagenesis so that protein translation initiates within the TK. Thefilled-in portion of thebox corresponds to the TK coding region. The Gfa2-TK2 transgene is essentially identical, except that the 3′ flanking region of the TK gene has been replaced by part of the mouse protamine-1 gene, which supplies an intron and polyadenylation site.
Transgenic mice were generated by microinjection of fertilized mouse eggs, and 18 offspring that carried either the TK1 or TK2 transgenes were identified by PCR analysis of tail DNA. Most of the transgenic males failed to transmit the gene to offspring, a phenotype documented previously in several other HSV-TK transgenics that results from expression of TK from an internal promoter in round spermatids of testis (Braun et al., 1990; Al-Shawi et al., 1991). Breeding lines were established from nine founders, and RNA from brain was analyzed for the presence of transgene transcripts by nuclease protection assay (Fig.2). Offspring from three of the female founders (Tg1.8, Tg60.8, and Tg76.3) produced RNA in brain that protected the expected length of the probe (Tg1.8 and Tg76.3 carry the Gfa2-TK1 transgene, and Tg60.8 carries the Gfa2-TK2 transgene). No protection was observed in a nontransgenic control or in the liver of a transgenic line. The Tg1.8 line has been designated TgN(GFAPTK)5Mes, according to the rules for standardized genetic nomenclature of transgenic animals (Committee on Transgenic Nomenclature, 1992), and is referred to as TgN5Mes hereafter in this report. As expected, the transgenic males in these lines fail to transmit the transgene to their offspring, and the lines have been maintained by mating transgenic females with nontransgenic FVB/N males. In the absence of drug treatment, the TK transgenic mice develop normally, and, except for the male infertility, seem phenotypically normal as adults.
Fig. 2.
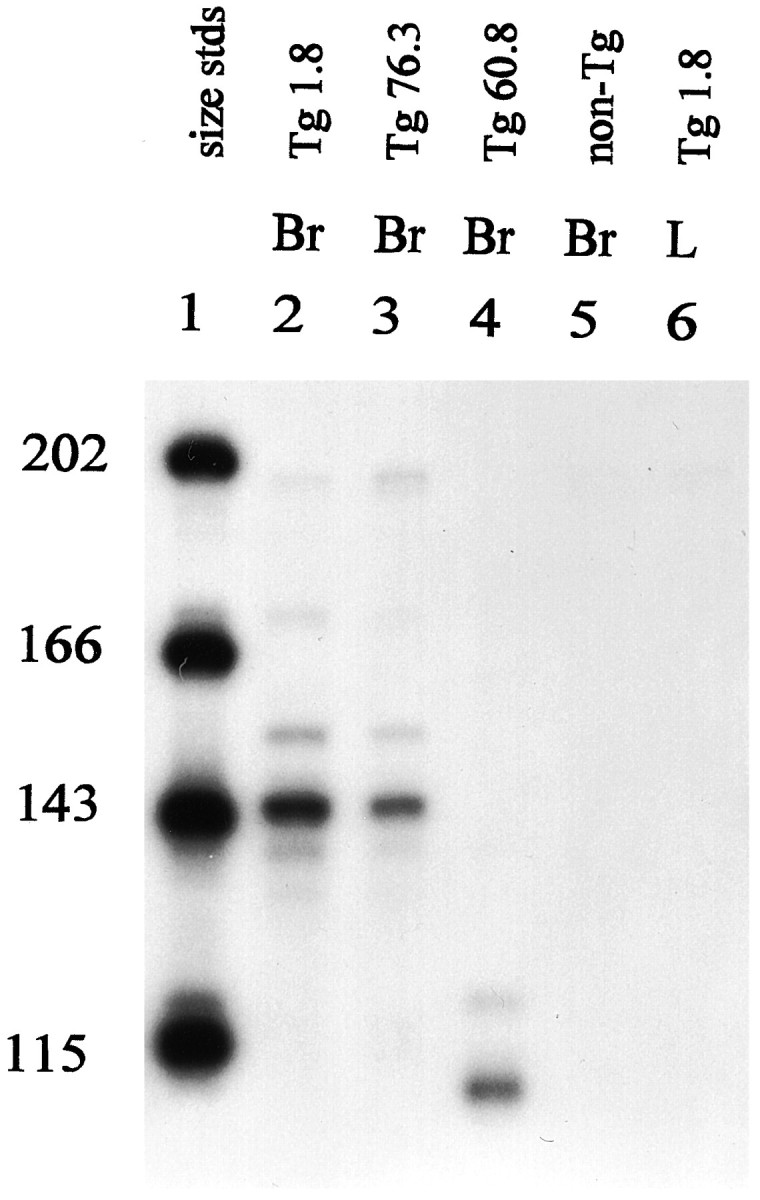
Nuclease protection assay. Representative mice of three transgenic lines were tested for production of transgene mRNA by a riboprobe protection assay, as described in Materials and Methods.Lane 1, Size standards; lane 2, Tg1.8 (referred to as TgN5Mes in this paper); lane 3, Tg76.3;lane 4, Tg60.8; lane 5, nontransgenic control; lane 6, Tg1.8. All RNA samples tested were from brain, except for lane 6, which was from liver. Either 10 μg of RNA purified by CsCl centrifugation (lanes 2,5, 6) or 20 μg of RNA purified by STAT-60 (Tel-Test “B,” Friendswood, TX) (lanes 3,4) was used. The predicted size of the riboprobe fragment protected by the Gfa2-TK1 transcript (lines Tg1.8 and Tg76.3) is 149 nucleotides, and that protected by the Gfa2-TK2 transcript (Tg60.8) is 113 nucleotides. Both probes were added to the assay of the nontransgenic control.
Phenotype of GCV-treated mice
In initial experiments, neonatal mice were given multiple injections of GCV during the first week after birth. However, these mice became very sick and often died by P10. To simplify our analysis, we then examined the effects of single drug treatments during the same early postnatal period. For each experiment, an entire litter of mouse pups received a single subcutaneous dose of GCV (see Materials and Methods) at either P1, P3, or P5. All litters were killed at P19. Because only one parent was transgenic in these matings, each litter contained both transgenic and nontransgenic pups, providing age-matched controls.
At the time of killing (P19), approximately one-half of the animals within each litter had developed characteristic motor deficits. They seemed ataxic and had difficulty standing up against the side of the cage and maintaining their balance on uneven surfaces. In all of the GCV-treated litters, every animal with ataxia was later confirmed as transgenic by PCR analysis, and every animal without ataxia was confirmed as nontransgenic (data not shown).
Histopathology of GCV-treated mice
On histological examination of the brain, the most dramatic lesions in GCV-treated TK transgenic mice were found in the cerebellum; consequently, our analysis has focused on this region. In all of the experiments described below, the lesions were most severe in animals treated at P1 and were progressively less severe with treatments at P3 and P5. In each case the overall size of the cerebellum was reduced (Fig. 3a–d). In addition, the number of granule cells and the width of the molecular layer was decreased markedly (Fig. 3e–h), particularly in transgenic animals treated with GCV at P1 (Fig. 3f). All characteristics examined (ataxia, cerebellar hypoplasia, and loss of granule cells after neonatal GCV treatments) were identical in all three lines of transgenic mice, indicating that it is independent of the integration site. The data presented below are from the TgN5Mes line of mice.
Fig. 3.
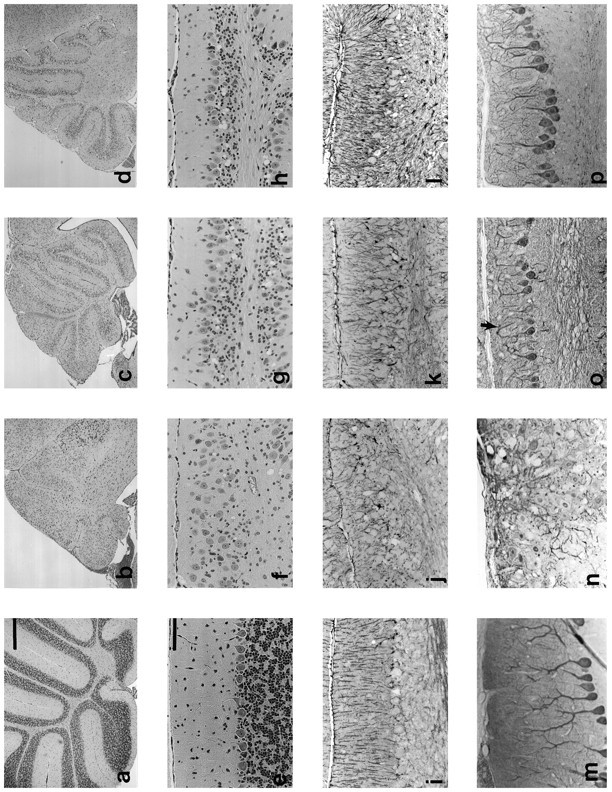
Ablation of astrocytes disrupts development of other cell types of the cerebellum. Cerebella of nontransgenic mice were treated with GCV at P1 [a, e, i, m (column 1)], or transgenic mice were treated at either P1 [b, f, j, n (column 2)], P3 [c, g, k, o (column 3)], or P5 [d, h, l, p (column 4)]. a–d, Low-power H&E paraffin sections show overall reduction in size of cerebellum of transgenic versus nontransgenic mice treated with GCV on P1, P3, or P5, with decreasing severity the later the treatment.e–h, H&E paraffin sections show loss of granule cells in transgenic mice. The Purkinje cells fail to align in a single layer in the animals treated at P1, and there is no distinct molecular layer. Both the granule cell loss and Purkinje cell disorganization become gradually less severe in animals treated at P3 and P5.i–l, GFAP immunocytochemistry displays disorganized radial Bergmann glia (especially in animals treated at P1) and apparent increase in GFAP immunoreactivity in animals treated at P3 and P5.m–p, Calbindin immunocytochemistry illustrates aberrant Purkinje cell dendritic morphology. In animals treated at P1, the dendrites project randomly rather than toward the pial surface. In animals treated at P3 or P5, the Purkinje cell primary dendrite projects toward the pial surface, but dendritic tree seems simplified, and many distal branches curve back and away from the pial surface (“weeping willow,” arrow in o). Scale bars: a–d, 500 μm; e–p, 100 μm.
Because the primary effect of GCV treatment was expected to involve astrocytes, we performed immunohistochemical staining for GFAP to visualize astrocyte morphology. As seen in Figure 3i–l, GFAP content of transgenics was not clearly diminished as the result of drug treatment and may even have increased in animals treated at P5 (Fig. 3k–l). However, there was marked disorganization of the radial fibers of Bergmann glia, especially in animals treated at P1, which had little or no molecular layer when examined at P19.
There was no decrease in the number of Purkinje cells (data not shown); however, their distribution and morphology were abnormal (Fig.3e–h). Purkinje cell bodies were distributed ectopically throughout the molecular layer in cerebella from transgenic animals treated at P1 (Fig. 3f,n). Purkinje cell dendritic morphology, evaluated by immunohistochemical detection of calbindin (Fig. 3m–p), was perturbed after all three treatment times. The primary dendrites in transgenics treated at P1 were multipolar and projected in random directions (Fig. 3n) instead of toward the pial surface. Animals treated at later stages had Purkinje cell dendrites with “weeping willow” appearances, with distal branches that turned back and away from the pial surface (Fig.3o,p).
Cerebellar histology seemed completely normal in untreated transgenic animals (data not shown) and in nontransgenic animals treated with the same dose of GCV (Fig. 3a,e,i,m). Some transgenic animals were examined at 6 months of age after a single GCV treatment at P3. These animals were still ataxic, and their cerebella showed the same loss of granule cells as in animals examined at P19, indicating a lack of regeneration (data not shown).
Localization of transgene expression
Because the most dramatic effects of GCV treatment of GFAP-TK transgenic mice were on developing granule cells, we considered the possibility that the transgene was expressed ectopically in the granule cell precursors that are mitotically active during the period of drug treatment. To localize sites of transgene expression in postnatal mice, we performed in situ hybridization with a riboprobe specific for the TK portion of the transcript. As a positive control, we used testis from the transgenic mice, which were presumed to express the transgene because of the male sterility (see above; Fig.4c). Nontransgenic littermates hybridized with the same antisense probe served as negative controls (Fig.4d–f). As shown in Figure 4 (a,b), neither the immature granule cell precursors in the external granule cell layer of the P1 mouse nor the mature granule cells in the internal granule cell layer of the adult mouse displayed detectable levels of labeling above background. The highest grain density was in the Purkinje cell layer, a localization consistent with expression in Bergmann glia, the cell bodies of which reside in this region. In addition, labeling over the white matter was higher in the transgenic mice, presumably reflecting expression in more dispersed populations of astrocytes throughout the cerebellum. This pattern of transgene expression is also consistent with that observed previously with the gfa2-lacZ transgene (Besnard et al., 1991). These data suggest that the granule cell loss is not the result of ectopic expression of the transgene in cells of the EGL but rather is a secondary effect of astrocyte and/or Bergmann glial expression.
Fig. 4.
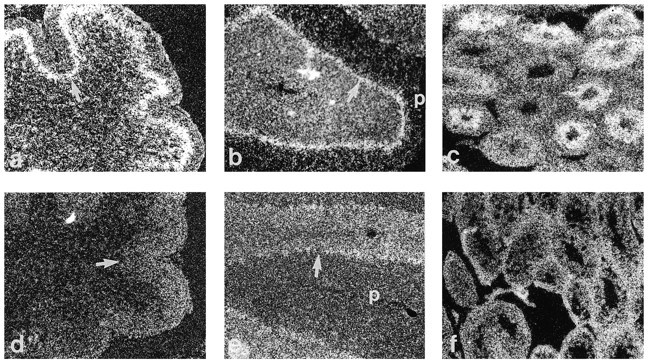
In situ hybridization showing localization of transgene expression by autoradiography and dark-field microscopy in nondrug-treated mice. a, d, P1 transgenic and nontransgenic cerebellum, respectively. b, e, Adult transgenic and nontransgenic cerebellum, respectively. c, f, Adult transgenic and nontransgenic testes, respectively. Note that there is no specific labeling in the external granule layer in the P1 transgenic cerebellum (a) nor in the internal granule cell layer of the adult transgenic cerebellum (b), as compared with the background level seen in nontransgenic controls (d, e). Specific labeling is highest in the Purkinje cell layer, which contains the cell bodies of Bergmann glia but is also apparent in white matter. Expression in postmitotic spermatids of the testis (c) has been observed previously in other TK transgenic models.p, Pial surface; arrow points to Purkinje cell layer in P1 and adult cerebella.
Evaluation of astrocyte loss
The premise of this project was that GCV treatment of the GFAP-TK transgenic mice would result in killing of dividing astrocytes. However, immunohistochemistry showed that there was no decrease, and perhaps even an increase, in GFAP staining in GCV-treated mice. As it was possible that a gliotic response in the treated animals obscured a decrease in cell numbers, we counted astrocytes by genetically marking their nuclei. This was accomplished by crossing the TgN5Mes mice with a previously described line, TgN3Mes (Brenner et al., 1994), which expresses a nuclear form of the lacZ reporter gene in astrocytes. Astrocytes then could be identified by their blue nuclei after histochemical staining.
In these experiments we focused on a single treatment with GCV at P3, because treatment at this time point yielded cerebella that still developed sufficiently to have clearly recognizable folia. Treatment of double transgenic TgN5Mes × TgN3Mes offspring with GCV at P3 yielded the same behavioral and histological phenotype described above for TgN5Mes single transgenic mice. To quantitate the effect of GCV treatment on astrocyte numbers, we counted X-gal-stained nuclei in vibratome sections prepared from lacZ single transgenic andlacZ-TK double transgenic mice, focusing on the central lobule and culmen. To minimize the potential artifact of counting off the midline where folia are shorter, only cells lying in the distal 500 μm of each folium were counted. Figure 5 shows the effects of GCV on lacZ expression in the central lobule (the second anterior folia of the mouse cerebellum) of a single transgeniclacZ transgenic mouse, which served as a control (Fig.5a), and the double transgenic lacZ-TK mouse (Fig. 5b). In the control cerebellum, there was an organized row of labeled nuclei in the Purkinje cell layer, which have previously been identified as Bergmann glia (Brenner et al., 1994). In addition, there were dispersed nuclei of astrocytes in the internal granule cell layer and the white matter. In the cerebellum from the double transgenic animal (Fig. 5b), there was a clear decrease in the number of astrocytes and no distinct Bergmann glial cell layer. Counts of the astrocyte nuclei revealed that the double transgenic mice contain approximately one-half the number of astrocytes of the singlelacZ transgenic mice (Fig. 6a). Additionally, the astrocyte density in the double transgenics was ∼19-40% less than that in single lacZ transgenic mice (Fig. 6b).
Fig. 5.
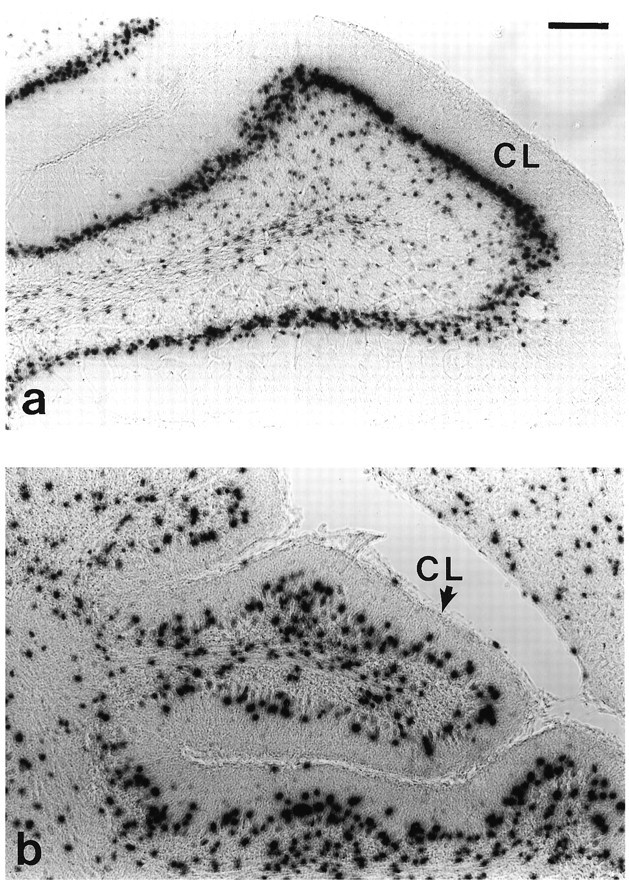
Histochemical detection of astrocyte nuclei with gfa2-nlacZ transgenic mice. a, Central lobule of cerebellum from nlacZ transgenic mouse (control) treated with GCV at P3. b, Central lobule of cerebellum from nlacZ-TK double transgenic mouse (experimental) treated with GCV at P3. Note the overall smaller size of the folia, the disorganization of the nuclei in the Purkinje cell layer, and the decrease in nlacZ-stained nuclei.CL, Central lobule; scale bar, 100 μm.
Fig. 6.
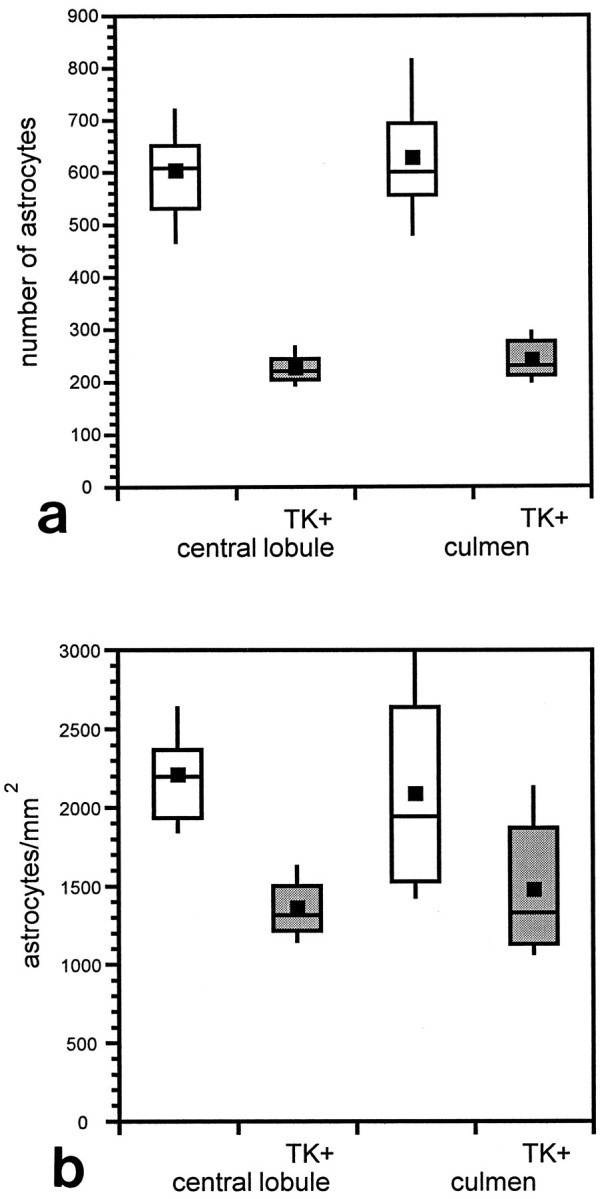
Statistical representation of astrocyte loss in TK transgenic mice. All counts were performed on 50 μm vibratome midline sagittal sections and included only the distal 500 μm of the folia. Shown are box plots of mean and variance of astrocyte number (a) or density (b) in the culmen and central lobule. Squares incenter of boxes represent the mean in that group of data. The second and third quartile are represented by the area above and below thecenter line, respectively. The barsextend to the minimum and maximum. a, With mean cell counts in the control folia of 601 and 625 for the central lobule and culmen, respectively, nested ANOVA suggested an average cell loss of 354 ± 54 in the central lobule and 360 ± 48 in the culmen (99.5% confidence interval). b, With mean density in the control of 2202 and 2081 astrocytes/mm2 for the central lobule and culmen, respectively, nested ANOVA suggested an average decrease in density of 876 ± 393 in the central lobule and 386 ± 116 in the culmen (99.7% confidence interval).
Rescue of granule cells
The presence of astrocytes has been shown to protect cultured neurons from the excitotoxic effects of glutamate (Rosenberg and Aizenman, 1989). It is thus possible that the loss of cerebellar granule cells in the transgenic mice could be, in part, attributable to an accumulation of glutamate resulting from a reduction in astrocyte numbers. Because activation of the NMDA receptor has been implicated in the glutamate toxicity of neurons (Choi, 1992), we examined the effect of the NMDA receptor antagonist MK-801 on granule cell survival. Wild-type and transgenic mice were treated with GCV in the presence or absence of MK-801 at P5, and their cerebella were examined 48 hr later. Figure 7a–c shows the base of the cerebellar folia between the culmen and the declive from a nontransgenic animal treated with GCV and MK-801, a transgenic animal treated with GCV and PBS, and a transgenic animal treated with GCV and MK-801, respectively. In the nontransgenic animal, the external granule cell layer (EGL) is 7–9 cells thick (Fig. 7a), whereas in the cerebellum of a transgenic animal treated with GCV and PBS, the EGL is reduced to 0–3 cells thick (Fig. 7b). In transgenic animals treated with GCV and MK-801, the EGL is 5–7 cells thick (Fig. 7c). Transgenic animals treated with GCV and CNQX (a non-NMDA receptor antagonist) have EGLs that are 0–3 cells thick, similar to animals treated with GCV and PBS (data not shown). These results suggest that NMDA receptor-mediated excitotoxicity is at least partially responsible for the death of cells in the EGL.
Fig. 7.
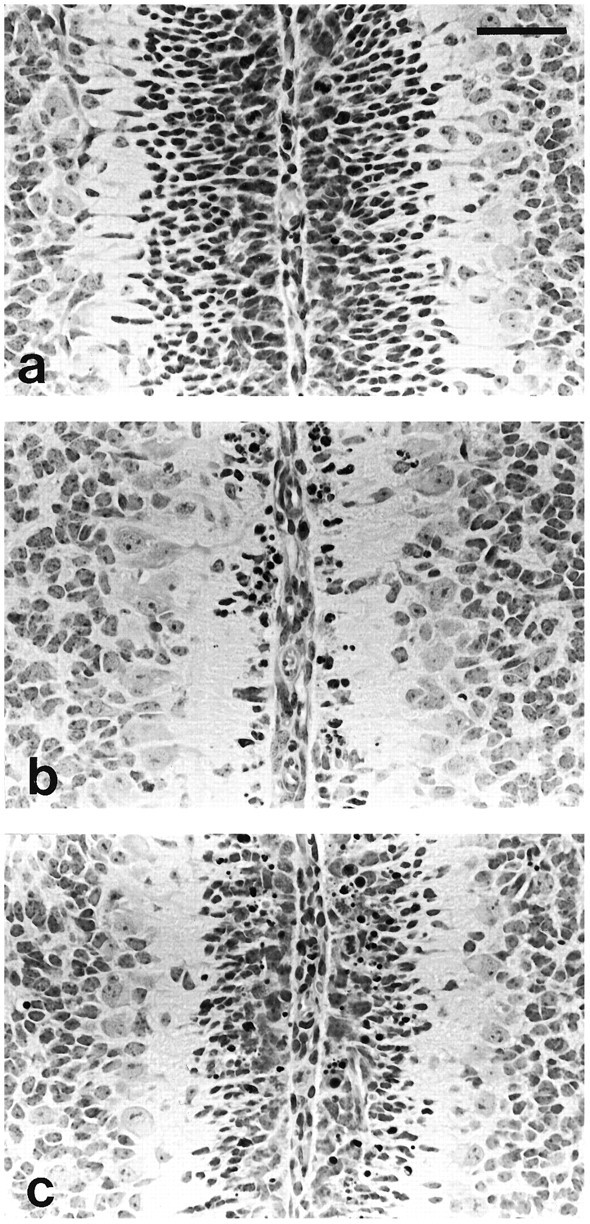
Effect of MK-801 on cell death in the EGL after GCV treatment. Animals were treated with GCV and either PBS or MK-801 at P5, and tissue was taken for analysis 48 hr later. Each panel shows a photomicrograph of an H&E-stained paraffin section through the base of the cerebellar folia between the culmen and declive, near the midline. a, Nontransgenic mouse treated with GCV and MK-801 at P5, 48 hr survival; the EGL is 7–9 layers thick.b, TgN5Mes treated with GCV and PBS; the EGL is 0–3 layers thick. c, TgN5Mes treated with GCV and MK-801; the EGL is 5–9 layers thick. Scale bar, 50 μm.
DISCUSSION
We have produced GFAP-TK transgenic mice so that dividing astrocytes can be ablated at selected times to evaluate their role in CNS function. In this initial study we have focused on the development of the cerebellum, which undergoes significant morphogenesis for several days after birth. GCV treatment of transgenic animals shortly after birth resulted in marked deficits in cerebellar development; Purkinje cells were ectopically distributed and had deranged dendritic trees, Bergmann glia were fewer in number and had disorganized processes, and granule cells were severely depleted. Although not the subject of the present study, GCV treatment also seemed to reduce myelination significantly (C.L. Delaney, M. Brenner, A. Messing, unpublished observations).
The effects of GCV treatment on cerebellar development progressively decreased as the interval between birth and drug administration increased. This declining effectiveness is consistent with the gradual decline in astrocyte proliferation in the cerebellum after birth (Korr, 1986). In adult animals, in which astrocyte mitosis is rare, even 100-fold higher doses of GCV than that used at P1–P5 produced no phenotype or histopathological effects (data not shown). These results are consistent with the toxicity of GCV being specific to dividing cells.
When evaluated at P19, a clear decrease in astrocyte numbers could be documented by counting nuclei genetically marked by expression of a GFAP-lacZ transgene. For technical reasons, the recorded decrease of ∼50% is probably an underestimate of the actual percentage of astrocytes that are lost. For instance, no correction was used for counting of partial nuclei in the sections. Because nuclei of reactive astrocytes are likely larger than those of normal astrocytes (Fedoroff et al., 1984), partial nuclei would be more likely in the GCV-transgenic mice than in controls. We also limited our cell counts to the distal portions of the folia to avoid artifacts from counting off the midline, where folia would be shorter than in the center. Because the GCV-transgenic group had a much smaller molecular layer than the control group and because this layer was relatively poor in astrocytes, the bias again would be toward minimizing the difference between the two groups. It is also possible that the loss of astrocytes produced by GCV treatment is compensated partially by a decrease in the extent of natural cell death that occurs during this period (Krueger et al., 1995). Additionally, because the half-life of GCV in blood is <1 hr (Paul and Dummer, 1992) and the half-life of the phosphorylated-GCV is slower but still <24 hr (Biron et al., 1985), there could be replenishment by proliferation of reactive astrocytes. These considerations suggest that the ∼50% decrease in astrocyte numbers observed at P19 in treated animals is a minimal estimate of the initial loss. Compounding the effects of astrocyte loss, some of the surviving astrocytes may have been dysfunctional. Such a sublethal effect was demonstrated recently in a transgenic study of TK expression in thyroid follicular cells (Wallace et al., 1994).
Although a clear decrease in astrocyte numbers by P19 was demonstrated, immunostaining for GFAP was not diminished at this time. This is likely attributable to the upregulation of GFAP levels and hypertrophy of the remaining astrocytes. Preliminary experiments involving additional drug treatments to ablate astrocytes more completely resulted in much higher mortality in the mice (unpublished observations). In several studies of the immediate post-treatment period (for example, Fig. 7b), we were unable to identify a significant increase in the numbers of dying astrocytes. However, it may have been difficult to observe GCV-mediated astrocyte death against the background of normal astrocyte loss that occurs during this period of development (Krueger et al., 1995) and the rapid clearing of apoptotic cells that typically takes place.
Perhaps the most striking observation in our study was the dramatic loss of granule cells and alteration of Purkinje cell dendrites after GCV treatment. Elaboration of Purkinje cell dendrites coincides with maturation of granule cells (Caddy and Herrup, 1990) and requires afferent input from granule cell parallel fibers (Bradley and Berry, 1976; Baptista et al., 1994). Therefore, the Purkinje cell defect is likely secondary to the loss of granule cells. We have considered several possible mechanisms by which the nearly complete ablation of granule cells could have occurred. A trivial explanation would be that the granule cells or their precursors also express the GFAP-TK transgene. Our in situ hybridization for TK mRNA did not detect any significant signal from nonastrocytic cells, nor, in extensive analyses of lacZ expression driven by the same GFAP promoter, have we seen any evidence for expression in granule cells or their precursors (Brenner et al., 1994). Thus, if these cells express the transgene, it is at levels lower than we can detect.
Another possibility is that granule cells or their precursors are being killed by a “bystander effect,” a phenomenon well documented in tumor models (Caruso et al., 1993; Freeman et al., 1993), in which cells in the vicinity of TK-expressing cells also are killed by antiherpes drugs. The mechanism generally accepted for this process is the transfer of the toxic phosphorylated GCV product to adjacent cells via gap junctions (Culver et al., 1992; Bi et al., 1993; Mesnil et al., 1996). This mechanism seems unlikely in this instance, because gap junctions have not been noted between astrocytes and granule cells or their precursors in any of multiple studies of cerebellar architecture (Rakic, 1971; Rakic and Sidman, 1973a). A caveat, however, is that the detection of such junctions was not the primary focus of these studies. An alternative but less favored mechanism for the bystander effect, phagocytosis of apoptotic bodies from the TK-expressing cells (Freeman et al., 1993), also cannot be dismissed.
The bystander effect can be excluded at least partially, however, on the basis of the protection of granule cells by the NMDA antagonist MK-801. The rescue of substantial numbers of granule cells by MK-801 indicates that excitotoxicity is a significant factor in the loss of these cells. The putative increase in extracellular glutamate triggering this loss could arise from one or a combination of sources, including release of glutamate from dying astrocytes, a reduced ability of surviving astrocytes to sequester glutamate, or release from dying granule cells.
The dramatic loss of granule cells after GCV treatment resembles the phenotypic effects of treatment with various toxins and in spontaneous mutants affecting the cerebellum (Hatten and Heintz, 1995). However, the roles that astrocytes play in these models has been difficult to establish. For instance, the antimetabolite 6-aminonicotinamide interrupts a metabolic pathway important in glial cells but damages developing granule neurons as well (Sotelo and Rio, 1980). Theweaver mutation originally was considered to cause granule cell loss secondary to a primary defect in Bergmann glia (Rakic and Sidman, 1973b) but recently has been identified as a potassium channel expressed in granule cells (Patil et al., 1995). It is interesting thatmeander tail mutants show loss of granule cells and disoriented astroglia and Purkinje cells, abnormalities that are very similar to those observed in the present study. The effects ofmeander tail, however, are limited to the anterior lobes of the cerebellum (Ross et al., 1990), and the molecular defect in this mutant has not yet been identified. Nevertheless, our studies suggest that meander tail may reflect a localized abnormality of astrocytes.
In conclusion, we have developed a mouse model that allows the selective ablation of dividing astrocytes. Postnatal ablations have suggested that astrocytes play an important role in the survival and maturation of neuronal populations examined in the cerebellum. This ablation model should also be useful for addressing other questions concerning the role of astrocytes during development and in response to injury throughout the mammalian CNS.
Footnotes
This work was supported by Grant RG 2487-A1 from the National Multiple Sclerosis Society to A.M. We thank M. Blonski, E. Galbreath, G. Lyons, H. Peickert, and D. Springman for technical assistance and R. Miller for performing the statistical analysis. We also thank Syntex Corporation for the gift of ganciclovir and Dr. R. D. Palmiter for the gift of a metallothionein-TK plasmid.
Correspondence should be addressed to Catherine L. Delaney, School of Veterinary Medicine, 2015 Linden Drive West, Madison, WI 53706.
REFERENCES
- 1.Al-Shawi R, Burke J, Wallace H, Jones C, Harrison S, Buxton D, Maley S, Chandley A, Bishop JO. The herpes simplex virus type 1-thymidine kinase is expressed in the testes of transgenic mice under the control of a cryptic promoter. Mol Cell Biol. 1991;11:4207–4216. doi: 10.1128/mcb.11.8.4207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Baptista CA, Hatten ME, Blazeski R, Mason CA. Cell–cell interactions influence survival and differentiation of purified Purkinje cells in vitro. Neuron. 1994;12:243–260. doi: 10.1016/0896-6273(94)90268-2. [DOI] [PubMed] [Google Scholar]
- 3.Behringer RR, Mathews LS, Palmiter RD, Brinster RL. Dwarf mice produced by genetic ablation of growth hormone-expressing cells. Genes Dev. 1988;2:453–461. doi: 10.1101/gad.2.4.453. [DOI] [PubMed] [Google Scholar]
- 4.Besnard F, Brenner M, Nakatani Y, Chao R, Purohit HJ, Freese E. Multiple interacting sites regulate astrocyte-specific transcription of the human gene for glial fibrillary acidic protein. J Biol Chem. 1991;266:18877–18883. [PubMed] [Google Scholar]
- 5.Bi W, Parysek L, Warnick R, Stambrook P. In vitro evidence that metabolic cooperation is responsible for the bystander effect observed with HSV-tk retroviral gene therapy. Hum Gene Ther. 1993;4:725–731. doi: 10.1089/hum.1993.4.6-725. [DOI] [PubMed] [Google Scholar]
- 6.Biron KK, Stanat SC, Sorrell JB, Fyfe JA, Keller PM, Lambe CU, Nelson DJ. Metabolic activation of the nucleoside analog 9-[(2-hydroxy-1-[hydroxymethyl]ethoxy)methyl]guanine in human diploid fibroblasts infected with human cytomegalovirus. Proc Natl Acad Sci USA. 1985;82:2473–2477. doi: 10.1073/pnas.82.8.2473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bogler O, Wren D, Barnett SC, Land H, Noble M. Cooperation between two growth factors promotes extended self-renewal and inhibits differentiation of oligodendrocyte-type-2 astrocyte (O-2A) progenitor cells. Proc Natl Acad Sci USA. 1990;87:6368–6372. doi: 10.1073/pnas.87.16.6368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Borrelli E, Heyman R, Hsi M, Evans RM. Targeting of an inducible toxic phenotype in animal cells. Proc Natl Acad Sci USA. 1988;85:7572–7576. doi: 10.1073/pnas.85.20.7572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Borrelli E, Heyman RA, Arias C, Sawchenko PE, Evans RM. Transgenic mice with inducible dwarfism. Nature. 1989;339:538–541. doi: 10.1038/339538a0. [DOI] [PubMed] [Google Scholar]
- 10.Borrett D, Becker LE. Alexander’s disease. A disease of astrocytes. Brain. 1985;108:367–385. doi: 10.1093/brain/108.2.367. [DOI] [PubMed] [Google Scholar]
- 11.Bradley P, Berry M. The effects of reduced climbing and parallel fibre input on Purkinje cell dendritic growth. Brain Res. 1976;109:133–151. doi: 10.1016/0006-8993(76)90384-x. [DOI] [PubMed] [Google Scholar]
- 12.Braun RE, Lo D, Pinkert CA, Widera G, Flavell RA, Palmiter RD, Brinster RL. Infertility in male transgenic mice: disruption of sperm development by HSV-tk expression in postmeiotic germ cells. Biol Reprod. 1990;43:684–693. doi: 10.1095/biolreprod43.4.684. [DOI] [PubMed] [Google Scholar]
- 13.Breitman ML, Clapoff S, Rossant J, Tsui LC, Glode LM, Maxwell IH, Bernstein A. Genetic ablation: targeted expression of a toxin gene causes microphthalmia in transgenic mice. Science. 1987;238:1563–1565. doi: 10.1126/science.3685993. [DOI] [PubMed] [Google Scholar]
- 14.Brenner M, Messing A (1996) GFAP transgenic mice. Methods, in press. [DOI] [PubMed]
- 15.Brenner M, Kisseberth WC, Su Y, Besnard F, Messing A. GFAP promoter directs astrocyte-specific expression in transgenic mice. J Neurosci. 1994;14:1030–1037. doi: 10.1523/JNEUROSCI.14-03-01030.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Brinster RL, Chen HY, Trumbauer M, Senear AW, Warren R, Palmiter RD. Somatic expression of herpes thymidine kinase in mice following injection of a fusion gene into eggs. Cell. 1981;27:223–231. doi: 10.1016/0092-8674(81)90376-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brinster RL, Chen HY, Trumbauer ME, Yagle MK, Palmiter RD. Factors affecting the efficiency of introducing foreign genes into mice by microinjecting eggs. Proc Natl Acad Sci USA. 1985;82:4438–4442. doi: 10.1073/pnas.82.13.4438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Butterworth RF. Portal-systemic encephalopathy: a disorder of neuron-astrocytic metabolic trafficking. Dev Neurosci. 1993;15:313–319. doi: 10.1159/000111350. [DOI] [PubMed] [Google Scholar]
- 19.Caddy KW, Herrup K. Studies of the dendritic tree of wild-type cerebellar Purkinje cells in lurcher chimeric mice. J Comp Neurol. 1990;297:121–131. doi: 10.1002/cne.902970109. [DOI] [PubMed] [Google Scholar]
- 20.Campbell IL, Abraham CR, Masliah E, Kemper P, Inglis JD, Oldstone MB, Mucke L. Neurologic disease induced in transgenic mice by cerebral overexpression of interleukin 6. Proc Natl Acad Sci USA. 1993;90:10061–10065. doi: 10.1073/pnas.90.21.10061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caruso M, Panis Y, Gagandeep S, Houssin D, Salzmann J-L, Klatzmann D. Regression of established macroscopic liver metastases after in situ transduction of a suicide gene. Proc Natl Acad Sci USA. 1993;90:7024–7028. doi: 10.1073/pnas.90.15.7024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Choi DW. Excitotoxic cell death. J Neurobiol. 1992;23:1261–1276. doi: 10.1002/neu.480230915. [DOI] [PubMed] [Google Scholar]
- 23.Committee on Transgenic Nomenclature. Standardized nomenclature for transgenic animals. ILAR News. 1992;34:45–50. [Google Scholar]
- 24.Culver KW, Ram Z, Wallbridge S, Ishii H, Oldfield EH, Blaese RM. In vivo gene transfer with retroviral vector-producer cells for treatment of experimental brain tumors. Science. 1992;256:1550–1552. doi: 10.1126/science.1317968. [DOI] [PubMed] [Google Scholar]
- 25.Edmondson JC, Hatten ME. Glial-guided granule neuron migration in vitro: a high-resolution time-lapse video microscopic study. J Neurosci. 1987;7:1928–1934. doi: 10.1523/JNEUROSCI.07-06-01928.1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fedoroff S, McAuley WA, Houle JD, Devon RM. Astrocyte cell lineage. V. Similarity of astrocytes that form in the presence of dBcAMP in cultures to reactive astrocytes in vivo. J Neurosci Res. 1984;12:14–27. doi: 10.1002/jnr.490120103. [DOI] [PubMed] [Google Scholar]
- 27.Freeman SM, Abboud CN, Whartenby KA, Packman CH, Koeplin DS, Moolten FL, Abraham GN. The “bystander effect”: tumor regression when a fraction of the tumor mass is genetically modified. Cancer Res. 1993;53:5274–5283. [PubMed] [Google Scholar]
- 28.Fujita S, Shimada M, Nakamura T. 3H-thymidine autoradiographic studies on the cell proliferation and differentiation in the external and internal granular layers of the mouse cerebellum. J Comp Neurol. 1966;128:191–208. doi: 10.1002/cne.901280206. [DOI] [PubMed] [Google Scholar]
- 29.Gage FH, Olejniczak P, Armstrong DM. Astrocytes are important for sprouting in the septohippocampal circuit. Exp Neurol. 1988;102:2–13. doi: 10.1016/0014-4886(88)90073-8. [DOI] [PubMed] [Google Scholar]
- 30.Galbreath E, Kim S-J, Park K, Brenner M, Messing A. Overexpression of TGF-b1 in the central nervous system of transgenic mice results in hydrocephalus. J Neuropathol Exp Neurol. 1995;54:339–349. doi: 10.1097/00005072-199505000-00007. [DOI] [PubMed] [Google Scholar]
- 31.Gao W-Q, Heintz N, Hatten ME. Cerebellar granule cell neurogenesis is regulated by cell–cell interaction in vitro. Neuron. 1991;6:705–715. doi: 10.1016/0896-6273(91)90168-y. [DOI] [PubMed] [Google Scholar]
- 32.Gomi H, Yokoyama T, Fujimoto K, Ideka T, Katoh A, Itoh T, Itohara S. Mice devoid of the glial fibrillary acidic protein develop normally and are susceptible to scrapie prions. Neuron. 1995;14:29–41. doi: 10.1016/0896-6273(95)90238-4. [DOI] [PubMed] [Google Scholar]
- 33.Hatten ME, Heintz N. Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci. 1995;18:385–408. doi: 10.1146/annurev.ne.18.030195.002125. [DOI] [PubMed] [Google Scholar]
- 34.Hatten ME, Liem RK. Astroglial cells provide a template for the positioning of developing cerebellar neurons in vitro. J Cell Biol. 1981;90:622–630. doi: 10.1083/jcb.90.3.622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heyman RA, Borrelli E, Lesley J, Anderson D, Richman DD, Baird SM, Hyman R, Evans RM. Thymidine kinase obliteration: creation of transgenic mice with controlled immune deficiency. Proc Natl Acad Sci USA. 1989;86:2698–2702. doi: 10.1073/pnas.86.8.2698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hutter H, Schnabel R. Specification of anterior–posterior differences within the AB lineage in the C. elegans embryo: a polarising induction. Development (Camb) 1995;121:1559–1568. doi: 10.1242/dev.121.5.1559. [DOI] [PubMed] [Google Scholar]
- 37.Kalderon N, Alfieri AA, Fuks Z. Beneficial effects of x-irradiation on recovery of lesioned mammalian central nervous tissue. Proc Natl Acad Sci USA. 1990;87:10058–10062. doi: 10.1073/pnas.87.24.10058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kawaja MD, Gage FH. Reactive astrocytes are substrates for the growth of adult CNS axons in the presence of elevated levels of nerve growth factor. Neuron. 1991;7:1019–1030. doi: 10.1016/0896-6273(91)90346-2. [DOI] [PubMed] [Google Scholar]
- 39.Khurgel M, Koo AC, Ivy GO. Selective ablation of astrocytes by intracerebral injections of α-aminoadipate. Glia. 1996;16:351–358. doi: 10.1002/(SICI)1098-1136(199604)16:4<351::AID-GLIA7>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 40.Kobayashi K, Morita S, Sawada H, Mizuguchi T, Yamada K, Nagatsu I, Fujita K, Kreitman RJ, Pastan I, Nagatsu T. Immunotoxin-mediated conditional disruption of specific neurons in transgenic mice. Proc Natl Acad Sci USA. 1995;92:1132–1136. doi: 10.1073/pnas.92.4.1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Korr H. Proliferation and cell cycle parameters of astrocytes. In: Fedoroff S, Vernadakis A, editors. Astrocytes: cell biology and pathology of astrocytes. Academic; New York: 1986. pp. 77–127. [Google Scholar]
- 42.Krueger BK, Burne JF, Raff MC. Evidence for large-scale astrocyte death in the developing cerebellum. J Neurosci. 1995;15:3366–3374. doi: 10.1523/JNEUROSCI.15-05-03366.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Largo C, Cuevas P, Somjen GG, Del Río RM, Herreras O. The effect of depressing glial function in rat brain in situ on ion homeostasis, synaptic transmission, and neuron survival. J Neurosci. 1996;16:1219–1229. doi: 10.1523/JNEUROSCI.16-03-01219.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lem J, Applebury ML, Falk JD, Flannery JG, Simon MI. Tissue-specific and developmental regulation of rod opsin chimeric genes in transgenic mice. Neuron. 1991;6:201–210. doi: 10.1016/0896-6273(91)90356-5. [DOI] [PubMed] [Google Scholar]
- 45.Lyons GE, Muhlebach S, Moser A, Masood R, Paterson BM, Buckingham ME, Perriard JC. Developmental regulation of creatine kinase gene expression by myogenic factors in embryonic mouse and chick skeletal muscle. Development (Camb) 1991;113:1017–1029. doi: 10.1242/dev.113.3.1017. [DOI] [PubMed] [Google Scholar]
- 46.MacVicar BA, Tse FW, Crichton SA, Kettenmann H. GABA-activated Cl− channels in astrocytes of hippocampal slices. J Neurosci. 1989;9:3577–3583. doi: 10.1523/JNEUROSCI.09-10-03577.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McCall MA, Gregg RG, Behringer RR, Brenner M, Delaney CL, Galbreath EJ, Zhang CL, Pearce RA, Chiu SY, Messing A. Targeted deletion in astrocyte intermediate filament (Gfap) alters neuronal physiology. Proc Natl Acad Sci USA. 1996;93:6361–6366. doi: 10.1073/pnas.93.13.6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McKinnon RD, Matsui T, Dubois-Dalcq M, Aaronson SA. FGF modulates the PDGF-driven pathway of oligodendrocyte development. Neuron. 1990;5:603–614. doi: 10.1016/0896-6273(90)90215-2. [DOI] [PubMed] [Google Scholar]
- 49.Mesnil M, Piccoli C, Tiraby G, Willecke K, Yamasaki H. Bystander killing of cancer cells by herpes simplex virus-thymidine kinase gene is mediated by connexins. Proc Natl Acad Sci USA. 1996;93:1831–1835. doi: 10.1073/pnas.93.5.1831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Messing A, Behringer RR, Hammang JP, Palmiter RD, Brinster RL, Lemke G. P0 promoter directs expression of reporter and toxin genes to Schwann cells in transgenic mice. Neuron. 1992;8:507–520. doi: 10.1016/0896-6273(92)90279-m. [DOI] [PubMed] [Google Scholar]
- 51.Miale IL, Sidman RL. An autoradiographic analysis of histogenesis in the mouse cerebellum. Exp Neurol. 1961;4:277–296. doi: 10.1016/0014-4886(61)90055-3. [DOI] [PubMed] [Google Scholar]
- 52.Mucke L, Oldstone MBA, Morris JC, Nerenberg MI. Rapid activation of astrocyte-specific expression of GFAP-lacZ transgene by focal injury. New Biol. 1991;3:465–474. [PubMed] [Google Scholar]
- 53.Mucke L, Masliah E, Johnson WB, Ruppe MD, Alford M, Rockenstein EM, Forss-Petter S, Pietropaolo M, Mallory M, Abraham CR. Synaptotrophic effects of human amyloid b protein precursors in the cortex of transgenic mice. Brain Res. 1994;666:151–167. doi: 10.1016/0006-8993(94)90767-6. [DOI] [PubMed] [Google Scholar]
- 54.Nirenberg S, Cepko C. Targeted ablation of diverse cell classes in the nervous system in vivo. J Neurosci. 1993;13:3238–3251. doi: 10.1523/JNEUROSCI.13-08-03238.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Noble M, Murray K, Stroobant P, Waterfield MD, Riddle P. Platelet-derived growth factor promotes division and motility and inhibits premature differentiation of the oligodendrocyte/type-2 astrocyte progenitor cell. Nature. 1988;333:560–562. doi: 10.1038/333560a0. [DOI] [PubMed] [Google Scholar]
- 56.Norenberg MD. Astrocyte responses to CNS injury. J Neuropathol Exp Neurol. 1994;53:213–220. doi: 10.1097/00005072-199405000-00001. [DOI] [PubMed] [Google Scholar]
- 57.Oh Y, Waxman SG. The b1 subunit mRNA of the rat brain Na+ channel is expressed in glial cells. Proc Natl Acad Sci USA. 1994;91:9985–9989. doi: 10.1073/pnas.91.21.9985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Olney JW, de Gubareff T, Collins JF. Stereospecificity of the gliotoxic and anti-neurotoxic actions of alpha-aminoadipate. Neurosci Lett. 1980;19:277–282. doi: 10.1016/0304-3940(80)90273-6. [DOI] [PubMed] [Google Scholar]
- 59.Palmiter RD, Behringer RR, Quaife CJ, Maxwell F, Maxwell IH, Brinster RL. Cell lineage ablation in transgenic mice by cell-specific expression of a toxin gene. Cell. 1987;50:435–443. doi: 10.1016/0092-8674(87)90497-1. [DOI] [PubMed] [Google Scholar]
- 60.Patil N, Cox DR, Bhat D, Faham M, Myers RM, Peterson AS. A potassium channel mutation in weaver mice implicates membrane excitability in granule cell differentiation. Nat Genet. 1995;11:126–129. doi: 10.1038/ng1095-126. [DOI] [PubMed] [Google Scholar]
- 61.Paul S, Dummer S. Topics in clinical pharmacology: ganciclovir. Am J Med Sci. 1992;304:272–277. doi: 10.1097/00000441-199210000-00010. [DOI] [PubMed] [Google Scholar]
- 62.Pekny M, Levéen P, Pekna M, Eliasson C, Berthold C-H, Westermark B, Betsholtz C. Mice lacking glial fibrillary acidic protein display astrocytes devoid of intermediate filaments but develop and reproduce normally. EMBO J. 1995;14:1590–1598. doi: 10.1002/j.1460-2075.1995.tb07147.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pippenger MA, Sims TJ, Gilmore SA. Development of the rat corticospinal tract through an altered glial environment. Dev Brain Res. 1990;55:43–50. doi: 10.1016/0165-3806(90)90104-7. [DOI] [PubMed] [Google Scholar]
- 64.Politis MJ, Houle JD. Effect of cytosine arabinofuranoside (AraC) on reactive gliosis in vivo. An immunohistochemical and morphometric study. Brain Res. 1985;328:291–300. doi: 10.1016/0006-8993(85)91041-8. [DOI] [PubMed] [Google Scholar]
- 65.Raff MC, Lillien LE, Richardson WD, Burne JF, Noble MD. Platelet-derived growth factor from astrocytes drives the clock that times oligodendrocyte development in culture. Nature. 1988;333:562–565. doi: 10.1038/333562a0. [DOI] [PubMed] [Google Scholar]
- 66.Rakic P. Neuron–glia relationship during granule cell migration in developing cerebellar cortex. A Golgi and electronmicroscopic study in Macacus rhesus. J Comp Neurol. 1971;141:283–312. doi: 10.1002/cne.901410303. [DOI] [PubMed] [Google Scholar]
- 67.Rakic P. Mode of cell migration to the superficial layers of fetal monkey neocortex. J Comp Neurol. 1972;145:61–84. doi: 10.1002/cne.901450105. [DOI] [PubMed] [Google Scholar]
- 68.Rakic P, Sidman RL. Sequence of developmental abnormalities leading to granule cell deficit in cerebellar cortex of weaver mutant mice. J Comp Neurol. 1973a;152:103–132. doi: 10.1002/cne.901520202. [DOI] [PubMed] [Google Scholar]
- 69.Rakic P, Sidman RL. Weaver mutant mouse cerebellum: defective neuronal migration secondary to abnormality of Bergmann glia. Proc Natl Acad Sci USA. 1973b;70:240–244. doi: 10.1073/pnas.70.1.240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Richardson WD, Pringle N, Mosley MJ, Westermark B, Dubois-Dalcq M. A role for platelet-derived growth factor in normal gliogenesis in the central nervous system. Cell. 1988;53:309–319. doi: 10.1016/0092-8674(88)90392-3. [DOI] [PubMed] [Google Scholar]
- 71.Rosenberg PA, Aizenman E. Hundred-fold increase in neuronal vulnerability to glutamate toxicity in astrocyte-poor cultures of rat cerebral cortex [Erratum (1990) 116:399]. Neurosci Lett. 1989;103:162–168. doi: 10.1016/0304-3940(89)90569-7. [DOI] [PubMed] [Google Scholar]
- 72.Ross ME, Fletcher C, Mason CA, Hatten ME, Heintz N. Meander tail reveals a discrete developmental unit in the mouse cerebellum. Proc Natl Acad Sci USA. 1990;87:4189–4192. doi: 10.1073/pnas.87.11.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rothstein JD, Martin L, Levey AI, Dykes-Hoberg M, Jin L, Wu D, Nash N, Kuncl RW. Localization of neuronal and glial glutamate transporters. Neuron. 1994;13:713–725. doi: 10.1016/0896-6273(94)90038-8. [DOI] [PubMed] [Google Scholar]
- 74.Ryder EF, Cepko CL. Migration patterns of clonally related granule cells and their progenitors in the developing chick cerebellum. Neuron. 1994;12:1011–1029. doi: 10.1016/0896-6273(94)90310-7. [DOI] [PubMed] [Google Scholar]
- 75.Sentry JW, Yang MM, Kaiser K. Conditional cell ablation in Drosophila. BioEssays. 1993;15:491–493. doi: 10.1002/bies.950150710. [DOI] [PubMed] [Google Scholar]
- 76.Shibuki K, Gomi H, Chen L, Bao SW, Kim JSK, Wakatsuki H, Fujisaki T, Fujimoto J, Katoh A, Ikeda T, Chen C, Thompson RF, Itohara S. Deficient cerebellar long-term depression, impaired eyeblink conditioning, and normal motor coordination in GFAP mutant mice. Neuron. 1996;16:587–599. doi: 10.1016/s0896-6273(00)80078-1. [DOI] [PubMed] [Google Scholar]
- 77.Smeyne RJ, Chu T, Lewin A, Bian F, Crisman SS, Kunsch C, Lira SA, Oberdick J. Local control of granule cell generation by cerebellar Purkinje cells. Mol Cell Neurosci. 1995;6:230–251. doi: 10.1006/mcne.1995.1019. [DOI] [PubMed] [Google Scholar]
- 78.Sotelo C, Rio JP. Cerebellar malformation obtained in rats by early postnatal treatment with 6-aminonicotinamide. Role of neuron–glia interactions in cerebellar development. Neuroscience. 1980;5:1737–1759. doi: 10.1016/0306-4522(80)90092-5. [DOI] [PubMed] [Google Scholar]
- 79.Stuart GW, Searle PF, Chen HY, Brinster RL, Palmiter RD. A 12-base-pair DNA motif that is repeated several times in metallothionein gene promoters confers metal regulation to a heterologous gene. Proc Natl Acad Sci USA. 1984;81:7318–7322. doi: 10.1073/pnas.81.23.7318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Toggas SM, Masliah E, Rockenstein EM, Rall GF, Abraham CR, Mucke L. Central nervous system damage produced by expression of the HIV-1 coat protein gp120 in transgenic mice. Nature. 1994;367:188–193. doi: 10.1038/367188a0. [DOI] [PubMed] [Google Scholar]
- 81.Wagner MJ, Sharp JA, Summers WC. Nucleotide sequence of the thymidine kinase gene of herpes simplex virus type 1. Proc Natl Acad Sci USA. 1981;78:1441–1445. doi: 10.1073/pnas.78.3.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wallace H, McLaren K, Al-Shawi R, Bishop JO. Consequences of thyroid hormone deficiency induced by the specific ablation of thyroid follicle cells in adult transgenic mice. J Endocrinol. 1994;143:107–120. doi: 10.1677/joe.0.1430107. [DOI] [PubMed] [Google Scholar]
- 83.Wyss-Coray T, Feng L, Masliah E, Ruppe MD, Lee HS, Toggas SM, Rockenstein EM, Mucke L. Increased central nervous system production of extracellular matrix components and development hydrocephalus in transgenic mice overexpressing transforming growth factor-b1. Am J Pathol. 1995;147:53–67. [PMC free article] [PubMed] [Google Scholar]
- 84.Yuasa S, Kawamura K, Ono K, Yamakuni T, Takahashi Y. Development and migration of Purkinje cells in the mouse cerebellar primordium. Anat Embryol (Berl) 1991;184:195–212. doi: 10.1007/BF01673256. [DOI] [PubMed] [Google Scholar]