

Abstract
Alström syndrome (OMIM #203800) is an autosomal recessive obesity ciliopathy caused by loss-of-function mutations in the ALMS1 gene. In addition to multi-organ dysfunction, such as cardiomyopathy, retinal degeneration and renal dysfunction, the disorder is characterized by high rates of obesity, insulin resistance and early-onset type 2 diabetes mellitus (T2DM). To investigate the underlying mechanisms of T2DM phenotypes, we generated a loss-of-function deletion of alms1 in the zebrafish. We demonstrate conservation of hallmark clinical characteristics alongside metabolic syndrome phenotypes, including a propensity for obesity and fatty livers, hyperinsulinemia and glucose response defects. Gene expression changes in β-cells isolated from alms1−/− mutants revealed changes consistent with insulin hypersecretion and glucose sensing failure, which were corroborated in cultured murine β-cells lacking Alms1. We also found evidence of defects in peripheral glucose uptake and concomitant hyperinsulinemia in the alms1−/− animals. We propose a model in which hyperinsulinemia is the primary and causative defect underlying generation of T2DM associated with alms1 deficiency. These observations support the alms1 loss-of-function zebrafish mutant as a monogenic model for mechanistic interrogation of T2DM phenotypes.
Introduction
Primary cilia are present on the majority of vertebrate cell types and act as cellular hubs for sensing and transducing signaling pathways (1). Deletions in ciliary genes—including basal body proteins, transition zone components and intraflagellar transport elements —are implicated in diseases termed ciliopathies, which exhibit a broad range of phenotypes (2,3). A unique subset among these disorders is obesity ciliopathies, which present with highly penetrant, early-onset obesity. Alström syndrome, one of the two major obesity ciliopathies, is associated with the most profound metabolic derangement among the disorders, including prominent early-onset truncal obesity, severe insulin resistance, hyperinsulinemia, early-onset type 2 diabetes mellitus (T2DM) and several other metabolic syndrome features (4,5). This rare autosomal recessive syndrome is caused by pathogenic variants in ALMS1, which localizes to the basal body of cilia (6), and may play a role in intracellular trafficking (7,8). The reported mutations include loss of function and truncations and a likely role in centrosome and basal body biology (4,6). However, the precise cellular function is unknown, although proposed roles include a structural or scaffolding role in centrosomal complexes (9), regulation of cytoskeletal components (7) and modulation of secretion and endosomal trafficking (7,8). Additional hallmark features of the disorder include cardiomyopathy, retinal degeneration and renal dysfunction. The unique metabolic features of Alström syndrome, however, offer the opportunity for generating models to better understand the cellular etiology underlying metabolic conditions, including T2DM, and to investigate the potential role of ciliary proteins.
Here, we present a zebrafish model of Alström syndrome. Zebrafish (Danio rerio) have long been used as models of human genetic disease. External embryonic development allows for whole-body knockdown and knockout studies, a translucent body plan permits high-resolution microscopy of whole animals, and high fecundity provides ease of mutant generation as well as large-scale omic studies (10–13). Zebrafish have also recently emerged as robust models of metabolic biology owing to conservation of basic physiology, including gastrointestinal function, endocrine regulation, lipid and glucose metabolism and central neuronal regulation of food intake, all present by 5 days post-fertilization (dpf) (14,15). We previously correlated β-cell proliferation to T2DM in obesity ciliopathies using knockdown models of two ciliopathies, Bardet–Biedl syndrome and Alström syndrome (8,16). While these transient knockdown studies informed the role of alms1 in regulation of β-cell production, the inherent limitations of transient knockdown preclude more large-scale studies and extensive investigation of later onset and adult phenotypes.
We generated a genomic Alström syndrome model by targeting the zebrafish alms1 gene using CRISPR/Cas9. The resulting heritable mutation ablated protein production and resulted in systemic defects that recapitulate the human syndrome, including defects in neurosensory, cardiac and renal systems. Metabolic phenotypes included highly penetrant hepatic steatosis, increased weight gain under high-fat (HF) feeding conditions, impaired glucose uptake, defective systemic responses to high-glucose conditions and early-onset hyperinsulinemia. To more closely examine glucose regulation in this monogenic, hyperinsulinemia-driven diabetic model, we introduced loss-of-function alms1 mutations into a transgenic β-cell reporter zebrafish line, isolated β-cells from the alms1−/− zebrafish and examined whole transcriptome data using RNA-seq. These data reveal a role for alms1 in modulation of β-cell insulin secretion through regulation of intracellular secretion and supported impaired glucose sensing. These findings indicate that β-cell-autonomous defects are a primary driver of early-onset hyperinsulinemia and T2DM, potentially providing insight into β-cell defects underlying more common forms of T2DM.
Results
Generation of a zebrafish alms1 loss-of-function mutant
We targeted zebrafish alms1 by injection of guide RNAs (gRNA) targeted to exon 4 of the gene along with Cas9 protein injection directly into one-cell stage embryos of Tg(insa:mCherry) transgenic zebrafish, in which β-cells express mCherry under control of the preproinsulin (insa) promoter (Fig. 1A) (17). This design allowed for whole-body phenotyping as well as examining β-cell-specific defects in mutants. In adult F1 animals, we identified a heritable 7 bp deletion (c.1086_1092del) in exon 4 that results in a frameshift and introduction of a premature stop codon at p.S364X (Fig. 1A). We identified multiple heterozygous F2 carriers of the deletion and in-crossed them to propagate a homozygous alms1−/− line. We noted non-Mendelian ratios in progeny of heterozygous parents, with homozygotes representing only 14% of genotyped adult fish (P = 0.007; chi-squared; Fig. 1B), suggesting an important developmental role for alms1. alms1−/− homozygous mutant animals survived to adulthood and could be mated to produce viable progeny. Their offspring also exhibited high rates of curly tail phenotype at 3 dpf, which is typical of zebrafish ciliary mutants (Fig. 1C) (18). alms1 RNA and Alms1 protein levels were dramatically decreased in alms1−/− animals (Fig. 1D and E). Intermediate RNA levels were identified in heterozygote animals, although no morphological abnormalities were observed (Supplementary Material, Fig. S1A), indicating non-hypomorphic penetrance of the mutation.
Figure 1.
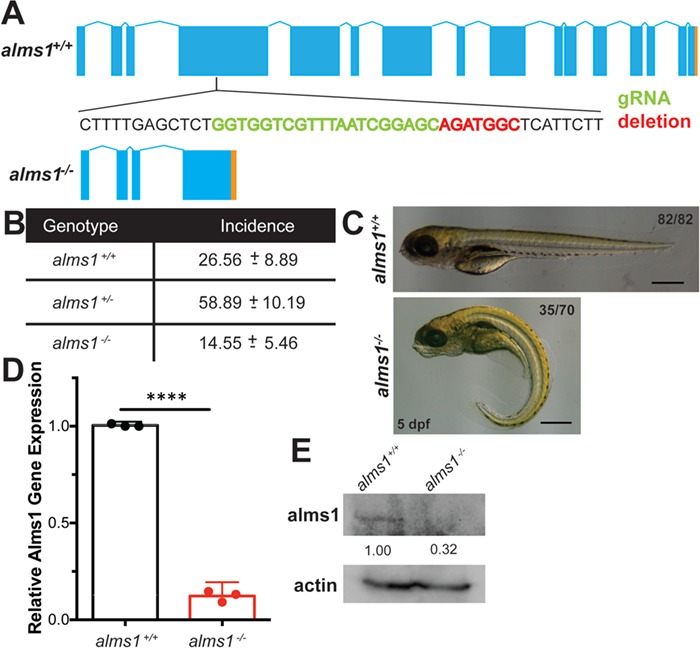
Generation of zebrafish alms1−/− line. (A) Schematic of alms1 genomic region with and without CRISPR/Cas9-induced deletion resulting in premature stop codon (orange). Sequence shown indicates region in exon 4 with sgRNA target (green) and deletion (red). (B) Ratios of identified mutants in heterozygous in-cross progeny indicating reduced rate of homozygous mutants relative to expected Mendelian ratios. (C) Representative images of alms1+/+ (n = 82) and alms1−/− (n = 70) larvae at 5 dpf. Numbers of alms1−/− larvae having ciliopathy body dysmorphogenesis shown. Scale bar, 1 mm. (D)alms1−/− larvae have reduced RNA expression levels compared to alms1+/+ at 5 dpf (n = 60–90 larvae). Statistics, Student's t-test, ****P < 0.0001. Dots, replicates; error bars, 95% confidence interval (CI). (E)alms1−/− larvae have reduced protein levels compared to alms1+/+ at 5 dpf.
alms1−/− zebrafish phenocopy Alström-induced cardiomyopathy, retinal dystrophy and renal defects
Having identified viable homozygous alms1 mutants, we examined broader phenotypes in alms1−/− fish. Alström syndrome patients and mouse mutant models exhibit a wide range of phenotypes across multiple organ systems (4,19).
Nearly two-thirds of Alström syndrome patients present with dilated cardiomyopathy and congestive heart failure (20). The earliest manifestation of cardiomyopathy in zebrafish is cardiac edema, which results from failed contractility, loss of vascular integrity or ventricular malformations (21). Compared to 2.5% of wild-type control animals generated from sibling clutch mates, 18% of alms1−/− embryos exhibited cardiac edema at 48 hours post-fertilization (hpf) (Fig. 2A). The frequency of this edema was consistent through 5 dpf in both genotypes (Supplementary Material, Fig. S1B), suggesting a lack of progressive cardiac defects. Although this reflected a 6-fold increase in edema (P < 0.0001; observed versus expected), the majority of alms1−/− embryos appeared largely normal (Fig. 2A). We observed smaller hearts in adult alms1−/− fish when compared with wild-type controls (Fig. 2B). A closer examination revealed loss of ventricular integrity as seen by the thickened walls in adult alms1−/− fish (Fig. 2C), expansion from 21.37 ± 2.7 μm in alms1+/+ adult hearts to 32.11 ± 3.4 μm (P = 0.026; Fig. 2C). These findings are consistent with cardiomyopathy in Alström, including reported variability in phenotypic presentation in patients (20).
Figure 2.
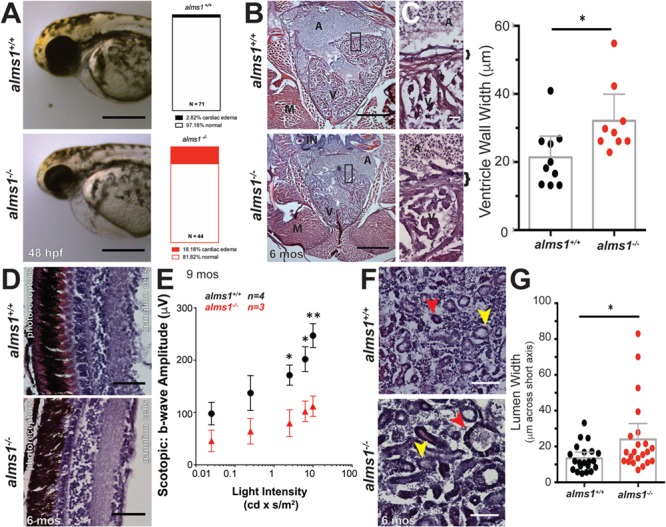
alms1−/− zebrafish display multiple defective organ systems. (A) Representative images of alms1+/+ and alms1−/− larvae showing severe cardiac edema at 48 h post-fertilization. Scale bar, 1 mm. Quantification of cardiac edema rates in alms1+/+ (n = 71) and alms1−/− (n = 44) larvae. Significance, chi-squared, ****P < 0.0001. (B) H&E sections from alms1+/+ and alms1−/− zebrafish at 6 months showing gross structure in the heart. Scale bar, 500 μm. A, atrium; V, ventricle; M, muscle; IN, intestine. (C) H&E sections from alms1+/+ and alms1−/− zebrafish showing high magnification of ventricular wall. Quantification of thickness of ventricular wall in alms1+/+ and alms1−/− animals (n = 4 animals). Bracket, ventricular wall. Scale bar, 25 μm. A, atrium; V, ventricle. Dots, individual measurements; error bars, 95% CI. (D) Representative images of H&E staining of retinal layers from alms1+/+ and alms1−/− zebrafish at 6 months showing degradation of multiple retinal layers in alms1−/− animals. Retina directionality indicated by photoreceptor and ganglion cell sides. Scale bar, 50 μm. (E) Quantification of scotopic b-wave amplitudes in response to light stimulation from alms1+/+ (n = 4) and alms−/− (n = 3) zebrafish at 9 months of age. Error bars, 95% CI. (F) Representative H&E images of kidneys in alms1+/+ and alms1−/− animals at demonstrating the abnormal shape and size of kidney tubules in alms1−/− animals. Distal tubule, yellow arrow; proximal tubule, red arrow. Scale bar, 100 μm. (G) Lumen width along short axis in kidneys from alms1+/+ and alms1−/− animals (n = 4 animals). Error bars, 95% CI. Where indicated, *P < 0.05, **P < 0.01.
Retinal degeneration is typical in Alström patients, with visual impairment appearing within a few weeks of birth and complete blindness before age 30 as a result of progressive photoreceptor loss (4,22). We assessed alms1+/+ adult retinas and found no impairment of retinal architecture, with the ganglion cell, plexiform and photoreceptor layers all intact (Fig. 2D; Supplementary Material, Fig. S1C). The alms1−/− adults exhibited reduced retinal integrity, most notably in the photoreceptor layer, which was visibly degraded (Fig. 2D). The overall retinal width and the photoreceptor layer were thinner in the alms1−/− retinas, supporting the reports of tissue degradation (Supplementary Material, Fig. S1C). These observations are consistent with the retinal degeneration and subsequent blindness observed in human patients. To assess functional impact of alms−/− on vision, we performed full-field electroretinograms (ERGs) on 9 month-old alms1+/+ and alms−/− zebrafish. Non-invasive in vivo scotopic ERGs showed attenuated b-wave amplitudes in alms−/− zebrafish as compared to controls (Fig. 2E), indicating reduced photoreceptor responses. Intriguingly, we noticed age-dependent reduced motility and decreased food-seeking behavior in alms1−/−mutants, consistent with severe vision loss (data not shown). Together with our histological findings, these data implicate Alms1 in maintenance of retinal sensory epithelia and visual function.
Nearly half of Alström patients have a broad range of renal defects, including total failure, calculi, interstitial fibrosis and glomerulosclerosis (4). We examined gross kidney morphology in H&E-stained sections of kidneys from alms1+/+ adults and observed well-formed tubule structure of varying intensities, indicating distal (lighter stain, yellow arrow) and proximal (darker stain, red arrow) tubules (Fig. 2F). alms1−/− kidney sections contained apparent interstitial degradation and dilation of distal and proximal tubules (Fig. 2F; colored arrows). Quantification of the tubule lumens, measuring across the shortest axis, indicates dilated tubules in the alms1−/− animals, increasing to 23.99 ± 4.3 μm, as compared to alms1+/+, an average of 13.43 ± 1.6 μm (P = 0.029; Fig. 2G). These observations are consistent with degradation of tubules and interstitial space and with previously reported phenotypes in Alms1−/− mice (19).
alms1−/− zebrafish exhibit systemic metabolic defects
Having confirmed prominent multi-organ features in alms1−/− zebrafish, consistent with reports in other organisms (4,19,20), we next assessed metabolic function in these animals. Alström patients exhibit high rates of early-onset obesity coupled with hyperinsulinemia and progression to frank T2DM, often as early as the second decade of life (4). To examine possible increased weight gain, we subjected adult wild-type or mutant fish clutch mates to an 8 week dietary regimen consisting of either regular pellet diet (control diet) or a 10-fold increased weight of pellet diet supplemented with 5-fold weight of egg yolk powder (HF diet) (23,24). alms1+/+ fish did not gain significant weight with control diet, but HF diet produced a 1.9-fold increase over starting weight (P = 0.033; two-way ANOVA; Fig. 3A and B). By comparison, alms1−/− fish gained weight under all dietary conditions. Control diet in alms1−/− fish resulted in a 1.6-fold increase over starting weight and HF diet produced a 3.2-fold increase in weight. Notably, significant weight gain in alms1−/− fish was apparent after only 3 weeks of HF feeding (P = 0.021; two-way ANOVA; Fig. 3A and B). We did not observe quantifiable differences in food intake at larval stages, suggesting that the obesity phenotype is unrelated to central regulation of satiety or hunger (Supplementary Material, Fig. S1D).
Figure 3.
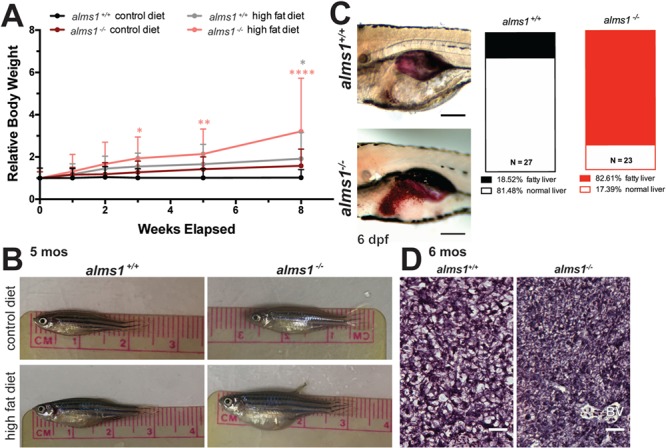
alms1−/− zebrafish exhibit increased weight gain and systemic metabolic defects. (A)alms1+/+ and alms1−/− zebrafish adults at 3 months were fed daily with set weights of either maintenance diet (control) or overfeeding with HF diet for 8 weeks (n = 4–6 animals per condition). Statistics, two-way ANOVA compared to alms1+/+ control diet. (B) Representative images of alms1+/+ and alms1−/− zebrafish after 8 weeks of either control diet or HF diet. (C) Representative images of Oil Red O staining in livers of alms1+/+ (n = 27) and alms1−/− (n = 23) larvae at 6 dpf. Scale bar, 500 μm. Quantification of Oil Red O positive livers. Significance, chi-squared. (D) Representative regions of sectioned H&E liver tissue from alms1+/+ and alms1−/− zebrafish at 6 months. Scale bar, 25 μm. BV, blood vessel.
Truncal obesity observed in Alström patients is accompanied by hepatic steatosis, a common hepatic manifestation of metabolic syndrome. Fatty liver has been reported in a substantial proportion of patients and mouse Alström models (4,19). To determine if mutant zebrafish exhibit abnormal accumulation of hepatic lipids, we stained whole-mount 6 dpf larvae with Oil Red O and examined hepatic lipid content (Fig. 3C). Only 18% of alms1+/+ embryos exhibited observable and dense Oil Red O staining in their livers, while 82% of alms1−/− embryos exhibited excess fat deposition (P < 0.0001; observed versus expected; Fig. 3C). In adult alms1−/− fish, this was accompanied by hepatopancreatic hypertrophy, demonstrated by increased cell density when compared to age- and section-matched wild-type animals (Fig. 3D).
alms1−/− zebrafish β-cells display abnormal glucose and insulin molecular responses
While hyperinsulinemia and insulin resistance provide the physiological foundation for T2DM, loss of β-cell mass and function is the hallmark of disease progression. We identified increased β-cell apoptosis and decreased β-cell proliferation after alms1 knockdown (16), indicating a β-cell deficiency in Alström-associated diabetes. Consistent with these data, alms1−/− adult pancreatic tissue had fewer insulin granule-positive areas, encompassed by the indicated regions, and lacked integrity, the gaps indicated by yellow arrowheads, when compared to location-matched control tissues (Fig. 4A). A total of 5 dpf alms1−/− larvae contained an average of 20.4 ± 1.3 β-cells per fish, compared to 31.9 ± 1.0 β-cells per fish in controls (P < 0.0001; Student's t-test; Fig. 4B). Given reduced β-cell number in alms1−/− animals at all stages examined, we hypothesized an inability to appropriately sense and regulate systemic glucose in alms1−/− animals.
Figure 4.
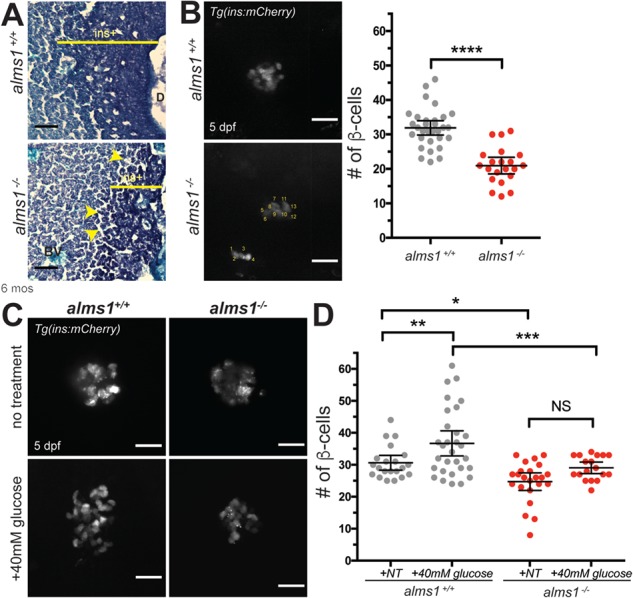
alms1−/− zebrafish islets have fewer β-cells and reduced glucose responsiveness. (A) Aldehyde fuchsin staining of sectioned zebrafish at 6 months showing aberrant islet structure (`ins+', dark purple regions) and tissue degradation (arrowheads) in alms1−/− as compared to alms1+/+. BV, blood vessel; D, secretory duct. Scale bar, 50 μm. (B)β-cell imaging and quantification (count method in bottom panel) of 5 dpf larvae in alms1+/+ (n = 31) and alms1−/− (n = 22) larvae via ins:mCherry reporter expression. Scale bar, 25 μm. Dots, individual larvae. Statistics, Student's t-test with Welch's Correction. (C) Representative images of β-cells with and without exposure to 40 mm glucose in alms1+/+ and alms1−/− larvae. Scale bar, 25 μm. (D) Quantification of β-cells at 5 dpf from alms1+/+ (NT = 22, Glu = 31) and alms1−/− (NT = 25, Glu = 18) larvae via ins:mCherry reporter expression. Statistics, one-way ANOVA. Dots, individual larvae. Error bars, 95% CI. Where indicated, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We evaluated the impact of this failed glucose sensing in the pancreas by examining β-cell numbers in larval fish, where high-glucose conditions are known to result in expansion (25). We evaluated this response using alms1+/− progeny treated with either glucose-free standard embryo media or media supplemented with high glucose (40 mm) starting at 24 hpf. A total of 5 dpf larvae were fixed and mCherry+ β-cells were imaged and counted prior to genotyping (Fig. 4C). This blinded analysis demonstrated β-cell expansion in wild-type larvae grown in 40 mm glucose media from an average of 30.6 ± 1.1 to 36.7 ± 1.9 β-cells per fish (P = 0.0049; one-way ANOVA; Fig. 4D) (16). The alms1−/− larvae exhibited reduced numbers of β-cells under control conditions, an average of 27.0 ± 1.1 β-cells, and failed to increase β-cell numbers after high-glucose exposure, an average of 29.1 ± 0.9 β-cells (P = 0.0671; one-way ANOVA; Fig. 4D), implicating Alms1 in β-cell response to elevated systemic glucose.
The β-cell deficits suggest a pathophysiology in which β-cells are unable to properly respond to exogenous cues. To better understand how loss of alms1 may impact β-cell function, we generated single-cell homogenates from 5 dpf larvae of either wild-type Tg(insa:mCherry) or Tg(insa:mCherry);alms1−/− and isolated mCherry+ cells via flow assisted cell sorting (FACS) sorting (Fig. 5A) (26). Using RNA from isolated cells, we carried out whole-transcriptome analysis via RNA-Seq (26,27). Using a cut off for differentially expressed genes of a fold change of greater than 1 or less than −1, we identified a total of 3880 up-regulated genes and 5531 down-regulated genes in alms1−/−β-cells (Fig. 5B). We confirmed the nature of these cells by identifying enriched expression of islet and β-cell markers (Fig. 5C). We then carried out pathway analysis of significantly changed genes and found that down-regulated genes in mutant β-cells strongly supported a generalized decrease in cellular activities, such as protein and RNA processing, consistent with previously reported significant apoptosis (Supplementary Material, Fig. S2 and Tables S1 and S2) (16). Over-represented terms in the up-regulated gene set were related to cellular transport and secretion (37 of 74 categories) and cell membrane ion transport (Supplementary Material, Table S1). These findings support a role for alms1 in regulating secretion and membrane depolarization in β-cells. We identified a group of significantly abundant pathways in up-regulated genes with known pancreatic functions (Supplementary Material, Table S1). These genes clustered into those implicated in monogenic diabetes, nutrient transport (mainly metal ion and amino acids carriers), insulin secretion (including calcium and potassium channels) and generalized pancreatic secretion (Fig. 5C). The intersections of these pancreas-related pathways contain a subset of six genes, including slc2a2/glut2, found in three pathways, and insulin, found in two pathways (red numbers; Fig. 5C).
Figure 5.
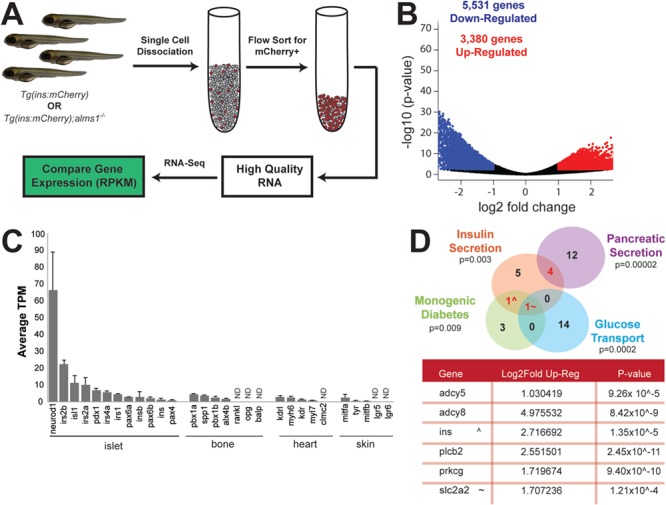
Transcriptomic analysis indicates dysregulation of insulin secretion and glucose sensing in zebrafish alms1−/−β-cells. (A) Schematic of experimental design for comparative gene expression in β-cell-enriched populations from age-matched control and alms1−/− larvae. (B) Volcano plot of significantly differentially expressed genes from GRZ10 between control and alms1−/− larvae. (C) Selected tissue specific genes identified in isolated alms1+/+ cells indicating high expression of pancreatic markers. Error bars, standard deviation. ND, not identified. (D) Subset of significantly up-regulated pathway nodes, identified by ConsensusPath DB, in alms1−/−β-cells. Genes within intersections are listed in table alongside the fold increase and significance compared to alms1+/+β-cells.
Alström syndrome represents a primary hyperinsulinemia diabetes model
Given the identified functional defects in alms1−/−β-cells and the implication of insulin secretion defects from transcriptomic data, we hypothesized that the diabetes phenotype may be linked to dysregulated glucose sensing or insulin secretion.
We evaluated the mechanism by which ALMS1 impacts insulin secretion using a simplified cultured mouse β-cell model, Beta-TC-6. Alms1 knockdown was accomplished via transient siRNA transfection (si-Alms1) with an average of 0.38 ± 0.04 Alms1 expression relative to control levels (P < 0.0001; Student's t-test; Supplementary Material, Fig. S1E). We examined the RNA-Seq implicated subset of β-cell function genes in cultured β-cells. When exposed to 16.7 mm glucose, physiological high-glucose conditions, control β-cells showed an increase in adenylyl cyclase 5 (Adcy5) and calcium/calmodulin-dependent protein kinase 2b (Camk2b), both known to transmit signals downstream of glucose transport in murine β-cells, along with moderate changes to Pklr, which initiates intracellular glucose processing and alters cellular metabolism, and Slc2a2, the main murine glucose transporter (Fig. 6A) (28). Irrespective of glucose levels, si-Alms1β-cells exhibited a dampened Adcy5 response and higher levels of Camk2b gene expression profiles (P = 0.002; two-way ANOVA; Fig. 6A). This appeared to be accompanied by a failure to alter Pklr and Slc2a2 expression in response to glucose (Fig. 6A). In fact, Pklr appeared to mimic high-glucose control conditions even in si-Alms1β-cells cultured in low-glucose conditions (Fig. 6A), suggesting an inability to properly sense external glucose conditions and regulate gene expression.
Figure 6.
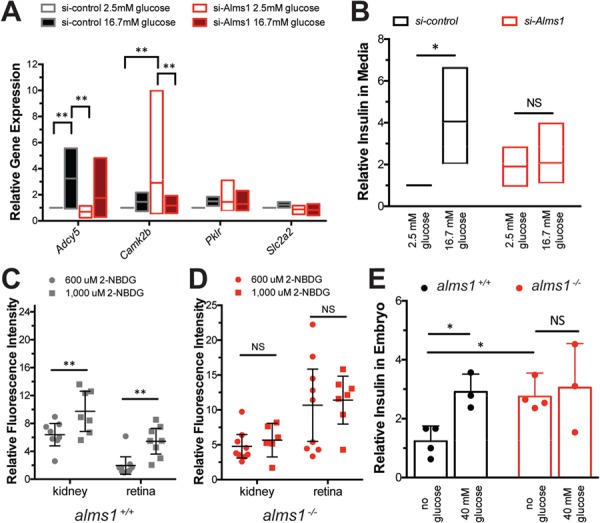
Hyperinsulinemia accompanied by defective glucose sensing with loss of Alms1. (A) Expression of glucose response genes in si-Alms1β-cells under basal (2.5 mm) and high-glucose (16.7 mm) conditions (n = 4). Cells were collected after 10 min after glucose stimulation. (B) Relative insulin by ELISA-based detection in culture media from β-cells after 30 min of exposure to 2.5 mm and 16.7 mm glucose (n = 3), normalized to basal si-control, shows failure of si-Alms1 cells to alter insulin secretion. (C and D) 2-NBDG at indicated concentrations was provided to (C) alms1+/+ and (D) alms1−/− larvae for 6 h. The kidney and retinal fluorescence intensity were quantified at each dose. Dots, individual larvae. (E) Relative insulin by ELISA-based detection under the indicated conditions in alms1+/+ (n = 30–50 per group) and alms1−/− (n = 30–50 per group) larvae at 5 dpf. All statistics, two-way ANOVA. Dots, replicates; error bars, 95% CI. Where indicated, *P < 0.05, **P < 0.01.
These gene expression data led us to hypothesize si-Alms1β-cells would exhibit inappropriate insulin secretion in response to high glucose. Secreted insulin upon stimulation with physiological high glucose in si-Alms1β-cells was evaluated using high-sensitivity ELISA. Control β-cells required 10 min to begin secreting insulin in response to high glucose and reached a total of 2.9-fold higher secreted insulin by 30 min (Fig. 6B). si-Alms1β-cells showed 1.8-fold higher levels of secreted insulin than control β-cells without glucose stimulation and after 30 min of high glucose, si-Alms1β-cells were at 2.0-fold of unstimulated control levels (P = 0.032; two-way ANOVA; Fig. 6B). These data suggest a modest hypersecretory basal state in unstimulated Alström β-cells accompanied by an impairment in glucose-stimulated insulin secretion.
We returned to our in vivo zebrafish model to evaluate glucose sensing and insulin secretion responses. First, we examined peripheral glucose uptake in wild-type zebrafish larvae via exposure to 2-(N-(7-nitrobenz-2-oxa-1,3-diazol-4-yl)amino)-2-deoxyglucose (2-NBDG), a fluorescently labeled glucose analog, and quantified fluorescence intensity in larval kidneys and retinas after 6 h (29). The kidney represents a highly glucose sensitive but non-insulin dependent tissue, which demonstrated a dose-dependent increase in fluorescence intensity, from 6.4-fold in 600 μm to 9.7-fold in 1000 μm 2-NBDG (P < 0.0072; two-way ANOVA; Fig. 6C). The retina exhibits insulin-dependent glucose uptake and also showed a dose-dependent increase in fluorescence, from 1.9-fold in 600 μm to 5.4-fold in 1000 μm 2-NBDG (P < 0.0033; two-way ANOVA; Fig. 6C). Unlike the wild-type animals, however, neither the kidney nor retina of alms1−/− animals increased fluorescence intensity at higher doses (NS, not significant; two-way ANOVA; Fig. 6D). Therefore, we conclude a loss of both mediated and passive glucose sensing and uptake in peripheral tissues of alms1−/− larvae.
While informative for assessing the capacity of alms1−/− animals to sense and regulate glucose homeostasis, the 2-NBDG compound does not address β-cell competency for insulin secretion. To assess β-cell responses, we challenged larvae with glucose and quantified total insulin in pooled 5 dpf larvae using a high-sensitivity insulin ELISA. Insulin increased to 3.55-fold over baseline in alms1+/+ grown in 40 mm glucose (P = 0.019; two-way ANOVA; Fig. 6E). However, the alms1−/− larvae showed a basal hyperinsulinemia, 3.57-fold that of untreated alms1+/+ (P = 0.022; one-way ANOVA), that was unaffected by high-glucose conditions (NS; two-way ANOVA; Fig. 6E). These data are suggestive of a primary hyperinsulinemic state that is decoupled from systemic glucose concentration.
Discussion
The studies presented here characterized the physiologic and phenotypic responses of a newly generated Alström model created in zebrafish using CRISPR/Cas9. This mutant line exhibits reduced Alms1 RNA and protein levels with known cilia-related body dysmorphogenesis (Fig. 1) (18,30) and recapitulates physiological defects of Alström patients and mouse Alström models (Fig. 2) (4,19). As obesity and related metabolic defects are defining characteristics of this ciliopathy, the propensity for excess weight gain and inappropriate lipid storage implicates Alms1 in zebrafish metabolic regulation (Fig. 3). These conserved effects underscore the utility of the model for interpretation of human phenotypes as zebrafish metabolic organs, includin adipose depot locations and regulating hormones, closely mimic human physiology (14,15,31,32).
Alström patients exhibit hyperinsulinemia, insulin resistance and T2DM (33). alms1 mutant zebrafish failed to respond to glucose challenge and showed overall reduced β-cell numbers (Fig. 4). RNA expression indicates that the remaining β-cells in alms1−/− animals are improperly functioning and likely susceptible to apoptosis. RNA-Seq gene expression analysis in cells isolated from alms1−/− animals show excess secretory activity and cellular transport, especially in insulin-related pathways, alongside an overall decrease in transcription and translation (Fig. 5; Supplementary Material, Fig. S2 and Tables S1 and S2). Of particular interest in these categories include ion channel transport genes—specifically 3 sodium-dependent glucose transporters (slc5a1, slc5a2 and slc5a9), 2 major calcium response elements (camk2a and camk2b), 24 potassium transport genes and another 15 genes involved in the transport of small sugars and amines. Consistent with these data indicating increased secretion, we found hyperinsulinemia in larvae and insulin hypersecretion in unstimulated cultured β-cells, along with impaired glucose-stimulated insulin secretion in both (Fig. 6). We conclude that unstimulated β-cells lacking alms1 are hypersecretory.
Based on these observations, we propose unstimulated β-cell hypersecretion as a primary driver of hyperinsulinemia in alms1−/− animals. Insight from RNA-Seq gene expression changes and observed phenotypic responses lead us to propose two aspects of β-cell function that are reliant upon Alms1: (1) control of insulin secretion in the absence of stimuli and (2) appropriate β-cell glucose sensing. We used a simplified β-cell culture system to examine these aspects individually. The knockdown of Alms1 resulted in excess secreted insulin without glucose stimulation, but failure to respond to high-glucose stimulation at the level of either insulin secretion or altered gene expression (Fig. 5). Epistatic analysis of glucose sensing and insulin response pathways in cultured β-cells will identify genetic pathway defects to be confirmed in alms1−/− animals. The molecular mechanism by which Alms1 impacts secretion is unknown, and such studies would inform the function of Alms1 protein in β-cells, potentially providing insight into its role in other cell types. Importantly, our observations provide additional evidence of a central role for β-cell cilia in sensing extracellular stimuli that directly impact insulin secretion. Ciliary localization of insulin receptors in β-cells is critical to normal function (34). Several adenylyl cyclases, including those encoded by ADCY5 and ADCY8, have also been linked to cilia in other cell types (35,36) and are critical for cAMP generation, a major regulator of glucose-stimulated insulin secretion (37). We propose that Alms1 impacts the ability to sense systemic insulin and glucose levels, resulting in dysregulated β-cell response to stimuli and subsequent secretory defects.
Early-onset insulin hypersecretion may contribute to systemic insulin resistance and eventual β-cell exhaustion. The primary role of β-cell dysfunction in T2DM versus a secondary defect to peripheral insulin resistance is a current topic of debate (38,39). Our data do not preclude some degree of insulin resistance in peripheral tissues at early life stages. Importantly, we observed hyperinsulinemia at larval stages in which islets are functional but prior to the onset of adipogenesis, potentially suggesting β-cell hypersecretion as a primary driver. Given the non-tissue specific nature of this mutant, individual contributions of adipose or muscle tissue to observed diabetes phenotypes cannot be evaluated. Generation of β-cell-specific deletion of alms1 will permit cell-autonomous analyses of Alms1. The utility of zebrafish and its high-fidelity recapitulation of human disease phenotypes support its utility in modeling of genetic diabetes and elucidating the root causes of β-cell failure and T2DM etiology.
Materials and Methods
Zebrafish husbandry and stocks
Experiments were performed using Tg(insa:mCherry) from the Zebrafish International Resource Center (ZIRC). Adult fish were screened and a 7 bp deletion in exon 4 of alms1 was identified, resulting in the Tg(insa:mCherry);alms1−/− line. Embryos were raised at 28.5°C for all experimental analyses. Adult zebrafish were housed and mated naturally, based upon standard protocols (ZIRC). All zebrafish work was conducted in accordance with the University of Maryland Baltimore Institutional Animal Care and Use Committee (IUCAC) guidelines.
Zebrafish analyses and imaging
For histological analyses, adult fish—~6 months in age—were sacrificed in 0.05% 3-amino benzoic acid ethyl ester (tricaine, Sigma-Aldrich, St Louis, MO). Samples were fixed for 24 h in Dietrich's solution (30% ethanol, 10% formalin and 2% glacial acetic acid), embedded in optimal cutting temperature compound (VWR, Radnor, PA) and sectioned into 10 μm thick transverse sections. Hematoxylin (Thermo Fisher Scientific, Waltham, MA) and eosin Y (95%, Sigma-Aldrich) staining was performed to evaluate overall tissue morphology. Aldehyde fuchsin staining with fast green counterstain (Sigma-Aldrich) was performed to examine insulin+ tissues.
Visual responses were examined in adult fish—~9 months old—that were dark adapted overnight. All following procedures were done under dim red light. Fish were anesthetized by submersion in tricaine solution. After anesthesia, zebrafish were transferred to wet Whatman filter paper stack and immobilized by injection of 10 μl of 25 mg/ml gallamine triethiodide (Enzo Life Sciences, Farmingdale, NY) below the gill. Anesthesia was maintained with continuous perfusion of tricaine-containing oxygenated system water. The reference AgCl electrode was placed near the eye, and the recording electrode was placed on the eye. The fish with attached electrodes was placed in a ganzfeld chamber and presented with increasing scotopic stimuli (0.025–9.952 cd × s/m2) at 10–60 s intervals using UTAS BigShot (LKC Technologies, Gaithersburg, MD). A minimum of three waveforms per intensity were averaged.
Genotyped clutch mates at 3 months of age were used in adult feeding studies. The volumes of food were modified from published protocols (23,24). Control diet volume was assessed prior to beginning the experiment as the amount of food consumed by wild-type fish in 5 min. The food volume was calculated after each weight measurement to account for weight gain throughout the study period. Fish were fed once a day with assigned dietary conditions, and the length and weight were quantified at indicated time points.
Embryonic feeding assays were performed as previously published (40). Briefly, equal amounts of BODIPY FL C12 (Life Technologies, Carlsbad, CA) egg yolk were provided to 7 dpf larvae for a period of 4 h at 28.5°C. The embryos were then lysed, and the total fluorescence at 510 nm was quantified.
Fat content in larval livers were examined after yolk absorption was complete, 5–7 dpf, via Oil Red O staining of whole animals fixed in 4% paraformaldehyde. Fatty liver was identified by Oil Red O accumulation in the liver area, found on the right side of yolk-depleted larvae.
β-cell analyses were performed at the indicated time points using previously published protocols (16,25,41). Tg(insa:mCherry);alms1+/− progeny were glucose challenged using medium supplemented with 40 mm glucose using previously published protocols (25,41). After β-cell counts were collected, the alms1 genomic identity of each larva was determined via polymerase chain reaction (PCR) genotyping.
2-NBDG in dimethyl sulfoxide was provided to 5 dpf larvae at 600 or 1000 μm in embryo medium at 28.5°C for 4 h. Glucose uptake was evaluated as the total fluorescence in the embryonic kidney or retina, normalized to untreated fluorescence within matched tissues. Larval insulin levels were evaluated in pooled larval lysates, mechanically dissociated in NP-40 protein buffer, using high-sensitivity insulin ELISA (Mercodia, Uppsala, Sweden).
All image processing and analyses were carried out using Fiji (42).
Cell culture
Beta-TC-6 cells (CRL11506; American Type Culture Collection, Manassas, VA) were cultured in DMEM-H (American Type Culture Collection) supplemented with 15% heat-inactivated fetal bovine serum and 1× penicillin/streptomycin (Sigma-Aldrich). Knockdowns were accomplished using Lipofectamine 3000 (Life Technologies) and either scrambled control or Alms1-targeted siRNA (Life Technologies). Efficacy of siRNA knockdown was evaluated via qRT-PCR.
Glucose stimulation of cultured β-cells was performed on cells plated at equal densities using 2.5 mm and 16.7 mm glucose as baseline and high-glucose concentrations, respectively. Insulin was assessed in media collected at the indicated time points using high-sensitivity insulin ELISA (Mercodia).
RNA-sequencing and analysis of isolated preparation β-cells
A total of 5 dpf larvae from Tg(insa:mCherry) and Tg(insa:mCherry);alms1−/− were dissociated into single cells and sorted via mCherry+ signal using a BD FACS Aria II (BD Biosciences, San Jose, CA) (26). RNA was extracted from isolated cell fraction via extraction kit (Qiagen, Hilden, Germany). RNA quantity and quality were assessed via 260/280 absorption. Samples were provided in duplicate for library preparation and quantitative analysis using next-generation sequencing and an Illumina HiSeq 2x150 PE (GENEWIZ, South Plainfield, NJ). Fragments were aligned to the GRZ10 genome with CLC Genomics Server program v10.0.1. The data discussed in this publication are accessible online (https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE125354) (43). The analysis of the identified list of significantly differentially expressed genes was performed as previously published (26). Pathway interactions were visualized using ConsensusPath DB (44,45), and ontology clusters were evaluated using the Gene Ontology Consortium toolset (46,47).
Western blots
A total of 5 dpf zebrafish larvae were homogenized in NP-40 buffer with protease and phosphatase inhibitors (Sigma-Aldrich), run via SDS-PAGE and blotted onto nitrocellulose. Membranes were incubated overnight with gt-αAlms1 (1:500; Abcam, Cambridge, United Kingdom) or rb-αActin (1:2000; Sigma-Aldrich), then 1 h at RT with species specific HRP-conjugated secondary antibodies (1:15 000; Jackson ImmunoResearch, Westgrove, PA) exposed with enhanced chemiluminescence (ECL) on film (Thermo Scientific). Protein intensity was normalized to actin and quantified via densitometry function in ImageJ.
Quantitative Real-Time PCR
RNA was extracted using RiboZOL RNA Extraction Reagent (VWR) and converted to cDNA via FirstStrand cDNA Synthesis (Thermo Fisher Scientific) per manufacturer protocols. Gene expression was determined on a LightCycler 480 (Roche) using 2X SYBR Green Master Mix (Roche, Basel, Switzerland) and compared by ΔΔCT. Actin and GADPH were used as controls in zebrafish and cultured cells, respectively. Primer sequences are available upon request.
Statistical analysis
All experiments represent a minimum of three replicates, with sample number (n) provided. Prismv6.0 (GraphPad, San Diego, CA) was used to determine appropriate analyses and statistical significance (indicated in figure legends).
Supplementary Material
Acknowledgements
J.E.N. designed and performed experiments, analyzed data and wrote the manuscript. T.L.H. generated the mutant line and performed the dietary experiments. C.C.L performed the Oil Red O experiments. R.M. performed embryonic dietary assays. S.L. assisted in mutant line generation. M.S.M. and R.H. performed cryopreservation and tissue sectioning. S.S. and Z.M.A. performed photoreceptor function tests. C.J.W. contributed the FACS sorting experiment. N.A.Z. contributed experimental design and data analysis and wrote the manuscript. This study has been previously published on the bioRxiv preprint server (http://biorxiv.org/cgi/content/short/439067v1).
The authors wish to thank the University of Maryland School of Medicine Center for Innovative Biomedical Resources, Confocal Microscopy Core in Baltimore, MD for equipment use and assistance with image acquisition. Charles Williams and Charles Hong's laboratory was instrumental in acquiring the glucose uptake images. Jonathan Van Ryzin and Margaret McCarthy's laboratory at the University of Maryland, Baltimore was invaluable in the acquisition of the histology images. NAZ is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Conflict of Interest statement. None declared.
Funding
National Institutes of Health; National Institutes of Diabetes, Digestive and Kidney Disorders (R01DK102001 to N.A.Z., P30DK072488 to N.A.Z. and R.M., T32DK098107 to J.N. and T.L.H., F31DK115179 to T.L.H.); National Institute on Deafness and Other Communication Disorders (R01DC013817 to R.H., R01DC016295 to Z.M.A., F31DC016218 to M.S.M.).
References
- 1. Goetz S.C. and Anderson K.V. (2010) The primary cilium: a signalling centre during vertebrate development. Nat. Rev. Genet., 11, 331–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Adams M., Smith U.M., Logan C.V. and Johnson C.A. (2008) Recent advances in the molecular pathology, cell biology and genetics of ciliopathies. J. Med. Genet., 45, 257–267. [DOI] [PubMed] [Google Scholar]
- 3. Lodh S., O'Hare E.A. and Zaghloul N.A. (2014) Primary cilia in pancreatic development and disease. Birth Defects Res. C Embryo Today, 102, 139–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Marshall J.D., Maffei P., Collin G.B. and Naggert J.K. (2011) Alström syndrome: genetics and clinical overview. Curr. Genomics, 12, 225–235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Alstrom C.H., Hallgren B., Nilsson L.B. and Asander H. (1959) Retinal degeneration combined with obesity, diabetes mellitus and neurogenous deafness: a specific syndrome (not hitherto described) distinct from the Laurence–Moon–Bardet–Biedl syndrome. Acta Psychiatr. Neurol. Scand. Suppl., 129, 1–35. [PubMed] [Google Scholar]
- 6. Hearn T., Spalluto C., Phillips V.J., Renforth G.L., Copin N., Hanley N.A. and Wilson D.I. (2005) Subcellular localization of ALMS1 supports involvement of centrosome and basal body dysfunction in the pathogenesis of obesity, insulin resistance, and type 2 diabetes. Diabetes, 54, 1581–1587. [DOI] [PubMed] [Google Scholar]
- 7. Collin G.B., Marshall J.D., King B.L., Milan G., Maffei P., Jagger D.J. and Naggert J.K. (2012) The Alström syndrome protein, ALMS1, interacts with α-actinin and components of the endosome recycling pathway. PLoS One, 7, e37925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Leitch C.C., Lodh S., Prieto-Echagüe V., Badano J.L. and Zaghloul N.A. (2014) Basal body proteins regulate Notch signaling through endosomal trafficking. J. Cell Sci., 127, 2407–2419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Knorz V.J., Spalluto C., Lessard M., Purvis T.L., Adigun F.F., Collin G.B., Hanley N.A., Wilson D.I. and Hearn T. (2010) Centriolar association of ALMS1 and likely centrosomal functions of the ALMS motif-containing proteins C10orf90 and KIAA1731. Mol. Biol. Cell, 21, 3617–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Mathavan S., Lee S.G.P., Mak A., Miller L.D., Murthy K.R.K., Govindarajan K.R., Tong Y., Wu Y.L., Lam S.H., Yang H. et al. (2005) Transcriptome analysis of zebrafish embryogenesis using microarrays. PLoS Genet., 1, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Irion U., Krauss J. and Nüsslein-Volhard C. (2014) Precise and efficient genome editing in zebrafish using the CRISPR/Cas9 system. Development, 141, 4827–4830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Vesterlund L., Jiao H., Unneberg P., Hovatta O. and Kere J. (2011) The zebrafish transcriptome during early development. BMC Dev. Biol., 11, 30. doi:10.1186/1471-213X-11-30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Howe K., Clark M.D., Torroja C.F., Torrance J., Berthelot C., Muffato M., Collins J.E., Humphray S., McLaren K., Matthews L. et al. (2013) The zebrafish reference genome sequence and its relationship to the human genome. Nature, 496, 498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Santoro M.M. (2014) Zebrafish as a model to explore cell metabolism. Trends Endocrinol. Metab., 25, 546–554. [DOI] [PubMed] [Google Scholar]
- 15. Seth A., Stemple D.L. and Barroso I. (2013) The emerging use of zebrafish to model metabolic disease. Dis. Model. Mech., 6, 1080–1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lodh S., Hostelley T.L., Leitch C.C., O'Hare E.A. and Zaghloul N.A. (2016) Differential effects on β-cell mass by disruption of Bardet–Biedl syndrome or Alström syndrome genes. Hum. Mol. Genet., 25, 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pisharath H., Rhee J.M., Swanson M.A., Leach S.D. and Parsons M.J. (2007) Targeted ablation of beta cells in the embryonic zebrafish pancreas using E. coli nitroreductase. Mech. Dev., 124, 218–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhao C. and Malicki J. (2007) Genetic defects of pronephric cilia in zebrafish. Mech. Dev., 124, 605–616. [DOI] [PubMed] [Google Scholar]
- 19. Collin G.B., Cyr E., Bronson R., Marshall J.D., Gifford E.J., Hicks W., Murray S.A., Zheng Q.Y., Smith R.S., Nishina P.M., and Naggert J.K. (2005) Alms1-disrupted mice recapitulate human Alström syndrome. Hum. Mol. Genet., 14, 2323–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brofferio A., Sachdev V., Hannoush H., Marshall J.D., Naggert J.K., Sidenko S., Noreuil A., Sirajuddin A., Bryant J., Han J.C. et al. (2017) Characteristics of cardiomyopathy in Alström syndrome: prospective single-center data on 38 patients. Mol. Genet. Metab., 121, 336–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Stainier D.Y., Fouquet B., Chen J.N., Warren K.S., Weinstein B.M., Meiler S.E., Mohideen M.A., Neuhauss S.C., Solnica-Krezel L., Schier A.F. et al. (1996) Mutations affecting the formation and function of the cardiovascular system in the zebrafish embryo. Development, 123, 285–292. [DOI] [PubMed] [Google Scholar]
- 22. Russell-Eggitt I., Harris C.M. and Kriss A. (1998) Delayed visual maturation: an update. Dev. Med. Child Neurol., 40, 130–136. [DOI] [PubMed] [Google Scholar]
- 23. Oka T., Nishimura Y., Zang L., Hirano M., Shimada Y., Wang Z., Umemoto N., Kuroyanagi J., Nishimura N. and Tanaka T. (2010) Diet-induced obesity in zebrafish shares common pathophysiological pathways with mammalian obesity. BMC Physiol., 10, 21. doi:10.1186/1472-6793-10-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Landgraf K., Schuster S., Meusel A., Garten A., Riemer T., Schleinitz D., Kiess W. and Körner A. (2017) Short-term overfeeding of zebrafish with normal or high-fat diet as a model for the development of metabolically healthy versus unhealthy obesity. BMC Physiol., 17, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Li M., Maddison L.A., Page-McCaw P. and Chen W. (2014) Overnutrition induces β-cell differentiation through prolonged activation of β-cells in zebrafish larvae. Am. J. Physiol. Endocrinol. Metab., 306, E799–E807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nesmith J.E., Hostelley T.L. and Zaghloul N.A. (2017) Sample preparation and analysis of RNASeq-based gene expression data from zebrafish. J. Vis. Exp., 56187. doi:10.3791/56187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Hostelley T.L., Lodh S. and Zaghloul N.A. (2016) Whole organism transcriptome analysis of zebrafish models of Bardet–Biedl syndrome and Alström syndrome provides mechanistic insight into shared and divergent phenotypes. BMC Genomics, 17, 318. doi: 10.1186/s12864-016-2679-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thorens B., Sarkar H.K., Kaback H.R. and Lodish H.F. (1988) Cloning and functional expression in bacteria of a novel glucose transporter present in liver, intestine, kidney, and beta-pancreatic islet cells. Cell, 55, 281–290. [DOI] [PubMed] [Google Scholar]
- 29. Lee J., Jung D.W., Kim W.H., Um J.I., Yim S.H., Oh W.K. and Williams D.R. (2013) Development of a highly visual, simple, and rapid test for the discovery of novel insulin mimetics in living vertebrates. ACS Chem. Biol., 8, 1803–1814. [DOI] [PubMed] [Google Scholar]
- 30. Wilkinson C.J., Carl M. and Harris W.A. (2009) Cep70 and Cep131 contribute to ciliogenesis in zebrafish embryos. BMC Cell Biol., 10, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Minchin J.E.N. and Rawls J.F. (2017) A classification system for zebrafish adipose tissues. Dis. Model. Mech., 10, 797–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Flynn E.J., Trent C.M. and Rawls J.F. (2009) Ontogeny and nutritional control of adipogenesis in zebrafish (Danio rerio). J. Lipid Res., 50, 1641–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Favaretto F., Milan G., Collin G.B., Marshall J.D., Stasi F., Maffei P., Vettor R. and Naggert J.K. (2014) GLUT4 defects in adipose tissue are early signs of metabolic alterations in alms1GT/GT, a mouse model for obesity and insulin resistance. PLoS One, 9, e109540. doi: 10.1371/journal.pone.0109540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gerdes J.M., Christou-Savina S., Xiong Y., Moede T., Moruzzi N., Karlsson-Edlund P., Leibiger B., Leibiger I.B., Östenson C.G., Beales P.L. and Berggren P.O. (2014) Ciliary dysfunction impairs beta-cell insulin secretion and promotes development of type 2 diabetes in rodents. Nat. Commun., 5, 5308 10.1038/ncomms6308. [DOI] [PubMed] [Google Scholar]
- 35. Vuolo L., Herrera A., Torroba B., Menendez A. and Pons S. (2015) Ciliary adenylyl cyclases control the hedgehog pathway. J. Cell Sci., 128, 2928–2937. [DOI] [PubMed] [Google Scholar]
- 36. Roger B., Papin J., Vacher P., Raoux M., Mulot A., Dubois M., Kerr-Conte J., Voy B.H., Pattou F., Charpentier G. et al. (2011) Adenylyl cyclase 8 is central to glucagon-like peptide 1 signalling and effects of chronically elevated glucose in rat and human pancreatic beta cells. Diabetologia, 54, 390–402. [DOI] [PubMed] [Google Scholar]
- 37. Hodson D.J., Mitchell R.K., Marselli L., Pullen T.J., Brias S.G., Semplici F., Everett K.L., Cooper D.M.F., Bugliani M., Marchetti P. et al . (2014) ADCY5 couples glucose to insulin secretion in human islets. Diabetes, 63, 3009–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Goel P. (2015) Insulin resistance or hypersecretion? The βIG picture revisited. J. Theor. Biol., 384, 131–139. [DOI] [PubMed] [Google Scholar]
- 39. Corkey B.E. (2012) Banting lecture 2011: hyperinsulinemia: cause or consequence? Diabetes, 61, 4–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Otis J.P. and Farber S.A. (2016) High-fat feeding paradigm for larval zebrafish: feeding, live imaging, and quantification of food intake. J. Vis. Exp., 116, 54735. doi:10.3791/54735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. O'Hare E.A., Yerges-Armstrong L.M., Perry J.A., Shuldiner A.R. and Zaghloul N.A. (2016) Assignment of functional relevance to genes at type 2 diabetes-associated loci through investigation of β-cell mass deficits. Mol. Endocrinol., 30, 429–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B. et al. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods, 9, 676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Edgar R., Domrachev M. and Lash A.E. (2002) Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kamburov A., Pentchev K., Galicka H., Wierling C., Lehrach H. and Herwig R. (2011) ConsensusPathDB: toward a more complete picture of cell biology. Nucleic Acids Res., 39, 712–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Kamburov A., Wierling C., Lehrach H. and Herwig R. (2009) ConsensusPathDB–a database for integrating human functional interaction networks. Nucleic Acids Res., 37, 623–628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Ashburner M., Ball C.A., Blake J.A., Botstein D., Butler H., Cherry J.M., Davis A.P., Dolinski K., Dwight S.S., Eppig J.T. et al. (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat. Genet., 25, 25–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Carbon S., Dietze H., Lewis S.E., Mungall C.J., Munoz-Torres M.C., Basu S., Chisholm R.L., Dodson R.J., Fey P., Thomas P.D. et al. (2017) Expansion of the gene ontology knowledgebase and resources: the gene ontology consortium. Nucleic Acids Res., 45, D331–D338. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.