

In this study, we investigated for the first time what roles, if any, innate lymphoid cells (ILCs) play in HSV-1 infection. Analysis of isolated ILCs in vitro revealed that all three subtypes could be infected with HSV-1 but that they were resistant to replication. The expression profiles of HSV-1-induced cytokines/chemokines and cellular and viral genes differed among the infected type 1, 2, and 3 ILCs in vitro. While ILCs play no role or a redundant role in the outcomes of latency-reactivation in infected mice, absence of type 1 and type 3, but not type 2, ILCs affects the survival of infected mice.
KEYWORDS: HSV-1, knockout, latency, infection, ocular, reactivation, replication
ABSTRACT
Innate lymphoid cells (ILCs) play important roles in host defense and inflammation. They are classified into three distinct groups based on their cytokine and chemokine secretion patterns and transcriptome profiles. Here, we show that ILCs isolated from mice can be infected with herpes simplex virus 1 (HSV-1) but that subsequent replication of the virus is compromised. After infection, type 2 ILCs expressed significantly higher levels of granulocyte colony-stimulating factor (G-CSF), interleukin 1α (IL-1α), IL-6, IL-9, RANTES, tumor necrosis factor alpha (TNF-α), CXCL1, CXCL2, CXCL10, CCL3, and CCL4 than infected type 1 or type 3 ILCs. Transcriptome-sequencing (RNA-seq) analysis of the ILCs 24 h after HSV-1 infection revealed that 77 herpesvirus genes were detected in the infected type 3 ILCs, whereas only 11 herpesvirus genes were detected in infected type 1 ILCs and 27 in infected type 2 ILCs. Compared with uninfected cells, significant upregulation of over 4,000 genes was seen in the HSV-1-infected type 3 ILCs, whereas 414 were upregulated in the infected type 1 ILCs and 128 in the infected type 2 ILCs. In contrast, in all three cell types, only a limited number of genes were significantly downregulated. Type 1, type 2, and type 3 ILC-deficient mice were used to gain insights into the effects of the ILCs on the outcome of ocular HSV-1 infection. No significant differences were found on comparison with similarly infected wild-type mice or on comparison of the three strains of deficient mice in terms of virus replication in the eyes, levels of corneal scarring, latency-reactivation in the trigeminal ganglia, or T-cell exhaustion. Although there were no significant differences in the survival rates of infected ILC-deficient mice and wild-type mice, there was significantly reduced survival of the infected type 1 or type 3 ILC-deficient mice compared with type 2 ILC-deficient mice. Adoptive transfer of wild-type T cells did not alter survival or any other parameters tested in the infected mice. Our results indicate that type 1, 2, and 3 ILCs respond differently to HSV-1 infection in vitro and that the absence of type 1 or type 3, but not type 2, ILCs affects the survival of ocularly infected mice.
IMPORTANCE In this study, we investigated for the first time what roles, if any, innate lymphoid cells (ILCs) play in HSV-1 infection. Analysis of isolated ILCs in vitro revealed that all three subtypes could be infected with HSV-1 but that they were resistant to replication. The expression profiles of HSV-1-induced cytokines/chemokines and cellular and viral genes differed among the infected type 1, 2, and 3 ILCs in vitro. While ILCs play no role or a redundant role in the outcomes of latency-reactivation in infected mice, absence of type 1 and type 3, but not type 2, ILCs affects the survival of infected mice.
INTRODUCTION
Innate lymphoid cells (ILCs) reside primarily at barrier surfaces. Although heterogeneous, they are derived from the same common lymphoid progenitor and lack antigen-specific receptors (1–4). They respond to changes in the tissue cytokine microenvironment by secreting specific effector cytokines and chemokines and have been shown to play key roles in the maintenance of mucosal homeostasis and protection against infection (1–4). Mature ILCs are divided into three major groups based on their cytokine secretion profiles and the transcription factors required for their development. These groups reflect the cytokine expression and transcription profiles of the classical CD4+ T helper (TH) cell subsets, with type 1 ILCs (ILC1) paralleling the profile of TH1 cells, type 2 ILCs paralleling the profile of TH2 cells, and type 3 ILCs paralleling the profile of TH17 cells (5–7). Type 1 ILCs produce interferon gamma (IFN-γ) in response to interleukin 12 (IL-12), IL-15, and IL-18, and type 2 ILCs secrete IL-5 and IL-13 in response to IL-25, IL-33, and thymic stromal lymphopoietin (TSLP), whereas type 3 ILCs produce IL-17, IL-22, and granulocyte-macrophage colony-stimulating factor (GM-CSF) when stimulated with IL-23 and/or IL-1β (7–9). The development of type 1 ILCs requires the T-box transcription factor T-bet (Tbx21), and the development of type 2 ILCs requires the transcription factor GATA3 or retinoic acid receptor-related orphan nuclear receptor α (RORα), whereas development of type 3 ILCs requires RORγt (10–12). Inbred C57BL/6 T-bet−/− (13), RoraFloxIL7RCre (14, 15), and Rorγt−/− mice (16) lack type 1 (17), type 2 (14), and type 3 ILCs (18), respectively, and were used as models for in vivo analysis of the effects of ILC deficiency.
ILCs are conserved in mice and humans (4, 10). They have been shown to play important roles in host defense, metabolic homeostasis, and tissue repair and can contribute to inflammatory diseases, such as asthma and colitis (19). Group 1 ILCs include type 1 ILCs and natural killer (NK) cells (3, 20). Similar to NK cells, type 1 ILCs function in the immune response to intracellular pathogens, including protozoan parasites, bacteria, and viruses (2, 21–23). Use of the T-bet−/− type 1 ILC-deficient mouse model has shown that type 1 ILCs limit Toxoplasma gondii replication in the intestine (24). Recently, it has been shown that human type 1 ILCs can be subcategorized into CD4+ and CD4− populations. CD4+ type 1 ILCs were efficiently infected by human immunodeficiency virus type 1 (HIV-1) in vitro and in vivo, while chronic infection with HIV-1 depleted both subpopulations and impaired their cytokine production (25).
Dysregulation of type 2 ILCs has been implicated in the pathology of several diseases, including allergies, asthma, dermatitis, and fibrosis, that have escalated in incidence in developed countries over the past decade (5, 7, 9, 11, 19, 26, 27). In terms of helminth infection, worm expulsion after Nippostrongylus brasiliensis infection is impaired in RoraFloxIL7RCre type 2 ILC-deficient mice (28), and the absence of major histocompatibility complex class II (MHC-II) reduced the ability of the type 2 ILCs to efficiently control helminth infection (29). In contrast, type 2 ILC deficiency had no effect on clearance of Citrobacter rodentium (28). Type 2 ILCs are the predominant ILC population in human and mouse lungs and are key initiators of allergen- and non-allergen-induced type 2 inflammation, as well as acting to promote airway tissue repair (1, 2, 30). Brain is also rich in type 2 ILCs, and it has been shown that mucosal neurons regulate type 2 inflammation by releasing neuromedin U (NMU), a neuropeptide that directly activates type 2 ILCs (31–33).
Type 3 ILCs are major regulators of inflammation and infection at mucosal barriers. Although they are present in small numbers in the intestinal tract, they have been shown to be important for controlling infection (34, 35), thereby providing defense against intestinal infections by various pathogenic bacteria, such as C. rodentium (36, 37), and fungi (2). Studies of mice with intact T cells have indicated that type 3 ILCs can have redundant roles in protection against enteropathogenic bacteria (38) but may function in the establishment and maintenance of a healthy gut microbial environment. A characteristic of the type 3 ILCs is their production of IL-17 (3, 4). Simian immunodeficiency virus (SIV) infection of macaques resulted in loss of IL-17-producing ILCs, especially in the jejunum (39) and in the intestinal mucosa (40). HIV-1 infection depleted type 3 ILCs during acute and chronic infection, and the numbers did not recover after resolution of peak infection (41, 42).
The published studies regarding the function of ILCs in infection and immunity suggest that ILCs have protective roles against certain diseases and infection, while their atypical activation is linked to pathogenesis. Their contributions to the immune response and pathogenesis can be influenced by external signals that can modulate or exacerbate their functional capabilities and, in some cases, generate novel ILC phenotypes. Currently, little is known regarding the role of ILCs in viral infection and virus-induced tissue damage in general and herpes simplex virus 1 (HSV-1) infection in particular. Recent findings that ILCs regulate adaptive T-cell responses (43, 44) led us to examine the regulatory potential of type 1, type 2, and type 3 ILCs in the context of HSV-1 infectivity in vitro and in vivo. Our approach included analysis of (i) virus replication in infected type 1, type 2, and type 3 ILCs; (ii) the effect of HSV-1 infection of ILCs on their expression of cytokines and chemokines; (iii) the transcriptomes of type 1, type 2, and type 3 ILCs before and after HSV-1 infection; and (iv) the effects of deficiency of ILCs on virus replication in the eye, survival, corneal scarring (CS), latency, T-cell exhaustion, and explant reactivation.
RESULTS
HSV-1 replication in infected ILCs.
To determine the susceptibility or resistance of the subtypes of mouse ILCs to HSV-1 infection, we isolated type 1 ILCs from the liver, type 2 ILCs from the lung, and type 3 ILCs from the intestine of Rag−/− mice with a C57BL/6 background. The cells were then incubated with 1 or 10 PFU/cell of wild-type (WT) HSV-1 strain McKrae. The kinetics of virus replication were quantitated by determining the amounts of infectious virus at various times postinfection (p.i.) using a standard rabbit skin (RS) plaque assay. At 1 PFU (Fig. 1A), the titers of HSV-1 in all the types of ILCs were less than that of the input virus. At 10 PFU, no increases in virus titers were observed over time (Fig. 1B). An increase in the titers of HSV-1 from 12 to 48 h p.i. at both 1 and 10 PFU was confirmed in the control RS cells (Fig. 1C). At all time points and both tested numbers of PFU, the amounts of infectious virus from all three types of ILCs were approximately 20% that of the input virus, suggesting that they do not support virus replication and that the detected virus was most likely residual input virus. These results suggest that, similar to our previous reports regarding dendritic cells (DCs) and macrophages (45), type 1, 2, and 3 ILCs from mice are susceptible to HSV-1 infection but do not support its replication.
FIG 1.
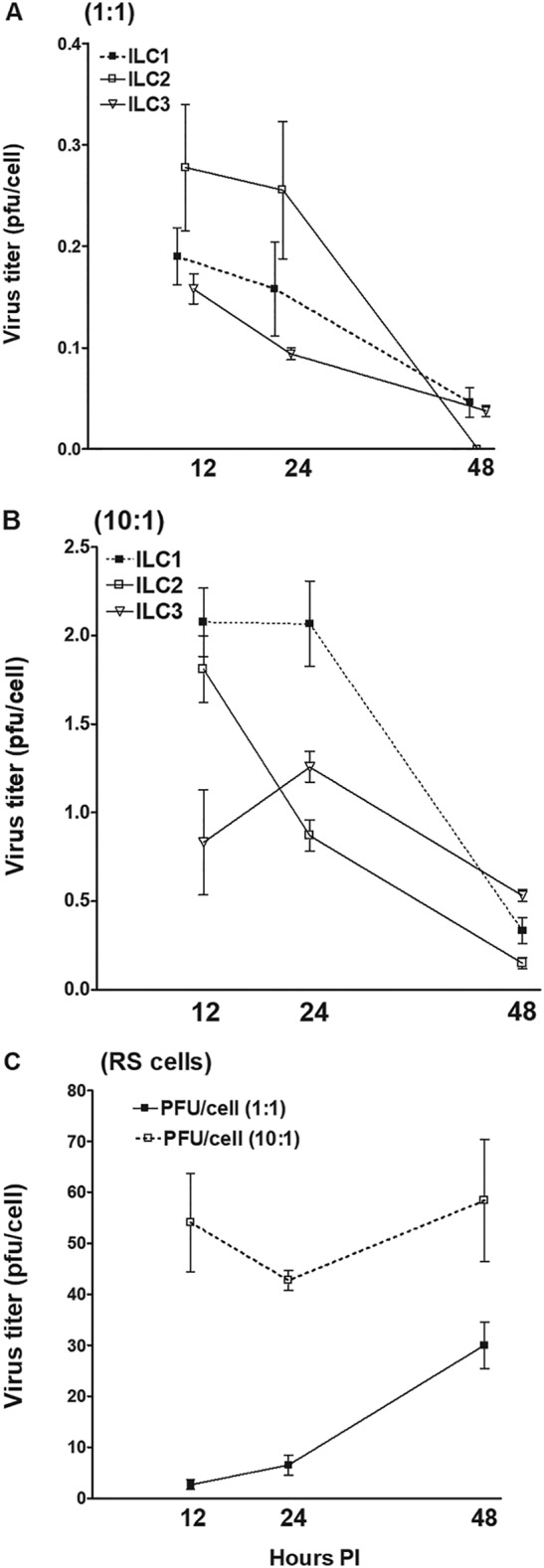
Virus replication in ILC1, -2, and -3. ILC1, -2, and -3 were isolated from Rag2−/− mice by cell sorting, as described in Materials and Methods. Each cell type was infected with 1 or 10 PFU/cell of WT McKrae virus for 12, 24, and 48 h. Total virus was harvested at the indicated times postinfection by two cycles of freeze-thawing. The amount of virus at each time for each cell was determined by standard plaque assays on RS cells. Each point represents the mean titers ± standard errors of the mean (SEM) from two independent experiments (n = 6). RS cells were similarly infected and used as controls. Due to the differences in the numbers of cells isolated per ILC subtype, the data are shown as virus titer per infected cell: 1 PFU/cell (A), 10 PFU/cell (B), and RS cells (C).
Differences in chemokine and cytokine expression in HSV-1-infected ILCs.
To determine if HSV-1 infection affects the production of cytokines and chemokines by the ILCs, the isolated ILCs were infected with HSV-1 strain McKrae (10 PFU/cell) as described above. Twenty-four hours later, the culture media from the wells containing the infected or uninfected cells were collected and the levels of cytokines and chemokines were determined using Illumina Mouse 32-Plex panels (Table 1). Since we added IL-2 and IL-7 to all cultures and IL-15 to the type 1 ILC cultures, these three cytokines were excluded from the analyses. Several factors were not detected in the supernatants from any of the uninfected ILC cell types, including the cytokines IL-1α, IL-3, IL-4, IL-9, IL-12p40, IL-12p70, IL-13, IL-17, leukemia inhibitory factor (LIF), granulocyte colony-stimulating factor (G-CSF), macrophage-CSF (M-CSF), IFN-γ, tumor necrosis factor alpha (TNF-α), and vascular endothelial growth factor (VEGF) and the chemokines CCL11, CCL24, CCL26, CXCL10 (IFN-γ-induced protein 10 [IP-10]), and CCL2 (MCP-1). Type 1 ILCs were unique in that IL-5 and IL-6 were not detectable in the supernatants from uninfected cells, and type 3 ILCs were unique in that GM-CSF was not detectable in the supernatants from uninfected cells. GM-CSF, RANTES, CXCL1 (KC), and CXCL9 (MIG) were not detectable in the supernatants from uninfected type 1 and type 3 ILCs, and IL-10 was not detectable in the supernatants from uninfected type 2 and type 3 ILCs.
TABLE 1.
Selected cytokine/chemokine levels in ILCs before and after infection with HSV-1
| Cytokine or chemokine | Level (pg/ml) in ILC typea
: |
|||||
|---|---|---|---|---|---|---|
| 1 |
2 |
3 |
||||
| Uninfected | Infected | Uninfected | Infected | Uninfected | Infected | |
| G-CSF | BDb | BD | BD | 16 ± 3c | BD | BD |
| GM-CSF | 4.1 ± 1.1 | BD | 10.5 ± 1.8 | 7.7 ± 0.6 | BD | 3.7 ± 0.7 |
| IFN-γ | BD | BD | BD | 3.5 ± 0.6 | BD | BD |
| IL-1α | BD | 2.6 ± 0.9 | BD | 13 ± 2c | BD | 1.1 ± 0.5 |
| IL-1β | 1.9 ± 1.4 | 6.6 ± 3.3 | 2.9 ± 2.4 | 0.5 ± 0.0 | 2.4 ± 1.9 | 5.8 ± 2.2 |
| IL-5 | BD | BD | 136 ± 3.0 | 111 ± 5c | 12.8 ± 1.8 | 18.1 ± 3.2 |
| IL-6 | BD | BD | 19.1 ± 0.1 | 67 ± 5c | 8 ± 2 | 17 ± 2 |
| IL-9 | BD | 58 ± 10c | BD | 12 ± 10c | BD | BD |
| IL-10 | 3.2 ± 0.2 | 3.8 ± 0.8 | BD | 4.0 ± 0.7 | BD | 3.9 ± 0.7 |
| CXCL5 (LIX) | 4.1 ± 0.6 | 4.8 ± 1.4 | 4.4 ± 0.3 | 5.1 ± 1.1 | 3.0 ± 0.5 | 4.9 ± 1.3 |
| CXCL10 (IP-10) | BD | BD | BD | 20 ± 3c | BD | BD |
| CXCL1 (KC) | BD | BD | 12.4 ± 3.7 | 213 ± 18c | BD | 8.6 ± 0.4 |
| CCL2 (MCP-1) | BD | 9.3 ± 4.3 | BD | 5.5 ± 0.5 | BD | 6.1 ± 1.1 |
| CCL3 (MIP-1α) | 7.7 ± 0.8 | 11.5 ± 2.8 | 15 ± 6 | 123 ± 3c | 9.5 ± 0.1 | 9.4 ± 1.3 |
| CCL4 (MIP-1β) | 16.4 ± 4.2 | 18.9 ± 6.4 | 22 ± 5 | 82 ± 7c | 12.4 ± 0.6 | 20 ± 0.8c |
| M-CSF | BD | BD | 5.9 ± 0.5 | 4.8 ± 1.8 | BD | BD |
| CXCL2 (MIP-2) | 19 ± 6 | 27 ± 10 | 87 ± 12 | 1195 ± 79c | 13 ± 6 | 34 ± 6c |
| CXCL9 (MIG) | BD | 2.5 ± 0.5 | 2.8 ± 0.8 | 2.3 ± 0.3 | BD | 2.5 ± 0.5 |
| RANTES | BD | BD | 4 ± 0.5 | 8 ± 0.5c | BD | BD |
| TNF-α | BD | BD | BD | 38 ± 3c | BD | BD |
Cytokine/chemokine levels in the culture media were analyzed using mouse 32-plex panels. Details of the experimental procedures are provided in Materials and Methods. Briefly, isolated type 1, type 2, and type 3 ILCs were infected with 10 PFU/cell of HSV-1 strain McKrae for 1 h at 37°C, washed with phosphate-buffered saline (PBS), and incubated for an additional 24 h in fresh medium. The results are presented as means ± SEM of the results from three experiments. No IL-3, IL-4, IL-12p40, IL-12p70, LIF, IL-13 IL-17, VEGF, or eotaxin was detected in any of the cell types before or after infection. As all three ILC cultures contained IL-2 and IL-7 and the type 1 ILC culture medium contained IL-15, these cytokines were excluded from the analyses. n = 3.
BD, below detection level.
Difference from the mock-infected counterpart is statistically significant.
Certain factors were not detectable in the supernatants of any of the infected ILCs, including IL-3, IL-4, IL-12p40, IL-12p70, IL-13, IL-17, LIF, M-CSF, and VEGF and the chemokines CCL11, CCL24, and CCL26. In contrast, certain factors were detectable in the supernatants of all three types of ILCs on infection, including IL-1α, IL-1β, and IL-10 and the chemokines CXCL2 (macrophage inflammatory protein 2 [MIP-2]), CXCL5 (LIX), CXCL9 (MIG), CCL2 (MCP-1), and CCL3 and CCL4 (MIP-1α and MIP-1β).
Type 1 ILCs were unique in that IL-5, IL-6, and the chemokine CXCL1 were not detectable in the supernatants of uninfected cells and GM-CSF was detectable in supernatants from uninfected but not infected cells. In these cells, IL-1β and IL-10 and the chemokines CXCL2, CXCL5, CCL3, and CCL4 were detected in the supernatants of both infected and uninfected cultures, but IL-9 was the only factor for which there was a statistically significant difference between the infected and uninfected cells (P < 0.0001) (Table 1). Similarly, in type 3 ILCs, there was a restricted pattern of altered expression on infection. Although IL-1β, IL-10, and the chemokines CXCL2, CXCL5, CCL3, and CCL4 were detected in the supernatants of both infected and uninfected type 3 ILCs, the only factors for which there were significant differences in levels between the infected and uninfected type 3 ILCs were CXCL2 and CCL4 (P < 0.05). The levels of cytokines and chemokines tended to be higher in the supernatants of the uninfected type 2 ILCs than in those of the uninfected type 1 and type 3 ILCs. Similarly, the levels of cytokines and chemokines tended to be higher in the supernatants of the infected type 2 ILCs than in those of the type 1 and type 3 ILCs. In common with type 1 ILCs, the infected type 2 ILCs exhibited significantly higher secretion of IL-9 than uninfected cells, and in common with the type 3 ILCs, they exhibited significantly higher secretion of CXCL2 and CCL4. However, the infected type 2 ILCS also secreted significantly higher levels of G-CSF, IL-1α, IL-6, TNF-α, RANTES, CXCL1, CXCL10, and CCL2, CCL3, and CCL4 than their uninfected counterparts, while the levels of IL-5 were significantly lower in the infected cells than in the uninfected cells (P < 0.05) (Table 1).
The above-mentioned results suggest that the patterns of cytokine and chemokine secretion in the uninfected type 1 and type 3 ILCs are similar and that HSV-1 infection induces similar changes in the profiles of these cell types, except for differences in the expression of IL-9, CCL4, and CXCL2. In contrast, the profile of cytokine and chemokine secretion in the uninfected type 2 ILCs differed considerably from that observed in the type 1 and type 3 ILCs, and HSV-1 infection of the type 2 ILCs resulted in significantly greater secretion of a much larger number of cytokines and chemokines.
Gene expression profiles of infected type 1, 2, and 3 ILCs.
To further characterize changes induced in the ILCs by HSV-1 infection, we used a transcriptome-sequencing (RNA-seq)-based transcriptome analysis approach. The ILCs were isolated and infected with HSV-1 as described above for analysis of cytokine expression. Total RNA was isolated using SMART-Seq V4 (Takara Bio USA, Inc., Mountain View, CA), and libraries were sequenced as described in Materials and Methods. As shown in Fig. 2, the transcriptome analysis of infected ILCs versus uninfected ILCs showed upregulation (>2-fold increase) of the expression of 414 genes in type 1 ILCs (Fig. 2a, right), 128 genes in type 2 ILCs (Fig. 2b, right), and 4,183 genes in type 3 ILCs (Fig. 2c, right). Downregulation (>2-fold decrease; P < 0.05) of the expression of 137 genes was found in type 1 ILCs (Fig. 2a, left), 110 genes in type 2 ILCs (Fig. 2b, left), and 127 genes in type 3 ILCs (Fig. 2c, left).
FIG 2.

The transcriptome in ILC1, -2, and -3 following infection with HSV-1. Type 1, 2, and 3 ILCs were isolated as for Fig. 1 and infected with 10 PFU/cell of WT McKrae for 24 h. The transcriptomes of infected type 1, type 2, and type 3 ILCs were compared with those of their corresponding uninfected counterparts. The x axis shows the fold change in gene expression in infected cells versus uninfected cells, and the y axis shows the statistical significance of the differences. The dots represent different genes. The red vertical lines represent a 2-fold change in gene expression, and the horizontal lines represent a P value of 0.05. (a) Type 1 ILCs. (b) Type 2 ILCs. (c) Type 3 ILCs.
GO of infected type 1, 2, and 3 ILCs.
Examination of the gene ontology (GO) terms enriched among the upregulated and downregulated genes in HSV-1-infected ILCs was carried out using the Database for Annotation, Visualization and Integrated Discovery (DAVID) v6.8. In the type 1 ILCs, the GO terms enriched among the upregulated genes included Immune Response, Immune System Process, Response to Virus Infection, Defense Response to Protozoan, and Positive Regulation of Interferon-γ Production (Fig. 3A). These GO terms are associated with genes involved in immune reactions, including Ltα, Ltβ, Ccl1, Ccl3, Gzmc, and Oas1c. The GO terms enriched among the downregulated genes in type 1 ILCs included Cellular Component Organization, Regulation of Interferon-γ Production, and Positive Regulation of Cell Migration (Fig. 3A). Type 2 ILCs also showed enrichment of GO terms associated with immune reactions, such as Chemotaxis, Immune Response Genes, Cellular Response to Lipopolysaccharide, and Inflammatory Response in upregulated genes. The genes associated with these GO terms are Cxcl2, Cxcl16, Ccr9, CTLA4, and Cxcl1. Thus, although these GO terms also are related to immune reaction genes, they appear to skew toward chemotaxis and inflammatory responses (Fig. 3B). GO terms enriched among downregulated genes in the type 2 ILCs included Lipid Transport, Bone Development, and Positive Regulation of Retinoic Acid (Fig. 3B). A broad range of GO terms that involved different cellular processes, such as Translation, RNA Splicing, mRNA Processing, and Protein Transport, were enriched in the genes that were upregulated in infected type 3 ILCs, which might reflect cellular proliferation induced by HSV-1 infection (Fig. 3C). In addition, the GO terms related to apoptosis and the MHC class I pathway were upregulated in infected type 3 ILCs (Fig. 3C). GO terms enriched among downregulated genes in the type 3 ILCs included Cell Adhesion, Bone Development, Meiotic Cell Cycle, and Extracellular Organization (Fig. 3C).
FIG 3.

GO term analysis of infected ILC1, -2, and -3. GO terms were enriched for genes that were upregulated (red) or downregulated (blue) in infected ILCs. The bars represent log-converted P values and minus log-converted P values for upregulated and downregulated genes, respectively, showing statistical significance. The GO terms were ranked according to statistical significance, and the top 15 GO terms for upregulated genes and the top 5 GO terms for downregulated genes are shown. (A) Type 1 ILCs. (B) Type 2 ILCs. (C) Type 3 ILCs.
Cellular gene expression in infected type 1, 2, and 3 ILCs.
To further examine the similarities and differences in the gene expression patterns induced in the ILCs by HSV-1 infection, we compared the gene sets showing significant changes in expression after infection (Fig. 4). There were overlaps in upregulated genes among the three types of ILCs (Fig. 4A). As shown in the Venn diagram, one gene, Tspan3, encoding tetraspanin 3, is upregulated in all three cell types (Fig. 4A). Tetraspanins are involved in multiple processes during virus infection and at various stages of infection, such as virus trafficking and exit. Among upregulated genes, there were 9 genes in common between type 1 and type 2 ILCs, 111 genes in common between type 1 and type 3 ILCs, and 22 genes in common between type 2 and type 3 ILCs (Fig. 4A); 293, 96, and 4,049 genes were uniquely upregulated in infected type 1 ILCs, type 2 ILCs, and type 3 ILCs, respectively (Fig. 4A). The downregulated genes in the infected ILCs were independent of each other except for three genes (Yam1, Zfp251, and Slc35e2) that were downregulated in both infected type 1 and type 3 ILCs (Fig. 4B).
FIG 4.

Cellular gene expression in infected ILCs. The Venn diagrams show the numbers of genes that are uniquely or commonly upregulated or downregulated upon infection of type 1, type 2, and type 3 ILCs (A and B), while the heat maps illustrate the genes with significant changes in expression upon HSV-1 infection (C to F). (A) upregulated genes in infected cells. (B) Downregulated genes in infected cells. (C) Heat map of type 2 ILCs versus type 3 ILCs showing genes that are significantly different from each other. (D) Heat map of type 1 ILCs versus type 2 ILCs showing genes that are significantly different from each other. (E) Heat map of type 1 ILCs versus type 3 ILCs showing genes that are significantly different from each other. (F) Heat map color key: red represents increase and blue represents decrease of gene expression upon infection.
Heat maps were used to identify genes that were either upregulated (red) or downregulated (blue) significantly upon HSV-1 infection in type 2 ILCs versus type 3 ILCs (Fig. 4C), type 1 ILCs versus type 2 ILCs (Fig. 4D), and type 1 ILCs versus type 3 ILCs (Fig. 4E). Besides the genes shown in the Venn diagrams, the heat maps show that eight genes are upregulated in type 2 ILCs but downregulated in type 3 ILCs (Cideb, Mylpf, Rad51b, Lbx2, Gm28151, Gpt, and CD38, and Fig4), and one gene was downregulated in type 2 ILCs but upregulated in type 3 ILCs (CD86) (Fig. 4C); three genes were upregulated in type 1 ILCs but downregulated in type 2 ILCs (3830403N18Rik, Ank, and Ssx2ip), and one gene was downregulated in type 1 ILCs but upregulated in type 2 ILCs (Adssl1) (Fig. 4D); 17 genes were upregulated in type 1 ILCs but downregulated in type 3 ILCs (Klf3, 2810021J22Rik, Usp53, Clybl, Zdhhc13, Ank, Pdlim5, Gmcl1, Ugdh, Mib1, Cpne3, Rnasel, Msh6, Nucks1, Kat2b, Abcb7, and Usp34), and one gene was downregulated in type 1 ILCs but upregulated in type 3 ILCs (CD40) (Fig. 4E).
HSV-1 gene expression in infected type 1, 2, and 3 ILCs.
Analysis of the expression of viral genes in infected ILCs (Fig. 5A) identified nine genes that were detected in all three cell types (ICP0, ICP4, UL15, UL19, UL28, UL36, gC, UL47, and gE genes). All of the HSV-1 genes detected in the type 1 and type 2 ILCs were also present in the type 3 ILCs, and an additional 48 genes were detected in the type 3 ILCs but not type 1 or type 2 ILCs (ICP34.5, gL, gM, UL2, UL3, UL4, UL7, UL11, UL12, UL14, UL16, UL18, UL19, UL20, UL21, gH, UL23, UL24, UL24, UL25, UL26, UL26.5, UL31, UL32, UL33, UL33, UL34, UL38, UL40, UL41, UL42, UL43, UL45, UL46, VP16, UL49, gN, UL50, UL51, gK, ICP27, UL55, UL56, ICP22, US2, gI, US8, and US9 genes). Eighteen genes were detected in type 2 and type 3 ILCs but not type 1 ILCs (UL5, UL6, UL8, UL9, UL13, UL17, gB, UL29, UL30, UL37, UL39, UL46, UL52, US10, US11, ICP47, gJ, and gD genes), and two genes (UL35 and US9 genes) were detected in type 1 and type 3 ILCs but not type 2 ILCs. Our results suggest that HSV-1 replication is curtailed greatly in type 1 ILCs and progresses slightly further in type 2 ILCs. In contrast, the process of replication is initiated in type 3 ILCs, although this does not culminate in a higher virus titer (Fig. 1). Detection of at least 77 HSV-1 genes in the infected type 3 ILCs is in line with the upregulation of a higher number of cellular genes in the infected type 3 ILCs than in infected type 1 or type 2 ILCs.
FIG 5.

Viral gene expression and RT-PCR in infected ILCs. (A) Venn diagram showing the numbers of HSV-1 genes uniquely or commonly detected in HSV-1-infected type 1, type 2, and type 3 ILCs. (B) Isolated ILC1, ILC2, and ILC3 were infected as for Fig. 1, and 24 h p.i., total RNA was isolated and qRT-PCRs for HSV-1 ICP4 and gB were performed as described in Materials and Methods. In each experiment, an estimated relative copy number for gB or ICP4 was calculated using standard curves generated from a plasmid containing the complete open reading frame (ORF) of gB or ICP4, respectively. GAPDH expression was used to normalize the relative expression of gB or ICP4 RNA. Each point represents the mean ± SEM from 3 infected ILC1, ILC2, or ILC3.
To confirm the RNA-seq results described above for HSV-1 gene expression in infected type 1, 2, and 3 ILCs, total RNA from infected cells was analyzed using quantitative real-time reverse transcription-PCR (qRT-PCR) with ICP4 or gB primers as described in Materials and Methods. We detected ICP4 and gB expression in all infected ILCs (Fig. 5B). The levels of both ICP4 and gB were significantly higher in infected ILC2 than in infected ILC1 or ILC3 (P < 0.001) (Fig. 5B). In contrast, the levels of ICP4 and gB expression in ILC1 and ILC3 cells were similar (Fig. 5B) (P > 0.05). Thus, similar to the RNA-seq results, the RT-PCR results showed differences in the level of HSV-1 gene expression between type 1, 2, and 3 ILCs.
Replication of HSV-1 in the eyes of ILC-deficient mice.
To assess the effects of ILCs on HSV-1 infection in vivo, we used inbred C57BL6 T-bet−/− (13), RoraFloxIL7RCre (14), and Rorγt−/− mice (16), which lack type 1 (17), type 2 (14), and type 3 (18) ILCs, respectively. In each of these experiments, groups of mice in which T cells from WT mice had been adoptively transferred to type 1 ILC-, type 2 ILC-, and type 3 ILC-deficient mice were included. No significant differences in any of the parameters that we tested were found between the mice that received T cells and those that did not. These results suggested that transfer of T cells to recipient type 1, type 2, and type 3 ILC-deficient mice did not alter their phenotypes with regard to HSV-1 infection. Correspondingly, the data presented (see Fig. 6 to 9) represent the data for mice that received T cells combined with the data generated using their respective counterparts that did not receive T cells.
FIG 6.
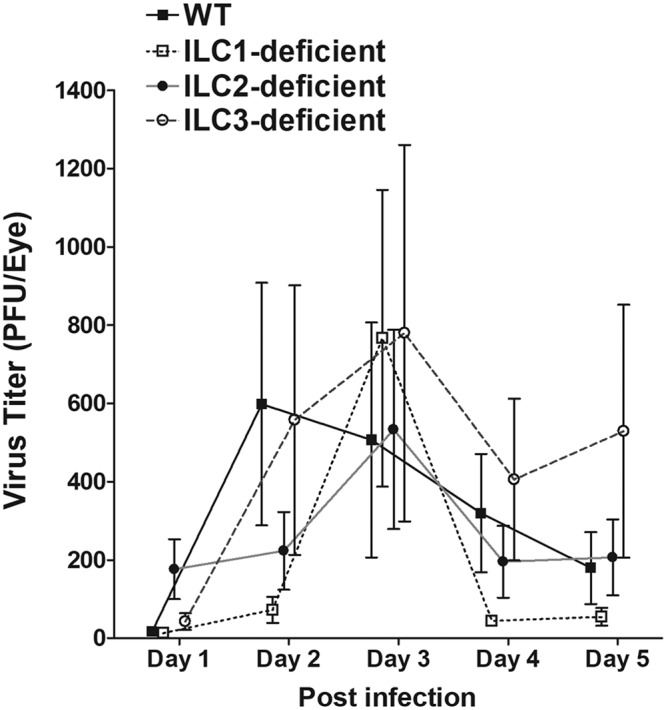
Virus titers in the eyes of infected mice. WT and type 1-, type 2-, and type 3 ILC-deficient mice were ocularly infected with 2 × 105 PFU/eye of HSV-1 strain McKrae as described in Materials and Methods. Tear films were collected on days 1 to 5, and virus titers were determined using standard plaque assays. Each point represents the mean titer ± SEM of 30 eyes for WT, 28 eyes for type 1 ILC-deficient, 26 eyes for type 2 ILC-deficient, and 80 eyes for type 3 ILC-deficient mice from 2 to 5 separate experiments.
FIG 9.
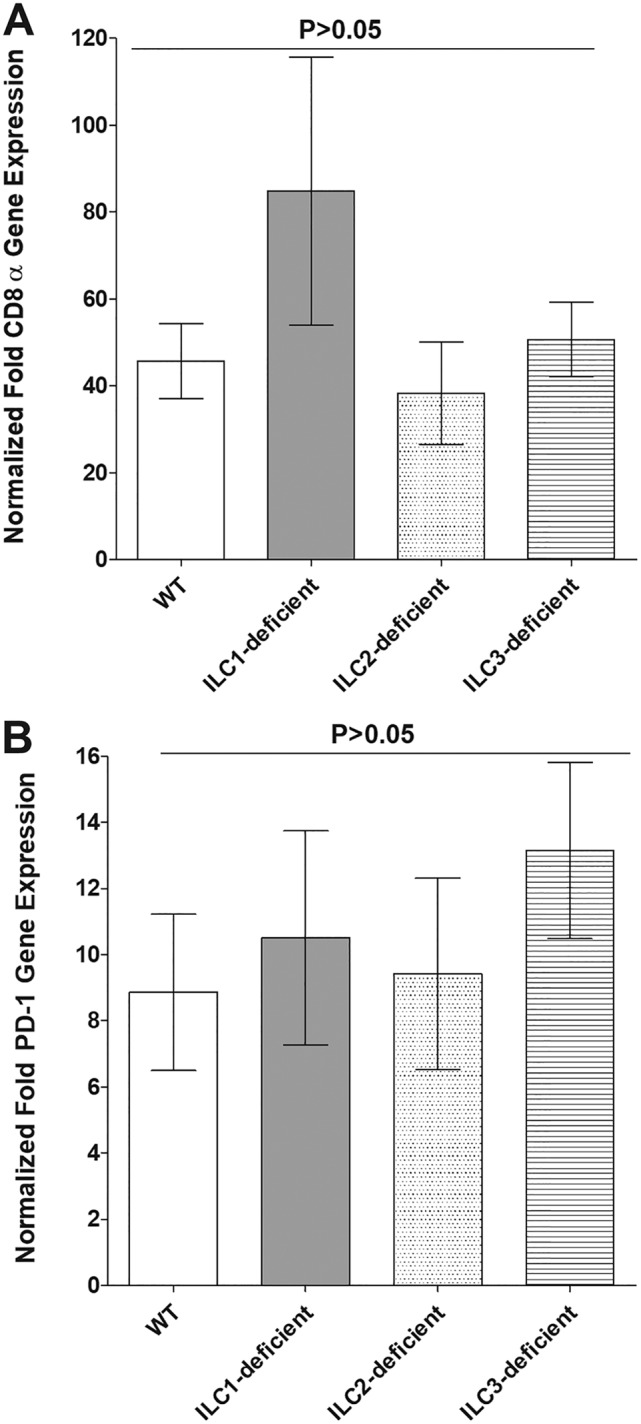
Roles of ILCs in CD8 and PD-1 expression in TG of latently infected mice. RNAs isolated from WT and type 1 ILC-, type 2 ILC-, and type 3 ILC-deficient mice as described in the legend to Fig. 8B were used to measure the expression of CD8α and PD-1 in latently infected TG. qRT-PCR was performed using total RNA, as described in Materials and Methods. CD8α and PD-1 expression in naive mice was used as a baseline control to estimate the relative expression of each transcript in TG of latently infected mice. GAPDH expression was used to normalize the relative expression of each transcript. Each point represents the mean ± SEM from 20, 10, 20, and 18 TG for WT and type 1 ILC-, type 2 ILC-, and type 3 ILC-deficient mice, respectively. The P value was determined using one-way ANOVA. (A) CD8α transcript. (B) PD-1 transcript.
We first tested the effects of deficiency of ILCs on ocular HSV-1 infection by ocularly infecting WT and ILC-deficient mice with 2 × 105 PFU/eye of HSV-1 strain McKrae. Tear films were collected from day 1 to day 5 p.i., and the titers of infectious virus in the tear films were determined using standard plaque assays. The virus titers in the eyes were similar among all four groups of mice between day 1 and day 5 p.i. (P > 0.05; analysis of variance [ANOVA]) (Fig. 6), suggesting that the absence of type 1, type 2, or type 3 ILCs did not affect HSV-1 replication in the eyes of the infected mice.
Virulence and corneal scarring in ocularly infected ILC-deficient mice.
Survival was monitored for 4 weeks in groups of WT and type 1 ILC-, type 2 ILC-, and type 3 ILC-deficient mice that had been infected in both eyes with 2 × 105 PFU/eye of strain McKrae. One mouse in the type 1 ILC-deficient group and three mice in the type 3 ILC-deficient group died after day 14 postinfection, and we did not include them in our survival figure. These deaths may or may not have been due to herpes encephalitis. As shown in Fig. 7A, consistent with previous reports, 84% of the WT mice (21 of 25) survived. All of the infected type 2 ILC-deficient mice (22/22) survived, 66% (8 of 12) of the infected type 1 ILC-deficient mice survived, and 58% (24 of 41) of the infected type 3-deficient mice survived. There were statistically significant differences between the type 3 ILC-deficient and type 1 ILC-deficient mice (P = 0.04; chi-square test) (Fig. 7A) and between the type 2 ILC-deficient and type 3 ILC-deficient mice (P = 0.0009; chi-square test) (Fig. 7A), which suggests that type 2 ILC-deficient mice are less susceptible to HSV-1 infection, while type 3 ILC-deficient mice are more susceptible.
FIG 7.
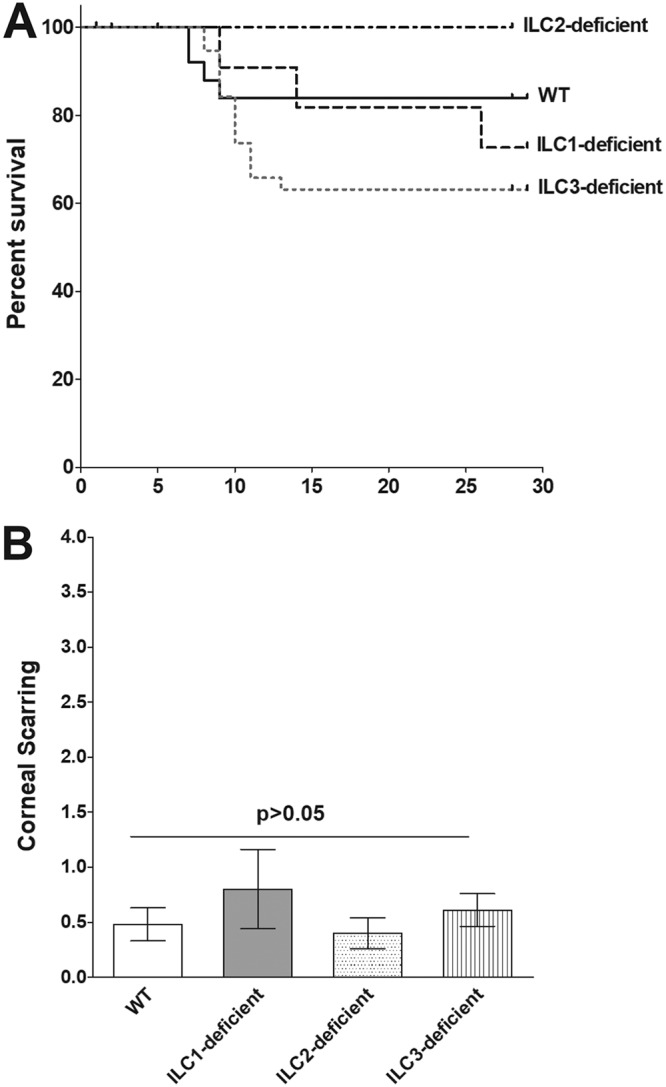
Survival of, and corneal scarring in, ocularly infected mice. Twelve to 41 mice per group were ocularly infected as for Fig. 6. Mouse survival was followed for 4 weeks. Survival analysis was based on 22 of 22 mice in the type 2 ILC-deficient group, 21 of 25 mice in the WT group, 8 of 12 mice in the type 1 ILC-deficient group, and 24 of 41 mice in the type 3 ILC-deficient group. The P value was determined using a chi-square test. CS was assessed in surviving mice on day 28 p.i. based on a scale of 0 (no scarring) to 4 (severe ulcer). CS was assessed in 42 eyes from WT mice, 16 eyes from type 1 ILC-deficient mice, 44 eyes from type 2 ILC-deficient mice, and 48 eyes from type 3 ILC-deficient mice. The CS score is presented as the mean ± SEM. The P value was determined using one-way ANOVA. (A) Survival in infected mice. (B) CS in surviving mice.
The eyes of the mice that survived ocular infection were examined for CS on day 28 p.i., and eye disease was scored on a 0-to-4 scale. We did not find significant differences in the levels of corneal scarring between each group of ILC-deficient mice and WT mice or among the groups of ILC-deficient mice (P > 0.05; ANOVA) (Fig. 7B). Thus, the absence of type 1, type 2, or type 3 ILCs neither ameliorated nor exacerbated the CS induced by ocular infection with HSV-1.
Time of reactivation is not affected in the TG of latently infected ILC-deficient mice.
In these experiments, WT and type 1 ILC-, type 2 ILC-, and type 3 ILC-deficient mice were infected ocularly with 2 × 105 PFU/eye of HSV-1 strain McKrae, and individual trigeminal ganglia (TG) from the surviving mice were isolated on day 28 p.i. Virus reactivation was analyzed using explanted individual TG from infected mice. We did not find significant differences in the times to explant reactivation between each group of ILC-deficient mice and WT mice or among the groups of ILC-deficient mice (P > 0.05; ANOVA) (Fig. 8A). Thus, the absence of type 1, type 2, or type 3 ILCs did not alter the time to explant reactivation in ocularly infected mice, suggesting that the cells do not play a role in virus reactivation.
FIG 8.

Duration of reactivation and level of latency in ocularly infected mice. For analysis of explant reactivation in infected mice, WT and type 1 ILC-, type 2 ILC-, and type 3 ILC-deficient mice were ocularly infected as described in the legend to Fig. 6. On day 28 p.i., TG from infected mice were harvested for explant reactivation. Each individual TG was incubated in 1.5 ml of tissue culture medium at 37°C, and the presence of infectious virus was monitored for 15 days. Reactivation is based on 20, 7, 17, and 22 TG for WT and type 1 ILC-, type 2 ILC-, and type 3 ILC-deficient mice, respectively. The average time that the TG from each group first showed CPE ± SEM is shown. The P value was determined using one-way ANOVA. For analysis of the levels of latency in the TG of latently infected mice, WT and type 1 ILC-, type 2 ILC-, and type 3 ILC-deficient mice were ocularly infected as described in the legend to Fig. 6. On day 28 p.i., TG were harvested from the latently infected mice. Quantitative RT-PCR was performed on the individual TG from each mouse. GAPDH expression was used to normalize the relative expression of LAT RNA in the TG. LAT copy numbers per TG were measured using pGEM5317, a LAT-containing plasmid, as we described previously (74). Latency was based on 20, 10, 20, and 18 TG for WT and type 1 ILC-, type 2 ILC-, and type 3 ILC-deficient mice, respectively. The P value was determined using one-way ANOVA. (A) Time of reactivation in TG of latently infected mice. (B) LAT expression in TG of latently infected mice.
The level of latency is not affected in the TG of latently infected ILC-deficient mice.
To determine if the absence ILCs modulates the levels of latency associated with ocular HSV-1 infection, WT and type 1 ILC-, type 2 ILC-, and type 3-ILC-deficient mice were infected ocularly with 2 × 105 PFU/eye of HSV-1 strain McKrae. Individual TG from surviving mice were isolated on day 28 p.i., and total RNA was isolated. Latency-associated transcript (LAT) RNA levels were quantitated using TaqMan RT-PCR. The combined data from two separate experiments are shown in Fig. 8B. We did not find significant differences in the amounts of LAT RNA during latency between type 1 ILC-, type 2 ILC-, or type 3 ILC-deficient mice and WT mice (P > 0.2; ANOVA) (Fig. 8B). Although the levels of LAT RNA in the type 2 ILC-deficient mice were twice those in WT and ILC1-deficient mice and three times those in ILC3-deficient mice, the differences were not statistically significant (P > 0.2) (Fig. 8B). These results suggest that ILC functions are not required for efficient latency in the TG of mice that have been ocularly infected with HSV-1.
Levels of CD8 and PD-1 mRNAs in TG of latently infected mice.
To investigate the effects of the absence of the ILCs on T-cell exhaustion in the TG of latently infected mice, the relative levels of CD8 and programmed cell death protein (PD-1; CD279) transcripts were determined in the TG of latently infected type 1 ILC-, type 2 ILC-, and type 3 ILC-deficient mice by RT-PCR of total TG RNA extracts, as depicted in Fig. 8B. The results are presented in Fig. 9 as “fold increase” in the infected type 1 ILC-, type 2 ILC-, and type 3 ILC-deficient mice compared to the baseline mRNA levels in the TG from their uninfected naive counterparts. The levels of CD8 (Fig. 9A) and PD-1 (Fig. 9B) mRNAs in the TG of the latently infected type 1 ILC-, type 2 ILC-, and type 3 ILC-deficient and WT mice were elevated compared with the levels in their uninfected counterparts; however, we did not find significant differences among the levels of CD8 and PD-1 mRNAs between the infected ILC-deficient mice and the infected WT mice or between the different groups of infected ILC-deficient mice (P > 0.05) (Fig. 9). The results were consistent with the previously observed increases in the numbers of CD8+ T cells and T-cell exhaustion, as evidenced by increased expression of the PD-1 markers in the TG of mice latently infected with HSV-1, and further suggest that the absence of type 1, type 2, or type 3 ILCs does not affect these parameters.
DISCUSSION
In the current study, we demonstrate that HSV-1 replication in type 1, type 2, and type 3 ILCs from WT mice is markedly lower than virus replication in RS cells. This result is similar to that of our previous studies, in which we found that both DCs and macrophages isolated from WT mice are refractory to HSV-1 infection (45). In the previous studies, we found that DCs and macrophages isolated from signal transducers and activators of transcription 1-deficient (STAT1−/−) mice were susceptible to HSV-1 replication and that the kinetics of replication were similar to those of HSV-1-infected RS. Our current results suggest that the refractory nature of ILCs to HSV-1 infection is not due to inefficient virus attachment. Similarly, our results suggest that the nonpermissiveness of ILCs to HSV-1 was not due to inefficient virus penetration, since viral transcripts were detected in all three types of ILCs. Despite the similarities in the levels of virus replication in type 1, type 2, and type 3 ILCs at 12, 24, and 48 h p.i. and at different levels (numbers of PFU) of infection, the patterns of gene expression as determined by RNA-seq differed significantly among the three cell types. More than 80 genes were found after efficient HSV-1 infection and replications; however, at 24 h p.i., only 11 HSV-1 genes were detected in the infected type 1 ILCs and 27 in the infected type 2 ILCs. In contrast, at least 77 HSV-1 genes were detected in the infected type 3 ILCs. Despite this detection of a large number of genes in the infected type 3 ILCs, the cells were refractory to HSV-1 replication. It is possible that the nonpermissive nature of the cells is immune mediated, as we have reported for DCs and macrophages (45).
RNA-seq analysis demonstrated that HSV-1 infection altered the expression of endogenous genes in ILCs and that there were substantial differences in the patterns of HSV-1-induced changes in gene expression among the type 1, type 2, and type 3 ILCs. Examination of the associated GO terms suggested preferential upregulation of immune response-associated genes in the type 1 and type 2 ILCs, and the higher levels of expression of these genes may have contributed to the low HSV-1 gene expression in these types of ILCs. In contrast, HSV-1 infection of the type 3 ILCs appears to upregulate the genes in a genomewide manner, given the very high numbers of significantly upregulated genes and the associated GO terms. The preferential association with the GO terms Translation, RNA Splicing, and mRNA Processing suggests that the virus is capable of hijacking the cellular machinery for RNA synthesis and protein production in infected ILCs and is consistent with the expression of a broad range of viral genes in these cells. We observed more cell death in the ILC3 group than in the other two infected-cell types. In infected ILC1, 20 apoptotic genes were upregulated, and in infected ILC2, no apoptotic gene was upregulated, while in infected ILC3, 140 apoptotic genes were upregulated. Thus, upregulated apoptosis genes in HSV-1-infected type 3 ILCs would make type 3 ILCs more vulnerable to virus infection. Although detection of most HSV-1 genes in infected type 3 ILCs is likely to be due to lower expression of immune-mediated genes, such as innate immune regulators that stimulate interferon genes. While reduced numbers of viral transcripts in type 1 and type 2 ILCs could be due to suppression of viral gene expression by higher immune-mediated responses, alternatively, it is possible that type 1 and type 2 ILCs are missing important cellular transcription factors.
Both T-cell-dependent immune responses and T-cell-independent immune responses have been implicated in protection against HSV-1 infection and manifestations of disease associated with infection (46–53). One of the goals of the present study was to determine if, similar to T cells and other innate cell types, the ILC-related immune responses contribute to protection against ocular HSV-1 infection or its disease manifestations. To determine the role of ILCs in HSV-1 infection, we used accepted ILC-deficient-mouse models; however, the absence of the specific transcription factors in these mice also may affect other immune-related responses. To address this issue, total T cells from WT mice were adoptively transferred into the ILC-deficient mice. We did not see any differences between mice that received T cells and their counterparts that did not receive T cells with regard to virus replication in the eye, eye disease, survival, latency, reactivation, or T-cell exhaustion. We did not observe any significant differences among the ILC-deficient mice in terms of the patterns of virus replication in the eye during primary infection or in the TG during latency, or in the time of reactivation or occurrence of eye disease. However, largely due to low abundance and lack of a good marker to detect different types of ILCs by immune staining or RT-PCR, it is not known if ILCs are present in healthy or diseased corneas. Compared with the other ILC-deficient mice, we did observe significantly reduced survival in the type 3 ILC-deficient RORγt mice. Although this reduced survival was not ameliorated by the adoptive transfer of WT T cells, it is possible that the profound effects of lack of RORγt on the immune system could affect survival independently of the lack of ILCs. For example, it has been shown previously that RORγt is required for thymocyte survival and lymphoid organ development (16).
Despite our in vitro findings that indicate that HSV-1 infection affects the biology of ILCs and the literature demonstrating their importance in autoimmunity, mucosal homeostasis, and certain infections (1–4), our in vivo studies suggest that ILCs do not play a significant role in determining the outcome of ocular HSV-1 infection, except for survival. Similar to our findings, it has been reported that the absence of ILCs had no effect in immunodeficient children after bone marrow transplantation and that ILC deficiencies in humans were not associated with any susceptibility to disease (54). The discrepancy between the effects of HSV-1 infection in vitro and in vivo may reflect differences in the microenvironmental cues but may also indicate that ILCs have redundant roles in the immune response to ocular HSV-1 infection with regard to some parameters, but not survival.
In summary, our results demonstrate that, similar to DCs and macrophages, ILCs are refractory to HSV-1 infection. Furthermore, the data presented here illustrate that the three different types of ILCs have very different responses to HSV-1 infection and control of HSV-1 replication. However, except for differences in survival among the three types of ILC-deficient mice, ILCs do not appear to affect the outcome of ocular HSV-1 infection. Based on our results, we propose a model in which ILC1 and ILC2 regulate HSV-1 via upregulation of immune response genes, whereas ILC3 may function via upregulation of apoptotic genes.
MATERIALS AND METHODS
Cells and virus.
RS cells were generated in our laboratory, prepared, grown in minimal essential medium (MEM) plus 5% fetal bovine serum (FBS), and used as described previously (55). The triple-plaque-purified virulent HSV-1 strain McKrae was grown in RS cell monolayers as described previously (56, 57).
Mice.
Inbred C57BL/6J T-bet−/− mice (13) lacking type 1 ILCs (17); Rorγt−/− mice (16) lacking type 3 ILCs (18); and Rag2−/− mice were obtained from the Jackson Laboratory (Bar Harbor, ME), while RoraFloxIL7RCre mice (lacking type 2 ILCs) were a gift from Andrew McKenzie (MRC Laboratory of Molecular Biology, Cambridge Biomedical Campus, Cambridge, United Kingdom) and have been described previously (14, 15). All the mice had a B6 background and were bred in-house. Both male and female (6- to 8-week-old) mice were used in the study. All animal procedures were performed in strict accordance with the Association for Research in Vision and Ophthalmology Statement for the Use of Animals in Ophthalmic and Vision Research and the NIH Guide for the Care and Use of Laboratory Animals (ISBN 0–309-05377-3). The animal research protocol was approved by the Institutional Animal Care and Use Committee of Cedars-Sinai Medical Center (protocols 5030 and 6134).
Isolation of type 1, type 2, and type 3 ILCs.
All cell isolations were carried out by cell sorting using a FACS Aria III instrument (BD Biosciences, Mountain View, CA) and the following antibodies: anti-CD11b (M1/70), anti-CD127 (A7R34), anti-Gr-1 (RB6-8C5), anti-IL-33Rα (DIH9), anti-NK1.1 (PK136), anti-NKp46 (29A1.4), anti-TER-119 (TER-119), and anti-TRAIL (N2B2) (all from BioLegend, San Diego, CA); anti-CD45 (30-F11; BioLegend or BD Biosciences, San Diego, CA); and anti-CD49b (DX5; eBioscience). For isolation of type 1 ILCs, livers from Rag2−/− mice were dissociated using a 70-μm cell strainer (Fisher Scientific, Waltham, MA) as described previously with minor modifications (58). Briefly, lymphocytes were enriched at the interface between gradients of 42% and 72% Percoll (GE Healthcare, Uppsala, Sweden). Type 1 ILCs were then sorted using a cocktail of antibodies to select for 7-aminoactinomycin D-negative (7AAD−) CD45+ CD49b− TRAIL+ cells. For isolation of type 2 ILCs, Rag2−/− mice were treated intranasally with 1 μg of mouse IL-33 (BioLegend) for 3 days in a row. The lungs were then removed aseptically, minced, and incubated in RPMI 1640 supplemented with 10% fetal calf serum (FCS), 250 U/ml of collagenase (CLS4; Worthington Biochemicals, Lakewood, NJ), and 25 U/ml DNase I (Sigma-Aldrich, St. Louis, MO) at 37°C for 20 min and then pressed through a 70-μm mesh filter, and the lymphocytes were enriched by centrifugation in 40% Percoll (GE Healthcare). Type 2 ILCs were sorted using a cocktail of antibodies to select for CD11b− Ter-119− Gr-1− NK1.1− 7AAD− CD45+ CD127+ IL33Rα+ cells, as described previously (59). Type 3 ILCs were isolated as described previously (60). Briefly, the intestines of Rag2−/− mice were removed and washed with Hanks balanced salt solution (HBSS) containing 2% FBS (Omega Bioscience, Tarzana, CA) and then opened and cut into 2-cm-long pieces that were incubated at 37°C for 20 min in HBSS containing 1 mM EDTA and 10 mM HEPES. The pieces were then further cut and incubated at 37°C for 20 min in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% FBS, 10 mM HEPES, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 500 U/ml collagenase IV, and 50 U/ml DNase I (Sigma-Aldrich). The solution containing digested tissue was passed through a 70-μm cell strainer, and the lymphocytes were enriched by centrifugation in 40% Percoll. The type 3 ILCs were sorted using a cocktail of antibodies to select for 7AAD− CD45+ NK1.1− NKp46+ cells. The average yields were 3 × 105 type 1 ILCs per mouse liver, 8 × 103 type 2 ILCs per mouse lung, and 3 × 104 type 3 ILCs per mouse intestine.
Virus titration in ILCs.
The isolated primary type 1, type 2, and type 3 ILCs were plated in 6-well plates and grown to 70 to 80% confluence. The cells were then infected with McKrae virus at 1 and 10 PFU/cell for 12, 24, and 48 h in RPMI 1640 medium supplemented with 10% FBS, 2 mM glutamine, 20 mM HEPES, 100 U/ml penicillin, 0.1 mg/ml streptomycin, 50 μM 2-mercaptoethanol, 10 ng/ml IL-2, and 10 ng/ml IL-7. In addition, 20 ng/ml IL-15 was added for type 1 ILCs, and 20 ng/ml IL-33 was added for type 2 ILCs (all the cytokines were from BioLegend). Virus was harvested at the indicated time points by two cycles of freeze-thawing of infected cells in medium. Virus titers were determined using a standard plaque assay on RS cells, as we described previously (61).
Luminex xMAP immunoassay (magnetic-bead kits).
Luminex assays were performed in the Immune Assessment Core at the University of California, Los Angeles (UCLA), as we described previously (62). Briefly, mouse 32-plex magnetic cytokine/chemokine kits were purchased from EMD Millipore (Billerica, MA) and used according to the manufacturer's instructions. Isolated type 1, type 2, and type 3 ILCs were infected with 10 PFU/cell of HSV-1 strain McKrae or left uninfected for 24 h as described above. Media from the infected and uninfected cells were collected, and 25 μl of 1:2-diluted samples were mixed with 25 μl magnetic beads and allowed to incubate overnight at 4°C with shaking. After washing the plates twice with wash buffer in a Biotek ELx405 washer, 25 μl of biotinylated detection antibody was added. The reaction mixture was incubated for 1 h at room temperature, and then streptavidin-phycoerythrin conjugate (25 μl) was added and the reaction mixture was incubated for another 30 min at room temperature. Following two washes, beads were resuspended in instrument sheath fluid buffer (Luminex Corp, Austin, TX), and fluorescence was quantified using a Luminex 200 instrument (Luminex Corp., Austin, TX).
Library preparation and sequencing.
Isolated type 1, type 2, and type 3 ILCs were infected with 10 PFU/cell of HSV-1 strain McKrae or left uninfected for 24 h as described above. Up to 10,000 infected or uninfected cells per sample in triplicate were pelleted, washed, flash-frozen in liquid nitrogen, and then stored at −80°C. Total RNA was isolated using a SMART-Seq V4 ultralow RNA input kit for sequencing (TaKaRa Bio USA, Inc., Mountain View, CA). The isolated RNA was used for reverse transcription and generation of double-stranded cDNA for subsequent library preparation using a Nextera XT library preparation kit (Illumina, San Diego, CA). Quantification of cDNA was performed using Qubit (Thermo Fisher Scientific). cDNA normalized to 80 pg/μl was fragmented, and the sequencing primers were added simultaneously. A limiting-cycle PCR added index 1 (i7) adapters, index 2 (i5) adapters, and sequences required for cluster formation on the sequencing flow cell. The indexed libraries were pooled and cleaned up, and the pooled library size was verified using a 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA) and quantified via Qubit. The libraries were sequenced using a NextSeq 500 (Illumina) with a single-end 75-bp read length and coverage of over 25 million reads/sample.
Data analysis.
The raw reads obtained were aligned to the transcriptome using STAR (version 2.5.0) (63) and RSEM (version 1.2.25) (64) with default parameters, using a custom mouse CRCm38 transcriptome reference, downloaded from http://www.gencodegenes.org, containing 92 External RNA Controls Consortium (ERCC) sequences, all protein-coding and long noncoding RNA genes based on Mouse GENCODE M8 annotation. Expression counts for each gene (transcripts per million [TPM]) in all samples were normalized by the sequencing depth. GO term analysis was performed using DAVID (v6.8) (65). Visualization of data was carried out using R statistical software (v3.5.1).
RNA extraction, cDNA synthesis, and TaqMan RT-PCR.
Isolated type 1, type 2, and type 3 ILCs were infected with 10 PFU/cell of HSV-1 strain McKrae or left uninfected for 24 h, and total RNA was isolated as described above. Following RNA extraction, the total RNA was reverse transcribed using random hexamer primers, as we have described previously (66). To confirm the RNA-seq results, the levels of HSV-1 ICP4 and gB in infected cells were evaluated using commercially available TaqMan gene expression assays (Applied Biosystems, Foster City, CA) with optimized primer and probe concentrations. The primer-probe sets consisted of two unlabeled PCR primers and the FAM (6-carboxyfluorescein) dye-labeled TaqMan MGB probe formulated into a single mixture. Additionally, all cellular amplicons included an intron-exon junction to eliminate signals from genomic-DNA contamination. The assays in this study used the following: (i) gB-specific primers (forward, 5′-AACGCGACGCACATCAAG-3′; reverse, 5′-CTGGTACGCGATCAGAAAGC-3′; and probe, 5′-FAM-CAGCCGCAGTACTACC-3′) and (ii) ICP4-specific primers (forward, 5′-GGCCGAGGGCTTCGA-3′; reverse, 5′-AGCTCGCGCAGCCA-3′; and probe, 5′-FAM-CCGCTTCCCCGCCGCC-3′). GAPDH (glyceraldehyde-3-phosphate dehydrogenase) (ABI Mm999999.15_G1) was used for normalization of transcripts.
Adoptive transfer of T cells.
To eliminate the possibility of an effect of the absence of each transcription factor on the development of T cells, WT T cells were adoptively transferred into type 1-, type 2-, and type 3-deficient mice before infection with HSV-1. Briefly, spleens from WT C57BL/6 mice were pooled, and single-cell suspensions were prepared as described previously (67). Total T cells were isolated using magnetic beads as described by the manufacturer (Miltenyi Biotec, San Diego, CA). Each recipient mouse was injected intraperitoneally once with T cells, equivalent to one donor mouse, suspended in MEM (300 μl). The control mice were injected intraperitoneally with MEM alone (300 μl). The mice were infected ocularly with McKrae virus 2 weeks after transfer of the T cells.
Ocular infection.
The mice deficient in type 1, type 2, or type 3 ILCs and the WT control mice were infected ocularly with 2 × 105 PFU of HSV-1 strain McKrae per eye in 2 μl of tissue culture medium as an eye drop without corneal scarification, as we have described previously (68).
Titration of virus in tears.
Tear films were collected from both eyes of infected mice on days 1 to 5 p.i. using a Dacron-tipped swab. Each swab was placed in tissue culture medium (1 ml), and the amount of virus in the medium was determined using a standard plaque assay on RS cells (69).
Evaluation of CS.
The severity of corneal scarring in surviving mice on day 28 p.i. was scored in a masked fashion on a scale of 0 to 4 (0, no disease; 1, 25% involvement; 2, 50% involvement; 3, 75% involvement; and 4, 100% involvement) as described previously (70).
In vitro explant reactivation assay.
Mice were sacrificed at 28 days p.i., and individual TG were removed and cultured in 1.5 ml tissue culture medium, as we described previously (71). Briefly, a 100-μl aliquot was removed from each culture daily for 15 days and used to infect RS cell monolayers. The RS cells were monitored daily for 5 days for the appearance of cytopathic effect (CPE) to determine the time of first appearance of reactivated virus from each TG. As the media from the explanted TG cultures were plated daily, the time at which reactivated virus first appeared in the explanted TG cultures could be determined.
RNA extraction, cDNA synthesis, and TaqMan RT-PCR.
TG were collected from naive mice and mice that had survived ocular infection on day 28 p.i., and the individual TG were immersed in RNAlater RNA stabilization reagent and stored at −80°C until they were processed. Tissue processing, total RNA extraction, and RNA yield determination were carried out as we have described previously (72, 73). Following RNA extraction, 1,000 ng of total RNA was reverse transcribed using random hexamer primers and murine leukemia virus (MuLV) reverse transcriptase from a high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA) in accordance with the manufacturer's recommendations. The levels of various RNAs were evaluated using commercially available TaqMan gene expression assays (Applied Biosystems, Foster City, CA) with optimized primer and probe concentrations. The primer-probe sets consisted of two unlabeled PCR primers and the FAM dye-labeled TaqMan MGB probe formulated into a single mixture. Additionally, all cellular amplicons included an intron-exon junction to eliminate signal from genomic DNA contamination. The assays used in this study were as follows: (i) CD8α, ABI assay Mn01182108_m1 (amplicon length, 67 bp); (ii) PD-1 (programmed death 1), ABI Mm00435532_m1 (amplicon size, 65 bp); and (iii) GAPDH, used for normalization of transcripts, ABI Mm999999.15_G1 (amplicon length, 107 bp).
The custom-made primer-probe set for LAT was as follows: forward primer, 5′-GGGTGGGCTCGTGTTACAG-3′; reverse primer, 5′-GGACGGGTAAGTAACAGAGTCTCTA-3′; and probe, 5′-FAM-ACACCAGCCCGTTCTTT-3′ (amplicon length, 81 bp, corresponding to LAT nucleotides 119553 to 119634). In each experiment, an estimated relative copy number for LAT was calculated using standard curves generated from pGem-LAT5317. Briefly, the plasmid DNA template was serially diluted 10-fold so that 5 μl contained from 103 to 1011 copies of the desired gene and then subjected to TaqMan PCR with the same set of primers as the test samples. By comparing the normalized threshold cycle of each sample to the threshold cycle of the standards, the copy number for each reaction was determined.
qRT-PCR was performed using the QuantStudio 5 system (Applied Biosystems, Foster City, CA) in 384-well plates, as we described previously (72, 73).
Statistical analyses.
Student's t test, ANOVA, and chi-square tests were performed using the computer program Instat (GraphPad, San Diego, CA). Results were considered statistically significant when the P value was <0.05.
ACKNOWLEDGMENTS
This study was supported by Public Health Service NIH grants R01EY029677, R01EY013615, and RO1EY026944.
REFERENCES
- 1.Artis D, Spits H. 2015. The biology of innate lymphoid cells. Nature 517:293–301. doi: 10.1038/nature14189. [DOI] [PubMed] [Google Scholar]
- 2.McKenzie ANJ, Spits H, Eberl G. 2014. Innate lymphoid cells in inflammation and immunity. Immunity 41:366–374. doi: 10.1016/j.immuni.2014.09.006. [DOI] [PubMed] [Google Scholar]
- 3.Fang D, Zhu J. 2017. Dynamic balance between master transcription factors determines the fates and functions of CD4 T cell and innate lymphoid cell subsets. J Exp Med 214:1861–1876. doi: 10.1084/jem.20170494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Vivier E, van de Pavert SA, Cooper MD, Belz GT. 2016. The evolution of innate lymphoid cells. Nat Immunol 17:790–794. doi: 10.1038/ni.3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Spits H, Di Santo JP. 2011. The expanding family of innate lymphoid cells: regulators and effectors of immunity and tissue remodeling. Nat Immunol 12:21–27. doi: 10.1038/ni.1962. [DOI] [PubMed] [Google Scholar]
- 6.Spits H, Artis D, Colonna M, Diefenbach A, Di Santo JP, Eberl G, Koyasu S, Locksley RM, McKenzie AN, Mebius RE, Powrie F, Vivier E. 2013. Innate lymphoid cells—a proposal for uniform nomenclature. Nat Rev Immunol 13:145–149. doi: 10.1038/nri3365. [DOI] [PubMed] [Google Scholar]
- 7.Tait Wojno ED, Artis D. 2016. Emerging concepts and future challenges in innate lymphoid cell biology. J Exp Med 213:2229–2248. doi: 10.1084/jem.20160525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rankin LC, Groom JR, Chopin M, Herold MJ, Walker JA, Mielke LA, McKenzie AN, Carotta S, Nutt SL, Belz GT. 2013. The transcription factor T-bet is essential for the development of NKp46+ innate lymphocytes via the Notch pathway. Nat Immunol 14:389–395. doi: 10.1038/ni.2545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salimi M, Barlow JL, Saunders SP, Xue L, Gutowska-Owsiak D, Wang X, Huang LC, Johnson D, Scanlon ST, McKenzie AN, Fallon PG, Ogg GS. 2013. A role for IL-25 and IL-33-driven type-2 innate lymphoid cells in atopic dermatitis. J Exp Med 210:2939–2950. doi: 10.1084/jem.20130351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eberl G, Colonna M, Di Santo JP, McKenzie AN. 2015. Innate lymphoid cells: a new paradigm in immunology. Science 348:aaa6566. doi: 10.1126/science.aaa6566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ebbo M, Crinier A, Vely F, Vivier E. 2017. Innate lymphoid cells: major players in inflammatory diseases. Nat Rev Immunol 17:665–678. doi: 10.1038/nri.2017.86. [DOI] [PubMed] [Google Scholar]
- 12.Diefenbach A, Colonna M, Koyasu S. 2014. Development, differentiation, and diversity of innate lymphoid cells. Immunity 41:354–365. doi: 10.1016/j.immuni.2014.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Finotto S, Neurath MF, Glickman JN, Qin S, Lehr HA, Green FH, Ackerman K, Haley K, Galle PR, Szabo SJ, Drazen JM, De Sanctis GT, Glimcher LH. 2002. Development of spontaneous airway changes consistent with human asthma in mice lacking T-bet. Science 295:336–338. doi: 10.1126/science.1065544. [DOI] [PubMed] [Google Scholar]
- 14.Halim TYF, Rana BMJ, Walker JA, Kerscher B, Knolle MD, Jolin HE, Serrao EM, Haim-Vilmovsky L, Teichmann SA, Rodewald HR, Botto M, Vyse TJ, Fallon PG, Li Z, Withers DR, McKenzie A. 2018. Tissue-restricted adaptive type 2 immunity is orchestrated by expression of the costimulatory molecule OX40L on group 2 innate lymphoid cells. Immunity 48:1195–1207. doi: 10.1016/j.immuni.2018.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rafei-Shamsabadi DA, van de Poel S, Dorn B, Kunz S, Martin SF, Klose CSN, Arnold SJ, Tanriver Y, Ebert K, Diefenbach A, Halim TYF, McKenzie ANJ, Jakob T. 2018. Lack of type 2 innate lymphoid cells promotes a type i-driven enhanced immune response in contact hypersensitivity. J Invest Dermatol 138:1962–1972. doi: 10.1016/j.jid.2018.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sun Z, Unutmaz D, Zou YR, Sunshine MJ, Pierani A, Brenner-Morton S, Mebius RE, Littman DR. 2000. Requirement for RORgamma in thymocyte survival and lymphoid organ development. Science 288:2369–2373. doi: 10.1126/science.288.5475.2369. [DOI] [PubMed] [Google Scholar]
- 17.Hoyler T, Connor CA, Kiss EA, Diefenbach A. 2013. T-bet and Gata3 in controlling type 1 and type 2 immunity mediated by innate lymphoid cells. Curr Opin Immunol 25:139–147. doi: 10.1016/j.coi.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 18.Montaldo E, Juelke K, Romagnani C. 2015. Group 3 innate lymphoid cells (ILC3s): origin, differentiation, and plasticity in humans and mice. Eur J Immunol 45:2171–2182. doi: 10.1002/eji.201545598. [DOI] [PubMed] [Google Scholar]
- 19.Zook EC, Kee BL. 2016. Development of innate lymphoid cells. Nat Immunol 17:775–782. doi: 10.1038/ni.3481. [DOI] [PubMed] [Google Scholar]
- 20.Spits H, Bernink JH, Lanier L. 2016. NK cells and type 1 innate lymphoid cells: partners in host defense. Nat Immunol 17:758–764. doi: 10.1038/ni.3482. [DOI] [PubMed] [Google Scholar]
- 21.Seillet C, Belz GT, Huntington ND. 2016. Development, homeostasis, and heterogeneity of NK cells and ILC1. Curr Top Microbiol Immunol 395:37–61. doi: 10.1007/82_2015_474. [DOI] [PubMed] [Google Scholar]
- 22.Abt MC, Buffie CG, Susac B, Becattini S, Carter RA, Leiner I, Keith JW, Artis D, Osborne LC, Pamer EG. 2016. TLR-7 activation enhances IL-22-mediated colonization resistance against vancomycin-resistant enterococcus. Sci Transl Med 8:27ra25. doi: 10.1126/scitranslmed.aad6663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Abt MC, McKenney PT, Pamer EG. 2016. Clostridium difficile colitis: pathogenesis and host defence. Nat Rev Microbiol 14:609–620. doi: 10.1038/nrmicro.2016.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klose CSN, Flach M, Mohle L, Rogell L, Hoyler T, Ebert K, Fabiunke C, Pfeifer D, Sexl V, Fonseca-Pereira D, Domingues RG, Veiga-Fernandes H, Arnold SJ, Busslinger M, Dunay IR, Tanriver Y, Diefenbach A. 2014. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 157:340–356. doi: 10.1016/j.cell.2014.03.030. [DOI] [PubMed] [Google Scholar]
- 25.Zhao J, Cheng L, Wang H, Yu H, Tu B, Fu Q, Li G, Wang Q, Sun Y, Zhang X, Liu Z, Chen W, Zhang L, Su L, Zhang Z. 2018. Infection and depletion of CD4+ group-1 innate lymphoid cells by HIV-1 via type-I interferon pathway. PLoS Pathog 14:e1006819. doi: 10.1371/journal.ppat.1006819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cording S, Medvedovic J, Aychek T, Eberl G. 2016. Innate lymphoid cells in defense, immunopathology and immunotherapy. Nat Immunol 17:755–757. doi: 10.1038/ni.3448. [DOI] [PubMed] [Google Scholar]
- 27.Hams E, Armstrong ME, Barlow JL, Saunders SP, Schwartz C, Cooke G, Fahy RJ, Crotty TB, Hirani N, Flynn RJ, Voehringer D, McKenzie AN, Donnelly SC, Fallon PG. 2014. IL-25 and type 2 innate lymphoid cells induce pulmonary fibrosis. Proc Natl Acad Sci U S A 111:367–372. doi: 10.1073/pnas.1315854111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Walker JA, Oliphant CJ, Englezakis A, Yu Y, Clare S, Rodewald HR, Belz G, Liu P, Fallon PG, McKenzie AN. 2015. Bcl11b is essential for group 2 innate lymphoid cell development. J Exp Med 212:875–882. doi: 10.1084/jem.20142224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oliphant CJ, Hwang YY, Walker JA, Salimi M, Wong SH, Brewer JM, Englezakis A, Barlow JL, Hams E, Scanlon ST, Ogg GS, Fallon PG, McKenzie AN. 2014. MHCII-mediated dialog between group 2 innate lymphoid cells and CD4(+) T cells potentiates type 2 immunity and promotes parasitic helminth expulsion. Immunity 41:283–295. doi: 10.1016/j.immuni.2014.06.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Monticelli LA, Sonnenberg GF, Abt MC, Alenghat T, Ziegler CG, Doering TA, Angelosanto JM, Laidlaw BJ, Yang CY, Sathaliyawala T, Kubota M, Turner D, Diamond JM, Goldrath AW, Farber DL, Collman RG, Wherry EJ, Artis D. 2011. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat Immunol 12:1045–1054. doi: 10.1031/ni.2131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klose CSN, Mahlakoiv T, Moeller JB, Rankin LC, Flamar AL, Kabata H, Monticelli LA, Moriyama S, Putzel GG, Rakhilin N, Shen X, Kostenis E, Konig GM, Senda T, Carpenter D, Farber DL, Artis D. 2017. The neuropeptide neuromedin U stimulates innate lymphoid cells and type 2 inflammation. Nature 549:282–286. doi: 10.1038/nature23676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cardoso V, Chesne J, Ribeiro H, Garcia-Cassani B, Carvalho T, Bouchery T, Shah K, Barbosa-Morais NL, Harris N, Veiga-Fernandes H. 2017. Neuronal regulation of type 2 innate lymphoid cells via neuromedin U. Nature 549:277–281. doi: 10.1038/nature23469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wallrapp A, Riesenfeld SJ, Burkett PR, Abdulnour RE, Nyman J, Dionne D, Hofree M, Cuoco MS, Rodman C, Farouq D, Haas BJ, Tickle TL, Trombetta JJ, Baral P, Klose CSN, Mahlakoiv T, Artis D, Rozenblatt-Rosen O, Chiu IM, Levy BD, Kowalczyk MS, Regev A, Kuchroo VK. 2017. The neuropeptide NMU amplifies ILC2-driven allergic lung inflammation. Nature 549:351–356. doi: 10.1038/nature24029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song C, Lee JS, Gilfillan S, Robinette ML, Newberry RD, Stappenbeck TS, Mack M, Cella M, Colonna M. 2015. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J Exp Med 212:1869–1882. doi: 10.1084/jem.20151403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pearson C, Thornton EE, McKenzie B, Schaupp AL, Huskens N, Griseri T, West N, Tung S, Seddon BP, Uhlig HH, Powrie F. 2016. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. Elife 5:e10066. doi: 10.7554/eLife.10066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guo X, Qiu J, Tu T, Yang X, Deng L, Anders RA, Zhou L, Fu YX. 2014. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity 40:25–39. doi: 10.1016/j.immuni.2013.10.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Pickard JM, Maurice CF, Kinnebrew MA, Abt MC, Schenten D, Golovkina TV, Bogatyrev SR, Ismagilov RF, Pamer EG, Turnbaugh PJ, Chervonsky AV. 2014. Rapid fucosylation of intestinal epithelium sustains host-commensal symbiosis in sickness. Nature 514:638–641. doi: 10.1038/nature13823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Basu R, O'Quinn DB, Silberger DJ, Schoeb TR, Fouser L, Ouyang W, Hatton RD, Weaver CT. 2012. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 37:1061–1075. doi: 10.1016/j.immuni.2012.08.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu H, Wang X, Liu DX, Moroney-Rasmussen T, Lackner AA, Veazey RS. 2012. IL-17-producing innate lymphoid cells are restricted to mucosal tissues and are depleted in SIV-infected macaques. Mucosal Immunol 5:658–669. doi: 10.1038/mi.2012.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li H, Richert-Spuhler LE, Evans TI, Gillis J, Connole M, Estes JD, Keele BF, Klatt NR, Reeves RK. 2014. Hypercytotoxicity and rapid loss of NKp44+ innate lymphoid cells during acute SIV infection. PLoS Pathog 10:e1004551. doi: 10.1371/journal.ppat.1004551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kloverpris HN, Kazer SW, Mjosberg J, Mabuka JM, Wellmann A, Ndhlovu Z, Yadon MC, Nhamoyebonde S, Muenchhoff M, Simoni Y, Andersson F, Kuhn W, Garrett N, Burgers WA, Kamya P, Pretorius K, Dong K, Moodley A, Newell EW, Kasprowicz V, Abdool Karim SS, Goulder P, Shalek AK, Walker BD, Ndung'u T, Leslie A. 2016. Innate lymphoid cells are depleted irreversibly during acute HIV-1 infection in the absence of viral suppression. Immunity 44:391–405. doi: 10.1016/j.immuni.2016.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Zhang Z, Cheng L, Zhao J, Li G, Zhang L, Chen W, Nie W, Reszka-Blanco NJ, Wang FS, Su L. 2015. Plasmacytoid dendritic cells promote HIV-1-induced group 3 innate lymphoid cell depletion. J Clin Invest 125:3692–3703. doi: 10.1172/JCI82124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Crome SQ, Lang PA, Lang KS, Ohashi PS. 2013. Natural killer cells regulate diverse T cell responses. Trends Immunol 34:342–349. doi: 10.1016/j.it.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 44.Gasteiger G, Rudensky AY. 2014. Interactions between innate and adaptive lymphocytes. Nat Rev Immunol 14:631–639. doi: 10.1038/nri3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mott KR, Underhill D, Wechsler SL, Town T, Ghiasi H. 2009. A role for the JAK-STAT1 pathway in blocking replication of HSV-1 in dendritic cells and macrophages. Virol J 6:56. doi: 10.1186/1743-422X-6-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hendricks RL, Janowicz M, Tumpey TM. 1992. Critical role of corneal Langerhans cells in the CD4- but not CD8-mediated immunopathology in herpes simplex virus-1-infected mouse corneas. J Immunol 148:2522–2529. [PubMed] [Google Scholar]
- 47.Ghiasi H, Roopenian DC, Slanina S, Cai S, Nesburn AB, Wechsler SL. 1997. The importance of MHC-I and MHC-II responses in vaccine efficacy against lethal herpes simplex virus type 1 challenge. Immunology 91:430–435. doi: 10.1046/j.1365-2567.1997.00261.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mercadal CM, Bouley DM, DeStephano D, Rouse BT. 1993. Herpetic stromal keratitis in the reconstituted scid mouse model. J Virol 67:3404–3408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tumpey TM, Cheng H, Yan XT, Oakes JE, Lausch RN. 1998. Chemokine synthesis in the HSV-1-infected cornea and its suppression by interleukin-10. J Leukoc Biol 63:486–492. doi: 10.1002/jlb.63.4.486. [DOI] [PubMed] [Google Scholar]
- 50.Oakes JE, Monteiro CA, Cubitt CL, Lausch RN. 1993. Induction of interleukin-8 gene expression is associated with herpes simplex virus infection of human corneal keratocytes but not human corneal epithelial cells. J Virol 67:4777–4784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Brandt CR, Salkowski CA. 1992. Activation of NK cells in mice following corneal infection with herpes simplex virus type-1. Invest Ophthalmol Vis Sci 33:113–120. [PubMed] [Google Scholar]
- 52.Ghiasi H, Cai S, Perng GC, Nesburn AB, Wechsler SL. 2000. The role of natural killer cells in protection of mice against death and corneal scarring following ocular HSV-1 infection. Antiviral Res 45:33–45. doi: 10.1016/S0166-3542(99)00075-3. [DOI] [PubMed] [Google Scholar]
- 53.Farooq AV, Shukla D. 2012. Herpes simplex epithelial and stromal keratitis: an epidemiologic update. Surv Ophthalmol 57:448–462. doi: 10.1016/j.survophthal.2012.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Weinkove R, Filbey K, Le Gros G. 2016. Immunity without innate lymphoid cells. Nat Immunol 17:1237–1238. doi: 10.1038/ni.3567. [DOI] [PubMed] [Google Scholar]
- 55.Perng GC, Dunkel EC, Geary PA, Slanina SM, Ghiasi H, Kaiwar R, Nesburn AB, Wechsler SL. 1994. The latency-associated transcript gene of herpes simplex virus type 1 (HSV-1) is required for efficient in vivo spontaneous reactivation of HSV-1 from latency. J Virol 68:8045–8055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ghiasi H, Slanina S, Nesburn AB, Wechsler SL. 1994. Characterization of baculovirus-expressed herpes simplex virus type 1 glycoprotein K. J Virol 68:2347–2354. doi: 10.1007/BF01379126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Allen SJ, Mott KR, Ghiasi H. 2014. Overexpression of herpes simplex virus glycoprotein K (gK) alters expression of HSV receptors in ocularly-infected mice. Invest Ophthalmol Vis Sci 55:2442–2451. doi: 10.1167/iovs.14-14013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Constantinides MG, McDonald BD, Verhoef PA, Bendelac A. 2014. A committed precursor to innate lymphoid cells. Nature 508:397–401. doi: 10.1038/nature13047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rigas D, Lewis G, Aron JL, Wang B, Banie H, Sankaranarayanan I, Galle-Treger L, Maazi H, Lo R, Freeman GJ, Sharpe AH, Soroosh P, Akbari O. 2017. Type 2 innate lymphoid cell suppression by regulatory T cells attenuates airway hyperreactivity and requires inducible T-cell costimulator-inducible T-cell costimulator ligand interaction. J Allergy Clin Immunol 139:1468–1477. doi: 10.1016/j.jaci.2016.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Weigmann B, Tubbe I, Seidel D, Nicolaev A, Becker C, Neurath MF. 2007. Isolation and subsequent analysis of murine lamina propria mononuclear cells from colonic tissue. Nat Protoc 2:2307–2311. doi: 10.1038/nprot.2007.315. [DOI] [PubMed] [Google Scholar]
- 61.Ghiasi H, Kaiwar R, Nesburn AB, Slanina S, Wechsler SL. 1994. Expression of seven herpes simplex virus type 1 glycoproteins (gB, gC, gD, gE, gG, gH, and gI): comparative protection against lethal challenge in mice. J Virol 68:2118–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lee DH, Ghiasi H. 2017. Roles of M1 and M2 macrophages in herpes simplex virus 1 infectivity. J Virol 91:e00578-17. doi: 10.1128/JVI.00578-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. 2013. STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Li B, Dewey CN. 2011. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics 12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Huang Da W, Sherman BT, Lempicki RA. 2009. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat Protoc 4:44–57. doi: 10.1038/nprot.2008.211. [DOI] [PubMed] [Google Scholar]
- 66.Wang S, Ljubimov AV, Jin L, Pfeffer K, Kronenberg M, Ghiasi H. 2018. Herpes simplex virus 1 latency and the kinetics of reactivation are regulated by a complex network of interactions between the herpesvirus entry mediator, its ligands (gD, BTLA, LIGHT, and CD160), and the latency-associated transcript. J Virol 92:e01451-18. doi: 10.1128/JVI.01451-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Ahmed R, King CC, Oldstone MB. 1987. Virus-lymphocyte interaction: T cells of the helper subset are infected with lymphocytic choriomeningitis virus during persistent infection in vivo. J Virol 61:1571–1576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Mott KR, Maazi H, Allen SJ, Zandian M, Matundan H, Ghiasi YN, Sharifi BG, Underhill D, Akbari O, Ghiasi H. 2015. Batf3 deficiency is not critical for the generation of CD8alpha(+) dendritic cells. Immunobiology 220:518–524. doi: 10.1016/j.imbio.2014.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ghiasi H, Cai S, Slanina SM, Perng GC, Nesburn AB, Wechsler SL. 1999. The role of interleukin (IL)-2 and IL-4 in herpes simplex virus type 1 ocular replication and eye disease. J Infect Dis 179:1086–1093. doi: 10.1086/314736. [DOI] [PubMed] [Google Scholar]
- 70.Mott KR, Bresee CJ, Allen SJ, BenMohamed L, Wechsler SL, Ghiasi H. 2009. Level of herpes simplex virus type 1 latency correlates with severity of corneal scarring and exhaustion of CD8+ T cells in trigeminal ganglia of latently infected mice. J Virol 83:2246–2254. doi: 10.1128/JVI.02234-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Mott KR, Ghiasi H. 2008. Role of dendritic cells in enhancement of herpes simplex virus type 1 latency and reactivation in vaccinated mice. Clin Vaccine Immunol 15:1859–1867. doi: 10.1128/CVI.00318-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mott KR, Perng GC, Osorio Y, Kousoulas KG, Ghiasi H. 2007. A recombinant herpes simplex virus type 1 expressing two additional copies of gK is more pathogenic than wild-type virus in two different strains of mice. J Virol 81:12962–12972. doi: 10.1128/JVI.01442-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mott KR, Osorio Y, Brown DJ, Morishige N, Wahlert A, Jester JV, Ghiasi H. 2007. The corneas of naive mice contain both CD4+ and CD8+ T cells. Mol Vis 13:1802–1812. [PubMed] [Google Scholar]
- 74.Allen SJ, Hamrah P, Gate DM, Mott KR, Mantopoulos D, Zheng L, Town T, Jones C, von Andrian UH, Freeman GJ, Sharpe AH, Benmohamed L, Ahmed R, Wechsler SL, Ghiasi H. 2011. The role of LAT in increased CD8+ T cell exhaustion in trigeminal ganglia of mice latently infected with herpes simplex virus type 1. J Virol 85:4184–4197. doi: 10.1128/JVI.02290-10. [DOI] [PMC free article] [PubMed] [Google Scholar]