

The nuclear factor (erythroid 2)-like 2 (NRF2 or NFE2L2) transcription factor regulates the expression of many genes that are critical in maintaining cellular homeostasis. Its deregulation has been implicated in many diseases, including cancer and metabolic and neurodegenerative diseases.
KEYWORDS: CRISPR, KEAP1, NFE2L2, NRF2, autophagy, gene reporters, genomics, molecular biology, oxidative stress, signal transduction
ABSTRACT
The nuclear factor (erythroid 2)-like 2 (NRF2 or NFE2L2) transcription factor regulates the expression of many genes that are critical in maintaining cellular homeostasis. Its deregulation has been implicated in many diseases, including cancer and metabolic and neurodegenerative diseases. While several mechanisms by which NRF2 can be activated have gradually been identified over time, a more complete regulatory network of NRF2 is still lacking. Here we show through a genome-wide clustered regularly interspaced short palindromic repeat (CRISPR) screen that a total of 273 genes, when knocked out, will lead to sustained NRF2 activation. Pathway analysis revealed a significant overrepresentation of genes (18 of the 273 genes) involved in autophagy. Molecular validation of a subset of the enriched genes identified 8 high-confidence genes that negatively regulate NRF2 activity irrespective of cell type: ATG12, ATG7, GOSR1, IFT172, NRXN2, RAB6A, VPS37A, and the well-known negative regulator of NRF2, KEAP1. Of these, ATG12, ATG7, KEAP1, and VPS37A are known to be involved in autophagic processes. Our results present a comprehensive list of NRF2 negative regulators and reveal an intimate link between autophagy and NRF2 regulation.
INTRODUCTION
The nuclear factor (erythroid 2)-like 2 (NRF2 or NFE2L2) transcription factor regulates the basal and stressor-inducible expression of genes containing an antioxidant response element (ARE) in the promoter region (1, 2). NRF2 activation promotes transcription of target genes involved in xenobiotic metabolism and efflux, thus making NRF2 a critical component of the cellular response to endogenous and exogenous toxicants and oxidants. Beyond its role in detoxification, NRF2 has pleiotropic roles in cancer chemoprevention (3–6), energy metabolism (7–11), proliferation and differentiation (11–14), iron and heme cycling (15, 16), inflammation (17), and apoptosis (18). NRF2 activation has shown promise in alleviating many complex ailments, including diabetes (19–22) or neurodegenerative diseases (23–27).
While NRF2 plays a pivotal role in many normal biological processes, its deregulation can contribute to pathologies such as cancer (18, 28–30). Constitutive NRF2 activation has been correlated with poor prognoses in many cancers, including cancers of the lung, gallbladder, esophagus, ovary, head and neck, and gastric systems (31–37). Indeed, NRF2 and its negative regulators are frequently mutated in cancer, and those mutations collectively result in sustained NRF2 activation (38, 39).
Given its multitude of beneficial and pathological roles, NRF2 activation is tightly regulated. The primary negative regulator of NRF2, Kelch-like ECH-associated protein 1 (KEAP1) (40), forms a ubiquitin ligase complex with the cullin-3 (CUL3) and ring-box 1 (RBX1) proteins (41). In this complex, two molecules of KEAP1 bind two amino acid motifs, DLG and ETGE motifs, which reside in the Nrf2-ECH homology domain 2 (Neh2) of NRF2 (42). When both motifs are bound, the KEAP1 complex can bring a single molecule of NRF2 into the E3 ubiquitin ligase machinery for ubiquitylation. Electrophilic and oxidative stress can modify redox-active cysteine residues on KEAP1 and antagonize KEAP1’s capacity to mediate NRF2 ubiquitylation. This allows newly synthesized NRF2 to accumulate, translocate to the nucleus, and promote transcription of target genes by binding to the ARE DNA sequence (43–45).
While the KEAP1 degradation route is regarded as the primary pathway for tuning NRF2 protein levels, other repressors have been added to the NRF2 regulatory network. The β-TrCP–SKIP1–CUL1–RBX1 E3 ubiquitin ligase complex can bind NRF2 at DSGIS or DSAPGS degrons found in the Neh6 domain of NRF2 and promote NRF2 ubiquitylation and degradation (46–48). Phosphorylation of the DSGIS motif by glycogen synthase kinase 3 (GSK-3) can enhance NRF2 degradation by this complex. Both the KEAP1 and β-TrCP degradation pathways require cullin substrate adaptor recycling mediated by CAND1 (49). Additionally, during activation of the endoplasmic reticulum (ER) stress response pathway, synoviolin 1 (SYVN1 or HRD1) was identified as an E3 ligase responsible for ubiquitylating NRF2 during liver cirrhosis (50).
While the KEAP1, β-TrCP, and SYVN1 pathways directly modulate the NRF2 protein level and activity, other signaling pathways indirectly affect NRF2. For example, arsenic inhibits autophagic flux and allows for accumulation of autophagosomes that sequester KEAP1 in a p62-dependent mechanism (51–53). p62 (sequestosome 1 or SQSTM1) is a cargo receptor for selective autophagy, where it can bind KEAP1 and several other proteins. The p62-KEAP1 complex can interact with autophagy-related protein 8 (ATG8), which is bound by phosphatidylethanolamine (PE) to the burgeoning autophagosome (54). This compartmentalizes KEAP1 into autophagosomes and away from cytosolic NRF2, which allows for newly synthesized NRF2 to accumulate, translocate to the nucleus, and promote transcription of its target genes. p62-dependent sequestration of KEAP1 into autophagosomes is enhanced by phosphorylation of p62 at S349, which increases its affinity for KEAP1 binding (55). This mechanism indicates a role for autophagy in NRF2 regulation; genetic depletion of the autophagy-related genes autophagy-related 5 (ATG5) (56), autophagy-related 7 (ATG7) (53, 57, 58), and beclin-1 (BECN1) (59) has been shown to activate NRF2.
Known negative regulators of NRF2 encompass diverse cellular functions. Aside from the aforementioned ubiquitylation and autophagy pathways, the metabolic enzyme fumarate hydratase (encoded by FH), histone-modifying enzyme SET domain containing 6 histone lysine methyltransferase (encoded by SETD6), and transcription factor BTB domain and CNC homolog 1 (encoded by BACH1) are all known to negatively regulate NRF2 activity under certain conditions (60–63).
To discover genes that negatively impact NRF2 activity in an unbiased manner, we conducted a genome-wide clustered regularly interspaced short palindromic repeat (CRISPR)/Cas9 screen that systematically knocked out 17,996 genes in conjunction with a reporter system for NRF2 activation. This system identified 273 genes that when knocked out activate NRF2 and alludes to autophagy as an NRF2-regulatory pathway.
RESULTS
CRISPR screen design and validation.
To uncover negative regulators of NRF2, we developed a human cell-based system that screens for cells harboring NRF2 activation. This system was based on a reporter construct, ARE-BSD-PEST, consisting of a transcriptional pause site followed by multiple ARE enhancer sequence for NRF2 binding and a minimal promoter controlling the transcription of a blasticidin S deaminase gene (BSD, a blasticidin resistance gene) with a PEST degron sequence (Fig. 1A). This construct allows for high expression of BSD only following sustained NRF2 activation and NRF2 binding to the ARE to enhance transcription.
FIG 1.
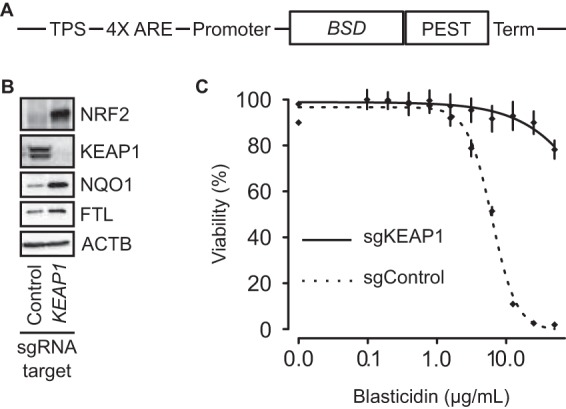
Design and validation of the ARE-BSD-PEST reporter. (A) ARE-BSD-PEST consists of a transcriptional pause site (TPS) followed by a synthetic sequence consisting of a triple antioxidant response sequence (4× ARE), a minimal promoter, an open reading frame for a blasticidin resistance gene with a PEST degron sequence (BSD-PEST), and a terminator; the BSD-PEST transcription is under the control of the NRF2 transcription factor. (B) Western blot showing effective KEAP1 knockout. The Western blot shows activation of NRF2 as well as NRF2 transcriptional targets NQO1 and FTL. β-Actin (ACTB) was used as a loading control. (C) HK2-BSD cells harboring KEAP1 knockout (HK2-BSD-sgKEAP1) are more resistant to blasticidin than control cells.
We stably transfected the ARE-BSD-PEST construct into HK2 cells, an immortalized, nonmalignant cell line derived from human kidney epithelia, to make HK2-BSD cells. To test the utility of this cell line in a pooled CRISPR screen, we transfected the HK2-BSD cells with single guide RNA (sgRNA) targeting KEAP1. The sgKEAP1-treated cells showed loss of KEAP1 protein, an increased level of NRF2 protein, and increased levels of the NRF2 transcription targets NQO1 and FTL compared to cells transfected with a nontargeting control sgRNA, sgControl (Fig. 1B). To understand whether the activated NRF2 was sufficient for blasticidin resistance, we treated HK2-BSD cells transfected with either sgKEAP1 or sgControl with various concentrations of blasticidin for 72 h. The dose response showed that the cells with sgKEAP1 were more resistant to blasticidin than those with the sgControl (Fig. 1C), allowing for the selection of cells with sustained NRF2 at a blasticidin concentration of 10 μg/ml.
CRISPR screen.
To systematically knock out all human genes and identify genes that negatively regulate NRF2, we utilized a pooled viral CRISPR sgRNA library in a scheme shown in Fig. 2A. We transduced HK2-BSD cells with the sgRNA library at a multiplicity of infection (MOI) of 0.1 at 1,000× library coverage. The sgRNA library virus vector confers puromycin resistance; therefore, positively transduced cells were selected with 2 μg/ml puromycin and expanded. Fifty percent of the expanded puromycin-resistant cells were used for genomic DNA isolation, whereby the isolated genomic DNA (gDNA) was used as a template for total library coverage control. The remaining 50% of cells were selected and expanded in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum (FBS), 4.5 g/liter glucose, and 10 μg/ml blasticidin in normoxic air enriched to 5% CO2; total genomic DNA isolated from these blasticidin-resistant cells was used as a template for samples with sustained NRF2 activation. Following sequencing and mapping of the sgRNA PCR amplicons, we identified 11,032 sgRNA species representing 7,957 genes that were present in the blasticidin-selected samples (Fig. 2B; see Data Set S1 in the supplemental material). Several known negative regulators of NRF2, including ATG5, ATG7, BECN1, FH, BACH1, SETD6, KEAP1, CUL3, and CAND1, were found in the blasticidin-selected samples (Fig. 2C); this suggests that the screen functioned as expected to select for negative regulators of NRF2. A total of 328 sgRNA species targeting 273 unique genes were statistically significantly enriched in the blasticidin-selected samples (Fig. 2D; see Data Set S2 in the supplemental material). This indicates that those genes, when knocked out, lead to sustained NRF2 activation. Again, known negative regulators of NRF2 passed the statistical threshold, including CUL3, KEAP1, CAND1, BECN1, and ATG7.
FIG 2.

CRISPR screen identifies negative regulators of NRF2. (A) HK2-BSD cells were transfected with the CRISPR libraries at a multiplicity of infection (MOI) of 0.1 to maximize the probability of 1 sgRNA species per cell. Cells were selected in 2 μg/ml puromycin to kill untransduced cells. Fifty percent of cells were collected for gDNA isolation of the unselected (total) samples, and 50% were selected for NRF2 activation with blasticidin. gDNA was collected for the blasticidin-selected samples. sgRNA sequences were amplified by PCR and sequenced using massively parallel sequencing. Genes were mapped to sgRNAs, and genes statistically significantly enriched in the blasticidin-selected samples were identified as the enriched samples. (B) Distribution of log10-transformed counts greater than 0 from the 11,032 sgRNA species representing 7,957 genes present in the blasticidin-selected samples. sgRNAs targeting previously described negative regulators of NRF2 activity and sgRNAs targeting NRF2 itself are identified by color. (C) Log10-transformed counts of all sgRNAs species targeting known negative regulators of NRF2 that have a count of greater than zero identified in the CRISPR screen. (D) Plot of –log10(adjusted P value) versus pseudo-log2(fold change) of statistically significantly enriched sgRNAs found in the blasticidin-selected samples. sgRNAs targeting well-described negative regulators of NRF2 activity are identified by color. INF, infinity. Values at INF representing known negative regulators of NRF2 were placed on separate lines for ease of interpretation. (E) Peptide sequence of the Neh2 degron domain of NRF2/NFE2L2 that harbored sgRNAs enriched in the blasticidin-selected samples; the enriched sgRNAs target genome regions near the negative regulatory domain of NRF2, allowing for possible activating mutations after Cas9 activity.
Interestingly, two sgRNAs targeting NRF2 (NFE2L2) were statistically enriched in the blasticidin-selected samples (Fig. 2B and D). Upon inspection, we identified that both these sgRNAs target the Neh2 domain of NRF2, which is required for KEAP1 binding (Fig. 2E). This suggests that the DNA repair following the CRISPR-mediated double-strand break may have resulted in activating mutations that disrupt NRF2-KEAP1 binding.
Pathway analyses.
We sought to identify signaling pathways involving the 273 genes enriched from the screen. Following gene ontology (GO) annotation, we identified a clear enrichment of 13 GO terms that were related to 18 of the 273 genes (Fig. 3A; see Data Set S3 in the supplemental material). GO annotation for the 273 genes can be found in Data Set S4 in the supplemental material. By back propagating the 13 enriched terms through the GO-directed acyclic graph, we identified autophagy as the converging biological pathway shared by the majority of those genes (Fig. 3B). This indicates that autophagy is a converging cellular process that regulates NRF2.
FIG 3.

Enriched genes are involved in autophagy pathways. (A) Gene ontology (GO) enrichment on 273 significantly enriched genes revealed enrichment of 13 GO terms. The red line indicates an adjusted P value of 0.05. (B) Directed acyclic graph of relationships between the 13 GO terms identified in panel A and the 18 of 273 genes annotated to these processes. Two additional GO terms, “autophagy of nucleus” and “autophagy,” were propagated from their child terms. (C) Twenty-five genes were targeted by at least two enriched sgRNAs. Black bars represent genes carried forward for validation. Genes labeled “A” were involved in the GO autophagy annotations in panel B.
Of the 273 significantly enriched genes, 25 were each targeted by at least two sgRNA species (Fig. 3C). The GO terms associated with these 25 genes are presented in Table 1. Again, many genes are associated with autophagy-related GO terms. Other general biological functions relevant to these 25 genes include stress response, proteostasis and protein modification, trafficking, growth and development, and immune system.
TABLE 1.
Gene ontology (GO) terms associated with enriched genes that were targeted by at least two sgRNA species
| Function(s) | GO term | Genesa |
|---|---|---|
| Autophagy | Late nucleophagy | ATG9A, ATG7 |
| Piecemeal microautophagy of the nucleus | RB1CC1, ATG7 | |
| Autophagy | ATG10, RB1CC1, ATG7 | |
| Autophagy of mitochondrion | RB1CC1, ATG12, ATG9A, ATG3, ATG7 | |
| Autophagosome assembly | RB1CC1, TMEM41B, ATG12, ATG9A, ATG3, ATG7 | |
| Macroautophagy | VPS37A, ATG10, RB1CC1, ATG12, ATG3, ATG7 | |
| Stress response | Cellular response to oxidative stress | TXNRD1, NFE2L2 |
| Cell redox homeostasis | TXNRD1, NFE2L2 | |
| Proteostasis and protein modification | Cellular protein modification process | ATG3, ATG7 |
| Proteasome-mediated ubiquitin-dependent protein catabolic process | NFE2L2, CUL3 | |
| Positive regulation of protein modification process | ATG10, ATG7 | |
| Protein modification by small-protein conjugation | ATG10, ATG7 | |
| Proteasomal ubiquitin-independent protein catabolic process | KEAP1, NFE2L2 | |
| Posttranslational protein modification | KEAP1, CAND1, CUL3 | |
| Protein ubiquitination | KEAP1, NFE2L2, ATG3, ATG7, CAND1, CUL3 | |
| Trafficking | Protein targeting to membrane | VPS37A, ATG3 |
| Transmembrane transport | SLC39A9, LRRC8A | |
| Intra-Golgi vesicle-mediated transport | RAB6A, GOSR1 | |
| Retrograde transport, endosome to Golgi apparatus | RAB6A, GOSR1 | |
| ER-to-Golgi vesicle-mediated transport | GOSR1, CUL3 | |
| Protein lipidation | ATG10, ATG7 | |
| C-terminal protein lipidation | ATG12, ATG7 | |
| Protein transport | ATG10, ATG9A, ATG7, GOSR1 | |
| Growth and development | Aging | NFE2L2, ATG7 |
| Immune system | Viral process | KEAP1, NFE2L2, RAB6A |
| Neutrophil degranulation | ATG7, RAB6A, CAND1, TUBB |
All genes were found in the blasticidin-selected samples and are statistically significantly enriched. TXNRD1, a bona fide NRF2 target gene, is in bold.
NRF2 target gene scouring.
It is worth noting that TXNRD1, a bona fide NRF2 target gene, is among these 25 genes. Hence, TXNRD1 may form a negative feedback loop, whereby NRF2 upregulates expression of TXNRD1, which in turn represses NRF2 signaling. To look holistically for known NRF2 target genes that could be participating in a feedback loop with NRF2, we broadened our search by looking at all genes in the blasticidin-selected samples instead of just the statistically significantly enriched sgRNA species. We identified 54 sgRNA species targeting 33 unique genes that are known NRF2 targets (Fig. 4); only sgRNA species targeting TXNRD1 were statistically significantly enriched (Fig. 4).
FIG 4.

Transformed counts of sgRNAs found in the blasticidin-selected samples that target genes known to be induced or inhibited following NRF2 activation. Functional areas are included. sgRNAs with black bars were statistically significantly enriched; sgRNAs with gray bars were found in the blasticidin-selected samples but were not statistically significantly enriched.
Gene validation.
We selected 19 genes for validation. Seventeen of these 19 genes were those with at least two significantly enriched sgRNA species in the blasticidin-selected samples (Fig. 3C), and the remaining two genes (COLEC10 and NRXN2) had one significantly enriched sgRNA. These 19 genes included known negative regulators of NRF2 as controls: KEAP1 and ATG7. Seven of these genes are involved in autophagy as defined by the GO analysis (Fig. 3B and C). Validation was performed in 4 different cell lines; these cell lines do not harbor any known gene mutations that could activate NRF2 and are derived from a variety of tissue/disease origins. They were MDA-MB-231 from breast cancer, HK2 from noncancerous kidney, BEAS-2B from noncancerous lung tissue, and NCI-H1299 from lung cancer.
Of the 19 genes tested, all showed increased protein levels of NRF2 and of select NRF2 transcription target genes (FTL, GCLM, or NQO1) in at least one of the four cell lines tested compared to control, nontargeting sgRNA (Fig. 5A to D). Loss of 17/19 genes showed NRF2 induction across all 4 cell lines; ATG10 and ATG12 did not show NRF2 induction in BEAS-2B cells (Fig. 5E). Knockout of 17 of the genes showed increases in at least two target genes across 2 of the 4 cell lines tested (Fig. 5F). Eight of the genes (ATG12, ATG7, GOSR1, IFT172, KEAP1, NRXN2, RAB6A, and VPS37A) showed target gene induction for all four cell lines, which we defined as “high confidence” negative regulators of NRF2. Intriguingly, many of the gene knockouts showed KEAP1 induction relative to control sgRNA (Fig. 5G). This indicates that knockout of these genes likely causes KEAP1 cysteine adduction, KEAP1 sequestration away from NRF2 (either by compartmentalization of KEAP1 or by induction of a protein that competes with NRF2 for KEAP1), posttranslational modification of NRF2 that prevents KEAP1 binding, or enhanced transcription or translation of NFE2L2 sufficient to outcompete KEAP1.
FIG 5.

Genes identified by CRISPR screen are negative regulators of NRF2 and NRF2 transcriptional targets. (A to D) Western blots of NRF2, KEAP1, p62, phospho-S349 p62, and NRF2 target genes (NQO1, GCLM, and FTL) following knockout of genes identified as negative regulators by the CRISPR screen. Genes were knocked out in the HK2 (A), NCI-H1299 (B), MDA-MB-231 (C), and BEAS-2B (D) cell lines. (E to G) Summary of Western blot results (A to D), showing NRF2 activation (E), target gene induction (F), and KEAP1 induction (G).
p62 dependency.
Given that many identified negative regulators of NRF2 are autophagy-related genes, we sought to determine whether these genes exert their effects on the NRF2-KEAP1 system through the autophagic cargo receptor p62. We found that the p62 level as well as the S349 phosphorylation status of p62 were inconsistently changed across sgRNA treatments and cell lines (Fig. 5A to D). This indicates that changes to levels of neither p62 nor phosphorylated p62 mediate NRF2 induction, signifying potential remaining knowledge gaps in the autophagic regulation of the NRF2-KEAP1 system.
While p62 and phosphorylated p62 levels may be relatively unchanged following gene knockout, it is possible that the processes are still p62 dependent: deletion of p62 in ATG7-deficient hepatocytes was shown to cancel NRF2 activation (53). We knocked out p62 in HK2 cells in conjunction with the aforementioned gene knockouts and then assessed the protein levels of NRF2 and its target genes (Fig. 6). Through these experiments we found that some of the newly identified negative regulators exert their effect through a p62-dependent mechanism (ATG7, ATG9A, ATG12, CELA2A, IFT172, LRRC8A, TMEM41B, and TXNRD1) and some through a p62-independent mechanism (ATG10, COLEC10, EDEM3, GOSR1, KEAP1, NRXN2, RAB6A, RB1CC1, SACM1L, SLC39A9, and VPS37A). Several studies have shown that p62 forms aggregates with KEAP1 upon autophagy impairment (52, 53, 58, 59, 64, 65). Thus, the p62-dependent genes, when knocked out, may impair autophagy and promote p62-KEAP1 aggregate formation. Our results also indicate that there are other mechanisms by which signals can be transduced to the NRF2-KEAP1 system in a p62-independent manner, signifying many venues for future exploration in the NRF2-KEAP1 pathway.
FIG 6.
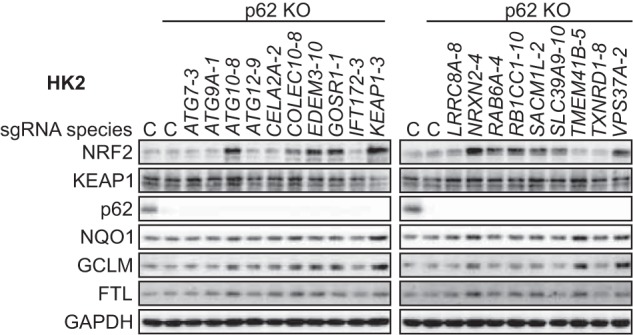
The newly identified negative regulators of NRF2 can be categorized into p62-dependent and p62-independent negative regulators. Western blots of NRF2, KEAP1, p62, and NRF2 target genes following combined knockout of p62 and genes identified as negative regulators by the CRISPR screen are shown.
Biological and pathological significance.
NRF2 activation is frequently found in cancer and is associated with poor prognoses. We sought to investigate whether the newly identified negative regulators of NRF2 are recurrently mutated in cancer using somatic mutation data from the Cancer Genome Atlas (TCGA) project. We focused on genes that were part of the autophagy pathway (Fig. 3B), were enriched for multiple sgRNAs (Fig. 3C), or were functionally validated as negative regulators of NRF2 (Fig. 4). Several genes were mutated in >5% of cases from certain tumor types (Fig. 7A). For example, RB1CC1 was frequently mutated in rectal adenocarcinoma and uterine corpus endometrial carcinoma, and NRXN2 was frequently mutated in colorectal adenocarcinoma, skin cutaneous melanoma, and uterine corpus endometrial carcinoma.
FIG 7.

Mutations to genes identified by CRISPR screen are found in cancer and may contribute to chemoresistance. (A) TCGA cases were stratified by tumor types for frequencies of mutations to genes identified by the CRISPR screen. Colors represent percentages of cases harboring mutations to that gene, within tumor type. (B) Frequency of amino acid mutations along the lengths of the top 12 most frequently mutated genes identified by CRISPR screen. Mutation spread along the length is indicative of a tumor suppressor. Positions harboring 4 or more mutations are annotated. (C) Twenty-four-hour cisplatin dose response of HK2 cells harboring various gene knockouts, as measured by MTS assay. (D) Cell viability assay of HK2 cells with various gene knockouts treated with 50 μM cisplatin, as measured by crystal violet assay. Error bars represent standard deviation of 3 biological replicates, and asterisks represent groups that were statistically significantly different from the control, as determined by analysis of variance (ANOVA) with the post hoc Tukey test.
We selected the 12 most frequently mutated genes and identified the frequency of mutations across the length of the protein. Oncogenes are repeatedly mutated at the same amino acid positions, while tumor suppressor genes are mutated throughout their lengths (66). Mutations of the negative regulators identified in this screen were spread throughout their lengths (Fig. 7B), which is indicative of a tumor suppressor role. Thus, loss of function of these genes may exhibit a cancer phenotype associated with sustained NRF2 activation.
A hallmark trait of NRF2-activated cancers is chemoresistance. Knockout of several of the negative regulators conferred resistance to various doses of cisplatin, a common chemotherapeutic (Fig. 7C). This was confirmed with a single administration of 50 μM cisplatin and assessment of cell viability relative to that of untreated cells by crystal violet staining (Fig. 7D). We conclude that mutation to many of the genes presented here may cause a cascade of loss of gene function, sustained NRF2 activation, and chemoresistance. Thus, identifying mutations to these genes in cancers may have clinical relevance.
DISCUSSION
The bulk of NRF2 regulation has been confined to a small number of degradation pathways mediated by relatively few key players: the KEAP1-CUL3-RBX1, β-TrCP–SKIP1–CUL1–RBX1, and SYNV1 pathways. Considering the role of NRF2 in a variety of physiological and pathological processes, identifying additional contributors to the NRF2 regulatory network will provide insight into the underlying mechanisms of various NRF2-related processes. We utilized a pooled CRISPR screen-based methodology to systematically identify 17 previously uncharacterized negative regulators of NRF2, with 8 of them, when knocked out, consistently upregulating NRF2 and NRF2 target genes across multiple cell lines. These 8 genes are “high-confidence” negative regulators of NRF2.
Using the CRISPR screen methodology, we found more than 11,000 sgRNAs present in the blasticidin-selected samples. Importantly, many of these sgRNAs targeted known negative regulators of NRF2, including ATG5, ATG7, BECN1, FH, BACH1, SETD6, KEAP1, CUL3, and CAND1. Of these, only BECN1, ATG7, KEAP1, CUL3, and CAND1 were deemed statistically significantly enriched despite all of them appearing in the blasticidin-selected samples. The absence of statistical enrichment for BACH1 may be explained by the unique mechanism by which BACH1 inhibits NRF2 activity. BACH1 does not directly inhibit NRF2; rather, it binds to the ARE to block NRF2 from binding. When BACH1 is knocked out, NRF2 levels do not increase beyond basal levels as they would with KEAP1 knockout; basal levels of NRF2 may have been insufficient to induce BSD transcription to a level high enough to confer adequate resistance that allowed a successful selection. Moreover, BACH1 affects only certain ARE-containing genes (67). Compared to many NRF2 target genes, such as AKR1C3, GCLC, TXNRD1, FTL, FTH1, and NQO1, BACH1 preferentially controls expression of HMOX-1 due to the presence of 12 putative AREs in its promoter region (61, 67). Indeed, repression by BACH1 has been proposed to be dependent on multiple AREs (67). Considering that our luciferase construct had 4 AREs, it is possible that stronger enrichment would have been identified with more AREs and weaker or nonexistent enrichment identified with fewer AREs.
The notable absence of the other genes could be due to slowed growth during the expansion phase of the experiment. For example, loss of ATG5 is associated with decreased proliferation in some cell types (68–72). Similarly, loss of SETD6 retards growth (73, 74). Loss of Krebs cycle function, such as that seen with FH inactivation, slows growth initially in most cell types while the population adapts to the new metabolic pathways activated for sufficient energy and anabolic processes. Indeed, FH is considered an “essential gene” in some cell systems (75). The study design and statistical methodology used here cannot account for different growth rates over the puromycin and blasticidin selection time periods. Additionally, our blasticidin selection stage was carried out under “normal” cell culture conditions. Several negative regulators may have more prominent roles in other cellular contexts that prevented their detection in this system. For example, GSK-3/β-TrCP negatively regulates NRF2 under conditions of low growth factor levels, and SYNV1 negatively regulates NRF2 under conditions of endoplasmic reticulum stress (50, 76, 77). Considering that neither β-TrCP nor SYNV1 was present in our screen, future investigations could attempt similar assays in other cellular contexts and further widen the NRF2 regulatory network. This could be particularly valuable under tissue conditions that are relevant to pathologies with known NRF2 dysregulation.
Despite the caveats of the statistical and study design methodologies, we narrowed the total set of 7,957 unique genes identified in our screen to 273 potential negative regulators of NRF2 by picking out those that were statistically enriched in the blasticidin-selected samples. Gene ontology analyses indicate that these genes were particularly enriched in autophagy processes. This is not entirely unexpected; NRF2 has been shown to be activated by dysfunctional autophagy previously. As mentioned, arsenic can inhibit autophagic flux, allowing for p62-dependent sequestration of KEAP1 and eventual NRF2 activation; additionally, silencing of autophagy genes ATG5, ATG7, and BECN1 is known to activate NRF2. Intriguingly, the NRF2 activator bis(2-hydoxybenzylidene)acetone induces autophagy more poorly in NRF2-deficient cells, suggesting that NRF2 plays a critical role in autophagy (78). This screen suggests that links between NRF2 and autophagy are more intimate and complicated than what has been discovered previously.
We identified autophagy as a converging regulatory point for NRF2. Several autophagy-related genes were found to negatively regulate NRF2 (Fig. 3B). Most of these genes are involved in autophagy nucleation or autophagosome elongation steps (Fig. 8).
FIG 8.

Negative regulators of NRF2 are found in autophagy. Negative regulators of NRF2 are found in autophagy pathways (red ovals). Signaling pathway diagrams were derived from references 116, to ,127.
Of the 8 high-confidence genes confirmed by functional validation, three of them (VPS37A, ATG7, and ATG12) are involved in autophagy. ATG5 (56), ATG7 (53, 57, 58), and BECN1 (59) were previously characterized as both autophagy related and negative regulators of NRF2. The other two autophagy genes identified in this screen, VPS37A and ATG12, were previously uncharacterized as negative regulators of NRF2. Despite no consistent changes in p62 or its phosphorylation level, p62 was still required for ATG12- and ATG7-mediated repression of NRF2. In contrast, VPS37A-mediated repression of NRF2 was p62 independent. Thus, autophagy genes may be negatively regulating NRF2 via at least two mechanisms, i.e., p62-dependent, and p62-independent mechanisms.
Intriguingly, several genes in this CRISPR screen only had one sgRNA species that was deemed significantly enriched by our statistical method. We highlighted that the sgRNAs targeting NRF2 may in fact be activating sgRNAs that mutate or knock out the degron required for KEAP1-mediated ubiquitylation. We hypothesize that some of the genes with only one sgRNA represented in the blasticidin-selected sample may be mutating to a variant that activates NRF2 (i.e., causing neomorphic mutations).
Of the 17 genes selected for validation (excluding KEAP1 and ATG7), some have shown relationships to the NRF2 pathway in the past. Most notably, TXNRD1 is a verified target gene of NRF2 with a known ARE enhancer (79, 80). Chronically TXNRD1-deficient hepatocytes show NRF2 induction (81), corroborating the efficacy of the screen presented here. Other works have demonstrated that both ATG10 and ATG7 have reactive thiols that are sensitive to oxidative stress, akin to KEAP1 (82). If any cross-signaling exists between NRF2 activation by oxidative insult and the ATG10/7 thiol-mediated stress response, it remains to be uncovered.
Several of the validated newly identified negative regulators of NRF2 (ATG12, IFT172, CELA2A, and TXNRD1) are themselves regulated by NRF2. Thus, some may serve as feedback loops to turn off NRF2 when its task is completed. For example, ATG12 was shown to be reduced in NRF2 knockdown cells (83). Similarly, in NRF2 knockout heart cells, IFT172 was shown to be downregulated (84). tert-Butyl-hydroquinone (TBHQ), a well-described NRF2 inducer, was shown to increase CELA2A mRNA (85), and, as mentioned above, TXNRD1 is a well-described NRF2 target. Despite these correlations between NRF2 activity and the induction of these genes, ATG12, IFT172, and CELA2A are not bona fide NRF2 target genes, so the mechanism by which NRF2 activates these genes remains unknown. For these genes and others from Fig. 4, their absence may increase oxidative or electrophilic stress and activate NRF2 via covalent modification of cysteine residues on KEAP1. Accordingly, TXNRD1 knockdown increased levels of NRF2 and oxidized (inactive) KEAP1 due to disulfide bridge formation between KEAP1-C226 and KEAP1-C613 (86). This has shown physiological relevance whereby the signaling gasotransmitter hydrogen sulfide stimulates production of the disulfide bond to inactivate KEAP1 and activate NRF2 (87). However, our results showed that TXNRD1 exerts its effect in a p62-dependent manner. It is possible that the disulfide bond formation does not disrupt KEAP1’s ability to mediate NRF2 ubiquitylation but instead facilitates p62-mediated KEAP1 sequestration. This mechanism potentially allows some negative regulators of NRF2 to form negative feedback loops by exploiting the autophagic regulation of the NRF2-KEAP1 system. Regardless of the mechanism, careful examination into the “off switches” of NRF2 is needed; current paradigms posit that (i) KEAP1 brings NRF2 back into the cytosol to turn off its signaling (88) and (ii) BACH1, a bona fide NRF2 target gene, competes with NRF2 for ARE binding at select ARE sequences, mitigating its signaling (89). It remains unknown whether ATG12, IF172, CELA2A, TXNRD1, or any of the target genes shown in Fig. 4 contribute to these OFF signaling paths or form part of a new, uncharacterized inhibitory feedback loop.
Due to its sustained activation in cancer, NRF2 contributes to many hallmarks of cancer (30). NRF2-activated tumors are notably resistant to therapeutics; we showed that several of the newly identified NRF2 negative regulators are mutated in cancer and upon depletion confer resistance to cisplatin. Loss of function of many of the negative regulators described here has been previously associated with poor prognoses in cancer. KEAP1 mutation or loss of KEAP1 is associated with poor patient prognosis in many cancers, including breast cancer (90, 91), glioma (92), and lung cancer (36, 93). Loss of RB1CC1 has been associated with poorer prognoses in breast cancer (94, 95), salivary gland cancer (96), prostate cancer (97), and bladder cancer (98). Low expression of NRXN2 is associated with shorter patient survival in ependymoma (99). Low expression of LRRC8A is associated with shorter patient survival in renal cancer (100). Low ATG9A is associated with lymph node involvement and lower patient survival in breast cancer (101, 102). Low NAE1 expression is unfavorable in thyroid cancer (100). Low COLEC10 expression leads to poorer prognoses in liver cancer (100, 103). Low SLC39A9 correlates with worse overall survival in gastric cancer and renal cancer (100, 104).
Identification of a potent and specific NRF2 inhibitor has been a longstanding goal in cancer biology, with many of the purported inhibitors showing significant off-target effects and there being a poor mechanistic understanding of their pharmacology (105). For example, the frequently used inhibitor brusatol is known to mitigate NRF2 activity through inhibition of protein translation (106). This mechanism is nonspecific and impedes the development of the compound for clinical use. Thus, there is an urgent need for improved NRF2 inhibitors. A better understanding of the mechanisms underlying NRF2-relevant functions of the 273 genes identified in our screen may augment current efforts to identify NRF2 inhibitors.
MATERIALS AND METHODS
Reagents.
The following compounds were used in this study: blasticidin (Gibco, A11139-03), hygromycin (Thermo Fisher, 10687010), puromycin (Gibco, A11138-03), and sulforaphane (LKT Labs, S8044). Insulin was purchased from Sigma (I6634). Cisplatin was purchased from Santa Cruz Biotechnology (sc-2008969) and made fresh for every use via dissolution in culture medium.
Cell culture and reagents.
All cell lines (BEAS-2B, MDA-MB-231, HK2, and NCI-H1299) were purchased from the ATCC. HK2, BEAS-2B, and NCI-H1299 cells and derivatives were cultured in Dulbecco’s modified Eagle’s medium (DMEM) with high glucose (4.5 g/liter) and no pyruvate and supplemented with heat-inactivated 10% fetal bovine serum (FBS). MDA-MB-231 was cultured in minimal essential medium (MEM) supplemented with 10% FBS and 6 ng/ml insulin. All cells were cultured at 37°C in atmospheric air enriched with 5% CO2.
Construction of ARE-BSD-PEST.
The open reading frame of a blasticidin resistance gene (BSD [encoding blasticidin S deaminase], obtained as a gene fragment from IDT) fused at the C terminus with a PEST sequence derived from the mouse ornithine decarboxylase gene (KLP RSH GFP PAV AAQ DDG TLP MSC AQE SGM DRH PAA CAS ARI NV) was cloned into pGL4.37 (Promega, E364A) in place of the endogenous luciferase gene. The resultant clone was named ARE-BSD-PEST (Fig. 1A). This system consists of a transcriptional pause site, a 4× ARE enhancer sequence, and a minimal promoter controlling the transcription of BSD-PEST (Fig. 1A). The transcriptional pause site, consisting of the sequence 5′-AAT CGA TAG TAC TAA CAT ACG CTC TCC ATC AAA ACA AAA CGA AAC AAA ACA AAC TAG CAA AAT AGG CTG TCC CCA GTG CAA GTG CAG GTG CCA GAA CAT TTC TCT-3′, was included to reduce background noise and prevent transcriptional interference (107–109). The quadruple ARE sequence consisted of 5′-TAG CTT GGA AAT GAC ATT GCT AAT GGT GAC AAA GCA ACT TTT AGC TTG GAA ATG ACA TTG CTA ATG GTG ACA AAG CAA CTT T-3′, where the canonical ARE sequence TGAYNNNGC is in bold.
Generation of HK2-BSD.
ARE-BSD-PEST was transfected into HK2 cells using Attractene transfection reagent (Qiagen). At 1 week after transfection, cells with stable incorporation of ARE-BSD-PEST were selected for with 300 μg/ml hygromycin. Single colonies were isolated and screened by reverse transcription-PCR (RT-PCR) for induction of BSD following treatment with 4 μM sulforaphane, a known NRF2 inducer; the colony showing the greatest induction of BSD following sulforaphane treatment was termed HK2-BSD.
Cell viability.
HK2-BSD was transfected using Attractene (Qiagen) with pSpCas9(BB)-2A-GFP (a gift from Feng Zhang; Addgene number 48138) (110), encoding sgRNA targeting no known gene in the human genome (control, 20-bp sequence GTA GCA CAT GGC GAC TCT TA), or a mixture of three sgRNAs targeting KEAP1 (20-bp sequences CCA GTT CAT GGC CCA CAA GG, GCT GCG GGA GCA GGG CAT GG, and GGC GCT CCA TGA CCT TGG GG) to make HK2-BSD-sgControl or HK2-BSD-sgKEAP1, respectively. KEAP1 knockout was validated by Western blotting. HK2-BSD-sgControl and HK2-BSD-sgKEAP1 were treated with various concentrations of blasticidin; 72 h later, viability was measured using the 3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)-2H-tetrazolium (MTS) assay (Promega) according to the manufacturer’s protocol. A blasticidin concentration of 10 μg/ml was selected as the optimized concentration for maximal cell death in non-NRF2-activated cells with minimal cytotoxicity to NRF2-activated cells.
sgRNA virus preparation.
The human activity-optimized CRISPR knockout plasmid library was purchased from Addgene (pooled library number 1000000067). This library was previously developed by the Sabatini and Lander laboratories (75). It consists of two pooled sublibraries, A and B, that collectively encode sgRNAs targeting all human genes for knockout. The libraries were electroporated into Endura electrocompetent cells and assessed for the number of CFU. After validating that the transformation efficiency was greater than 20-fold the library size, plasmids were isolated and prepared for virus packaging. For sgRNAs targeting individual genes, the requisite primers (see Data Set S4 in the supplemental material) were annealed, phosphorylated, and cloned into the pL-CRISPR.EFS.GFP vector (a gift from Benjamin Ebert; Addgene number 57818) (111). Lentivirus for all plasmids was produced, and titers were determined, using the ViraPower lentivirus expression system (Thermo Fisher) according to the manufacturer’s protocols.
sgRNA screen and sequencing.
sgRNA libraries were transduced into 2.1 × 109 HK2-BSD cells (1.05 × 109 cells per library) at a multiplicity of infection (MOI) of 0.1, which achieves an approximate 1,000× coverage of the total library. At 72 h postransduction, cells were selected with 2 μg/ml of puromycin to kill off untransduced cells. The surviving cells were expanded back to 2.1 × 109 cells in medium containing 2 μg/ml puromycin. Fifty percent of the surviving cells were used for genomic DNA isolation, and the isolated DNA was used as the template for total library coverage controls. The remaining 50% of cells were selected in medium containing 10 μg/ml blasticidin. Surviving cells were expanded back to ∼2.1 × 109 cells in medium containing 10 μg/ml blasticidin. Total genomic DNA was isolated from the blasticidin-selected cells and was used as the template for samples with sustained NRF2 activation. We used High Fidelity Q5 polymerase (NEB) to amplify the sgRNA cassette from the isolated genomic DNA templates. PCR amplicons were gel purified and subjected to next-generation sequencing using HiSeq 2500 with 10% PhiX spike-in.
Identification of enriched genes.
Sequencing fastq files were quality trimmed and processed into bins containing different sgRNA sequences using an in-house C++-implemented hash map. Statistical evaluation of the significance of enrichment for sgRNA species was performed by comparing the Poisson means for each sgRNA in the control to that in the sustained NRF2 activation samples according to the Etest (112). The test statistic was defined as , where c = t2/t1, t1 is the total number of counts in the unselected samples (also known as library coverage), and t2 is the total number of counts in the enriched sample (also known as enriched sample coverage). λ1 and λ2 represent the Poisson mean for each sgRNA in the total library and in the enriched sample, respectively. Known NRF2 targets were compiled from recent reviews (28, 30).
GO analysis.
Gene ontology (GO) enrichment analyses were conducted on the enriched genes in the R statistical environment (113). Gene-to-GO annotations defining relationships between genes and GO terms were accessed from NCBI. Significance testing was performed using the hypergeometric test to assess enrichment for the number of genes found in the blasticidin-selected samples for a specific GO term compared to the number of genes found in the unselected (total) samples for a specific GO term, with Holm correction for multiple testing. GO terms that were significantly enriched can be found in Data Set S3 in the supplemental material. Only GO terms with at least 2 genes enriched were taken forward for further analysis. We then took the set of enriched genes and GO terms and propagated the GO terms forward to their parental terms until we reached a converging biologically relevant term, “autophagy.” Directed acyclic graphs were generated using DiagrammeR in the R statistical environment (114). All GO terms for biological processes associated with the statistically enriched samples were tallied (Table 1; Data Set S4).
NRF2 induction validation by gene knockout.
Cells were plated at 30% confluence and transduced with relevant sgRNAs (see Data Set S5 in the supplemental material for primers that were used to generate the sgRNA species). For the MDA-MB-231, BEAS-2B, and NCI-H1299 cell lines, cells were harvested for Western blotting 5 to 7 days later. HK2 cells were harvested at approximately 3 weeks postransduction.
p62 knockout.
Two sgRNAs targeting p62 (5′-AGG GCT TCT CGC ACA GCC GC-3′ and 5′-CGT GGG CTC CAG TTT CCT GG-3′) were cloned into lentiCRISPR v2 (Addgene number 52961, a gift from Feng Zhang) (115). Lentivirus was produced as described above, and the viruses harboring the two sgRNAs were mixed together. Cells were infected with virus for 72 h, and positively transduced cells were selected with 2 μg/ml puromycin.
Western blotting and antibodies.
Cells were harvested in Laemmli sample buffer. Lysates were boiled, sonicated, resolved by SDS-PAGE, and then subjected to immunoblot analysis. Primary antibodies against the following proteins were used: ACTB (Sigma A1978, 1:10,000), FTL (Santa Cruz Biotechnology sc-74513, 1:1,000), GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (Santa Cruz Biotechnology sc-32233, 1:3,000), GCLM (Santa Cruz Biotechnology sc-55586, 1:1,000), KEAP1 (Santa Cruz Biotechnology sc-15246, 1:1,000), NQO1 (Santa Cruz Biotechnology sc-32793, 1:1,000), NRF2 (Santa Cruz Biotechnology sc-13032, 1:1,000), p62 (Santa Cruz Biotechnology sc-28359, 1:1,000), and phospho-p62 (anti-phospho-p62-S349, Abcam ab211324). ACTB or GAPDH was used as a loading control for all Western blots.
Cisplatin dose-response viability assay.
Cells were seeded at 6,500 cells per well on a 96-well plate and treated with various concentrations of cisplatin. After 24 h of cisplatin treatment, cell viability was measured using the CellTiter 96 AQueous One Solution assay (Promega, Madison, WI).
Crystal violet staining.
Cells were seeded at 100,000 cells per well on a 24-well plate and treated with 50 μM cisplatin. After 24 h of cisplatin treatment, cells were fixed in 100% ethanol for 10 min and stained with crystal violet solution (0.5% [wt/vol] crystal violet in 20% [vol/vol] methanol). Following washing and drying, stained cells were solubilized in 1% SDS. Absorbance was measured at 590 nm.
TCGA analysis.
Somatic mutations were downloaded from the Cancer Genome Atlas (TCGA) (http://cancergenome.nih.gov/) on 2 March 2017. Somatic mutation data from 33 tumor types identified using 4 different somatic mutation-calling algorithms (MuSE, MuTect2, SomaticSniper, and VarScan2) were utilized in the analyses as previously described (38). Protein sequences were retrieved from NCBI.
Data availability and ethical considerations.
Fastq files for the amplicon sequencing are available through the NCBI short-read archive (NCBI BioProject ID PRJNA540301). The mutation data sets analyzed in this study were generated by the TCGA Research Network and are freely available from The Cancer Genome Atlas (TCGA) consortium (http://cancergenome.nih.gov/). Only deidentified, publicly available data were downloaded and used for this study. Patients’ consent and institutional review board approval for the collection of the original data were obtained by TCGA. All methods were carried out in accordance with relevant guidelines and regulations.
Supplementary Material
ACKNOWLEDGMENTS
We declare that we have no conflicts of interest.
This work was supported by grant R21ES027920 (A.O.) from the National Institute of Environmental Health Sciences. A.O. was supported in part by R01CA226920. D.D.Z. was supported in part by R01DK109555, R01ES026845, and P42ES004940. Both D.D.Z. and A.O. are members of the P30ES006694-funded center. M.J.K. is supported by the National Science Foundation Graduate Research Fellowship Program under grant no. DGE-1746060.
Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.
We thank the UCLA TCGB sequencing core under the direction of Xinmin Li for technical advice.
Footnotes
Supplemental material for this article may be found at https://doi.org/10.1128/MCB.00037-19.
REFERENCES
- 1.Itoh K, Chiba T, Takahashi S, Ishii T, Igarashi K, Katoh Y, Oyake T, Hayashi N, Satoh K, Hatayama I, Yamamoto M, Nabeshima Y. 1997. An Nrf2/small Maf heterodimer mediates the induction of phase II detoxifying enzyme genes through antioxidant response elements. Biochem Biophys Res Commun 236:313–322. doi: 10.1006/bbrc.1997.6943. [DOI] [PubMed] [Google Scholar]
- 2.Kuosmanen SM, Viitala S, Laitinen T, Peräkylä M, Pölönen P, Kansanen E, Leinonen H, Raju S, Wienecke-Baldacchino A, Närvänen A, Poso A, Heinäniemi M, Heikkinen S, Levonen A-L. 2016. The effects of sequence variation on genome-wide NRF2 binding—new target genes and regulatory SNPs. Nucleic Acids Res 44:1760–1775. doi: 10.1093/nar/gkw052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Baird L, Dinkova-Kostova AT. 2011. The cytoprotective role of the Keap1–Nrf2 pathway. Arch Toxicol 85:241–272. doi: 10.1007/s00204-011-0674-5. [DOI] [PubMed] [Google Scholar]
- 4.Hayes JD, McMahon M, Chowdhry S, Dinkova-Kostova AT. 2010. Cancer chemoprevention mechanisms mediated through the Keap1-Nrf2 pathway. Antioxid Redox Signal 13:1713–1748. doi: 10.1089/ars.2010.3221. [DOI] [PubMed] [Google Scholar]
- 5.Kwak M-K, Kensler TW. 2010. Targeting NRF2 signaling for cancer chemoprevention. Toxicol Appl Pharmacol 244:66–76. doi: 10.1016/j.taap.2009.08.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lau A, Villeneuve NF, Sun Z, Wong PK, Zhang DD. 2008. Dual roles of Nrf2 in cancer. Pharmacol Res 58:262–270. doi: 10.1016/j.phrs.2008.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Chartoumpekis DV, Wakabayashi N, Kensler TW. 2015. Keap1/Nrf2 pathway in the frontiers of cancer and non-cancer cell metabolism. Biochem Soc Trans 43:639–644. doi: 10.1042/BST20150049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Holmström KM, Baird L, Zhang Y, Hargreaves I, Chalasani A, Land JM, Stanyer L, Yamamoto M, Dinkova-Kostova AT, Abramov AY. 2013. Nrf2 impacts cellular bioenergetics by controlling substrate availability for mitochondrial respiration. Biol Open 2:761–770. doi: 10.1242/bio.20134853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ludtmann MH, Angelova PR, Zhang Y, Abramov AY, Dinkova-Kostova AT. 2014. Nrf2 affects the efficiency of mitochondrial fatty acid oxidation. Biochem J 457:415–424. doi: 10.1042/BJ20130863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mitsuishi Y, Taguchi K, Kawatani Y, Shibata T, Nukiwa T, Aburatani H, Yamamoto M, Motohashi H. 2012. Nrf2 redirects glucose and glutamine into anabolic pathways in metabolic reprogramming. Cancer Cell 22:66–79. doi: 10.1016/j.ccr.2012.05.016. [DOI] [PubMed] [Google Scholar]
- 11.Kuosmanen SM, Kansanen E, Kaikkonen MU, Sihvola V, Pulkkinen K, Jyrkkänen H-K, Tuoresmäki P, Hartikainen J, Hippeläinen M, Kokki H, Tavi P, Heikkinen S, Levonen A-L. 2018. NRF2 regulates endothelial glycolysis and proliferation with miR-93 and mediates the effects of oxidized phospholipids on endothelial activation. Nucleic Acids Res 46:1124–1138. doi: 10.1093/nar/gkx1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murakami S, Motohashi H. 2015. Roles of Nrf2 in cell proliferation and differentiation. Free Radic Biol Med 88:168–178. doi: 10.1016/j.freeradbiomed.2015.06.030. [DOI] [PubMed] [Google Scholar]
- 13.Homma S, Ishii Y, Morishima Y, Yamadori T, Matsuno Y, Haraguchi N, Kikuchi N, Satoh H, Sakamoto T, Hizawa N, Itoh K, Yamamoto M. 2009. Nrf2 enhances cell proliferation and resistance to anticancer drugs in human lung cancer. Clin Cancer Res 15:3423–3432. doi: 10.1158/1078-0432.CCR-08-2822. [DOI] [PubMed] [Google Scholar]
- 14.Kerins MJ, Vashisht A, Liang B-T, Duckworth SJ, Praslicka BJ, Wohlschlegel JA, Ooi A. 2017. Fumarate mediates a chronic proliferative signal in fumarate hydratase inactivated cancer cells by increasing transcription and translation of ferritin genes. Mol Cell Biol 37:e00079-17. doi: 10.1128/MCB.00079-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kerins MJ, Ooi A. 2018. The roles of NRF2 in modulating cellular iron homeostasis. Antioxid Redox Signal 29:1756–1773. doi: 10.1089/ars.2017.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kasai S, Mimura J, Ozaki T, Itoh K. 2018. Emerging regulatory role of Nrf2 in iron, heme, and hemoglobin metabolism in physiology and disease. Front Vet Sci 5:242. doi: 10.3389/fvets.2018.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mills EL, Ryan DG, Prag HA, Dikovskaya D, Menon D, Zaslona Z, Jedrychowski MP, Costa ASH, Higgins M, Hams E, Szpyt J, Runtsch MC, King MS, McGouran JF, Fischer R, Kessler BM, McGettrick AF, Hughes MM, Carroll RG, Booty LM, Knatko EV, Meakin PJ, Ashford MLJ, Modis LK, Brunori G, Sévin DC, Fallon PG, Caldwell ST, Kunji ERS, Chouchani ET, Frezza C, Dinkova-Kostova AT, Hartley RC, Murphy MP, O'Neill LA. 2018. Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556:113. doi: 10.1038/nature25986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kansanen E, Kuosmanen SM, Leinonen H, Levonen A-L. 2013. The Keap1-Nrf2 pathway: mechanisms of activation and dysregulation in cancer. Redox Biol 1:45–49. doi: 10.1016/j.redox.2012.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Uruno A, Furusawa Y, Yagishita Y, Fukutomi T, Muramatsu H, Negishi T, Sugawara A, Kensler TW, Yamamoto M. 2013. The Keap1-Nrf2 system prevents onset of diabetes mellitus. Mol Cell Biol 33:2996–3010. doi: 10.1128/MCB.00225-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zheng H, Whitman SA, Wu W, Wondrak GT, Wong PK, Fang D, Zhang DD. 2011. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes 60:3055–3066. doi: 10.2337/db11-0807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chartoumpekis DV, Kensler TW. 2013. New player on an old field; the keap1/Nrf2 pathway as a target for treatment of type 2 diabetes and metabolic syndrome. Curr Diabet Rev 9:137–145. doi: 10.2174/1573399811309020005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Uruno A, Yagishita Y, Yamamoto M. 2015. The Keap1-Nrf2 system and diabetes mellitus. Arch Biochem Biophys 566:76–84. doi: 10.1016/j.abb.2014.12.012. [DOI] [PubMed] [Google Scholar]
- 23.Buendia I, Michalska P, Navarro E, Gameiro I, Egea J, León R. 2016. Nrf2–ARE pathway: an emerging target against oxidative stress and neuroinflammation in neurodegenerative diseases. Pharmacol Ther 157:84–104. doi: 10.1016/j.pharmthera.2015.11.003. [DOI] [PubMed] [Google Scholar]
- 24.de Vries HE, Witte M, Hondius D, Rozemuller AJM, Drukarch B, Hoozemans J, van Horssen J. 2008. Nrf2-induced antioxidant protection: a promising target to counteract ROS-mediated damage in neurodegenerative disease?. Free Radic Biol Med 45:1375–1383. doi: 10.1016/j.freeradbiomed.2008.09.001. [DOI] [PubMed] [Google Scholar]
- 25.Johnson DA, Johnson JA. 2015. Nrf2—a therapeutic target for the treatment of neurodegenerative diseases. Free Radic Biol Med 88:253–267. doi: 10.1016/j.freeradbiomed.2015.07.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yamazaki H, Tanji K, Wakabayashi K, Matsuura S, Itoh K. 2015. Role of the Keap1/Nrf2 pathway in neurodegenerative diseases. Pathol Int 65:210–219. doi: 10.1111/pin.12261. [DOI] [PubMed] [Google Scholar]
- 27.Dinkova‐Kostova AT, Kostov RV, Kazantsev AG. 2018. The role of Nrf2 signaling in counteracting neurodegenerative diseases. FEBS J 285:3576–3590. doi: 10.1111/febs.14379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Praslicka BJ, Kerins MJ, Ooi A. 2016. The complex role of NRF2 in cancer: a genomic view. Curr Opin Toxicol 1:37–45. doi: 10.1016/j.cotox.2016.09.003. [DOI] [Google Scholar]
- 29.Jaramillo MC, Zhang DD. 2013. The emerging role of the Nrf2-Keap1 signaling pathway in cancer. Genes Dev 27:2179–2191. doi: 10.1101/gad.225680.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rojo de la Vega M, Chapman E, Zhang DD. 2018. NRF2 and the hallmarks of cancer. Cancer Cell 34:21–43. doi: 10.1016/j.ccell.2018.03.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Qian Z, Zhou T, Gurguis CI, Xu X, Wen Q, Lv J, Fang F, Hecker L, Cress AE, Natarajan V, Jacobson JR, Zhang DD, Garcia JGN, Wang T. 2015. Nuclear factor, erythroid 2-like 2-associated molecular signature predicts lung cancer survival. Sci Rep 5:16889. doi: 10.1038/srep16889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang J, Zhang M, Zhang L, Cai H, Zhou S, Zhang J, Wang Y. 2010. Correlation of Nrf2, HO-1, and MRP3 in gallbladder cancer and their relationships to clinicopathologic features and survival. J Surg Res 164:e99–e105. doi: 10.1016/j.jss.2010.05.058. [DOI] [PubMed] [Google Scholar]
- 33.Konstantinopoulos PA, Spentzos D, Fountzilas E, Francoeur N, Sanisetty S, Grammatikos AP, Hecht JL, Cannistra SA. 2011. Keap1 mutations and Nrf2 pathway activation in epithelial ovarian cancer. Cancer Res 71:5081–5089. doi: 10.1158/0008-5472.CAN-10-4668. [DOI] [PubMed] [Google Scholar]
- 34.Shibata T, Kokubu A, Saito S, Narisawa-Saito M, Sasaki H, Aoyagi K, Yoshimatsu Y, Tachimori Y, Kushima R, Kiyono T, Yamamoto M. 2011. NRF2 mutation confers malignant potential and resistance to chemoradiation therapy in advanced esophageal squamous cancer. Neoplasia 13:864–873. doi: 10.1593/neo.11750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hu X-F, Yao J, Gao S-G, Wang X-S, Peng X-Q, Yang Y-T, Feng X-S. 2013. Nrf2 overexpression predicts prognosis and 5-FU resistance in gastric cancer. Asian Pac J Cancer Prev 14:5231–5235. doi: 10.7314/APJCP.2013.14.9.5231. [DOI] [PubMed] [Google Scholar]
- 36.Solis LM, Behrens C, Dong W, Suraokar M, Ozburn NC, Moran CA, Corvalan AH, Biswal S, Swisher SG, Bekele BN, Minna JD, Stewart DJ, Wistuba II. 2010. Nrf2 and Keap1 abnormalities in non-small cell lung carcinoma and association with clinicopathologic features. Clin Cancer Res 16:3743–3753. doi: 10.1158/1078-0432.CCR-09-3352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Namani A, Rahaman MM, Chen M, Tang X. 2018. Gene-expression signature regulated by the KEAP1-NRF2-CUL3 axis is associated with a poor prognosis in head and neck squamous cell cancer. BMC Cancer 18:46. doi: 10.1186/s12885-017-3907-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kerins MJ, Ooi A. 2018. A catalogue of somatic NRF2 gain-of-function mutations in cancer. Sci Rep 8:12846. doi: 10.1038/s41598-018-31281-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hayes JD, McMahon M. 2009. NRF2 and KEAP1 mutations: permanent activation of an adaptive response in cancer. Trends Biochem Sci 34:176–188. doi: 10.1016/j.tibs.2008.12.008. [DOI] [PubMed] [Google Scholar]
- 40.Itoh K, Wakabayashi N, Katoh Y, Ishii T, Igarashi K, Engel JD, Yamamoto M. 1999. Keap1 represses nuclear activation of antioxidant responsive elements by Nrf2 through binding to the amino-terminal Neh2 domain. Genes Dev 13:76–86. doi: 10.1101/gad.13.1.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McMahon M, Thomas N, Itoh K, Yamamoto M, Hayes JD. 2006. Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism a two-site interaction model for the Nrf2-Keap1 complex. J Biol Chem 281:24756–24768. doi: 10.1074/jbc.M601119200. [DOI] [PubMed] [Google Scholar]
- 42.Yamamoto M, Kensler TW, Motohashi H. 2018. The KEAP1-NRF2 system: a thiol-based sensor-effector apparatus for maintaining redox homeostasis. Physiol Rev 98:1169–1203. doi: 10.1152/physrev.00023.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McMahon M, Lamont DJ, Beattie KA, Hayes JD. 2010. Keap1 perceives stress via three sensors for the endogenous signaling molecules nitric oxide, zinc, and alkenals. Proc Natl Acad Sci U S A 107:18838–18843. doi: 10.1073/pnas.1007387107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Dinkova-Kostova AT, Holtzclaw WD, Cole RN, Itoh K, Wakabayashi N, Katoh Y, Yamamoto M, Talalay P. 2002. Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc Natl Acad Sci U S A 99:11908–11913. doi: 10.1073/pnas.172398899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang DD, Hannink M. 2003. Distinct cysteine residues in Keap1 are required for Keap1-dependent ubiquitination of Nrf2 and for stabilization of Nrf2 by chemopreventive agents and oxidative stress. Mol Cell Biol 23:8137–8151. doi: 10.1128/MCB.23.22.8137-8151.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rada P, Rojo AI, Evrard-Todeschi N, Innamorato NG, Cotte A, Jaworski T, Tobón-Velasco JC, Devijver H, García-Mayoral MF, Van Leuven F. 2012. Structural and functional characterization of Nrf2 degradation by the GSK-3/β-TrCP axis. Mol Cell Biol 88:147–157. doi: 10.1128/MCB.00180-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Suzuki T, Yamamoto M. 2015. Molecular basis of the Keap1-Nrf2 system. Free Radic Biol Med 88:93–100. doi: 10.1016/j.freeradbiomed.2015.06.006. [DOI] [PubMed] [Google Scholar]
- 48.Rada P, Rojo AI, Chowdhry S, McMahon M, Hayes JD, Cuadrado A. 2011. SCF (beta-TrCP) promotes glycogen synthase kinase-3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol Cell Biol 31:1121–1133. doi: 10.1128/MCB.01204-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lo S-C, Hannink M. 2006. CAND1-mediated substrate adaptor recycling is required for efficient repression of Nrf2 by Keap1. Mol Cell Biol 26:1235–1244. doi: 10.1128/MCB.26.4.1235-1244.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wu T, Zhao F, Gao B, Tan C, Yagishita N, Nakajima T, Wong PK, Chapman E, Fang D, Zhang DD. 2014. Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev 28:708–722. doi: 10.1101/gad.238246.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jiang T, Harder B, De La Vega MR, Wong PK, Chapman E, Zhang DD. 2015. p62 links autophagy and Nrf2 signaling. Free Radic Biol Med 88:199–204. doi: 10.1016/j.freeradbiomed.2015.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lau A, Zheng Y, Tao S, Wang H, Whitman SA, White E, Zhang DD. 2013. Arsenic inhibits autophagic flux, activating the Nrf2-Keap1 pathway in a p62-dependent manner. Mol Cell Biol 33:2436–2446. doi: 10.1128/MCB.01748-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Komatsu M, Kurokawa H, Waguri S, Taguchi K, Kobayashi A, Ichimura Y, Sou Y-S, Ueno I, Sakamoto A, Tong KI, Kim M, Nishito Y, Iemura S-i, Natsume T, Ueno T, Kominami E, Motohashi H, Tanaka K, Yamamoto M. 2010. The selective autophagy substrate p62 activates the stress responsive transcription factor Nrf2 through inactivation of Keap1. Nat Cell Biol 12:213. doi: 10.1038/ncb2021. [DOI] [PubMed] [Google Scholar]
- 54.Katsuragi Y, Ichimura Y, Komatsu M. 2016. Regulation of the Keap1-Nrf2 pathway by p62/SQSTM1. Curr Opin Toxicol 1:54–61. doi: 10.1016/j.cotox.2016.09.005. [DOI] [Google Scholar]
- 55.Ichimura Y, Waguri S, Sou YS, Kageyama S, Hasegawa J, Ishimura R, Saito T, Yang Y, Kouno T, Fukutomi T, Hoshii T, Hirao A, Takagi K, Mizushima T, Motohashi H, Lee MS, Yoshimori T, Tanaka K, Yamamoto M, Komatsu M. 2013. Phosphorylation of p62 activates the Keap1-Nrf2 pathway during selective autophagy. Mol Cell 51:618–631. doi: 10.1016/j.molcel.2013.08.003. [DOI] [PubMed] [Google Scholar]
- 56.Ni H-M, Boggess N, McGill MR, Lebofsky M, Borude P, Apte U, Jaeschke H, Ding W-X. 2012. Liver-specific loss of Atg5 causes persistent activation of Nrf2 and protects against acetaminophen-induced liver injury. Toxicol Sci 127:438–450. doi: 10.1093/toxsci/kfs133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Riley BE, Kaiser SE, Shaler TA, Ng ACY, Hara T, Hipp MS, Lage K, Xavier RJ, Ryu K-Y, Taguchi K, Yamamoto M, Tanaka K, Mizushima N, Komatsu M, Kopito RR. 2010. Ubiquitin accumulation in autophagy-deficient mice is dependent on the Nrf2-mediated stress response pathway: a potential role for protein aggregation in autophagic substrate selection. J Cell Biol 191:537. doi: 10.1083/jcb.201005012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Inami Y, Waguri S, Sakamoto A, Kouno T, Nakada K, Hino O, Watanabe S, Ando J, Iwadate M, Yamamoto M, Lee M-S, Tanaka K, Komatsu M. 2011. Persistent activation of Nrf2 through p62 in hepatocellular carcinoma cells. J Cell Biol 193:275–284. doi: 10.1083/jcb.201102031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lau A, Wang X-J, Zhao F, Villeneuve NF, Wu T, Jiang T, Sun Z, White E, Zhang DD. 2010. A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol Cell Biol 30:3275. doi: 10.1128/MCB.00248-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ooi A, Wong J-C, Petillo D, Roossien D, Perrier-Trudova V, Whitten D, Min BWH, Tan M-H, Zhang Z, Yang XJ, Zhou M, Gardie B, Molinié V, Richard S, Tan PH, Teh BT, Furge KA. 2011. An antioxidant response phenotype shared between hereditary and sporadic type 2 papillary renal cell carcinoma. Cancer Cell 20:511–523. doi: 10.1016/j.ccr.2011.08.024. [DOI] [PubMed] [Google Scholar]
- 61.Reichard JF, Motz GT, Puga A. 2007. Heme oxygenase-1 induction by NRF2 requires inactivation of the transcriptional repressor BACH1. Nucleic Acids Res 35:7074–7086. doi: 10.1093/nar/gkm638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chen A, Feldman M, Vershinin Z, Levy D. 2016. SETD6 is a negative regulator of oxidative stress response. Biochim Biophys Acta 1859:420–427. doi: 10.1016/j.bbagrm.2016.01.003. [DOI] [PubMed] [Google Scholar]
- 63.Sun J, Hoshino H, Takaku K, Nakajima O, Muto A, Suzuki H, Tashiro S, Takahashi S, Shibahara S, Alam J, Taketo MM, Yamamoto M, Igarashi K. 2002. Hemoprotein Bach1 regulates enhancer availability of heme oxygenase‐1 gene. EMBO J 21:5216–5224. doi: 10.1093/emboj/cdf516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kageyama S, Sou YS, Uemura T, Kametaka S, Saito T, Ishimura R, Kouno T, Bedford L, Mayer RJ, Lee MS, Yamamoto M, Waguri S, Tanaka K, Komatsu M. 2014. Proteasome dysfunction activates autophagy and the Keap1-Nrf2 pathway. J Biol Chem 289:24944–24955. doi: 10.1074/jbc.M114.580357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Fan W, Tang Z, Chen D, Moughon D, Ding X, Chen S, Zhu M, Zhong Q. 2010. Keap1 facilitates p62-mediated ubiquitin aggregate clearance via autophagy. Autophagy 6:614–621. doi: 10.4161/auto.6.5.12189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vogelstein B, Papadopoulos N, Velculescu VE, Zhou S, Diaz LA Jr, Kinzler KW. 2013. Cancer genome landscapes. Science 339:1546–1558. doi: 10.1126/science.1235122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.MacLeod AK, Higgins LG, McMahon M, Hayes JD, Plummer SM, Penning TM, Igarashi K. 2009. Characterization of the cancer chemopreventive NRF2-dependent gene battery in human keratinocytes: demonstration that the KEAP1-NRF2 pathway, and not the BACH1-NRF2 pathway, controls cytoprotection against electrophiles as well as redox-cycling compounds. Carcinogenesis 30:1571–1580. doi: 10.1093/carcin/bgp176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yu Y, Zhang J, Jin Y, Yang Y, Shi J, Chen F, Han S, Chu P, Lu J, Wang H, Guo Y, Ni X. 2018. MiR-20a-5p suppresses tumor proliferation by targeting autophagy-related gene 7 in neuroblastoma. Cancer Cell Int 18:5. doi: 10.1186/s12935-017-0499-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhu J, Li Y, Tian Z, Hua X, Gu J, Li J, Liu C, Jin H, Wang Y, Jiang G, Huang H, Huang C. 2017. ATG7 overexpression is crucial for tumorigenic growth of bladder cancer in vitro and in vivo by targeting the ETS2/miRNA196b/FOXO1/p27 axis. Mol Ther Nucleic Acids 7:299–313. doi: 10.1016/j.omtn.2017.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Pua HH, Dzhagalov I, Chuck M, Mizushima N, He Y-W. 2007. A critical role for the autophagy gene Atg5 in T cell survival and proliferation. J Exp Med 204:25–31. doi: 10.1084/jem.20061303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Vuppalapati KK, Bouderlique T, Newton PT, Kaminskyy VO, Wehtje H, Ohlsson C, Zhivotovsky B, Chagin AS. 2015. Targeted deletion of autophagy genes Atg5 or Atg7 in the chondrocytes promotes caspase‐dependent cell death and leads to mild growth retardation. J Bone Miner Res 30:2249–2261. doi: 10.1002/jbmr.2575. [DOI] [PubMed] [Google Scholar]
- 72.Jia W, He M-X, McLeod IX, Guo J, Ji D, He Y-W. 2015. Autophagy regulates T lymphocyte proliferation through selective degradation of the cell-cycle inhibitor CDKN1B/p27Kip1. Autophagy 11:2335–2345. doi: 10.1080/15548627.2015.1110666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mukherjee N, Cardenas E, Bedolla R, Ghosh R. 2017. SETD6 regulates NF-κB signaling in urothelial cell survival: implications for bladder cancer. Oncotarget 8:15114. doi: 10.18632/oncotarget.14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.O'Neill DJ, Williamson SC, Alkharaif D, Monteiro ICM, Goudreault M, Gaughan L, Robson CN, Gingras A-C, Binda O. 2014. SETD6 controls the expression of estrogen-responsive genes and proliferation of breast carcinoma cells. Epigenetics 9:942–950. doi: 10.4161/epi.28864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang T, Birsoy K, Hughes NW, Krupczak KM, Post Y, Wei JJ, Lander ES, Sabatini DM. 2015. Identification and characterization of essential genes in the human genome. Science 350:1096–1101. doi: 10.1126/science.aac7041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Hayes JD, Ebisine K, Sharma RS, Chowdhry S, Dinkova-Kostova AT, Sutherland C. 2016. Regulation of the CNC-bZIP transcription factor Nrf2 by Keap1 and the axis between GSK-3 and β-TrCP. Curr Opin Toxicol 1:92–103. doi: 10.1016/j.cotox.2016.10.003. [DOI] [Google Scholar]
- 77.Chowdhry S, Zhang Y, McMahon M, Sutherland C, Cuadrado A, Hayes JD. 2013. Nrf2 is controlled by two distinct beta-TrCP recognition motifs in its Neh6 domain, one of which can be modulated by GSK-3 activity. Oncogene 32:3765–3781. doi: 10.1038/onc.2012.388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Naidu SD, Dikovskaya D, Gaurilcikaite E, Knatko EV, Healy ZR, Mohan H, Koh G, Laurell A, Ball G, Olagnier D. 2017. Transcription factors NRF2 and HSF1 have opposing functions in autophagy. Sci Rep 7:11023. doi: 10.1038/s41598-017-11262-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hintze KJ, Keck A-S, Finley JW, Jeffery EH. 2003. Induction of hepatic thioredoxin reductase activity by sulforaphane, both in Hepa1c1c7 cells and in male Fisher 344 rats. J Nutr Biochem 14:173–179. doi: 10.1016/S0955-2863(02)00282-6. [DOI] [PubMed] [Google Scholar]
- 80.Hintze KJ, Wald KA, Zeng H, Jeffery EH, Finley JW. 2003. Thioredoxin reductase in human hepatoma cells is transcriptionally regulated by sulforaphane and other electrophiles via an antioxidant response element. J Nutr 133:2721–2727. doi: 10.1093/jn/133.9.2721. [DOI] [PubMed] [Google Scholar]
- 81.Suvorova ES, Lucas O, Weisend CM, Rollins MF, Merrill GF, Capecchi MR, Schmidt EE. 2009. Cytoprotective Nrf2 pathway is induced in chronically txnrd 1-deficient hepatocytes. PLoS One 4:e6158. doi: 10.1371/journal.pone.0006158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Frudd K, Burgoyne T, Burgoyne JR. 2018. Oxidation of Atg3 and Atg7 mediates inhibition of autophagy. Nat Commun 9:95. doi: 10.1038/s41467-017-02352-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bao L-J, Jaramillo MC, Zhang Z-B, Zheng Y-X, Yao M, Zhang DD, Yi X-F. 2014. Nrf2 induces cisplatin resistance through activation of autophagy in ovarian carcinoma. Int J Clin Exp Pathol 7:1502. [PMC free article] [PubMed] [Google Scholar]
- 84.He X, Ma Q. 2012. Redox regulation by Nrf2: gate-keeping for the basal and diabetes-induced expression of thioredoxin interacting protein. Mol Pharmacol 112:081133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Yi YW, Oh S. 2015. Comparative analysis of NRF2-responsive gene expression in AcPC-1 pancreatic cancer cell line. Genes Genomics 37:97–109. doi: 10.1007/s13258-014-0253-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Fourquet S, Guerois R, Biard D, Toledano MB. 2010. Activation of NRF2 by nitrosative agents and H2O2 involves KEAP1 disulfide formation. J Biol Chem 285:8463–8471. doi: 10.1074/jbc.M109.051714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hourihan JM, Kenna JG, Hayes JD. 2013. The gasotransmitter hydrogen sulfide induces nrf2-target genes by inactivating the keap1 ubiquitin ligase substrate adaptor through formation of a disulfide bond between cys-226 and cys-613. Antiox Redox Signal 19:465–481. doi: 10.1089/ars.2012.4944. [DOI] [PubMed] [Google Scholar]
- 88.Sun Z, Wu T, Zhao F, Lau A, Birch CM, Zhang DD. 2011. Importin α7 (KPNA6)-mediated nuclear import of Keap1 represses the Nrf2-dependent antioxidant response. Mol Cell Biol 31:1800–1811. doi: 10.1128/MCB.05036-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Jyrkkänen H-K, Kuosmanen S, Heinäniemi M, Laitinen H, Kansanen E, Mella-Aho E, Leinonen H, Ylä-Herttuala S, Levonen A-L. 2011. Novel insights into the regulation of antioxidant-response-elementmediated gene expression by electrophiles: induction of the transcriptional repressor BACH1 by Nrf2. Biochem J 440:167–174. doi: 10.1042/BJ20110526. [DOI] [PubMed] [Google Scholar]
- 90.Barbano R, Muscarella LA, Pasculli B, Valori VM, Fontana A, Coco M, la Torre A, Balsamo T, Poeta ML, Marangi GF, Maiello E, Castelvetere M, Pellegrini F, Murgo R, Fazio VM, Parrella P. 2013. Aberrant Keap1 methylation in breast cancer and association with clinicopathological features. Epigenetics 8:105–112. doi: 10.4161/epi.23319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Lee DF, Kuo HP, Liu M, Chou CK, Xia W, Du Y, Shen J, Chen CT, Huo L, Hsu MC, Li CW, Ding Q, Liao TL, Lai CC, Lin AC, Chang YH, Tsai SF, Li LY, Hung MC. 2009. KEAP1 E3 ligase-mediated downregulation of NF-kappaB signaling by targeting IKKbeta. Mol Cell 36:131–140. doi: 10.1016/j.molcel.2009.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Muscarella LA, Barbano R, D’Angelo V, Copetti M, Coco M, Balsamo T, la Torre A, Notarangelo A, Troiano M, Parisi S, Icolaro N, Catapano D, Valori VM, Pellegrini F, Merla G, Carella M, Fazio VM, Parrella P. 2011. Regulation of KEAP1 expression by promoter methylation in malignant gliomas and association with patient's outcome. Epigenetics 6:317–325. doi: 10.4161/epi.6.3.14408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Takahashi T, Sonobe M, Menju T, Nakayama E, Mino N, Iwakiri S, Nagai S, Sato K, Miyahara R, Okubo K, Hirata T, Date H, Wada H. 2010. Mutations in Keap1 are a potential prognostic factor in resected non-small cell lung cancer. J Surg Oncol 101:500–506. doi: 10.1002/jso.21520. [DOI] [PubMed] [Google Scholar]
- 94.Chano T, Ikebuchi K, Tomita Y, Jin Y, Inaji H, Ishitobi M, Teramoto K, Ochi Y, Tameno H, Nishimura I, Minami K, Inoue H, Isono T, Saitoh M, Shimada T, Hisa Y, Okabe H. 2010. RB1CC1 together with RB1 and p53 predicts long-term survival in Japanese breast cancer patients. PLoS One 5:e15737. doi: 10.1371/journal.pone.0015737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang L, Yao L, Zheng Y-Z, Xu Q, Liu X-P, Hu X, Wang P, Shao Z-M. 2015. Expression of autophagy-related proteins ATG5 and FIP200 predicts favorable disease-free survival in patients with breast cancer. Biochem Biophys Res Commun 458:816–822. doi: 10.1016/j.bbrc.2015.02.037. [DOI] [PubMed] [Google Scholar]
- 96.Tameno H, Chano T, Ikebuchi K, Ochi Y, Arai A, Kishimoto M, Shimada T, Hisa Y, Okabe H. 2012. Prognostic significance of RB1-inducible coiled-coil 1 in salivary gland cancers. Head Neck 34:674–680. doi: 10.1002/hed.21797. [DOI] [PubMed] [Google Scholar]
- 97.Li X, Wan X, Chen H, Yang S, Liu Y, Mo W, Meng D, Du W, Huang Y, Wu H, Wang J, Li T, Li Y. 2014. Identification of miR-133b and RB1CC1 as independent predictors for biochemical recurrence and potential therapeutic targets for prostate cancer. Clin Cancer Res 20:2312–2325. doi: 10.1158/1078-0432.CCR-13-1588. [DOI] [PubMed] [Google Scholar]
- 98.Andrew AS, Gui J, Hu T, Wyszynski A, Marsit CJ, Kelsey KT, Schned AR, Tanyos SA, Pendleton EM, Ekstrom RM, Li Z, Zens MS, Borsuk M, Moore JH, Karagas MR. 2015. Genetic polymorphisms modify bladder cancer recurrence and survival in a USA population-based prognostic study. BJU Int 115:238–247. doi: 10.1111/bju.12641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lukashova-V Zangen I, Kneitz S, Monoranu CM, Rutkowski S, Hinkes B, Vince GH, Huang B, Roggendorf W. 2007. Ependymoma gene expression profiles associated with histological subtype, proliferation, and patient survival. Acta Neuropathol 113:325–337. doi: 10.1007/s00401-006-0190-5. [DOI] [PubMed] [Google Scholar]
- 100.Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, Sanli K, von Feilitzen K, Oksvold P, Lundberg E, Hober S, Nilsson P, Mattsson J, Schwenk JM, Brunnström H, Glimelius B, Sjöblom T, Edqvist P-H, Djureinovic D, Micke P, Lindskog C, Mardinoglu A, Ponten F. 2017. A pathology atlas of the human cancer transcriptome. Science 357:eaan2507. doi: 10.1126/science.aan2507. [DOI] [PubMed] [Google Scholar]
- 101.Zhang X, Li C, Wang D, Chen Q, Li CL, Li HJ. 2016. Aberrant methylation of ATG2B, ATG4D, ATG9A and ATG9B CpG island promoter is associated with decreased mRNA expression in sporadic breast carcinoma. Gene 590:285–292. doi: 10.1016/j.gene.2016.05.036. [DOI] [PubMed] [Google Scholar]
- 102.Nunes J, Zhang H, Angelopoulos N, Chhetri J, Osipo C, Grothey A, Stebbing J, Giamas G. 2016. ATG9A loss confers resistance to trastuzumab via c-Cbl mediated Her2 degradation. Oncotarget 7:27599–27612. doi: 10.18632/oncotarget.8504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Zhang B, Wu H. 2018. Decreased expression of COLEC10 predicts poor overall survival in patients with hepatocellular carcinoma. Cancer Manag Res 10:2369–2375. doi: 10.2147/CMAR.S161210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Ding B, Lou W, Xu L, Li R, Fan W. 2019. Analysis the prognostic values of solute carrier (SLC) family 39 genes in gastric cancer. Am J Transl Res 11:486–498. [PMC free article] [PubMed] [Google Scholar]
- 105.Zhu J, Wang H, Chen F, Fu J, Xu Y, Hou Y, Kou HH, Zhai C, Nelson MB, Zhang Q, Andersen ME, Pi J. 2016. An overview of chemical inhibitors of the Nrf2-ARE signaling pathway and their potential applications in cancer therapy. Free Radic Biol Med 99:544–556. doi: 10.1016/j.freeradbiomed.2016.09.010. [DOI] [PubMed] [Google Scholar]
- 106.Harder B, Tian W, La Clair JJ, Tan AC, Ooi A, Chapman E, Zhang DD. 2017. Brusatol overcomes chemoresistance through inhibition of protein translation. Mol Carcinog 56:1493–1500. doi: 10.1002/mc.22609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Eggermont J, Proudfoot NJ. 1993. Poly(A) signals and transcriptional pause sites combine to prevent interference between RNA polymerase II promoters. EMBO J 12:2539–2548. doi: 10.1002/j.1460-2075.1993.tb05909.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Gromak N, West S, Proudfoot NJ. 2006. Pause sites promote transcriptional termination of mammalian RNA polymerase II. Mol Cell Biol 26:3986–3996. doi: 10.1128/MCB.26.10.3986-3996.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Shearwin KE, Callen BP, Egan JB. 2005. Transcriptional interference—a crash course. Trends Genet 21:339–345. doi: 10.1016/j.tig.2005.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, Zhang F. 2013. Genome engineering using the CRISPR-Cas9 system. Nat Protoc 8:2281. doi: 10.1038/nprot.2013.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Heckl D, Kowalczyk MS, Yudovich D, Belizaire R, Puram RV, McConkey ME, Thielke A, Aster JC, Regev A, Ebert BL. 2014. Generation of mouse models of myeloid malignancy with combinatorial genetic lesions using CRISPR-Cas9 genome editing. Nat Biotechnol 32:941. doi: 10.1038/nbt.2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Krishnamoorthy K, Thomson J. 2004. A more powerful test for comparing two Poisson means. J Stat Planning Inference 119:23–35. doi: 10.1016/S0378-3758(02)00408-1. [DOI] [Google Scholar]
- 113.R Core Team. 2018. R: a language and environment for statistical computing. R Foundation for Statistical Computing, Vienna, Austria: http://www.R-project.org. [Google Scholar]
- 114.Iannone R. 2018. DiagrammeR: graph/network visualization. https://cran.r-project.org/package=DiagrammeR. Accessed 2018.
- 115.Sanjana NE, Shalem O, Zhang F. 2014. Improved vectors and genome-wide libraries for CRISPR screening. Nat Methods 11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Klionsky DJ. 2005. The molecular machinery of autophagy: unanswered questions. J Cell Sci 118:7–18. doi: 10.1242/jcs.01620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Popovic D, Akutsu M, Novak I, Harper JW, Behrends C, Dikic I. 2012. Rab GTPase-activating proteins in autophagy: regulation of endocytic and autophagy pathways by direct binding to human ATG8 modifiers. Mol Cell Biol 32:1733–1744. doi: 10.1128/MCB.06717-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Velazquez AFC, Jackson WT. 2018. So many roads: the multifaceted regulation of autophagy induction. Mol Cell Biol 38:e00303-18. doi: 10.1128/MCB.00303-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Helgason GV, Holyoake TL, Ryan KM. 2013. Role of autophagy in cancer prevention, development and therapy. Essays Biochem 55:133–151. doi: 10.1042/bse0550133. [DOI] [PubMed] [Google Scholar]
- 120.Morita K, Hama Y, Izume T, Tamura N, Ueno T, Yamashita Y, Sakamaki Y, Mimura K, Morishita H, Shihoya W, Nureki O, Mano H, Mizushima N. 2018. Genome-wide CRISPR screen identifies TMEM41B as a gene required for autophagosome formation. J Cell Biol 217:3817–3828. doi: 10.1083/jcb.201804132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Noda T. 2017. Autophagy in the context of the cellular membrane-trafficking system: the enigma of Atg9 vesicles. Biochem Soc Trans 45:1323–1331. doi: 10.1042/BST20170128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Huang J, Birmingham CL, Shahnazari S, Shiu J, Zheng YT, Smith AC, Campellone KG, Heo WD, Gruenheid S, Meyer T, Welch MD, Ktistakis NT, Kim PK, Klionsky DJ, Brumell JH. 2011. Antibacterial autophagy occurs at PI(3)P-enriched domains of the endoplasmic reticulum and requires Rab1 GTPase. Autophagy 7:17–26. doi: 10.4161/auto.7.1.13840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Shoemaker CJ, Huang TQ, Weir NR, Polyakov N, Schultz SW, Denic V. 2019. CRISPR screening using an expanded toolkit of autophagy reporters identifies TMEM41B as a novel autophagy factor. PLoS Biol 17:e2007044. doi: 10.1371/journal.pbio.2007044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Bento CF, Puri C, Moreau K, Rubinsztein DC. 2013. The role of membrane-trafficking small GTPases in the regulation of autophagy. J Cell Sci 126:1059–1069. doi: 10.1242/jcs.123075. [DOI] [PubMed] [Google Scholar]
- 125.Kern A, Dikic I, Behl C. 2015. The integration of autophagy and cellular trafficking pathways via RAB GAPs. Autophagy 11:2393–2397. doi: 10.1080/15548627.2015.1110668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Cuif MH, Possmayer F, Zander H, Bordes N, Jollivet F, Couedel‐Courteille A, Janoueix‐Lerosey I, Langsley G, Bornens M, Goud B. 1999. Characterization of GAPCenA, a GTPase activating protein for Rab6, part of which associates with the centrosome. EMBO J 18:1772–1782. doi: 10.1093/emboj/18.7.1772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Shoemaker CJ, Huang TQ, Weir NR, Polyakov N, Denic V. 2017. A new CRISPR screening approach for identifying novel autophagy-related factors and cytoplasm-to-lysosome trafficking routes. BioRxiv doi: 10.1101/229732. [DOI]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Fastq files for the amplicon sequencing are available through the NCBI short-read archive (NCBI BioProject ID PRJNA540301). The mutation data sets analyzed in this study were generated by the TCGA Research Network and are freely available from The Cancer Genome Atlas (TCGA) consortium (http://cancergenome.nih.gov/). Only deidentified, publicly available data were downloaded and used for this study. Patients’ consent and institutional review board approval for the collection of the original data were obtained by TCGA. All methods were carried out in accordance with relevant guidelines and regulations.