

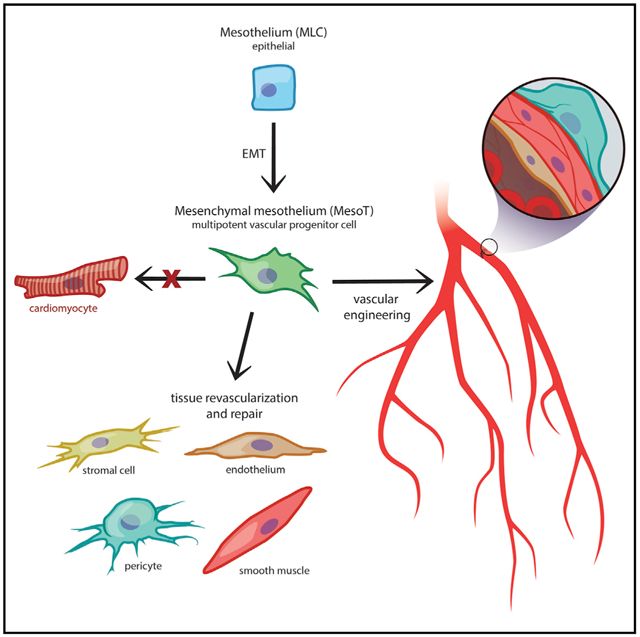
SUMMARY
In this report we describe a human pluripotent stem cell-derived vascular progenitor (MesoT) cell of the mesothelium lineage. MesoT cells are multipotent and generate smooth muscle cells, endothelial cells, and pericytes and self-assemble into vessel-like networks in vitro. MesoT cells transplanted into mechanically damaged neonatal mouse heart migrate into the injured tissue and contribute to nascent coronary vessels in the repair zone. When seeded onto decellularized vascular scaffolds, MesoT cells differentiate into the major vascular lineages and self-assemble into vasculature capable of supporting peripheral blood flow following transplantation. These findings demonstrate in vivo functionality and the potential utility of MesoT cells in vascular engineering applications.
Graphical Abstract
INTRODUCTION
Coelomic organs, including the heart, spleen, lungs, liver, and gut, are lined on their outer surface by a thin layer of cells with epithelial characteristics known as visceral mesothelium (Mutsaers and Wilkosz, 2007). During early development, mesothelium is highly dynamic and critical for growth and maintenance of the underlying tissue. Following the formation of the mesothelial layer, a subpopulation of these cells undergo an epithelial-to-mesenchymal transition (EMT) and invade the underlying tissue. Here, they transition through a mesenchymal progenitor intermediate and in response to local signals they differentiate into vascular lineages, which contribute to a nascent vascular network (Asahina et al., 2009; Cano et al., 2013; Dixit et al., 2013; Que et al., 2008; Rinkevich et al., 2012; Smith et al., 2011; Wilm et al., 2005; Zangi et al., 2013). Mesothelium-derived progenitor cells with mesenchymal characteristics have been described in the heart (Chong et al., 2011; Rinkevich et al., 2012; Zangi et al., 2013), gut, lungs, and liver (Rinkevich et al., 2012) and contribute to vascularization of these organs during embryonic development and possibly during tissue regeneration (Kikuchi et al., 2011; Smart et al., 2011). Numerous reports have also highlighted the broad potential of mesothelium and mesothelium-derived cells in neo-vascularization and tissue regeneration (Elmadbouh et al., 2005; Lucas, 2007; Mutsaers and Wilkosz, 2007; Shelton and Bader, 2012). For example, the epicardium and its derivatives have been implicated in regenerative responses following myocardial infarction (Elmadbouh et al., 2005; Smart et al., 2011; van Wijk et al., 2012) and mechanical injury (Porrello et al., 2011).
Immediately following ischemic events, re-establishment of blood flow is crucial to prevent further tissue damage and organ functional decline. Endogenous repair mechanisms rely on the resident vascular progenitor population, including mesothelium, to achieve this. Recently, considerable progress has been made to generate vascular lineages (Cheung et al., 2012; James et al., 2010; Liu et al., 2014; Patsch et al., 2015) in vitro. While smooth muscle cells (SMCs) (Cheung et al., 2012; Liu et al., 2014; Patsch et al., 2015) and endothelial cells (ECs) (James et al., 2010; Patsch et al., 2015) have been directly generated from human pluripotent stem cell (hPSC)-derived mesoderm, a multipotent progenitor equivalent to a tissue resident vascular precursor has not been reported. The availability of such a progenitor cell could have utility in revascularization of damaged tissue or potentially in vascular engineering (Colunga and Dalton, 2018). Thus, we attempted to recapitulate the developmental program of mesothelium in order to identify such a cell type in vitro.
In this report, we describe the generation of a multipotent vascular progenitor cell of the mesothelium lineage (MesoT) from hPSCs. MesoT cells exhibit the ability to generate all the required vascular lineages such as SMCs, ECs, and pericytes, which is consistent with reported tissue resident progenitor cells of the coelomic organs. We also report the ability of MesoT cells to revascularize damaged tissue and exhibit its potential utility in tissue engineering contexts. This is of significance because current tissue engineering approaches for generating transplantable blood vessels have been largely limited by the availability and reliability of vascular cells with proven utility. The availability of hPSC-derived multipotent vascular progenitors of the mesothelium lineage opens up opportunities for the advancement of tissue engineering and regenerative medicine.
RESULTS
An hPSC-Derived Cell of the Mesothelium Lineage with Vascular Characteristics
Splanchnic mesoderm (SplM) is a transient progenitor population that arises during embryonic heart formation. Developmentally, SplM is specified via Wnt and bone morphogenetic protein 4 (BMP4) signaling dynamics and is the primitive precursor cell type of epicardium and cardiovascular lineages (Klaus et al., 2012). Equivalent cells can be generated at high efficiency from hPSCs in chemically defined media (CDM) supplemented with Wnt3a and BMP4 (Figures 1A and S1A–S1D). These cells have epithelial characteristics (ZO1+, αSMA−, VIM−) (Figure 1B), express transcription factors that are characteristic of SplM, including ISL1, NKX2.5, and GATA4 (Figures 1B, 1C, S1A, and S1B), and show decreased levels of pluripotency markers NANOG, OCT4, and SOX2 (Figures S1C and S1D).
Figure 1. hPSC-Derived MesoT Displays Molecular Characteristics of Primary Mesothelium.

(A) Sources of cells used for RNA-seq analysis. hPSC-derived MesoT cells and tdTomato+ mesothelium isolated from mouse embryonic gut, liver, heart, and lungs (embryonic day 15.5 [E15.5]) were compared to RNA-seq data in the public domain.
(B) Addition of Wnt3a, BMP4, and retinoic acid (RA) to SplM (ISL1+, NKX2.5+, ZO1+) efficiently generates MesoT (WT1+, αSMA+, VIM+) with a mesenchymal phenotype. Scale bars, 50 μm.
(C) qRT-PCR data showing fold-change of transcript levels for markers of SplM (ISL1, NKX2.5, and GATA4) and MesoT (WT1, TBX18, and TCF21) following directed differentiation of human embryonic stem cells (hESCs, WA09). TaqMan assays for each transcript were performed in technical triplicate and fold-change shown relative to untreated hESCs (WA09) after normalization with 18S RNA.
(D) Left: hierarchical clustering (Euclidean distance, complete linkage) of all human (black box) and mouse (red box) RNA-seq samples according to mouse and human orthologs. Data from both species were log transformed and scaled to mean = 0 and standard deviation = 1 prior to clustering. Right: zoomed-in section of the highlighted portion of the array tree dendrogram (left) showing that hPSC-derived MesoT cells are closely related to human (h) and adult (a) epicardium (m, mouse mesothelium; f, fetal mesothelium). Replicate numbers from independent experiments are indicated. Error bars ± standard deviation. See also Figures S1 and S2 and Table S4.
Retinoic acid (RA) signaling and synthesis plays a crucial role in the specification and identification of mesothelium (Iyer et al., 2015; Kikuchi et al., 2011; Pérez-Pomares and de la Pompa, 2011; Witty et al., 2014) from hPSC-derived mesoderm as well as during development. While characterizing our hPSC-derived SplM, we found that treatment with all-trans RA promoted a morphological transformation (Figure 1B). RA treatment downregulated SplM markers (ISL1, NKX2.5) (Figures 1B and 1C) and promoted an EMT, as shown by loss of ZO1 and increased vimentin and αSMA expression (Figure 1B). The RNA sequencing (RNA-seq) signature of RA-treated cells was then compared to that of human and mouse tissues to identify the lineage of these cells (Figure 1A). Hierarchical clustering analysis of RNA-seq data showed that RA-treated SplM clustered with primary human epicardium and mouse mesothelium isolated from heart, liver, lung, and gut (Figure 1D), suggesting that it belongs to the mesothelium lineage (MesoT). Although MesoT cells exhibit characteristics of embryonic mesothelium at the molecular level such as the expression of transcription factors WT1, TBX18, and TCF21 (Figures 1B, 1C, and S1E–S1G) they also have mesenchymal characteristics (αSMA+, VIM+, ZO1−) (Figure 1B). This contrasts with the typical epithelial characteristics of mesothelium but is reminiscent of mesothelium-derived mesenchymal cells that invade the underlying tissue during organogenesis (Asahina et al., 2009; Que et al., 2008; Smith et al., 2011; Wilm et al., 2005).
To determine whether MesoT cells are descendants of visceral mesothelium, we repeated the differentiation of SplM in CDM supplemented with Wnt3a, BMP4, and RA but in the absence of factors known to promote EMT (Activin A and Fgf2) (Figure S2A). This set of conditions generated epithelial cells that expressed mesothelium markers (Figures S2B and S2C) and were designated as “mesothelium-like cells” (MLCs). Once Activin A and Fgf2 signaling was restored, MLCs transitioned through an EMT and toward a phenotype reminiscent of MesoT cells at the molecular and cellular level (Figure S2C). These results are consistent with the development of hPSC-derived SplM along the mesothelium lineage (Nagai et al., 2013; Tian et al., 2015); first through an epithelial state (MLCs) followed by a migratory state (MesoT cells).
Since mesothelium-derived cells have been implicated in vascular development during embryogenesis (Rinkevich et al., 2012; Zangi et al., 2013), we sought to obtain corroborative evidence that MesoT cells have vascular potential by characterizing their epigenetic signature. We identified a MesoT-specific CpG methylation signature that is non-overlapping with corresponding signatures for SplM, hPSC-derived cardiomyocytes (Laflamme et al., 2007), and hPSCs. A cohort of 1,846 methylated CpGs were identified that fulfilled this condition (Figure S3A). This signature was used to screen an expanded panel of DNA methylation datasets including 30 primary human tissues and primary cell samples. This approach showed that primary SMCs, primary ECs, and umbilical cord cells have a similar methylation signature to MesoT cells (Figure 2A). This indicates that MesoT cells have epigenetic marks consistent with being part of the vascular lineage.
Figure 2. Epigenetic and Transcript Profiles of MesoT Are Similar to Vascular Cell Types.

(A) Hierarchical clustering (Euclidean distance, complete linkage) of human tissue and hESC-derived samples according to beta values for the 1,846 cytosines comprising module 9 of the DNA methylation profile. Array tree dendrograms and the distribution of beta values for these cytosines are presented in heatmap form (top) and as box and whisker plots (bottom).
(B) Cartoon depicting the epigenetic landscape at primed and activated enhancers as MesoT cells transition to a vascular fate. Top portion depicts vascular genes “primed” in MesoT with the presence of K4me1 on histone H3 at enhancer sites. Bottom portion depicts the primed enhancers for vascular genes being activated by addition of K27ac as they differentiate to smooth muscle cells (SMCs) or endothelial cells (ECs).
(C) Enhancer and gene ontology discovery pipeline for data in (D) (Figures S3B–S3D).
(D) Heatmaps of H3K4me1 (red) and H3K27ac (blue) ChIP-seq data for enhancers linked to upregulated genes in primary smooth muscle and ECs, compared to MesoT cells.
(E) Principal component analysis of the top 50% of highly expressed genes in hESC-derived mesothelium cells(MesoT and MLC), primary fetal tissue (liver, heart, brain, spinal cord, and hindbrain), and primary cells (Endo and SMC). MesoT and MesoT (FCS) (self-renewing) cells cluster tightly with vascular cell types (Endo and SMC) and hESC-derived mesothelium (MLC).
If MesoT cells are precursors for either SMCs or ECs, it would be anticipated that enhancers required for specification of these vascular lineages would be in a H3K4me1high H3K27aclow “primed” state (Creyghton et al., 2010; Rada-Iglesias et al., 2011) in MesoT cells (Figure 2B). To address this, the epigenetic state of active enhancers (H3K27achigh) linked to transcriptionally upregulated gene sets in primary SMCs and ECs were analyzed in MesoT cells by chromatin immunoprecipitation sequencing (ChIP-seq) (Figure 2C; Table S1). These results show that active enhancers linked to vascular transcriptional programs are in a primed state in MesoT cells, in contrast to the active state in SMCs and ECs (Figure 2D). A similar analysis was performed where cardiomyocytes (CMs) were compared to MesoT cells. Here, CM-specific enhancers were not primed in MesoT cells (Figure S3B), indicating MesoT cells are not competent for CM differentiation. Gene ontology analysis shows that epigenetically primed (H3K4me1high H3K27aclow) enhancers in MesoT cells are linked to genes that are significantly enriched for functions in vascular development and activity (Figures S3C and S3D). Since epigenetic analysis pointed toward MesoT cells being part of the vascular lineage, a more focused RNA-seq analysis was also performed. Principal component analysis (PCA) of RNA-seq data shows that MesoT transcriptome is not only similar to MLCs but also clusters with vascular cells such as SMCs and endothelial progenitor cells (EPCs) (Figure 2E). Together, these results are consistent with the hypothesis that MesoT cells are of the vascular lineage and are precursors for SMCs and ECs.
MesoT Cells Are Multipotent Vascular Progenitor Cells
With global transcript and epigenetic analysis suggesting MesoT cells as vascular progenitors of mesothelial origin, we next sought to evaluate the differentiation potential of these cells in vitro. MesoT cells were treated with platelet derived growth factor-BB (PDGF-BB) or with a combination of vascular endothelial growth factor (VEGF) and the transforming growth factor β (TGFβ) superfamily type I activin receptor-like kinase inhibitor SB431542, conditions known to promote the differentiation of mesoderm to SMC (Mellgren et al., 2008; Rinkevich et al., 2012) and EC (James et al., 2010) identities, respectively. PDGF-BB treatment of MesoT cells yielded cultures in which ~85% cells expressed the SMC markers calponin, alpha smooth muscle actin (αSMA), and myosin heavy chain 11 (MYH11) (Figures 3A and 3B). The amplification in cell number as hPSCs transitioned to SMCs through a MesoT progenitor state is approximately 30-fold (Figure S4A). Although MesoT cells are αSMA+, they are distinguished from their downstream derivates, SMCs and fibroblasts, based on their differential expression of markers such as calponin, MYH11, DDR2, and WT1 (Figures 3A, S4B, and S4C). Treatment with the acetylcholine receptor agonist carbachol and the membrane depolarizing agent potassium chloride (KCl) both triggered cell contraction within 30 min (Figures 3C and 3D), responses expected of SMCs. In contrast, human embryonic stem cells (hESCs) showed no contractile activity under these conditions (Figure S4D). These observations indicate that MesoT cells are capable of efficiently generating functional SMCs when treated with PDGF-BB.
Figure 3. MesoT Cells Efficiently Differentiate to Smooth Muscle and ECs.

(A) MesoT cells treated with PDGF-BB (50 ng/ml) in CDM (–Activin A) for 12 days were probed with antibodies for alpha smooth muscle actin (αSMA), calponin, and myosin heavy chain 11 (MYH11). Scale bars, 100 μm (left) and 50 μm (right).
(B) MesoT cells grown in CDM (–A) supplemented with PDGF-BB or VEGF-A165 and SB431542 were quantified based on expression of lineage specific markers for SMCs and ECs. All experiments in biological triplicate.
(C) 3,3’-dioctadecyloxacarbocyanine perchlorate (DiO)-labeled SMCs as shown in (A) were treated with 100 μM carbachol or 50 mM KCl to stimulate functional contraction. Scale bar, 200 μm.
(D) SMC surface area was measured after treatment (C). Contraction is shown as the % change in cell surface area for individual cells. Each treatment group was compared to corresponding control time point to determine statistical significance. b = 20.
(E and F) MesoT cells treated with CDM (–A), VEGF-A165, and SB431542 for 12 days were fixed and probed with antibodies for CD31, vWF, and DAPI (E) or characterized by flow cytometry with VE-cadherin (blue) or isotype control (red) (F). Scale bar, 50 μm.
(G) Trans-endothelial electrical resistance (TEER) was measured after culturing MesoT cells in CDM (–A) supplemented with VEGF-A165 alone (+VEGF) or with SB431542 (+VEGF +SB) after 14, 21, and 28 days and compared against primary dermal microvascular endothelium (1° ECs). n = 3.
(H) Barrier integrity was tested by measuring FITC-dextran (40 kDa) perfusion from the apical to basolateral side. Control represents the absence of cells. No statistical significance was determined when comparing cells to 1° ECs.
(I) Schematic of the bioreactor culture system used in (G) and (H).
(J) Immunofluorescence of cell monolayer as in (I) showing expression of tight junction marker ZO1 (top) and H&E staining (bottom). Scale bars, 100 μm.
(K) Transmission electron microscopy image of MesoT-derived endothelium. Red arrows depict tight cell junctions. Inset (top) is depicted on bottom. Scale bars, 500 nm and 200 nm, respectively.
**p = 0.0027 for two-way ANOVA. ****p < 0.0001 for one-tailed t test. Error bars ± SEM. See also Figure S4.
Treatment of MesoT cells with VEGF and SB431542 uniformly increased the expression of endothelial markers von Willebrand factor (vWF), cluster of differentiation 31 (CD31), and vascular endothelial (VE)-cadherin (Figures 3E and 3F). EC markers such as vWF and CD31 are not expressed in MesoT cells or in SMCs (Figures S4B, S4E, and S4F). Functional characterization of VEGF-SB431542 (SB)-treated cells was first evaluated using trans-endothelial electrical resistance (TEER) assays after plating cells on a transwell system coated with a collagen IV-fibronectin matrix. Here, TEER of MesoT cells treated with SB-VEGF increased over 28 days and was comparable or greater than that observed for primary human microvascular ECs (Figure 3G). Fluorescein isothiocyanate (FITC)-dextran diffusion was assessed across cell monolayers treated with VEGF-SB. It was expected that if VEGF-SB-treated MesoT cells generated selectively permeable ECs, they would resist diffusion of FITC-dextran (40 kDa) across the monolayer. As expected, primary EC monolayers allowed only low levels of label to diffuse through the cellular layer. The extent of FITC-dextran retention was even greater in VEGF alone or VEGF-SB-treated MesoT cells (Figure 3H), consistent with formation of a tight epithelial barrier. When cultured on a collagen IV-fibronectin-coated transwell insert (Figure 3I), ECs generated from MesoT cells formed a layer of cells expressing the tight junction protein ZO1 (Figures 3J and S4G). When examined by transmission electron microscopy, cells exhibited typical endothelial architecture with tight junctions (Figure 3K). Together, these data show that functional ECs can be generated from MesoT progenitors at high efficiency. The overall cell amplification as hPSCs differentiate to ECs through a MesoT intermediate is ~50-fold (Figure S4A). The ~30-fold amplification to SMCs and 50-fold amplification to ECs indicates that differentiation of MesoT progenitor cells results in significant amplification of vascular lineages.
To obtain evidence that MesoT cells are multipotent vascular progenitors, it was necessary to establish culture conditions in which MesoT cells could be amplified for approximately 15–20 days to allow for clonal amplification. CDM did not support maintenance and proliferation of MesoT cells over this time frame, but supplementation with 10% fetal bovine serum supported maintenance for up to 9 passages. During this period, MesoT cells retained their proliferative capacity (Figure 4A), cell cycle properties (Figures 4B–4D), and global transcriptome profile (Figure 2E). By interrogating RNA-seq data, we identified several cell surface markers expressed on the surface of MesoT cells that were absent on SplM, making these markers potentially useful for cell purification and single cell analysis. To confirm the utility of these markers, which include CD44, CD73, and CD105, triple CD44+/CD73+/CD105+ MesoT cells were isolated, sorted into 96-well dishes, and, following amplification, differentiated under smooth muscle (+PDGF-BB) and EC (+VEGF) conditions (Figures 4E and 4F). All 14 amplified clones showed SMC (MYH11+) and EC (vWF+) differentiation capacity under the respective conditions, although individual clones showed different potencies (Figure 4G). We conclude that MesoT cells are multipotent vascular progenitor cells, confirming predictions made from the earlier molecular analysis (Figures 2 and 3).
Figure 4. MesoT Cells Are Multipotent Vascular Progenitor Cells.

(A) Population doubling of MesoT (FBS) cells. Time = 0 is when cells are first passed into serum containing media. Simple linear regression analysis (blue dashes) applied to data shows a tightly fitted regression line with a coefficient of determination (R2) = 0.9774 and slope of 0.8282+/−0.0563. Experiment n = 3 performed in technical triplicate.
(B) Cell cycle analysis of self-renewing MesoT (FBS) cells at passages 4 and 9 (p4 and p9) using Life Technologies Click-iT Plus EdU Alexa Fluor flow cytometry assay kit. n = 4 for p4 and n = 3 for p9, all in technical triplicate.
(C and D) Representative two-dimensional flow plots for each passage showing gating strategy to determine percentage of cells in each cell cycle phase for (B).
(E) Clonal assay strategy to determine multipotency of self-renewing MesoT (FBS).
(F) Fluorescence-activated cell sorting (FACS) gating strategy to obtain single cells for clonal analysis. Triple positive single cells (CD44+/CD73+/CD105+) were sorted onto a 96-well plate for amplification and downstream lineage analysis. Cyan are isotype controls.
(G) After amplification, 14 individual clones were selected for downstream lineage analysis. MesoT (FBS) cells were treated with 2% FBS +VEGF or +PDGF-BB for endothelial or smooth muscle cell differentiation, respectively. Cells were fixed and probed with antibodies against vWF (endothelium) or MYH11 (SMC). > 45 cells in 3 separate images per clone were quantified using ImageJ to determine percentage of cells that give rise to each lineage.
(H) MesoT in CDM (–Activin A) treated with VEGF generate mixtures of ECs (vWF+) and SMCs (calponin+). Scale bar, 50 μm.
(I and J) Cells as in (H) were cultured on Matrigel for 12 days. Bright-field images (I) of resulting vessel structures were probed with antibodies for αSMA and vWF and the nuclei counter stained with DAPI (J). Scale bars, 50 μm.
Error bars ± SEM. See also Figure S4.
We reasoned that multipotent MesoT cells could give rise to mixed populations of SMCs and ECs. By this rationale, we determined that CDM (–Activin A) supplemented with VEGF was sufficient to support the differentiation of MesoT cells into mixtures of SMCs and ECs (Figure 4H). Under these conditions, mixtures of SMCs and ECs were generated. When cultured under these conditions for >12 days, mixed populations of SMCs and ECs self-assembled into vessel-like networks (Figure 4I). The composition and organization of such “vessels” was evaluated by immunofluorescence (IF) and were shown to be composed of vWF+ ECs and αSMA+ SMC-pericyte-like cells (Figure 4J). vWF+ cells formed the core of these structures, while αSMA+ cells were generally located around the periphery. This is an interesting observation because it establishes conditions in which a single progenitor cell (MesoT cells) can concurrently give rise to SMCs and ECs that self-assemble into a simple vessel structure.
MesoT Cells Contribute to Neo-vascularization during Tissue Repair
To investigate the vascular potential of MesoT cells in a tissue injury context, we used a murine neonatal heart repair model (Porrello et al., 2011). Following resection of the ventricular apex, the neonatal mouse heart undergoes endogenous repair including partial muscle regeneration and neo-vascularization with variable fibrosis (Andersen et al., 2014; Bryant et al., 2015; Porrello et al., 2011). Work in zebrafish suggests that mesothelium-derived cells may be part of such a response by contributing to peri-vascular cells (Kikuchi et al., 2011; Lepilina et al., 2006). To determine whether MesoT cells have capacity for vascular repair, they were injected into the pericardial space of post-natal day 0.5 (P0.5) mouse pups, adjacent to the resected tissue (Figures 5A, 5B, S5A, and S5B). 30 days after transplantation, mice were sacrificed and cardiac tissue was analyzed by immunohistochemistry on cryosections. Mice receiving saline alone (n = 11) or MesoT cells (n = 13) revascularized the injured heart. In each heart receiving MesoT cells, a major contribution to coronary vessels in repair zones was made by hGolgi+ CD31+ human cells (Figure 5C). Hearts that did not receive MesoT cells repaired as expected but were negative for hGolgi antigen (Figure S5C). These vessels were connected to the host vasculature as indicated by the presence of erythrocytes (Figure S5D). Further analysis showed that hGolgi+ CD31+ ECs and hGolgi αSMA+ SMC-pericyte-like cells made significant contributions to the neo-vasculature in repair zones (Figure 5D). These results show that MesoT progenitor cells can differentiate and assemble into functional vessels in this tissue injury model and suggests that they may be useful in a broader range of regenerative approaches.
Figure 5. MesoT Cells Incorporate into Newly Formed Blood Vessels in a Neonatal Mouse Heart Injury Model.

(A) Surgery on P0.5 mouse pups that includes ventricle apex resection.
(B) After resection, the damaged heart was overlaid with a 2 μL suspension of 1 × 106 DiO-labeled MesoT cells followed by suturing of the rib cage and chest wall. Micron bar, 1 mm.
(C) Cryo-section (10 μm) of a repaired mouse heart, 30 days post-injury. Tissue was probed with anti-bodies for αSMA, CD31, human Golgi antigen, and DAPI. Scale bars, 50 μm.
(D) Repair zone showing a submesothelial blood vessel (bv) comprised of human μSMA+ cells and human CD31+ cells. Scale bars: top left, 20 μm; other panels, 10 μm.(E) Magnified images of panels shown in (E) showing incorporation of human endothelial and smooth muscle cells into nascent vessels. Scale bar, 10 μm.
See also Figure S5.
Vascularization of Biological Scaffolds by MesoT Cells: In Vitro and In Vivo Function
The cell culture and in vivo experiments presented suggest that MesoT cells may be a viable alternative to existing approaches for the recellularization of vascular scaffolds used for tissue engineering. To investigate this possibility, we used explanted, decellularized rat jejunal segments (Figure 6A) as a biological scaffold on which to seed MesoT cells in a bioreactor (Figure S6A). In this model, the rat vasculature remains intact and can be perfused using the afferent artery and efferent vein (Figures 6B and 6C). Following the introduction of MesoT cells through the lumen of the vascular bed, constructs were perfused with medium supporting differentiation to SMCs and ECs (Figures 4H–4J). Perfusion was pulsatile and increased gradually to a physiological pressure of 80–120 mmHg to promote vessel maturation. Constructs were then perfused with 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT), a substrate used to evaluate the assembly of metabolically active cells in the vascular network (Figure 6D). This shows that viable cells broadly lined the jejunal scaffold and that these cells were of human origin (Figure 6E). No MTT staining was observed in constructs that were not recellularized (Figure S6B). Analysis of vessels by IF on cryo-sections showed that capillaries were lined with human CD31+ cells and that medium (~100–150 μm) and larger (>150 μm) vessels were lined with CD31+ cells and αSMA+ cells. The organization of these cells was as expected, with CD31+ ECs forming a contiguous layer on the lumenal surface and αSMA+ SMCs forming an adjacent layer on the ablumenal side (Figures 6F–6H). The perfusion of decellularized vascular beds with MesoT cells supports their differentiation and self-assembly into vascular structures; this is reminiscent of their behavior in two-dimensional culture (Figures 4H–4J).
Figure 6. Reperfusion of a Decellularized Biological Scaffold with MesoT Vascular Progenitor Cells Repopulates the Vascular Network and Forms Functional Invested Vessels when Transplanted In Vivo.

(A-C) Image of a decellularized rat jejunum (A) injected with phenol red to contrast the vasculature before cell perfusion (B and C).
(D) MesoT cells were perfused through the arterial and venous cannulas with (+VEGF) media. MTT assay imaging depicts reseeded vasculature with metabolically active cells after 28 days. Right is blowup of inset on left. Red lines mark representative vessels of various sizes. Micron bar, 1 mm.
(E) Vasculature derived by seeding MesoT cells was stained with human nuclear antigen (hNA) and DAPI.
(F–H) Vessels of small (F), medium (G), and large (H) diameter as in (D) were stained with antibodies against CD31 or αSMA. Scale bars, 100 μm.
(I) Vascular barrier integrity testing by perfusion with FITC-dextran. Left: bright-field image before perfusion. Middle: intravital microscopy image of FITC-dextran retention after repeated perfusion and washing of the vascular network. Right: uptake of acetylated low-density lipoprotein (LDL, red). Nuclei were visualized with NucBlue Live ReadyProbes. Scale bars, 100 μm.
(J) Light sheet microscopy image of vessel networks after fixation and staining with CD31 antibody. Scale bar, 400 μm.
(K) Reseeded vascular constructs were transplanted into 8-week-old immunodeficient female rats and anastomosed (white arrow) with the host circulatory system.
(L) Gross anatomical image of a transplanted graft after harvesting, showing the presence of host oxygenated blood and the absence of occlusion or leakage.
(M–O) Harvested grafts stained with antibodies for CD31 (red) and αSMA (green). Small (M), medium (N), and (O) large blood vessels are shown. Scale bars, 100 μm.
To evaluate the barrier function of MesoT-derived cells in the vascular structures, cellularized constructs were perfused with FITC-dextran and monitored by time lapse imaging. We observed that MesoT-derived vessels retained FITC-dextran (Figure 6I; Video S1) in contrast to constructs that were sparsely seeded where rapid diffusion and leakage was observed (Figure S6C). Scaffolds without cells are not shown because label leaks immediately at the site of infusion and cannot be visualized. EC function was verified by demonstrating the uptake of acetylated low-density lipoprotein (LDL) (Figure 6I). To determine the extent to which MesoT-derived endothelium lined the vascular tree, we stained perfused constructs with human-specific anti-CD31 antibody. Light sheet microscopy (LSM) showed widespread incorporation of these cells into the vasculature (Figures 6J, S6D, and S6E; Video S2), as expected from our analysis of the frozen sections (Figures 6F–6H). LSM also showed incorporation of NG2+ pericyte-like cells into vascular networks (Figure S6F). This indicates that MesoT cells can contribute to three vascular lineages.
To investigate the functionality of MesoT-populated vascular constructs under physiological conditions, recellularized jejunal scaffolds were connected to the rat circulation by anastomosis (Figure 6K) for 3 days. Analysis of anastomosed constructs showed that vessels retained gross morphologic integrity with no indication of blood leakage or occlusion (Figures 6L and S6G). Further analysis of these vessels by IF following cryo-sectioning showed maintenance of capillaries and larger vessels, in composition and architecture, after anastomosis (Figures 6M–6O). These observations show that MesoT cells can populate biological scaffolds with cells that assemble into vessels that withstand physiological pressures and conditions, at least over short periods of time.
DISCUSSION
Several reports have described mesenchymal progenitor descendants of the epicardial lineage (Chong et al., 2011; Gittenberger-de Groot et al., 2010; Rinkevich et al., 2012; Singh and Epstein, 2012); however, embryonic mesothelial layers of different coelomic organs have similar molecular and cellular characteristics (Rinkevich et al., 2012; Winters et al., 2014) (Figure 1D). Lineage-marked descendants of mesothelium from multiple organs have been isolated and shown to have a similar potency to epicardium-derived cells (EPDCs) (Rinkevich et al., 2012). While there is some debate about the degree to which EPDCs contribute to the endothelium of the coronary vasculature (Pennisi, 2016), several studies indicate some contribution of the epicardium lineage to arterial vessels with the remainder coming from other sources (Cano et al., 2016; Chen et al., 2014a; Katz et al., 2012; Palmquist-Gomes et al., 2018; Pérez-Pomares and de la Pompa, 2011). Whether this endothelial contribution comes from EPDCs or another epicardial subtype is currently not known.
While “epicardium” from hPSCs (Iyer et al., 2015; Witty et al., 2014) are potentially generated along similar developmental pathways as MesoT cells, they differ in key respects. First, epicardium cells are epithelial and develop through a “pro-epicardium” stage (Witty et al., 2014). Second, hPSC-derived epicardium has a propensity for SMC and fibroblast differentiation, with no multipotency or endothelial potential reported (Gittenberger-de Groot et al., 2010). It has been proposed that the pro-epicardium and epicardium are heterogeneous in cell composition (Cao et al., 2016; Katz et al., 2012), making different contributions to the coronary vasculature. It is possible that hPSC-derived epicardium and the mesothelium described here are representative of different mesothelial subtypes, but because the developmental pathway for each has not been clearly defined, it is not possible to unequivocally assign MLCs and MesoT cells to mesothelium from a specific anatomical location (e.g., the heart). In the absence of specific information to address this issue, we have designated MLCs and MesoT cells as being of “mesothelial origin.” We hypothesize from a developmental standpoint that MLCs represent mesothelium and that MesoT cells are their vasculogenic mesenchymal descendants (Chong et al., 2011; Rinkevich et al., 2012; Singh and Epstein, 2012).
The differentiation of hPSCs along the mesothelium lineage described here is consistent with the generation of this cell type during embryonic development. The approach described in this report shows that SplM progenitors are efficiently specified along the mesothelium lineage by a combination of BMP, Wnt, and RA signaling. The mechanism of lineage specification by these signaling pathways is not understood and will be a subject of future investigation. In the future, it will be necessary to evaluate the relationship between MLCs and MesoT cells further. For example, is the developmental potential of MesoT cells also exhibited by MLCs, and if so, which will be better suited to tissue engineering and regenerative medicine? The incorporation of MesoT-derived pericytes into vascular constructs suggests that they can be directly generated from MesoT cells in culture. Further work will be required to establish if MesoT cells can generate pericytes at high efficiency by directed differentiation.
Following the plating of MesoT cells under specific signaling conditions, a mixture of SMCs and ECs are generated that self-assemble into tube-like networks. This assay is reminiscent of tube-forming assays used to characterize EC progenitors (Alphonse et al., 2015; Prasain et al., 2014). Here, ECs migrate over a two-dimensional matrix and assemble into tube-like structures. In contrast to EC progenitors, which give rise to tubes comprised solely of ECs, MesoT cells generate invested structures composed of SMCs and ECs. It is assumed that part of the tube assembly mechanism described in this report is similar to that described for EC progenitors with the additional complexity of SMCs superimposed into the system. This general process has been described previously where mixed populations of SMCs and ECs are plated simultaneously and give rise to invested tube-like structures (Marchand et al., 2014). Understanding the assembly of MesoT-derived SMCs and ECs into tube structures will be important to harness the full utility of MesoT cells for translational purposes. The self-assembly of MesoT-derived cells into tube structures in a two-dimensional culture system suggested that this may also occur in a three-dimensional system, supported by shear stress from perfusion. This was confirmed using decellularized vascular scaffolds that were shown to be stable and functional for over 28 days. In the future, it will be necessary to optimize numerous variables in vessel construction using MesoT cells, including perfusion rates and pressures, seeding density, media composition, and scaffold composition.
In this report, we have characterized a mesenchymal cell of the mesothelium lineage that has multipotent vascular potential. Even though extensive efforts were made, CMs could not be generated from MesoT cells, indicating that this progenitor does not have broad spectrum cardiovascular lineage potential. This is supported by epigenetic analysis in this study. Epigenetic characterization supports our conclusions that MesoT cells are a multipotent vascular progenitor based on the “priming” signature observed at enhancers used by vascular lineages and by the DNA methylation profile linking them to vascular cell types. The epigenetic signature of MesoT cells is consistent with them being vascular progenitor cells and is supported by clonal analysis performed in this study. These factors rule out the possibility that MesoT cells are proliferating vascular cells. We are not aware of any other reported cell type from the mesothelium lineage that has the multipotent vascular progenitor characteristics of that described here for MesoT cells. hPSC- and tissue-derived vascular cell types, including SMCs and ECs, have been used for the development of cell therapeutics and tissue engineering (Doi et al., 2017; Ren et al., 2015), providing proof of concept for this strategy. However, barriers to using these cell types in vascular repair scenarios exist and need to be considered if PSC-derived vascular cells are to become clinically viable. For example, fully differentiated SMCs and ECs have not yet been shown to efficiently incorporate into new vasculature in a tissue repair model following transplantation. This could be related to limitations of mature cells to integrate into remodeling tissue or an inability to assemble into neo-vessels in a repair environment. In tissue engineering, combinations of SMCs, ECs, and mesenchymal stem cells (MSCs) are frequently used to seed vascular scaffolds, but this process is complicated, and the long-term efficacy of such constructs are unclear (Drews et al., 2017; Lee et al., 2016; Ren et al., 2015; Villalona et al., 2010). Our results suggest that vascular progenitor cells may be a solution to the problems associated with using mature vascular cell types. In support of this idea, circulating EPCs first identified in vivo (Alphonse et al., 2015; Asahara et al., 1997), but also recently derived from hPSCs, show functional efficacy by contributing to new vasculature in a diabetic retinopathy model (Prasain et al., 2014). Although EPCs have promising clinical utility, their potency is restricted to the endothelial lineage. These limitations do not apply to MesoT cells because of their vasculogenic, multipotent properties.
To develop strategies for use of MesoT cells in vascular engineering it will be necessary to apply them to vessels that have utility in a transplantation context. Instead of using a vascular tree, decellularized vessels or fabricated scaffolds will be utilized on which to seed MesoT cells. Fine-tuning of seeding and perfusion conditions will then be required to obtain vessels with suitable characteristics for transplantation purposes, for example, the generation of venous and arterial vessels. The properties of MesoT cells described in this report indicate that they offer an important option for vessel engineering in a wide range of contexts.
STAR ★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Stephen Dalton (sdalton@uga.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell Culture and Differentiations
Maintenance of pluripotent cells (Cliff et al., 2017) was as previously described. Briefly, WA09 (Sex: female, WiCell, NIHhESC-10-0062), WA07 (Sex: male, WiCell, NIHhESC-10-0061), WA01 (Sex: male, WiCell, NIHhESC-10-0043), TE03 (Sex: female, WiCell, NIHhESC-13-0204) human embryonic stem cells (hESCs) and K3 human induced pluripotent stem cells (hiPSCs), a gift from Stephen Duncan (Si-Tayebet al., 2010), were maintained in chemically-defined media (CDM) (Wang et al., 2007), seeded at 5 × 104 cells/cm2 on Geltrex (Thermo Fisher, A1413302) coated plates at 1:200 dilution in DMEM/F12 w/o glutamine (Corning, 15-090-CM), and passaged with Accutase (Innovative Cell Technologies, AT104) upon confluency.
CDM consists of DMEM/F12 w/o glutamine supplemented with 1x nonessential amino acids (Corning, 25-025-CI), 1x antimycotic/antibiotic (Corning, 30-004-Cl), 1x trace elements A (Corning, 25-021-CI), 1x trace elements B (Corning, 25-022-CI), 1x trace elements C (Corning, 25-023-CI), 2 mM L-alanyl-L-glutamine (Corning, 25-015-CI), 10 μg/ml transferrin (HOLO), human plasma, tissue culture grade (Athens Research and Technology, 16-16-032001-LEL), 2% Probumin® bovine serum albumin life science grade (EMD Millipore, 821005), 0.1 mM β-mercaptoethanol (GIBCO, 21985023), 50 μg/mL ascorbic acid (Sigma-Aldrich, A8960), 10 ng/ml rhHeregulin β-1 (Peprotech, AF-100-03), 200 ng/ml LONG® R3 IGF-I human (Sigma-Aldrich, 85580C), 10 ng/ml rhActivin A (R&D Systems, 338-AC) and 8 ng/ml rhFGF basic (R&D Systems, 4114-TC).
To generate splanchnic mesoderm (SplM) (Berger et al., 2016), hPSCs were passaged as above and reseeded at 5 × 104 cells/cm2 (WA09) or 1 × 105 cells/cm2 (WA01, WA07, TE03 and K3) onto Geltrex coated plates into CDM supplemented with 25 ng/ml rhWNT3a (R&D Systems, 5036-WN) and 100 ng/ml rhBMP4 (R&D Systems, 314-BP) for 4 days with media changed daily.
Mesothelium-like cells (MLCs) were generated by passaging SplM with TrypLE select (Thermo Fisher, A1217701) diluted to 1x in a salt balanced solution consisting of PBS (Corning, 21-031), sodium chloride (1.85 g/L, Fisher Scientific, S640), and 0.5 M EDTA (1ml/L, Thermo Fisher, AM9260G). Cells were reseeded at 1.5 × 105 cells/cm2 in CDM without FGF2 or Activin A, and with 25 ng/ml rhWNT3a, 50 ng/ml BMP4, 20 μM SB431542 (Tocris, 1614), 4 μM all-trans retinoic acid (Sigma-Aldrich, R2625) for 18 days with media changed daily.
Migratory mesothelium (MesoT) is generated from SplM and does not require passaging and reseeding of cells. On day 4 of SplM induction, CDM is merely supplemented with 25 ng/ml rhWNT3a, 50 ng/ml rhBMP4 and 4 μM all-trans retinoic acid for an additional 14-18 days. To generate MesoT cells from MLCs, cells are passaged with Tryp LE select and reseeded at 1.5 × 105 cells/cm2 in CDM supplemented with 25 ng/ml rhWNT3a, 50 ng/ml rhBMP4 and 4 μM all-trans retinoic acid for an additional 18 days. To self-renew MesoT (FBS), cells were passaged with collagenase type IV (800 units/ml, Thermo Fisher, 17104-019) followed by 1x TrypLE Select and cultured with 10% fetal bovine serum (Atlanta Biologicals, S10250), Minimal Essential Media, Alpha (Corning, 15-012-CV), 1x antibiotic/antimycotic, 2 mM L-alanyl-L-glutamine and 8 ng/ml FGF2 at an initial seeding density of 5 × 104/cm2 on plastic polystyrene plates then 1 × 104/cm2 for subsequent passages using 1x TrypLE Select. Media was changed daily.
Downstream lineages were generated by passaging MesoT cells as above and reseeding at a density of 1.5 × 105/cm2 on Geltrex coated plates. Activin A was removed from CDM and supplemented with the following: 50 ng/ml rhVEGF-A165 (R&D Systems, 293-VE) and 20 μM SB431542 for EC differentiations; 50 ng/ml PDGF-BB (R&D Systems, 220-BB) for smooth muscle cell (SMC) differentiations, and 50 ng/ml PDGF-AA (R&D Systems, 221-AA) for fibroblasts. Each differentiation was allowed to proceed for 12 days with media changed every other day. Removal of SB431542 from EC media was used to generate mixtures of ECs and smooth muscle cells that can self-assemble in vitro into tube structures.
Animal Use and Welfare Compliance
Animal studies and care were conducted in accordance with the Institutional Animal Care and Use Committee (IACUC) at the University of Georgia and FELASA, WHO (WHO-TRS978 Annex3), and FDA (FDA-OCTGT Preclinical Guidance) after approval from the institutional animal protection board (registration reference number #2532-2-12, Ethics Committee of the District of Unterfranken, Wurzburg, Germany). Male and female mice were used for all experiments where possible at an age most appropriate for the experiment being performed. Details are provided in the respective sections of the STAR Methods.
METHOD DETAILS
Immunofluorescence Analysis of Fixed Cells
Immunofluorescence analysis was performed on 4% paraformaldehyde (VWR, 15170) fixed cells (10 min) in the presence of 10% donkey serum (Equitech-Bio, SD30) and 0.25% Triton X-100 (Fisher Scientific, BP151) followed by visualization using 2.5% donkey serum in PBS with fluorescent conjugated secondary antibodies. Primary antibodies were incubated overnight at 4°C followed by conjugated secondary antibodies for 1 hour at room temperature (RT) in the dark. Nuclei were counterstained with 4′,6-Diamidino-2-phenylindole dihydrochloride (DAPI, Sigma-Aldrich, D9542) for 5 min and coverslips were affixed with ProLong Diamond Antifade (Thermo Fisher, P36961). Fluorescent cells were visualized on a Leica DM6000 B and BioTek Lionheart FX. Confocal images were obtained on an Olympus FV1200 laser scanning confocal microscope. All antibodies are listed in Table S2.
qRT-PCR
mRNA was isolated using the E.Z.N.A.® Total RNA Kit I (Omega Bio-Tek, R6834) followed by quantification using a Biotek Synergy 2 plate reader. cDNA was made using iSCRIPT cDNA kit (Bio-Rad, 1708841). qRT-PCR was performed using TaqMan Universal PCR Master Mix No AmpErase UNG (Thermo Fisher, 4324020) and TaqMan Primers on a ViiA7 Real-Time PCR System (Life Technologies). TaqMan probe mixes are listed in Table S3.
Flow Cytometry, Cell Cycle Analysis and Clonal Analysis
Cells were collected as single cell suspensions following removal from the culture as noted above then analyzed by flow cytometry using a CyAn ADP (Beckman Coulter, Hialeah, Florida). Cells were stained with specific flow antibodies per manufacturer recommendations with isotype controls. Intracellular markers were analyzed using Flow Cytometry Permeabilization/Wash Buffer I (1x, R&D Systems, FC009). Cell cycle analysis of MesoT cells was performed using a Click-iT Plus EdU Alexa Fluor 647 Flow Cytometry Assay Kit (Invitrogen, C10634) with 1-hour incubation. Population doublings were calculated using the following equation (Takeuchi et al., 2009):
| (1) |
For clonal analysis, CD44+/CD73+/CD105+ MesoT cells were single cell sorted onto 96 well plates (VWR) using 50/50 fresh and pre-conditioned media with a MoFlo XDP sorter (Beckman Coulter, Hialeah, Florida). After amplification, cells were passaged to down-stream lineages using 2% fetal bovine serum, Minimal Essential Media, Alpha, 1x antibiotic/antimycotic, 2 mM L-alanyl-L-glutamine and 50 ng/ml rhVEGF-A165 (endothelium) or 50 ng/ml PDGF-BB (smooth muscle). Multipotency was assessed by confocal imaging on an Olympus FV1200 confocal microscope and quantified with ImageJ software (NIH https://imagej.nih.gov/ij/). All flow and sorting analysis used FlowJo® cell analysis software (FlowJo, LLC, Ashland, Oregon).
Isolation of Embryonic Mouse Mesothelium
Male and female Wt1creERT2/+ Rosa26tdT/+ embryonic mice were generated by crossing wt1creERT2/+ male mice (Zhou et al., 2008) with a knock-in of tamoxifen inducible cre-recombinase in the Wt1 locus (Jackson Laboratory, 010912) and female Rosa26tdT/tdT (Jackson Laboratory, 007914) mice with a loxP-flanked STOP cassette preventing transcription of a CAG promoter-driven red fluorescent protein variant, tdTomato (Madisen et al., 2010). At E12.5, pregnant dams were injected with 300 μL of 10 mg/ml tamoxifen (Sigma-Aldrich, T5648) in corn oil solution (Sigma-Aldrich, 8267) intra-peritoneally at 24-hour intervals for three consecutive days. Embryos were harvested at E15.5 and the heart, lung, liver and gut were carefully isolated using sterile technique. The outer mesothelial layer was digested by exposing intact organs to a dissociation buffer containing 1 mg/ml collagenase IV and 0.05% trypsin-EDTA (Thermo Fisher, 25300054) as described previously (Zhou and Pu, 2012). Briefly, the intact organs were repeatedly digested (7-8 times) in collagenase IV-trypsin dissociation buffer at 6-7 min intervals on a 37°C shaker and the supernatants from each digest neutralized with horse serum (Atlanta Biologicals, S12150), pooled and filtered through a 70 μM nylon mesh. Cells isolated from nine embryos were pooled for analysis in order to obtain sufficient material. Digested samples were suspended in FACS buffer (0.2% Probumin® in PBS). tdTomato+ cell fractions were sorted using a MoFlo XDP and RNA was extracted for differential gene expression analysis.
RNA-seq Analysis
RNA from tdTomato+ fractions along with RNA from hESC-derived MesoT and MLCs were extracted with Trizol (Invitrogen, 15596-026) and purified with RNeasy mini kit (QIAGEN, 74104) according to the manufacturer’s instructions. RNA yield was determined using the NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies) and purified RNA (1 μg) from each sample was submitted to the Hudson Alpha Institute (Huntsville, Alabama) for polyA+ RNA sequencing (HiSeq v4 50 PE, 25 million reads per sample) and deposited under GSE113090. Additional raw data for human adult and fetal tissues were downloaded from the Sequence Read Archive (ERP003613, SRP001371) while additional mouse adult and fetal tissues were downloaded from ENCODE and the Sequence Read Archive (SRP049248, SRP018511). A summary of all samples analyzed by RNA-seq in this study is listed in Table S4. When necessary, *.sra formatted data were converted to the *.fastq format using the NCBI SRA Toolkit (version 2.4.1). Data quality was assessed using FastQC (version 0.10.1) before being mapped to known Ensemble genes (GRCh37/NCBIM37) using Tophat2 (version 2.0.13) (Kim et al., 2013). Raw read counts for each gene were obtained using Subread (version 1.4.2) (Liao et al., 2013). Manipulation of raw sequence data was executed on an 8-core UNIX node with 48G RAM. All ftp data accessions and Tophat2/Subread mapping statistics are provided in Table S4A.
Raw mouse and human read count data were read into R (R Core Team, 2013) and combined into species specific data frames for differential expression analyses using linear modeling strategies. The mouse sample set was comprised of four FACS-sorted mesothelium samples and replicated samples from 18 mouse tissues (40 total samples). The human sample set was comprised of 2 replicates of hPSC-derived MLCs, 4 replicates of hPSC-derived MesoT, and replicated samples from 25 human tissues (62 total samples). Sample group annotations required for normalization and differential expression analyses are provided in Table S4B. The RNA-seq expression sets were filtered to include genes having at least 5 reads in 2 or more samples (nmouse = 18836, nhuman = 21761). Raw read counts were normalized using the trimmed mean of M-values (TMM) method (Robinson and Oshlack, 2010) and precision weights were calculated using voom (Law et al., 2014) prior to differential analysis using the Limma empirical Bayes analysis pipeline (Smyth, 2004).
Principal Component Analysis
For principal component analysis, each sample has two biological replicates. MesoT (CDM & FCS) and mesothelium (MLC) were deposited under GSE113090. WA09 hESCs, human coronary artery endothelium, human coronary artery smooth muscle cells, fetal liver, heart and brain, fetal hindbrain and spinal cord datasets were obtained from GSE101655, PRJDB4498, GSE51878, GSE63634, GSM2229922. Raw data were aligned to human reference genome (hg19) using HIAST2 (version 2.1.0) with default parameters. Aligned reads were sorted with Samtools (version 1.3.1) (Li et al., 2009)and the number of reads mapping to each gene were counted by HTseq (version 0.6.1p1). All counting files from HTseq were imported into R studio (version 1.1.423) (RStudioTeam, 2015). In order to generate the PCA plot, data were pre-processed using Limma edgeR (McCarthy et al., 2012; Robinson et al., 2010) to normalize sequencing depth and gene expression distributions as well as to remove low counts. The PCA plot was made by Glimma (Su et al., 2017) using the top 50% of genes.
DNA Methylation Analysis
Bisulfite conversion was performed using the EZ DNA Methylation Kit (Zymo Research, D5001) according to the manufacturer’s instructions. Whole genome amplification, fragmentation and preparation of the DNA for hybridization were performed using the Infinium HumanMethylation450 BeadChip kit (Illumina, WG-314-1001) as described in the manufacturer’s protocol. Illumina IDATS were read directly into Rand normalized using Subset-quantile within array normalization (Maksimovic et al., 2012). A summary of all DNA methylation samples analyzed in this study is provided in Table S4D and deposited under GSE116754. Public datasets were downloaded from GSE31848. Dynamic methylation analyses were performed using Limma in R (Ritchie et al., 2015). Briefly, triplicate MesoT samples were fit to a linear model along with undifferentiated hPSC, hPSC-derived SplM and hPSC-derived CMs (all biological duplicates). Dynamic methylation loci were identified by selecting probes with FDR ≤ 0.01 in a targeted t test comparing MesoT to undifferentiated hPSCs. These loci were further filtered by removing probes where the maximum difference in Beta values was < 0.3 between any of the sample group averages used to fit the linear model (n = 22561). DNA methylation modules were identified using weighted gene correlation network analysis in R (WGCNA) (Langfelder and Horvath, 2008). The filtered set of significant differentially methylated loci and methylation module assignments are provided in Table S4E.
ChIP-seq and Gene Ontology Analysis
ChIP-seq was performed as previously described (Singh et al., 2015). Briefly, MesoT cells were chemically crosslinked with 1% formaldehyde (Thermo Scientific, 28906) for 10 min at room temperature, followed by quenching with 2.5 M glycine (Sigma-Aldrich, G8898) for 5 min. Samples were rinsed with cold 1x PBS once, harvested by centrifugation and stored at −80°C before use. Cross linked DNA was fragmented by sonication on a Covaris S220 with 140V peak power, a duty factor of 5 and 200 cycles for 10 min. H3K4 mono-methylation (Abcam, ab8895) and H3K27 acetylation (Abcam, ab4729) antibodies were incubated with 50 μL Protein G Dynabeads (Invitrogen, 10004D) on a shaker at 4°C for 2 hours. Fragmented DNA, antibodies and beads were then incubated together on a shaker at 4°C overnight, approximately 5 million cells per reaction. Cross-linking was reversed by adding 20 mg/ml Proteinase K (QIAGEN, 19131) at 55°C for 30-40 min followed by incubation at 65°C for 8 hours. The final library was submitted to Hudson Alpha Institute for 50bp single-end sequencing. For each sample, ~25 M reads were obtained, and input samples were used as controls. MesoT H3K4me1 and H3K27ac ChIP-seq data was deposited under GEO accession GSE113090.
All ChIP-seq raw data were aligned to UCSC human reference genome (hg19) using Bowtie2 software (version2.2.9) (Langmead et al., 2009) and only uniquely aligned reads were retained. Samtools was used to sort aligned reads into genomic order for further analysis. For visualization, sorted BAM files were indexed with Samtools and genome coverage tracks (bigwig files) were generated using Deeptools (version 2.3.1) (Ramirez et al., 2016) with parameters of–binsize 10,–extendReads 200. Normalization was performed by reads per genome content (RPGC). H3K4me1 peaks, H3K27ac peaks and differentially regulated H3K27ac regions between MesoT, human coronary artery smooth muscle cells (HCASMC) and human umbilical vein ECs (HUVEC) and were detected using MACS2 (version 2.1.1) by calling broad peaks and differential binding events. All reads were extended from 5′ to 3′ direction to a final length of 200 bp. The enriched H3K4me1 and H3K27ac peaks were defined as the regions with significant enrichment of FDR < 0.01 relative to respective input control reads. Differentially regulated H3K27ac sites were obtained with a cutoff of log10 likelihood ratio between two samples. The overlapping regions of H3K4me1 and H3K27ac sites and peaks annotation were analyzed by ChIPseek (Chen et al., 2014b). Potential primed enhancers were defined as sites which have highly enriched H3K27ac signals in HCASMC and HUVEC compared to MesoT and were also marked with H3K4me1 in MesoT. A list of lineage specific genes with primed enhancers regions in MesoT were obtained in R by intersecting HCASMC- and HUVEC-specific genes (identified as fold change > 4, compared to MesoT, from RNA seq analysis) with annotation tables from potential primed enhancers. The H3K4me1 and H3K27ac heatmaps were generated with Deeptools. The matrix was built on a list of genes having primed enhancer along with their corresponding H3K4me1 peak regions and signal files of MesoT H3K4me1 bigwig file and HCASMC and HUVEC H3K27ac bigwig files with bin size of 50 bp. The signals were restricted to 5kb around the central region of H3K4me1 peaks. The Gene Ontology enrichment analysis was performed on HCASMC- and HUVEC-specific genes with primed enhancers in MesoT cells with Panther (Mi et al., 2017). The GEO accession numbers for HCASMC and HUVEC H3K27ac ChIP-seq are GSM1876036 and GSM1009635, respectively.
Contraction, tube formation, transwell barrier assays
Day 12 SMCs differentiated from MesoT were labeled with DiO (Thermo Fisher, V22886) per manufacturer’s instructions then replated at 3 × 105 cells per cm2 on Geltrex coated 12 well plates (Corning) for 24 hours. Cells were treated with 100 μM carbachol (Iyer et al., 2015) (Sigma-Aldrich, 212385), 50 mM KCl (Motherwell et al., 2017) (Fisher Scientific, P333-500) or left untreated (control) and monitored for 30 min using a BioTek Lionheart FX. Surface area change for 20 selected cells was measured with ImageJ. For tube formation assays, MesoT cells were cultured on Matrigel (VWR, 47743-715) in CDM (-Activin A) supplemented with 50 ng/ml rhVEGF-A165 at a density of 1.5 × 105 cells/cm2. Media was changed every other day. Tubes were fixed and probed with antibodies as above.
To conduct the endothelial barrier assay, ThinCert Cell Culture Inserts (pore size 0.4 μm, Greiner Bio-One International, 662641) were coated with collagen IV (Sigma-Aldrich, C5533) and fibronectin (Thermo Fisher, 33016015). MesoT cells were seeded onto the PET membrane and cultured at 37°C and 5% CO2 using media to generate ECs or EC/SMC mixtures, as noted above, for 28 days under static conditions in a standard well-plate. Primary dermal microvascular endothelium, a gift from Heike Walles (Kress et al., 2018), cultured in VascuLife® VEGF-Mv Endothelial Complete Kit (Lifeline Cell Technology, LL-0005) were used as a control. Barrier integrity of cells was examined by trans-endothelial electrical resistance (TEER) measurements and a FITC-dextran (40 kDa, Sigma-Aldrich, FD40S) permeability assay. TEER values of the barrier separating the apical and basolateral compartment were determined using a hand-electrode (Millicell ERS-2, Millipore). Permeability of FITC-dextran was measured after application into the apical compartment on an orbital shaker (77 rpm) for 30 min. Cell junctions were visualized by osmium tetroxide (Sigma-Aldrich, 75633) contrasting followed by imaging on a JEM-2100 transmission electron microscope (JEOL, Tokyo, Japan).
Transplantation of MesoT Cells in a Neonatal Mechanical Injury Model
Male and female Wt1creERT2/+;Rosa26tdT/+ and Wt1+/+;Rosa26tdT/+ neonatal mice were generated by crossing male Wt1creERT2/+ and female Rosa26tdT/tdT mice and given an intragastric injection of tamoxifen (1 μg) dissolved in corn oil on day of birth (D0.5). Six hours after injection, neonatal mice were separated from their mothers and placed on ice for 5 min to induce hypothermia-based anesthesia (Wixson and Smiler, 1997). Hearts of neonatal mice were mechanically injured as previously described (Porrello et al., 2011). Briefly, a lateral thoracotomy in the 4th intercostal space was performed to expose the apex of the heart. In the sham operated mice, after the heart was exposed, the heart was gently placed back into the chest cavity and the rib cage and chest wall were surgically sewn up using 6-0 non-absorbable Prolene sutures. In mechanically injured mice, the heart was resected using iridectomy scissors. In mice receiving treatment with MesoT, 1 million cells labeled with DiO cell labeling solution were suspended in 2 μL of Ca2+ and Mg2+-free PBS and injected into the pericardial cavity before sewing up the rib cage and chest wall. Once surgery was complete, neonatal pups were allowed to recover by rapid warming and then returned to their mother. Hearts from each treatment condition were harvested at various times up to 30 days post-injury. Following removal, collected hearts were washed in PBS, fixed in 4% paraformaldehyde for 2 hours then cryo-protected overnight in 30% sucrose solution (Sigma-Aldrich, S0389) (both at 4°C) and embedded in OCT (Fisher Scientific, 14-373-65). To assess the human cellular contribution to the injury site, cryo-sections of 10 mm thickness were probed with antibodies for human-specific Golgi (TGN46, AbD Serotec, AHP500G) or human nuclear antigen (Millipore, MAB1281) and cell lineage markers.
Recellularization of Vascular Scaffolds with MesoT Cells
Jejunal scaffolds were isolated as described previously (Kress etal., 2018). Briefly, 6-8 week old anesthetized Lewis rats were subject to a median laparotomy to isolate a jejenal segment containing its arterial and venous pedicle. The animal was systemically perfused with heparin (100 IE/kg) with the draining vein and feeding artery cannulated with a 20-Guage catheter and flushed with phosphatebuffered saline. Decellularization of the jejenum was performed by perfusion with deionized water at 4oC for 24 hr followed by perfusion with 4% sodium deoxycholate at room temperature for 4 hr and then 1 mg/ml DNase I for a further 3 hr at room temperature. Each step was followed by a PBS wash.
MesoT cells were injected through the arterial and venous cannulas into the vascular tree of a decellularized rat jejenum. Optimization of recellularization was achieved by injecting a bolus of cells on three consecutive days. On day 1, 0.5 - 1 × 106 cells in 0.5 mL cell culture medium were injected per cannula with an infusion rate of 4 ml/min. Following a one-hour static incubation, allowing for cellular adherence, the injection and static incubation were repeated. To promote functional maturation of the cellular layer on the vascular bed, the scaffold was connected to a bioreactor system to mimic physiological blood flow conditions by perfusion culture (Kress et al., 2018). Cell injection and static adherence were repeated once per arterial and venous cannula per day with subsequent perfusion culture overnight for three consecutive days. After the third day of injection, media was perfused for up to 28 days in the bioreactor. Temperature and CO2 were maintained at 37°C and 5%, respectively, with a pressure of 120/80 mmHg. Primary human microvascular ECs (1° ECs) cultured with VascuLife Endothelial Medium served as control and were loaded as per the first day injection for MesoT cells.
Intravital microscopy was performed to validate recellularized scaffold integrity using a Zeiss inverted microscope after perfusion with carbogen-gassed PBS at 37°C. FITC-dextran retention and vascular integrity was detected using real-time fluorescence as previously described (Kress et al., 2018). Functional endothelium lining the vascular tree was confirmed via acetylated LDL uptake (Invitrogen, L3484); nuclei were counterstained with NucBlue Live ReadyProbes (Invitrogen, R37605). Metabolically active cells lining the scaffold vasculature were identified by injecting 1mg/ml of MTT (Serva, 20395) in cell culture media for 90 min followed by aspiration and scaffold washing. Recellularized scaffolds were prepared for immunofluorescence analysis by fixation in 4% paraformaldehyde at RT for 2 hours then embedded in paraffin. Deparaffinized sections (5 uM) were dehydrated and stained with specific antibodies. All primary antibodies were incubated over night at 4°C followed by conjugated secondary antibodies for 1 hour at RT. Fluorescence was visualized on a Keyence BZ-9000 fluorescence microscope. All antibodies are listed in Table S2. For light sheet microscopy (LSM) of cellularized scaffolds, tissue was fixed and then stained with primary antibodies for CD31 (Dako, M0823) and NG2 (Abcam, ab83508) at 4°C overnight. Conjugated secondary antibodies were then incubated at room temperature for 1 hour followed by optical clearing as previously described (Brede et al., 2012).
Graft Implantation by Anastomosis
Graft implantation into the abdominal region was conducted in female NIH-Foxn1rnu rats (age: 8 weeks, obtained from Charles River). Anesthesia was induced via isoflurane with the intraoperative analgesia Rimadyl® (5 mg/kg subcutaneously, Zoetis, 779-358). After the abdominal cavity was opened by a median laparotomy, the infrarenal aorta abdominalis and the infrahepatic vena cava were isolated from surrounding tissues. Proximal and distal vessels were clamped and side-to-end anastomosis of the scaffold artery to the aorta abdominalis and vein to the vena cava was performed. Following patency examination and perfusion of blood from the host circulatory system, the abdominal cavity was closed occluding the abdominal musculature and skin was sutured. Grafts were harvested after 3 days for further analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
qRT-PCR samples were done in technical triplicate and error bars calculated in Microsoft Excel. All other assays and statistical measurements were done in GraphPad Prism version 7.0b for Mac (GraphPad Software, La Jolla California USA). SMC contraction assay used one-tailed t test, Holm-Sidak method to determine statistical significance with alpha = 0.05. For carbachol versus control, the t ratios for 10, 20 and 30 min, respectively, were 5.551, 5.46 and 5.165 with df = 38. For KCl versus control, the t ratios for 10, 20 and 30 min, respectively, were 7.62, 7.318 and 5.581 with df = 38. p values were all < 0.0001. All error bars are standard error of the mean (SEM) with n = 20 for all time points and groups. TEER significance was determined by comparison against 1° ECs using two-way analysis of variance (ANOVA). Dunnett’s test corrected for multiple comparisons with 95% confidence interval (CI), alpha = 0.05, F-value = 9.376, df = 2. Error bars are SEM with n = 3 for all measurements. For +VEGF +SB d28 sample versus 1° ECs, p value = 0.0027. FITC diffusion significance was determined via two-way ANOVA with 1° ECs acting as control for measurements. Control (no cells) were graphed only for visualization. Dunnett’s test corrected for multiple comparisons with 95% CI, alpha = 0.05, F-value = 1.605, df = 2. No statistical significance was determined. Errors bars are SEM with n = 3 for all measurements. For population doublings, simple linear regression analysis showed a coefficient of determination (R2) = 0.9774. Experiment n = 3 done in technical triplicate. Error bars are SEM. Cell cycle analysis for p4 (n = 4) and p9 (n = 3) were all done in technical triplicate; error bars are SEM. Clonal multipotency analysis was determined in technical triplicate by counting positive cells with ImageJ and plotting the average (n = 14).
DATA AND SOFTWARE AVAILABILITY
All the sequencing datasets generated for this paper have been deposited into GEO under the accession numbers GSE113090 for RNA-seq and ChIP-seq, GSE116754 for DNA methylation.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Goat Polyclonal anti-Islet-1 | R&D systems | Cat#AF1837; RRID: AB_2126324 |
| Mouse monoclonal anti-CD105 (clone SN6) | Abcam | Cat#ab11414; RRID: AB_298019 |
| Rabbit polyclonal anti-VE-Cadherin(CD144) | Signma-Aldrich | Cat#V1514; RRID: AB_477609 |
| Mouse monoclonal anti-fast myosin skeletal heavy chain antibody (Clone MY32) | Abcam | Cat#ab51263; RRID: AB_2297993 |
| Mouse Monoclonal anti-Nkx2.5 (Clone 259416) | R&D Systems | Cat#MAB2444; RRID: AB_2151378 |
| Mouse Monoclonal anti-TBX18 (Clone 635305) | R&D Systems | Cat#MAB63371; RRID: AB_10892533 |
| Rabbit Monoclonal anti-Wilms Tumor Protein (Clone CAN-R9(IHC)-56-2) | Abcam | Cat#ab89901; RRID: AB_2043201 |
| Goat Polyclonal anti-E-cadherin | R&D Systems | Cat#AF648; RRID: AB_355504 |
| Mouse Monoclonal anti-ZO-1 (Clone 1) | BD Biosciences | Cat#610966; RRID: AB_398279 |
| Mouse Monoclonal anti-alpha Smooth Muscle Actin (Clone 1A4) | Abcam | Cat#ab7817; RRID: AB_262054 |
| Rabbit Polyclonal anti-alpha Smooth Muscle Actin | Abcam | Cat#ab5694; RRID: AB_2223021 |
| Mouse Monoclonal anti-smooth muscle Myosin heavy chain 11 antibody (Clone 1G12) | Abcam | Cat#ab683; RRID: AB_2235569 |
| Rabbit Polyclonal anti-von Willebrand Factor | Dako | Cat#A0082; RRID: AB_2315602 |
| Rabbit Polyclonal anti-CD31 | Abcam | Cat#ab28364; RRID: AB_726362 |
| Mouse Monoclonal anti-CD31 (Clone JC70A) | Dako | Cat#M0823; RRID: AB_2114471 |
| Mouse Monoclonal anti-CD31 BD Horizon BV421 Conjugated (Clone WM59) | BD Biosciences | Cat#564089; RRID: AB_2714010 |
| Mouse Monoclonal anti-CD105 BD Horizon BV421 Conjugated (Clone 266) | BD Biosciences | Cat#563920; RRID: AB_2722606 |
| Mouse Monoclonal anti-NG2 | Abcam | Cat#ab83508; RRID: AB_2087616 |
| Mouse Monoclonal anti-NG2/MCSP Phycoerythrin Conjugated (Clone LHM-2) | R&D Systems | Cat#FAB2585P; RRID: AB_2087615 |
| Mouse Monoclonal anti-CD73 Phycoerythrin Conjugated (Clone AD2) | BD Biosciences | Cat#550257; RRID: AB_393561 |
| Rat Monoclonal anti-CD44 Allophyocyanin Conjugated (Clone IM7) | eBioscience | Cat#17-0441-82; RRID: AB_469390 |
| Mouse Monoclonal IgG1 kappa Isotype Control BD Horizon BV421 Conjugated (Clone X40) | BD Biosciences | Cat#562438; RRID: AB_11207319 |
| Rat IgG2b kappa Isotype Control Allophycocyanin Conjugated (Clone eB149/10H5) | eBioscience | Cat#17-4031-82; RRID: AB_470176 |
| Mouse Monoclonal IgG1 kappa Isotype Control Phycoerythrin Conjugated (Clone MOPC-21) | BD Biosciences | Cat#555749; RRID: AB_396091 |
| Mouse IgG1 kappa Isotype Control Allophycocyanin Conjugated (Clone P3.6.2.8.1) | eBioscience | Cat# 17-4714-42, RRID:AB_1603315 |
| Mouse IgG1 Isotype Control Allophycocyanin Conjugated (Clone 11711) |
R&D Systems | Cat#IC002A; RRID: AB_357239 |
| Sheep Polyclonal anti-Human TGN46 (Human Golgi) | AbD Serotec | Cat#AHP500G; RRID: AB_323104 |
| Goat Polyclonal anti-SOX2 (Clone Y-17) | Santa Cruz Biotechnology | Cat#sc-17320; RRID: AB_2286684 |
| Rabbit Polyclonal anti-Flk1-1 | Acris Antibodies GmbH | Cat# AP02618PU-S; RRID: AB_1624459 |
| Goat Polyclonal anti-DDR2 (N-20) | Santa Cruz Biotechnology | Cat#sc-7555; RRID: AB_639054 |
| Mouse Monoclonal anti-Calponin | Sigma-Aldrich | Cat#C2687; RRID: AB_476840 |
| Rabbit Polyclonal anti-VE-cadherin | Abcam | Cat#ab33168; RRID: AB 870662 |
| Rabbit Monoclonal anti-Vimentin (Clone EPR3776) | Abcam | Cat#ab92547; RRID: AB_10562134 |
| Mouse Monoclonal anti-Nuclei (Human Nuclear Antigen) (Clone 235-1) | Millipore | Cat#MAB1281; RRID: AB_94090 |
| Donkey Polyclonal anti-Mouse IgG (H+L) Alexa Fluor 488 Conjugated | Thermo Fisher | Cat#:A21202; RRID: AB_141607 |
| Donkey Polyclonal anti-Rabbit IgG (H+L) Alexa Fluor 488 Conjugated | Thermo Fisher | Cat#A21206; RRID: AB_141708 |
| Donkey Polyclonal anti-Goat IgG (H+L) Alexa Fluor 555 Conjugated | Thermo Fisher | Cat#A21432; RRID: AB_141788 |
| Donkey Polyclonal anti-Mouse IgG (H+L) Alexa Fluor 555 Conjugated | Thermo Fisher | Cat#A31570; RRID: AB_2536180 |
| Donkey Polyclonal anti-Rabbit IgG (H+L) Alexa Fluor 555 Conjugated | Thermo Fisher | Cat#A31572; RRID: AB_162543 |
| Donkey Polyclonal anti-Sheep IgG (H+L) Alexa Fluor 647 Conjugated | Thermo Fisher | Cat#A21448; RRID: AB_2535865 |
| Donkey Polyclonal anti-Goat IgG (H+L) Alexa Fluor 647 Conjugated | Thermo Fisher | Cat#A21447; RRID: AB_141844 |
| Donkey Polyclonal anti-Rabbit IgG (H+L) Alexa Fluor 647 Conjugated | Thermo Fisher | Cat#A31573; RRID: AB_2536183 |
| Anti-Histone H3 (mono methyl K4) antibody – ChIP Grade | Abcam | Cat#ab8895; RRID: AB_306847 |
| Anti-Histone H3 (acetyl K27) antibody – ChIP Grade | Abcam | Cat#ab4729; RRID: AB_2118291 |
| Biological Samples | ||
| rBioVaSc – Lewis rat jejunal scaffolds | Laboratory of Heike Walles | Kress et al., 2018 |
| Human fetal brain, fetal pancreas, fetal umbilical cord, fetal small intestine, and fetal thymus | Laboratory of Mana Parast | N/A |
| Human adult brain | University of California, Irvine | N/A |
| Human pancreatic islet | Laboratory or Jeanne Loring | N/A |
| Human adult diaphragm and duodenum | Laboratory of Chris Barry | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Probumin Life Science Grade | EMD Millipore | Cat#821005 |
| Animal-Free Recombinant Human Heregulinβ-1 | Peprotech | Cat#AF-100-03 |
| LONG® R3 IGF-I human | Sigma-Aldrich | Cat#85580C |
| Recombinant Human FGF basic, 145 aa (TC Grade) Protein, CF | R&D Systems | Cat#4114-TC |
| Recombinant Human/Mouse/Rat Activin A Protein, CF | R&D Systems | Cat#338-AC |
| Recombinant Human Wnt-3a Protein | R&D Systems | Cat#5036-WN |
| Recombinant Human BMP-4 Protein, CF | R&D Systems | Cat#314-BP |
| L-Ascorbic acid 2-phosphate sesquimagnesium salt hydrate ≥ 95% | Sigma-Aldrich | Cat#A8960 |
| Transferrin (HOLO), Human Plasma, Tissue Culture Grade | Athens Research & Technology | Cat#16-16-032001-LEL |
| Glutagro supplement (200mM L-alanyl-L-Glutamine) | Corning | Cat#25-015-CI |
| Antibiotic Antimyotic Solution 100X | Corning | Cat#30-004-CI |
| MEM Nonessential Amino Acids | Corning | Cat#25-025-CI |
| Trace Elements A | Corning | Cat#25-021-CI |
| Trace Elements B | Corning | Cat#25-022-CI |
| Trace Elements C | Corning | Cat#25-023-CI |
| 2-Mercaptoethanol | GIBCO | Cat#21985023 |
| Dulbecco’s Modification of Eagle’s Medium/Ham’s F-12 50/50 Mix |
Corning | Cat#15-090-CM |
| Recombinant Human VEGF 165 Protein | R&D Systems | Cat#293-VE |
| Minimal Essential Media, Alpha | Corning | Cat#15-012-CV |
| Fetal Bovine Serum – Embryonic Stem Cell Qualified | Atlanta Biolgoicals | Cat#S10250 |
| Recombinant Human PDGF-BB Protein, CF | R&D Systems | Cat#220-BB |
| Recombinant Human PDGF-AA Protein, CF | R&D Systems | Cat#221-AA |
| SB 431542 | Tocris | Cat#1614 |
| Geltrex LDEV-Free, hESC-Qualified, Reduced Growth Factor Basement Membrane Matrix | Thermo Fisher | Cat#A1413302 |
| Matrigel | VWR | Cat#47743-715 |
| Collagenase, Type IV, powder | Thermo Fisher | Cat#17104-019 |
| TrypLE Select Enzyme (10X), no phenol red | Thermo Fisher | Cat#A1217701 |
| Retinoic acid ≥ 98% (HPLC), powder | Sigma-Aldrich | Cat#R2625 |
| DPBS, 1X without calcium or magnesium | Corning | Cat#21-031 |
| Accutase | Innovative Cell Technologies | Cat#AT104 |
| Tamoxifen | Sigma-Aldrich | Cat#T5648 |
| Donkey Serum Sterile Filtered | Equitech-Bio | Cat#SD30 |
| Triton X-100 | Fisher Scientific | Cat#BP151 |
| Glycine | Sigma-Aldrich | Cat#G8898 |
| 4′,6-Diamidino-2-phenylindole dihydrochloride | Sigma-Aldrich | Cat#D9542 |
| ProLong Diamond Antifade Reagent | Invitrogen | Cat#P36961 |
| TaqMan Universal PCR Master Mix No AmpErase UNG | Thermo Fisher | Cat#4324020 |
| EDTA (0.5 M), pH 8.0 | Thermo Fisher | Cat#AM9260G |
| Sucrose | Sigma-Aldrich | Cat#S0389 |
| Sodium Chloride | Fisher Scientific | Cat#S640 |
| Paraformaldehyde, 16% Solution, EM Grade. | VWR | Cat#15170 |
| methanol-free formaldehyde | Thermo Scientific | Cat#28906 |
| Protein G Dynabeads | Invitrogen | Cat#10004D |
| Proteinase K | QIAGEN | Cat#19131 |
| Corn Oil Solution | Sigma-Aldrich | Cat#8267 |
| Trypsin-EDTA (0.05%), phenol red | Thermo Fisher | Cat#25300054 |
| Trizol | Invitrogen | Cat#15596-026 |
| Horse Serum | Atlanta Biologicals | Cat#S12150 |
| Fluorescein isothiocyanate–dextran | Sigma-Aldrich | Cat#FD40S |
| Osmium tetroxide solution | Sigma-Aldrich | Cat#75633 |
| Rimadyl® | Zoetis | Cat#779-358 |
| 3-(4,5-Dimethyl-2-thiazolyl)-2,5-diphenyl-2H-tetrazolium· bromide research grade (MTT, Thiazolyl blue) |
SERVA | Cat#20395 |
| Low Density Lipoprotein from Human Plasma, Acetylated, Dil complex (Dil AcLDL) |
Invitrogen | Cat#L3484 |
| VascuLife® VEGF-Mv Endothelial Complete Kit | Lifeline Cell Technology | Cat#LL-0005 |
| NucBlue Live ReadyProbes Reagent | Invitrogen | Cat#R37605 |
| Collagen Type 4 | Sigma-Aldrich | Cat#C5533 |
| Fibronectin Human Protein, Plasma | Thermo Fisher | Cat#33016015 |
| Vybrant DiO Cell-Labeling Solution | Thermo Fisher | Cat#V22886 |
| Carbachol - CAS 51-83-2 - Calbiochem | Sigma-Aldrich | Cat#212385 |
| Potassium Chloride | Fisher Scientific | CatP333-500 |
| Critical Commercial Assays | ||
| E.Z.N.A.® Total RNA Kit I | Omega Bio-Tek | Cat#R6834 |
| iScript Reverse Transcription Supermix | Bio-Rad | Cat#1708841 |
| RNeasy Mini Kit | QIAGEN | Cat#74104 |
| EZ DNA Methylation Kit | Zymo Research | Cat#D5001 |
| Infinium HumanMethylation450 BeadChip Kit | Illumina | Cat#WG-314-1001 |
| Flow Cytometry Fixation & Permeabilization Buffer Kit I | R&D Systems | Cat#FC009 |
| Click-iT® Plus EdU Alexa Fluor® 647 Flow Cytometry Assay Kit | Invitrogen | Cat#C10634 |
| Deposited Data | ||
| RNA-seq of WA09-derived MesoT, MLC, MesoT FBS, SplM-derived SMC, Mouse E15.5 mesothelium of lung, liver, heart, gut | This paper | GenBank: GSE113090 |
| ChIP-seq of WA09-derived MesoT K4me1 and MesoT K27ac |
This paper | GenBank: GSE113090 |
| Methylation data of WA09 hESCs, SplM, MesoT, WA07-derived CMs, E15.5 mouse heart/lung/liver/gut mesothelium, Primary human samples | This paper | GenBank: GSE116754 |
| Experimental Models: Cell Lines | ||
| WA09 hESCs (NIH approval number NIHhESC-10-0062) | WiCell | RRID: CVCL_9773 |
| WA07 hESCs (NIH approval number NIHhESC-10-0061) | WiCell | RRID: CVCL_9772 |
| WA01 hESCs (NIH approval number NIHhESC-10-0043) | WiCell | RRID: CVCL_9771 |
| TE03 hESCs (NIH approval number NIHhESC-13-0204) | WiCell | RRID: CVCL_C376 |
| K3 hiPSCs | Laboratory of Stephen Duncan | Si-Tayeb et al., 2010 |
| Human: dermal microvascular ECs | Laboratory of Heike Walles | Kress et al., 2018 |
| Experimental Models: Organisms/Strains | ||
| Mouse: Wt1tm2(cre/ERT2)Wtp/J | The Jackson Laboratory | JAX:010912; RRID: IMSR_JAX:010912 |
| Mouse: B6.Cg-Gt(ROSA)26Sortm14(CAG-tdTomato)Hze/J | The Jackson Laboratory | JAX:007914; RRID: IMSR_JAX:007914 |
| Rat: NIH-Foxn1rnu | Charles River | CrkNIH-Foxn1rnu |
| Oligonucleotides | ||
| POU5F1/OCT4 TaqMan® Probe for RT-qPCR | Thermo Fisher | Assay ID: Hs04260367_gH |
| SOX2 TaqMan® Probe for RT-qPCR | Thermo Fisher | Assay ID: Hs01053049_s1 |
| NANOG TaqMan® Probe for RT-qPCR | Thermo Fisher | Assay ID: Hs02387400_g1 |
| ISL1 TaqMan® Probe for RT-qPCR | Thermo Fisher | Assay ID: Hs00158153_m1 |
| NKX2.5 TaqMan® Probe for RT-qPCR | Thermo Fisher | Assay ID: Hs00231763_m1 |
| GATA4 TaqMan® Probe for RT-qPCR | Thermo Fisher | Assay ID: Hs00171403_m1 |
| WT1 TaqMan® Probe for RT-qPCR | Thermo Fisher | Assay ID: Hs01103751_m1 |
| TBX18 TaqMan® Probe for RT-qPCR | Thermo Fisher | Assay ID: Hs01385457_m1 |
| TCF21 TaqMan® Probe for RT-qPCR | Thermo Fisher | Assay ID: Hs00162646_m1 |
| MSLN TaqMan® Probe for RT-qPCR | Thermo Fisher | Assay ID: Hs00245879_m1 |
| RNA18S5 TaqMan® Probe for RT-qPCR | Thermo Fisher | Assay ID: Hs03928985_g1 |
| Software and Algorithms | ||
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
| Slidebook 6 | Intelligent Imaging Solutions | N/A |
| FlowJo v10 | FlowJo, LLC | N/A |
| GraphPad Prism v7.0b | https://www.graphpad.com/ | N/A |
| SRA Toolkit v2.4.1 | https://trace.ncbi.nlm.nih.gov/Traces/sra/sra.cgi?view=software | N/A |
| FastQC v0.10.1 | http://www.bioinformatics.babraham.ac.uk/projects/fastqc/ | N/A |
| Tophat2 v2.0.13 | Kim et al., 2013 | https://ccb.jhu.edu/software/tophat/index.shtml |
| Subread v1.4.2 | Liao et al., 2013 | https://sourceforge.net/projects/subread/files/subread-1.4.2/ |
| R | http://www.R-project.org/ | N/A |
| R studio v1.1.423 | https://www.rstudio.com/ | N/A |
| R package Limma | Ritchie et al., 2015 | https://bioconductor.org/packages/release/bioc/html/limma.html |
| Samtools v.1.3.1 | Li et al., 2009 | http://samtools.sourceforge.net/ |
| HTseq V0.6.1P1 | https://pypi.org/project/HTSeq/ | N/A |
| R package edgeR | Robinson et al., 2010; McCarthy et al., 2012 | N/A |
| R package Glimma v1.4 | Su et al., 2017 | https://bioconductor.org/packages/release/bioc/html/Glimma.html |
| Bowtie2 v2.2.9 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| Deeptools v2.3.1 | Ramírez et al., 2016 | https://deeptools.readthedocs.io/en/latest/index.html |
| MACS2 v2.1.1 | https://pypi.org/project/MACS2/ | N/A |
| ChIPSeek | Chen et al., 2014b | http://chipseek.cgu.edu.tw/index.py |
| Panther | Mi et al., 2017 | http://pantherdb.org |
| Other | ||
| Stericup-GP, 0.22 μm, polyethersulfone, 500 mL, radio-sterilized | EMD Millipore | Cat#SCGPU05RE |
| Steriflip Sterile Disposable Vacuum Filter Units | EMD Millipore | Cat#SE1M 179 M6 |
| Fisherbrand Sterile Cell Strainers | Fisher Scientific | Cat#22-363-547 |
| ThinCert Cell Culture Inserts | Greiner Bio-One International | Cat#662641 |
| Andwin Scientific Tissue-Tek CRYO-OCT Compound | Fisher Scientific | Cat#14-373-65 |
Highlights.
Multipotent vascular progenitor (MesoT) cells generated from pluripotent stem cells
MesoT cells generate vascular lineages including smooth muscle cells and endothelial cells
hPSC-derived MesoT cells have potential utility in tissue engineering and tissue repair
ACKNOWLEDGMENTS
Work in S.D. laboratory was supported by NIH grants P01 HL089471 and P01 GM75334; J.F.L. and K.L.N. were supported by the California Institute for Regenerative Medicine (RT1-01108, RT3-07655, TR1-01250, TR3-05603, and CL1-00502) and NIH (R01NS092042); and S.K. and M.M. were supported by the Bavarian Research Foundation (AZ-1044-12) and Bavarian FIT Program. We thank Andy Wessels for many useful discussions and Julie Nelson of the UGA CTEGD Cytometry Shared Resources Laboratory for continued help with flow cytometry and cell sorting supported by NIH grant 1S10RR027814.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.016.
DECLARATION OF INTERESTS
Patents have been awarded or are pending for intellectual property described in this report (D.M.R. and S.D.).
REFERENCES
- Alphonse RS, Vadivel A, Zhong S, McConaghy S, Ohls R, Yoder MC, and Thébaud B (2015). The isolation and culture of endothelial colony-forming cells from human and rat lungs. Nat. Protoc. 10, 1697–1708. [DOI] [PubMed] [Google Scholar]
- Andersen DC, Ganesalingam S, Jensen CH, and Sheikh SP (2014). Do neonatal mouse hearts regenerate following heart apex resection? Stem Cell Reports 2, 406–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, and Isner JM (1997). Isolation of putative progenitor endothelial cells for angiogenesis. Science 275, 964–967. [DOI] [PubMed] [Google Scholar]
- Asahina K, Tsai SY, Li P, Ishii M, Maxson RE Jr., Sucov HM, and Tsukamoto H (2009). Mesenchymal origin of hepatic stellate cells, submesothelial cells, and perivascular mesenchymal cells during mouse liver development. Hepatology 49, 998–1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger RP, Sun YH, Kulik M, Lee JK, Nairn AV, Moremen KW, Pierce M, and Dalton S (2016). ST8SIA4-Dependent Polysialylation is Part of a Developmental Program Required for Germ Layer Formation from Human Pluripotent Stem Cells. Stem Cells 34, 1742–1752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brede C, Friedrich M, Jordán-Garrote A-L, Riedel SS, Bäuerlein CA, Heinze KG, Bopp T, Schulz S, Mottok A, Kiesel C, et al. (2012). Mapping immune processes in intact tissues at cellular resolution. J. Clin. Invest. 122, 4439–4446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryant DM, O’Meara CC, Ho NN, Gannon J, Cai L, and Lee RT (2015). Asystematic analysis of neonatal mouse heart regeneration after apical resection. J. Mol. Cell. Cardiol. 79, 315–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano E, Carmona R, and Muñoz-Chápuli R (2013). Wt1-expressing progenitors contribute to multiple tissues in the developing lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 305, L322–L332. [DOI] [PubMed] [Google Scholar]
- Cano E, Carmona R, Ruiz-Villalba A, Rojas A, Chau YY, Wagner KD, Wagner N, Hastie ND, Muñoz-Chápuli R, and Pérez-Pomares JM (2016). Extracardiac septum transversum/proepicardial endothelial cells pattern embryonic coronary arterio-venous connections. Proc. Natl. Acad. Sci. USA 113, 656–661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao J, Navis A, Cox BD, Dickson AL, Gemberling M, Karra R, Bagnat M, and Poss KD (2016). Single epicardial cell transcriptome sequencing identifies Caveolin 1 as an essential factor in zebrafish heart regeneration. Development 143, 232–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HI, Sharma B, Akerberg BN, Numi HJ, Kivelä R, Saharinen P, Aghajanian H, McKay AS, Bogard PE, Chang AH, et al. (2014a). The sinus venosus contributes to coronary vasculature through VEGFC-stimulated angiogenesis. Development 141, 4500–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen T-W, Li H-P, Lee C-C, Gan R-C, Huang P-J, Wu TH, Lee C-Y, Chang Y-F, and Tang P (2014b). ChIPseek, a web-based analysis tool for ChIP data. BMC Genomics 15, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung C, Bernardo AS, Trotter MW, Pedersen RA, and Sinha S (2012). Generation of human vascular smooth muscle subtypes provides insight into embryological origin-dependent disease susceptibility. Nat. Biotechnol. 30, 165–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chong JJ, Chandrakanthan V, Xaymardan M, Asli NS, Li J, Ahmed I, Heffernan C, Menon MK, Scarlett CJ, Rashidianfar A, et al. (2011). Adult cardiac-resident MSC-like stem cells with a proepicardial origin. Cell Stem Cell 9, 527–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cliff TS, Wu T, Boward BR, Yin A, Yin H, Glushka JN, Prestegaard JH, and Dalton S (2017). MYC Controls Human Pluripotent Stem Cell Fate Decisions through Regulation of Metabolic Flux. Cell Stem Cell 21, 502–516.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colunga T, and Dalton S (2018). Building Blood Vessels with Vascular Progenitor Cells. Trends Mol. Med. 24, 630–641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Creyghton MP, Cheng AW, Welstead GG, Kooistra T, Carey BW, Steine EJ, Hanna J, Lodato MA, Frampton GM, Sharp PA, et al. (2010). Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci. USA 107, 21931–21936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixit R, Ai X, and Fine A (2013). Derivation of lung mesenchymal lineages from the fetal mesothelium requires hedgehog signaling for mesothelial cell entry. Development 140, 4398–4406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doi R, Tsuchiya T, Mitsutake N, Nishimura S, Matsuu-Matsuyama M, Nakazawa Y, Ogi T, Akita S, Yukawa H, Baba Y, et al. (2017). Transplantation of bioengineered rat lungs recellularized with endothelial and adipose-derived stromal cells. Sci. Rep. 7, 8447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drews JD, Miyachi H, and Shinoka T (2017). Tissue-engineered vascular grafts for congenital cardiac disease: Clinical experience and current status. Trends Cardiovasc. Med. 27, 521–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elmadbouh I, Chen Y, Louedec L, Silberman S, Pouzet B, Meilhac O, and Michel JB (2005). Mesothelial cell transplantation in the infarct scar induces neovascularization and improves heart function. Cardiovasc. Res. 68, 307–317. [DOI] [PubMed] [Google Scholar]
- Gittenberger-de Groot AC, Winter EM, and Poelmann RE (2010). Epicardium-derived cells (EPDCs) in development, cardiac disease and repair of ischemia. J. Cell. Mol. Med. 14, 1056–1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer D, Gambardella L, Bernard WG, Serrano F, Mascetti VL, Pedersen RA, Talasila A, and Sinha S (2015). Robust derivation of epicardium and its differentiated smooth muscle cell progeny from human pluripotent stem cells. Development 142, 1528–1541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- James D, Nam HS, Seandel M, Nolan D, Janovitz T, Tomishima M, Studer L, Lee G, Lyden D, Benezra R, et al. (2010). Expansion and maintenance of human embryonic stem cell-derived endothelial cells by TGFbeta inhibition is Id1 dependent. Nat. Biotechnol. 28, 161–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz TC, Singh MK, Degenhardt K, Rivera-Feliciano J, Johnson RL, Epstein JA, and Tabin CJ (2012). Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev. Cell 22, 639–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kikuchi K, Holdway JE, Major RJ, Blum N, Dahn RD, Begemann G, and Poss KD (2011). Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev. Cell 20, 397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, and Salzberg SL (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klaus A, Müller M, Schulz H, Saga Y, Martin JF, and Birchmeier W (2012). Wnt/β-catenin and Bmp signals control distinct sets of transcription factors in cardiac progenitor cells. Proc. Natl. Acad. Sci. USA 109, 10921–10926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kress S, Baur J, Otto C, Burkard N, Braspenning J, Walles H, Nickel J, and Metzger M (2018). Evaluation of a miniaturized biologically vascularized scaffold in vitro and in vivo. Sci. Rep. 8, 4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laflamme MA, Chen KY, Naumova AV, Muskheli V, Fugate JA, Dupras SK, Reinecke H, Xu C, Hassanipour M, Police S, et al. (2007). Cardiomyocytes derived from human embryonic stem cells in pro-survival factors enhance function of infarcted rat hearts. Nat. Biotechnol. 25, 1015–1024. [DOI] [PubMed] [Google Scholar]
- Langfelder P, and Horvath S (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9, 559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law CW, Chen Y, Shi W, and Smyth GK (2014). voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 15, R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y-U, Mahler N, Best CA, Tara S, Sugiura T, Lee AY, Yi T, Hibino N, Shinoka T, and Breuer C (2016). Rational design of an improved tissue-engineered vascular graft: determining the optimal cell dose and incubation time. Regen. Med. 11, 159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lepilina A, Coon AN, Kikuchi K, Holdway JE, Roberts RW, Burns CG, and Poss KD (2006). A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 127, 607–619. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). The Subread aligner: fast, accurate and scalable read mapping by seed-and-vote. Nucleic Acids Res. 41, e108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Q, Huang X, Oh JH, Lin RZ, Duan S, Yu Y, Yang R, Qiu J, Melero-Martin JM, Pu WT, and Zhou B (2014). Epicardium-to-fat transition in injured heart. Cell Res. 24, 1367–1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lucas PA (2007). Stem cells for mesothelial repair: an understudied modality. Int. J. Artif. Organs 30, 550–556. [DOI] [PubMed] [Google Scholar]
- Madisen L, Zwingman TA, Sunkin SM, Oh SW, Zariwala HA, Gu H, Ng LL, Palmiter RD, Hawrylycz MJ, Jones AR, et al. (2010). A robust and high-throughput Cre reporting and characterization system for the whole mouse brain. Nat. Neurosci. 13, 133–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maksimovic J, Gordon L, and Oshlack A (2012). SWAN: Subset-quantile within array normalization for illumina infinium HumanMethylation450 BeadChips. Genome Biol. 13, R44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand M, Anderson EK, Phadnis SM, Longaker MT, Cooke JP, Chen B, and Reijo Pera RA (2014). Concurrent generation of functional smooth muscle and endothelial cells via a vascular progenitor. Stem Cells Transl. Med. 3, 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy DJ, Chen Y, and Smyth GK (2012). Differential expression analysis of multifactor RNA-Seq experiments with respect to biological variation. Nucleic Acids Res. 40, 4288–4297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mellgren AM, Smith CL, Olsen GS, Eskiocak B, Zhou B, Kazi MN, Ruiz FR, Pu WT, and Tallquist MD (2008). Platelet-derived growth factor receptor beta signaling is required for efficient epicardial cell migration and development of two distinct coronary vascular smooth muscle cell populations. Circ. Res. 103, 1393–1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, and Thomas PD (2017). PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res. 45 (D1), D183–D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motherwell JM, Azimi MS, Spicer K, Alves NG, Hodges NA, Breslin JW, Katakam PVG, and Murfee WL (2017). Evaluation of Arteriolar Smooth Muscle Cell Function in an Ex Vivo Microvascular Network Model. Sci. Rep. 7, 2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutsaers SE, and Wilkosz S (2007). Structure and Function of Mesothelial Cells In Peritoneal Carcinomatosis: A Multidisciplinary Approach, Ceelen WP and Boston MA, eds. (Springer; ), pp. 1–19. [DOI] [PubMed] [Google Scholar]
- Nagai H, Chew SH, Okazaki Y, Funahashi S, Namba T, Kato T, Enomoto A, Jiang L, Akatsuka S, and Toyokuni S (2013). Metamorphosis of mesothelial cells with active horizontal motility in tissue culture. Sci. Rep. 3, 1144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmquist-Gomes P, Guadix JA, and Pérez-Pomares JM (2018). Avian embryonic coronary arterio-venous patterning involves the contribution of different endothelial and endocardial cell populations. Dev. Dyn. 247, 686–698. [DOI] [PubMed] [Google Scholar]
- Patsch C, Challet-Meylan L, Thoma EC, Urich E, Heckel T, O’Sullivan JF, Grainger SJ, Kapp FG, Sun L, Christensen K, et al. (2015). Generation of vascular endothelial and smooth muscle cells from human pluripotent stem cells. Nat. Cell Biol. 17, 994–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pennisi DJ (2016). Towards consensus on coronary vessel development: Coronary arterial endothelial cells derive primarily from the sinus venosus during embryogenesis. Circ. Res. 118, 1861–1862. [DOI] [PubMed] [Google Scholar]
- Pérez-Pomares JM, and de la Pompa JL (2011). Signaling during epicardium and coronary vessel development. Circ. Res. 109, 1429–1442. [DOI] [PubMed] [Google Scholar]
- Porrello ER, Mahmoud AI, Simpson E, Hill JA, Richardson JA, Olson EN, and Sadek HA (2011). Transient regenerative potential of the neonatal mouse heart. Science 331, 1078–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasain N, Lee MR, Vemula S, Meador JL, Yoshimoto M, Ferkowicz MJ, Fett A, Gupta M, Rapp BM, Saadatzadeh MR, et al. (2014). Differentiation of human pluripotent stem cells to cells similar to cord-blood endothelial colony-forming cells. Nat. Biotechnol. 32, 1151–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Que J, Wilm B, Hasegawa H, Wang F, Bader D, and Hogan BLM (2008). Mesothelium contributes to vascular smooth muscle and mesenchyme during lung development. Proc. Natl. Acad. Sci. USA 105, 16626–16630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Core Team (2013). R: A language and environment for statistical computing (R Foundation for Statistical Computing). http://www.R-project.org/
- Rada-Iglesias A, Bajpai R, Swigut T, Brugmann SA, Flynn RA, and Wysocka J (2011). A unique chromatin signature uncovers early developmental enhancers in humans. Nature 470, 279–283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Moser PT, Gilpin SE, Okamoto T, Wu T, Tapias LF, Mercier FE, Xiong L, Ghawi R, Scadden DT, et al. (2015). Engineering pulmonary vasculature in decellularized rat and human lungs. Nat. Biotechnol. 33, 1097–1102. [DOI] [PubMed] [Google Scholar]
- Rinkevich Y, Mori T, Sahoo D, Xu PX, Bermingham JR Jr., and Weissman IL (2012). Identification and prospective isolation of a mesothelial precursor lineage giving rise to smooth muscle cells and fibroblasts for mammalian internal organs, and their vasculature. Nat. Cell Biol. 14, 1251–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RStudio Team (2015). RStudio: Integrated Development for R (RStudio). [Google Scholar]
- Shelton EL, and Bader DM (2012). Thymosin β4 mobilizes mesothelial cells for blood vessel repair. Ann. N Y Acad. Sci. 1269, 125–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Si-Tayeb K, Noto FK, Sepac A, Sedlic F, Bosnjak ZJ, Lough JW, and Duncan SA (2010). Generation of human induced pluripotent stem cells by simple transient transfection of plasmid DNA encoding reprogramming factors. BMC Dev. Biol. 10, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh MK, and Epstein JA (2012). Epicardium-derived cardiac mesenchymal stem cells: expanding the outer limit of heart repair. Circ. Res. 110, 904–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh AM, Sun Y, Li L, Zhang W, Wu T, Zhao S, Qin Z, and Dalton S (2015). Cell-Cycle Control of Bivalent Epigenetic Domains Regulates the Exit from Pluripotency. Stem Cell Reports 5, 323–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smart N, Bollini S, Dubé KN, Vieira JM, Zhou B, Davidson S, Yellon D, Riegler J, Price AN, Lythgoe MF, et al. (2011). De novo cardiomyocytes from within the activated adult heart after injury. Nature 474, 640–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CL, Baek ST, Sung CY, and Tallquist MD (2011). Epicardialderived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 108, e15–e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smyth GK (2004). Linear models and empirical bayes methods for assessing differential expression in microarray experiments. Stat. Appl. Genet. Mol. Biol. 3, Article3. [DOI] [PubMed] [Google Scholar]
- Su S, Law CW, Ah-Cann C, Asselin-Labat ML, Blewitt ME, and Ritchie ME (2017). Glimma: interactive graphics for gene expression analysis. Bioin-formatics 33, 2050–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi M, Takeuchi K, Ozawa Y, Kohara A, and Mizusawa H (2009). Aneuploidy in Immortalized Human Mesenchymal Stem Cells with Non-Random Loss of Chromosome 13 in Culture. Vitro Cell Dev. Anim. 45, 290–299. [DOI] [PubMed] [Google Scholar]
- Tian X, Pu WT, and Zhou B (2015). Cellular origin and developmental program of coronary angiogenesis. Circ. Res. 116, 515–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Wijk B, Gunst QD, Moorman AFM, and van den Hoff MJB (2012). Cardiac regeneration from activated epicardium. PLoS ONE 7, e44692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villalona GA, Udelsman B, Duncan DR, McGillicuddy E, Sawh-Martinez RF, Hibino N, Painter C, Mirensky T, Erickson B, Shinoka T, and Breuer CK (2010). Cell-seeding techniques in vascular tissue engineering. Tissue Eng. Part B Rev. 16, 341–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang L, Schulz TC, Sherrer ES, Dauphin DS, Shin S, Nelson AM, Ware CB, Zhan M, Song C-Z, Chen X, et al. (2007). Self-renewal of human embryonic stem cells requires insulin-like growth factor-1 receptor and ERBB2 receptor signaling. Blood 110, 4111–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilm B, Ipenberg A, Hastie ND, Burch JB, and Bader DM (2005). The serosal mesothelium is a major source of smooth muscle cells of the gut vasculature. Development 132, 5317–5328. [DOI] [PubMed] [Google Scholar]
- Winters NI, Williams AM, and Bader DM (2014). Resident progenitors, not exogenous migratory cells, generate the majority of visceral mesothelium in organogenesis. Dev. Biol. 391, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witty AD, Mihic A, Tam RY, Fisher SA, Mikryukov A, Shoichet MS, Li RK, Kattman SJ, and Keller G (2014). Generation of the epicardial lineage from human pluripotent stem cells. Nat. Biotechnol. 32, 1026–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wixson SK, and Smiler KL (1997). Anesthesia and Analgesia in Rodents. In Anesthesia and Analgesia in Laboratory Animals, Kohn DF, ed. (Academic Press; ), pp. 165–203. [Google Scholar]
- Zangi L, Lui KO, von Gise A, Ma Q, Ebina W, Ptaszek LM, Später D, Xu H, Tabebordbar M, Gorbatov R, et al. (2013). Modified mRNA directs the fate of heart progenitor cells and induces vascular regeneration after myocardial infarction. Nat. Biotechnol. 31, 898–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B, and Pu WT (2012). Isolation and characterization of embryonic and adult epicardium and epicardium-derived cells In Cardiovascular Development. Methods in Molecular Biology (Methods and Protocols), Peng X and Antonyak M, eds. (Humana Press; ), pp. 155–168. [DOI] [PubMed] [Google Scholar]
- Zhou B, Ma Q, Rajagopal S, Wu SM, Domian I, Rivera-Feliciano J, Jiang D, von Gise A, Ikeda S, Chien KR, and Pu WT (2008). Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 454, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All the sequencing datasets generated for this paper have been deposited into GEO under the accession numbers GSE113090 for RNA-seq and ChIP-seq, GSE116754 for DNA methylation.