

Abstract
As part of a project aimed at the development of chiral processive catalysts that can write information on a polymer chain we describe the synthesis of two optically active porphyrin macrocycles, which are prepared in 3 steps from an achiral precursor compound. Fluorescence and 1H‐NMR studies show that one of the macrocycles displays selectivity in the binding of chiral viologen guest molecules.
Keywords: Host–guest systems, Chirality, Porphyrinoids, Threading, Pseudo‐rotxanes, Cage compounds
Introduction
The work described in this paper is part of a larger project aimed at encoding information into polymers with the help of chiral cage catalysts that thread onto a polymer chain and, while moving along it unidirectionally, write chiral (R,R)‐ or (S,S)‐epoxides (digital codes 0 and 1) on this chain (Figure 1)1, 2, 3, 4, 5 In a previous paper, we reported on the facile, one‐step synthesis of a chiral porphyrin cage compound, i.e. (Rp,Sp)‐H22 (for notation of chirality, see below) from the achiral derivative H21.6 The manganese complex of compound H22 may be an interesting catalyst for the above‐mentioned writing process. As a first step in this direction we report here on the reduction of (Rp,Sp)‐H22 to its amine, (Rp,Sp)‐H23, and the resolution of the Mosher derivatives of this compound into two diastereomers, i.e. compounds (R,Sp*)‐H24 and (R,Rp*)‐H25. Furthermore, we show that (R,Sp*)‐H24 and (R,Rp*)‐H25 have different affinities for chiral viologen guest molecules. (for recent other chiral cage compounds, see refs.7, 8, 9, 10, 11, 12, 13, 14, 15, 16). These experiments will help us decide what chiral polymeric guests should be used in the future threading and epoxidation experiments needed to develop a catalytic machine that can write chiral information on a polymer chain.
Figure 1.
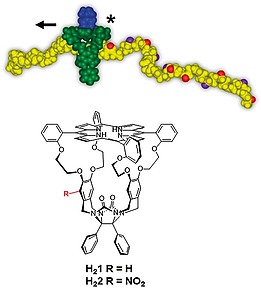
(Top) Chiral porphyrin cage catalyst writing chemical information ((R,R)‐ and (S,S)‐epoxides; red and purple balls, respectively) on a polymer chain while moving along it. (Bottom) Chemical structures of achiral porphyrin cage compound H21 and its chiral derivative (Rp,Sp)‐H22.
Results and Discussion
Racemic compound (Rp,Sp)‐H22 was synthesized from H21 by nitration with HNO3 in chloroform at –40 °C (isolated yield 75 %).6 Reduction of the nitro function of (Rp,Sp)‐H22 to its amine was initially tried with palladium on carbon under hydrogen pressure (40 bar) at 40 °C. This resulted in the desired conversion, but also in the reduction of one of the β‐pyrrole double bonds of the porphyrin moiety (as shown by e.g. Maldi‐TOF: m/z 1362 instead of 1360). Compound (Rp,Sp)‐H23 could be obtained, however, by using SnCl2 in dioxane/HCl as the reducing agent in a Schlenk bomb.
Reaction of the amine with enantiopure (R)‐(–)‐α‐methoxy‐α‐(trifluoromethyl)phenylacetyl chloride (Mosher's acid chloride) in dichloromethane using triethylamine (TEA) as a base, readily provided the diastereomeric amides (R,Sp*)‐H24 and (R,Rp*)‐H25 in 40 % yield over two steps (Scheme 1). These diastereomers could be separated by conventional column chromatography (60H silica, eluent CHCl3/MeOH). Compounds H24 and H25 display planar chirality, for which the notations Sp and Rp can be derived, as indicated in Scheme 1 (see ref.17 for nomenclature of compounds displaying planar chirality). In addition to the chirality of the Mosher substituent, which is (R), the cage compounds each contain two chiral centers of which the chirality is coupled to the planar chirality of the particular molecule. These chiral centers are also indicated in Scheme 1. Hence, the notations for the two diastereomeric compounds are: (R,Sp,R,S)‐H24 and (R,Rp,S,R)‐H25. Since we do not know which absolute chirality should be assigned to H24 and H25 (this requires X‐ray structures of these compounds), we will denote this uncertainty by adding an asterisk: (R,Sp*)‐H24 and (R,Rp*)‐H25, in which the first letter R refers to the chirality of the Mosher substituent and the second letter to the unknown chirality of the porphyrin cage compound.
Scheme 1.
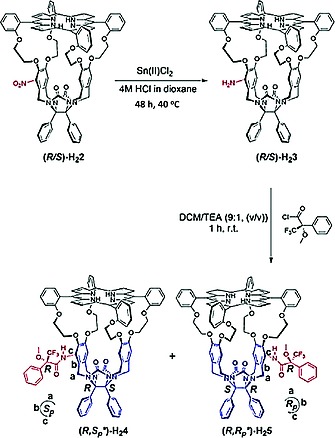
Synthesis of cavity containing porphyrin compounds (R,Sp*)‐H24 and (R,Rp*)‐H25. Reaction conditions: (i) Sn(II)Cl2, 4 M HCl in 1,4‐dioxane, (ii) (R)‐(–)‐α‐methoxy‐α‐(trifluoromethyl)phenylacetyl chloride, chloroform, TEA, 40 % yield over 2 steps.
The structures of the newly synthesized compounds were confirmed by MALDI‐TOF and 1D and 2D NMR spectroscopy. The 1H‐NMR spectra of (R,Sp*)‐H24 and (R,Rp*)‐H25 showed different peaks as can be expected for diastereomeric compounds. 19F‐NMR spectroscopy was used as a tool to check the diastereomeric purity of the compounds. The spectra of the reaction mixture of (R,Sp*)‐H24 and (R,Rp*)‐H25 and that of pure (R,Sp*)‐H24 after separation are shown in Figure 2a as examples. Circular dichroism revealed that the two isolated diastereo‐pure compounds had Cotton effects with opposite sign (Figure 2b).
Figure 2.
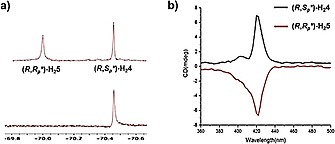
(a) 19F‐NMR spectra of the reaction mixture of (R,Sp*)‐H24 and (R,Rp*)‐H25 (top) and that of pure (R,Sp*)‐H24 after separation (bottom). (b) CD spectra of (R,Sp*)‐H24 and (R,Rp*)‐H25.
Having the diastereomeric pure hosts (R,Sp*)‐H24 and (R,Rp*)‐H25 in hand we investigated whether these compounds could bind the previously studied guest N,N′‐dimethylviologen dihexafluorophosphate (V1) and the chiral guest molecules (S,S)‐V2 and (R,R)‐V2 (Scheme 2). Furthermore, we were interested to see whether the diastereomeric hosts could discriminate between the latter two chiral guests. To this end, fluorescence titrations were performed in a solvent mixture of acetonitrile and chloroform (1:1, v/v). On binding of the guest, the fluorescence of the porphyrin is quenched, and this process can be followed as a function of the amount of added guest by recording the intensities of the maxima at 653 and 719 nm in the fluorescence spectra. The results are presented in Table 1.
Scheme 2.
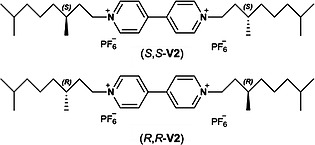
Structures of chiral guest molecules (S,S)‐ and (R,R)‐V2.
Table 1.
Association constants and Gibbs free energies of binding for various host–guest complexesa
| Host | (R,Sp*)‐H24 | (R,Sp*)‐H25 | ||||
|---|---|---|---|---|---|---|
| Guest | V1 | (R,R)‐V2 | (S,S)‐V2 | V1 | (R,R)‐V2 | (S,S)‐V2 |
| K a/M–1 | 0.883 × 106 | 1.388 × 106 | 3.88 × 106 | 1.29 × 106 | 8.0 × 106 | 7.3 × 106 |
| Error K a /M–1 | 0.089 × 106 | 0.045 × 106 | 0.26 × 106 | 0.21 × 106 | 1.4 × 106 | 0.37 × 106 |
| ΔG° /kJ mol–1 | –33.9 | –35.0 | –37.6 | –34.9 | –39.4 | –39.1 |
Measurements were performed in duplicate or triplicate in acetonitrile‐chloroform (1:1. v/v). The errors in the Ka values are indicated in the table. The errors in the fitting of the data points are given in the Supporting Information. λexc. 420 nm, λmeas. 653 and 719 nm.
V1 binds in H21 with a binding constant of Ka = 0.60 × 106 M–1 18 and in (R,Sp*)‐H24 and (R,Rp*)‐H25 with binding constants of K a = 0.88 × 106 and 1.39 × 106 M–1, respectively. The somewhat higher affinities of V1 for the latter two hosts compared to H21 may be attributed to the additional van der Waals interactions that are possible with the Mosher substituent present on the side‐walls of (R,Sp*)‐H24 and (R,Rp*)‐H25. Table 1 shows that the chiral guests (S,S)‐V2 and (R,R)‐V2 all bind stronger to the two hosts than V1. This must be the result of the longer tails in V2, which allow for additional binding interactions with the hosts. Binding of the chiral guests is lower in (R,Sp*)‐H24 than in (R,Rp*)‐H25, suggesting that the substituent in the former diastereomer shields the entrance of the cavity more than the substituent in the latter diastereomer. Interestingly, host (R,Sp*)‐H24 displays selectivity towards the guest (S,S)‐V2 (factor of 2.8), whereas (R,Rp*)‐H25 does not discriminate between (S,S)‐V2 and (R,R)‐V2, within experimental error. The fact that compound (R,Sp*)‐H24 displays selectivity is promising because the chiral centers in the guests are quite far away from the viologen binding moieties, making that the non‐bonded interactions with the host that lead to chiral discrimination will not be optimal. Guests containing bulkier chiral substituents can be expected to be more efficient and experiments in this direction are underway.
In order to find out what the orientations of the chiral substituents in hosts H24 and H25 are and to see to what extent they block the entrance of the cavities, 2D‐NMR experiments were carried out. In compound (R,Sp*)‐H24, 1H‐19F nOe correlations are found between the trifluoromethyl group and the out‐proton 32a (for numbering see Scheme 3) of one of the benzylic methylene groups of the most nearby side‐wall (red arrow in Figure 3 left). Additionally, in the 2D‐ROESY spectrum nOe contacts are present between the methoxy protons of the Mosher amide substituent and the same out‐proton of the afore‐mentioned methylene group (blue arrow in Figure 3 left). 3D‐modelling (Spartan '14) revealed that such interactions are only possible when the phenyl group of the Mosher substituent is pointing upwards, i.e. in the direction of the meso‐phenyl‐substituted porphyrin ring. In line with this, nOe contacts are observed between the phenyl protons of the Mosher amide group at 7.22 ppm and the peripheral β‐pyrrole protons 3 and 4 of the porphyrin at 8.94 ppm (green arrow in Figure 3, left). Furthermore, an unusual upfield shift from 7.40 to 6.64 ppm was seen for the ortho‐proton 25 of the meso‐phenyl group of the porphyrin ring and for both upper methylene protons 27 of the oxyethylene spacer (from ca. 4.5–3.9 to 3.7 and 3.4 ppm) on the side to which the Mosher amide group is attached.
Scheme 3.
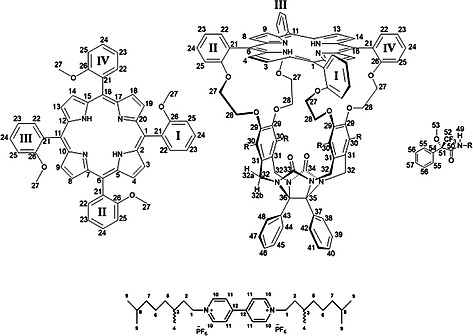
Proton and carbon numbering in host and guest compounds. For protons 27, 28, and 32 two different signals are present, but only for the CH2‐32 protons, a distinction was made between the proton pointing upwards (32a) and the proton pointing downwards (32b).
Figure 3.
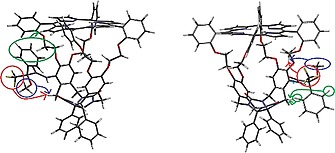
3D calculated models (Spartan '14) of (R,Sp*)‐H24 (left) and (R,Rp*)‐H25 (right). The colored circles and arrows indicate nOe contacts observed in the 1H‐19F and 1H‐1H correlation spectra, see text for details.
Diastereomer (R,Sp*)‐H25, showed 1H‐19F nOe correlations between the trifluoro group of the Mosher amide substituent and one of the protons 27 (4.48 ppm) of the oxyethylene spacer (Figure 3, right, red arrow). Additionally, in the 2D‐ROESY spectrum, nOe contacts were present between the methoxy protons of the Mosher amide group and the same proton 27 of the oxyethylene spacer (Figure 3, right, blue arrow). These observations indicate that the phenyl group of the Mosher amide is oriented downwards, i.e. in the direction of the diphenylglycoluril part of the cage compound. Further evidence for this orientation comes from nOe contacts between the ortho‐phenyl protons 55 of the Mosher phenyl group and the out‐proton 32a of the benzylic methylene group of one of the side‐walls (Figure 3, right, green arrow).
The upward orientation of the Mosher phenyl group in (R,Sp*)‐H24 allows the formation of a hydrogen bond between the Mosher amide proton and the nearby carbonyl group (33) of the glycoluril framework of the cage compound. According to the calculations we performed, such a hydrogen bond is not possible for compound (R,Rp*)‐H25. The chemical shift of the amide proton in (R,Sp*)‐H24 is 9.17 ppm and in (R,Rp*)‐H25 8.13 ppm. The hydrogen bond in the former compound is expected to have a length of circa 2.5 Å,19 resulting in a 1 ppm downfield shift compared to the non‐hydrogen bonded amide proton in the latter compound. For comparison we also measured the chemical shift of the amide proton in the model compound (R)‐N‐(2,6‐dimethylphenyl)‐3,3,3‐trifluoro‐2‐methoxy‐2‐phenylpropanamide (5), which in diluted form cannot form a hydrogen bond. It was found to be located at 8.17 ppm, which is similar to the shift value in (R,Rp*)‐H25. Based on the results above in combination with the calculated structures of the two compounds (see Figure 3) we tentatively propose that compound H24 has the (R,Sp*)‐ configuration and compound H25 the (R,Rp*)‐one, but further studies, e.g. elucidation of the X‐ray structures of the compounds, are needed to substantiate these assignments.
With the above information on the structures of the diastereomeric host compounds available, we may further interpret the measured binding constants (Table 1). Due to the upward orientation of the phenyl group in (R,Sp*)‐H24 and the possibility of a hydrogen bond between the Mosher amide group and the carbonyl group of the diphenylglycoluril framework in this compound, fixating this upward orientation, the cavity of this host is partly blocked, making it less accessible for viologen guests than the cavity of (R,Rp*)‐H25. This orientation may also lead to the observed chiral discrimination between the two enantiomeric guests (R,R)‐V2 and (S,S)‐V2.
In subsequent experiments, we further analyzed the structures of the complexes between hosts H24 and H25 and guests V1 and V2 by NMR. Complexation of V1 to H21 has been described in our previous paper.18 This guest has two pyridinium rings that are slightly rotated with respect to each other. According to our previous NMR studies, these pyridinium rings of V1 are sandwiched between the side‐walls of H21 and this orientation is stabilized by π‐π stacking interactions of the guest with the o‐xylylene rings and electrostatic interactions with the crown ether‐like rings. Complexation of V1 to (R,Sp*)‐H24 results in upfield shifts of most of the signals of the protons lining the inside of the cage, e.g. the side‐wall protons 30, the oxyethylene protons 28, and the pyrrole NH protons.18 Downfield shifts were observed for the signal of the amide protons 49 and the methoxy protons 53 of the Mosher substituent. From the various CISVs. (Table S1 of SI) it can be concluded that V1 is located unsymmetrically between the walls, i.e. more pushed towards the wall opposite to that where the Mosher substituent is located (compare the CISVs. of the signals of the side‐wall protons 30: +0.02, –0.18, and –0.29 ppm).
Binding of guests (S,S)‐ and (R,R)‐V2 to host (R,Sp*)‐H24 yields similar, but different spectra. The porphyrin pyrrole NH protons signal displayed similar shifts and CIS values as observed for the complex with V1 indicating that the guests are also positioned perpendicular to the porphyrin planes. The CISVs. of the signals of the oxyethylene protons 27 and 28 were significantly different and from the shift values (see Table S2 of SI) it can be concluded that the guests are oriented asymmetrically in the complexes with (R,Sp*)‐H24, i.e. more to the back side of the host (under phenyl rings III and IV), where no substituent is present. This means that the centers of the guests are not aligned with the center of the cavity of (R,Sp*)‐H24, which can also be concluded from the CISVs. of the signals of the guest protons, which are different for the two sides of the guests (see Table S2 of SI). The amide proton signal of the Mosher substituent in (R,Sp*)‐H24 was shifted downfield in both complexes with V2, but this shift was larger for the complex with the (S,S)‐guest (+0.31 ppm, see Table S2 in SI) than for the complex with the (R,R)‐guest (0.07 ppm). This indicates that the positive charge of the viologen moiety is closer to the Mosher amide group in the former case than in the latter, indicating that the (S,S)‐guest binds lower (more to the bottom) in the cavity of (R,Sp*)‐H24 than the (R,R)‐guest (Figure 4). The methyl protons 4 of the latter guest have nOe interactions with the β‐pyrrole protons 3 and 4 of the porphyrin ring, i.e. the protons that are located at the side of the Mosher substituent. This indicates that this guest is positioned higher (more to the top) in the cage than the (S,S)‐guest, in line with what was concluded above. The lower location of the (S,S)‐guest was further checked with a 1D‐ROESY experiment, in which the protons of the methyl groups 4 of the (R,R)‐ and (S,S)‐guests inside the cavity, were excited and the amount of magnetization transfer to the methoxy group of the Mosher substituent of (R,Sp*)‐H24 was measured and compared (see Figure S72 of SI). From this experiment it appeared that more magnetization was transferred from the (S,S)‐guest to the Mosher methoxy substituent (1.06 %) than from the (R,R)‐guest (0.36 %), suggesting that the former is situated closer to the substituent and hence located lower in the cavity than the latter. It should be noted, however, that this is a qualitative experiment because the amount of NOE transfer might be dependent on the exchange rate, which could be different for the two guests.
Figure 4.
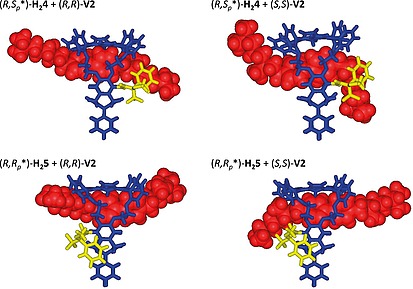
Calculated structures (Spartan '14) of the diastereomeric host–guest complexes based on the NMR experiments. The viologens guests are indicated space‐filling in red, the Mosher's substituent in yellow.
The same types of NMR experiments were carried out with guests V1 and V2 and the diastereomeric host (R,Rp*)‐H25. In this host V1 is also located unsymmetrically, i.e. more in the direction of the side wall where the Mosher substituent is not located (see CISVs. of the side wall protons 30 in Table S1 of SI). For the complexes with V2 also upfield shifts of the signals of the pyrrole NH protons were observed, indicating a perpendicular orientation of the guests with respect to the porphyrin plane. The oxyethylene protons 27 and 28 displayed a similar shift pattern as observed for host (R,Sp*)‐H24, again suggesting an asymmetric binding of the guests, i.e. more to the backside of the host and away from the Mosher substituent (see Table S3 of SI). The protons at the lower part of the cage all became deshielded upon binding of the guest molecules. The largest (downfield) CIS values were found for the Mosher amide proton signal, i.e. +1.24 for the complex with the (R,R)‐guest and +1.11 ppm for the complex with the (S,S)‐guest. The methoxy protons of the Mosher substituent became only slightly deshielded after binding of the guests, but they did not show any nOe interactions with these molecules, in contrast to what was observed for the binding of the guests in the diastereomeric (R,Sp*)‐H24 host. The CIS values of the protons of the guest molecules indicate that they are bound in an asymmetric fashion in the cavity, as could also be concluded from the CISVs. of the host oxyethylene protons 27 and 28 (see above). The pyrrole NH protons and the β‐pyrrole protons 3/4 and 13/14 of the porphyrin ring displayed nOe interactions with the protons 10 and 11 of the viologen moieties, which indicates that both enantiomers of the V2 guest reside in the upper part of host (R,Rp*)‐H25.
In conclusion, the NMR studies on the 4 diastereomeric host–guest complexes reveal that guests V2 are bound higher, i.e. more in the upper part of the host, in (R,Rp*)‐H25 than in (R,Sp*)‐H24, hence they are farther away from the chiral Mosher substituent (Figure 4). This is probably the result of the fact that the phenyl group of the Mosher substituent in (R,Rp*)‐H25 points downwards, making room for binding at a higher location in the cage. This binding in the upper part of the host may explain why (R,R*)‐H25 does not discriminate well between the (R,R)‐guest and the (S,S)‐guest. The NMR studies revealed that the V2 guests bind lower in host (R,Sp*)‐H24 than in (R,Rp*)‐H25, hence, they will feel the influence of the Mosher substituent more strongly, leading to the observed discrimination between the (R,R)‐ and (S,S)‐forms. The studies further show that the (S,S)‐guest binds lower in the cavity of (R,Sp*)‐H24 and is closer to the Mosher substituent than the (R,R)‐one, leading to stronger π‐π‐stacking and van der Waals interactions and hence to a higher binding constant (Table 1).
Conclusion
We have synthesized the new chiral porphyrin cage compounds (R,Sp*)‐H24 and (R,Rp*)‐H25, which are derived from the achiral diphenylglycoluril derivative H21. The reaction sequence involved a regioselective nitration, followed by a reduction of the formed nitro‐compound to the amine, and functionalization of the latter with the chiral derivatizing agent Mosher's acid chloride. 1H‐NMR studies revealed that the Mosher group attached to the side‐wall of the toroidal‐shaped host compounds has different orientations in (R,Sp*)‐H24 and (R,Rp*)‐H25, leading to different binding affinities for chiral viologen guest molecules. Compound (R,Sp*)‐H24 is capable of discriminating between enantiomeric guests (by a factor of 2.8), whereas compound (R,Rp*)‐H25 does not have this capability. Current work is directed towards threading experiments with chiral polymers with the objective to realize unidirectional movement of the chiral porphyrin cage compounds on the polymer chains. In subsequent studies these cage compounds will be provided with manganese centers to realize writing on chiral polymers chains in the form of (R,R)‐ and (S,S)‐epoxides (digits 1 and 0).
Experimental Section
General Information: All chemicals were commercially obtained and used without further purification unless stated otherwise. 1H, 13C and 19F NMR spectra were collected on a Bruker 500 MHz Avance III spectrometer equipped with a Prodigy BB cryoprobe, a Bruker 400 MHz Avance III HD nanobay spectrometer equipped with a BBFO probe, or a Agilent 400 MHz INOVA spectrometer equipped with a dual‐channel inverse probe. All spectra were obtained at room temperature. Manual phase correction and automatic baseline correction were applied to all spectra. NMR data is presented as follows: chemical shift (in ppm relative to internal trimethylsilane), multiplicity, coupling constant in Hz, integration, assignment. Abbreviations used are: s=singlet, d=doublet, dd=doublet of doublets, t=triplets, dt=doublet of triplets, td=triplet of doublets and m=multiplet. All NMR signals were assigned on the basis of 1H NMR, 13C NMR, 1H‐1H COSY, 1H‐13C HSQC, 1H‐15N HSQC, 1H‐13C HMBC, 1H‐15N HMBC, 1H‐1H ROESY and 1H TOCSY experiments. Mass spectra were recorded on a Thermo Finnigan LCQ Advantage Max mass spectrometer (MS) or on a Bruker Microflex LRF Maldi‐tof system in the reflective mode employing dithranol as a matrix.
Synthesis. H21 and H22: These compounds were synthesized and characterized as reported previously.6, 18
(R,Sp*)‐H24 and (R,Rp*)‐H25: Compound H22 (33.7 mg, 24 µmol) and SnCl2 (184 mg, 969 µmol) were dissolved in HCl in dioxane (4 mL, 4 M) in a Schlenk‐bomb under an inert atmosphere. Concentrated HCl (10 µL) was added and the Schlenk‐bomb was closed. The suspension was stirred vigorously at 45 °C for 40 hours, after which it was quenched with aqueous NaOH (30 mL, 3 M). The aqueous layer was extracted with CHCl3 (2 × 20 mL). The combined organic layers were washed with aqueous NaOH (20 mL, 3 M), saturated aqueous NaHCO3 (20 mL), brine (20 mL), and subsequently evaporated to dryness. The residue was re‐dissolved in freshly distilled CHCl3 (1 mL), after which triethylamine (0.07 mL, 0.5 mmol) and (R)‐(‐)‐α‐methoxy‐α‐(trifluoromethyl)phenylacetyl chloride (12 µL, 64 µmol) were added. The mixture was stirred at 20 °C for 1 h and subsequently quenched with saturated aqueous NaHCO3 (20 mL). The aqueous layer was extracted with CHCl3 (10 mL). The organic layer was washed with saturated aqueous NaHCO3 (25 mL), brine (25 mL), and the solvents evaporated to dryness. The residue was purified by silica gel flash column chromatography (60H Silica, CHCl3/MeOH, 99.73:0.27, v/v) and subsequently by precipitation from CH2Cl2 and n‐heptane yielding (R,Sp*)‐H24 (12.1 mg, 7.7 µmol, 32 %) and (R,Rp*)‐H25 (6.2 mg, 3.9 µmol, 16 %) as purple solids.
(R,Sp*)‐H24: 1H NMR (500 MHz, [D]chloroform). The respective protons are indicated in bold. For numbering see Scheme 3. δ 9.17 (s, 1H, 49), 8.95 (d, J = 4.7 Hz, 1H, 4), 8.80 (d, J = 4.8 Hz, 1H, 3), 8.79–8.75 (m, 2H, 8, 9, 13, 14, 18 or 19), 8.70–8.66 (m, 3H, 8, 9, 13, 14, 18 or 19), 8.63 (d, J = 4.8 Hz, 1H, 8, 9, 13, 14, 18 or 19), 8.28 (dd, J = 7.3, 1.8 Hz, 1H, 22(II)), 8.18 (dd, J = 7.3, 1.7 Hz, 1H, 22(I,III or IV)), 8.14 (dd, J = 7.4, 1.7 Hz, 1H, 22(I,III or IV)), 8.10 (dd, J = 7.4, 1.7 Hz, 1H, 22(I,III or IV)), 7.80–7.71 (m, 3H, 24(I,III, IV)), 7.66–7.59 (m, 1H, 24(II)), 7.48 (d, J = 7.6 Hz, 2H, 55), 7.45–7.33 (m, 7H, 23, 25(I,III,IV)), 7.27–7.18 (m, 3H, 56, 57), 7.05–6.86 (m, 9H, 39–42, 44–48), 6.74 (dd, J = 8.1, 1.8 Hz, 1H, 38), 6.64 (d, J = 8.2 Hz, 1H, 25(II)), 6.28 (s, 1H, 30(IV)), 6.13 (s, 1H, 30(III)), 6.11 (s, 1H, 30(I)), 4.41 (d, J = 16.3 Hz, 1H, 32a(II)), 4.27 (d, J = 15.7 Hz, 1H 32a(III)), 4.22 (d, J = 15.6 Hz, 1H, 32a(I)), 4.25–4.10 (m, 3H, 27(I), 27a(IV)), 4.17 (d, J = 15.9 Hz, 1H 32a(IV)), 4.00–3.87 (m, 2H, 27b(IV), 28a(II)), 3.78 (d, J = 15.6 Hz, 1H, 32b(III)), 3.73 (d, J = 15.8 Hz, 1H, 32b(I)) 3.72 (d, J = 15.9 Hz, 1H, 32b(IV)), 3.81–3.54 (m, 4H, 27a(II,III) or 28a(I,III)), 3.57 (d, J = 16.3 Hz, 1H, 32b(II)), 3.45–3.37 (m, 3H, 27b(II), 28b(I,II)), 3.25 (s, 4H, 27b(III), 53), 3.20 (t, J = 5.7 Hz, 2H, 28(IV)), 3.09–3.02 (m, 1H, 28b(III)), –2.69 (s, 2H, pyr‐NH). 13C NMR (126 MHz, [D]chloroform) δ 165.00 (50), 159.71 (26), 159.05 (26), 158.98 (26), 158.93 (26), 157.71 (33), 156.91 (34), 156.10, 150.49(29(III)), 147.57 (29(I)), 146.40 (29(IV)), 141.97 (29(II)), 135.94 (22), 135.90 (22), 135.80 (22), 135.37 (22), 133.85 (37 or 43), 133.22 (37 or 43), 132.73 (21(I,III or IV)), 132.51 (31(III)), 132.14 (21(I,III or IV)), 131.99 (21(I,III or IV)), 131.32 (54), 130.81 (31(I) or 21(II)), 130.74 (31(I) or 21(II)), 129.71, 129.67, 129.57, 129.54, 129.43, 129.35 (31(IV)), 128.70, 128.66, 128.62, 128.51, 128.40, 128.38, 128.33, 127.83, 127.59, 127.36 (30(II)), 124.84 (31(II)), 120.77 (23(I,III or IV)), 120.13 (23(I,III or IV)), 119.93 (23(I,III or IV)), 119.43 (23(II)), 117.99 (30(IV)), 115.61 (6), 115.46 (1, 11, 16 or 25), 115.33 (1, 11, 16 or 25), 115.27 (1, 11, 16 or 25), 114.96 (1, 11, 16 or 25), 114.79 (30(I)), 112.91 (25), 112.17 (1, 11, 16 or 25 or 30 (III)), 112.14 (1, 11, 16 or 25 or 30 (III)), 112.05 (1, 11, 16 or 25 or 30 (III)), 85.29 (35), 84.64 (36), 72.26, 71.14 (27 or 28), 69.26 (27 or 28), 68.58 (27 or 28), 68.43 (27 or 28), 67.59 (27 or 28), 67.41 (27 or 28), 67.12 (27 or 28), 61.90, 55.02 (53), 44.59 (32(I,III or IV)), 44.22 (32(I,III or IV)), 39.23 (32(II)). 19F NMR (471 MHz, [D]chloroform) δ –69.22 (52). MALDI‐TOF (m/z): [M + H]+ calculated for C94H72F3N9O12+H+, 1577.54; found 1577.355. HRMS (m/z): [M + Na]+ calcd. for C94H72F3N9Na1O12, 1598.51502; found 1598.51431. UV/Vis: λ max = 420 nm. Fluorescence: λ = 653 and 719 nm (c = 3 µM in CHCl3/MeCN, 1:1, v/v, T = 295 K, λex = 420 nm). M.p. 292 °C. No optical rotation could be measured because solutions of this compound were too colored. For CD‐spectrum, see Figure 2.
(R,Rp*)‐H25: 1H NMR (500 MHz, [D]chloroform) δ 8.78 (d, J = 4.8 Hz, 1H, 3, 4, 8, 9, 13, 14, 18 or 19), 8.76–8.70 (m, 4H, 3, 4, 8, 9, 13, 14, 18 or 19), 8.69–8.61 (m, 3H, 3, 4, 8, 9, 13, 14, 18 or 19), 8.28 (dd, J = 7.4, 1.7 Hz, 1H, 22), 8.16 (dd, J = 7.4, 1.8 Hz, 1H, 22), 8.13 (s, 1H, 49), 8.11 (dd, J = 7.5, 1.6 Hz, 1H, 22), 8.07 (dd, J = 7.4, 1.7 Hz, 1H, 22), 7.80–7.73 (m, 4H, 24), 7.60–7.56 (m, 2H, 55), 7.48–7.25 (m, 11H, 23, 25, 56, 57), 6.99–6.86 (m, 6H, 39, 40, 41, 45, 46, 47), 6.83–6.79 (m, 1H, 38), 6.76 (d, J = 8.1 Hz, 1H, 44 or 48), 6.72 (dd, J = 7.5, 2.0 Hz, 1H, 44 or 48), 6.57–6.52 (m, 1H, 42), 6.22 (s, 1H, 30(III)), 6.16 (s, 1H, 30(IV)), 6.13 (s, 1H, 30(II)), 4.49 (td, J = 9.9, 2.5 Hz, 1H, 27(I)), 4.29–4.09 (m, 3H, 27(II), 27a(III)), 4.21 (d, J = 15.6 Hz, 1H, 32a(IV)), 4.15 (d, J = 15.9 Hz, 1H, 32a(II or III)), 4.15 (d, J = 15.7 Hz, 1H, 32a(II or III)), 4.04 (d, J = 9.9 Hz, 1H, 27b(I)), 4.02–3.86 (m, 3H, 27a(IV), 27b(III), 28a(I)), 3.78–3.50 (m, 6H, 27b(IV), 28a(III,IV), 28b(I), 32a(I), 32b(II,III,IV), 3.46–3.38 (m, 1H, 28b(II)), 3.30–3.22 (m, 4H, 32b(I), 53), 3.22–3.14 (m, 2H), 3.14–3.08(m, 1H, 28b(IV)), –2.69 (s, 2H). 13C NMR (126 MHz, [D]chloroform) δ 165.46 (50), 159.48 (26), 159.04 (26), 158.79 (26), 157.21 (33), 156.86 (34), 149.76 (29(IV)), 147.45 (29(II)), 146.44 (29(III), 142.71, 142.21 (29(I)), 135.96 (22), 135.91 (22), 135.40 (22), 133.83, 133.53, 133.23, 132.48, 132.09, 132.00, 131.87 (31(IV)), 131, 13 130.60 (31(II)), 129.87, 129.65, 129.36 (31(III)), 129.11, 128.62, 128.55, 128.51, 128.50, 128.46, 128.36, 128.28, 128.22, 128.17, 127.98, 127.76, 127.31 (55), 126.62 (30(I)), 125.43 (31(I)), 120.43 (23), 120.06 (23), 119.90 (23), 119.71 (23), 115.50 (1, 6, 11 or 16), 115.21 (1, 6, 11 or 16), 115.13 (1, 6, 11 or 16), 115.01 (1, 6, 11 or 16), 114.94, 114.90 (30(II)), 114.12 (25), 112.77 (25 or 30(IV)), 112.61 (25 or 30(IV)), 112.09 (25), 111.71 (25), 85.09 (36), 84.41 (35), 71.26 (28(I)), 68.60 (27(IV)), 68.52 (27(I)), 68.43 (28(III)), 67.39 (27(III)), 67.33 (27(I)), 67.21 (28(II)), 67.11 (28(III)), 55.51 (53), 44.62 (32(III)), 44.40 (32(II)), 44.22 (32(IV)), 38.42 (32(I)). 19F NMR (471 MHz, [D]chloroform) δ –68.42 (52). MALDI‐TOF: m/z: 1576.562 [M + H]+; calculated for C94H72F3N9O12 +H+: m/z: 1576.53. HRMS: (m/z): [M + H]+ calcd. for C94H73F3N9O12, 1576.53308; found 1576.53376. UV/Vis: λ max = 420 nm. Fluorescence:λ = 653 and 719 nm (c = 3 µM in CHCl3/MeCN, 1:1, v/v, T = 295 K, λex = 420 nm). M.p. 150 °C. No optical rotation could be measured because solutions of this compound were too colored. For CD‐spectrum, see Figure 2.
(R)‐N‐(2,6‐dimethylphenyl)‐3,3,3‐trifluoro‐2‐methoxy‐2‐phenylpropanamide (Mosher model): A Schlenk flask was evacuated three times and 2,6‐dimethylaniline (51 µL, 0.41 mmol) and (R)‐3,3,3‐trifluoro‐2‐methoxy‐2‐phenylpropanoyl chloride (0.19 mL, 1.0 mmol) were dissolved in dry chloroform (10 mL) and this solution was added to the flask. Triethylamine (1 mL) was added dropwise and the resulting mixture was stirred at room temperature for 16 hours. The reaction was quenched by adding saturated aqueous NaHCO3 and the mixture was extracted with chloroform. The organic layer was washed with NaHCO3 (2 ×) and brine (2 ×). The organic layers were combined, dried with NaSO4, and concentrated in vacuo to give the desired product as a white powder. Yield 0.11 g (0.31 mmol, 76 %). M.p. 124 °C.

1H NMR (500 MHz, [D]Chloroform) δ 8.17 (s, 1H, 5), 7.69–7.62 (m, 2H, 3), 7.47–7.39 (m, 3H, 1, 2), 7.12–7.07 (m, 1H), 7.09–7.03 (m, 3H, 7, 8), 3.53 (q, J = 1.6 Hz, 3H, 4), 2.18 (s, 6H, 6). HRMS (m/z): [M + H]+ calcd. for C18H18F3NNaO2, 360.11873; found 360.11848.
Synthesis of guest compounds
Compound V1 was a commercial product. Compounds (R,R)‐ and (S,S)‐V2 were synthesized as shown in Scheme 4 for the (R,R)‐ derivative.
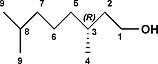
Scheme 4.
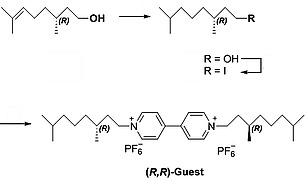
Synthesis of (R,R)‐V2.
(R)‐3,7‐dimethyloctan‐1‐ol: Palladium on carbon (35 mg, 33 µmol, 10 wt.‐%) was submerged in CH2Cl2 before MeOH (10 mL) was added. (R)‐3,7‐dimethyl‐6‐octen‐1‐ol (0.12 mL, 0.66 mmol) was added under inert atmosphere and the mixture was bubbled through with H2 at 20 °C for 4 hours, after which the suspension was filtered through Celite. The filtrate was concentrated in vacuo, yielding (R)‐3,7‐dimethyloctan‐1‐ol (0.11 g, 0.68 mmol, quantitative yield) as a colourless oil. 1H NMR (500 MHz, [D]chloroform) δ 3.77–3.62 (m, 2H, 1), 1.65–1.47 (m, 3H, 2a, 3, 8), 1.42–1.20 (m, 4H, 2b, 5a, 6a, 7a), 1.20–1.07 (m, 4H, 5b, 6b, 7b), 0.91–0.84 (m, 9H, 4, 9). 13C NMR (126 MHz, [D]chloroform) δ 61.29 (1), 40.02 (2), 39.26 (7), 37.37 (5), 29.51 (3), 27.97 (8), 24.69 (6), 22.70 (9), 22.60 (9), 19.65 (4).21
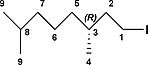
(R)‐1‐iodo‐3,7‐dimethyloctane: PPh3 (1.03 g, 3.9 mmol) and imidazole (0.39 g, 5.7 mmol) were added to a degassed solution of (R)‐3,7‐dimethyloctan‐1‐ol (0.31 g, 2.0 mmol) in MeCN (3 mL) and Et2O (3 mL). Iodine (0.63 g, 2.5 mmol) was added under inert atmosphere at 0 °C. The mixture was stirred for 30 min at 0 °C and for 2 hours at 20 °C, after which it was diluted with n‐heptane (20 mL). The organic layer was washed with a saturated aqueous solution of NaS2O3 (20 mL), a saturated aqueous solution of CuSO4 (20 mL), brine (20 mL), dried with MgSO4, and concentrated in vacuo. The residue was purified by silica gel flash column chromatography (eluent n‐heptane/EtOAc, 9:1, v/v) yielding (R)‐1‐iodo‐3,7‐dimethyloctane (0.39 g, 1.5 mmol, 74 %) as a colourless oil. 1H NMR (500 MHz, [D]chloroform) δ 3.25 (ddd, J = 9.6, 8.6, 5.7 Hz, 1H, 1a), 3.17 (ddd, J = 9.5, 8.3, 7.2 Hz, 1H, 1b), 1.87 (dddd, J = 13.9, 8.5, 7.2, 5.4 Hz, 1H, 2a), 1.64 (dtd, J = 13.9, 8.0, 5.7 Hz, 1H, 2b), 1.60–1.45 (m, 2H, 3, 8), 1.38–1.06 (m, 6H, 5, 6, 7), 0.91–0.82 (m, 9H, 4,9). 13C NMR (126 MHz, [D]chloroform) δ 41.13 (2), 39.32 (7), 36.62 (5), 34.03 (3), 28.09 (8), 24.67 (6), 22.84 (9), 22.74 (9), 18.89 (4), 5.50 (1).21
1,1′‐Bis(R)‐3,7‐dimethyloctyl‐4,4′‐bipyridi‐1,1′‐diium dihexafluorophosphate (R,R)‐V2: 4,4′‐bipyridine (0.14 g, 0.90 mmol) and (R)‐1‐iodo‐3,7‐dimethyloctane (0.96 g, 3.6 mmol) were suspended in DMF (4 mL). The mixture was stirred at 100 °C for 40 hours. After cooling the reaction mixture Et2O (10 mL) was added and the formed precipitate was filtered off. The orange solid was washed with Et2O (10 mL) and dried on the air. The orange solid was dissolved in the smallest possible amount of boiling MeCN, after which a saturated aqueous solution of NH4PF6 (5 mL) was added dropwise. The layers were separated and the yellow MeCN layer was concentrated in vacuo. The yellow precipitate was filtered off and recrystallized from MeOH yielding 1,1′‐bis((R)‐3,7‐dimethyloctyl‐4,4′‐bipyridi‐1,1′‐diium di hexafluorophosphate (0.18 g, 0.25 mmol, 28 %) as a nearly white solid. M.p. 279 °C (decomposition). [α]20 D –0.018° (c 2.451, CH3CN). 1H NMR (500 MHz, [D3]acetonitrile) (for proton numbering see Scheme 3). δ 8.93–8.88 (m, 4H, 10), 8.38–8.34 (m, 4H, 11), 4.71–4.58 (m, 4H, 1), 2.07–1.98 (m, 2H, 2a), 1.84 (dddd, J = 13.7, 11.6, 8.8, 4.8 Hz, 2H, 2b), 1.63–1.48 (m, 4H, 3, 8), 1.42–1.13 (m, 12H, 5, 6, 7), 1.01 (d, J = 6.7 Hz, 6H, 4), 0.88 (d, J = 6.6 Hz, 12H, 9). 13C NMR (126 MHz, [D3]acetonitrile) δ 150.81 (12), 146.59 (10), 128.17 (11), 61.64 (1), 39.83 (7), 39.13 (2), 37.48 (5), 31.28 (3), 28.71 (8), 25.25 (6), 22.93 (9), 22.83 (9), 19.48 (4). HRMS (m/z): [M – PF6]+ calcd. for C30H50F6N2P1, 583.36029; found 583.36093.
1,1′‐Bis(S)‐3,7‐dimethyloctyl‐4,4′‐bipyridi‐1,1′‐diium dihexafluorophosphate (S,S)‐V2: this compound was prepared as described for the (R,R)‐ derivative starting from (S)‐3,7‐dimethyloctan‐1‐ol. M.p. 283 °C (decomposition). [α]20 D +0.019 (c 2.453, CH3CN). 1H NMR (500 MHz, [D3]acetonitrile) (for proton numbering see Scheme 3). δ 8.93 (d, J = 6.9 Hz, 4H, 10), 8.39 (d, J = 6.4 Hz, 4H, 11), 4.71–4.58 (m, 4H, 1), 2.09–1.99 (m, 2H, 2a), 1.85 (dddd, J = 13.6, 10.1, 8.0, 5.9 Hz, 2H, 2b), 1.62–1.50 (m, 4H, 3, 8), 1.42–1.14 (m, 12H, 5, 6, 7), 1.01 (d, J = 6.6 Hz, 6H, 4), 0.88 (d, J = 6.6 Hz, 12H, 9). 13C NMR (126 MHz, [D3]acetonitrile) δ 150.82 (12), 146.59 (10), 128.17 (11), 61.62 (1), 39.84 (7), 39.13 (2), 37.49 (5), 31.28 (3), 28.71 (8), 25.24 (6), 22.94 (9), 22.83 (9), 19.48 (4). HRMS (m/z): [M – PF6]+ calcd. for C30H50F6N2P1, 583.36029; found 583.36029.
Host–guest complexes
(R,Sp*)‐H24 with (R,R)‐V2: 1H NMR (500 MHz, [D3]acetonitrile: [D]chloroform, 1:1). Guest peaks are marked red: δ 9.29 (d, J = 4.8 Hz, 1H, 4), 9.18 (d, J = 4.7 Hz, 1H, 8 or 9), 9.15 (d, J = 4.8 Hz, 1H, 3), 9.14 (d, J = 4.7 Hz, 1H, 8 or 9), 9.05 (s, 1H, 49), 8.92 (s, 0H, V10 unbound), 8.86 (d, J = 4.8 Hz, 2H, 8, 9, 18 or 19), 8.83–8.75 (m, 2H, 8, 9, 18 or 19), 8.40 (s, 1H, V11 unbound), 8.32 (dd, J = 7.4, 1.7 Hz, 1H, 22(II)), 8.22 (dd, J = 7.4, 1.7 Hz, 1H, 22(I)), 8.15–8.08 (m, 2H, 22(III,IV)), 7.91–7.81 (m, 4H, 24), 7.63–7.45 (m, 7H, 23, 25(I,III,IV)), 7.47–7.25 (m, 8H, 55, 56, 57, V10 out), 7.19–6.96 (m, 9H, 39–42, 44–48), 6.95 (d, J = 8.2 Hz, 1H, 38), 6.91 (d, J = 8.3 Hz, 1H, 25(II)), 6.08 (s, 1H, 30(IV)), 6.07 (s, 1H, 30(III)), 5.88 (s, 1H, 30(I)), 5.21 (s, 2H, V10 in), 4.93 (s, 2H, V11 out), 4.63 (d, J = 23.9 Hz, 1H, V1 unbound), 4.38 (d, J = 16.0 Hz, 1H, 32a(III)), 4.33–4.20 (m, 5H, 27a(III,IV), 32a(I,II,IV)), 4.15 (t, J = 9.1 Hz, 2H, 27a(I), V1a out), 4.09–3.96 (m, 4H, 27b(I,III,IV), V1b out), 3.90 (d, J = 16.1 Hz, 1H, 32b(III)), 3.87 (d, J = 16.4 Hz, 1H, 32b(I)), 3.83 (t, J = 16.2 Hz, 1H, 32b(IV)), 3.80–3.74 (m, 1H, 27a(I)), 3.68 (d, J = 16.5 Hz, 1H, 32b(II)), 3.65–3.46 (m, 5H, 27b(I), V11 in), 3.40–3.23 (m, 9H, 28a, 53, V1a in), 3.09–3.02 (m, 1H, 28b(II)), 2.96–2.86 (m, 1H, V1b in), 2.84–2.76 (m, 1H, 28b(I)), 2.65 (t, J = 10.3 Hz, 1H, 28b(III)), 2.46 (t, J = 10.0 Hz, 1H, 28b(IV)), 1.72–0.82 (m, 50H), 0.77 (d, J = 6.7 Hz, 3H, V4 in), 0.74–0.68 (m, 1H, V2a in), 0.39 (s, 1H, V2b in), –2.81 (s, 2H, pyr‐NH). 13C NMR (126 MHz, [D3]acetonitrile: [D]chloroform, 1:1) δ 165.72, 160.11, 159.97, 159.66, 159.60, 158.69, 158.36, 151.03, 148.16, 147.31, 145.91, 145.76, 145.05, 142.95, 142.93, 142.91, 142.52, 136.76, 136.63, 136.43, 134.76, 133.93, 133.47, 132.94, 132.46, 132.20, 131.87, 131.75, 131.70, 131.61, 131.44, 131.40, 131.30, 130.89, 130.81, 130.41, 130.29, 130.26, 129.91, 129.86, 129.81, 129.61, 129.07, 129.02, 128.94, 128.37, 128.34, 125.28, 123.08, 121.99, 121.78, 121.66, 117.70, 117.61, 117.36, 117.19, 114.58, 114.22, 114.05, 113.85, 113.83, 97.19, 87.15, 86.42, 71.14, 70.06, 69.30, 68.84, 68.28, 61.52, 61.40, 56.24, 45.56, 45.18, 44.74, 40.28, 40.07, 39.87, 38.91, 37.81, 37.36, 32.85, 31.86, 30.62, 30.24, 29.01, 25.65, 25.42, 23.65, 23.61, 23.58, 23.49, 23.37, 23.30, 19.84, 19.49, 14.85. 19F NMR (471 MHz, [D3]acetonitrile: [D]chloroform, 1:1) δ –69.15 (52), –73.78 (d, J = 708.1 Hz, PF6).
(R,Sp*)‐H24 with (S,S)‐V2: 1H NMR (500 MHz, [D3]acetonitrile: [D]chloroform, 1:1). Guest protons are marked red: δ 9.31 (d, J = 4.7 Hz, 1H, 4), 9.29 (s, 1H, 49), 9.21 (d, J = 4.7 Hz, 1H, 8 or 9), 9.15 (d, J = 4.7 Hz, 1H, 8 or 9), 9.13 (d, J = 4.8 Hz, 1H, 3), 8.94 (d, J = 6.3 Hz, 2H, V10 unbound), 8.87 (d, J = 4.8 Hz, 2H, 8, 18 or 19), 8.79 (d, J = 4.8 Hz, 1H, 9), 8.76 (d, J = 4.9 Hz, 1H, 18 or 19), 8.42 (d, J = 6.2 Hz, 2H, V11 unbound), 8.30 (dd, J = 7.4, 1.7 Hz, 1H, 22(II)), 8.21 (dd, J = 7.4, 1.7 Hz, 1H, 22(I)), 8.15–8.09 (m, 2H, 22(III,IV)), 7.94–7.79 (m, 6H, 24, V10 out), 7.63–7.31 (m, 12H, 23, 25(I,III,IV), 55, 56, 57), 7.18–6.98 (m, 9H, 39–42, 44–48), 6.97–6.92 (m, 1H, 38), 6.88 (d, J = 8.3 Hz, 1H, 25(II)), 6.09 (s, 1H, 26(IV)), 6.04 (s, 1H, 26(III)), 5.93 (s, 1H, 26(I)), 5.46 (s, 2H, V11 out), 4.73–4.48 (m, 3H, V1 unbound, V10 in), 4.39 (d, J = 16.0 Hz, 1H, 32a(III)), 4.38–4.28 (m, 2H, V1 out), 4.28–4.19 (m, 6H, 27a(III,IV), 32a(I,II,IV)), 4.15 (t, J = 9.3 Hz, 1H, 27a(I)), 4.05–3.94 (m, 3H, 27b(I,III,IV)), 3.90 (d, J = 15.9 Hz, 1H, 32b(III)), 3.87 (d, J = 15.9 Hz, 1H, 32b(I)), 3.83 (d, J = 15.9 Hz, 1H, 32b(IV)), 3.79 (d, J = 10.2 Hz, 1H, 27a(II)), 3.70 (d, J = 16.5 Hz, 1H, 32b(II)), 3.68–3.57 (m, 2H, 27b(II)), 3.40–3.30 (m, 5H, 28b, V11 in), 3.28 (s, 3H, 53), 3.27–3.16 (m, 1H, V1a in), 3.11 (d, J = 9.8 Hz, 1H, 28b(II)), 2.90 (d, J = 10.9 Hz, 1H, 28b(I)), 2.52 (t, J = 10.4 Hz, 1H, 28b(III)), 2.49–2.36 (m, 1H, V1b in), 2.34–2.26 (m, 1H, 28b(IV)), 1.90–0.77 (m, 44H), 0.69 (d, J = 6.5 Hz, 3H, V4 in), 0.52–0.37 (m, 1H, V2a in), 0.01 to –0.12 (m, 1H, V2b in), –2.81 (s, 2H, pyr‐NH). 13C NMR (126 MHz, [D3]acetonitrile: [D]chloroform, 1:1) δ 165.29, 159.74, 159.45, 159.32, 159.16, 159.03, 158.72, 150.57, 146.81, 146.24, 146.17, 146.16, 146.10, 145.59, 145.02, 141.90, 141.47, 141.22, 137.87, 136.22, 136.06, 135.97, 135.91, 134.20, 133.34, 132.83, 132.44, 131.76, 131.47, 131.38, 131.12, 131.08, 131.00, 130.92, 130.76, 130.71, 130.22, 129.86, 129.71, 129.68, 129.33, 129.27, 129.22, 129.03, 128.52, 128.42, 128.33, 127.89, 127.80, 125.28, 122.17, 122.15, 121.39, 121.08, 121.01, 119.58, 117.08, 116.54, 116.34, 113.21, 113.14, 113.07, 86.61, 85.88, 69.23, 68.46, 67.59, 67.44, 61.33, 60.97, 60.74, 55.52, 45.01, 44.58, 44.17, 39.71, 39.55, 39.49, 39.26, 39.09, 38.94, 37.49, 37.18, 31.28, 31.21, 31.00, 28.42, 28.39, 28.34, 25.35, 25.02, 24.90, 23.08, 23.02, 22.91, 22.83, 22.73, 19.39, 19.31, 18.52. 19F NMR (471 MHz, [D3]acetonitrile: [D]Chloroform, 1:1) δ –69.14 (52), –73.76 (d, J = 708.4 Hz, PF6).
(R,Rp*)‐H25 with (R,R)‐V2: 1H NMR (500 MHz, [D3]acetonitrile: [D]chloroform, 1:1). Guest protons are marked red: δ 9.31 (s, 1H, 49), 9.21 (d, J = 4.7 Hz, 1H, 13 or 14), 9.20 (d, J = 4.7 Hz, 1H, 3 or 4), 9.13 (d, J = 4.7 Hz, 1H, 13 or 14), 9.03 (d, J = 4.8 Hz, 1H, 3 or 4), 8.93 (d, J = 4.7 Hz, 3H, 19, V10 unbound), 8.83 (d, J = 4.7 Hz, 1H, 8 or 9), 8.80 (d, J = 4.8 Hz, 1H, 18), 8.76 (d, J = 4.8 Hz, 1H, 8 or 9), 8.44–8.39 (m, 1H, V11 unbound), 8.35 (dd, J = 7.4, 1.7 Hz, 1H, 22), 8.15 (dd, J = 7.2, 1.8 Hz, 2H, 22), 8.10 (dd, J = 7.5, 1.8 Hz, 1H, 22), 7.93–7.82 (m, 4H, 24), 7.70–7.57 (m, 2H, V10 out), 7.54–7.35 (m, 28H, 23, 25, 55, 56, 57, CHCl3), 7.25–6.97 (m, 9H, 38–42, 45–48), 6.96–6.91 (m, 1H, 44), 6.07 (s, 1H, 30(III)), 6.04 (s, 1H, 30(IV)), 5.96 (s, 1H, 30(II)), 5.14 (s, 2H, V11 out), 4.93 (s, 2H, V10 in), 4.75–4.54 (m, 1H, V1 unbound), 4.38 (d, J = 16.0 Hz, 1H, 32a(IV)), 4.32–4.11 (m, 9H, 27a, 32a(I,II,III), V1 out), 4.06–3.98 (m, 3H, 27b(II,III,IV)), 3.97–3.88 (m, 2H, 27b(I), 32b(IV)), 3.86 (d, J = 13.2 Hz, 1H, 32b(II)), 3.83 (d, J = 16.0 Hz, 1H, 32b(III)), 3.75 (d, J = 16.3 Hz, 1H, 32b(I)), 3.49–3.40 (m, 1H, V1a in), 3.39–3.30 (m, 7H, 28a, V11 in), 3.27 (s, 3H, 53), 3.17 (d, J = 26.5 Hz, 1H, 28b(I)), 2.91 (d, J = 8.7 Hz, 1H, 28b(II)), 2.66 (s, 1H, V1b in), 2.52 (t, J = 10.5 Hz, 1H, 28b(IV)), 2.35 (t, J = 9.8 Hz, 1H, 28b(III)), 1.70–0.86 (m, 76H), 0.86 (d, J = 6.6 Hz, 3H, V4 in), 0.62 (d, J = 11.8 Hz, 1H, V2a in), 0.20–0.04 (m, 1H, V2b in), –2.83 (s, 2H, pyr‐NH). 13C NMR (126 MHz, [D3]acetonitrile: [D]chloroform, 1:1) δ 165.01, 159.61, 159.50, 159.39, 158.96, 155.70, 150.78, 147.49, 145.04, 145.02, 142.29, 136.50, 136.37, 136.19, 134.31, 133.71, 133.17, 131.69, 131.50, 131.45, 131.27, 131.21, 131.14, 131.12, 131.02, 130.95, 130.59, 130.10, 130.04, 129.75, 129.64, 129.60, 129.53, 129.42, 129.40, 129.14, 128.75, 128.72, 128.70, 125.29, 122.44, 121.70, 121.41, 121.39, 117.43, 113.64, 113.30, 87.02, 86.24, 68.70, 68.56, 68.12, 67.67, 64.15, 62.73, 62.67, 62.52, 61.27, 40.23, 40.08, 39.85, 39.58, 37.86, 37.56, 31.79, 31.63, 28.83, 28.80, 28.73, 28.53, 26.42, 25.77, 25.43, 23.53, 23.44, 23.32, 23.29, 23.18, 19.78, 18.68, 14.71, 14.65. 19F NMR (471 MHz, [D3]acetonitrile: [D]chloroform, 1:1) δ –68.80 (52), –73.79 (d, J = 708.6 Hz, PF6).
(R,Rp*)‐H25 with (S,S)‐V2: 1H NMR (500 MHz, [D3]acetonitrile: [D]chloroform, 1:1). Guest protons are marked red: δ 9.25–9.15 (m, 3H, 3, 4, 13 or 14, 49), 9.12 (d, J = 4.7 Hz, 1H, 3, 4, 13 or 14), 9.04 (d, J = 4.7 Hz, 1H, 3, 4, 13 or 14), 8.95 (d, J = 6.3 Hz, 3H, V10 unbound), 8.93–8.89 (m, 1H, 19), 8.84–8.74 (m, 3H, 8, 9, 18), 8.44 (d, J = 6.2 Hz, 3H, V11 unbound), 8.35 (dd, J = 7.4, 1.7 Hz, 1H, 22), 8.18–8.11 (m, 2H, 22), 8.09 (dd, J = 7.3, 1.8 Hz, 1H, 22), 7.93–7.82 (m, 4H, 24), 7.62–7.36 (m, 34H, 23, 25, 55, 56, 57CHCl3), 7.17 (d, J = 6.0 Hz, 2H, V10 out), 7.17–6.97 (m, 9H, 38–42, 45–48), 6.93 (d, J = 7.9 Hz, 1H, 44), 6.11 (s, 1H, 30(IV)), 6.10 (s, 1H, 30(III)), 5.97 (s, 1H, 30(II)), 5.52 (s, 2H, V10 in), 4.75 (s, 2H, V11 out), 4.73–4.56 (m, 2H, V1 unbound), 4.39 (d, J = 16.0 Hz, 1H, 32a(IV)), 4.34–4.16 (m, 7H, 27a, 32a(I,II,III)), 4.09–3.98 (m, 8H, 27b(I,II,III), V1a out), 3.99–3.78 (m, 5H, 27b(IV), 32b(II,III,IV), V1b out), 3.78–3.50 (m, 7H, 32b(I), V11 in, V1a in), 3.43–3.34 (m, 4H, 28a), 3.29 (s, 3H, 53), 3.26–3.14 (m, 1H, 28b(I)), 3.16–3.04 (m, 1H, V1b in), 2.86 (s, 1H, 28b(II)), 2.69 (s, 1H, 28b(IV)), 2.50 (t, J = 9.6 Hz, 1H, 28b(III)), 1.92–1.09 (m, 41H), 1.04–0.95 (m, 19H), 0.90–0.87 (m, 9H), 0.84 (d, J = 6.6 Hz, 3H, V4 in), 0.49 (s, 1H, V2b in). 13C NMR (126 MHz, [D3]acetonitrile: [D]chloroform, 1:1) δ 157.92, 157.71, 157.62, 157.17, 146.27, 146.11, 145.64, 144.81, 143.54, 142.78, 134.84, 134.66, 134.44, 132.66, 131.96, 131.42, 130.78, 130.33, 130.06, 129.84, 129.80, 129.72, 129.50, 129.36, 129.30, 128.92, 128.82, 128.35, 128.33, 128.29, 128.11, 128.00, 127.89, 127.85, 127.75, 127.74, 127.63, 127.38, 126.95, 126.46, 122.95, 121.11, 121.10, 121.03, 119.93, 119.79, 119.74, 116.62, 112.15, 111.95, 111.78, 111.69, 85.26, 84.46, 69.34, 69.05, 67.85, 67.83, 67.14, 67.03, 66.44, 66.15, 60.01, 59.46, 54.54, 54.16, 53.64, 43.58, 43.24, 42.83, 38.33, 38.19, 38.13, 37.84, 37.62, 37.24, 35.84, 35.33, 29.88, 29.78, 29.66, 27.11, 26.98, 23.72, 23.55, 23.47, 21.74, 21.61, 21.55, 21.53, 21.42, 17.99, 17.94, 17.50, 12.99. 19F NMR (471 MHz, [D3]acetonitrile: [D]chloroform, 1:1) δ –68.92 (52), –73.93 (d, J = 708.1 Hz, PF6).
Host–guest complexation experiments: The solutions for the UV/Vis and fluorescence titration experiments were prepared in a mixture of MeCN and CHCl3 (1:1, v/v). The MeCN was freshly distilled prior to use. The anhydrous CHCl3 was bought from Acros Organics and filtered through anhydrous K2CO3 prior to use to prevent protonation of the porphyrin cage compounds during the titrations. The stock solutions of the hosts (ca. 0.1 mM) were prepared by dissolving circa 1–2 mg of the respective compound in the solvent mixture. From this stock solution, three measuring solutions were prepared containing the host in a concentration of 3.0 µM. The guest stock solution (2.0 mM) was prepared by dissolving 14.56 mg of the guest in the solvent mixture. From both stock solutions, two titration solutions were prepared both containing the host at a concentration of 3.0 µM and the guest at a concentration of 0.80 mM and of 80 µM. Prior to the fluorescence titrations, the excitation wavelength (λ ca. 420 nm) was determined by measuring the maximum absorption wavelength in the UV/Vis spectrum. The decays of fluorescence intensities as a result of the addition of small amounts of guests were recorded. The resulting data was uploaded to a web application to fit the data and calculate the association constants.20
Supporting information
Supporting Information
Acknowledgements
R. J. M. N. acknowledges support from the European Research Council (ERC Advanced Grant ENCOPOL‐74092) and from the Dutch National Science Organization NWO (Gravitation program 024.001.035).
Contributor Information
Johannes A. A. W. Elemans, Email: J.Elemans@science.ru.nl, https://www.ru.nl/science/molecularnanotechnology/
Roeland J. M. Nolte, Email: r.nolte@science.ru.nl, https://www.ru.nl/science/molecularnanotechnology/.
References
- 1. Thordarson P., Bijsterveld E. J. A., Rowan A. E. and Nolte R. J. M., Nature, 2003, 424, 915–918. [DOI] [PubMed] [Google Scholar]
- 2. Deutman A. B. C., Monnereau C., Elemans J. A. A. W., Ercolani G., Nolte R. J. M. and Rowan A. E., Science, 2008, 322, 1668–1671. [DOI] [PubMed] [Google Scholar]
- 3. Monnereau C., Ramos P. H., Deutman A. B. C., Elemans J. A. A. W., Nolte R. J. M. and Rowan A. E., J. Am. Chem. Soc., 2010, 132, 1529–1531. [DOI] [PubMed] [Google Scholar]
- 4. Varghese S., Elemans J. A. A. W., Rowan A. E. and Nolte R. J. M., Chem. Sci., 2015, 6, 6050–6058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rutten M. G. T. A., Vaandrager F. W., Elemans J. A. A. W. and Nolte R. J. M., Nature Rev. Chem., 2018, 2, 365–381. [Google Scholar]
- 6. Varghese S., Spierenburg B., Swartjes A., White P. B., Tinnemans P., Elemans J. A. A. W. and Nolte R. J. M., Org. Lett., 2018, 20, 3719–3722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bako P., Keglevich G., Rapi Z. and Toke Z. L., Curr. Org. Chem., 2012, 16, 297–304. [Google Scholar]
- 8. Suzuki T., Cerón‐Carrasco J. P., Tamaoki H., Ishigaki Y., Katoono R., Fukushima T. and Pérez‐Sánchez H., Heterocycles, 2017, 94, 1123–1132. [Google Scholar]
- 9. Huang Q., Jiang L., Liang W., Gui J., Xu D., Wu W., Nakai Y., Nishijima M., Fukuhara G., Mori T., Inoue Y. and Yang C., J. Org. Chem., 2016, 81, 3430–3434. [DOI] [PubMed] [Google Scholar]
- 10. Allenmark S., Chirality, 2003, 15, 409–422. [DOI] [PubMed] [Google Scholar]
- 11. Dıaz Gomez E., Brotin T. and Duddeck H., Tetrahedron: Asymmetry, 2007, 18, 2155–2164. [Google Scholar]
- 12. Chen C.‐F. and Han Y., Acc. Chem. Res., 2018, 51, 2093–2106. [DOI] [PubMed] [Google Scholar]
- 13. Tlustý M., Slavík P., Kohout M., Eigner V. and Lhoták P., Org. Lett., 2017, 19, 2933–2936. [DOI] [PubMed] [Google Scholar]
- 14. Aav R., Shmatova E., Reile I., Borissova M., Topi F. and Rissanen K., Org. Lett., 2013, 15, 3786–3789. [DOI] [PubMed] [Google Scholar]
- 15. Chen Y., She N., Meng X., Yin G., Wu A. and Isaacs L., Org. Lett., 2007, 9, 1899–1902. [DOI] [PubMed] [Google Scholar]
- 16. Cakmak Y., Erbas‐Cakmak S. and Leigh D. A., J. Am. Chem. Soc., 2016, 138, 1749–1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Testa B., Helv. Chim. Acta, 2013, 96, 360–366. [Google Scholar]
- 18. Elemans J. A. A. W., Claase M. B., Aarts P. P. M., Rowan A. E., Schenning A. P. H. J. and Nolte R. J. M., J. Org. Chem., 1999, 64, 7009–7016. [Google Scholar]
- 19. Jeffrey G. A., in: An introduction to hydrogen bonding, 1997. [Google Scholar]
- 20.See http://app.supramolecular.org/bindfit/.
- 21. Chen C. Y., Nagumo S. and Akita H., Chem. Pharm. Bull., 1996, 44, 2153–2156. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Information