

Abstract
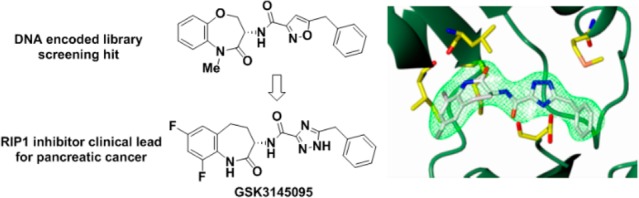
RIP1 regulates cell death and inflammation and is believed to play an important role in contributing to a variety of human pathologies, including immune-mediated inflammatory diseases and cancer. While small-molecule inhibitors of RIP1 kinase have been advanced to the clinic for inflammatory diseases and CNS indications, RIP1 inhibitors for oncology indications have yet to be described. Herein we report on the discovery and profile of GSK3145095 (compound 6). Compound 6 potently binds to RIP1 with exquisite kinase specificity and has excellent activity in blocking RIP1 kinase-dependent cellular responses. Highlighting its potential as a novel cancer therapy, the inhibitor was also able to promote a tumor suppressive T cell phenotype in pancreatic adenocarcinoma organ cultures. Compound 6 is currently in phase 1 clinical studies for pancreatic adenocarcinoma and other selected solid tumors.
Keywords: RIP1, type III kinase inhibitors, pancreatic cancer
Receptor interacting proteins (RIPs) 1 and 3 are known to form a complex termed necrosome that leads to a form of cell death called necroptosis and inflammatory cytokine production.1 Necroptosis can drive an inflammatory response through release of damage-associated molecular patterns,1 and the necrosome is also thought to contribute to neuronal cell loss in neurodegenerative diseases.2 Several RIP1 inhibitors are now progressing in the clinic for the treatment of inflammatory diseases, such as psoriasis, rheumatoid arthritis, and ulcerative colitis,3 as well for CNS indications such as ALS and Alzheimer’s disease.2 Additional functions of the RIP1 continue to be discovered. Recently Seifert and Miller have reported that the necrosome can also promote an immune suppression in pancreatic tumors.4 Additionally, Miller’s lab, in collaboration with our group, have shown that RIP1 kinase inhibition resulted in T cell differentiation toward a tumor suppressive phenotype leading to tumor-immunity in mice and in models of human pancreatic cancer.5 Importantly, RIP1 kinase inhibition sensitized tumors to checkpoint blockade, offering the potential for combination with PD-1 immunotherapy as a new option for the treatment of pancreatic cancer. We report in this Letter on the identification of a new clinical RIP1 inhibitor for pancreatic cancer and other solid tumors.
The first reported RIP1 inhibitors discovered by Degterev et al., exemplified by indole-hydantoin 1 (see Figure 1), were shown by cocrystallized in the RIP1 kinase domain to occupy an allosteric lipophilic pocket at the back of the ATP binding site.6,7 This type III binding mode, resulted in an excellent kinase selectivity profile.8 Our screening efforts against DNA-encoded libraries led to the identification of a benzoxazepinone RIP1 inhibitor 2 (GSK’481), whose RIP1 cocrystal structure showed it occupied the same allosteric binding pocket as 1.9 Subsequent lead-optimization resulted in selection of development candidate 3 (GSK2982772), which is currently under evaluation in phase 2a clinical trials in psoriasis, rheumatoid arthritis, and ulcerative colitis patients.10 Recently, Yoshikawa et al. have disclosed a benzoxazepinone analog 4 with a higher brain penetration and efficacy in a mouse experimental autoimmune encephalomyelitis (EAE) model of multiple sclerosis.11
Figure 1.

Structure of RIP1 kinase inhibitors.
We recently disclosed the efficacy of RIP1 inhibitor 5 (known as GSK’547) in an oncology mouse model of pancreatic cancer in both reducing tumor size and activating an immune cell response.5 Co-treatment of 5 with either a PD-1 antibody, to block immune down-regulation, or an ICOS antibody, to stimulate immune signaling, further reduced tumor size. Although 5 was an excellent tool for exploring RIP1 inhibition in mouse oncology models, it did not possess the required preclinical profile to support development as a clinical lead, most notable suffering from high turnover in human hepatocytes. Herein, we describe the identification of a new RIP1 inhibitor clinical candidate (S)-5-benzyl-N-(7,9-difluoro-2-oxo-2,3,4,5-tetrahydro-1H-benzo[b]azepin-3-yl)-1H-1,2,4-triazole-3-carboxamide 6 (GSK3145095), structurally related to benzoxazepinone 3. Although 3 is in clinical development for chronic immunoinflammatory disorders, development of a separate RIP1 inhibitor for oncology indications with their distinct safety tolerance and dosing criteria was required. The high RIP1 potency, monokinase selectivity, and excellent preclinical pharmacokinetic and developability profile of benzazepinone 6 led to its selection for clinical development and it recently initiated phase 1 evaluation in pancreatic cancer patients.
We have recently detailed the lead optimization of the DNA-encoded library derived hit benzoxazepinone 2 that led to identification of 3, the first RIP1 inhibitor to enter clinical evaluation.10 Since the SAR observations identified during the discovery of benzoxazepinone 3 also apply to benzazepinone 6, only a brief synopsis of some of the key modifications is warranted here. As a benchmark, benzoxazepinone 2 possessed excellent RIP1 in vitro potency, as measured by both an ADP-Glo biochemical assay and a human monocytic U937 cellular assay, but suffered from high lipophilicity and suboptimal pharmacokinetic (PK) profile (Table 1).9 The corresponding NH lactam analog 7 possessed a better rat PK profile but at the cost of lower RIP1 potency. This was a general trend observed in this series: methylation of the amide increased RIP1 potency by up to 10-fold, as the methyl group occupies a small pocket on the face of the β-sheet defined by Leu90-Met92 and Ile43-Lys45.10 However, metabolic demethylation of the N-methyl lactam can increase turnover; thus, NH lactams tended to have better in vivo exposures. Replacement of the isoxazole of both 2 and 7 with a triazole (analogs 3 and 8) maintained comparable RIP1 potencies with significantly reduced lipophilicity and improved rat oral exposures. Replacement of the benzoxazepinone heterocycle for benzazepinone 9 and 10 showed comparable in vitro potencies. Finally, introduction of 7,9-difluoro substitution at the benzazepinone yielded analogs 11 and 6. The increase in potency of 6 compared to 10 could be attributed to the two electron withdrawing fluorines decreasing the pKa of the lactam nitrogen, thus strengthening its H-bond to the backbone of Leu90 (Figure 2). The NH-lactam of 6 predictably increased rat exposure compared to NMe-lactam 11. The optimal potency and rat oral PK profile of benzazepinone 6 led to further preclinical evaluation as detailed below.
Table 1. SAR Summary of Key Compounds.

| Cpd | ADP-Gloa IC50 (nM) | U937a IC50 (nM) | CHI log Db | rat AUC0-∞c (μg·h/mL) |
|---|---|---|---|---|
| 2 | 1.6 | 7.9 | 5.9 | 0.38 |
| 7 | 32 | 200 | 5.1 | 2.2 |
| 3 | 1.0 | 6.3 | 3.8 | 2.3 |
| 8 | 32 | 100 | 3.0 | 2.5 |
| 9 | 5.0 | 10 | 3.8 | 0.31 |
| 10 | 50 | 63 | 3.1 | nd |
| 11 | 6.3 | 6.3 | 4.3 | 0.16 |
| 6 | 6.3 | 10 | 3.4 | 1.1 |
Assay protocols are described in Supporting Information; IC50 values are the average of at least two determinations.
CHI (chromatographic hydrophobicity index) log D at pH 7.4 was calculated from the retention time (tR) observed in a fast gradient reverse-phase HPLC.
Rat oral exposure at 2 mg/kg.
Figure 2.
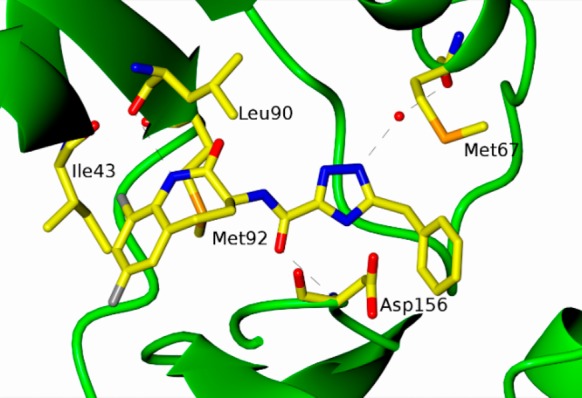
Cocrystal structure of RIP1 (1–294, C34A, C127A, C233A, and C240A) and benzazepinone 6.
Cocrystallization of benzazepinone 6 was achieved with the RIP1 construct (1–294, C34A, C127A, C233A, and C240A). The resulting X-ray structure was refined to resolution of 2.87 Å (see Supporting Information). In this structure, benzazepinone 6 was observed to be buried deep in the pocket between the N-terminal and C-terminal domains with no interaction with the hinge residues, making this a type III class of kinase inhibitor (see Figure 2). The triazole and benzyl functionality of 6 occupies the same allosteric lipophilic pocket at the back of the ATP binding site as Necrostatin-4. The benzoxazepine ring occupies space where the α-phosphate of ATP would reside, so the inhibition is not strictly allosteric. The amide carbonyl attached to the triazole makes a direct hydrogen bond interaction with the backbone amide NH of Asp156. The triazole nitrogen makes a water-mediated hydrogen bond to the carbonyl oxygen of Met67. The lactam nitrogen shows a possible long hydrogen bond interaction to the backbone carbonyl of Leu90, and the fluorine at the 9-position of the benzazepinone occupies a pocket between Met92 and Ile43. Details on cocrystallography are available in the Supporting Information section.
The type III binding mode observed for this pharmacophore in RIP1 results in complete kinase selectivity for RIP1 as exemplified by the kinase profile of 6 against both a P33 radiolabeled assay screen at Reaction Biology Corp (359 kinases) and a competition binding assay KINOMEscan at DiscoveRx Corp (456 kinases). In both assays, benzazepinone 6 was tested at a concentration of 10 μM and showed no inhibition of any kinase other than RIP1, representing a >1500-fold selectivity window based on the RIP1 ADP-Glo potency of 6.3 nM. Details are available in the Supporting Information.
Although benzazepinone 6 is binding in an allosteric pocket at the back of the ATP binding site, it is also partially occupying the region where the α-phosphate of ATP would reside, making them mutually exclusive, and ATP competitive inhibition is indeed observed (see Supporting Information). The binding kinetics are similar to benzoxazepinones recently profiled, with a moderate on-rate constant (kon = 2.5 × 104 M–1 s–1), accompanied by a slow off-rate constant koff = 1.2 × 10–4 s–1 (t1/2 = 99 min), measured by stopped-flow kinetics and fluorescence polarization competitive binding, respectively (see Supporting Information). In addition to efficacy against the immortalized U937 human monocyte cell line, activity of 6 was also assessed in primary neutrophils isolated from human whole blood. As previously described, in this assay TNF is coincubated with both the caspase inhibitor QVD-Oph and the SMAC mimetic RMT 5265, which block the apoptosis and NF-κB pathways, respectively, driving the TNF response down the necrosis pathway.10 Inhibitor 6 was able to potently block this response as shown by determination of overall cell viability as measured by cellular ATP levels (IC50 = 1.6 nM), cell death as measured by LDH release (IC50 = 0.5 nM), and RIP1-dependent inflammatory cytokine MIP-1β production, either as absolute levels for protein or fold changes in mRNA expression (IC50 = 0.4 nM).
A human whole blood stimulation assay was previously developed in which the necroptosis pathway is activated through stimulation with TNF coincubated with the caspase inhibitor QVD-Oph or zVAD.fmk, and the SMAC mimetic RMT 5265.10 In this assay, benzazepinone 6 was also shown to be very potent as measured by inhibition of cytokine MIP-1β ((IC50 = 5 nM). In a similar monkey whole blood stimulation assay, benzazepinone 6 exhibited an IC50 of 16 nM.
A distinctive feature of RIP1 type III inhibitors is a significant reduction in potency against nonprimate RIP1.9 Compound 6 is over 380-fold less potent against nonprimate RIP1 compared to primate RIP1 in biochemical assays (see Supporting Information) and shows a 340-fold reduction in cellular potency for blockage of necrotic death in mouse fibrosarcoma L929 cells (IC50 = 1.3 μM) compared to human U937 cells (IC50 = 6.3 nM). This reduced RIP1 mouse potency precludes evaluation of 6 in rodent oncology models. However, 6 was evaluated in human ex vivo tumor cultures. For these studies, patient derived organotypic spheroids (PDOTS) were prepared from freshly resected tumors from pancreatic adenocarcinoma, colorectal, breast, and gastric cancer patients, as previously described.12 PDOTS were treated with 6 or vehicle, and following 3 days incubation, they were harvested and analyzed by flow cytometry (see Figure 3). PDOTS treated with 6 developed significantly more effector-memory T cells (CD44+) and immunogenic CD4+ T cells (IFNγ+) compared to vehicle-treated samples. A trend toward increase CD8+ cytolytic T cells and TNFα expression was also observed (see Supporting Information); however, those markers failed to reach statistical significance in this study. Patients with increased T-cell infiltration and activation have a better prognosis with longer survival and response to immunotherapy agents.13 This agrees with earlier results in the orthotopic mouse model of pancreatic cancer where RIP1 inhibition leads to an immunogenic tumor microenvironment.5
Figure 3.

Individual PDOTS were prepared from resected patient tumors and incubated for 3 days with vehicle or compound 6 (0.5 nM in culture medium). Cells were labeled with antibodies to detect CD3 (pan-T cells), CD4 (T helper cells), CD44 (effector-memory cells), and IFNγ (marker of T cell activation) then analyzed by flow cytometry. The percentage of cells expressing the indicated marker in vehicle treated samples was set to 1 and the fold change in the presence of compound 6 determined.
Benzazepinone 6 displayed a good free fraction in blood of rat (13%), dog (12%), monkey (12%), and human (8.1%). In vitro evaluation of the metabolic stability of 6 in hepatocytes indicated moderate turnover in the rat and low turnover in monkey and human (see Supporting Information). A metabolite identification study in rat, monkey, and human hepatocytes highlighted that 6 was metabolized via both phase I and phase II biotransformation pathways, as shown in Figure 4. The primary metabolic pathways for 6 included hydroxylation and glucuronidation. No human specific metabolites of 6 were detected in these hepatocyte incubations, but trace levels of GSH conjugates were detected only in rat and monkey, not in human hepatocyte incubations. A pharmacokinetic evaluation of 6 in rat, dog, and monkey demonstrated a low clearance (≤35% liver blood flow) in all species (see Table 2). The volume of distribution was moderate along with a relatively short to moderate terminal half-life. Good oral bioavailability was observed across these preclinical species. The tissue distribution of 6 in rat was evaluated following iv infusion over 4 h (see Supporting Information). This study showed that compared to blood 6 distributed at greater concentrations (7–8-fold) in liver and kidney, and comparable concentrations (0.7–3-fold) in colon, heart, and skin. In contrast, 6 had low brain penetration (6%) despite possessing moderate cell permeability (6.7 × 10–6 cm/s), which is likely due to active extrusion from brain via the efflux drug transporter P-glycoprotein (P-gp) (see Supporting Information).
Figure 4.

Proposed metabolic pathways of compound 6 in rat (r), monkey (m), and human (h) hepatocytes.
Table 2. Pharmacokinetic Parameters of 6 Following Administration to Rat, Dog, and Monkey.
| route | parameter | rat | dog | monkey |
|---|---|---|---|---|
| iva | dose (mg/kg) | 1.0 | 1.1 | 0.93 |
| Cl (mL/min/kg) (%LBF) | 27 ± 5 (35) | 9.8 ± 1.8 (18) | 6.4 ± 0.5 (15) | |
| Vdss (L/kg) | 1.8 ± 0.3 | 1.1 ± 0.2 | 1.8 ± 0.1 | |
| t1/2 (h) | 2.2 ± 0.8 | 1.7 ± 0.02 | 4.2 ± 0.6 | |
| pob | dose (mg/kg) | 2.1 | 2.0 | 1.9 |
| Tmax (h) | 0.83 ± 0.29 | 0.75 ± 0.43 | 1.5 ± 0.0 | |
| Cmax (μg/mL) | 320 ± 79 | 910 ± 130 | 770 ± 99 | |
| AUC (μg·h/mL) | 1.1 ± 0.2 | 2.5 ± 0.49 | 4.4 ± 0.5 | |
| bioavailability (%) | 84 ± 8 | 78 ± 3 | 88 ± 13 |
The vehicle for iv studies was 20% Cavitron, 5% DMSO in PBS.
The vehicle for po studies was 6% Cavitron, 5% DMSO in PBS.
Allometric scaling and in vitro to in vivo extrapolations were used to generate predictions of human PK parameters. These showed a high level of correlation and predicted 6 to have high bioavailability, low clearance, moderate volume, and a terminal half-life in the order of 3.3 h. The average clearance and volume values were then used to reconstruct a monoexponential predicted human blood concentration time profile, as shown in Figure 5, for once and twice daily dosing. Doses were scaled and modeled to target maintaining concentrations above the human whole blood IC90 over 24 h. Inhibitor 6 is predicted to require doses of either 273 mg daily or 40 mg twice daily to maintain 90% inhibition, with an AUC of 11.7 or 1.7 μg·h/mL, respectively. The predicted human PK/PD was modeled using this predicted human PK profile at these doses, along with human whole blood activity, maintaining 90% RIP1 inhibition levels over 24 h, as shown in the Supporting Information.
Figure 5.
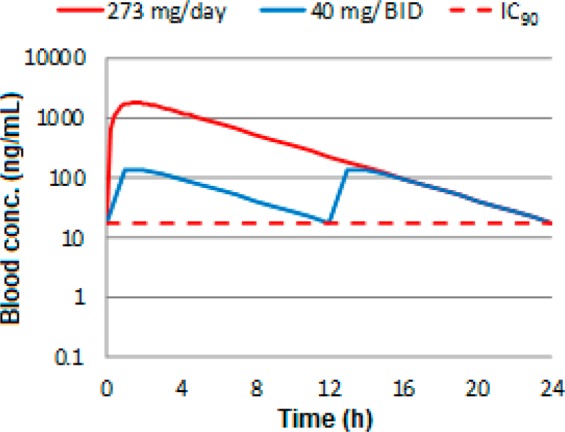
Predicted once and twice daily human blood concentration time profile of benzazepinone 6 overlaid with the human whole blood inhibition IC90 concentrations.
The emerging biological understanding of the multiple roles of RIP1 kinase has opened up a number of exciting opportunities to investigate this target for drug intervention in a number of diseases. Our recent findings that RIP1 signaling in macrophages regulates immune tolerance in pancreatic cancer and confers resistance to immunotherapy offers a strong rationale that RIP1 kinase inhibition could help address this most significant unmet need in oncology. Benzazepinone 6 was optimized from a DNA-encoded library screen with both high in vitro RIP1 potency and complete kinase selectivity. It has recently initiated phase 1 evaluation in pancreatic cancer patients, the results of which will be reported in future publications.
Acknowledgments
The authors are indebted to Nicolas Faucher, Nicolas George, Frederic Donche, and Alain Daugan from the former GSK group at Les Ulis, France for the design of the in vivo tool inhibitor 5 (GSK’547).
Glossary
ABBREVIATIONS USED
- LDH
lactate dehydrogenase
- MIP
macrophage inflammatory protein
- PDOTS
patient derived organotypic spheroids
- RIP
receptor interacting protein
- QVD-OPh
(3S)-5-(2,6-difluorophenoxy)-3-[[(2S)-3-methyl-1-oxo-2-[(2-quinolinylcarbonyl)amino]butyl]amino]-4-oxopentanoic acid hydrate
- SMAC
second mitochondrial-derived activator of caspases
- TNF
tumor necrosis factor
- zVAD.fmk
carbobenzoxy-valyl-alanyl-aspartyl-[O-methyl]-fluoromethylketone
Biography
Philip Harris received his degree in Chemistry (1985) and a Ph.D. degree in Organic Synthesis (1988) at the University of Manchester (UK). After postdoctoral training at the University of Florida, he joined Burroughs Wellcome (a legacy company of GlaxoSmithKline) in 1992 as a medicinal chemist mostly focused around kinase inhibitors. Philip was a part of drug discovery teams that resulted in two currently marketed drugs: Votrient (VEGFR inhibitor, renal cancer) and Tafinlar (b-Raf inhibitor, melanoma). During the last decade his focus has been on RIP1 kinase inhibitors, two of which are currently in clinical trials for oncology and inflammation indications.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsmedchemlett.9b00108.
Details on preparation of compounds 2, 3, and 6–11, in vitro assays, and RIP1 (1–375) preparation. The following data specific to compound 6 are included: mode of inhibition, enzyme kinetics, ortholog inhibition profile, kinase selectivity profile, RIP1 cocrystallization, rat tissue distribution, permeability and P-gp substrate evaluation, hepatocyte turnover, and human PK/PD predictions, and additional PDOTS evaluation for CD8+ T cells and TNFα expression (PDF)
Accession Codes
Coordinates and structure factors for the cocrystal structure of RIP1 (1–294, C34A, C127A, C233A, C240A) and benzoxazepinone 6 have been deposited in the Protein Data Bank with the accession number 6RLN.
The human biological samples were sourced ethically, and their research use was in accord with the terms of the informed consents under an IRB/EC approved protocol. All animal studies were conducted in accordance with the GSK Policy on the Care, Welfare and Treatment of Laboratory Animals and were reviewed the Institutional Animal Care and Use Committee either at GSK or by the ethical review process at the institution where the work was performed.
The authors declare the following competing financial interest(s): All authors, with the exception of George Miller, Wei Wang, Anirudh Chirala, and Dongling Wu, are current employees and stockholders of GlaxoSmithKline.
Supplementary Material
References
- Pasparakis M.; Vandenabeele P. Necroptosis and its role in inflammation. Nature 2015, 517, 311–20. 10.1038/nature14191. [DOI] [PubMed] [Google Scholar]
- Yuan J.; Amin P.; Ofengeim D. Necroptosis and RIPK1-mediated neuroinflammation in CNS diseases. Nat. Rev. Neurosci. 2019, 20, 19–33. 10.1038/s41583-018-0093-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisel K.; Scott N. E.; Tompson D. J.; Votta B. J.; Madhavan S.; Povey K.; Wolstenholme A.; Simeoni M.; Rudo T.; Richards-Peterson L.; Sahota T.; Wang J. G.; Lich J.; Finger J.; Verticelli A.; Reilly M.; Gough P. J.; Harris P. A.; Bertin J.; Wang M. L. Randomized clinical study of safety, pharmacokinetics, and pharmacodynamics of RIPK1 inhibitor GSK2982772 in healthy volunteers. Pharmacol. Res. Perspect. 2017, 5, e00365 10.1002/prp2.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert L.; Miller G. Molecular Pathways: The Necrosome - A Target for Cancer Therapy. Clin. Cancer Res. 2017, 23, 1132–1136. 10.1158/1078-0432.CCR-16-0968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W.; Marinis J. M.; Beal A. M.; Savadkar S.; Wu Y.; Khan M.; Taunk P. S.; Wu N.; Su W.; Wu J.; Ahsan A.; Kurz E.; Chen T.; Yaboh I.; Li F.; Gutierrez J.; Diskin B.; Hundeyin M.; Reilly M.; Lich J. D.; Harris P. A.; Mahajan M. K.; Thorpe J. H.; Nassau P.; Mosley J. E.; Leinwand J.; Kochen Rossi J. A.; Mishra A.; Aykut B.; Glacken M.; Ochi A.; Verma N.; Kim J. I.; Vasudevaraja V.; Adeegbe D.; Almonte C.; Bagdatlioglu E.; Cohen D. J.; Wong K. K.; Bertin J.; Miller G. RIP1 Kinase Drives Macrophage-Mediated Adaptive Immune Tolerance in Pancreatic Cancer. Cancer Cell 2018, 34, 757–774. 10.1016/j.ccell.2018.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degterev A.; Hitomi J.; Germscheid M.; Ch’en I. L.; Korkina O.; Teng X.; Abbott D.; Cuny G. D.; Yuan C.; Wagner G.; Hedrick S. M.; Gerber S. A.; Lugovskoy A.; Yuan J. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie T.; Peng W.; Liu Y.; Yan C.; Maki J.; Degterev A.; Yuan J.; Shi Y. Structural basis of RIP1 inhibition by necrostatins. Structure 2013, 21, 493–499. 10.1016/j.str.2013.01.016. [DOI] [PubMed] [Google Scholar]
- Christofferson D. E.; Li Y.; Hitomi J.; Zhou W.; Upperman C.; Zhu H.; Gerber S. A.; Gygi S.; Yuan J. A novel role for RIP1 kinase in mediating TNFα production. Cell Death Dis. 2012, 3, e320 10.1038/cddis.2012.64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris P. A.; King B. W.; Bandyopadhyay D.; Berger S. B.; Campobasso N.; Capriotti C. A.; Cox J. A.; Dare L.; Dong X.; Finger J. N.; Grady L. C.; Hoffman S. J.; Jeong J. U.; Kang J.; Kasparcova V.; Lakdawala A. S.; Lehr R.; McNulty D. E.; Nagilla R.; Ouellette M. T.; Pao C. S.; Rendina A. R.; Schaeffer M. C.; Summerfield J. D.; Swift B. A.; Totoritis R. D.; Ward P.; Zhang A.; Zhang D.; Marquis R. W.; Bertin J.; Gough P. J. DNA-Encoded Library Screening Identifies Benzo[b][1,4]oxazepin-4-ones as Highly Potent and Monoselective Receptor Interacting Protein 1 Kinase Inhibitors. J. Med. Chem. 2016, 59, 2163–2178. 10.1021/acs.jmedchem.5b01898. [DOI] [PubMed] [Google Scholar]
- Harris P. A.; Berger S. B.; Jeong J. U.; Nagilla R.; Bandyopadhyay D.; Campobasso N.; Capriotti C. A.; Cox J. A.; Dare L.; Dong X.; Eidam P. M.; Finger J. N.; Hoffman S. J.; Kang J.; Kasparcova V.; King B. W.; Lehr R.; Lan Y.; Leister L. K.; Lich J. D.; MacDonald T. T.; Miller N. A.; Ouellette M. T.; Pao C. S.; Rahman A.; Reilly M. A.; Rendina A. R.; Rivera E. J.; Schaeffer M. C.; Sehon C. A.; Singhaus R. R.; Sun H. H.; Swift B. A.; Totoritis R. D.; Vossenkämper A.; Ward P.; Wisnoski D. D.; Zhang D.; Marquis R. W.; Gough P. J.; Bertin J. Discovery of a First-in-Class Receptor Interacting Protein 1 (RIP1) Kinase Specific Clinical Candidate (GSK2982772) for the Treatment of Inflammatory Diseases. J. Med. Chem. 2017, 60, 1247–1261. 10.1021/acs.jmedchem.6b01751. [DOI] [PubMed] [Google Scholar]
- Yoshikawa M.; Saitoh M.; Katoh T.; Seki T.; Bigi S. V.; Shimizu Y.; Ishii T.; Okai T.; Kuno M.; Hattori H.; Watanabe E.; Saikatendu K. S.; Zou H.; Nakakariya M.; Tatamiya T.; Nakada Y.; Yogo T. Discovery of 7-Oxo-2,4,5,7-tetrahydro-6 H-pyrazolo[3,4- c]pyridine Derivatives as Potent, Orally Available, and Brain-Penetrating Receptor Interacting Protein 1 (RIP1) Kinase Inhibitors: Analysis of Structure-Kinetic Relationships. J. Med. Chem. 2018, 61, 2384–2409. 10.1021/acs.jmedchem.7b01647. [DOI] [PubMed] [Google Scholar]
- Jenkins R. W.; Aref A. R.; Lizotte P. H.; Ivanova E.; Stinson S.; Zhou C. W.; Bowden M.; Deng J.; Liu H.; Miao D.; He M. X.; Walker W.; Zhang G.; Tian T.; Cheng C.; Wei Z.; Palakurthi S.; Bittinger M.; Vitzthum H.; Kim J. W.; Merlino A.; Quinn M.; Venkataramani C.; Kaplan J. A.; Portell A.; Gokhale P. C.; Phillips B.; Smart A.; Rotem A.; Jones R. E.; Keogh L.; Anguiano M.; Stapleton L.; Jia Z.; Barzily-Rokni M.; Cañadas I.; Thai T. C.; Hammond M. R.; Vlahos R.; Wang E. S.; Zhang H.; Li S.; Hanna G. J.; Huang W.; Hoang M. P.; Piris A.; Eliane J. P.; Stemmer-Rachamimov A. O.; Cameron L.; Su M. J.; Shah P.; Izar B.; Thakuria M.; LeBoeuf N. R.; Rabinowits G.; Gunda V.; Parangi S.; Cleary J. M.; Miller B. C.; Kitajima S.; Thummalapalli R.; Miao B.; Barbie T. U.; Sivathanu V.; Wong J.; Richards W. G.; Bueno R.; Yoon C. H.; Miret J.; Herlyn M.; Garraway L. A.; Van Allen E. M.; Freeman G. J.; Kirschmeier P. T.; Lorch J. H.; Ott P. A.; Hodi F. S.; Flaherty K. T.; Kamm R. D.; Boland G. M.; Wong K. K.; Dornan D.; Paweletz C. P.; Barbie D. A. Ex Vivo Profiling of PD-1 Blockade Using Organotypic Tumour Spheroids. Cancer Discovery 2018, 8, 196–215. 10.1158/2159-8290.CD-17-0833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridman W. H.; Zitvogel L.; Sautes-Fridman C.; Kroemer G. The immune contexture in cancer prognosis and treatment. Nat. Rev. Clin. Oncol. 2017, 14, 717–734. 10.1038/nrclinonc.2017.101. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.