

Sulfide-to-metal electroreduction likely harnessed the reducing power of hydrothermal systems for the autotrophic origin of life.
Abstract
One of the most plausible scenarios of the origin of life assumes the preceding prebiotic autotrophic metabolism in sulfide-rich hydrothermal vent environments. However, geochemical mechanisms to harness the reductive power provided by hydrothermal systems remain to be elucidated. Here, we show that, under a geoelectrochemical condition realizable in the early ocean hydrothermal systems, several metal sulfides (FeS, Ag2S, CuS, and PbS) undergo hour- to day-scale conversion to the corresponding metals at ≤−0.7 V (versus the standard hydrogen electrode). The electrochemically produced FeS-Fe0 assemblage promoted various reactions including certain steps in the reductive tricarboxylic acid cycle with efficiencies far superior to those due to pure FeS. The threshold potential is readily generated in the H2-rich alkaline hydrothermal systems that were probably ubiquitous on the Hadean seafloor. Thus, widespread metal production and metal-sustained primordial metabolism were likely to occur as a natural consequence of the active hydrothermal processes on the Hadean Earth.
INTRODUCTION
A recent in situ electrochemical survey of the Okinawa Trough hydrothermal fields observed spontaneous generation of electricity widespread in deep-sea vent chimneys and mineral deposits (1). Field and laboratory investigations have verified that electrons are catalytically provided by the oxidation of reductive hydrothermal fluid chemicals (for example, H2S and H2) at the fluid-mineral interface and are transported to the mineral-seawater interface via electrically conductive sulfide rocks across the redox gap between the fluid and seawater (Fig. 1A) (1, 2). Considering the ubiquity of sulfide deposits in the present-day and the early ocean hydrothermal environments (3), together with ever-existing redox disequilibrium between Earth’s surface and the interior (4), the geoelectrochemical systems must have occurred extensively on the seafloor throughout Earth’s history (5). We have previously demonstrated efficient CO2 electroreduction to carbon monoxide (CO) on some metal sulfides (for example, CdS and Ag2S) simulating early ocean hydrothermal vent environments (6). The reaction conditions favorable for the CO production were consistent with the conditions assumed in Wächtershäuser’s abiotic organic synthesis starting from CO (7–9); thereby, the early ocean alkaline hydrothermal systems were suggested to have favored the prebiotic CO2 fixation and the subsequent evolution of primordial metabolism toward the origin of life (6).
Fig. 1. Geoelectrochemical metal production in the early ocean alkaline hydrothermal systems.
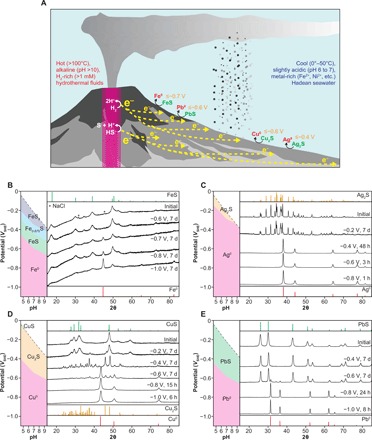
(A) At the vent-seawater interface, metal sulfides precipitated through mixing between the ancient seawater rich in metal cations (for example, Fe2+) and the alkaline hydrothermal fluid containing HS− were exposed to a negative electric potential and were electroreduced to the corresponding metals (for example, Fe0) with the reactivity depending on the potential and the nature of sulfides. (B to E) X-ray diffraction (XRD) patterns of FeS, Ag2S, CuS, and PbS before and after the electrolysis, respectively. The small peaks noted by asterisks (*) in (B) represent NaCl signals. The XRD data for the other sulfides are presented in fig. S3. The potential/pH diagrams of the relevant metal-sulfide systems are shown in the left columns. The colors represent the thermodynamically predicted stability regions of metals (red) and sulfides (green, orange, and blue refer to the sulfides with the metal/sulfur ratio of 1, >1, and <1, respectively) in the aqueous condition examined in the present study.
Most anabolic pathways in extant organisms go through five universal intermediates (acetyl–coenzyme A, pyruvate, oxaloacetate, succinate, and α-ketoglutarate) as starting points for the synthesis of entire metabolome (10). Autotrophic theories of the origin of life postulate that these precursors were produced nonenzymatically via an incomplete version of the reductive tricarboxylic acid (rTCA) cycle in sulfide-rich hydrothermal environments (11). Recent biochemical studies even suggested that a mixotrophic bidirectional TCA cycle could be the most ancient enzymatic metabolism (12). Although possible geochemical mechanisms to harness the disequilibrium energy provided by hydrothermal systems have been discussed over the past three decades (11, 13), experimental evidences that support the scenario remain meager.
Two recent studies suggest that metals are favorable for the nonenzymatic protometabolic reactions (14, 15). Muchowska et al. (14) have shown that 6 of the 11 steps in the TCA cycle are promoted in the reductive direction by metallic iron (Fe0) in the presence of Zn2+ and Cr3+. The same group also reported that zerovalent metals (Fe0, Co0, and Ni0) selectively reduced CO2 to acetate and pyruvate (15). Native metals including Fe0, Ag0, Au0, Cu0, and Ni0 are produced sustainably through hydrothermal alterations of ultramafic rocks (for example, olivine) under deep, high-temperature, and H2-rich conditions (16–19). Fe0 may also be generated by high-temperature (>1000°C) alterations of basaltic iron in the presence of organic carbon as a reducing agent (20) and by the extraterrestrial input of meteorite-derived reducing power (21). However, the first process is unlikely to have contributed substantially to prebiotic chemistry because of the difficulty in fluid-mediated material transport from the hot reaction zone to the early ocean floor. The latter two processes cannot provide a continuous supply of fresh Fe0 at a certain surface environment. Furthermore, because the oxidation state of the Hadean upper mantle has been estimated to be similar to the present state (22), the sustained availability of reactive native metals in the Hadean environment still poses a problem.
Iron was a major dissolved metal species in the Hadean ocean with a valence state of +2 (23). By forming a complex with sulfide anion (S2−), Fe2+ undergoes a geometric change in the electron orbital from octahedral to tetrahedral that considerably facilitates a 2e− uptake of Fe2+ to Fe0 (24). A voltammetric analysis of the Fe2+/Fe0 redox couple in the presence of HS− observed a reductive signal due to Fe2+ in iron sulfide complexes at around −0.85 V [versus standard hydrogen electrode (SHE)] and a signal due to free Fe2+ at −1.15 to −1.2 V under the present-day seawater condition at pH 8.1 and 25°C (24). The −0.85 V is close to the FeS/Fe0 equilibrium potential in the examined seawater condition (−0.68 V versus SHE) and corresponds to a geoelectrochemical potential generable in moderately active hydrothermal settings (for example, 1 mmol kg−1 H2 has the oxidation potential of −0.85 V versus SHE at pH 11.1, 150°C, and 500 bar). Even less negative potentials are expected to suffice for the FeS-to-Fe0 electroreduction at lower pH, owing to the positive shift of FeS/Fe0 redox equilibrium with decreasing pH (Fig. 1B). Considering the slightly acidic (pH 6 to 7) (25) and Fe2+-rich (10−2 to 10−4 M) (23) character of ancient seawater, the precipitation of seawater Fe2+ as sulfide (FeS) and its subsequent electroreduction to Fe0 must have occurred in alkaline hydrothermal vent environments on the early ocean floor (Fig. 1A). FeS can promote, albeit only slightly, certain steps in the rTCA cycle at neutral pH (26) and the keto acid–to–amino acid reductive amination at alkaline pH (8 to 10) (27). Thus, the geoelectrochemical processes should have facilitated FeS to drive organic reactions and enabled the operation of primordial metabolism under aqueous conditions consistent with the early ocean alkaline hydrothermal systems.
Here, we present a laboratory demonstration of day-scale electroreduction of FeS to Fe0 under a simulated early ocean condition and show that the resultant FeS-Fe0 assemblage considerably promotes some prebiotically important reactions at neutral to alkaline pH. To the best of our knowledge, this study provides the first experimental evidence of the solid-phase FeS-to-Fe0 electroreduction (28), although formation of amalgamated iron from dissolved/colloidal FeS species has been characterized by voltammetric analyses with a mercury working electrode (24, 29). We also examined the reduction behaviors of various metal sulfides (Ag2S, CdS, CoS, CuS, MnS, MoS2, NiS, PbS, WS2, and ZnS). These metals have been observed both in the present-day and Paleozoic hydrothermal sulfide deposits (3, 30) and have been inferred to have played key roles in prebiotic chemistry (30). In addition to FeS, Ag2S, CuS, and PbS exhibited even more rapid conversions to the corresponding metals under geochemically feasible potential conditions. Ag0, Cu0, and Pb0 are known to work as catalysts or reducing agents for various organic reactions (31–33). Thus, taking the reactivity of transition metals into account, we propose a new scheme, electroreduction of metal sulfides, to provide reductive energy and catalytic surfaces to support the autotrophic origin of life in the Hadean ocean floor.
RESULTS
The metal sulfide electrolysis was conducted in an H-type cell that had two compartments separated by a proton exchange membrane, with a Ag/AgCl reference electrode and a carbon working electrode placed at one side and a platinum counter electrode at the other side (fig. S1). Laboratory-prepared (Ag2S, CdS, CoS, CuS, FeS, MnS, NiS, PbS, and ZnS) or commercially obtained (MoS2 and WS2) sulfides were deposited on the carbon electrode and exposed to a constant potential for up to 7 days in 100 mM NaCl aqueous solution saturated with 1 atm of CO2 containing 4 parts per million (ppm) of H2S at pH 6 ± 0.25 and room temperature (23° to 25°C). Although the ionic strength and pressure conditions adopted in our electrochemical experiment are likely different from the ancient deep-sea hydrothermal settings (34, 35), a thermodynamic calculation indicates that these differences have no significant influence on the sulfide/metal equilibrium potentials (see Materials and Methods). The sulfide samples were then separated from the supernatant solution, dried under vacuum, and measured for x-ray diffraction (XRD) pattern.
The Fe0 formation from FeS (FeS + 2H+ + 2e− → Fe0 + H2S) was demonstrated after the 7-day experiment at −1.0 V (versus SHE) (Fig. 1B); broad but recognizable signals appeared around 44.7°, 65.0°, and 82.3° that are assigned to the α-form of Fe0 with a body-centered cubic structure while signal intensities due to mackinawite decreased. The Fe0 signals grew in proportion to the duration of −1.0-V electrolysis (fig. S2). The highest 44.7° peak was also observed at −0.8 and −0.7 V (versus SHE), whereas no appreciable signal except for those of mackinawite was observed at −0.6 V (versus SHE). Metal was also identified from Ag2S, CuS, and PbS after the electrolysis (Fig. 1, C to E), although the other sulfides exhibited no metal XRD signal under the examined conditions (≥−1.0 V versus SHE, ≤7 days) (Fig. 2 and fig. S3). Nearly complete reductions were achieved within 48 hours at ≤−0.4, ≤−0.8, and ≤−0.8 V (versus SHE) for Ag2S, CuS, and PbS, respectively (Fig. 1, C to E). Detectable amounts of Cu0 and Pb0 were produced at −0.6 V (versus SHE) after longer experimental duration (7 days) but not at −0.4 V (versus SHE). The threshold potentials of the sulfide-to-metal electroreduction are close to the respective equilibrium potentials (−0.55, −0.23, −0.49, and −0.53 V versus SHE for iron, silver, copper, and lead, respectively), indicating existence of low overpotentials less than 0.2 V. Note that the total charges built up during the electrolysis were, in many cases, much more than those required for the metal productions (fig. S4). Electrons were consumed largely by H2 generation (2H+ + 2e− → H2) except for the case of Ag2S with an applied potential of ≤− 0.9 V (versus SHE), where CO evolved with up to 30% Faraday efficiency (CO2 + 2H+ + 2e− → CO + H2O) (6). The observed rapid Ag2S-to-Ag0 electroreduction under these potential conditions (Fig. 1C) suggests that the CO production was promoted by Ag0 rather than by Ag2S (36).
Fig. 2. Summary of the electroreduction behaviors of metal sulfides under a simulated early ocean condition.
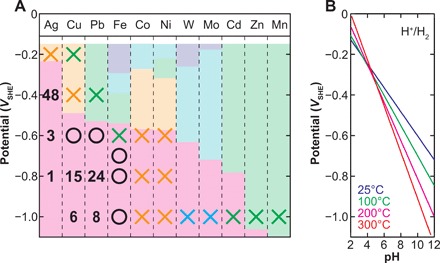
(A) A circle located at a potential and a metal indicates that a detectable amount of the metal was generated during the 7-day electrolysis with the potential. Analogously, crosses show that no metal production was observed in our experiment. The color of the cross denotes the dominant sulfide stoichiometry seen after the 7-day electrolysis (see Fig. 1 legend for the color convention). The numbers indicate the duration in hours of experiments required by the complete sulfide-to-metal conversions. (B) A redox calculation for 1 mmol kg−1 H2 [H2(aq) → 2H+ + 2e−] as a function of temperature and pH indicates a geoelectrochemically feasible potential range of 0 to −1.1 V versus SHE.
To examine whether the electrochemically reduced FeS [hereafter described as FeS_PERM (FeS partially electroreduced to metal)] facilitates abiotic reactions of prebiotic importance, we electrolyzed FeS at −0.7 V (versus SHE) for 7 days, dried under vacuum, sealed in a serum bottle with an aqueous solution of organic or inorganic chemicals, and agitated at room temperature (23° to 25°C) for 2 days without externally imposing electric potential. Solution pH was buffered at near neutral (pH 6.5) by adding CO2 gas into the bottle for reduction experiments of oxaloacetate, fumarate, and nitrate (5 mM for each) while maintained at pH 9.6 by ammonia (0.5 M) for the reductive aminations of glyoxylate, pyruvate, oxaloacetate, and α-ketoglutarate (5 mM for each). The electroreduction markedly enhanced the FeS’s capability of facilitating target reactions (Fig. 3). Around 37% of oxaloacetate was converted to malate in the presence of the FeS_PERM, while pyruvate and acetate were the sole identified products when as-prepared FeS was used (fig. S9). Malate was not observed when a commercially obtained pure Fe0 was used instead of the FeS_PERM (fig. S9) because pure Fe0 can promote the reaction only in highly acidic solutions (1 M HCl) (14). Another abiotic mechanism for the oxaloacetate-to-malate conversion proposed so far is ultraviolet (UV)–induced photoelectroreduction on ZnS colloidal semiconductor (37) that is unavailable in deep-sea environments. Recently, both the oxidative and reductive directions of TCA cycle were identified to be driven nonenzymatically using sulfate radicals generated from peroxydisulfate homolysis (S2O82− → 2SO4·−) (38). The presence of these highly oxidized sulfur species with a concentration sufficient to sustain protometabolism on the Hadean Earth is unlikely.
Fig. 3. Nonenzymatic reactions in the presence of as-prepared FeS (blue) and the FeS electrochemically reduced at −0.7 V (versus SHE) for 7 days (FeS_PERM) (red).
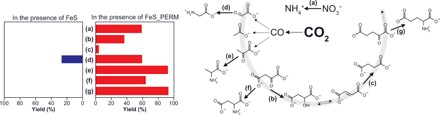
The yields were calculated relative to the initial amounts of starting materials of respective reactions. A full dataset for the identified and quantified products are presented in fig. S9 and tabulated in table S1 together with the results under the following conditions: with H2 gas, with FeCl2, with pure Fe0, and without reductant. The right diagram shows the reactions examined in the present study (a to g) and those demonstrated previously by simulating hydrothermal vent environments on the early Earth [CO2 → CO in (6) and CO → C2 and C3 compounds in (7, 9)].
Malate then links to fumarate in the rTCA cycle through reversible dehydration catalyzed by Zn2+ or Cr3+ (14). The fumarate reduction to succinate was poorly assisted by the FeS_PERM (yield, 4%; Fig. 3), but at 80°C, under otherwise identical condition, 22% of fumarate converted to succinate (fig. S10). Similar reduction behaviors as observed for the C4 compounds are likely seen in the C6 intermediates of the TCA cycle (from oxalosuccinate to citrate) (14). We did not examine its possibility because the C6 steps may be a biological invention retrofitted with a preceding incomplete rTCA cycle (39, 40).
At alkaline pH (9.6), pyruvate and α-ketoglutarate reacted nearly completely with ammonia to generate alanine and glutamate, respectively, with the aid of FeS_PERM (Fig. 3). Ammonia can be made by the FeS_PERM-promoted reduction of nitrate (Fig. 3). Nitrate possibly dissolved in the early ocean with micro- to millimolar concentrations (41). Alternatively, ammonia could have been provided directly from fluids from hydrothermal vents as a consequence of nitrogen dissolution into magma ocean during Earth’s formation process, followed by water-rock interactions beneath the ocean floor (42). The FeS_PERM also led to excellent yields in the glycine and aspartate syntheses from glyoxylate and oxaloacetate (59 and 64%, respectively). The present study succeeded in driving the oxaloacetate-to-aspartate conversion without UV irradiation using ammonia as a nitrogen source (27). Although this reaction was recently demonstrated in the presence of hydroxylamine and pure Fe0 at 100°C (43), the availability of hydroxylamine on the Hadean Earth is unclear. Pure Fe0 also promoted the aspartate formation from oxaloacetate and ammonia (fig. S9). However, pyruvate and its derived amino acid alanine were formed in comparable amounts via oxaloacetate decarboxylation. Consequently, the selectivity for aspartate synthesis by pure Fe0 was lower than by the FeS_PERM.
We also examined the reductive amination of four keto acids (1.25 mM for each) in one serum bottle under the condition described above. All corresponding amino acids were generated with the yields higher than 50% (fig. S11). Alanine and glutamate were formed preferentially over the others.
DISCUSSION
Our experiments revealed that electrolysis is an effective process of producing Fe0 from FeS (Fig. 1B) and markedly enhances FeS’s capability of driving certain reactions (Fig. 3). The selectivities for target reactions controlled by FeS_PERM are comparable with, or even superior to, those by pure Fe0 in all examined steps except for the fumarate reduction (fig. S9). Our prepared FeS has a poorly crystalline structure (Fig. 1B) possibly reflecting the generation and aggregation of nanoparticle iron sulfide precursors that precede the mackinawite precipitation (44). The metastable character and high specific surface area (28) should facilitate the FeS-to-Fe0 electroreduction and the FeS_PERM-promoted reductions of organic/inorganic molecules. It is also conceivable that the electrochemically generated Fe0 is present not only as a poorly crystalline aggregate identifiable by XRD (Fig. 1B) but also as a molecular-level admixture with FeS. The Fe0-FeS interface may provide a synergy between the Fe0’s reducing power and the FeS’s capability of catalyzing electron uptake of chemisorbed molecules (11), thereby boosting the efficiencies of target reactions.
At the vent-seawater interface in early ocean hydrothermal systems, metal sulfide precipitates must have been exposed to a sustained negative electric potential by the geoelectrochemical processes (Fig. 1A). The potential level required for the FeS-to-Fe0 electroreduction (≤−0.7 V versus SHE) is attainable under moderately hot, H2-rich, and alkaline hydrothermal conditions (for example, at 100°C and pH 10 in the presence of 1 mmol kg−1 H2), as observed even in the present-day hydrothermal systems (45). Alkaline hydrothermal fluids generate a steep pH gradient, as well as highly negative potentials, across the metal/mineral precipitates where a wide range of pH conditions are created, favoring both certain steps in the rTCA cycle (pH 6.5) and in the keto acid–to–amino acid reductive amination (pH 9.6). The vent precipitates also provide three-dimensional microcompartments within which organic products could have been retained (46). The Fe0 formation proceeds with the time scale much shorter than the decay time scale of most rTCA intermediates and amino acids under ancient oceanic temperatures [0° to 50°C; (25)] (47–50). Although oxaloacetate decarboxylates with a half-life of hours (51), it can quickly convert to more stable compounds (malate and aspartate) with the aid of FeS_PERM. Together, coupling of oxidation of metals generated by sulfide reduction with the reductive organic reactions can be a very potent general means to harness the reducing power of hydrothermal systems. Considering much greater thermal convection (52) and wider and denser distribution of ultramafic rocks in the Hadean ocean crust (53) than today, widespread metal-rich conditions containing Fe0, Ag0, Cu0, and Pb0 and their sustained primordial metabolism were likely to have occurred on the Hadean ocean floor.
The letters from Moran and colleagues (14, 15) suggest that zerovalent metals were advantageous to the protometabolic reactions. However, we must point out that a lot of future work is needed to complete the autotrophic scenario of the origin of life. In Muchowska et al. (14), all experiments were conducted in highly acidic solutions (1 M HCl or 1 M H2SO4) to couple the Fe0 oxidation with the reductions of TCA intermediates. This acidic condition required hydrazine (N2H4) as a nitrogen source for the reductive amination of α-keto acids to the corresponding α-amino acids. The exploration of more realistic reaction settings is definitely needed. Varma et al. (15) reported CO2 conversion to several C2 to C3 compounds (for example, acetate and pyruvate) on commercial Fe0, but we did not observe organic molecule formation from CO2 in the presence of FeS_PERM even at elevated temperatures (but ≤100°C) in a range of experimental duration ≤7 days. The difference in the CO2 reactivity may be due to insufficient FeS-to-Fe0 electroreduction that prevents CO2 from binding to Fe atoms with a surface density enough to make C─C bonds between adsorbed CO2 molecules (15). An alternative mechanism of abiotic CO2 fixation in the deep-sea hydrothermal vent environments is the CO2 electroreduction to CO on CdS at <0.8 V (versus SHE) (6) and the subsequent CO fixations, as demonstrated by Huber and Wächtershäuser (7–9). The CO2 activation may also be achieved on the FeS_PERM by doping impurities (for example, NiFe3S4 by Ni doping). Multielement-containing FeS and Fe0 typically exhibit higher CO2 reduction efficiencies than the pure counterparts by offering reaction sites for intermediates to be stabilized (54). This strategy is adopted in various biological CO2 fixation pathways (4). Further investigation of the optimal composition of iron and other metal sulfides that enhances the C─C bond formation based on the extant metabolic strategy is desirable. Catalytic and reductive functions of Ag2S_PERM, CuS_PERM, and PbS_PERM in the protometabolic reactions must also be addressed to elucidate the role of hydrothermal systems in the origin and early evolution of life.
The hour- to day-scale electroreduction of several sulfides to the corresponding metals (Fe0, Ag0, Cu0, and Pb0) and the capability of the resultant FeS-Fe0 assemblage for driving abiotic reactions demonstrated in the present study may have a great impact not only on the emergence of life on Earth but also possible lives on exoplanetary systems. Ongoing ground- and space-based observations have discovered thousands of exoplanetary systems (55), among which two systems were identified to host Earth-size rocky planets that potentially maintain liquid water on their surfaces (56, 57). Deep-sea hydrothermal systems have been proposed to be one of the most plausible cradles of life on Earth and other rocky planets (4), while the preceding key geo- and astrochemical processes remain largely unknown. The precipitation and subsequent electroreduction of sulfides in hydrothermal mixing zones (Fig. 1A) can be predicted from the chemical compositions of hydrothermal fluids and seawater, all of which are also predictable from the physical and chemical conditions of water-rock interaction beneath the ocean floor. Thus, understanding of the electrochemical behaviors of sulfides and their prebiotic/biotic functions must be a highly interesting direction of extraterrestrial life studies.
MATERIALS AND METHODS
Preparation of metal sulfides
All metal sulfides except for WS2 and MoS2 were prepared by a drop-wise addition of 100 mM Na2S (or 50 mM Na2S for the case of Ag2S) into a 100 mM aqueous solution of the corresponding metal chloride (AgCl, CdCl2, CoCl2, CuCl2, FeCl2, MnCl2, NiCl2, PbCl2, or ZnCl2) under vigorous stirring with a final volume ratio of 1:1. Solid precipitates were then separated from the supernatant solution by centrifugation and were dried under vacuum. To prevent oxidation by atmospheric O2, the sample preparation was conducted in a globe box filled with N2 gas (>99.99995%), with 4% H2 being added (the COY system). All chemicals were purchased from Wako as reagent grade. Deaerated Milli-Q water (18.2 megohms) was used as the solvent. WS2 and MoS2 were obtained from Sigma-Aldrich (99%, ~90 nm) and were used without further purification.
Although our prepared copper sulfide is covellite (CuS), chalcosite (Cu2S) was likely more prevalent in the early ocean hydrothermal vent environments because the Hadean seawater is expected to have had the redox potential (Eh = −0.27 V versus SHE; see below) lower than the Cu2+/Cu+ equilibrium potential (0.15 V versus SHE). However, CuS electroreduction to Cu0 always occurred via Cu2S (fig. S12). The activation energy barrier of the CuS-to-Cu2S conversion is low because the reaction nearly completed at −0.4 V (versus SHE) and occurred even at −0.2 V (versus SHE) by the 7-day electrolysis (Fig. 1E). Thus, the use of CuS instead of Cu2S does not influence the observed threshold potential of the Cu0 formation and hence does not alter the conclusion of the present study.
Electrochemical experiments
The Hadean ocean was rich in Fe2+ (10−2 to 10−4 M) (23), mildly cool (0° to 50°C) (25), and slightly acidic (pH 6 to 7) (25) under CO2-rich (0.1 to 1 bar) (42, 58), anoxic but H2 poor (<0.01 bar) (22, 59) atmosphere. The seawater volume was likely larger than the present level (60). Shibuya et al. (34) assumed a 5-km water depth (500 bar) in their thermodynamic modeling of the Hadean deep-sea hydrothermal processes. Seawater H2S concentration has not been determined precisely, but setting a saturation with respect to pyrite (FeS2) leads to a provisional value of 0.1 to 1 μmol kg−1 (35). For example, an equilibrium between FeS2 and 10−4 mol kg−1 Fe2+ (Fe2+ + 2HS− → FeS2 + H2) in deep seawater at pH 6, 25°C, 500 bar, and with the ionic strength of 0.5 in the presence of 0.001 bar of atmospheric H2 results in the dissolved H2S plus HS− concentrations of 0.5 μmol kg−1 (see “Thermodynamic calculation” section below).
To simulate the metal sulfide electroreduction under the early ocean condition summarized above, 100 to 400 mg of metal sulfide was deposited on the carbon working electrode (5.7 cm2; fig. S1), immersed in a deaerated aqueous solution of 100 mM NaCl and 0 to 15 mM NaOH (60 ml), and exposed to a flow of CO2 gas (>99.995%) containing 4 ppm of H2S (20 ml min−1). No binder material was used in the sulfide deposition to avoid potential adverse influences. The CO2 and H2S gas buffered the solution pH to slightly acidic pH (6 ± 0.25) and supplied H2S and HS− into the solution with the equilibrium total concentration of 0.5 μmol kg−1. The pH value was monitored by a portable pH meter (Seven2Go Pro, Mettler Toledo), whereas dissolved H2S and HS− concentrations were predicted by thermodynamic calculation. Although the NaCl concentration used in this experiment is lower than the salinity of the early ocean that would have been comparable with the modern level (61), NaCl is likely ineffective for the sulfide electroreduction reactivity (24). The same would be true of dissolved free Fe2+ (24, 62). Other ionic species that would have dissolved in the Hadean seawater were not considered because of a lack of definitive constraint for their concentrations. Note that the differences in ionic strength and pressure between the experimental condition and the deep-sea hydrothermal settings do not significantly influence the sulfide/metal equilibrium potentials. For example, the mackinawite/Fe0 equilibrium potential at 25°C and pH 6 in the presence of 0.5 μmol kg−1 H2S is −0.545 V (versus SHE) at I = 0.1 and 1 bar, whereas −0.550 V (versus SHE) at I = 0.5 and 500 bar.
While keeping the CO2 and H2S gas flow that was started at least 1 hour before each experiment, a constant potential was applied on the carbon electrode for up to 7 days by using a multi-potentiostat (PS-08; Toho Technical Research). In the course of the electrolysis, a fraction of metal sulfide floated with electrochemically generated gas bubbles (for example, H2) and deposited onto a cell compartment out of the electrode. The percentage remaining on the electrode after the 7-day experiment was around 50% at −0.7 V (versus SHE) and around 30% at −1.0 V (versus SHE) for the case of FeS. All potentials were measured against an Ag/AgCl reference electrode in saturated KCl and were converted to the SHE scale by the following equation
| (1) |
After the electrolysis, the electrochemical cell was immediately transferred into a globe box filled with N2 and H2 gases (volume ratio, 96:4). The solid sample was separated from the supernatant solution, dried under vacuum, and measured for XRD pattern using an x-ray diffractometer with Cu Kα radiation (MiniFlex 600, Rigaku). All the runs were conducted with 2θ ranging from 10° to 90° using 0.02° 2θ step with a scan rate of 1° min−1 except for the runs for FeS and CoS, which were done with a scan rate of 0.1° min−1. To prevent oxidation by atmospheric O2 during the measurement, the solid samples were shielded in an air-sensitive sample holder (Rigaku). Peak identifications were made on the basis of the reference patterns reported in the Power Diffraction File published by the International Centre for Diffraction Data. The reference patterns are presented in Fig. 1 (B to E) and fig. S3.
Reactions with the electrochemically reduced FeS
Two types of experiments were conducted with the electrochemically reduced FeS at −0.7 V (versus SHE) for 7 days. For comparison, the same procedures were followed with as-prepared FeS, with pure Fe0 obtained commercially, and without reductant. FeCl2 and pure H2 gas were also examined for their reducing capabilities. Pure Fe0 was purchased from EM Japan (catalog no. NP-FE-2-25). The reported purity, mean particle diameter, and specific surface area are >99.5%, 95 to 105 nm, and 4 to 6 m2 g−1, respectively. XRD measurement confirmed that the commercial Fe0 is in the α phase (fig. S13) that is the same structure as the Fe0 formed by the FeS electroreduction at ≤−0.7 V versus SHE (Fig. 1B). Each experiment was performed at least twice to ensure reproducibility. The product yields of multiple runs are summarized in table S1 as the averaged values with ±SD.
Experiment 1. Reductions of NO3−, oxaloacetate, and fumarate
A serum bottle (30 ml) was charged with 100 mg of FeS_PERM and sealed with a butyl rubber cap and an aluminum stopper in a globe box filled with N2 and H2 gases (volume ratio, 96:4). The bottle was then filled with pure CO2 gas (>99.995%) by flowing the CO2 through a stainless needle at a rate of 50 ml min−1 over 10 min. This was followed by the addition of 1.5 ml of deaerated aqueous solution of NaNO3, oxaloacetic acid, or fumaric acid (5 mM for each). After rotating the bottle at 60 rpm min−1 for 48 hours at room temperature (23° to 25°C), the sample suspension was filtered with a polytetrafluoroethylene membrane filter (pore size, 0.2 μm) and measured for pH by a portable pH meter (Seven2Go Pro, Mettler Toledo) (table S1). The filtrate was mixed with a reagent grade 1 mM NaOH aqueous solution to precipitate out dissolved iron as hydroxides and filtered again before the product analysis (see below). This experiment was also conducted with pure H2 gas (>99.9999%) or with 0.75 ml of aqueous solution of NaNO3, oxaloacetic acid, or fumaric acid (10 mM for each) plus 0.75 ml of aqueous FeCl2 solution (100 mM). No solid reductant was added in the latter two systems.
Experiment 2. Reductive aminations of glyoxylate, pyruvate, oxaloacetate, and α-ketoglutarate
A serum bottle (30 ml) was charged with 100 mg of FeS_PERM and sealed with a butyl rubber cap and an aluminum stopper in a globe box filled with N2 and H2 gases (volume ratio, 96:4). The bottle was then filled with pure N2 gas (>99.99995%) by flowing the N2 through a stainless needle at a rate of 50 ml min−1 over 10 min. This was followed by the addition of 0.75 ml of deaerated aqueous solution of 1 M NH4Cl plus 10 mM glyoxylic acid, sodium pyruvate, oxaloacetic acid, or α-keto glutaric acid and the subsequent addition of 0.75 ml of deaerated 0.5 M NaOH. After rotating the bottle at 60 rpm min−1 for 48 hours at room temperature (23° to 25°C), the sample suspension was filtered and measured for pH (table S1). The filtration was done again before the sample analysis. This experiment was also conducted with pure H2 gas (>99.9999%) or with 0.75 ml of aqueous solution of NH4Cl (1 M) plus glyoxylic acid, sodium pyruvate, oxaloacetic acid, or α-keto glutaric acid (10 mM for each), 0.375 ml of aqueous FeCl2 solution (200 mM), plus 0.375 ml of NaOH (1 M). No solid reductant was added in the latter two systems.
Sample analysis
Organic and inorganic species in the sample solutions were characterized by high-performance liquid chromatography (HPLC). Product identifications for organic compounds were also made by proton nuclear magnetic resonance (1H NMR) spectroscopy. In many cases, the total amounts of identified compounds were less than the initial amounts of starting materials probably because of condensation into insoluble macromolecules and/or adsorption on solid surfaces (63).
Organic acids were quantified by a Shimadzu HPLC system equipped with an electric conductivity detector and an anion exchange column (Shim-pack SCR-102H, Shimadzu) set at 40°C. The p-toluenesulfonic acid aqueous solution (5 mM) was used as the eluent at a rate of 1.6 ml min−1. The reproducibility of the data was within ±5%, which was estimated from the peak area-concentration relationship of each compound under at least four different concentrations (fig. S5).
The same instrument was used for quantifying inorganic anions (for example, NO3− and NO2−). An anion exchange column (SI-90 4E, Shodex) was used at 25°C. Na2CO3 (1.8 mM) plus 1.7 mM NaHCO3 aqueous solution was used as the eluent at a rate of 1.5 ml min−1. The reproducibility of the data was within ±5% (fig. S6).
1H NMR measurements were conducted with a Bruker Avance III spectrometer (400 MHz) at the sample temperature of 303.0 K. Typically, 0.45 ml of sample solution was mixed with 0.05 ml of D2O (99.9%; Merck Millipore) containing 5 mM 3-(trimethylsilyl)-1-propanesulfonic acid-d6 sodium (DSS-d6; Sigma-Aldrich) and was placed in an NMR tube (5 mm outside diameter; Wilmad-LabGlass). DSS-d6 was used for the calibration of the 0-ppm position and to estimate the product and reactant concentrations as an internal standard. A solvent suppression was run to minimize the solvent signal.
Ammonia and amino acids were analyzed by a Jasco HPLC system equipped with post-column derivatization with o-phthalaldehyde and a fluorescence detector operated at 345 nm for excitation and at 455 nm for emission. Five citrate buffer solutions of different citrate concentrations and pH values were used as eluents in a stepwise condition. A cation exchange column (AApak Na II-S2, Jasco) was used at 50°C. The reproducibility of the data was within ±5% (fig. S7).
The gas headspaces of serum bottles were analyzed by a GC-2010 Plus (Shimadzu) equipped with a BID-2010 Plus detector (Shimadzu) and a MICROPACKED-ST column (Shinwa). He (>99.99995%) was used as the carrier gas at a flow rate of 17 ml min−1. The column temperature was initially kept at 35°C for 2.5 min, raised to 250°C at a rate of 20°C min−1, and then raised to 265°C at a rate of 4°C min−1. H2, CO, CH4, and C2H6 were quantified within ±10% error (figs. S8 and S14 and table S2). All chromatograms showed the O2, N2, and CO2 signals (partly) due to the intrusion of air into the gas chromatography (GC) system at the sample injections (fig. S14).
Thermodynamic calculation
The equilibrium potentials of electrochemical reactions were calculated by using the Nernst equation
| (2) |
In that equation, T, R, and F stand for temperature in kelvin, the gas constant (8.31447 J mol−1 K−1), and the Faraday constant (96485 J mol−1 V−1), respectively. αi represents the activity of the species i that was calculated either using the extended Debye-Hückel equation (64) for aqueous ionic species or setting the activity coefficient to unity for aqueous neutral species and solid materials. z signifies the number of electrons transferred in the reaction. denotes the standard redox potential at the temperature and pressure of interest. The values of were calculated by combining the standard Gibbs energies of formation (∆fGo) of the individual compounds involved in the reaction
| (3) |
The value of ∆fGo for aqueous species at desired temperature and pressure was calculated according to the revised Helgeson-Kirkham-Flowers (HKF) equations of state (65) by using the thermodynamic data and the revised HKF parameters reported by Shock et al. (66, 67). The temperature and pressure dependences of ∆fGo for solid materials were calculated as follows
| (4) |
where and , respectively, represent the standard molar Gibbs energy and entropy at the reference temperature (Tr = 298.15 K) and pressure (Pr = 1 bar). represents the standard molar heat capacity at Pr, and denotes the standard molar volume at the temperature of interest. The values of , , , and as a function of temperature for metal sulfides used in the present study are listed in table S3 (68–75). The thermodynamic data for pure metals are all taken from (68).
The potential/pH diagrams of the sulfide-metal systems (Figs. 1 and 2 and fig. S3) were computed using the Act2 program in Geochemist’s Workbench version 10.0.5 using the thermodynamic dataset calculated by the above procedures.
Supplementary Material
Acknowledgments
We thank S. McGlynn and Y. Li for discussions about prebiotic chemistry in the early ocean hydrothermal systems. Funding: This research was supported by JSPS KAKENHI (grant nos.16H04074, 16K13906, 17H06105, 18H04456, and 18H05328) and the Astrobiology Center Program of NINS (grant no. AB292004). Author contributions: N.K. and Y.O. conceived the whole project on the basis of the principled approach (76), and N.K. realized the possibility of metal sulfide reduction. N.K. and R.N. established the setup for electrochemical experiment. M.Y. and K.T. provided information for the present and ancient deep-sea hydrothermal systems. N.Y. provided analytical instruments. N.K. performed all experiments and prepared all figures and tables. All authors contributed to writing the paper. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav7848/DC1
Fig. S1. A schematic of the electrochemical cell.
Fig. S2. XRD patterns of FeS electrolyzed at −1.0 V versus SHE for different durations.
Fig. S3. XRD patterns of metal sulfides before and after the 7-day electrolysis.
Fig. S4. Total charges build up during the electrolysis.
Fig. S5. Calibration curves for organic acids by the LC-electric conductivity detector system.
Fig. S6. Calibration curves for NO3− and NO2− by the LC-electric conductivity detector system.
Fig. S7. Calibration curves for amino acids and ammonia by the LC-fluorescence detector system.
Fig. S8. Calibration curves for H2, CO, CH4, and C2H6 by the GC-BID detector system.
Fig. S9. Nonenzymatic reactions in the presence of pure H2 gas, FeCl2, FeS, FeS_PERM, and Fe0 and those examined in the absence of reductant.
Fig. S10. Analytical results of fumarate (5 mM, 1.5 ml) incubated with the FeS_PERM (100 mg) at 80°C for 2 days.
Fig. S11. Reductive amination of four keto acids promoted by the FeS_PERM in one serum bottle.
Fig. S12. XRD patterns of CuS electrolyzed at −0.8 and −1.0 V (versus SHE) for short durations.
Fig. S13. XRD patterns of pure Fe0 used in the present study.
Fig. S14. GC chromatograms of the gas headspaces of serum bottles measured after the reduction experiments of organic/inorganic compounds.
Table S1. Summary of the reduction experiments of organic/inorganic compounds.
Table S2. Amounts of H2, CO, CH4, and C2H6 in the serum bottles (30 ml) after the reduction experiments of organic/inorganic compounds.
Table S3. Thermodynamic data for sulfide minerals.
REFERENCES AND NOTES
- 1.Yamamoto M., Nakamura R., Kasaya T., Kumagai H., Suzuki K., Takai K., Spontaneous and widespread electricity generation in natural deep-sea hydrothermal fields. Angew. Chem. Int. Ed. 56, 5725–5728 (2017). [DOI] [PubMed] [Google Scholar]
- 2.Nakamura R., Takashima T., Kato S., Takai K., Yamamoto M., Hashimoto K., Electrical current generation across a black smoker chimney. Angew. Chem. Int. Ed. 49, 7692–7694 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Maslennikov V. V., Maslennikova S. P., Large R. R., Danyushevsky L. V., Herrington R. J., Ayupova N. R., Zaykov V. V., Lein A. Y., Tseluyko A. S., Melekestseva I. Y., Tessalina S. G., Chimneys in Paleozoic massive sulfide mounds of the Urals VMS deposits: Mineral and trace element comparison with modern black, grey, white and clear smokers. Ore Geol. Rev. 85, 64–106 (2017). [Google Scholar]
- 4.Russell M. J., Barge L. M., Bhartia R., Bocanegra D., Bracheer P. J., Branscomb E., Kidd R., McGlynn S., Meier D. H., Nitschke W., Shibuya T., Vance S., White L., Kanik I., The drive to life on wet and icy worlds. Astrobiology 14, 308–343 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakashima S., Kebukawa Y., Kitadai N., Igisu M., Matsuoka N., Geochemistry and the origin of life: From extraterrestrial processes, chemical evolution on Earth, fossilized life’s records, to natures of the extant life. Life 8, E39 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kitadai N., Nakamura R., Yamamoto M., Takai K., Li Y., Yamaguchi A., Gilbert A., Ueno Y., Yoshida N., Oono Y., Geoelectrochemical CO production: Implications of the autotrophic origin of life. Sci. Adv. 4, eeao7265 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Huber C., Wächtershäuser G., Activated acetic acid by carbon fixation on (Fe,Ni)S under primordial conditions. Science 276, 245–247 (1997). [DOI] [PubMed] [Google Scholar]
- 8.Huber C., Wächtershäuser G., Peptides by activation of amino acids with CO on (Ni,Fe)S surfaces: Implications for the origin of life. Science 281, 670–672 (1998). [DOI] [PubMed] [Google Scholar]
- 9.Huber C., Wächtershäuser G., α-hydroxy and α-amino acids under possible Hadean, volcanic origin-of-life conditions. Science 314, 630–632 (2006). [DOI] [PubMed] [Google Scholar]
- 10.Braakman R., Smith E., The compositional and evolutionary logic of metabolism. Phys. Biol. 10, 011001 (2013). [DOI] [PubMed] [Google Scholar]
- 11.Camprubi E., Jordan S. F., Vasiliadou R., Lane N., Iron catalysis at the origin of life. IUBMB Life 69, 373–381 (2017). [DOI] [PubMed] [Google Scholar]
- 12.Nunoura T., Chikaraishi Y., Izaki R., Suwa T., Sato T., Harada T., Mori K., Kato Y., Miyazaki M., Shimamura S., Yanagawa K., Shuto A., Ohkouchi N., Fujita N., Takaki Y., Atomi H., Takai K., A primordial and reversible TCA cycle in a facultatively chemolithoautotrophic thermophile. Science 359, 559–563 (2018). [DOI] [PubMed] [Google Scholar]
- 13.Wächtersäuser G., Evolution of the first metabolic cycles. Proc. Natl. Acad. Sci. U.S.A. 87, 200–204 (1990). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Muchowska K. B., Varma S. J., Chevallot-Beroux E., Lethuiller-Karl L., Li G., Moran J., Metals promote sequences of the reverse Krebs cycle. Nat. Ecol. Evol. 1, 1716–1721 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Varma S. J., Muchowska K. B., Chatelain P., Moran J., Native iron reduces CO2 to intermediates and end-products of the acetyl-CoA pathway. Nat. Ecol. Evol. 2, 1019–1024 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Dekov V., Native nickel in the TAG hydrothermal field sediments (Mid-Atlantic Ridge, 26°N): Space trotter, guest from mantle, or a widespread mineral, connected with serpentinization? J. Geophys. Res. 111, B05103 (2006). [Google Scholar]
- 17.Hannington M. D., Thompson G., Rona P. A., Scott S. D., Gold and native copper in supergene sulphides from the Mid-Altantic ridge. Nature 333, 64–66 (1988). [Google Scholar]
- 18.Kanellopoulos C., Valsami-Jones E., Voudouris P., Stouraiti C., Mortiz R., Mavrogonatos C., Mitropoulos P., A new occurrence of terrestrial native iron in the Earth’s surface: The Ilia thermogenic travertine case, northwestern Euboea, Greece. Geosciences 8, 287 (2018). [Google Scholar]
- 19.Staude S., Wagner T., Markl G., Mineralogy, mineral compositions and fluid evolution at the Wenzel hydrothermal deposit, southern Germany: Implications for the formation of Kongsberg-type silver deposits. Can. Mineral. 45, 1147–1176 (2007). [Google Scholar]
- 20.Pernet-Fisher J. F., Day J. M. D., Howarth G. H., Ryabov V. V., Taylor L. A., Atmospheric outgassing and native-iron formation during carbonaceous sediment-basalt melt interactions. Earth Planet. Sci. Lett. 460, 201–212 (2017). [Google Scholar]
- 21.Genda H., Brasser R., Mojzsis S. J., The terrestrial late veneer from core disruption of a lunar-sized impactor. Earth Planet. Sci. Lett. 480, 25–32 (2017). [Google Scholar]
- 22.Trail D., Watson E. B., Tailby N. D., The oxidation state of Hadean magmas and implications for early Earth’s atmosphere. Nature 480, 79–82 (2011). [DOI] [PubMed] [Google Scholar]
- 23.Song H., Jiang G., Poulton S. W., Wignall P. B., Tong J., Song H., An Z., Chu D., Tian L., She Z., Wang C., The onset of widespread marine red beds and the evolution of ferruginous oceans. Nat. Commun. 8, 399 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Theberge S. M., Luther G. W., Determination of the electrochemical properties of a soluble aqueous FeS species present in sulfidic solutions. Aquat. Geochem. 3, 191–211 (1997). [Google Scholar]
- 25.Krissansen-Totton J., Arney G. N., Catling D. C., Constraining the climate and ocean pH of the early Earth with a geological carbon cycle model. Proc. Natl. Acad. Sci. U.S.A. 115, 4105–4110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Blöchl E., Keller M., Wächtershäuser G., Stetter K. O., Reactions depending on iron sulfide and linking geochemistry with biochemistry. Proc. Natl. Acad. Sci. U.S.A. 89, 8117–8120 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Huber C., Wächtershäuser G., Primordial reductive amination revisited. Tetrahedron Lett. 44, 1695–1697 (2003). [Google Scholar]
- 28.Wolthers M., van der Gaast S. J., Rickard D., The structure of disordered mackinawite. Am. Mineral. 88, 2007–2015 (2003). [Google Scholar]
- 29.Krznaric D., Ciglenecki I., Voltammetric study of an FeS layer on a Hg electrode in supersaturated FeS chloride solution. Environ. Chem. 12, 123–129 (2015). [Google Scholar]
- 30.Li Y., Kitadai N., Nakamura R., Chemical diversity of metal sulfide minerals and its implications for the origin of life. Life 8, E46 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mitsudome T., Mikami Y., Ebata K., Mizugaki T., Jitsukawa K., Kaneda K., Copper nanoparticles on hydrotalcite as a heterogeneous catalyst for oxidant-free dehydrogenation of alcohols. Chem. Commun. 39, 4804–4806 (2008). [DOI] [PubMed] [Google Scholar]
- 32.Mitsudome T., Noujima A., Mikami Y., Mizugaki T., Jitsukawa K., Kaneda K., Supported gold and silver nanoparticles for catalytic deoxygenation of epoxides into alkanes. Angew. Chem. Int. Ed. 49, 5545–5548 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Srinivasa G. R., Abiraj K., Gowda D. C., Lead-catalyzed synthesis of azo compounds by ammonium acetate reduction of aromatic nitro compounds. Synth. Commun. 33, 4221–4227 (2003). [Google Scholar]
- 34.Shibuya T., Russell M. J., Takai K., Free energy distribution and hydrothermal mineral precipitation in Hadean submarine alkaline vent systems: Importance of iron redox reactions under anoxic conditions. Geochim. Cosmochim. Acta 175, 1–19 (2016). [Google Scholar]
- 35.J. P. Amend, T. M. McCollom, Energetics of biomolecule synthesis on early Earth, in Chemical Evolution II: From the Origins of Life to Modern Society, L. Zaikowski, J. M. Friedrich, S. R. Seidel, Eds. (American Chemical Society, 2009), pp. 63–94.
- 36.Hori Y., Kikuchi K., Suzuki S., Production of CO and CH4 in electrochemical reduction of CO2 at metal electrodes in aqueous hydrogencarbonate solution. Chem. Lett. 14, 1695–1698 (1985). [Google Scholar]
- 37.Zhang X. V., Martin S. T., Driving parts of Krebs cycle in reverse through mineral photochemistry. J. Am. Chem. Soc. 128, 16032–16033 (2006). [DOI] [PubMed] [Google Scholar]
- 38.Keller M. A., Kampjut D., Harrison S. A., Ralser M., Sulfate radicals enable a non-enzymatic Krebs cycle precursor. Nat. Ecol. Evol. 1, 0083 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.G. Fuchs, Alternative pathways of autotrophic CO2 fixation, in Autotrophic Bacteria, H. G. Schlegel, B. Bowien, Eds. (Science Tech Publishers, 1989), pp. 365–382.
- 40.P. G. Simpson, W. B. Whitman, Anabolic pathways in methanogens, in Methanogenesis: Ecology, Physiology, Biochemistry & Genetics, J. G. Ferry, Ed. (Chapman & Hall, 1993), pp. 445–472.
- 41.Wong M. L., Charnay B. D., Gao P., Yung Y. L., Russell M. J., Nitrogen oxides in early Earth’s atmosphere as electron acceptors for life’s emergence. Astrobiology 17, 975–983 (2017). [DOI] [PubMed] [Google Scholar]
- 42.Lammer H., Zerkle A. L., Gebauer S., Tosi N., Noack L., Scherf M., Pilat-Lohinger E., Gudel M., Grenfell J. L., Godolt M., Nikolaou A., Origin and evolution of the atmospheres of early Venus, Earth and Mars. Astron. Astrophys. Rev. 26, 2 (2018). [Google Scholar]
- 43.Muchowska K. B., Varma S. J., Moran J., Synthesis and breakdown of the universal precursors to biological metabolism promoted by ferrous iron. Nature 569, 104–107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Matamoros-Veloza A., Cespedes O., Johnson B. R. G., Stawski T. M., Terranova U., de Leeuw N. H., Benning L. G., A highly reactive precursor in the iron sulfide system. Nat. Commun. 9, 3125 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morrill P. L., Kuenen J. G., Johnson O. J., Suzuki S., Rietze A., Sessions A. L., Fogel M. L., Nealson K. H., Geochemistry and geobiology of a present-day serpentinization site in California: The Cedars. Geochim. Cosmochim. Acta 109, 222–240 (2013). [Google Scholar]
- 46.Baaske P., Weinert F. M., Duhr S., Lemke K. H., Russell M. J., Braun D., Extreme accumulation of nucleotides in simulated hydrothermal pore systems. Proc. Natl. Acad. Sci. U.S.A. 104, 9346–9351 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bada J. L., Miller S. L., Kinetics and mechanism of the nonenzymatic reversible deamination of aspartic acid. J. Am. Chem. Soc. 91, 3946–3948 (1969). [DOI] [PubMed] [Google Scholar]
- 48.Palmer D. A., Drummond S. E., Thermal decarboxylation of acetate. Part I. The kinetics and mechanism of reaction in aqueous solution. Geochim. Cosmochim. Acta 50, 813–823 (1986). [Google Scholar]
- 49.Belsky A. J., Maiella P. G., Brill T. B., Spectroscopy of hydrothermal reactions 13. Kinetics and mechanisms of decarboxylation of acetic acid derivatives at 100–260 °C under 275 bar. J. Phys. Chem. A 103, 4253–4260 (1999). [Google Scholar]
- 50.Lee N., Foustoukos D. I., Sverjensky D. A., Cody G. D., Hazen R. M., The effects of temperature, pH and redox state on the stability of glutamic acid in hydrothermal fluids. Geochim. Cosmochim. Acta 135, 66–86 (2014). [Google Scholar]
- 51.Wolfenden R., Lewis C. A. Jr., Yuan Y., Kinetic challenges facing oxalate, malonate, acetoacetate, and oxaloacetate decarboxylases. J. Am. Chem. Soc. 133, 5683–5685 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lowell R. P., Keller S. M., High-temperature seafloor hydrothermal circulation over geologic time and Archean banded iron formations. Geophys. Res. Lett. 30, 1391 (2003). [Google Scholar]
- 53.Shibuya T., Yoshizaki M., Sato M., Shimizu K., Nakamura K., Omori S., Suzuki K., Takai K., Tsunakawa H., Maruyama S., Hydrogen-rich hydrothermal environments in the Hadean ocean inferred from serpentinization of komatiites at 300 °C and 500 bar. Prog. Earth Planet. Sci. 2, 46 (2015). [Google Scholar]
- 54.Guan G., Kida T., Ma T., Kimura K., Abe E., Yosida A., Reduction of aqueous CO2 at ambient temperature using zero-valent iron-based composites. Green Chem. 5, 630–634 (2003). [Google Scholar]
- 55.Winn J. N., Fabrycky D. C., The occurrence and architecture of exoplanetary systems. Annu. Rev. Astron. Astrophys. 53, 409–447 (2015). [Google Scholar]
- 56.Anglada-Escude G., Amado P. J., Barnes J., Berdinas Z. M., Butler R. P., Coleman G. A. L., de la Cueva I., Dreizler S., Endl M., Giesers B., Jeffers S. V., Jenkins J. S., Jones H. R. A., Kiraga M., Kürster M., López-González M. J., Marvin C. J., Morales N., Morin J., Nelson R. P., Ortiz J. L., Ofir A., Paardekooper S. J., Reiners A., Rodriguez E., Rodríguez-López C., Sarmiento L. F., Strachan J. P., Tsapras Y., Tuomi M., Zechmeister M., A terrestrial planet candidate in a temperate orbit around Proxima Centauri. Nature 536, 437–440 (2016). [DOI] [PubMed] [Google Scholar]
- 57.Gillon M., Triaud A. H. M. J., Demory B. O., Jehin E., Agol E., Deck K. M., Lederer S. M., de Wit J., Burdanov A., Ingalls J. G., Bolmont E., Leconte J., Raymond S. N., Selsis F., Turbet M., Barkaoui K., Burgasser A., Burleigh M. R., Carey S. J., Chaushev A., Copperwheat C. M., Delrez L., Fernandes C. S., Holdsworth D. L., Kotze E. J., Van Grootel V., Almleaky Y., Benkhaldoun Z., Magain P., Queloz D., Seven temperate terrestrial planets around the nearby ultracool dwarf star TRAPPIST-1. Nature 542, 456–460 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Charnay B., Hir G. L., Fluteau F., Forget F., Catling D. C., A warm or a cold early Earth? New insights from a 3-D climate-carbon model. Earth Planet. Sci. Lett. 474, 97–109 (2017). [Google Scholar]
- 59.Kuramoto K., Umemoto T., Ishiwatari T. M., Effective hydrodynamic hydrogen escape from an early Earth atmosphere inferred from high-accuracy numerical simulation. Earth Planet. Sci. Lett. 375, 312–318 (2013). [Google Scholar]
- 60.Genda H., Origin of Earth’s oceans: An assessment of the total amount, history and supply of water. Geochem. J. 50, 27–42 (2016). [Google Scholar]
- 61.Marty B., Avice G., Bekaert D. V., Broadley M. W., Salinity of the Archaean oceans from analysis of fluid inclusions in quartz. C. R. Geosci. 350, 154–163 (2018). [Google Scholar]
- 62.Rickard D., The solubility of FeS. Geochim. Cosmochim. Acta 70, 5779–5789 (2006). [Google Scholar]
- 63.Novikov Y., Copley S. D., Reactivity landscape of pyruvate under simulated hydrothermal vent conditions. Proc. Natl. Acad. Sci. U.S.A. 110, 13283–13288 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Helgeson H. C., Kirkham D. H., Flowers G. C., Theoretical prediction of the thermodynamic behavior of aqueous electrolytes at high pressures and temperatures: IV. Calculation of activity coefficients, osmotic coefficients, and apparent molal and standard and relative partial molal properties to 600°C and 5 kB. Am. J. Sci. 281, 1249–1516 (1981). [Google Scholar]
- 65.Shock E. L., Oelkers E. H., Johnson J. W., Sverjensky D. A., Helgeson H. C., Calculation of the thermodynamic properties of aqueous species at high pressures and temperatures. Effective electrostatic radii, dissociation constants and standard partial molal properties to 1000 °C and 5 kbar. J. Chem. Soc. Faraday Trans. 88, 803–826 (1992). [Google Scholar]
- 66.Shock E. L., Helgeson H. C., Sverjensky D. A., Calculation of the thermodynamic and transport properties of aqueous species at high pressures and temperatures: Standard partial molal properties of inorganic neutral species. Geochim. Cosmochim. Acta 53, 2157–2183 (1989). [Google Scholar]
- 67.Shock E. L., Sassani D. C., Willis M., Sverjensky D. A., Inorganic species in geologic fluids: Correlations among standard molal thermodynamic properties of aqueous ions and hydroxide complexes. Geochim. Cosmochim. Acta 61, 907–950 (1997). [DOI] [PubMed] [Google Scholar]
- 68.R. A. Robie, B. S. Hemingway, “Thermodynamic Properties Of Minerals And Related Substances At 298.15 K And 1 Bar (105 Pascals) Pressure And At Higher Temperatures” (U.S. Geological Survey Bulletin 2131, U.S. Geological Survey, 1995).
- 69.M. W. Chase, “NIST-JANAF Thermochemical Tables,” J. Phys. Chem. Ref. Data Monogr. 9, 1–1951 (1998).
- 70.K. C. Mills, Thermodynamic Data for Inorganic Sulphides, Selenides and Tellurides (Butterworth-Heinemann, 1974).
- 71.Ning J., Zheng Y., Brown B., Young D., Nešić S., A thermodynamic model for the prediction of mild steel corrosion products in an aqueous hydrogen sulfide environment. Corrosion 71, 945–960 (2015). [Google Scholar]
- 72.Rajamani V., Prewitt C. T., Refinement of the structure of Co9S8. Can. Mineral. 13, 75–78 (1975). [Google Scholar]
- 73.J. R. Rumble, D. R. Lide, T. J. Bruno, CRC Handbook of Chemistry and Physics (CRC Press, ed. 98, 2017), pp. 4-37–4-89.
- 74.Schutte W. J., Disselborg F., DeBoer J. L., Determination of the two-dimensional incommensurately modulated structure of Mo2S3. Acta Cryst. 49, 787–794 (1993). [Google Scholar]
- 75.J. R. Smyth, T. C. McCormick, Crystallographic data for minerals, in Mineral Physics and Crystallography: A Handbook of Physical Constants, T. J. Ahrens, Ed. (American Geophysical Union, 1995), pp. 1–17.
- 76.Aono M., Kitadai N., Oono Y., A principled approach to the origin problem. Orig. Life. Evol. Biopsh. 45, 327–338 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/6/eaav7848/DC1
Fig. S1. A schematic of the electrochemical cell.
Fig. S2. XRD patterns of FeS electrolyzed at −1.0 V versus SHE for different durations.
Fig. S3. XRD patterns of metal sulfides before and after the 7-day electrolysis.
Fig. S4. Total charges build up during the electrolysis.
Fig. S5. Calibration curves for organic acids by the LC-electric conductivity detector system.
Fig. S6. Calibration curves for NO3− and NO2− by the LC-electric conductivity detector system.
Fig. S7. Calibration curves for amino acids and ammonia by the LC-fluorescence detector system.
Fig. S8. Calibration curves for H2, CO, CH4, and C2H6 by the GC-BID detector system.
Fig. S9. Nonenzymatic reactions in the presence of pure H2 gas, FeCl2, FeS, FeS_PERM, and Fe0 and those examined in the absence of reductant.
Fig. S10. Analytical results of fumarate (5 mM, 1.5 ml) incubated with the FeS_PERM (100 mg) at 80°C for 2 days.
Fig. S11. Reductive amination of four keto acids promoted by the FeS_PERM in one serum bottle.
Fig. S12. XRD patterns of CuS electrolyzed at −0.8 and −1.0 V (versus SHE) for short durations.
Fig. S13. XRD patterns of pure Fe0 used in the present study.
Fig. S14. GC chromatograms of the gas headspaces of serum bottles measured after the reduction experiments of organic/inorganic compounds.
Table S1. Summary of the reduction experiments of organic/inorganic compounds.
Table S2. Amounts of H2, CO, CH4, and C2H6 in the serum bottles (30 ml) after the reduction experiments of organic/inorganic compounds.
Table S3. Thermodynamic data for sulfide minerals.