

Abstract
Cells are continuously subjected to an array of reactive/toxic chemical species which are produced both endogenously through metabolic pathways and taken up exogenously by diet and exposure to drugs or toxins. As a result, proteins often undergo non-enzymatic covalent modifications (NECMs) by these species, which can alter protein structure, function, stability, and binding partner affinity. NECMs accumulate over time and are linked to various diseases such as Alzheimer’s disease, cancer, and diabetes. In the cellular proteome, histones have some of the longest half-lives, making them prime targets for NECMs. In addition, histones have emerged as key regulators of transcription, a function that is primarily controlled by modification of their tails. These modifications are usually installed or removed enzymatically, but recent evidence suggests that some may also occur non-enzymatically. Despite the vast knowledge detailing the relationship between histone modifications and gene regulation, NECMs on histones remain poorly explored. A major reason for this difference stems from the fact that, unlike their enzymatically installed counterparts, NECMs are difficult to both control and test in vivo. Here, we review advances in our understanding of the effect non-enzymatic covalent modifications (NECMs) have on the epigenetic landscape, cellular fate, and their implications in disease. Cumulatively, this illustrates how the epigenetic code is directly toxified by chemicals and detoxified by corresponding eraser enzymes.
TOC graphic
1. Introduction
The cellular environment contains a range of metabolites that can chemically react with nucleophilic functional groups within a given protein depending on the sequence, structure, microenvironment, and half-life.(1)These chemicals are either endogenously generated or taken up from the cellular microenvironment.(2) The accumulation of toxic modifications occurs under normal physiological conditions, but can also be stimulated by various changes in the cellular environment such as redox and metabolic states.(3) The main types of known non-enzymatic covalent modifications (NECMs) include glycation, oxidation, cross-linking, deamidation, and lipidation (Figures 1 and 2). Identified in numerous intra- and extra-cellular proteins, NECMs have been shown to have a detrimental effect on protein structure, function, and stability by either altering key amino acids or changing the protein backbone structure entirely.(4−6) Indeed, high levels of NECM-modified proteins were found to correlate with aging as well as an array of pathologies including Alzheimer’s disease, cancer, cardiovascular disease, and diabetes.(7−9)
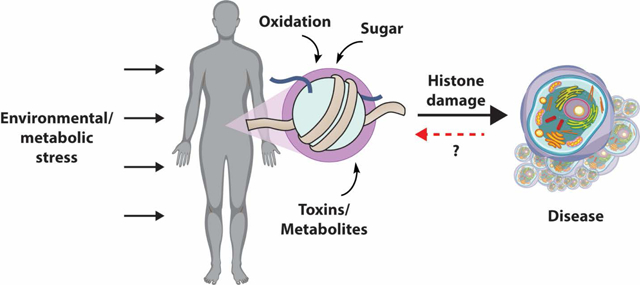
Figure 1.
General schematic of NECM exposure and effect on chromatin. Environmental exposure to reactive metabolites through nutrition, toxins (automobile gas, pollution, cigarette smoke), damaging radiation (UV, ionizing), or drugs changes the cellular microenvironment and introduces highly reactive species. These toxins react with various proteins in the cell, but accumulate on histones, which have a very long half-life and highly accessible nucleophilic tails. Non-enzymatic modification of histones competes with other regulatory enzymatic modifications and changes chromatin architecture.
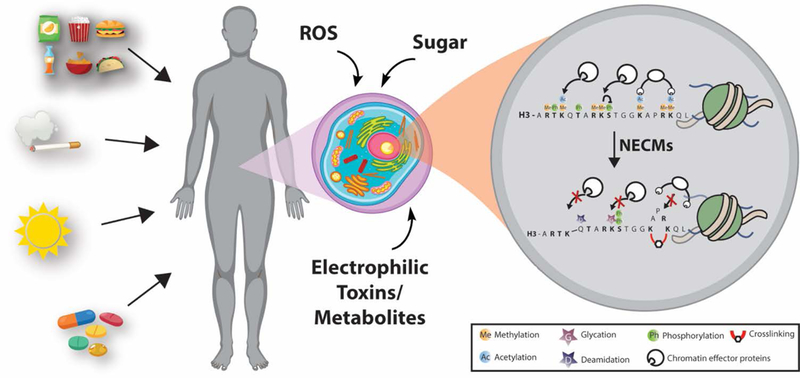
Figure 2.
Chemical structures of reactive species responsible for NECMs and their resulting protein adducts. A vast array of non-enzymatic modifications have been shown to occur in vitro and in vivo. Histone proteins have been shown to experience a majority of described NECMs (red boxes), though others remain to be shown (gray).
The core histone proteins, which include H2A, H2B, H3, and H4, have long half-lives that can reach months in nonproliferating cells.(10) Histones compose the nucleosome functional unit of eukaryotic chromatin, where an octamer of two copies of each core histone is wrapped by ∼150bp of DNA. An additional linker histone, H1, is deposited between two nucleosomes and aids in the higher-order structural organization of chromatin.(11)Histones have an irregular distribution in their amino acid composition, with an over-representation of basic amino acids such as lysines and arginines. These residues primarily populate the histones’ unstructured tails, which extend away from the nucleosome and undergo a variety of regulatory post-translational modifications (PTMs), including acetylation, methylation, and ubiquitination.(12) These modifications can directly affect the degree to which chromatin compacts, and thus the accessibility of a genomic region, by disrupting the electrostatic interaction between positively charged lysines and arginines and the negatively charged DNA, as in the case of acetylation.(13,14) Other histone PTMs such as methylation, which does not change the net charge of the residue side chain, primarily affect chromatin structure by recruiting dedicated protein “readers” that bind and elicit change in the local epigenetic landscape.(12) Extensive efforts have been invested to understand the biochemical cascades coupling histone PTMs to transcription control in both healthy and diseased cells.(15,16) Specific marks have been identified as gene activators (e.g., H3K4me3 and H2BK120Ub) or deactivators (e.g., H3K9me3 and H3K27me3). Their misregulation can lead to aberrant cellular functions and a variety of malignancies, including cancer.(17)
Specific histone PTMs are installed by a network of tightly regulated “writer” enzymes that often utilize metabolic cofactors, which provide one link between metabolism and epigenetic regulation.(18) For example, glucose influx perturbs the levels of acetyl-CoA and S-adenosylmethionine (SAM), both key cofactors of histone PTM writers. Cellular acetyl-CoA concentration was shown to directly affect the levels of histone acetylation in multiple genomic regions, establishing an important layer of epigenetic landscapes and affecting transcription levels.(19) Similarly, SAM production is directly affected by glucose influx, which then changes its occupation in many histone methyltransferases.(20) Moreover, many “eraser” enzymes that regulate PTMs through their removal also utilize metabolic cofactors.(18,19) An example is the JMJ histone demethylase family whose functions rely on α-ketoglutarate, a key intermediate of the TCA cycle.(21)
Despite our deep understanding of these enzymatically installed histone PTMs and their assorted functions, little is known about the role that NECMs play in transcription regulation.(22) Histone proteins are prime candidates for NECMs because of their long-lived nature, sensitivity to chemical modification, and the enrichment of nucleophilic lysine and arginine residues on their tails. However, despite the observation of various non-enzymatic modifications existing on histone tails, there have been few mechanistic analyses of their regulation and direct functional consequences or downstream effects of their installation. With recent strong evidence of a breadth of non-enzymatic modifications on histones (Figure 2), new interest arises for understanding their effect on cellular fate.(23) Here we review this novel mechanism of epigenetic regulation and its implication in disease.
2. Acylation
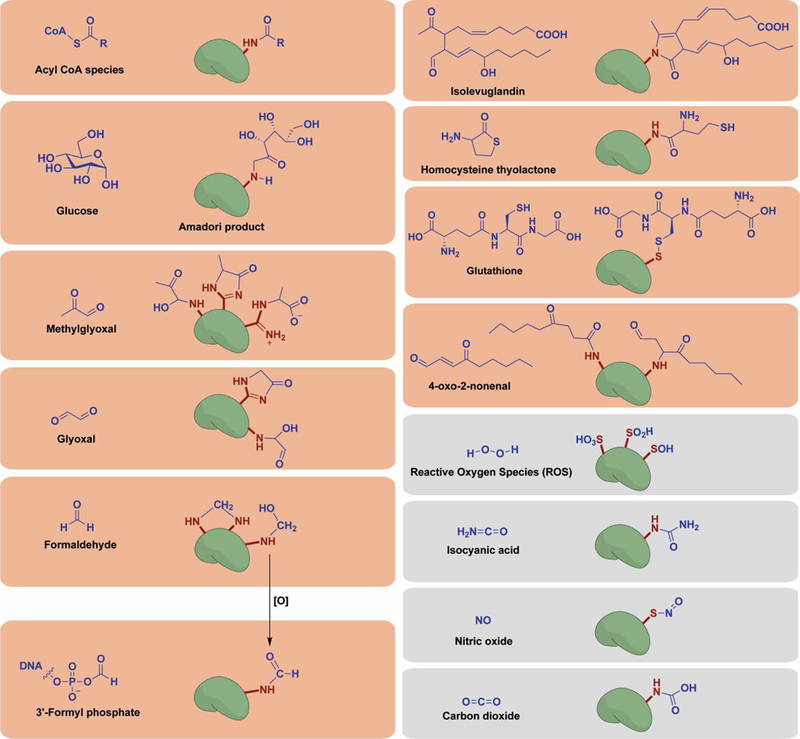
Lysine acetylation on histones plays a critical role in maintaining the open-chromatin configuration through capping of the positive charge of the ε-amine, subsequently abolishing the salt bridge between the formerly positive amine and negatively charged phosphate backbone of DNA. This highly dynamic modification is regulated by the antagonistic activity of histone acetyl-transferases (HATs) and deacetylases (HDACs).(24)However, the reactive thioester of the acetyl-CoA cofactor has long been speculated to react non-enzymatically with the target amine on lysines (Figures 2 and 3). In fact, its ability to non-enzymatically acetylate histones in vitro has been known for nearly half a century,(25) though whether this reaction occurred physiologically was unknown until more recently.
Figure 3.
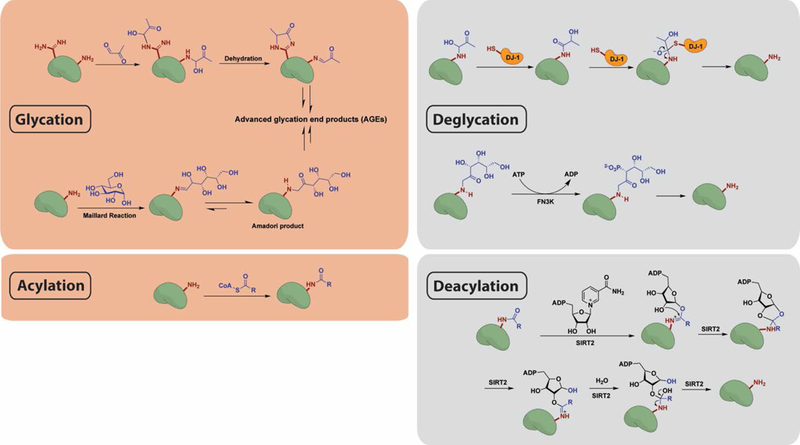
Reaction mechanisms for chemical modification and enzymatic removal of glycation and acylation marks. (Top) Lysine or arginine residues condense with aldehyde moieties in reducing sugars and can then undergo further rearrangement to yield AGEs. DJ-1 functions as a deglycase on early stage glycation adducts by participating in a nucleophilic addition–elimination reaction. FN3K phosphorylates the Amadori product of reducing sugar adducts, resulting in its destabilization and ultimate collapse. (Bottom) Lysine residues react with acyl-CoA thioesters via nucleophilic addition–elimination. By an NAD-dependent mechanism, SIRT2 removes acyl groups from the modified proteins.
Analyses performed by Marmorstein and colleagues have suggested that the local microenvironment may poise certain lysines to be hyperactive toward such NECMs,(26) and the Meier group has used chemical biology techniques to probe and identify targets of non-enzymatic acylation, including histones.(27) Further cellular assays and proteomics experiments have illustrated that an even wider range of non-enzymatic acylations occurs on histone proteins stemming from a variety of acyl-donors such as propionyl-, butyryl-, crotonyl-, malonyl-, succinyl-, β-hydroxybutyryl-, and glutaryl-CoA.(28) In addition to short- and medium-chain acylation of lysine residues, an unbiased proteomics screen by Hang and colleagues came to the surprising finding that the sole conserved cysteine residue in histones, C110 in the core histone H3, undergoes S-palmitoylation.(29) However, separating correlation and causality with regard to these modifications is crucial in understanding their cellular functions. Due to the nature of the proteomic and metabolomic experiments that originally identified many of these non-enzymatic acylation marks on histones, their distinguishing functional roles from enzymatic acetylation, if any exist, remain unclear and are the subjects of ongoing work in many laboratories.
Akin to acetylation, some of these marks may be subject to enzymatic installation. For example, one of the earliest identified histone acylation marks, crotonylation, has been shown to have an evolutionarily conserved function. Specifically, through a distinct genomic localization pattern, crotonylation marks were localized to active sex-chromosome linked genes.(30) The existence of a conserved function, as well as crotonyl-specific “reader” proteins, but no dedicated “writer” (although p300 was shown to be able to utilize it in vitro) raises the question of whether crotonylation is purely non-enzymatic in nature.(28,31) Furthermore, crotonylation has emerged as a link between the gut microbiota and epigenetic signaling, strengthening the possibility that it may serve as an NECM.(32)
Finally, sodium benzoate, an FDA-approved drug and food preservative, was recently reported to induce the generation of benzoyl-CoA, which in turn modifies histones through an unknown mechanism and can be removed by the deacylase SIRT2. Utilizing chromatin immunoprecipitation-sequencing (ChIP-seq) and RNA-seq analyses, Zhao and colleagues demonstrated that benzoylation marks have relevance in active transcription and a distinct pattern to canonical acetylation.(33) Together these evidence suggest an abundance of non-enzymatic acylations occurring on histones that may compete against or enhance acetylation or have a completely different epigenetic function.
3. Glycation/Glycoxidation
Glycation is one of the most prevalent NECMs and is characterized by the condensation of the aldehyde form of monosaccharides (mainly glucose and fructose) with reactive groups (mainly lysines and arginines), forming stable adducts (Figures 2 and 3). The initial glycation product can be further oxidized and subsequently rearranged to form a series of products generally referred to as advanced glycation end-products (AGEs), which themselves can undergo additional chemical transformation including the ability to form cross-links.(34) In diabetes, hemoglobin glycation (HbA1c) is routinely used as a diagnostic marker, and glycation levels were shown to correlate with the severity of various complications including neuropathy, nephropathy, and retinopathy.(5) Moreover, the accumulation of AGE amyloids has been proposed to play a role in age-related neurodegenerative disorders such as Alzheimer’s and Parkinson’s disease.(35) Oxidative stress due to increase in reactive oxygen species (ROS) enhances the formation of AGEs, which in turn increases the presence of ROS in a positive feedback loop termed glycoxidation.(36)
An early and low-resolution analysis of histones extracted from diabetic murine liver cells indicated a significant increase in AGE levels compared to normal mice liver cells.(37) Additionally, in vitro analysis of histone glycation has been performed using purified recombinant H2B and H1 incubated with high levels of glucose and subjected to mass spectrometry analysis.(38) Several sites on both histones were modified with various AGEs, including sites known to carry enzymatically added PTMs. Moreover, placing these glycated histones under oxidative stress enhanced the accumulation of oxidation on the histone over nonglycated histones, illustrating that histones can undergo glycoxidation.
4. Glycation by reactive carbonyl species (RCS)
In addition to evidence of glycation by common monosaccharides, multiple recent reports have identified the glycation of histones by reactive carbonyls. These species can be taken up from the environment (via nutrition or alcohol consumption) but can also be generated as byproducts of cellular metabolism, as in the case of the toxic glycolysis byproduct methylglyoxal (MGO) (Figure 3).(39) RCS can spontaneously modify the amine and guanidino side groups in histones via the Maillard reaction, first forming Schiff base intermediates and then proceeding toward the development of AGEs (Figures 2 and 3). The reaction kinetics of RCS such as MGO are also often more rapid than for monosaccharides like glucose or ribose.
Recent findings indicate that histones are not only specific targets of methylglyoxal glycation in healthy cells,(40) but also that such glycation marks pathologically accumulate in cancer.(41) Metabolomics analyses suggest that these adducts occur on specific sites on histones that are known to undergo regulatory PTMs, and possibly even prevent enzymatic PTMs from being installed.(40,41) Moreover, mechanistic investigation revealed that MGO glycation can also directly change chromatin architecture by perturbing histone-DNA contacts and promoting cross-linking.(41) Curiously, the abrogation of chromatin architecture appears to take place in a two-step process, whereby chromatin first relaxes (likely due to glycation events quenching the positive charges on lysine and arginine residues) and then hyper-compacts (likely due to the formation of AGEs and cross-links between histones and DNA). These findings are supported by yet another recent paper, illustrating a direct effect of aldehydes on chromatin resulting in abrogation of stem cells.(42) While the precise mechanism by which this takes place is not known, aldehydes can react with either DNA, histones, or both, to induce this DNA damage, which provides a direct link between alcohol consumption and disruption of the chromatin landscape.
5. Oxidation by reactive oxygen species (ROS)
Reactive oxygen species are chemical moieties primarily generated endogenously, as byproducts of normal oxygen metabolism, and play an important role in cellular signaling. ROS concentrations can vastly increase during environmental stress conditions, which have damaging effects on both DNA and proteins in what is known as oxidative stress. The most abundant damage occurs in the form of guanine lesions on DNA and oxidation of key protein residues (primarily cysteine and methionine) (Figure 2).(43−45) This damage can be lethal to cells and induce apoptotic death either via the DNA damage response (DDR) or unfolded protein response (UPR) and is associated with aging, cardiovascular diseases, and neurodegeneration.(46) Exogenous uptake of various toxins (including environmental chemicals, cigarette smoke, and various prescription and recreational drugs) can also induce ROS formation through the generation of free radicals, which have been suggested to promote various diseases including cancer.(47) However, cellular responses to oxidative stress could also be critical for inducing positive signaling cascades in immune response and inflammation, for example, when leukocytes are recruited to sites of injury following the release of ROS by platelets.(48)
Despite their low incidence, all the core and linker histones contain multiple methionine residues, and the canonical H3 variant contains two cysteine residues. Further studies are needed to determine if these residues experience oxidation under hypoxic conditions, though existing studies suggest that it is possible. In fact, the H3 cysteines were shown to be involved in redox sensing through S-glutathionylation, leading to a more open chromatin structure.(49,50) Importantly, the levels of S-glutathionylation increase with cellular proliferation and decrease during aging, which are regulated through changes in cellular transcription, potentially involving this NECM.
6. Electrophilic Lipid Peroxidation End Products
Through their free-radical form, ROS can also generate toxic bioreactive electrophiles by endogenous peroxidation mechanisms or reacting with toxins taken up exogenously. This highly reactive family of molecules modifies both proteins and nucleic acids and was shown to disrupt cellular homeostasis, although the precise mechanism is not well understood. For example, olefin aldehyde compounds, such as 4-oxo-2-noneal (4-ONE), react with lysine residues in histones via Michael addition on key regulatory sites, which disrupts nucleosome assembly and three-dimensional structure (Figure 2).(51) Interestingly, this histone NECM can be reversed by the SIRT2 deacylase.(52) Moreover, isolevuglandins and other products of free radical-induced oxidation of arachidonates were shown to react with H4 in macrophages and lung carcinoma cells following stimulation, suggesting a role in the inflammatory response.(53) Beyond these, histones were shown to be targets of other electrophilic species, including acrolein,(54) which disrupt both nucleosome stability and susceptibility to undergo other PTMs, suggesting these could change cellular function through epigenetic mechanisms.
7. Formylation and Hydroxymethylation
Formaldehyde exists within the human body at concentrations up to 100 μM. This highly reactive species is generated as a byproduct of enzymatic reactions in diverse pathways including one-carbon metabolism, lysine demethylation, and alcohol metabolism.(55,56) While the ability of formaldehyde to react with the ε-amine of lysine residues within proteins has been recognized for several decades,(57) the earliest reports of histone formyllysine formation date back just over 10 years. In fact, histone formylation was first discovered as a secondary product resulting from oxidative DNA damage. ROS or DNA-modifying agents can react with DNA to produce 3′-formalphosphate species, whose labile phosphate thioesters are easily attacked by nucleophilic lysine side chains (Figure 2).(58) In addition to this canonical formylation pathway, formaldehyde can react directly with lysine side chains within proteins to form a hydroxymethyl lysine structure. This unstable intermediate can be either subsequently oxidized into formyllysine or cross-linked to nearby lysine residues if they are present (Figure 2). Formaldehyde has been used extensively in this way as a cross-linking reagent in mass spectrometry and ChIP methodologies.(59)
All four core histones and the linker H1 histone were shown to undergo extensive formylation in cells following a treatment with the enediyne antiproliferative drug neocarzinostat.(59) Furthermore, formylation was found to occur not only on the histones’ unstructured tails but also within their core globular domains.(60) Moreover, formylation was shown to compete with enzymatically installed PTMs at key residues for signaling as well as to take place on histone residues shown to directly contact DNA.(58–60)
One mechanistic study went further to investigate the source of the endogenous formaldehyde behind the modification of histone lysine residues. Despite the fact that lysine demethylation reactions form a carbinolamine intermediate that can spontaneously collapse to yield formaldehyde, metabolic labeling found that such reactions contribute negligibly, if at all, to histone formylation.(59) Instead, formaldehyde produced either endogenously from one-carbon metabolism or exogenously via the addition of DNA oxidizing agents must be the source of lysine formylation. However, further studies are needed to determine whether histone formylation is (patho-) physiological in nature or if such marks are merely adventitious.
8. Homocysteinylation
Homocysteine thiolactone (HTL), an intramolecular thioester of homocysteine (Hcy), is generated in human cells by methionyl-tRNA synthetase in an error-editing reaction that averts translational incorporation of homocysteine into proteins.(61) Thus, HTL synthesis is directly affected by the levels of cellular Hcy, which occurs both as a result of a high-methionine diet as well as under an array of pathological conditions.(62) HTL is known to react with lysine residues on various proteins with detrimental effects to their structure and function and has been implicated in diseases such as neurodegeneration (Figure 2).(61,62)
Multiple studies have not only identified Hcy modification of histone residues but also ascribed them pathophysiological roles. First, mass spectrometry analyses found homocysteinylation to occur throughout the N-terminal tails of histones extracted from cells treated with pathophysiological concentrations of HTL.(62)Furthermore, Hcy modification of histone H3 was found to correlate with both decreased methylation of key H3 residues, including K9 and K27, as well as increased acetylation on H3 residues K18 and K23. These data thus suggest a role for histone homocysteinylation in regulating crosstalk of canonical enzymatic PTMs.
Another mechanistic report identified a deleterious effect of homocysteinylation on histones. Specifically, all four core histones were shown to be modified by homocysteine on multiple residues in neuronal tissue, with H3K79 becoming a major target following elevated Hcy levels. Using ChIP-seq and RNA-seq assays, Zhang and colleagues demonstrated that an increase in H3K79Hcy level down-regulates the expression of selected neuronal-tube closure-related genes.(63) This provides a potential mechanistic explanation to the known correlation between high maternal Hcy levels and developmental neuronal tube defects. Thus, a growing body of evidence has identified this non-enzymatic modification as a new link between dietary intake, toxic sulfur metabolic intermediates, and developmental regulation through epigenetic mechanisms.
9. Deamidation
Protein deamidation is an intramolecular process that occurs through cyclization of the amide side-chain functionality of asparagine (or glutamine) residues onto the amide backbone, forming a reactive succinimide ring. This reactive intermediate then hydrolyzes to form either an aspartate or isoaspartate in place of the native asparagine residue (Figure 4).(64) These alterations both introduce a side-chain negative charge and, in the case of isoaspartate, the disruption of the amide backbone through a one-carbon homologation. The deamidation of proteins is known to be dependent on both the protein’s sequence and three-dimensional conformation. For example, proteins containing Asn-Gly junctions are known to have a half-life of deamidation of as little as 30 h under physiological conditions.(65)
Figure 4.
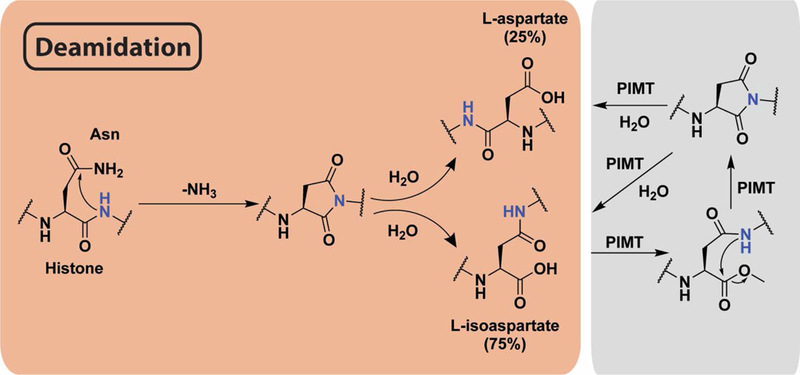
Mechanism of asparagine deamidation and its reversal. Peptide backbone nitrogen attacks the side chain amide, releasing ammonia to form a succinimide ring. Upon hydrolysis, either aspartate or isoaspartate are formed. PIMT methylates the resulting carboxylate, forming an ester that is spontaneously hydrolyzed by intramolecular attack from backbone amide nitrogen to reform the succinimide ring. Successive rounds of this mechanism may be required to fully repair deamidation.
The structural alterations imposed by this NECM contribute to protein misfolding and inactivation, although recent evidence suggests it regulates normal cellular functions and may act as an internal clock for protein aging.(66) In fact, eukaryotic cells have developed a mechanism to moderate the accumulation of isoaspartate through the activity of protein l-isoaspartyl-O-methyltransferase (PIMT), a highly conserved enzyme which promotes conversion of isoaspartate to aspartic acid. Decreased activity of PIMT has been associated with aging, Alzheimer’s disease, and epilepsy.(67) Despite PIMT activity, isoaspartate still accumulates in several long-lived proteins and was linked to cell senescence and cancer.(68) In the context of histones, Aswad and colleagues used a specific PIMT inhibitor and a PIMT knockout mouse model to identify isoaspartatyl-H2B accumulation in cells, suggesting the deamidation contributes to altered gene regulation and ultimately shorter life span.(69) In addition, deamidation of Asn3 of H1.0 was shown to accumulate in vivo in an age-dependent manner in both liver and brain tissues.(70) Taken together, these findings support a model wherein histone asparagine deamidation not only serves to mark protein aging but also alters histone function in transcriptional regulation.
10. DNA-Histone Crosslinking
As mentioned above, the dynamic nature of chromatin allows for the rapid cellular response to stimuli through changes in the accessibility of certain genetic regions, resulting in the adjustment of the transcriptional program. However, the intimate bonding between the DNA polymer and the associated histone proteins, which crucially enables the formation of an organized chromatin structure, also makes both biomolecules susceptible to chemical cross-linking with detrimental consequences. Indeed, a variety of exogenous environmental and endogenous metabolic toxins, as well as harmful radiation (ionizing radiation, UV light), were shown to induce chemical DNA-histone cross-linking under disease states (Figure 5). One example is the carcinogen 1,2,3,4-diepoxybutane (DEB), produced from the metabolic activation of 1,3-butadiene (BD), which is taken up from air polluted from automobile exhaust, urban air, and cigarette smoke. DEB can alkylate adenine or guanine bases to produce 2-hydroxy-3,4-epoxybut-1-yl (HEB) monoadducts, which rearrange to form DNA-DNA cross-linking.(71,72) Alternatively, it can react with nucleophilic amino acid side chains of adjacent proteins, forming DNA-protein cross-linking. Tretyakova and colleagues used high-resolution proteomics to survey all the DNA-protein cross-linking events which are induced by exposure to DEB and found that among the 152 proteins identified, all four-core histones and linker histone H1 contain these DNA-protein cross-links.(73)
Figure 5.
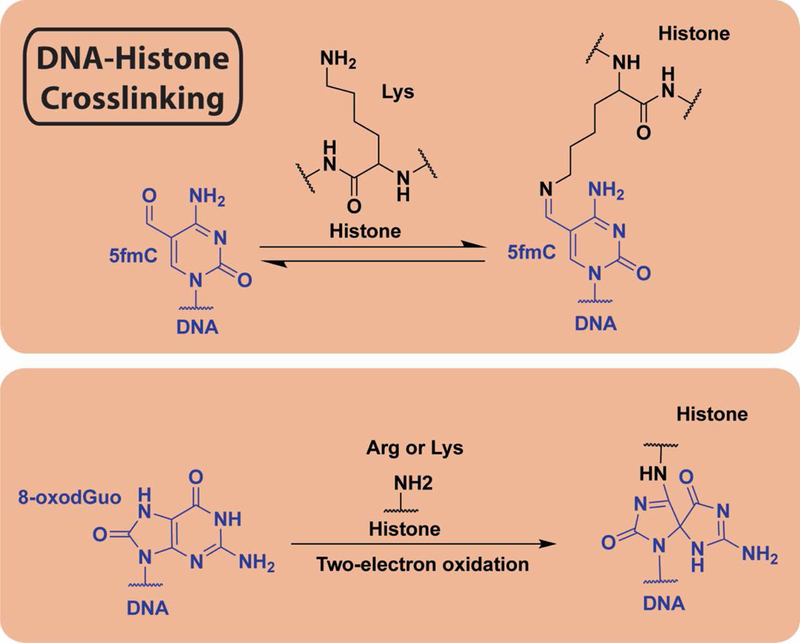
Mechanisms of DNA-histone cross-linking. (Top) Covalent bond formation between histone and DNA via lysine ε-amine condensation with 5-formylmethyl cytosine aldehyde to produce a Schiff base. (Bottom) Two electron oxidation of 8-oxodGuo, a common primary product of cellular oxidative damage, followed by nucleophilic attack by side chain arginine or lysine forms covalent adducts between damaged DNA and histone side chains.
N7-Methyl-2′-deoxyguanosine (MdG), which is the major product of a variety of alkylating agents used for research and for cancer therapy, is a long-lived toxin that was shown to directly react with histones and form cross-links with the DNA both in vitro and in cells.(56) This was suggested to be interfaced by the positive charge on the histone tails as illustrated using histone mutagenesis.(74) In an attempt to understand other DNA-cross-linking events induced by malondialdehyde, which is an abundant product of lipid peroxidation, Zhitkovich and colleagues performed an in vitro analysis to find that DNA-histone cross-links were readily generated and stable for days under physiological conditions.(75) Additional in vitro analysis illustrated that one of the most common products of oxidative DNA damage, 8-oxo-7,8-dihydro-2′-deoxyguanosine (8-oxodGuo) forms quantitative DNA-histone cross-linking involving the OGox trapping by the N-terminal amine of histones (Figure 5). These cross-links, although not observed in cells, were stable under physiological conditions.(45)
Finally, it was recently shown that MGO glycation (see Glycation/Glycoxidation) rearranges to form both histone-histone and DNA-histone cross-linking products in a cellular-metabolic state-dependent manner (Figure 6).(41) These observed constraining cross-linking events limit the decompaction range of chromatin fibers in vitro as illustrated by several biophysical analyses, including optical tweezers.(41) Moreover, it was observed to disrupt chromatin architecture in cells, which affects the accessibility of transcription start sites. Analogous to the MGO-protein Schiff base adduct, 5-formylcytosines (5fCs) react with lysine residues with similar chemistry. DNA-histone cross-links formed by 5fCs were shown to associate with tissue-specific nucleosome organization and increased nucleosome occupancy in vitro and in vivo.(76) Permanent disruption of the dynamic DNA-protein interactions likely has serious biological consequences on cellular transcription, replication, cell fate, and, ultimately, survival.(77)
Figure 6.
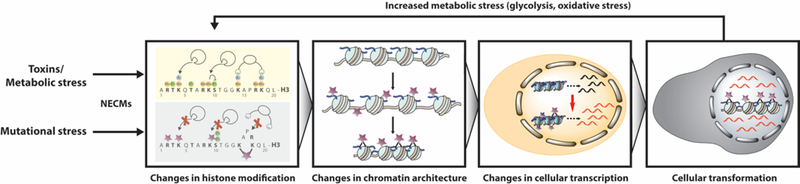
Protein NECMs as a new mechanistic link between microenvironmental stress and cell fate. Abrogated metabolism and mutational stress (for example, in NECM repair enzymes) promote accumulation of NECMs on histones. These adducts disrupt histones’ other regulatory enzymatic modifications, altering the epigenetic landscape, thus leading to changes in the cellular transcriptome and inducing transformation. Diseased cells, in turn, increase metabolic stress (through increased glycolysis in cancer, for example), generating reactive metabolites and oxygen species that further drive this cycle.
11. Enzymatic Regulation of NECMs
While all the modifications described herein occur through uncatalyzed chemical reactions between small molecules and active histone residues or as internal backbone modifications, cells have evolved enzymatic mechanisms to control them. The regulation is generally divided into three classes (1) removal, (2) destabilization, and (3) scavenging of reactive molecule by small molecules or converting enzymes (see Table1). For example, although deemed irreversible, new findings indicate that early intermediates of glycation can be removed from modified histones and DNA by DJ-1 (primarily MGO adducts) or destabilized through phosphorylation by FN3 kinase (primarily glucose adducts) (Figure 3).(41) To regulate MGO levels and avoid its accumulation, cells utilize a cytosolic chemical quencher, carnosine,(78) as well as the GLO1/GLO2 enzymes that convert MGO to d-lactate.(79)
Table 1: Summary of the enzymatic regulators of NECMs.
Indicated in the table are all known enzymatic regulators of NECMs, listed with their functions, any associated cofactors, and disease relevance.
| Eraser/Regulator | Detoxifying Function | Cofactor | Disease correlation |
|---|---|---|---|
| SIRT2 | Deacylation | NAD | Cancer |
| DJ-1 (PARK7) | Deglycation (GO and MGO) |
None | Parkinson’s disease, cancer |
| FN3K | Deglycation (Glucose) |
ATP | Cancer |
| Sulfiredoxin (SRX) | Desulfination | ATP | Parkinson’s disease |
| Denitrosylase | Denitrosylation | None | Innate immunity |
| PIMT | Reverse the formation of isoaspartate | SAM | Cancer, Alzheimer’s disease |
| GLO1 | Scavenge MGO and GO | GSH | Anxiety, cancer |
| Formaldehyde dehydrogenase (S-nitrosoglutathione reductase, GSNOR) |
Scavenge formaldehyde | NAD | Respiratory diseases (asthma) |
| Alcohol dehydrogenase (ADH) |
Convert alcohol to aldehyde | NAD | Alcoholism, drug dependence |
| Aldehyde dehydrogenase (ALDH) |
Convert aldehyde to carboxylic acid | NAD | Alcoholism, drug dependence, neurodegeneration |
| Aldo-keto reductase (AKR) |
Convert aldehyde to alcohol | NADPH | Alcoholism, drug dependence, cancer |
In the case of the non-enzymatic acylation, although acetylation can be reversed by multiple enzyme families, including both HDACs and sirtuins,(80) other non-enzymatic acylations were shown to be reversed by only a single deacylase, SIRT2 (Figure 3). One exception is crotonylation, which can be removed by HDACs 1, 2, and 3.(32) Formylation, on the other hand, has shown resistance to removal by both HDACs and sirtuins, despite its similarities to acetylation.(59) Sulfiredoxin, a sulfinic acid reductase, reduces cysteine sulfinylation and acts on multiple proteins including DJ-1, which was shown to have key regulatory cysteines sensitive to oxidation stress.(81) Despite the backbone rearrangement that takes place, deamidation damage can be reversed through the enzymatic activity of PIMT, which, as mentioned previously, converts the damaged asparagine residue to aspartic acid.
With most of the NECMs described here being heavily associated with aging as well as an array of diseases, it is not surprising that many of their regulatory enzymes have driver mutations in the same conditions (Table1). For example, both DJ-1 and FN3K have been indicated as oncogenes due to their overexpression in cancer. It is speculated that cancer cells depend on their overexpression as a mechanism to overcome glycation damage which is a byproduct of the Warburg effect (i.e., dependence on rapid glycolysis).(41) DJ-1, also named PARK7, has conversely also been implicated in neurodegeneration as a site-specific mutation in a noncatalytic residue of DJ-1 induces familial Parkinson through an unknown mechanism.(82) Histone deacetylaces are major targets in both neurodegeneration and cancer and have FDA-approved drugs to both conditions. With more investigation of the long-term effects of NECMs on cellular transcription and fate, targeting these enzymes for therapeutic purposes, either through inhibition, activation, or induction, will become more prevalent. Having these drugs in hand will also help researchers advance the understanding of their mechanism of action as well as role in maintaining cellular homeostasis in health and disease.
12. Outlook
While NECMs have been known to exist for decades, only in recent years has a better understanding of their extent as well as cellular ramifications been established. Histone NECMs are now known to occur under normal cellular conditions, but also to substantially accumulate in disease states. Moreover, there are still multiple NECMs that have never been explored in the context of histones or chromatin (Figure 2).
It is still not known whether NECM accumulation directly drives disease or is simply a result or biomarker of an abrogated cellular state. One of the constraints in understanding the causal role of NECMs is that it is difficult to distinguish enzymatic and non-enzymatic modifications (as in the case of acetylation/acylation) or uncouple the general metabolic/toxic effect from the non-enzymatic addition to chromatin of these reactive molecules. One way to address this technical barrier is through the development of methodologies that allow for the direct and site-specific manipulation of histones in vivo.(83,84)
A commonly proposed mechanism for the epigenetic effect of NECMs is their direct competition with enzymatic modifications (Figure 6). Indeed, many NECMs occur on prime regulatory sites such as H3K4, H3K9, H3R8, and H4K12.(40,41) For instance, a counter-correlation between histone glycation and acetylation has been observed.(40,41) Since acetylation is an abundant and dynamic modification, it has been suggested that acetylation acts as a protective cap for histone lysine residues, which can be readily removed in response to a change in cellular signaling.(41) This alteration in histone PTM pattern can affect the binding of specific effector proteins such as readers and remodelers and thus change the overall activity at the site of modification (Figure 6). Since many histone PTM readers rely on multivalency,(85) many times a disruption of a single modification could drastically lower the binding affinity to induce an effect.
Strong evidence for histone-DNA cross-linking also suggests that NECMs can affect cellular transcriptional response by modulating chromatin architecture and dynamics. Indeed, cross-linked DNA in glycated cells shows a substantial effect on the accessibility of transcriptional start sites, which can explain the robust change of the transcriptional program.(40,41) In addition, NECMs may also impair the three-dimensional topology and transcriptional function of chromatin by tampering with the electrostatic interaction between histones and DNA, which are essential for maintaining chromatin structure. This could take place by modifying the histone or DNA charges, as in the case of lysine and arginine glycation. Although poorly characterized, NECMs on the linker histone H1 detected before(86) might alter its binding affinity or capacity to induce chromatin compaction. Finally, NECMs may perturb the binding of architectural protein complexes such as cohesin, condensin, or CTCF, which will alter chromatin configuration. Studies utilizing chromatin conformation capture (3C)-based approaches will be required to examine these possibilities further.
Because NECM levels directly correlate with changes in the cellular microenvironment and information from chromatin is a major upstream determinant of cell fate, it is easy to envision NECMs on histones as a key factor in linking the two. Moreover, since epigenetic changes are heritable, NECMs have potential implications in processes such as embryonic development. Finally, since many exogenous toxicants and drugs either contain reactive electrophiles or are metabolized into them, their potential reactivity toward histones could serve to explain some of their physiological effects. In support, a recent thorough proteomics analysis of nonsteroidal anti-inflammatory drugs and their cellular interactome profile found that the most significant and specific targets are in fact H2A and H2B.(87)
In conclusion, despite a vast and ever-growing knowledge of the notion of enzymatic PTMs forming a “histone code”, that determine the cellular transcriptional program, it is possible that histone NECMs represent a missing chapter that may explain many inherited epigenetic effects. Future efforts will determine the full scope of such phenomena and tease out their direct roles on cellular response to the environment.
Acknowledgements
Work in the David lab is supported by grants R21 DA044767 and CCSG core grant P30 CA008748 from the National Institutes of Health. NA.P. is supported by the NIH Chemistry-Biology Interface training grant T32 GM115327-Tan and the National Science Foundation Graduate Research Fellowship 2017239554. Y.D. is a Josie Robertson Young Investigator. Vectors graphics used in Figure 1 were designed by Freepik.
Author Biographies
Dr. Qingfei Zheng obtained his B.S. from Tsinghua University in 2012, majoring in chemical biology and research training in organic chemistry, epigenetics, and neurobiology. Qingfei then moved to pursue his Ph.D. at Shanghai Institute of Organic Chemistry (SIOC), Chinese Academy of Sciences (CAS) where he focused on the biochemical and genetic basis of microbial metabolite biosynthesis. Qingfei joined the David lab at MSKCC in 2017 as a postdoctoral research fellow. He is now working on noncanonical histone post-translational modifications induced by reactive metabolites or toxins as a new link between metabolic disorders, environmental stress, and epigenetic regulation.

Nicholas A. Prescott obtained his B.S. in biochemistry from the University of Texas at Austin, where he worked in the laboratory of Yan Jessie Zhang. He is currently an NSF Graduate Research Fellow in the Tri-Institutional Program in Chemical Biology at Memorial Sloan Kettering Cancer Center. His research in the David lab focuses on perturbations to epigenetic signaling in disease states.

Igor Maksimovic is a graduate student in the Tri-Institutional Program in Chemical Biology at Memorial Sloan Kettering Cancer Center. He obtained his B.S. in Chemistry from New York University where he focused on the rational design of triazolamer-based peptidomimetic scaffolds in the laboratory of Paramjit Arora. His current research in the David lab is focused on developing chemical probes towards understanding the glycation and deglycation of histones and target these pathways for therapeutics.

Dr. Yael David is an Assistant Member at the Chemical Biology Program at Memorial Sloan Kettering Cancer Center since 2016. Her laboratory is predominantly interested in understanding signaling pathways linking histone modifications and transcriptional regulation. For that, they develop and apply chemical tools towards the site-specific manipulation of histones in vitro and in vivo. Before starting at MSKCC, Yael received her Ph.D. in biochemistry from The Weizmann Institute in Israel and performed her postdoctoral research with Prof. Tom Muir at Princeton University. Yael is a Josie Robertson Young Investigator and was recognized as ACS “Future of Biochemistry” among others.

Footnotes
The authors declare no competing financial interest.
References
- 1.Cloos PA, and Christgau S (2002) Non-enzymatic covalent modifications of proteins: mechanisms, physiological consequences and clinical applications. Matrix Biol. 21 (1), 39–52. [DOI] [PubMed] [Google Scholar]
- 2.DeBerardinis RJ, and Thompson CB (2012) Cellular metabolism and disease: what do metabolic outliers teach us? Cell 148 (6), 1132–1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wellen KE, and Thompson CB (2010) Cellular metabolic stress: Considering how cells respond to nutrient excess. Mol. Cell 40 (2), 323–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Grimsrud PA, Xie H, Griffin TJ, and Bernlohr DA (2008) Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 283 (32), 21837–21841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huebschmann AG, Regensteiner JG, Vlassara H, and Reusch JE (2006) Diabetes and advanced glycoxidation end products. Diabetes Care 29 (6), 1420–1432. [DOI] [PubMed] [Google Scholar]
- 6.Robinson NE (2002) Protein deamidation. Proc. Natl. Acad. Sci U. S. A. 99 (8), 5283–5288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Soskic V, Groebe K, and Schrattenholz A (2008) Nonenzymatic posttranslational protein modifications in ageing. Exp. Gerontol. 43 (4), 247–257. [DOI] [PubMed] [Google Scholar]
- 8.Zhao R, Yang FT, and Alexander DR (2004) An oncogenic tyrosine kinase inhibits DNA repair and DNA-damage-induced Bcl-xL deamidation in T cell transformation. Cancer Cell 5 (1), 37–49. [DOI] [PubMed] [Google Scholar]
- 9.Nilsson MR, Driscoll M, and Raleigh DP (2002) Low levels of asparagine deamidation can have a dramatic effect on aggregation of amyloidogenic peptides: implications for the study of amyloid formation. Protein Sci. 11 (2), 342–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Commerford SL, Carsten AL, and Cronkite EP (1982) Histone turnover within nonproliferating cells. Proc. Natl. Acad. Sci U. S. A. 79 (4), 1163–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Osunsade A, Prescott NA, Hebert JM, Ray DM, Jmeian Y, Lorenz IC, and David Y (2018) A robust method for the purification and characterization of recombinant human histone H1 variants. Biochemistry DOI: 10.1021/acs.biochem.8b01060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jenuwein T, and Allis CD (2001) Translating the histone code. Science 293 (5532), 1074–1080. [DOI] [PubMed] [Google Scholar]
- 13.Bartholomew B (2014) Regulating the chromatin landscape: structural and mechanistic perspectives. Annu. Rev. Biochem. 83, 671–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Karch KR, Denizio JE, Black BE, and Garcia BA (2013) Identification and interrogation of combinatorial histone modifications. Front. Genet. 4, 264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soldi M, Bremang M, and Bonaldi T (2014) Biochemical systems approaches for the analysis of histone modification readout. Biochim. Biophys. Acta 1839 (8), 657–668. [DOI] [PubMed] [Google Scholar]
- 16.Frederiks F, Stulemeijer IJ, Ovaa H, and van Leeuwen F (2011) A modified epigenetics toolbox to study histone modifications on the nucleosome core. Chembiochem 12 (2), 308–313. [DOI] [PubMed] [Google Scholar]
- 17.Chi P, Allis CD, and Wang GG (2010) Covalent histone modifications--miswritten, misinterpreted and mis-erased in human cancers. Nat. Rev. Cancer 10 (7), 457–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schvartzman JM, Thompson CB, and Finley LWS (2018) Metabolic regulation of chromatin modifications and gene expression. J. Cell Biol. 217 (7), 2247–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Etchegaray JP, and Mostoslavsky R (2016) Interplay between Metabolism and Epigenetics: A Nuclear Adaptation to Environmental Changes. Mol. Cell 62 (5) 695–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Teperino R, Schoonjans K, and Auwerx J (2010) Histone methyl transferases and demethylases; can they link metabolism and transcription? Cell Metab. 12 (4) 321–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gut P, and Verdin E (2013) The nexus of chromatin regulation and intermediary metabolism. Nature 502 (7472), 489–498. [DOI] [PubMed] [Google Scholar]
- 22.Harmel R, and Fiedler D (2018) Features and regulation of non-enzymatic post-translational modifications. Nat. Chem. Biol. 14 (3), 244–252. [DOI] [PubMed] [Google Scholar]
- 23.Galligan JJ, and Marnett LJ (2017) Histone adduction and its functional impact on epigenetics. Chem. Res. Toxicol. 30 (1), 376–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Carrozza MJ, Utley RT, Workman JL, and Côté J (2003) The diverse functions of histone acetyltransferase complexes. Trends Genet. 19 (6), 321–329. [DOI] [PubMed] [Google Scholar]
- 25.Paik WK, Pearson D, Lee HW, and Kim S (1970) Nonenzymatic acetylation of histones with acetyl-CoA. Biochim. Biophys. Acta 213 (2), 513–522. [DOI] [PubMed] [Google Scholar]
- 26.Olia AS, Barker K, McCullough CE, Tang HY, Speicher DW, Qiu J, LaBaer J, and Marmorstein R (2015) Nonenzymatic protein acetylation detected by NAPPA protein arrays. ACS Chem. Biol. 10 (9), 2034–2047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kulkarni RA, Worth AJ, Zengeya TT, Shrimp JH, Garlick JM, Roberts AM, Montgomery DC, Sourbier C, Gibbs BK, Mesaros C, Tsai YC, Das S, Chan KC, Zhou M, Andresson T, Weissman AM, Linehan WM, Blair IA, Snyder NW, and Meier JL (2017) Discovering targets of non-enzymatic acylation by thioester reactivity profiling. Cell Chem. Biol. 24 (2), 231–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Simithy J, Sidoli S, Yuan ZF, Coradin M, Bhanu NV, Marchione DM, Klein BJ, Bazilevsky GA, McCullough CE, Magin RS, Kutateladze TG, Snyder NW, Marmorstein R, and Garcia BA (2017) Characterization of histone acylations links chromatin modifications with metabolism. Nat. Commun. 8 (1), 1141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wilson JP, Raghavan AS, Yang YY, Charron G, and Hang HC (2011) Mol. Cell Proteomics 10 (3), M110.001198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, Lu Z, Ye Z, Zhu Q, Wysocka J, Ye Y, Khochbin S, Ren B, and Zhao Y (2011) Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146 (6):1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Li Y, Sabari BR, Panchenko T, Wen H, Zhao D, Guan H, Wan L, Huang H, Tang Z, Zhao Y, Roeder RG, Shi X, Allis CD, and Li H (2016) Molecular Coupling of Histone Crotonylation and Active Transcription by AF9 YEATS Domain. Mol. Cell 62 (2), 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fellows R, Denizot J, Stellato C, Cuomo A, Jain P, Stoyanova E, Balázsi S, Hajnády Z, Liebert A, Kazakevych J, Blackburn H, Corrêa RO, Fachi JL, Sato FT, Ribeiro WR, Ferreira CM, Perée H, Spagnuolo M, Mattiuz R, Matolcsi C, Guedes J, Clark J, Veldhoen M, Bonaldi T, Vinolo MAR, and Varga-Weisz P (2018) Microbiota derived short chain fatty acids promote histone crotonylation in the colon through histone deacetylases. Nat. Commun. 9 (1), 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Huang H, Zhang D, Wang Y, Perez-Neut M, Han Z, Zheng YG, Hao Q, and Zhao Y (2018) Lysine benzoylation is a histone mark regulated by SIRT2. Nat. Commun. 9 (1):3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hellwig M, and Henle T (2014) Baking, ageing, diabetes: a short history of the Maillard reaction. Angew. Chem. Int. Ed. Engl. 53 (39), 10316–10329. [DOI] [PubMed] [Google Scholar]
- 35.Stroo E, Koopman M, Nollen EA, and Mata-Cabana A (2017) Cellular regulation of Amyloid formation in aging and disease. Front. Neurosci. 11, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ott C, Jacobs K, Haucke E, Navarrete Santos A, Grune T, and Simm A (2014) Role of advanced glycation end products in cellular signaling. Redox Biol. 2, 411–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gugliucci A, and Bendayan M (1995) Histones from diabetic rats contain increased levels of advanced glycation end products. Biochem. Biophys. Res. Commun. 212 (1), 56–62. [DOI] [PubMed] [Google Scholar]
- 38.Guedes S, Vitorino R, Domingues MR, Amado F, and Domingues P (2011) Glycation and oxidation of histones H2B and H1: in vitro study and characterization by mass spectrometry. Anal. Bioanal. Chem. 399 (10), 3529–3539. [DOI] [PubMed] [Google Scholar]
- 39.Allaman I, Bélanger M, and Magistretti PJ (2015) Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 9, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Galligan JJ, Wepy JA, Streeter MD, Kingsley PJ, Mitchener MM, Wauchope OR, Beavers WN, Rose KL, Wang T, Spiegel DA, and Marnett LJ (2018) Methylglyoxal-derived posttranslational arginine modifications are abundant histone marks. Proc. Natl. Acad. Sci. U. S. A. 115 (37), 9228–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zheng Q, Omans ND, Leicher R, Osunsade A, Agustinus AS, Finkin-Groner E, D’Ambrosio H, Liu B, Chandarlapaty S, Liu S, and David Y (2019) Reversible histone glycation is associated with disease-related changes in chromatin architecture. Nat. Commun. DOI: 10.1038/s41467-019-09192-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Garaycoechea JI, Crossan GP, Langevin F, Mulderrig L, Louzada S, Yang F, Guilbaud G, Park N, Roerink S, Nik-Zainal S, Stratton MR, and Patel KJ (2018) Nature 553 (7687), 171–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bin P, Huang R, and Zhou X (2017) Oxidation resistance of the sulfur amino acids: methionine and cysteine. Biomed. Res. Int. 2017, 9584932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jena NR (2012) DNA damage by reactive species: Mechanisms, mutation and repair. J. Biosci. 37 (3), 503–517. [DOI] [PubMed] [Google Scholar]
- 45.Bai J, Zhang Y, Xi Z, Greenberg MM, and Zhou C (2018) Oxidation of 8-oxo-7,8-dihydro-2’-deoxyguanosine leads to substantial DNA-histone cross-links within nucleosome core particles. Chem. Res. Toxicol. 31 (12), 1364–1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kietzmann T, Petry A, Shvetsova A, Gerhold JM, and Görlach A (2017) The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 174 (12), 1533–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lobo V, Patil A, Phatak A, and Chandra N (2010) Free radicals, antioxidants and functional foods: Impact on human health. Pharmacogn. Rev. 4 (8), 118–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rossaint J, Margraf A, and Zarbock A (2018) Role of Platelets in Leukocyte Recruitment and Resolution of Inflammation. Front. Immunol. 9, 2712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.García-Giménez JL, Òlaso G, Hake SB, Bönisch C, Wiedemann SM, Markovic J, Dasí F, Gimeno A, Pérez-Quilis C, Palacios O, Capdevila M, Viña J, and Pallardó FV (2013) Histone H3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Antioxid. Redox Signal. 19 (12), 1305–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hake SB, and Allis CD (2006) Histone H3 variants and their potential role in indexing mammalian genomes: the “H3 barcode hypothesis”. Proc. Natl. Acad. Sci. U. S. A. 103 (17), 6428–6435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galligan JJ, Rose KL, Beavers WN, Hill S, Tallman KA, Tansey WP, and Marnett LJ (2014) J. Am. Chem. Soc. 136 (34), 11864–11866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cui Y, Li X, Lin J, Hao Q, and Li XD (2017) Histone Ketoamide Adduction by 4-Oxo-2-nonenal Is a Reversible Posttranslational Modification Regulated by Sirt2. ACS Chem. Biol. 12 (1), 47–51. [DOI] [PubMed] [Google Scholar]
- 53.Carrier EJ, Zagol-Ikapitte I, Amarnath V, Boutaud O, and Oates JA (2014) Levuglandin forms adducts with histone H4 in a cyclooxygenase-2-dependent manner, altering its interaction with DNA. Biochemistry 53 (15) 2436–2441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fang L, Chen D, Yu C, Li H, Brocato J, Huang L, and Jin C (2016) Mechanisms underlying acrolein-mediated inhibition of chromatin assembly. Mol. Cell. Biol. 36 (23), 2995–3008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Burgos-Barragan G, Wit N, Meiser J, Dingler FA, Pietzke M, Mulderrig L, Pontel LB, Rosado IV, Brewer TF, Cordell RL, Monks PS, Chang CJ, Vazquez A, and Patel KJ (2017) Mammals divert endogenous genotoxic formaldehyde into one-carbon metabolism. Nature 548 (7669), 549–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Nakamura J, Shimomoto T, Collins LB, Holley DW, Zhang Z, Barbee JM, Sharma V, Tian X, Kondo T, Uchida K, Yi X, Perkins DO, Willis MS, Gold A, and Bultman SJ (2017) Evidence that endogenous formaldehyde produces immunogenic and atherogenic adduct epitopes. Sci. Rep. 7 (1), 10787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Trézl L, Rusznák I, Tyihák E, Szarvas T, and Szende B (1983) Spontaneous N epsilon-methylation and N epsilon-formylation reactions between L-lysine and formaldehyde inhibited by L-ascorbic acid. Biochem. J. 214 (2), 289–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Jiang T, Zhou X, Taghizadeh K, Dong M, and Dedon PC (2007) N-formylation of lysine in histone proteins as a secondary modification arising from oxidative DNA damage. Proc. Natl. Acad. Sci. U. S. A. 104 (1), 60–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Edrissi B, Taghizadeh K, and Dedon PC (2013) Quantitative analysis of histone modifications: formaldehyde is a source of pathological n(6)-formyllysine that is refractory to histone deacetylases. PLoS Genet. 9 (2), e1003328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wiśniewski JR, Zougman A, and Mann M (2008) Nε-formylation of lysine is a widespread post-translational modification of nuclear proteins occurring at residues involved in regulation of chromatin function. Nucleic Acids Res. 36 (2), 570–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Sharma GS, Kumar T, and Singh LR (2014) N-homocysteinylation induces different structural and functional consequences on acidic and basic proteins. PLoS One 9 (12), e116386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Xu L, Chen J, Gao J, Yu H, and Yang P (2015) Crosstalk of homocysteinylation, methylation and acetylation on histone H3. PLoS One 9 (12), e116386. [DOI] [PubMed] [Google Scholar]
- 63.Zhang Q, Bai B, Mei X, Wan C, Cao H, Dan Li., Wang S, Zhang M, Wang Z, Wu J, Wang H, Huo J, Ding G, Zhao J, Xie Q, Wang L, Qiu Z, Zhao S, and Zhang T (2018) Elevated H3K79 homocysteinylation causes abnormal gene expression during neural development and subsequent neural tube defects. Nat. Commun. 9 (1), 3436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Catak S, Monard G, Aviyente V, and Ruiz-López MF (2009) Deamidation of asparagine residues: direct hydrolysis versus succinimide-mediated deamidation mechanisms. J. Phys. Chem. A 113 (6), 1111–1120. [DOI] [PubMed] [Google Scholar]
- 65.Robinson NE, and Robinson AB (2001) Deamidation of human proteins. Proc. Natl. Acad. Sci. U. S. A. 98 (22), 12409–12413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson NE, and Robinson AB (2001) Molecular clocks. Proc. Natl. Acad. Sci. U. S. A. 98 (3), 944–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.David CL, Szumlanski CL, DeVry CG, Park-Hah JO, Clarke S, Weinshilboum RM, and Aswad DW (1997) Human erythrocyte protein L-isoaspartyl methyltransferase: heritability of basal activity and genetic polymorphism for thermal stability. Arch. Biochem. Biophys. 346 (2), 277–286. [DOI] [PubMed] [Google Scholar]
- 68.Corti A, and Curnis F (2011) Isoaspartate-dependent molecular switches for integrin-ligand recognition. J. Cell Sci. 124 (Pt 4), 515–522. [DOI] [PubMed] [Google Scholar]
- 69.Young AL, Carter WG, Doyle HA, Mamula MJ, and Aswad DW (2001) Structural integrity of histone H2B in vivo requires the activity of protein L-isoaspartate O-methyltransferase, a putative protein repair enzyme. J. Biol. Chem. 276 (40), 37161–37165. [DOI] [PubMed] [Google Scholar]
- 70.Lindner H, Sarg B, Hoertnagl B, and Helliger W (1998) The microheterogeneity of the mammalian H1(0) histone. Evidence for an age-dependent deamidation. J. Biol. Chem. 273 (21), 13324–13330. [DOI] [PubMed] [Google Scholar]
- 71.Goggin M, Swenberg JA, Walker VE, and Tretyakova N (2009) Molecular dosimetry of 1,2,3,4-diepoxybutane-induced DNA-DNA cross-links in B6C3F1 mice and F344 rats exposed to 1,3-butadiene by inhalation. Cancer Res. 69 (6), 2479–2486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Michaelson-Richie ED, Loeber RL, Codreanu SG, Ming X, Liebler DC, Campbell C, and Tretyakova NY (2010) DNA-protein cross-linking by 1,2,3,4-diepoxybutane. J. Proteome Res. 9 (9), 4356–4367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Gherezghiher TB, Ming X, Villalta P, Campbell C, and Tretyakova NY (2013) 1,2,3,4-Diepoxybutane-induced DNA-protein cross-linking in human fibrosarcoma (HT1080) cells. J. Proteome Res. 12 (5), 2151–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang K, Park D, Tretyakova NY, and Greenberg MM (2018) Histone tails decrease N7-methyl-2’-deoxyguanosine depurination and yield DNA-protein cross-links in nucleosome core particles and cells. Proc. Natl. Acad. Sci. U. S. A. 115 (48), E11212–E11220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Voitkun V, and Zhitkovich A (1999) Analysis of DNA-protein crosslinking activity of malondialdehyde in vitro. Mutat. Res. 424 (1–2), 97–106. [DOI] [PubMed] [Google Scholar]
- 76.Raiber EA, Portella G, Martínez Cuesta S, Hardisty R, Murat P, Li Z, Iurlaro M, Dean W, Spindel J, Beraldi D, Liu Z, Dawson MA, Reik W, and Balasubramanian S (2018) 5-Formylcytosine organizes nucleosomes and forms Schiff base interactions with histones in mouse embryonic stem cells. Nat. Chem. 10 (12), 1258–1266. [DOI] [PubMed] [Google Scholar]
- 77.Tretyakova NY, Groehler A 4th, and Ji S (2015) DNA-protein cross-links: Formation, structural identities, and biological outcomes. Acc. Chem. Res. 48 (6), 1631–1644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kalra J, Balion C, Massey KL, and Laxdal VA (1988) Regulation of carnosine metabolism: the subcellular localization of carnosinase in liver. Clin. Biochem. 21 (5), 315–318. [DOI] [PubMed] [Google Scholar]
- 79.Maessen DE, Stehouwer CD, and Schalkwijk CG (2015) The role of methylglyoxal and the glyoxalase system in diabetes and other age-related diseases. Clin. Sci. (Lond) 128 (12), 839–861. [DOI] [PubMed] [Google Scholar]
- 80.Seto E, and Yoshida M (2014) Erasers of histone acetylation: the histone deacetylase enzymes. Cold Spring Harb. Perspect. Biol. 6 (4), a018713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Akter S, Fu L, Jung Y, Conte ML, Lawson JR, Lowther WT, Sun R, Liu K, Yang J, and Carroll KS (2018) Chemical proteomics reveals new targets of cysteine sulfinic acid reductase. Nat. Chem. Biol. 14 (11), 995–1004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ariga H, Takahashi-Niki K, Kato I, Maita H, Niki T, and Iguchi-Ariga SM (2013) Neuroprotective function of DJ-1 in Parkinson’s disease. Oxid. Med. Cell Longev. 2013, 683920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.David Y, Vila-Perelló M, Verma S, and Muir TW (2015) Chemical tagging and customizing of cellular chromatin states using ultrafast trans-splicing inteins. Nat. Chem. 7 (5), 394–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.David Y, and Muir TW (2017) Emerging chemistry strategies for engineering native chromatin. J. Am. Chem. Soc. 139 (27), 9090–9096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Ruthenburg AJ, Li H, Patel DJ, and Allis CD (2007) Multivalent engagement of chromatin modifications by linked binding modules. Nat. Rev. Mol. Cell. Biol. 8 (12), 983–994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Pashikanti S, Boissonneault GA, and Cervantes-Laurean D (2011) Ex vivo detection of histone H1 modified with advanced glycation end products. Free Radic. Biol. Med. 50 (10), 1410–1416. [DOI] [PubMed] [Google Scholar]
- 87.Gao J, Mfuh A, Amako Y, and Woo CM (2018) Small molecule interactome mapping by photoaffinity labeling reveals binding site hotspots for the NSAIDs. J. Am. Chem. Soc. 140 (12), 4259–4268. [DOI] [PubMed] [Google Scholar]