

Abstract
Background
Pathophysiological mechanisms that connect obesity to cardiovascular disease are incompletely understood. FSP27 (Fat‐specific protein 27) is a lipid droplet‐associated protein that regulates lipolysis and insulin sensitivity in adipocytes. We unexpectedly discovered extensive FSP27 expression in human endothelial cells that is downregulated in association with visceral obesity. We sought to examine the functional role of FSP27 in the control of vascular phenotype.
Methods and Results
We biopsied paired subcutaneous and visceral fat depots from 61 obese individuals (body mass index 44±8 kg/m2, age 48±4 years) during planned bariatric surgery. We characterized depot‐specific FSP27 expression in relation to adipose tissue microvascular insulin resistance, endothelial function and angiogenesis, and examined differential effects of FSP27 modification on vascular function. We observed markedly reduced vasodilator and angiogenic capacity of microvessels isolated from the visceral compared with subcutaneous adipose depots. Recombinant FSP27 and/or adenoviral FSP27 overexpression in human tissue increased endothelial nitric oxide synthase phosphorylation and nitric oxide production, and rescued vasomotor and angiogenic dysfunction (P<0.05), while siRNA‐mediated FSP27 knockdown had opposite effects. Mechanistically, we observed that FSP27 interacts with vascular endothelial growth factor‐A and exerts robust regulatory control over its expression. Lastly, in a subset of subjects followed longitudinally for 12±3 months after their bariatric surgery, 30% weight loss improved metabolic parameters and increased angiogenic capacity that correlated positively with increased FSP27 expression (r=0.79, P<0.05).
Conclusions
Our data strongly support a key role and functional significance of FSP27 as a critical endogenous modulator of human microvascular function that has not been previously described. FSP27 may serve as a previously unrecognized regulator of arteriolar vasomotor capacity and angiogenesis which are pivotal in the pathogenesis of cardiometabolic diseases linked to obesity.
Keywords: angiogenesis, endothelial dysfunction, endothelial function, obesity, vasodilation
Subject Categories: Angiogenesis, Endothelium/Vascular Type/Nitric Oxide, Metabolism, Translational Studies
Short abstract
See Editorial Mineo
Clinical Perspective
What Is New?
FSP27 (Fat‐specific protein 27) was thought to be expressed mainly in adipocytes regulating lipolysis; however, we discovered its expression in human endothelial cells where it has additional key functions.
FSP27 restores vascular insulin sensitivity, vasodilator function, and angiogenesis in dysfunctional microvessels of obese subjects.
Bariatric surgical weight loss improves metabolism and augments FSP27 expression.
What Are the Clinical Implications?
FSP27 is a newly identified modulator of vascular function in human disease that exerts regulatory control over vascular endothelial growth factor‐A in the human vasculature.
Obesity‐related downregulation of FSP27 may link obesity to cardiometabolic diseases.
Obesity represents our largest preventable healthcare problem with the majority of Americans currently categorized as overweight or obese.1, 2, 3 The American Medical Association labeled obesity as a disease highlighting its critical negative impact on public health,4 and over two thirds of deaths related to obesity worldwide are from cardiovascular causes.5 Excess fat, particularly accumulation of intra‐abdominal visceral fat, has been linked to lipotoxicity, adipose tissue dysfunction, and inflammation that are implicated in the development of insulin resistance, endothelial dysfunction, and cardiovascular disease;6, 7, 8, 9, 10 however, pathophysiological mechanisms are incompletely understood.11, 12
The endothelium plays a critical role in vascular homeostasis and many of its functions are governed by the basal and stimulated release of endothelium‐derived nitric oxide (NO) that maintains arterial tone, inhibits inflammation, promotes fibrinolysis, and modulates reparative angiogenesis.13, 14, 15 Studies have shown that insulin resistance not only disrupts metabolic pathways and organs, but also adversely impacts the vasculature.13 Insulin normally regulates blood flow through activation of endothelial NO synthase (eNOS) by binding to its receptor and subsequent activation of eNOS via PI3‐K/Akt.16 Defective insulin signaling leads to vascular inflammation, vasoconstriction, plaque progression, and ischemia. Endothelium‐specific deletion of the insulin receptor in animal models causes atherosclerosis.17, 18 Thus, understanding signaling pathways that regulate endothelial function are pivotal for atherosclerosis initiation and progression.
FSP27 (Fat‐specific protein 27) is a lipid droplet‐associated protein that regulates lipolysis and insulin sensitivity in adipocytes, and plays a key role in governing whole body metabolism.19, 20, 21, 22 Experimental models showed that FSP27 deletion augments basal lipolysis and promotes insulin‐resistance under high‐fat diet conditions.23 Human genetic mutations of FSP27 are associated with lipodystrophy, hypertriglyceridemia, and insulin‐resistance.24 While cellular actions of FSP27 have been well characterized almost exclusively in adipocytes, we unexpectedly discovered that it is expressed abundantly in human vascular endothelial cells, yet downregulated in association with visceral/central obesity. As lipid storage and breakdown are generally not viewed as a primary function of endothelial cells, we sought to examine whether FSP27 controls vascular phenotype by mechanisms beyond regulation of lipid metabolism and serves as a previously unrecognized regulator of arteriolar vasomotor function and angiogenesis which are important in mechanisms of ischemic cardiovascular disease.25, 26, 27
Materials and Methods
The authors declare that all supporting data are available within the article and its online supplementary files.
Study Subjects
Consecutive obese men and women (body mass index ≥35 kg/m2, aged ≥18 years), with severe long‐standing obesity undergoing elective bariatric weight‐loss surgery at Boston Medical Center were recruited into the study. Subjects with unstable medical conditions or pregnancy were not eligible for bariatric surgery and thus excluded. The study was approved by the Boston University Medical Center Institutional Review Board, and written consent was obtained from all participants.
Anthropometric and Biochemical Measures
During a presurgical outpatient and subsequent postoperative follow‐up visit, clinical characteristics including blood pressure, height, weight, and waist circumference were measured, along with recording of cardiovascular risk factors. Fasting blood was taken via an antecubital vein for biochemical parameters including lipids, glucose, insulin, glycosylated hemoglobin, and high‐sensitivity C‐reactive protein. Homeostasis model assessment was used as the index of insulin sensitivity. All biochemical analyses were performed by the Boston Medical Center clinical chemistry laboratory.
Adipose Tissue Collection and Percutaneous Adipose Tissue Biopsy
Subcutaneous and visceral adipose tissue were collected intra‐operatively from the lower abdominal wall and greater omentum, during planned bariatric surgery, as previously described.28, 29, 30 Each subject provided one biopsy specimen from the subcutaneous and one specimen from the visceral fat depot. For the longitudinal study component after weight loss, follow‐up peri‐umbilical subcutaneous fat biopsies were performed percutaneously during a postoperative visit as previously described.31 Briefly, subjects were placed in supine position with sterile draping of the abdominal region. Local skin anesthesia was performed with subcutaneous lidocaine injection and a small superficial 0.5 cm skin incision made lateral to the umbilicus with a tiny scalpel, which allows for both aspiration of fat using a large‐bore cannula and several punch biopsies providing specimens of intact adipose tissue. The anatomic layer and qualitative yield of this procedure is the same as the intra‐operative baseline collection. The superficial skin incision was then closed with self‐absorbing sutures.
Adipose Vessel Preparation and Assessment of Adipose Arteriolar Function
Adipose tissue specimens were placed immediately in cold HEPES buffer solution, pH 7.51 (American Bioanalytical, Natick, MA). Small adipose arterioles (75–350‐μmol/L internal diameter) were carefully removed of surrounding fat and connective tissue. Arterioles were cannulated securely with glass micropipettes within an organ chamber as previously described.32, 33, 34 The chamber was then mounted onto a stage of an inverted microscope (×200 magnification) and video camera monitor (model VIA‐100; Boeckler Instruments, Inc, Tuscon, AZ) for vascular diameter measurements using videomicroscopy. Arterioles were pressure equilibrated in continuously perfused with Krebs buffer aerated with a gas mixture of 5% O2, 21% CO2 and 74% N2. The internal arterial diameter of each vessel was initially measured at a steady state followed by administration of endothelin‐1 (ET‐1, 2×10−6 mol/L; Sigma‐Aldrich, St. Louis, MO) to pre‐constrict vessels to 50% to 70% of their internal resting diameter. Insulin‐mediated, endothelial nitric oxide synthase (eNOS)‐dependent vasodilation was determined by measuring the change in diameter in response to insulin (10−5 mol/L; Sigma‐Aldrich) at 5‐minute intervals over a time course of 30 minutes in visceral fat microvessels from obese subjects as previously described.33 Vessels viability and maximum dilator capacity were examined using the endothelial‐independent vasodilator papaverine (Pap, 2×10−4 mol/L; Sigma‐Aldrich). To assess the effect of FSP27, isolated microvessels were exposed to vehicle control or 5‐μmol/L recombinant FSP27 (rFSP27) in vessel chambers for 60 minutes followed by vasodilation studies. The effect of rFSP27 on insulin‐induced vasodilation was examined in the presence or absence of eNOS inhibitor Nω‐nitro‐L‐arginine methyl ester (L‐NAME, 3×10−4 mol/L, Sigma‐Aldrich).
Endothelial Cell Isolation From Adipose Tissue and Insulin Stimulation
Endothelial cells were isolated from human fat as described previously.29, 30 Briefly, subcutaneous and visceral fat tissue samples collected during surgery were placed immediately in cold DMEM (catalogue # 11885‐084, Gibco Life Technology, Grand Island, NY) supplemented with penicillin, and streptomycin and 0.5% serum. Tissue was cut, minced and digested in collagenase I (catalogue # C130, 1 mg/mL, Sigma‐Aldrich, St. Louis, MO) for 1 hour in a 37°C water bath at 100 rpm rotation. Undigested tissue was removed by passing through 100‐μmol/L filters. Filtrate was then centrifuged at 60g at 4°C for 10 minutes to separate adipocytes (top layer). Red blood cells were lysed using 1X RBC lysis buffer (Catalogue # WL1000, R&D Systems, Minneapolis, MN) and remaining cells passed through 40‐μmol/L filters and washed with DMEM. Cells were labeled with CD31 microbeads (catalogue # 130‐091‐935, Miltenyi Biotech, Auburn, CA) before being loaded into the auto MACS Pro Separator. Isolated CD31 positive endothelial cells were plated on fibronectin (catalogue # NC0702888, Fisher Scientific, Pittsburg, PA) coated 8‐well chamber slides (catalogue # 354104 and 354108BD, BD Bioscience, San Jose, CA). Cells were allowed to settle for 4 hours and serum starved (0.5% serum media) overnight. Slides were then fixed in 4% paraformaldehyde and stored in −80°C for quantitative immunofluorescence staining.
Treatment With Recombinant FSP27 and Insulin Stimulation of Adipose Tissue
Freshly collected visceral fat was cut into 1 to 2 mm pieces and treated with 100‐μmol/L rFSP27 (Catalogue # ab188449, Abcam, Cambridge, MA) for 24 hours. In the last 5 hours of incubation, tissue was serum starved (0.5% serum) in endothelial basal media‐2 (EBM‐2) without growth factors (catalogue # CC5036, Lonza, Hopkinton, MA). At the end of 24 hours treatment, tissue was then treated with vehicle (control) or 100‐nmol/L insulin for 30 minutes, snap frozen in liquid nitrogen, and stored at −80°C for protein analysis.
Biological Inhibition of FSP27 in Adipose Tissue and Endothelial Cells
Biological inhibition of FSP27 using siRNA (small interfering RNA) in adipose tissue was performed using previously described techniques.29 Briefly, freshly collected adipose tissue was washed with PBS under sterile condition and ≈70 to 80 mg of tissue was cut into 1‐ to 2‐mm pieces and suspended in 200‐μL Opti‐MEM (catalogue # 31985‐062, Gibco) with 100‐nmol/L siRNA (FSP27) (catalogue # Sc‐78016, Santa Crutz) or scrambled siRNA (catalogue # Sc‐37007, Santa Crutz) in electroporation cuvette (0.4 cm, USA Scientific) and applied 16 shocks of 50 V and 950 μF for 30 milliseconds on a Bio‐Rad Gene Pulser II system. Tissue was then incubated at 37°C for 55 hours, in EBM‐2 media with 5% serum supplemented with 50‐μg/mL streptomycin and 50‐U/mL penicillin. Media were changed after 2 hours initially and every 24 hours thereafter. At the end of the experiment, tissue was serum starved for 5 hours and treated with 100‐nmol/L of insulin or vehicle control for 30 minutes, and froze in liquid nitrogen, and stored at −80°C for protein analysis.
siRNA‐mediated depletion of FSP27 in cultured human aortic endothelial cells (HAEC) was performed according to the manufacturer's protocol for reverse transfection using lipofectamine RNAiMAX (catalogue # 13778030, Thermo Fisher). Briefly, RNA‐lipid complexes were made using RNAiMAX and siRNA (scrambled‐siRNA and FSP27, 100‐nmol/L) in Opti‐MEM and added to the culture flask. Trypsinized human aortic endothelial cells (HAEC) were then added and allowed to settle with RNA‐lipid complex for 4 hours, EBM‐2 media were added, and cells were allowed to grow for 48 hours. For Western immunoblot experiments, cytosolic proteins were collected using protein lysis buffer with protease and phosphate inhibitors and subjected to Western immunoblot. For NO production experiments, cells were starved for 5 hours and stimulated with insulin for 30 minutes and followed by NO measurement protocols.
Endothelial Cell Quantitative Immunofluorescence
We quantified protein FSP27 expression in isolated endothelial cells from subcutaneous and visceral fat. Briefly, fixed samples were rehydrated with 50‐mmol/L glycine (Sigma‐Aldrich, St. Louis, MO), permeabilized with 0.1% Triton‐X and blocked with 0.5% bovine serum albumin. Slides were incubated overnight at 37°C with primary antibodies against FSP27 (1:250 dilution, catalogue # PA5‐30793, Thermo Fisher), and CD31 (1:300 dilution, catalogue # MA3100, Thermo Fisher) to select endothelial cells. We used analogous Alexa Fluor‐488 and Alexa Fluor‐594 antibodies (1:200 dilution; Invitrogen, Carlsbad, CA) for secondary antibodies. Cells were mounted under glass coverslips with VECTASHIELD (catalogue # H1500, Vector Laboratories, Burlingame, CA) containing DAPI to identify nuclei. Slides were imaged using a fluorescent microscope (×20 magnification, Nikon Eclipse TE2000‐E) and digital images were captured using a Photometric CoolSnap HQ2 Camera (Photometrics, Tucson, AZ). Exposure time was kept constant and fluorescent intensity (corrected for background fluorescence) was quantified by NIS Elements AR Software (Nikon Instruments Inc, Melville, NY). Fluorescence intensity was quantified in 20 cells from each depot/subject and averaged. To control for batch‐to‐batch staining variability, fluorescence intensity for each sample was normalized to the intensity of HAEC staining performed simultaneously. Data are expressed in arbitrary units (au) calculated by dividing the average fluorescence intensity of the subject sample by the intensity of the HAEC sample ×100, as previously described and validated.28, 29 Confocal microscopy for primary endothelial cells was performed for FSP27 and VEGF‐A (using Leica Sp5 (Leica microsystem, Buffalo Grove, IL) with an ×63 oil immersion objective. Images were processed using Image J.
For 3‐dimensional (3D) immunohistochemistry images of fibroblasts (which do not express any endogenous FSP27), we exposed cells to rFSP27 for 2 hours and performed 3‐dimensional immunohistochemistry using standard measures. Immunostaining was performed using anti‐FSP27 polyclonal antibodies followed by labeling with the fluorescent secondary antibody. Labeled cells were washed 3 times with PBS and mounted on slides in media with DAPI (Vector Laboratories). Confocal microscopy was performed using Nikon A1Rsi confocal microscope (Tokyo, Japan) with a ×100 oil immersion objective. Z‐Slices of 0.3‐μm thickness were captured from top to bottom of the nucleus. The 3‐dimensional image was constructed from the Z‐slices using Nikon software.
Adenovirus Transduction in Primary Endothelial Cells
Overexpression of human FSP27‐HA‐tagged adenovirus was performed as described previously.20 Briefly, adenovirus, either control or adenovirus overexpressing FSP27, was added at a multiplicity of infection of 100 to primary endothelial cells isolated from visceral fat from obese subjects, Cells were grown for 48 hours under both experimental conditions, serum starved for 5 hours, stimulated with 100‐nmol/L insulin for 30 minutes and then processed for NO production as described below.
Endothelial Cell Nitric Oxide Production
Endothelial NO production was measured using a sensitive and specific fluorescent probe by utilizing the membrane permeable probe 4,5‐diaminofluorescein diacetate (DAF‐2DA, Enzo, Framingdale, NY) as described previously.29 Briefly, isolated endothelial cells were grown for 1 to 2 days, and either FSP27‐overexpressed or ‐silenced for 48 hours. Cells were then serum starved and subsequently treated with vehicle (control) or 100‐nmol/L insulin and NO probe DAF‐2DA for 30‐minutes. For the last 10 minutes of the experiment, Hoechst (Molecular probes, Grand Island, NY) was added for nuclear staining. Slides were imaged using a fluorescent microscope (×20 magnification, Nikon Eclipse TE2000‐E) and digital images were captured using a Photometric CoolSnap HQ2 Camera (Photometrics, Tucson, AZ). Exposure time was kept constant and fluorescent intensity (corrected for background fluorescence) was quantified by NIS Elements AR Software (Nikon Instruments Inc, Melville, NY) as described above.
Western Immunoblot Analyses
Proteins were extracted from adipose tissue by homogenization in liquid nitrogen. Ice‐cold 1X lysis buffer (Cell Signaling, Danvers, MA) supplemented with protease inhibitor cocktail and phosphatase inhibitor II and III (Sigma Aldrich, St. Louis, MO) were added. For cell culture samples, including macrophages (human peripheral blood macrophages, Stemcell technologies, MA), ice‐cold lysis buffer with inhibitors were directly added to cells. All samples were assayed for protein content using Bradford's method. Thirty‐five micrograms of protein was subjected to electrophoresis in SDS‐polyacrylamide gel under reducing conditions and blotted to a nitrocellulose membrane using the Bio‐Rad Transblot Turbo Transfer system. For co‐immunoprecipitation (co‐IP) 500 μg of protein was incubated with antibody overnight, washed with PBS and ran immunoblot as described above. The membranes were blocked in 5% bovine serum albumin 0.1% Tween‐20 in TBS for 1 hour at room temperature, and then incubated overnight at 4°C with primary anti‐human antibodies (1:500–1000). Membranes were then washed off using TBS and incubated with horseradish peroxidase‐conjugated secondary anti‐rabbit IgG (R&D System, Minneapolis, MN) for 1 hour at room temperature, immune complexes were detected with the enhanced chemiluminescence ECL detection system (Bio‐Rad). Densitometric analysis of bands was performed using ImageQuant™ LAS 4000 biomolecular imaging system (GE Healthcare, Pittsburg, PA). Proteins of interests were FSP27 (Catalogue #PA5‐30793, Thermofisher); p‐eNOS at serine 1177 (Catalogue #ab184154, Abcam); pAKT (protein kinase B) at serine 473 (Catalogue #44‐609G, Thermofisher); VEGF‐A (Catalogue #PA5‐16754, Thermofisher), and GAPDH as a loading control. Intensity of bands for each protein was normalized to control band GAPDH.
In Vitro Transcription and Translation co‐IP Immunoblot Assays
In vitro transcription and translation constructs were generated following the guidelines of Pierce's polymerase chain reaction (PCR) protocol for generating optimized templates. A 600‐bp T7‐IRES‐Kozak sequence containing a T7 promoter, internal ribosome entry site, and Kozak sequence was amplified from pT7CFE1 vectors (Catalogue #88860, Pierce) with T7 sense and Kozak anti‐sense primers. Next, an overlap sense primer (overlap with Kozak sequence) and an overlap anti‐sense primer (overlap with gene ORF or fragments) containing a 30‐nt poly‐A sequence were used to amplify the gene ORF or gene fragment of FSP27 and VEGF‐A. These 2 fragments were combined for overlap extension PCR with T7 sense and overlap anti‐sense primers. PCR cycles were performed according to the guidelines. The resulting PCR templates were purified, and the identities of the templates were confirmed by restriction enzyme analysis. One‐microgram of template DNA was used for 50‐μL of transcription‐translation reaction mixture with a 1‐Step Human Coupled IVT Kit according to manufacturer's recommendations (Thermo‐Scientific). A sample without added DNA was used as a negative control; GFP (green fluorescent protein) template DNA was used as a positive control. Translation reaction time was optimized for protein length to reduce truncated proteins attributable to material exhaustion. Following translation, total protein concentration was calculated and small portions were subjected to Western blotting to confirm protein integrity or placed in −80°C storage for co‐IP.
Angiogenic Sprout and Tube Formation Assays
For angiogenic sprout assays, fat‐biopsy samples were collected at baseline and after weight‐loss surgery. Samples were immediately placed in sterile endothelial basal media‐2 (EBM‐2, Lonza) and angiogenic capacity quantified as described previously.30, 35 Briefly, fat samples were finely minced and digested with 1 mg/mL collagenase (Catalogue #LS004194, Worthington) for 30 minutes at 37°C. Digested tissue was passed through 100‐μmol/L nylon filters, and washed in EBM‐2 media. One mm2 digested piece were embedded on ice cold 150‐μL growth factor depleted matrigel (Catalogue #354230, BD Discovery) on a 48 well plate. The plate was incubated for 30 minutes in 37°C to let the matrigel polymerize. At the end of incubation, wells were covered with 500‐μL of EBM‐2 supplemented with growth factors and cultured at 37°C for 7 days. Half of the media was removed and replaced with fresh media every other day. Sprouts growing from the fat explants along the perimeter of each sample were counted under ×100 magnification by a masked investigator, as previously described.35
For angiogenic tube formation assays, primary endothelial cells isolated from visceral fat were grown for 7 days in culture to generate enough cells numbers. On day 7 of culture, tube formation assay was performed according to protocol previously described (nature protocol 2010).36 Briefly, cells were trypsinized and ≈5×105 cells were plated in growth factor depleted gelled matrigel (polymerized) in duplicates. Cells were allowed to organize and coalesce into capillary‐like structures for 6 hours with and without 100‐nmol/L rFSP27. Branch formation was quantified according to company's quantification protocol (Chemicon/Invitrogen: angiogenic assay protocol) by counting each branch point formation per condition per subject.
Adipose VEGF‐A Secretion
Visceral fat was incubated with and without human rFSP27 for 24 hours in serum‐free media (0.5%, described above). For subcutaneous fat, tissue was subjected to either control or FSP27 siRNA for 48 hours as previously described.29 Media were collected from all experimental conditions and VEGF‐A concentration measured using an ELISA kit from R&D system (Catalogue # DVE00), and expressed as picograms of VEGF‐A in milliliters of media, per milligrams of total protein in tissue.
Gene Expression
Immediately following adipose tissue collection, tissue samples were stored in RNAlater (Sigma‐Aldrich) solution at −80°C. Total RNAs were isolated from homogenized whole adipose tissues using the QIAzol reagent and RNeasy Mini kits (Qiagen). RNA (0.5–1.5 μg) was retrotranscribed with High Capacity cDNA Synthesis Kits (Life Technologies). Quantitative real‐time PCR reactions were performed using TaqMan gene expression assays in a ViiA7 PCR system (Life Technologies). Results were analyzed with the ∆∆Ct method using GAPDH as a reference.
Statistical Analysis
All statistical analyses were performed using GraphPad Prism 6.0 software unless otherwise mentioned. Clinical characteristics of subjects were presented as mean±SD or percentage. Differences in clinical characteristics and adipose tissue gene expression between baseline and follow‐up visits were examined using Student paired t tests. Inter‐depot differences in FSP27 expression, eNOS and AKT activation, and VEGF‐A production were analyzed with Student paired t tests. Differences in tube formation angiogenesis were analyzed with Student paired t tests. Repeated measures ANOVA was used to compare vascular response to rFSP27 treatments. Four‐group differences in NO production were analyzed using ANOVA. Area under the curve of the plot for cumulative angiogenic growth (quantified as capillary sprout count) over the time period of 7 days was examined with Origin v6.1. Spearman correlation analysis was performed to examine associations between FSP27 gene expression and angiogenic capacity. Statistical significance was defined as P<0.05.
Results
Clinical Characteristics
A total of 61 subjects undergoing planned bariatric surgery were recruited for the study, each providing paired subcutaneous and visceral adipose tissue samples at baseline which were partitioned for different experiments. Clinical characteristics of all subjects are displayed in Table 1 and are consistent with those of the bariatric population at our medical center. For the longitudinal component, we followed a subset of subjects for up to 12 months (range 4–15 months) after bariatric surgery and their clinical characteristics are displayed in Table 2.
Table 1.
Clinical Characteristics of the Study Population
| Parameter | Obese (n=61) |
|---|---|
| Age, y | 48±4 |
| Female, % | 86.9 |
| BMI, kg/m2 | 44±8 |
| Waist circumference, cm | 114±14 |
| Weight, kg | 121±25 |
| Insulin, mIU/mL | 18.8±15 |
| Glucose, mg/dL | 119±69 |
| HbA1c, % | 6.1±1.8 |
| HOMA‐IR | 5.1±6 |
| hsCRP, mg/dL | 10.8±8.8 |
| Triglycerides, mg/dL | 115±54 |
| Total cholesterol, mg/dL | 183±41 |
| HDL‐C, mg/dL | 45±9 |
| LDL‐C, mg/dL | 115±37 |
| Systolic BP, mm Hg | 131±15 |
| Diastolic BP, mm Hg | 76±12 |
| Diabetes mellitus, % | 30 |
| Hypertension, % | 36 |
| Hypercholesterolemia, % | 30 |
Data are mean±SD. BMI indicates body mass index; BP, blood pressure; HbA1c, hemoglobin A1c; HDL‐C, high‐density lipoprotein cholesterol; HOMA IR, homeostatic model assessment of insulin resistance; hsCRP, high‐sensitivity C‐reactive protein; LDL‐C, low‐density lipoprotein cholesterol.
Table 2.
Clinical Effects of Weight Loss
| Parameter | Baseline (n=7) | Weight Loss (n=7) | P Value |
|---|---|---|---|
| Age, y | 41.5±8 | 42.3±8 | 0.07 |
| Female, % | 71 | 71 | |
| BMI, kg/m2 | 46±8 | 32±5 | <0.05 |
| Waist circumference, cm | 132±11 | 106±19 | <0.05 |
| Weight, kg | 127±19 | 89±13 | <0.05 |
| Insulin, mIU/mL | 10.4±7 | 5.7±1.4 | 0.06 |
| Glucose, mg/dL | 199±131 | 104±24 | 0.13 |
| HbA1c, % | 8.2±3.8 | 6.1±1.5 | 0.1 |
| hsCRP, mg/dL | 10.5±7.4 | 1.6±1 | 0.05 |
| Triglycerides, mg/dL | 202±78 | 96±41 | <0.05 |
| HDL‐C, mg/dL | 40±7 | 47±14 | 0.86 |
| LDL‐C, mg/dL | 141±34 | 107±16 | 0.14 |
| Systolic BP, mm Hg | 133±13 | 122±10 | 0.11 |
| Diastolic BP, mm Hg | 77±13 | 78±11 | 0.54 |
| Diabetes mellitus, % | 42 | 28 | 0.06 |
| Hypertension, % | 50 | 28 | 0.05 |
| Hypercholesterolemia, % | 18 | 14 | 0.5 |
Data are mean±SD. BMI indicates body mass index; BP, blood pressure; HbA1c, hemoglobin A1c; HDL‐C, high‐density lipoprotein cholesterol; HOMA IR, homeostatic model assessment for insulin resistance; hsCRP, high‐sensitivity C‐reactive protein; LDL‐C, low‐density lipoprotein cholesterol.
FSP27 Protein Expression in Primary Endothelial Cells
Using cellular immunofluorescence, we observed significantly lower FSP27 protein expression (red signal) in endothelial cells isolated from visceral versus subcutaneous depots (Figure 1A and 1B, n=14, P<0.01).
Figure 1.
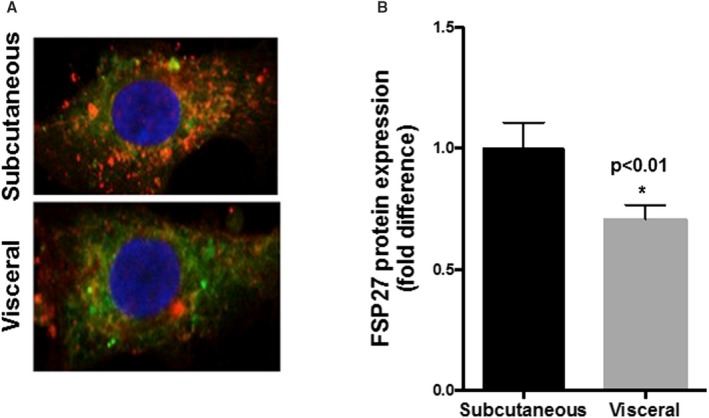
Comparison of FSP27 (fat‐specific protein 27) protein in primary endothelial cells from subcutaneous vs visceral fat of obese subjects. A, Representative immunofluorescence image of isolated endothelial cells demonstrating significantly reduced expression of FSP27 protein (red=FSP27, green=CD31, endothelial cell marker, blue=DAPI, a nuclear stain) in endothelial cells isolated from visceral compared with subcutaneous depots from obese subjects. B, Quantification of FSP27 protein expression in endothelial cells isolated from subcutaneous and visceral fat (n=14, P<0.01). Data are presented as arbitrary units and as mean±SEM, indexed to 1 for the subcutaneous depot. *indicates statistical significance, such as P < 0.05.
FSP27 Modulates Insulin‐Mediated Vasodilation
We have previously shown that insulin‐mediated endothelium dependent vasodilation in arterioles isolated from visceral fat is impaired compared with arterioles from subcutaneous fat in human obesity.33 Consequently, we examined insulin‐mediated vasomotor function of arterioles isolated from visceral fat, with and without human recombinant FSP27 (rFSP27) treatment in paired samples from each subject. We established an optimal dose of rFSP27 for arterioles by performing dose response experiments (data not displayed). Exposing arterioles to 5‐nmol/L rFSP27 for an hour significantly improved insulin‐mediated vasodilation (Figure 2, n=10, P<0.01). Endothelium‐independent vasodilation in response to papavarine was intact, and the positive response was nearly abolished by eNOS inhibitor Nw‐nitro‐L‐arginine methyl ester (L‐NAME, 300‐μmol/L) suggesting that the beneficial mechanism was related primarily to improved endothelial NO bioaction.
Figure 2.
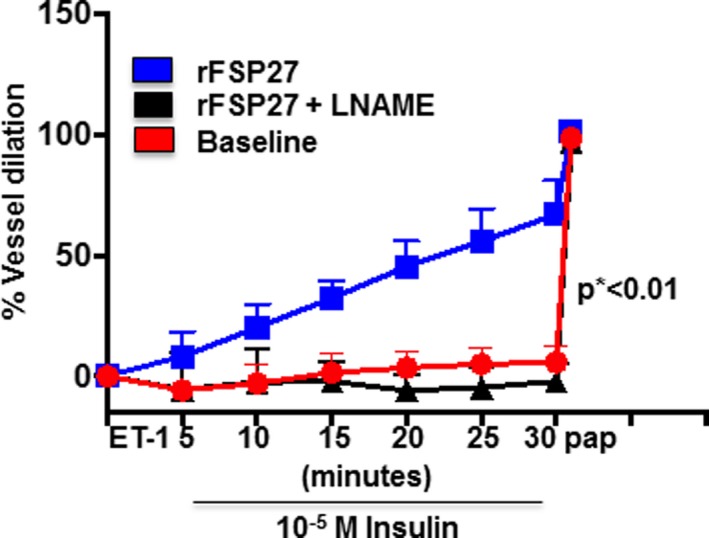
Endothelium‐dependent, insulin‐mediated vasodilation of adipose arterioles from visceral fat. Endothelium‐dependent, insulin‐mediated vasodilation was severely impaired in arterioles from visceral depot (circle). Within an hour of treatment with human recombinant FSP27 (5‐nmol/L) endothelium‐dependent, insulin‐mediated vasodilation of arterioles from the visceral depot improved significantly (square). The improvement in vasodilation with recombinant FSP27 was completely blocked by N(ω)‐nitro‐L‐arginine methyl ester (L‐NAME) (triangle). (n=10 vessels, by repeated measures ANOVA P<0.01). Data are presented as mean±SEM. ET indicates endothelin; pap, papaverine. *indicates statistical significance, such as P < 0.05.
Recombinant Human FSP27 Improves Insulin‐Mediated Activation of eNOS
To build on our observation in Figure 2, we exposed visceral fat to rFSP27 (100‐nmol/L) and examined insulin‐mediated AKT and eNOS activation at serine 473 and 1177, respectively. As shown in Figure 3A through 3C (n=10, P<0.05), rFSP27 significantly increased insulin‐mediated AKT and eNOS activation, suggesting that FSP27 modulates vascular function, in part, by augmenting eNOS bioaction.
Figure 3.
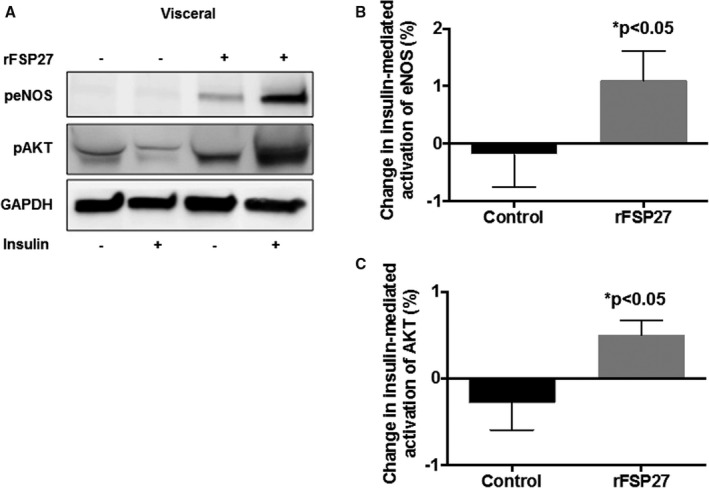
Insulin‐mediated activation of eNOS (endothelial NO synthase) and AKT (protein Kinase B) in response to recombinant FSP27 in visceral depot. A, Representative visceral adipose tissue immunoblot demonstrating severe impairment in insulin‐mediated activation of eNOS and AKT in visceral fat. After 24 hours of rFSP27 (recombinant FSP27) exposure, insulin‐mediated activation is restored. B, Quantification of percent change in insulin‐mediated activation of eNOS at baseline and after 24 hours of treatment with rFSP27 in the visceral depot. C, Quantification of percent change in insulin‐mediated activation of AKT at baseline and after 24 hours of treatment with rFSP27 in the visceral depot (n=10, P<0.05). Data are presented as arbitrary units (au) and as mean±SEM. rFSP27 indicates recombinant fat‐specific protein 27. *indicates statistical significance, such as P < 0.05.
Overexpression of FSP27 Improves Nitric Oxide Production
We overexpressed human FSP27, for the first time to our knowledge, in human primary endothelial cells isolated from visceral fat using an adenoviral transfer method. As shown in Figure 4A, using an adenoviral vector fusion construct that specifically overexpressed FSP27 intracellularly, we were able to increase cellular FSP27 protein expression by >2‐fold as measured by immunofluorescence. This significantly increased insulin‐mediated NO production, as measured by DAF‐2DA (Figure 4B, n=5, P<0.05), complementing our functional data.
Figure 4.
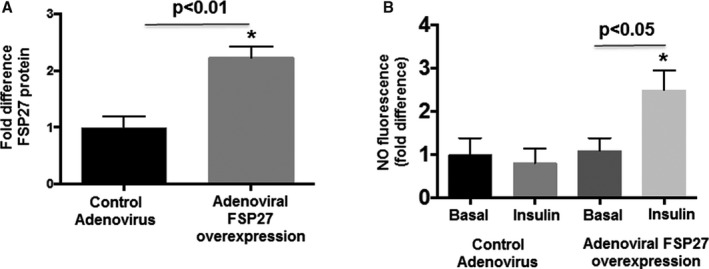
Effect of FSP27 overexpression of FSP27 and insulin on nitric oxide (NO) production in primary endothelial cells isolated from visceral fat. A, Human adenovirus construct of FSP27 and control adenovirus was transfected into primary endothelial cells isolated from visceral fat (n=5, P<0.01). B, Quantification of insulin‐mediated production of NO in control virus vs FSP27 over‐expressed primary endothelial cells isolated from visceral fat (n=5, P<0.05). Data are presented as arbitrary units (au) and as mean±SEM. FSP27 indicates fat‐specific protein 27. *indicates statistical significance, such as P < 0.05.
Inhibition of FSP27 Via siRNA Impairs Insulin‐Mediated Activation of eNOS and Nitric Oxide Production
Conversely, we silenced FSP27 using siRNA (≈50%–60% reduction) in subcutaneous fat from paired samples where visceral fat was used in experiments for Figure 3A through 3C. To support our findings, FSP27 silencing had the opposite effect and impaired insulin‐mediated activation of eNOS and AKT (Figure 5A through 5C, n=10, P<0.05). Additionally, we inhibited FSP27 in cultured HAECs and expectedly observed actions opposite to rFSP27 in primary cells. As displayed in Figure S1A and S1B, biological inhibition of FSP27 impaired insulin‐mediated NO production (Figure S1A n=3, P<0.01, Figure S1B n=3, P<0.05).
Figure 5.
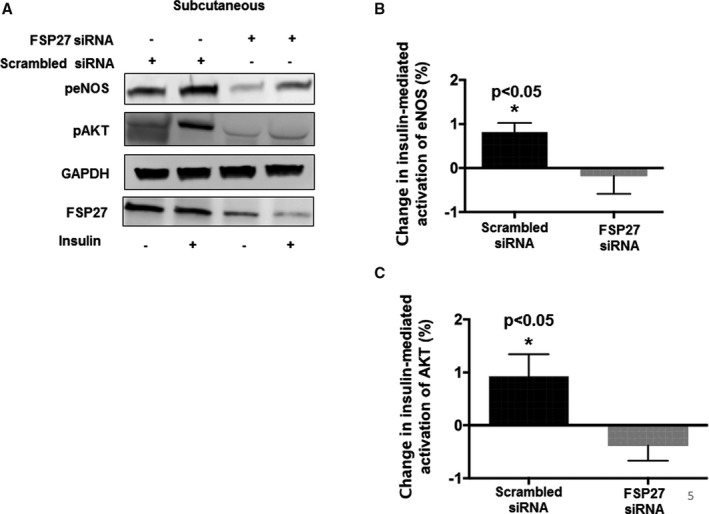
Insulin‐mediated activation of eNOS (endothelial NO synthase) and AKT (protein Kinase B) in response to siRNA‐mediated knockdown of FSP27 in the subcutaneous depot. A, Representative immunoblot demonstrating insulin‐mediated activation of eNOS and AKT in subcutaneous fat under scrambled siRNA (small interfering RNA) conditions and after knockdown of FSP27 by siRNA. B, Quantification of percent change in insulin‐mediated activation of eNOS at baseline and after siRNA‐mediated knockdown of FSP27 in the subcutaneous fat depot. C, Quantification of percent change in insulin‐mediated activation of AKT at baseline and after siRNA‐mediated knockdown of FSP27 in subcutaneous fat depot (n=10, P<0.05). Data are presented as arbitrary units and as mean±SEM. FSP27 indicates fat‐specific protein 27. *indicates statistical significance, such as P < 0.05.
FSP27 Modulates Angiogenesis
We then explored whether FSP27 influences broader functions of blood vessels beyond vasodilation and focused on angiogenesis. We and others have shown that the visceral depot of obese humans display defective angiogenesis35, 37 which has been linked to insulin resistance.29 We used a method of angiogenic assessment involving a tube formation assay that quantifies the capacity of endothelial cells to organize and coalesce into capillary‐like structures forming a rudimentary lumen within hours.36 As shown in Figure 6A and 6B (n=7, P<0.05), isolated primary endothelial cells from obese subjects from the visceral depot exhibited weak vasculogenic behavior (control), whereas rFSP27 significantly improved channel formation at 6 hours. Correspondingly, we repeated these experiments with siRNA‐mediated inhibition of FSP27 in HAECs which decreased the capacity of endothelial cells to form capillary‐like structures (Figure S1C, n=3, P<0.01).
Figure 6.

In‐vitro angiogenic capacity of primary endothelial cells from visceral fat in response to recombinant FSP27 exposure. A, Representative images of primary endothelial cells isolated from visceral fat that exhibited increased angiogenic branch formation when exposed to rFSP27 (recombinant FSP27) compared with control. B, Quantification of in‐vitro angiogenic capacity of primary endothelial, cells with and without rFSP27 treatment for up to 6 hours (n=7, P<0.05). rFSP27 indicates recombinant fat‐specific protein 27. *indicates statistical significance, such as P < 0.05.
FSP27 Regulates Vascular Function Via VEGF‐A
To understand the mechanism of a potential cell autonomous action of FSP27 on vascular function, we considered a potential role of vascular endothelial growth factor‐A (VEGF‐A), a master regulator of neovascularization, and also stimulus for nitric oxide synthesis and release.38, 39 We probed for an interaction between FSP27 and VEGF‐A using co‐immunoprecipitation and found an interaction between the 2 proteins (Figure 7A). In vitro transcription and translation co‐IP immunoblot assay confirmed direct interaction of FSP27 and VEGF‐A (Figure 7B), suggesting a functional relationship between these proteins.
Figure 7.
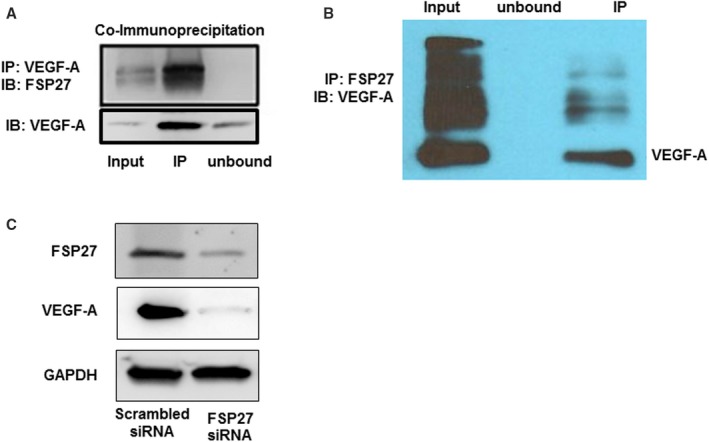
Interaction of FSP27 and VEGF‐A. A, Co‐immunoprecipitation of FSP27 followed by immunoblot of VEGF‐A in cultured human aortic endothelial cells demonstrating an interaction between FSP27 and VEGF‐A. B, In‐vitro transcription translation of FSP27 and VEGF‐A followed by immunoblot of FSP27 depicting an interaction between FSP27 and VEGF‐A. C, Western blot showing that siRNA (small interfering RNA)‐mediated inhibition of FSP27 nearly fully abolished VEGF‐A in human aortic endothelial cells. Experiments were repeated 3 times. FSP27 indicates fat‐specific protein 27; IP, immunoprecipitation; IB, immunoblot; VEFG‐A, vascular endothelial growth factor‐A.
Further, using cultured HAECs we knocked‐down FSP27 using siRNA, and measured VEGF‐A protein to determine whether alteration in FSP27 modulates it. As displayed in Figure 7C, FSP27 silencing nearly abolished VEGF‐A expression, and both proteins co‐localized in primary endothelial cells isolated from human fat (Figure S2). Additionally, we found that exposure of visceral fat to rFSP27 (100‐nmol/L) for 24 hours significantly increased adipose tissue VEGF‐A elaboration (Figure S3A, n=11, P<0.01), while knockdown of FSP27 in subcutaneous fat reduced VEGF‐A secretion (Figure S3B, n=10, P<0.01). Our observation of reduced VEGF‐A elaboration rather than complete shutdown in fat with FSP27 knockdown is likely attributed to VEGF‐A production from other cell lines such as macrophages not under FSP27 regulatory control. To support this, as depicted in Figure S4, we observed abundant VEGF‐A in cultured human macrophages but absence of FSP27. These data suggest that FSP27 bioaction in endothelial cells may be regulated via control of VEGF‐A. Moreover, to explore the mode of action of rFSP27 at the cellular level, we exposed cultured fibroblast (which do not express any endogenous FSP27) to rFSP27 for 2 hours and performed 3‐dimensional immunohistochemistry. As shown in Figure S5, exogenous rFSP27 enters and localizes internally in the cytosol rather than organizing around the cell membrane, suggesting that mode of bioaction of FSP27 may not be through cell surface receptors.
Effect of Weight Loss on FSP27
We explored the effects of bariatric surgery on FSP27 gene expression and adipose microvascular vascular function. A subset of subjects (n=7) who participated in the follow‐up study were followed longitudinally for up to 12±3 months after the operation and provided subcutaneous biopsy samples after 30% weight loss. Their characteristics are displayed in Table 2. Following extensive weight loss, angiogenic capacity of blood vessels in the subcutaneous depot significantly increased (Figure 8A, n=7, P<0.05). In the same‐paired samples, we observed parallel upregulation of FSP27 mRNA expression (Figure 8B, n=7, P<0.05), which correlated positively with capillary sprouting despite the relatively small sample (r=0.79, P<0.05).
Figure 8.
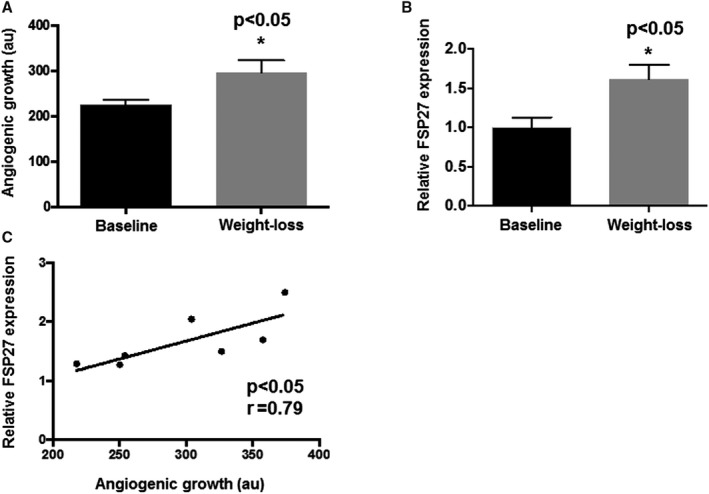
Angiogenic capacity and FSP27 mRNA expression of subcutaneous fat after weight‐loss. A, Longitudinally, paired fat tissue explants demonstrated increased angiogenic growth in subcutaneous depots after weight‐loss (B) mRNA expression of FSP27 increased following weight loss and (C) positively correlated with angiogenic sprouting (n=7, P<0.05). au indicates arbitrary units; FSP27, fat‐specific protein 27. *indicates statistical significance, such as P < 0.05.
Discussion
In the present study, we identified to our knowledge for the first time, FSP27 as a novel regulator of vascular function that is downregulated in association with visceral obesity. We used several complementary approaches harnessing physiological studies of vascular insulin resistance, endothelial vasodilator function, and angiogenesis in live microvessels, as well as gain‐and‐loss of function biological methods exclusively using human samples to characterize the central role of FSP27 in the control of vascular phenotype. Specifically, recombinant FSP27 and/or targeted adenoviral endothelial FSP27 overexpression increased eNOS phosphorylation, NO production, and rescued defective angiogenic and vasodilator responses in dysfunctional human microvessels. Conversely, FSP27 siRNA‐blunted insulin‐mediated AKT/eNOS activation and impaired angiogenesis. Additionally, we observed that surgical weight loss bolstered adipose tissue FSP27 expression and augmented angiogenic capacity. From a mechanistic standpoint, we identified FSP27 as a previously unrecognized modulator of VEGF‐A, a master regulator of angiogenesis and stimulus for NO production.38, 39 Strikingly, FSP27 silencing in endothelial cells nearly fully abolished VEGF‐A expression, and its knock‐down in whole adipose tissue reduced VEGF‐A elaboration. Collectively, these data strongly support a key role and functional significance of FSP27 as an endogenous modulator of vascular function in human obesity that has not been previously described that may play a key role in the pathogenesis of vascular disease.
It had been generally considered that FSP27 functions primarily as a regulator of lipolysis in adipocytes and hence its designation as a “fat specific” protein. The existing literature exclusively describes its functions in relationship to metabolism, with experimental models showing that FSP27 deletion augments basal lipolysis and promotes insulin‐resistance and hepatosteatosis under high‐fat diet conditions.23 Overexpression of FSP27 in human adipocytes protects from fatty acid induced insulin resistance, whereas its silencing impairs insulin‐stimulated activation of AKT.19 A major finding of our study is the discovery of FSP27 in vascular endothelial cells that is quantifiable and its expression downregulated in endothelial cells isolated from visceral compared with subcutaneous fat depots. We previously demonstrated that the visceral microenvironment is associated with several microvascular perturbations linked to vasodilator dysfunction and defective angiogenesis.29, 33, 35 Clinically, accumulation of visceral fat has been most closely linked to the development of cardiometabolic disease and while underlying mechanisms are not fully understood, adipose tissue dysfunction and overproduction of pro‐atherogenic adipocytokines has been a leading hypothesis. Among a large cadre of candidate mediators that have been put forth, FSP27 may be unique as it regulates insulin sensitivity and is differentially expressed in both adipocytes and endothelial cells in varying adipose depots and under conditions of obesogenic stress. While adipocyte FSP27 deficiency could negatively impact the vasculature by stimulating free fatty acid induced lipotoxicity, our data suggest that FSP27 also has distinct cell autonomous functions in endothelial cells. Recent studies demonstrate endothelial presence of lipid droplets as well as trans‐differentiation with adipocytes; however, their cellular functions are not well known.40, 41, 42, 43
In this regard, we demonstrated that FSP27 modulation in endothelial cells produced striking changes in their phenotype. We considered the role of VEGF‐A since we identified a direct interaction between FSP27 and VEGF‐A by in vitro transcription and translation, and VEGF‐A is known to exert control over angiogenesis and NO‐dependent processes.38, 39, 44 Modulation of FSP27 bioaction, either by rFSP27 or siRNA, induced changes in VEGF‐A secretion from human adipose tissue, and siRNA knockdown of FSP27 in cultured endothelial cells induced a profound decrease in VEGF‐A protein expression. Within the adipose microenvironment, adipocytes, and macrophages are likely the major sources of VEGF‐A.45, 46 We did not observe any significant FSP27 expression in cultured human macrophages which supports our finding that adipose FSP27 silencing in fat did not completely shut off VEGF production. Animal models of FSP2747 or VEGF‐A knockout48 induce directionally similar metabolic abnormalities and insulin resistance. FSP27 may regulate VEGF‐A at several stages including transcription translation as we observed changes in VEGF‐A protein within 24 hours of FSP27 modulation (both with rFSP27 and siRNA), or potentially direct effects as vasomotor function improved rapidly with rFSP27 exposure.
In the present study, we also explored the role of bariatric surgery in a subset of subjects that provided repeat adipose tissue after profound weight loss. Bariatric surgery has emerged as an effective treatment for obesity and its cardiometabolic comorbidities, as it improves cardiac risk factors, remits diabetes mellitus, and improves long‐term total and cardiovascular mortality.49, 50, 51, 52 The effect of weight change on FSP27 are limited, as gene expression in the liver fell,53 while increased in subcutaneous fat with weight loss.54 We similarly observed FSP27 upregulation in human fat after 30% weight loss that was correlated with increased angiogenic sprouting in adipose tissue explants. The clinical importance of increased angiogenic capacity in human fat is an open question; however, capillary rarefaction and local tissue pseudo‐hypoxia have been linked to mechanisms of adipose tissue dysfunction locally,55 and consequently whole‐body metabolic dysfunction.
Our current study has several limitations. First, our study consists of severely obese subjects undergoing bariatric surgery, with a relatively small sample size in some experiments, and findings may not be applicable to individuals with milder degrees of obesity. Second, the experimental set‐up involved ex vivo manipulations that may not fully recapitulate in vivo conditions. Third, the majority of study populations were women, which reflects general clinical practice and known sex differences in population that seek weight loss treatments. Fourth, many of our findings are in adipose tissue microvessels and may not necessarily apply to the systemic vasculature which merits further investigation. However, this is counterbalanced by our work with primary tissue from living subjects that brings high translational value. We also emphasize that our ability to directly access human samples and examine live dysfunctional blood vessels within minutes after surgical biopsy provides us with major opportunities to discover novel regulatory networks. Mechanisms learned from the fat microenvironment could have clinical impact and provide translational clues to systemic disease or drugable targets. Adipose arteriolar function correlates with systemic endothelial function, cerebrovascular responses, and tracks cardiovascular risk factors.56 Fifth, while we explored regulation of VEGF‐A by direct interaction with FSP27, the mode of action of FSP27 has not been fully elucidated and mapping out binding domains of these proteins was beyond the scope of the current paper that will be a focus of our future studies. Lastly, follow‐up biopsies were limited to subcutaneous fat depots since these were the only reachable sites without requiring repeat surgery for visceral access that could not be justified for solely research purposes.
Conclusions
In conclusion, we believe we have identified a previously unrecognized, and important regulator of arteriolar vasomotor function and angiogenesis. Obesity‐related downregulation of FSP27 may link obesity to cardiometabolic risk. Further studies are needed to provide functional frameworks to understand the unique features of FSP27 signaling and it pathological impact on the human vascular system.
Sources of Funding
Dr Karki is supported by National Institutes of Health (NIH) grant K01DK114897, and Dr Farb is supported by NIH grant K23HL135394. Dr Apovian is supported by NIH grant P30DK46200, and Dr Puri is supported by NIH grant R01DK101711, R01HL140836, MERCK‐GGI (2018) and funds from Osteopathic Heritage Foundation's Vision 2020 to Heritage college of Osteopathic medicine at Ohio University. Dr Gokce is supported by NIH grants R01HL126141 and R01HL140836.
Disclosures
Dr Apovian has participated on advisory boards for Nutrisystem, Zafgen, Sanofi‐Aventis, Orexigen, EnteroMedics, GI Dynamics, Scientific Intake, Gelesis, Novo Nordisk, SetPoint Health, Xeno Biosciences, Rhythm Pharmaceuticals, Eisai and Takeda; she has received research funding from Aspire Bariatrics, GI Dynamics, Orexigen, Takeda, the Vela Foundation, Gelesis, Energesis and Coherence Lab; she has participated on the Takeda Speakers Bureau for the medication Contrave; and she has owned stock in Science‐Smart LLC. The remaining authors have no disclosures to report.
Supporting information
Figure S1. Inhibition of fat‐specific protein 27 in cultured human arterial endothelial cells and measurement of vascular function. A, Quantification of siRNA mediated knockdown of fat‐specific protein 27 in cultured human arterial endothelial cells. B, siRNA mediated inhibition of fat‐specific protein 27 in human arterial endothelial cells impairs insulin‐mediated nitric oxide production. C, Quantification of in‐vitro angiogenic capacity of human arterial endothelial cells after siRNA‐mediated knockdown of fat‐specific protein 27. Experiments were repeated 3 times.
Figure S2. Co ‐localization of VEGF‐A and FSP27 in primary endothelial cells isolated from human fat. A, Merged image of FSP27 and VEGF‐A. B, Image of VEGF‐A alone. C, Image of FSP27 alone. Green=VEGF‐A, red=FSP27, blue=DAPI, a nuclear stain.
Figure S3. Secretion of VEGF‐A from human visceral and subcutaneous fat in response to recombinant human FSP27 and FSP27 siRNA. A, Exposure of visceral fat to rFSP27 increased VEGF‐A secretion (n=11, P<0.01). B, Conversely, siRNA FSP27 knockdown significantly decreased VEGF‐A secretion from subcutaneous fat. (n=10, P<0.01). Data are expressed as picogram of VEGF‐A in millilters of media per milligrams of total protein in tissue.
Figure S4. Protein levels of VEGF‐A and FSP27 in human aortic endothelial cells and human peripheral blood macrophages. Western blot showing prominent protein expression of FSP27 and VEGF‐A in commercially available human aortic endothelial cells while FSP27 expression was essentially absent in macrophages.
Figure S5. Three‐dimensional immunohistochemistry of fibroblasts demonstrating intracellular/cytosol localization of rFSP27. Green‐rFSP27, blue=nucleus.
(J Am Heart Assoc. 2019;8:e011431 DOI: 10.1161/JAHA.118.011431.)
References
- 1. Ogden CL, Carroll MD, Fryar CD, Flegal KM. Prevalence of obesity among adults and youth: United States, 2011–2014. NCHS Data Brief. 2015;219:1–8. [PubMed] [Google Scholar]
- 2. Flegal KMCM, Kit BK, Ogden CL. Prevalence of obesity and trends in the distribution of body mass index among US adults. JAMA. 2012;307:491–497. [DOI] [PubMed] [Google Scholar]
- 3. Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, Abraham JP, Abu‐Rmeileh NME, Achoki T, AlBuhairan FS, Alemu ZA, Alfonso R, Ali MK, Ali R, Guzman NA, Ammar W, Anwari P, Banerjee A, Barquera S, Basu S, Bennett DA, Bhutta ZQ, Blore J, Cabral N, Nonato IC, Chang J, Chowdhury R, Courville KJ, Criqui MH, Cundiff DK, Dabhadkar KC, Dandona L, Davis A, Dayama A, Dharmaratne SD, Ding EL, Durrani AM, Esteghamati A, Farzadfar F, Fay DFJ, Feigin VL, Flaxman A, Forouzanfar MH, Goto A, Green MA, Gupta R, Hafezi‐Nejad N, Hankey GJ, Harewood HC, Havmoeller R, Hay S, Hernandez L, Husseini A, Idrisov BT, Ikeda N, Islami F, Jahangir E, Jassal SK, Jee SH, Reys MJ, Jonas JB, Kabagambe EK, Khalifa SEAH, Kengne AP, Khader YS, Khang Y, Kim D, Kimokoti RW, Kinge JM, Kokubo Y, Kosen S, Kwan G, Lai T, Leinsalu M, Li Y, Liang X, Liu S, Logroscino G, Lotufo PA, Lu Y, Ma J, Mainoo NK, Mensah GA, Merriman TR, Mokdad AH, Moschandreas J, Naghavi M, Naheed A, Nand D, Narayan KMV, Nelson EL, Neuhouser ML, Nisar MI, Ohkubo T, Oti SO, Pedroza A, Prabhakaran D, Roy N, Sampson U, Seo H, Sepanlou SG, Shibuya K, Shiri R, Shiue I, Singh GM, Singh JA, Skirbekk V, Stapelberg NJC, Sturua L, Sykes BL, Tobias M, Tran BX, Trasande L, Toyoshima H, Vijver SVD, Vasankari TJ, Veerman L, Velasquez‐Melendez G, Vlassov VV, Vollset SE, Vos T, Wang C, Wang X, Weiderpass E, Werdecker A, Wright JL, Yang YC, Yatsuya H, Yoon J, Yoon S, Zhao Y, Zhou M, Zhu S, Lopez AD, Murray CJL, Gakidou E. Global, regional, and national prevalence of overweight and obesity in children and adults during 1980–2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet. 2014;384:766–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Jensen MD, Ryan DH, Apovian CM, Ard JD, Comuzzie AG, Donato KA, Hu FB, Hubbard VS, Jakicic JM, Kushner RF, Loria CM, Millen BE, Nonas CA, Pi‐Sunyer FX, Stevens J, Stevens VJ, Wadden TA, Wolfe BM, Yanovski SZ; American College of Cardiology/American Heart Association Task Force on Practice Guidelines and Obesity Society . 2013 AHA/ACC/TOS guideline for the management of overweight and obesity in adults: a report of the American College of Cardiology/American Heart Association Task Force on Practice Guidelines and The Obesity Society. Circulation. 2014;129:S102–S138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. GBD 2015 Obesity Collaborators , Afshin A, Forouzanfar MH, Reitsma MB, Sur P, Estep K, Lee A, Marczak L, Mokdad AH, Moradi‐Lakeh M, Naghavi M, Salama JS, Vos T, Abate KH, Abbafati C, Ahmed MB, Al‐Aly Z, Alkerwi A, Al‐Raddadi R, Amare AT, Amberbir A, Amegah AK, Amini E, Amrock SM, Anjana RM, Ärnlöv J, Asayesh H, Banerjee A, Barac A, Baye E, Bennett DA, Beyene AS, Biadgilign S, Biryukov S, Bjertness E, Boneya DJ, Campos‐Nonato I, Carrero JJ, Cecilio P, Cercy K, Ciobanu LG, Cornaby L, Damtew SA, Dandona L, Dandona R, Dharmaratne SD, Duncan BB, Eshrati B, Esteghamati A, Feigin VL, Fernandes JC, Fürst T, Gebrehiwot TT, Gold A, Gona PN, Goto A, Habtewold TD, Hadush KT, Hafezi‐Nejad N, Hay SI, Horino M, Islami F, Kamal R, Kasaeian A, Katikireddi SV, Kengne AP, Kesavachandran CN, Khader YS, Khang YH, Khubchandani J, Kim D, Kim YJ, Kinfu Y, Kosen S, Ku T, Defo BK, Kumar GA, Larson HJ, Leinsalu M, Liang X, Lim SS, Liu P, Lopez AD, Lozano R, Majeed A, Malekzadeh R, Malta DC, Mazidi M, McAlinden C, McGarvey ST, Mengistu DT, Mensah GA, Mensink GBM, Mezgebe HB, Mirrakhimov EM, Mueller UO, Noubiap JJ, Obermeyer CM, Ogbo FA, Owolabi MO, Patton GC, Pourmalek F, Qorbani M, Rafay A, Rai RK, Ranabhat CL, Reinig N, Safiri S, Salomon JA, Sanabria JR, Santos IS, Sartorius B, Sawhney M, Schmidhuber J, Schutte AE, Schmidt MI, Sepanlou SG, Shamsizadeh M, Sheikhbahaei S, Shin MJ, Shiri R, Shiue I, Roba HS, Silva DAS, Silverberg JI, Singh JA, Stranges S, Swaminathan S, Tabarés‐Seisdedos R, Tadese F, Tedla BA, Tegegne BS, Terkawi AS, Thakur JS, Tonelli M, Topor‐Madry R, Tyrovolas S, Ukwaja KN, Uthman OA, Vaezghasemi M, Vasankari T, Vlassov VV, Vollset SE, Weiderpass E, Werdecker A, Wesana J, Westerman R, Yano Y, Yonemoto N, Yonga G, Zaidi Z, Zenebe ZM, Zipkin B, Murray CJL. Health effects of overweight and obesity in 195 countries over 25 years. N Engl J Med. 2017;377:13–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Britton KA, Fox CS. Ectopic fat depots and cardiovascular disease. Circulation. 2011;124:e837–e841. [DOI] [PubMed] [Google Scholar]
- 7. Shah RV, Murthy VL, Abbasi SA, Blankstein R, Kwong RY, Goldfine AB, Jerosch‐Herold M, Lima JA, Ding J, Allison MA. Visceral adiposity and the risk of metabolic syndrome across body mass index: the MESA study. JACC Cardiovasc Imaging. 2014;7:1221–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Neeland IJ, Poirier P, Despres JP. Cardiovascular and metabolic heterogeneity of obesity: clinical challenges and implications for management. Circulation. 2018;137:1391–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Unger RH, Clark GO, Scherer PE, Orci L. Lipid homeostasis, lipotoxicity and the metabolic syndrome. Biochem Biophys Acta. 2010;1801:209–214. [DOI] [PubMed] [Google Scholar]
- 10. Shulman GI. Ectopic fat in insulin resistance, dyslipidemia, and cardiometabolic disease. N Engl J Med. 2014;371:1131–1141. [DOI] [PubMed] [Google Scholar]
- 11. Nakamura K, Fuster JJ, Walsh K. Adipokines: a link between obesity and cardiovascular disease. J Cardiol. 2014;63:250–259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Van Gaal LF, Mertens IL, De Block CE. Mechanisms linking obesity with cardiovascular disease. Nature. 2006;444:875–880. [DOI] [PubMed] [Google Scholar]
- 13. Muniyappa R, Montagnani M, Koh KK, Quon MJ. Cardiovascular actions of insulin. Endocr Rev. 2007;28:463–491. [DOI] [PubMed] [Google Scholar]
- 14. Widlansky ME, Gokce N, Keaney JF Jr, Vita JA. The clinical implications of endothelial dysfunction. J Am Coll Cardiol. 2003;42:1149–1160. [DOI] [PubMed] [Google Scholar]
- 15. Deanfield JE, Halcox JP, Rabelink TJ. Endothelial function and dysfunction: testing and clinical relevance. Circulation. 2007;115:1285–1295. [DOI] [PubMed] [Google Scholar]
- 16. Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, Franke TF, Papapetropoulos A, Sessa WC. Regulation of endothelium‐derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gage MC, Yuldasheva NY, Viswambharan H, Sukumar P, Cubbon RM, Galloway S, Imrie H, Skromna A, Smith J, Jackson CL, Kearney MT, Wheatcroft SB. Endothelium‐specific insulin resistance leads to accelerated atherosclerosis in areas with disturbed flow patterns: a role for reactive oxygen species. Atherosclerosis. 2013;230:131–139. [DOI] [PubMed] [Google Scholar]
- 18. Duncan ER, Crossey PA, Walker S, Anilkumar N, Poston L, Douglas G, Ezzat VA, Wheatcroft SB, Shah AM, Kearney MT. Effect of endothelium‐specific insulin resistance on endothelial function in vivo. Diabetes. 2008;57:3307–3314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Grahn TH, Kaur R, Yin J, Schweiger M, Sharma VM, Lee MJ, Ido Y, Smas CM, Zechner R, Lass A, Puri V. Fat‐specific protein 27 (FSP27) interacts with adipose triglyceride lipase (ATGL) to regulate lipolysis and insulin sensitivity in human adipocytes. J Biol Chem. 2014;289:12029–12039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Singh M, Kaur R, Lee MJ, Pickering RT, Sharma VM, Puri V, Kandror KV. Fat‐specific protein 27 inhibits lipolysis by facilitating the inhibitory effect of transcription factor Egr1 on transcription of adipose triglyceride lipase. J Biol Chem. 2014;289:14481–14487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Gong J, Sun Z, Wu L, Xu W, Schieber N, Xu D, Shui G, Yang H, Parton RG, Li P. Fsp27 promotes lipid droplet growth by lipid exchange and transfer at lipid droplet contact sites. J Cell Biol. 2011;195:953–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Kim JY, Liu K, Zhou S, Tillison K, Wu Y, Smas CM. Assessment of fat‐specific protein 27 in the adipocyte lineage suggests a dual role for FSP27 in adipocyte metabolism and cell death. Am J Physiol Endocrinol Metab. 2008;294:E654–E667. [DOI] [PubMed] [Google Scholar]
- 23. Tanaka N, Takahashi S, Matsubara T, Jiang C, Sakamoto W, Chanturiya T, Teng R, Gavrilova O, Gonzalez FJ. Adipocyte‐specific disruption of fat‐specific protein 27 causes hepatosteatosis and insulin resistance in high‐fat diet‐fed mice. J Biol Chem. 2015;290:3092–3105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rubio‐Cabezas O, Puri V, Murano I, Saudek V, Semple RK, Dash S, Hyden CS, Bottomley W, Vigouroux C, Magre J, Raymond‐Barker P, Murgatroyd PR, Chawla A, Skepper JN, Chatterjee VK, Suliman S, Patch AM, Agarwal AK, Garg A, Barroso I, Cinti S, Czech MP, Argente J, O'Rahilly S, Savage DB; Consortium LDS . Partial lipodystrophy and insulin resistant diabetes in a patient with a homozygous nonsense mutation in CIDEC. EMBO Mol Med. 2009;1:280–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Bonetti PO, Lerman LO, Lerman A. Endothelial dysfunction: a marker of atherosclerotic risk. Arterioscler Thromb Vasc Biol. 2003;23:168–175. [DOI] [PubMed] [Google Scholar]
- 26. Versari D, Daghini E, Virdis A, Ghiadoni L, Taddei S. Endothelial dysfunction as a target for prevention of cardiovascular disease. Diabetes Care. 2009;32:S314–S321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Simons M, Bonow RO, Chronos NA, Cohen DJ, Giordano FJ, Hammond HK, Laham RJ, Li W, Pike M, Sellke FW, Stegmann TJ, Udelson JE, Rosengart TK. Clinical trials in coronary angiogenesis: issues, problems, consensus: an expert panel summary. Circulation. 2000;102:E73–E86. [DOI] [PubMed] [Google Scholar]
- 28. Farb MG, Tiwari S, Karki S, Ngo DT, Carmine B, Hess DT, Zuriaga MA, Walsh K, Fetterman JL, Hamburg NM, Vita JA, Apovian CM, Gokce N. Cyclooxygenase inhibition improves endothelial vasomotor dysfunction of visceral adipose arterioles in human obesity. Obesity (Silver Spring). 2014;22:349–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Karki S, Farb MG, Ngo DT, Myers S, Puri V, Hamburg NM, Carmine B, Hess DT, Gokce N. Forkhead box O‐1 modulation improves endothelial insulin resistance in human obesity. Arterioscler Thromb Vasc Biol. 2015;35:1498–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Karki S, Ngo DTM, Farb MG, Park SY, Saggese SM, Hamburg NM, Carmine B, Hess DT, Walsh K, Gokce N. WNT5A regulates adipose tissue angiogenesis via antiangiogenic VEGF‐A165b in obese humans. Am J Physiol Heart Circ Physiol. 2017;313:H200–H206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Karki S, Farb MG, Myers S, Apovian C, Hess DT, Gokce N. Effect of bariatric weight loss on the adipose lipolytic transcriptome in obese humans. Mediators Inflamm. 2015;2015:106237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Farb MG, Ganley‐Leal L, Mott M, Liang Y, Ercan B, Widlansky ME, Bigornia SJ, Fiscale AJ, Apovian CM, Carmine B, Hess DT, Vita JA, Gokce N. Arteriolar function in visceral adipose tissue is impaired in human obesity. Arterioscler Thromb Vasc Biol. 2012;32:467–473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Farb MG, Karki S, Park SY, Saggese SM, Carmine B, Hess DT, Apovian C, Fetterman JL, Breton‐Romero R, Hamburg NM, Fuster JJ, Zuriaga MA, Walsh K, Gokce N. WNT5A‐JNK regulation of vascular insulin resistance in human obesity. Vasc Med. 2016;21:489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Farb MG, Park SY, Karki S, Gokce N. Assessment of human adipose tissue microvascular function using videomicroscopy. J Vis Exp. 2017;127:56079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ngo DT, Farb MG, Kikuchi R, Karki S, Tiwari S, Bigornia SJ, Bates DO, LaValley MP, Hamburg NM, Vita JA, Hess DT, Walsh K, Gokce N. Antiangiogenic actions of vascular endothelial growth factor‐A165b, an inhibitory isoform of vascular endothelial growth factor‐A, in human obesity. Circulation. 2014;130:1072–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5:628–635. [DOI] [PubMed] [Google Scholar]
- 37. Gealekman O, Guseva N, Hartigan C, Apotheker S, Gorgoglione M, Gurav K, Tran KV, Straubhaar J, Nicoloro S, Czech MP, Thompson M, Perugini RA, Corvera S. Depot‐specific differences and insufficient subcutaneous adipose tissue angiogenesis in human obesity. Circulation. 2011;123:186–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Gelinas DS, Bernatchez PN, Rollin S, Bazan NG, Sirois MG. Immediate and delayed VEGF‐mediated NO synthesis in endothelial cells: role of PI3K, PKC and PLC pathways. Br J Pharmacol. 2002;137:1021–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Daher Z, Boulay PL, Desjardins F, Gratton JP, Claing A. Vascular endothelial growth factor receptor‐2 activates ADP‐ribosylation factor 1 to promote endothelial nitric‐oxide synthase activation and nitric oxide release from endothelial cells. J Biol Chem. 2010;285:24591–24599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kuo A, Lee MY, Yang K, Gross RW, Sessa WC. Caveolin‐1 regulates lipid droplet metabolism in endothelial cells via autocrine prostacyclin‐stimulated, cAMP‐mediated lipolysis. J Biol Chem. 2018;293:973–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kuo A, Lee MY, Sessa WC. Lipid droplet biogenesis and function in the endothelium. Circ Res. 2017;120:1289–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Tran KV, Gealekman O, Frontini A, Zingaretti MC, Morroni M, Giordano A, Smorlesi A, Perugini J, De Matteis R, Sbarbati A, Corvera S, Cinti S. The vascular endothelium of the adipose tissue gives rise to both white and brown fat cells. Cell Metab. 2012;15:222–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cao Y. Angiogenesis and vascular functions in modulation of obesity, adipose metabolism, and insulin sensitivity. Cell Metab. 2013;18:478–489. [DOI] [PubMed] [Google Scholar]
- 44. Felmeden DC, Blann AD, Lip GY. Angiogenesis: basic pathophysiology and implications for disease. Eur Heart J. 2003;24:586–603. [DOI] [PubMed] [Google Scholar]
- 45. Cho CH, Koh YJ, Han J, Sung HK, Jong Lee H, Morisada T, Schwendener RA, Brekken RA, Kang G, Oike Y, Choi TS, Suda T, Yoo OJ, Koh GY. Angiogenic role of LYVE‐1‐positive macrophages in adipose tissue. Circ Res. 2007;100:e47–e57. [DOI] [PubMed] [Google Scholar]
- 46. Mick GJ, Wang X, McCormick K. White adipocyte vascular endothelial growth factor: regulation by insulin. Endocrinology. 2002;143:948–953. [DOI] [PubMed] [Google Scholar]
- 47. Zhou L, Park SY, Xu L, Xia X, Ye J, Su L, Jeong KH, Hur JH, Oh H, Tamori Y, Zingaretti CM, Cinti S, Argente J, Yu M, Wu L, Ju S, Guan F, Yang H, Choi CS, Savage DB, Li P. Insulin resistance and white adipose tissue inflammation are uncoupled in energetically challenged Fsp27‐deficient mice. Nat Commun. 2015;6:5949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sung HK, Doh KO, Son JE, Park JG, Bae Y, Choi S, Nelson SM, Cowling R, Nagy K, Michael IP, Koh GY, Adamson SL, Pawson T, Nagy A. Adipose vascular endothelial growth factor regulates metabolic homeostasis through angiogenesis. Cell Metab. 2013;17:61–72. [DOI] [PubMed] [Google Scholar]
- 49. Bigornia SJ, Mott MM, Hess DT, Apovian CM, McDonnell ME, Duess MA, Kluge MA, Fiscale AJ, Vita JA, Gokce N. Long‐term successful weight loss improves vascular endothelial function in severely obese individuals. Obesity (Silver Spring). 2010;18:754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Adams TD, Mehta TS, Davidson LE, Hunt SC. All‐cause and cause‐specific mortality associated with bariatric surgery: a review. Curr Atheroscler Rep. 2015;17:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Carlsson LM, Peltonen M, Ahlin S, Anveden A, Bouchard C, Carlsson B, Jacobson P, Lonroth H, Maglio C, Naslund I, Pirazzi C, Romeo S, Sjoholm K, Sjostrom E, Wedel H, Svensson PA, Sjostrom L. Bariatric surgery and prevention of type 2 diabetes in Swedish obese subjects. N Engl J Med. 2012;367:695–704. [DOI] [PubMed] [Google Scholar]
- 52. Hallersund P, Sjostrom L, Olbers T, Lonroth H, Jacobson P, Wallenius V, Naslund I, Carlsson LM, Fandriks L. Gastric bypass surgery is followed by lowered blood pressure and increased diuresis—long term results from the Swedish Obese Subjects (SOS) study. PLoS One. 2012;7:e49696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hall AM, Brunt EM, Klein S, Finck BN. Hepatic expression of cell death‐inducing DFFA‐like effector C in obese subjects is reduced by marked weight loss. Obesity (Silver Spring). 2010;18:417–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Moreno‐Navarrete JM, Ortega F, Serrano M, Rodriguez‐Hermosa JI, Ricart W, Mingrone G, Fernandez‐Real JM. CIDEC/FSP27 and PLIN1 gene expression run in parallel to mitochondrial genes in human adipose tissue, both increasing after weight loss. Int J Obes (Lond). 2014;38:865–872. [DOI] [PubMed] [Google Scholar]
- 55. Fuster JJ, Ouchi N, Gokce N, Walsh K. Obesity‐induced changes in adipose tissue microenvironment and their impact on cardiovascular disease. Circ Res. 2016;118:1786–1807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Dharmashankar K, Welsh A, Wang J, Kizhakekuttu TJ, Ying R, Gutterman DD, Widlansky ME. Nitric oxide synthase‐dependent vasodilation of human subcutaneous arterioles correlates with noninvasive measurements of endothelial function. Am J Hypertens. 2012;25:528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Inhibition of fat‐specific protein 27 in cultured human arterial endothelial cells and measurement of vascular function. A, Quantification of siRNA mediated knockdown of fat‐specific protein 27 in cultured human arterial endothelial cells. B, siRNA mediated inhibition of fat‐specific protein 27 in human arterial endothelial cells impairs insulin‐mediated nitric oxide production. C, Quantification of in‐vitro angiogenic capacity of human arterial endothelial cells after siRNA‐mediated knockdown of fat‐specific protein 27. Experiments were repeated 3 times.
Figure S2. Co ‐localization of VEGF‐A and FSP27 in primary endothelial cells isolated from human fat. A, Merged image of FSP27 and VEGF‐A. B, Image of VEGF‐A alone. C, Image of FSP27 alone. Green=VEGF‐A, red=FSP27, blue=DAPI, a nuclear stain.
Figure S3. Secretion of VEGF‐A from human visceral and subcutaneous fat in response to recombinant human FSP27 and FSP27 siRNA. A, Exposure of visceral fat to rFSP27 increased VEGF‐A secretion (n=11, P<0.01). B, Conversely, siRNA FSP27 knockdown significantly decreased VEGF‐A secretion from subcutaneous fat. (n=10, P<0.01). Data are expressed as picogram of VEGF‐A in millilters of media per milligrams of total protein in tissue.
Figure S4. Protein levels of VEGF‐A and FSP27 in human aortic endothelial cells and human peripheral blood macrophages. Western blot showing prominent protein expression of FSP27 and VEGF‐A in commercially available human aortic endothelial cells while FSP27 expression was essentially absent in macrophages.
Figure S5. Three‐dimensional immunohistochemistry of fibroblasts demonstrating intracellular/cytosol localization of rFSP27. Green‐rFSP27, blue=nucleus.