

Abstract
Duchenne muscular dystrophy (DMD) is a lethal, muscle degenerative disease causing premature death of affected children. DMD is characterized by mutations in the dystrophin gene that result in a loss of the dystrophin protein. Loss of dystrophin causes an associated reduction in proteins of the dystrophin glycoprotein complex, leading to contraction-induced sarcolemmal weakening, muscle tearing, fibrotic infiltration and rounds of degeneration and failed regeneration affecting satellite cell populations. The α7β1 integrin has been implicated in increasing myogenic capacity of satellite cells, therefore restoring muscle viability, increasing muscle force and preserving muscle function in dystrophic mouse models. In this study, we show that a Food and Drug Administration (FDA)-approved small molecule, Sunitinib, is a potent α7 integrin enhancer capable of promoting myogenic regeneration by stimulating satellite cell activation and increasing myofiber fusion. Sunitinib exerts its regenerative effects via transient inhibition of SHP-2 and subsequent activation of the STAT3 pathway. Treatment of mdx mice with Sunitinib demonstrated decreased membrane leakiness and damage owing to myofiber regeneration and enhanced support at the extracellular matrix. The decreased myofiber damage translated into a significant increase in muscle force production. This study identifies an already FDA-approved compound, Sunitinib, as a possible DMD therapeutic with the potential to treat other muscular dystrophies in which there is defective muscle repair.
Introduction
Duchenne muscular dystrophy (DMD) is one of the most common X-linked neuromuscular diseases, with an incidence of 1 in 5000, leading to premature death of affected children (1). DMD is characterized by the loss of dystrophin, a 427 kDa protein found at the sarcolemma of skeletal, cardiac and vascular smooth muscle (2,3). Structurally, dystrophin is essential to anchor intracellular actin filaments and sarcolemmal proteins to promote myofiber stability (4,5). The loss of dystrophin in DMD leads to the absence of dystrophin-associated proteins that results in altered muscle cell signaling, contraction-induced muscle degeneration and replacement with fibrotic and fatty tissue (6–8). Clinical features of DMD include gross motor delays, loss of ambulation leading to wheelchair confinement, respiratory insufficiency requiring ventilator assistance and dilated cardiomyopathy beginning during the second decade of life and premature death (1,9).
Currently, there are limited treatment options available for DMD patients. Therapeutic options include corticosteroids (Prednisone) and glucocorticoids (Deflazacort), both used to decrease inflammation and suppress the immune response (10,11). Recently, the exon skipping drug Eteplirsen (Exondys 51) was given Food and Drug Administration (FDA) approval (12). Although showing promising results, eteplirsen is only applicable to 14% of the DMD population with specific exon 51 mutations. Therefore, it is still essential to develop therapies targeting pathways viable to all DMD patients, regardless of mutation.
One of the hallmarks of DMD pathology is cycles of muscle degeneration and failed muscle regeneration that occur in the absence of dystrophin (6). This faulty regeneration has been attributed to a number of factors, one of them being decreased satellite cell (SC) capacity. SCs are the essential precursors to myogenesis and in DMD; while increased in number, they have been shown to be impaired due to faulty asymmetric division and interrupted SC niches, leading to improper muscle regeneration (13–16). The signal transducer and activator of transcription 3 (STAT3) activation via the interleukin-6 (IL-6) cytokine is important for SC proliferation and self-renewal in response to resistance exercise and muscle injury (17–22). Activated STAT3 can directly affect the expression of the myogenic regulatory marker MyoD1 and promote myoblast differentiation (23–26). Additionally, STAT3 inhibition promotes enhanced symmetric SC proliferation and increased SC engraftment into injured muscle (27,28), suggesting that transient STAT3 activation is important for both proliferation of SCs and differentiation into mature myofibers.
Several studies have identified the α7β1 integrin as a positive modifier of DMD disease pathology in different mouse models utilizing transgenic and AAV delivery techniques (29–39). More recently, our laboratory demonstrated that treatment with an α7β1 integrin enhancing small molecule, SU9516, increased muscle force generation and decreased disease pathology in mdx mice (40).
Sunitinib is structurally related to SU9516 and is currently used as an FDA-approved, multi receptor tyrosine kinase (RTK) inhibitor for the treatment of renal cell carcinoma (RCC; 41,42). Sunitinib has also been implicated in modulating the STAT3 pathway in cancer (43). Treatment with Sunitinib promotes SC activation and myogenic regeneration, leading to significantly improved muscle disease pathology and functional skeletal muscle force production. Together, our results provide evidence that Sunitinib can be repurposed into the first small molecule therapy targeting muscle regeneration for the treatment of DMD.
Results
Sunitinib treatment increases α7β1 levels via MyoD1 and Myog transcription factors
A previous study demonstrated the beneficial effects of treatment with the small molecule SU9516 on DMD pathology via enhanced α7B integrin expression (40). Unfortunately, during study progression, SU9516 was determined to have toxicity levels that would prevent its use in patients. Sunitinib is an FDA-approved, structurally related compound of SU9516 (Fig. 1A) expected to have similar effects on DMD disease progression. Optimal Sunitinib treatment dose was determined by performing a dosing curve consisting of 5 day treatment in mdx mice followed by assessment of α7B integrin levels in diaphragm muscle (Fig. 1B). A maximum ~1.5-fold increase in α7B integrin expression was observed at 1 mg/kg Sunitinib (Fig. 1B) when compared to vehicle-treated muscle. This effective concentration was significantly lower than its structurally related compound SU9516 (effective at 5 mg/kg; 40). To determine the effect of Sunitinib on disease pathology, the treatment schedule developed consisted of 3 days on 1 mg/kg Sunitinib and 4 days off, weekly treatments, beginning at 4 weeks of age and ending at 12 weeks in the mdx mouse model of DMD (Fig. 1C). This treatment schedule was developed based on the robust and rapid increase in α7B integrin protein expression (Fig. 1B) and on potential downstream targets. Post 8 week treatment assessment shows the 1 mg/kg Sunitinib treatment produced a significant increase in α7B integrin protein expression in mdx diaphragm muscle compared to vehicle-treated muscle (Fig. 1D and E). Additionally, transcript levels of α7B integrin and two α7B integrin regulating transcription factors, MyoD1 and Myog (44,45), were also significantly up-regulated in the tibialis anterior (TA) muscle of Sunitinib-treated mdx mice (Fig. 1G and H). The TA muscle was used for transcript analysis as the diaphragm muscle was required for protein expression and histological analysis as well as muscle ex vivo contractility studies.
Figure 1.
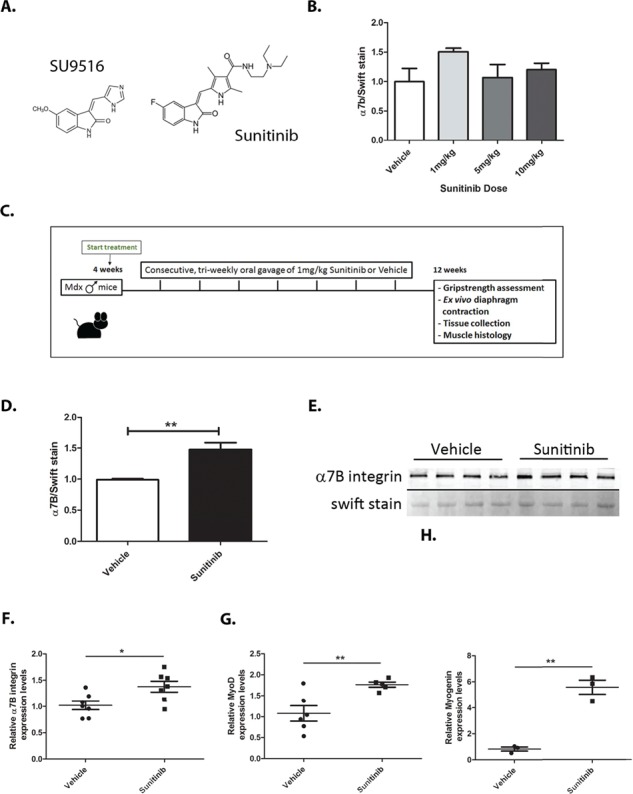
Sunitinib treatment increases α1;7B integrin via activation of Myod1 and Myog transcription factors. (A) Structural similarities between the known integrin α7 enhancing molecule SU9516 and Sunitinib. (B) Western blot dose response curve performed on N = 4 mdx mice with daily 1 mg/kg–10 mg/kg Sunitinib treatment for a total of 5 days showing optimal α7B integrin enhancing dose at 1 mg/kg. (C) Schematic representation of the 8 week Sunitinib treatment plan and final muscle assessments. (D and E) Western blot analysis and quantification of diaphragm α7B integrin levels showing a 1.5-fold increase in α7B expression with Sunitinib treatment. (F) RT-qPCR analysis of α7B integrin transcript levels showing ~1.4-fold increase with Sunitinib treatment. (G and H) RT-qPCR analysis of α7B upstream transcription factors and differentiation markers MyoD1 and Myog showing a ~1.8-fold and ~5.8-fold transcript increase, respectively, with Sunitinib treatment. Statistical significance of mean ± SEM; *P < 0.05, **P < 0.01.
Sunitinib improves mdx diaphragm muscle function
Loss of dystrophin in DMD renders muscles fragile and unable to exert normal amounts of force. Regeneration of muscle fibers is expected to yield an increase in muscle force production. To test whether Sunitinib treatment improved muscle contractility, ex vivo isolated muscle isometric contraction studies were performed on mdx diaphragm muscle. Isolated, isometric twitch (1 Hz) force production in vehicle-treated mdx diaphragm muscle (2.722 ± 0.75 mN/mg) is considerably lower than that of wild-type muscle (6.014 ± 0.93 mN/mg; Fig. 2A). Sunitinib treatment produced a significant increase in twitch force (3.689 ± 0.29 mN/mg) compared to vehicle (Fig. 2A). Specific tetanic force, measured at peak 150 Hz stimulation, was also increased with Sunitinib (13.02 ± 1.38 mN/mg) compared to vehicle-treated diaphragm (10.47 ± 2.31 mN/mg; Fig. 2B). Sunitinib enhanced specific force production at increasing stimulations ranging from 50 to 150 Hz when compared to vehicle-treated muscle (Fig. 2C). To assess overall muscle strength, forelimb grip strength was performed post 8 week Sunitinib or vehicle treatment. Results show that mdx forelimb muscle force significantly decreased from wild-type in the last two grip strength trials, whereas none of the trials shows significant difference in force between wild-type and Sunitinib-treated mice (Fig. 2D). Sunitinib-treated mdx forelimb strength was significantly increased compared to mdx forelimb strength in the last three grip strength trials (Fig. 2D). These results suggest Sunitinib treatment shows significant improvements in muscle strength and function shown by isolated muscle force contraction studies and ameliorates forelimb muscle fatigue observed in mdx mice.
Figure 2.
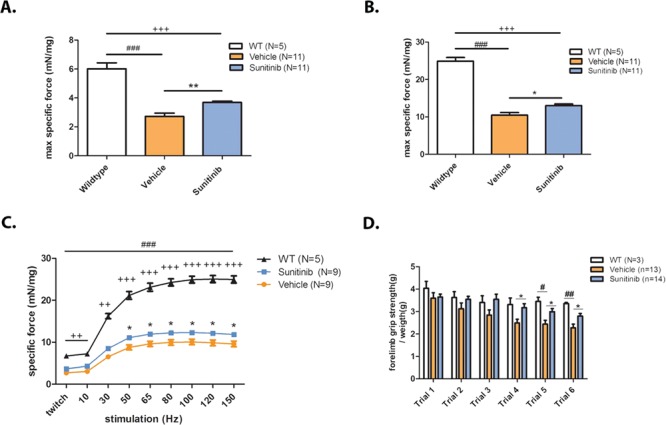
Sunitinib improves specific muscle contractile function and overall muscle strength. Muscle isometric contractility and strength assessments performed on 12-week-old mdx mice. (A) Single 1 Hz pulses generated isometric twitch force outputs showing the severe decline in diaphragm muscle twitch between WT (N = 5) and mdx vehicle-treated muscle (N = 11); Sunitinib-treated muscle (N = 11) twitch is significantly increased compared to vehicle treated. (B) Isometric tetanic stimuli performed at 100 Hz on diaphragm muscle showing a significant decline in tetanic force output between WT (N = 5) and vehicle-treated muscle (N = 11), Sunitinib treatment (N = 11) increased isometric tetanic force compared to vehicle treatment. (C) Isometric force performed at increasing stimulation frequencies; Sunitinib treatment increased force production in the 50–150 Hz frequency stimulations when compared to vehicle-treated muscle. (D) Muscle exhaustion depicted as decreased forelimb grip strength is apparent in vehicle-treated mdx mice compared to WT mice at trials 5 and 6. No significant change in forelimb force is observed between WT and Sunitinib-treated mdx mice during the six trials; Sunitinib-treated mdx mice are stronger than the vehicle treated in trials 4–6 suggesting less muscle exhaustion. Data assessed for significance using one-way ANOVA and statistical significance of mean ± SEM; WT versus mdx vehicle treatment #P < 0.05, ##P < 0.01, ###P < 0.001; WT versus mdx-Sunitinib treatment ++P < 0.01, +++P < 0.001; mdx vehicle treatment versus mdx-Sunitinib treatment *P < 0.05, **P < 0.01.
Sunitinib promotes myogenesis in the mdx mouse
We next determined whether the increase in α7B integrin observed with Sunitinib treatment translated into improved DMD disease pathology. Diaphragm muscle sections from 8 week, Sunitinib-treated and vehicle-treated mdx mice were analyzed for expression of a developmental form of myosin heavy chain (MyHC). Embryonic myosin heavy chain (eMyHC) is transiently expressed in developing fetal muscle fibers and after muscle injury, in newly regenerated, adult fibers (46). Our results show that vehicle-treated mdx diaphragm contained ~4% eMyHC expressing fibers (Fig. 3A and B) due to the hallmark bouts of degeneration/regeneration in DMD muscle. Expression of eMyHC was increased to 12% in Sunitinib-treated diaphragm muscle, indicating that Sunitinib treatment is capable of promoting regeneration of muscle fibers in vivo. Diaphragm muscle was also assessed for centrally located nuclei (CLN) as further evidence of muscle regeneration. Sunitinib treatment significantly increased CLNs by ~5% (Fig. 3C and D). Minimum Feret’s diameter showed a shift in fiber size. Diaphragms from vehicle-treated mice show a distribution consisting of a higher percentage of smaller fibers in the 20–25 μm range compared to Sunitinib fiber diameters that show a higher percentage of larger fibers in the 30–50 μm range (Fig. 3E). Newly incorporated fibers would expectedly decrease the amount of total muscle damage. To assess the amount of leakiness in a whole mdx muscle, an Evans blue dye (EBD) assay was performed. Results from the EBD assay show that Sunitinib treatment decreased the amount of muscle damage as represented by decreased numbers of positive EBD fibers in a diaphragm muscle section and quantified in whole gastrocnemius (GA) muscle of treated mdx mice (Fig. 3F). Muscle damage allows for robust infiltration of fibrotic and fatty tissues that become indicators of DMD disease progression (7,47). The amount of fibrotic infiltration in whole muscle, quantified via hydroxyproline colorimetric assay, was also shown to be decreased in GA muscle with Sunitinib treatment (Fig. 3G).
Figure 3.
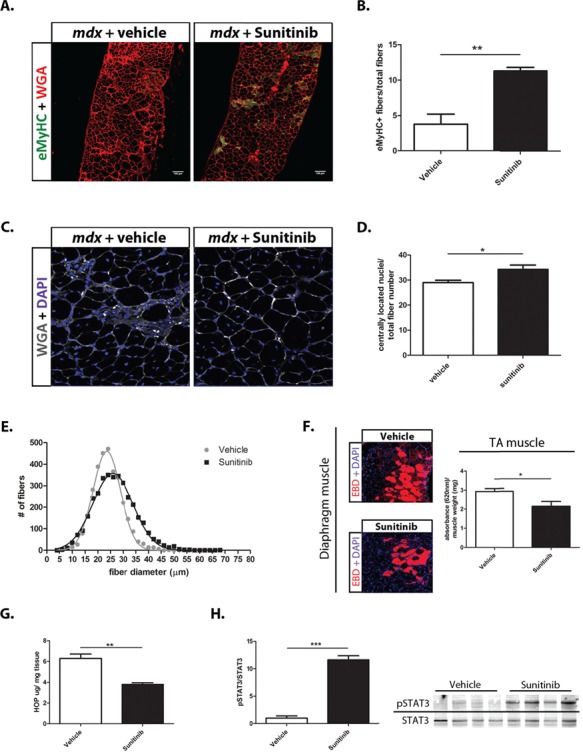
Sunitinib promotes muscle repair and improves markers of DMD disease progression. (A) Representative section of immunohistochemistry performed on 10 μm diaphragm muscle sections showing increased number of eMyHC (green) positive fibers in Sunitinib-treated muscle. WGA (red); scale bar, 100 μm. (B) Quantification of eMyHC positive (+) muscle fibers normalized to total fiber numbers of a whole mdx diaphragm muscle section (N = 4); Sunitinib treatment causes ~8% increase in eMyHC+ fibers compared to vehicle treatment. (C and D) CLN counts performed on WGA (grey) and DAPI (blue) immunolabeled 10 μm diaphragm muscle sections (N = 6); Sunitinib treatment increased CLNs by ~5% compared to vehicle treated. (E) Myofiber size distribution of Sunitinib- and vehicle-treated whole 10 μm sections diaphragm sections, measured using minimum Feret’s diameter (N = 4); Sunitinib-treated muscle shows a shift toward higher percentage of large fiber sizes compared to vehicle treated. (F) Representative sections of EBD (red) infiltration in Sunitinib-treated diaphragm muscle. EBD quantification performed on total GA muscle shows decreased EBD infiltration compared to vehicle treated (N = 4). (G) Hydroxyproline assay was performed to quantify the amount of fibrotic infiltration in whole GA muscle (N = 4); Sunitinib treatment decreases fibrosis as shown by a 2-fold decrease in collagen content in the mdx GA muscle, compared to vehicle treated. (H) Western blot analysis showing the STAT3 pathway is activated in response to Sunitinib treatment 1 h post final dose (6 week total dosing) in mdx diaphragm muscle. Data assessed for significance using unpaired t-test and statistical significance of mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
To elucidate the pathway by which Sunitinib promoted muscle regeneration, we next performed protein analysis on diaphragm tissue 1 h post-treatment on pathways postulated to be regulated by Sunitinib. Our results indicated that Sunitinib promoted the activation of the STAT3 pathway, in vivo, in the diaphragm muscle of mdx-treated mice (Fig. 3H). Taken together these results show that Sunitinib treatment promotes muscle regeneration in vivo through STAT3 activation, thus decreasing muscle fiber damage and replacement of muscle with fibrotic/fatty tissue.
Sunitinib promotes SC proliferation and myofiber fusion
To determine the mechanism by which Sunitinib promotes muscle fiber regeneration, we next analyzed SC populations in dystrophic muscle. Pax7 is a transcription factor expressed in quiescent and activated SCs (48,49). Pax7 transcript was significantly increased in Sunitinib-treated TA muscles compared to vehicle-treated animals (Fig. 4B). Accordingly, Pax7 labeling in diaphragm muscle sections showed Sunitinib-treated mice exhibit increased numbers of SCs compared to muscle treated with vehicle (Fig. 4A and C). Due to the lack of availability of diaphragm muscle, as it has been used for several other experiments, transcript levels were not assessed in this muscle. Together, increased Pax7 transcript in the TA and protein in the diaphragm muscle suggests Sunitinib treatment is promoting the proliferation of SCs required for myogenic regeneration.
Figure 4.
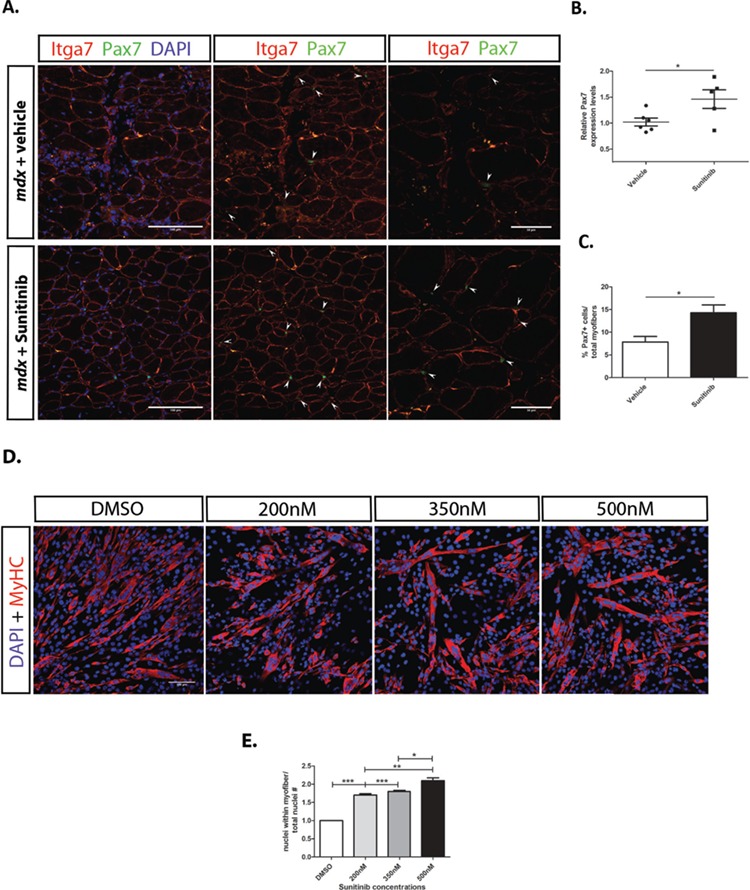
Sunitinib treatment promotes SC proliferation and myoblast fusion. (A) Immunohistochemistry performed on diaphragm muscle sections of mdx mice treated with vehicle or Sunitinib. Higher numbers of SCs are present in Sunitinib-treated diaphragm; SCs are identified (arrows) by their location around myofibers [α7B integrin (red)], Pax7+ cells (green) and co-localization with nuclear DAPI (blue) stain. Middle panels without DAPI staining for easier identification of SCs. Panels to the right were taken at 60× magnification, also to better identify SCs. (B) Increased Pax7 transcript levels observed in Sunitinib-treated TA whole muscle. (C) Quantification of SC numbers, determined by counting Pax7+ cells per total fiber counts (10 panels per tissue, magnification 40×, N = 3); Sunitinib treatment significantly increased SC numbers in diaphragm and TA muscles. (D) C2C12 myoblasts under differentiating conditions 48 h post DMSO or Sunitinib treatment at varying concentrations immunolabeled for MHC (red) and nuclear DAPI (blue). (E) Fusion index determined by counting the number of nuclei within MHC+ fibers and normalizing to total nuclei counts per 10× panel (N = 3); Increased myofiber fusion observed at all three Sunitinib concentrations with 500 nm showing the highest fusion index compared to DMSO-vehicle-treated cells. Data assessed for significance using one-way ANOVA and statistical significance of mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001
Next we determined whether Sunitinib treatment promoted myofiber fusion. To measure fusion, C2C12 myoblasts were treated under differentiating conditions with increasing doses of Sunitinib. Treatment with Sunitinib results in larger fibers containing increased numbers of nuclei (Fig. 4D). Sunitinib doses as low as 200 nm are capable of promoting ~1.7-fold increase in the myofiber fusion with maximum increase observed at 500 nm (Fig. 4E). Together these results show that Sunitinib treatment promotes terminal differentiation of myotubes without depleting the SC pool.
Sunitinib inhibits SHP-2–ERK1/2 activation and promotes STAT3 phosphorylation
To elucidate the mechanism by which Sunitinib acts to promote myogenic differentiation, the C2C12 cell line was used. A previous study showed Sunitinib can modulate the STAT3 pathway in cancer (43). Additionally, studies have shown a direct, inhibitory interaction between activated ERK1/2 and STAT3 (50,51). ERK1/2 is a downstream target of activation of SHP-2 by Jak2. C2C12 myoblasts were treated with Sunitinib and cells harvested at varying time points after treatment. Our results show treatment with 500 nm Sunitinib caused inhibition of SHP-2 activation within 5 min post-treatment, continuing for up to 1 h (Fig. 5A and B). ERK1/2 activation is decreased starting at 15 min post Sunitinib treatment and lasting up to 2 h (Fig. 5C and D). STAT3 activation is observed starting at 1.5 h post Sunitinib treatment and lasting at least up to 3 h (Fig. 5E). Additionally, α7B integrin expression was increased in myoblasts treated with Sunitinib after 48 h (Fig. 5F). Next we assessed whether an optimal dose of 5 μm SU9516 led to inhibition of ERK1/2 phosphorylation and activation of STAT3 in C2C12 myoblasts. Contrary to our expectations, we did not observe an alteration in either pSTAT3 (Supplementary Material, Fig. S1A and B) or pERK1/2 (Supplementary Material, Fig. S1C and D).
Figure 5.
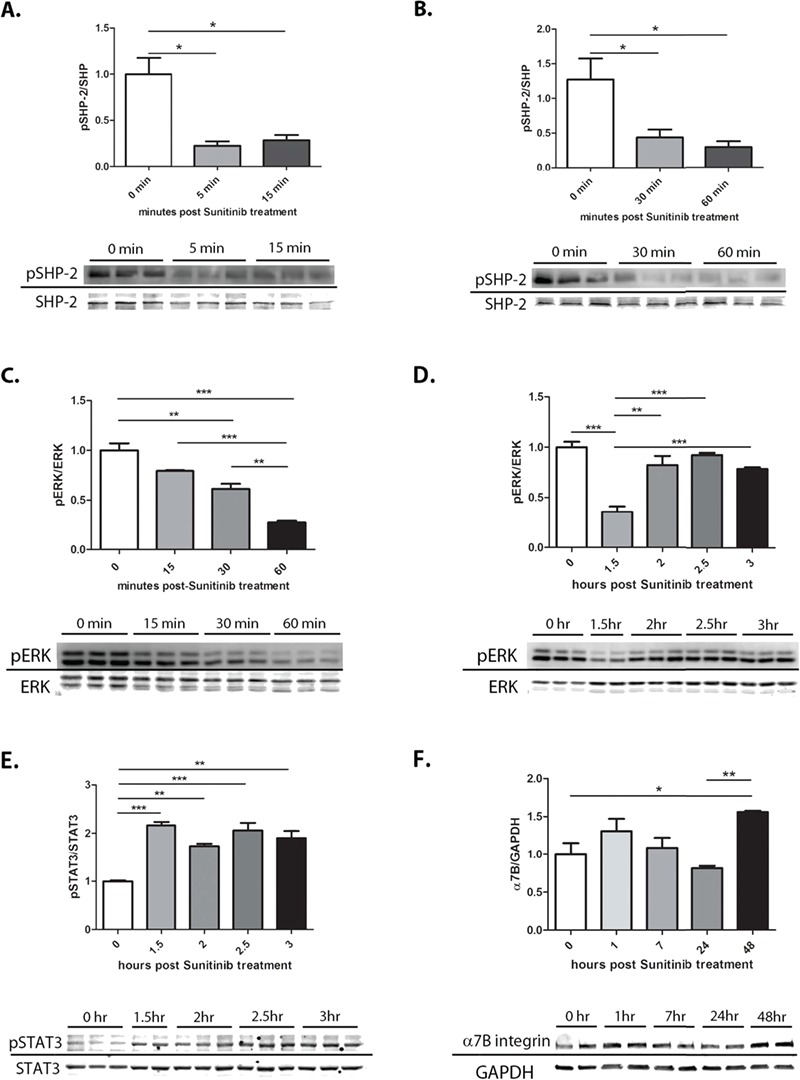
Sunitinib inhibits SHP-2–ERK1/2 and activates the STAT3 pathway in C2C12 cell line. C2C12 myoblasts treated with 500 nm Sunitinib in triplicate (N = 3) and taken at different time points to assess SHP-2–ERK1/2 inhibition and STAT3 activation. (A) Quantification of SHP-2 phosphorylation shows significant inhibition at 5 min lasting up to 15 min. (B) Quantification of SHP-2 phosphorylation shows significant inhibition continuing from 30 min to 1 h. (C) Quantification of ERK1/2 phosphorylation shows significant inhibition starting at 15 min post-treatment and lasting up to 1 h. (D) Quantification of ERK1/2 phosphorylation showing continued inhibition at 1.5 h with recuperating phosphorylation after 2 h of treatment. (E) Quantification of STAT3 phosphorylation showing activation after 1.5 h of treatment. (F) Quantification of α7B integrin shows a 1.5-fold increase in expression 48 h post-treatment. Data assessed for significance using one-way ANOVA and statistical significance of mean ± SEM; *P < 0.05, **P < 0.01, ***P < 0.001.
Together, these results further support the idea that Sunitinib exerts its effects predominately on early myogenic cells such as SCs and myoblasts by inhibition of SHP-2–ERK1/2 signaling followed by activation of STAT3.
Discussion
DMD is a progressive neuromuscular disease characterized by rounds of muscle degeneration and regeneration with eventual failure of muscle repair. In the present study, we provide evidence of halted DMD disease progression in mdx mice that received long term treatment with Sunitinib. Marketed under the name SUTENT®, Sunitinib was originally developed for the treatment of RCC and gastrointestinal stromal tumors. Sunitinib is a multi-RTK inhibitor with binding affinities for c-KIT, FLT3, RET, VEGFR and PDGFR (42). The optimal anti-tumor dosing schedule for Sunitinib during human clinical trials was determined to be 50 mg/day. These levels are required to reach circulating plasma levels of 50–100 ng/ml for highest RTK inhibition with manageable side effects; these levels are equivalent to 40 mg/kg/day in mice (41,52,53). Studies have been conducted on pediatric patients with refractory solid tumors demonstrating that a 15 mg/day is a more suitable dose for children (54,55). For the purposes of our study, initial dosing studies with α7B integrin protein expression as the outcome measure determined the optimal dose (1 mg/kg) to be 40-fold lower than the anti-tumor dose. This dose is lower than both the adult and pediatric recommended doses for cancer; severe toxicity, while not to be ruled out, is unlikely. These results also suggest the myogenic effects observed are possibly due to targeting of a novel pathway not previously described for Sunitinib.
DMD disease pathology presents with lower SC capacity due to interrupted SC niches, resulting in impaired muscle regeneration (14,16,56,57). Therapeutic interventions aimed at enhancing the proliferative capacity of endogenous SCs could prove significantly beneficial to DMD pathology. Numerous studies have indicated that STAT3 activation via the IL-6 cytokine is important for SC proliferation and self-renewal in response to resistance exercise and muscle injury (17–22,58). Activated STAT3 can directly affect the expression of the myogenic regulatory marker MyoD1 and promote myoblast differentiation (23–26). Additionally, STAT3 inhibition was shown to promote enhanced symmetric SC proliferation and increased SC engraftment into injured muscle (27,28). These studies indicate that transient STAT3 activation might allow controlled cycles of SC proliferation and differentiation.
Small molecule therapeutics are ideal for the transient activation of a specific pathway. In the current study, we provide evidence of STAT3 activation concurrent with Sunitinib treatment in a myogenic cell line and in skeletal muscle of treated mdx mice. Phosphorylation of STAT3 allows for its dimerization and translocation into the nucleus where phospho-STAT3 dimers directly promote MyoD1 expression (Fig. 6B; 24,25). Analysis of transcript levels in the TA muscle of treated mdx mice indicated significant up-regulation of MyoD1 and Myog transcripts, suggesting significant myogenic remodeling is occurring during Sunitinib treatment. The direct interaction of MyoD1 and myogenin with the α7 integrin promotor has been determined to positively regulate α7 integrin transcription (44,45). Additionally, α7 integrin has emerged as an important, positive modifier of DMD disease progression (29–32,34,38,39,59), potentially due to promoting enhanced myogenic capacity and skeletal muscle hypertrophy (33,60). This suggests that activation of the STAT3 pathway can directly lead to up-regulation of α7 integrin levels and myogenic hypertrophy.
Figure 6.
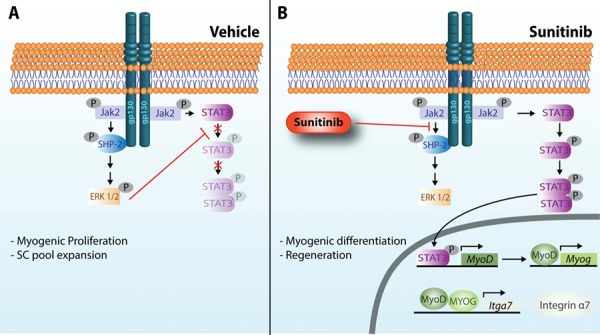
Proposed mechanism of SHP2–ERK1/2 inhibition and STAT3 activation by Sunitinib. In response to muscle injury, gp130 mediated by Jak2 can activate both ERK1/2 and STAT3. (A) Upon gp130 receptor dimerization in response to growth factor binding, constitutively bound Jak2 is activated and trans-phosphorylates gp130 at several tyrosine residues (Y). P-Y759 recruits SHP-2 that is phosphorylated by Jak2 and signals via Grb2-Sos-Ras–Raf and MEK to activate ERK1/2. Once activated, ERK1/2 has been shown to directly phosphorylate STAT3 at a serine residue, thus preventing phosphorylation at tyrosine residues and nuclear translocation (50). ERK1/2 becomes the predominant signaling pathway promoting the proliferation of myogenic cells. (B) Sunitinib inhibits the activation of ERK1/2, potentially by (1) preventing the association of SHP-2 with gp130 and (2) preventing the phosphorylation of SHP-2 by Jak2. Non-phosphorylated ERK1/2 no longer phosphorylates STAT3 at its serine residue, thus allowing tyrosine phosphorylation and activation of STAT3 by Jak2. Dimerized STAT3 translocates into the nucleus where it promotes the transcription of the transcription factor MyoD1 that can subsequentially promote the transcription of myogenin (Myog). Both MyoD1 and MYOG can then promote the transcription of integrin α7 (Itga7). Integrin α7 promotes differentiation and fusion of myofibers, enhancing muscle regeneration.
To elucidate this pathway in vivo, α7 integrin transcript and protein expression levels in the mdx mouse skeletal muscle were analyzed and determined to be significantly up-regulated. Assessment of protein expression for MyoD and myogenin was attempted via skeletal muscle tissue western blotting and immunolabeling. However, antibody recognition of the epitopes was unsuccessful. To associate the increase in α7 integrin with enhanced hypertrophy, markers of muscle regeneration were analyzed. Diaphragm muscle fibers of Sunitinib-treated mice expressed higher levels of eMyHC, a type of myosin found both developmentally and during adult myogenesis (46). Fiber diameters were increased in the diaphragm muscle of mdx mice treated with Sunitinib, suggesting muscle hypertrophy. We hypothesized that the observed increase in myogenic regeneration occupied the interstitial space left behind by degenerating fibers before the precocious deposition of fibrotic and fatty tissue. This was supported by a decrease in fibrotic content in Sunitinib-treated mdx GA and reduced leaky muscle fibers as evidenced by decreased EBD infiltration. Enhanced muscle fiber stability supports the increase in force production of the isolated diaphragm muscle and increased forelimb strength of treated mdx mice.
The activation of STAT3 in response to muscle injury and resistance exercise occurs due to the production of the IL-6 cytokine (17,19,20). The canonical pathway for IL-6 involves binding to gp130 receptors, recruitment and auto-phosphorylation of JAK2, in turn phosphorylating downstream targets SHP-2–ERK1/2 and STAT3 (Fig. 6; 61). Due to Sunitinib’s inherent nature as a kinase inhibitor, a direct interaction with STAT3 is unlikely. Activated ERK1/2 was shown to directly interact with STAT3 by promoting phosphorylation at a STAT3 serine residue, thus preventing the tyrosine phosphorylation that activates and dimerizes STAT3 (50,51). We observed STAT3 activation after only 1.5 h of Sunitinib treatment. This activation of STAT3 can be explained by the rapid and prolonged SHP-2 and ERK1/2 inhibition that prevents phospho-ERK1/2’s phosphorylation of STAT3 at its inhibitory serine residue, thus allowing phosphorylation on tyrosine residues required for STAT3 activation (Fig. 6B). This demonstrates the direct effect of Sunitinib on skeletal muscle cells via STAT3 activation and increased α7β1 integrin.
Studies were performed to determine if Sunitinib and SU9516 share the same mechanism of action. C2C12 myoblasts treated with 5 μm SU9516 did not show activation of the STAT3 pathway (Supplementary Material, Fig. S1A and B) and no inhibition of the ERK1/2 pathway was observed (Supplementary Material, Fig. S1C and D). These results suggest that while structurally similar and both capable of α7 integrin enhancement, Sunitinib and SU9516 exert their effects via different mechanisms of action. This experiment also provides strong evidence that Sunitinib itself promotes the inhibition of SHP-2–ERK1/2 and activation of STAT3, not its breakdown metabolite SU12662 (42).
The effect of Sunitinib treatment on SC populations was examined. Our results indicate higher numbers of SCs are present in the diaphragm of Sunitinib-treated mdx mice as well as increased Pax7 transcript levels in the TA muscle. This can be explained by the transient nature of the treatment schedule in this study. The 4 days of off time (no treatment) allowed for the inhibition of ERK1/2 to be reversed, which in turn blocks STAT3 activation and allows cells to re-enter the proliferative cell cycle. STAT3 has also been implicated in the proliferation of SCs in numerous studies; therefore, it is also possible that a threshold of STAT3 phosphorylation is required to remove SCs from the cell cycle, allowing symmetric division of SCs to occur prior to cell cycle withdrawal. We conclude that Sunitinib treatment not only promotes myogenic differentiation but also allows SC proliferation and self-renewal. This suggests that the transient nature of our dosing schedule allows for the re-population of SC pools, asymmetric cell division and subsequent differentiation into mature myofibers.
Together our results show that the FDA-approved drug Sunitinib is capable of promoting muscle regeneration via transient activation of STAT3 in the mdx mouse model of DMD. The significantly lower, 1 mg/kg tri-weekly Sunitinib dose is not postulated to produce the same side effects observed with the 40 mg/kg/day cancer doses and would therefore be deemed sufficiently safer for pediatric use. In the future, it is important to determine the exact target of Sunitinib that is causing the inhibition of SHP-2 and consequential activation of STAT3, whether it is a direct inhibition of SHP-2 or an upstream target (62,63). A case study has determined that a gain of function mutation in PTPN11 (SHP-2 in humans) is sufficient to promote DMD muscle pathology in an otherwise Becker’s muscular dystrophy patient, suggesting a detrimental role of constitutive PTPN11 activation (64). Here we identify Sunitinib as a potent skeletal muscle regenerative drug capable of mitigating disease progression that could be fast tracked into clinical trials for DMD as well as other muscular dystrophies in which regeneration of muscle would be beneficial.
Materials and Methods
Cell culture
The C2C12 cell line was originally purchased from ATCC and grown in media containing Dulbecco's Modified Eagle Medium (DMEM) (Life Technologies, Carlsbad, CA) supplemented with 20% fetal bovine serum (Atlanta Biologicals, Flowery Branch, GA), 1% penicillin/streptomycin (GIBCO, Gaithersburg, MD) and 1% L-glutamine (GIBCO, Gaithersburg, MD). Myoblasts were grown until 70% confluent and differentiated into myotubes in media containing DMEM (Life Technologies, Carlsbad, CA) supplemented with 2% horse serum (Atlanta Biologicals, Flowery Branch, GA) and 1% insulin–transferrin–selenium (GIBCO, Gaithersburg, MD) with media changed daily for 120 h; cells were incubated at 37°C with 5% CO2. Cells were treated with varying concentrations of Sunitinib (Sigma, St. Louis, MO) diluted in 1% dimethyl sulfoxide -DMSO (Sigma, St. Louis, MO) or 1% DMSO alone with a maximum concentration of DMSO at 0.1%.
Animals
Previous studies (43) showed Sunitinib is soluble in a solution of 0.1% methylcellulose (Sigma, St. Louis, MO) at 25 mm. Initial, 5 day dosing study determined optimal Sunitinib dose for α7B integrin enhancement to be 1 mg/kg. The 4-week-old male mdx mice were administered Sunitinib via oral gavage at 1 mg/kg, three times per week until 12 weeks of age. All animals were treated according to the rules and regulations specified in the University of Nevada Reno Institutional Animal Care and Use Committee. At the end of the study, the mice were euthanized by cervical dislocation under anesthesia and all skeletal muscles were harvested; the diaphragms were harvested for contractile studies. All studies conducted using skeletal muscle tissue were done so in a blinded fashion including all contraction studies, grip strength, immunofluorescence and immunoblotting.
Immunoblotting
Protein was extracted from cell pellets or whole tissue using radioimmunoprecipitation assay buffer containing 1:500 dilution of protease inhibitor cocktail and 1:100 dilution of NaF and Na3VO4 phosphatase inhibitors. Protein quantification was performed via bicinchoninic acid assay (BCA) (Thermo Scientific, Waltham, MA), separated by sodium dodecyl sulfate- polyacrylamid gel electrophoresis (SDS-PAGE) and transferred onto nitrocellulose membrane. Detection of α7B was performed as previously described (59). pSTAT3 (D3A7; 1:250), STAT3 (79D7; 1:1000), pERK (1:1000), ERK (1:1000), pSHP-2 (3751; 1:250) and SHP-2 (D50F2; 1:1000) (Cell Signaling, Danvers, MA) were incubated overnight and followed by Alexa Fluor 680 or 800 conjugated goat anti-rabbit (Invitrogen, Carlsbad, CA) or horseradish peroxidase (HRP) - conjugated anti rabbit (Cell Signaling, Danvers, MA) detected using pierce enhanced chemiluminescence (ECL) western blotting substrate (Thermo Scientific, Waltham, MA). Protein quantity was normalized to GAPDH (1:1000, Cell Signaling, Danvers, MA) or swift stain (G-biosciences, St. Louis, MO) and imaged using LI-COR imaging system. Protein quantification was performed using Fiji (ImageJ, image processing software).
Immunofluorescence
Cells
C2C12 cells were grown in chamber slides, fixed with 4% paraformaldehyde and blocked in 1% BSA containing 0.1% Tween and 0.05% TritonX-100 for 1 h at room temperature. Cells were then incubated overnight with MF20 monoclonal antibody [1:40, Developmental Studies Hybridoma Bank (DSHB), Iowa City, IA] against MyHC, followed by a 1 h incubation with fluorescein isothiocyanate (FITC)-anti mouse secondary (1:200, BD Biosciences, Billerica, MA). Nuclei DAPI label contained in Vectashield (Vector Labs, Burlingham, CA) used to mount chamber slides.
Tissue
Post-harvest fresh hemi-diaphragms were embedded in a 2:3 (v/v) optimum cutting temperature compound to 30% sucrose/PBS and cryosectioned. The 10 μm sections were immunostained for α7 integrin as previously described (35) followed by 1 h incubation with FITC-anti rabbit (1:200, BD Biosciences, Billerica, MA). Sections were labeled with Alexa Fluor 647 conjugated wheat germ agglutinin (WGA, 1:250) for 20 min and mounted with vectashield containing DAPI for CLN. Immunostaining for eMyHC (1:40, DSHB) and Pax7 (1:50, DSHB—30 min permeabilization in 0.2% triton) were performed using Mouse on Mouse detection kit (Vector Labs, Burlingham, CA), followed by 1 h incubation with FITC-anti mouse and 20 min incubation with WGA-647. All tissues were imaged using the Olympus Fluoview FV 1000 laser confocal microscope.
Ex vivo contractility
Mice were put under anesthesia using isoflurane (1.5% isoflurane and 400 ml O2/min) and placed supine on a temperature-controlled platform to maintain their body temperature at 37°C. Diaphragm muscle strip was dissected in oxygenated physiological salt solution (pH 7.6, 30°C) and strung up with rib cage portion facing up on computer-controlled dual mode Aurora Scientific, Inc. 300B servomotor as previously described (65). The following protocol was performed: (1) determination of optimum length (L0) following three isometric twitches and three tetani (150 Hz). (2) Force frequency protocol will follow frequencies of 1, 30, 50, 80, 100, 120 and 150 Hz for 900 ms each with 4 min intervals. After all protocols are completed muscle was be stripped of rib cage and tendons and blotted on paper to remove excess moisture before weighing. Analysis of force contraction was normalized to weight for consistency as the diaphragm is cut into a strip and cross-sectional area is variable.
Forelimb grip strength
Forelimb grip strength measurements were obtained using a computerized grip strength meter (Colombus Instruments, Colombus, OH) according to Treat-NMD guidelines. Measurements were performed once after the 8 week treatment regime to prevent adaptation.
Whole muscle EBD assay
The EBD assay was performed as previously described (66). Briefly, EBD (Sigma, St. Louis, MO) was dissolved in phosphate buffered saline (PBS) at 10 mg/ml and injected intraperitoneally at 5 ul/g. Tissue was collected 24 h post-injection, weighed and incubated for 2 h at 55°C in 1 ml formamide (Sigma, St. Louis, MO). Absorbance was read at 620 nm by spectrophotometer.
TaqMan reverse transcription- polymerase chain reaction (RT-PCR)
TA muscle was homogenized in Trizol reagent (Ambion, Foster City, CA) and processed for RNA extraction. First-strand cDNA synthesis was performed using VILO reaction mix (ThermoFisher, Waltham, MA) and real-time qPCR reactions were performed on a 96 well plate using TaqMan Fast Advanced Master Mix and TaqMan Gene Expression Assays for Itga7 (Mm00434400_m1), MyoD1 (Mm00440387_m1), Myog (Mm00446194_m1) Pax7 (Mm01354484_m1). Levels normalized to ribosomal RNA 18S transcript. Reactions were run on the Applied Biosystems 7900HT Fast System in triplicate.
Statistical analysis
GraphPad Prism software was used for statistical calculations. Student’s t-test was used to compare means between two groups and one-way ANOVA was used to compare means between three or more groups. All ANOVA calculations were followed by Tukey’s post hoc test. Means considered statistically significant when P < 0.05.
Supplementary Material
{kind=link}
Acknowledgements
T.M.F. and P.B.F. were supported by Mick Hitchcock Scholarships.
Conflict of Interest statement. The University of Nevada, Reno has a patent pending on the therapeutic use of Sunitinib for the treatment of muscle disease. The University of Nevada, Reno has licensed this technology to StrykaGen Corp., a biopharmaceutical company owned by Drs Burkin and Wuebbles. The University of Nevada, Reno has a small equity share in this company.
Funding
National Institutes of Health (R01AR064338-01A1, R01AR053697, R21NS58429-01A1, R21NS58429S); Muscular Dystrophy Association (MDA238981).
References
- 1. Yiu E.M. and Kornberg A.J. (2015) Duchenne muscular dystrophy. J. Paediatr. Child Health, 51, 759–764. [DOI] [PubMed] [Google Scholar]
- 2. Hoffman E.P., Brown R.H. and Kunkel L.M. (1987) Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell, 51, 919–928. [DOI] [PubMed] [Google Scholar]
- 3. Bonilla E., Samitt C.E., Miranda A.F., Hays A.P., Salviati G., DiMauro S., Kunkel L.M., Hoffman E.P. and Rowland L.P. (1988) Duchenne muscular dystrophy: deficiency of dystrophin at the muscle cell surface. Cell, 54, 447–452. [DOI] [PubMed] [Google Scholar]
- 4. Durbeej M. and Campbell K.P. (2002) Muscular dystrophies involving the dystrophin–glycoprotein complex: an overview of current mouse models. Curr. Opin. Genet. Dev., 12, 349–361. [DOI] [PubMed] [Google Scholar]
- 5. Le Rumeur E., Winder S.J. and Hubert J.F. (2010) Dystrophin: more than just the sum of its parts. Biochim. Biophys. Acta Proteins Proteom., 1804, 1713–1722. [DOI] [PubMed] [Google Scholar]
- 6. Goldstein J. and McNally E.M. (2010) Mechanisms of muscle weakness in muscular dystrophy. J. Gen. Physiol., 136, 29–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Klingler W., Jurkat-Rott K., Lehmann-Horn F. and Schleip R. (2012) The role of fibrosis in Duchenne muscular dystrophy Werner. Acta Myol., 31, 184–195. [PMC free article] [PubMed] [Google Scholar]
- 8. Ostrovidov S., Shi X., Sadeghian R.B., Salehi S., Fujie T., Bae H., Ramalingam M. and Khademhosseini A. (2015) Stem cell differentiation toward the myogenic lineage for muscle tissue regeneration: a focus on muscular dystrophy, Stem Cell Rev and Rep., 11, 866–884. [DOI] [PubMed] [Google Scholar]
- 9. Flanigan K.M. (2014) Duchenne and Becker muscular dystrophies. Neurol. Clin., 32, 671–688. [DOI] [PubMed] [Google Scholar]
- 10. Biggar W.D., Harris V.A., Eliasoph L. and Alman B. (2006) Long-term benefits of deflazacort treatment for boys with Duchenne muscular dystrophy in their second decade, Neuromuscular Disorders, 16, 249–255. [DOI] [PubMed] [Google Scholar]
- 11. Goto M., Komaki H., Takeshita E., Abe Y., Ishiyama A., Sugai K., Sasaki M., Goto Y.-I. and Nonaka I. (2016) Long-term outcomes of steroid therapy for Duchenne muscular dystrophy in Japan, Brain & Development, 38, 785–791. [DOI] [PubMed] [Google Scholar]
- 12. Lim K.R., Maruyama R. and Yokota T. (2017) Eteplirsen in the treatment of Duchenne muscular dystrophy. Drug Des. Devel. Ther., 11, 533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Dumont N.A., Wang Y.X., Von Maltzahn J., Pasut A., Bentzinger C.F., Brun C.E. and Rudnicki M.A. (2015) Dystrophin expression in muscle stem cells regulates their polarity and asymmetric division. Nat. Med., 21, 1455–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Thomas K. Extracellular matrix regulation in the muscle satellite cell niche. Connect Tissue Res., 56, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Almada A.E. and Wagers A.J. (2016) Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat. Rev. Mol. Cell Biol., 17, 267–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chang N.C., Chevalier F.P. and Rudnicki M.A. (2016) Satellite cells in muscular dystrophy—lost in polarity. Trends Mol. Med., 22, 479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Serrano A.L., Baeza-Raja B., Perdiguero E., Jardí M. and Muñoz-Cánoves P. (2008) Interleukin-6 is an essential regulator of satellite cell-mediated skeletal muscle hypertrophy. Cell Metab., 7, 33–44. [DOI] [PubMed] [Google Scholar]
- 18. Mckay B.R., De Lisio M., Johnston A.P.W., O ‘reilly C.E., Phillips S.M., Tarnopolsky M.A., Parise G. and Hotchin N. (2009) Association of interleukin-6 signalling with the muscle stem cell response following muscle-lengthening contractions in humans. Plosone, 4, doi: 10.1371/journal.pone.0006027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Toth K.G., McKay B.R., De Lisio M., Little J.P., Tarnopolsky M.A. and Parise G. (2011) IL-6 induced STAT3 signalling is associated with the proliferation of human muscle satellite cells following acute muscle damage. PLoS One, 6, e17392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Naë Lle Begue G., Douillard A., Galbes O., Rossano B., Vernus B., Candau R. and Py G. (2013) Early activation of rat skeletal muscle IL-6/STAT1/STAT3 dependent gene expression in resistance exercise linked to hypertrophy Plosone, 8, doi: 10.1371/journal.pone.0057141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kurosaka M. and Machida S. (2013) Interleukin-6-induced satellite cell proliferation is regulated by induction of the JAK2/STAT3 signalling pathway through cyclin D1 targeting. Cell Prolif., 46, 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zhu H., Xiao F., Wang G., Ip N.Y., Cheung T.H., Wu Z., Wei X., Jiang L., Chen Y., Zhu L. et al. (2016) STAT3 Regulates self-renewal of adult muscle satellite cells during injury-induced muscle regeneration. Cell Rep., 16, 2102–2115. [DOI] [PubMed] [Google Scholar]
- 23. Kami K. and Senba E. (2002) In vivo activation of STAT3 signaling in satellite cells and myofibers in regenerating rat skeletal muscles. J. Histochem. Cytochem., 50, 1579–1589. [DOI] [PubMed] [Google Scholar]
- 24. Yang Y., Xu Y., Li W., Wang G., Song Y., Yang G., Han X., Du Z., Sun L. and Ma K. (2009) STAT3 induces muscle stem cell differentiation by interaction with myoD. Cytokine, 46, 137–141. [DOI] [PubMed] [Google Scholar]
- 25. Hoene M., Runge H., Häring H.U., Schleicher E.D. and Weigert C.. Interleukin-6 promotes myogenic differentiation of mouse skeletal muscle cells: role of the STAT3 pathway. Am J Physiol Cell Physiol., 304, C128–C136. [DOI] [PubMed] [Google Scholar]
- 26. Liu J., Jing X., Gan L. and Sun C.. The JAK2/STAT3 signal pathway regulates the expression of genes related to skeletal muscle development and energy metabolism in mice and mouse skeletal muscle cells, doi: 10.1271/bbb.120324. [DOI] [PubMed]
- 27. Price F.D., Maltzahn J., Bentzinger C.F., Dumont N.A., Yin H., Chang N.C., Wilson D.H., Frenette J. and Rudnicki M.A. (2014) Inhibition of JAK-STAT signaling stimulates adult satellite cell function. Nat. Med., 20, 1174–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Tierney M.T., Aydogdu T., Sala D., Malecova B., Gatto S., Puri P.L., Latella L. and Sacco A. (2014) STAT3 signaling controls satellite cell expansion and skeletal muscle repair. Nat. Med., 10, 1182–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hodges B.L., Hayashi Y.K., Nonaka I., Wang W., Arahata K. and Kaufman S.J. (1997) Altered expression of the α7β1 integrin in human and murine muscular dystrophies. J. Cell Sci., 110, 2873–2881. [DOI] [PubMed] [Google Scholar]
- 30. Hayashi Y.K., Chou F.L., Engvall E., Ogawa M., Matsuda C., Hirabayashi S., Yokochi K., Ziober B.L., Kramer R.H., Kaufman S.J. et al. (1998) Mutations in the integrin alpha7 gene cause congenital myopathy. Nat. Genet., 1, 94–97. [DOI] [PubMed] [Google Scholar]
- 31. Burkin D.J. and Kaufman S.J. (1999) The α7β1 integrin in muscle development and disease Cell Tissue Res ., 296, 182–190. [DOI] [PubMed] [Google Scholar]
- 32. Burkin D.J., Wallace G.Q., Nicol K.J., Kaufman D.J. and Kaufman S.J. (2001) Enhanced expression of the alpha 7 beta 1 integrin reduces muscular dystrophy and restores viability in dystrophic mice. J. Cell Biol., 152, 1207–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Burkin D. (2005) Transgenic expression of alpha7beta1 integrin (2005). Am. J. Pathol., 166, 253–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Guo C., Willem M., Werner A., Raivich G., Emerson M., Neyses L. and Mayer U. (2006) Absence of a7 integrin in dystrophin-deficient mice causes a myopathy similar to Duchenne muscular dystrophy. Hum. Mol. Genet., 15, 989–998. [DOI] [PubMed] [Google Scholar]
- 35. Rooney J.E., Welser J.V., Melissa A., Dechert N.L.F.-D., Kaufman S.J. and Burkin D.J. (2006) Severe muscular dystrophy in mice that lack dystrophin and 7 integrin J. Cell Sci., 119, 2185–2195. [DOI] [PubMed] [Google Scholar]
- 36. Marshall J.L., Chou E., Jennifer O., Kwok A., Burkin D.J. and Crosbie-Watson R.H. (2012) Dystrophin and utrophin expression require sarcospan: loss of a7 integrin exacerbates a newly discovered muscle phenotype in sarcospan-null mice. Hum. Mol. Genet., 21, 4378–4393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hakim C.H., Burkin D.J. and Duan D. (2013) Alpha 7 integrin preserves the function of the extensor digitorum longus. J. Appl. Physiol., 115, 1388–1392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Heller K.N. (2013) AAV-mediated overexpression of human α7 integrin leads to histological and functional improvement in dystrophic mice. Mol. Ther., 21, 520–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Heller K.N., Montgomery C.L., Shontz K.M., Clark K.R., Mendell J.R. and Rodino-Klapac L.R. (2015) Human a7 integrin gene (ITGA7) delivered by adeno-associated virus extends survival of severely affected dystrophin/utrophin-deficient mice. Hum. Gene Ther., 26, 647–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Sarathy A., Wuebbles R.D., Fontelonga T.M., Tarchione A.R., Mathews Griner L.A., Heredia D.J., Nunes A.M., Duan S., Brewer P.D., Van Ry T. et al. (2017) SU9516 Increases α7β1 integrin and ameliorates disease progression in the mdx mouse model of Duchenne muscular dystrophy. Mol. Ther., 25, 1394–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Faivre S., Delbaldo C., Vera K., Robert C., Lozahic S., Lassau N., Bello C., Deprimo S., Brega N., Massimini G. et al. Safety, pharmacokinetic, and antitumor activity of SU11248, a novel oral multitarget tyrosine kinase inhibitor, in patients with cancer. J. Clin. Oncol., 24, 25–35. [DOI] [PubMed] [Google Scholar]
- 42. Izzedine H., Buhaescu I., Rixe O. and Deray G. (2007) Sunitinib malate. Cancer Chemother. Pharmacol., doi: 10.1007/s00280-006-0376-5. [DOI] [PubMed] [Google Scholar]
- 43. Pretto F., Ghilardi C., Moschetta M., Bassi A., Rovida A., Scarlato V., Talamini L., Fiordaliso F., Bisighini C., Damia G. et al. (2014) Sunitinib prevents cachexia and prolongs survival of mice bearing renal cancer by restraining STAT3 and MuRF-1 activation in muscle. Oncotarget, 6, 3043–3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ziober B.L. and Kramer R.H.. Identification and characterization of the cell type-specific and developmentally regulated α7 integrin gene promoter. The Journal of Biol Chem., 271, 22915–22922. [DOI] [PubMed] [Google Scholar]
- 45. Jethanandani P. and Kramer R.H. (2005) α7 Integrin expression is negatively regulated by δEF1 during skeletal myogenesis, doi: 10.1074/jbc.M508698200. [DOI] [PubMed]
- 46. Schiaffino S., Rossi A.C., Smerdu V., Leinwand L.A. and Reggiani C.. Developmental myosins: expression patterns and functional significance, doi: 10.1186/s13395-015-0046-6. [DOI] [PMC free article] [PubMed]
- 47. Kharraz Y., Guerra J., Pessina P., Serrano A. and Muñoz-Cánoves P. (2014) Understanding the process of fibrosis in Duchenne muscular dystrophy. Biomed. Res. Int., 2014, 965631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yin H., Price F. and Rudnicki M.A. (2013) Satellite cells and the muscle stem cell niche. Physiol. Rev., 93, 23–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Dumont N.A. and Rudnicki M.A. (2017) Characterizing satellite cells and myogenic progenitors during skeletal muscle regeneration Methods Mol. Biol., 1560, 179–188. [DOI] [PubMed] [Google Scholar]
- 50. Chung J., Uchida E., Grammer T.C. and Blenis J. (1997) STAT3 serine phosphorylation by ERK-dependent and -independent pathways negatively modulates its tyrosine phosphorylation. Mol and Cell Biol., 17, 6508–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Sengupta T.K., Talbot E.S., Scherle P.A., Ivashkiv L.B. and Darnell J.E. (1998) Rapid inhibition of interleukin-6 signaling and Stat3 activation mediated by mitogen-activated protein kinases. Biochemistry, 95, 11107–11112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kim A., Balis F.M. and Widemann B.C.. Sorafenib and Sunitinib learning objectives. The Oncologist, 14, 800–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Zhang L., Smith K.M., Chong A.L., Stempak D., Yeger H., Marrano P., Thorner P.S., Irwin M.S., Kaplan D.R. and Baruchel S. (2009) In vivo antitumor and antimetastatic activity of Sunitinib in preclinical neuroblastoma mouse model 1. Neoplasia, 11, 426–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Dubois S.G., Shusterman S., Ingle A.M., Ahern C.H., Reid J.M., Wu B., Baruchel S., Glade-Bender J., Ivy P., Grier H.E. et al. Phase I and pharmacokinetic study of Sunitinib in pediatric patients with refractory solid tumors: a Children’s Oncology Group study. Clin. Cancer Res., 17, 5113–5122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Dubois S.G., Shusterman S., Reid J.M., Ingle A.M., Ahern C.H., Baruchel S., Glade-Bender J., Ivy P., Adamson P.C. and Blaney S.M. (2012) Tolerability and pharmacokinetic profile of a Sunitinib powder formulation in pediatric patients with refractory solid tumors: a Children’s Oncology Group study. Cancer Chemother. Pharmacol., 69, 1021–1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Motohashi N. and Asakura A. (2014) Muscle satellite cell heterogeneity and self-renewal. Front. Cell Dev. Biol., 2, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Almada A.E. and Wagers A.J. (2016) Molecular circuitry of stem cell fate in skeletal muscle regeneration, ageing and disease. Nat. Publ. Gr., 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Spangenburg E.E. and Booth F.W.. Multiple signaling pathways mediate LIF-induced skeletal muscle satellite cell proliferation, Am J Physiol Cell Physiol., 283, C204–C211. [DOI] [PubMed] [Google Scholar]
- 59. Rooney J.E., Welser J.V., Dechert M.A., Flintoff-Dye N.L., Kaufman S.J. and Burkin D.J. (2006) Severe muscular dystrophy in mice that lack dystrophin and alpha7 integrin J Cell Sci. England, 119, 2185–2195. [DOI] [PubMed] [Google Scholar]
- 60. Lueders T.N., Zou K., Huntsman H.D., Meador B., Mahmassani Z., Abel M., Valero M.C., Huey K.A. and Boppart M.D. (2011) The α7β1-integrin accelerates fiber hypertrophy and myogenesis following a single bout of eccentric exercise. Am. J. Physiol. Cell Physiol., 301, C938–C946. [DOI] [PubMed] [Google Scholar]
- 61. Belizário J.E., Fontes-oliveira C.C., Borges J.P., Kashiabara J.A. and Vannier E. (2016) Skeletal muscle wasting and renewal: a pivotal role of myokine IL-6. Springer Plus, 5, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Fairlie W.D., De Souza D., Nicola N.A. and Baca M. (2003) Negative regulation of gp130 signalling mediated through tyrosine-757 is not dependent on the recruitment of SHP2. Biochem. J., 372, 495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Li J., Reed S.A. and Johnson S.E.. Hepatocyte growth factor (HGF) signals through SHP2 to regulate primary mouse myoblast proliferation. Exp Cell Res., 315, 2284–2292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Dinopoulos A., Papadopoulou A., Manta P., Kekou K., Kanelopoulos T., Fretzayas A. and Kitsiou S. Coinheritance of Noonan syndrome and Becker muscular dystrophy. Neuromuscular Disorders, 20, 61–63. [DOI] [PubMed] [Google Scholar]
- 65. Moorwood C., Liu M., Tian Z. and Barton E.R. (2013) Isometric and eccentric force generation assessment of skeletal muscles isolated from murine models of muscular dystrophies video link. J. Vis. Exp., 71, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Heydemann A., Swaggart K.A., Kim G.H., Holley-Cuthrell J., Hadhazy M. and McNally E.M.. The superhealing MRL background improves muscular dystrophy. Skelet. Muscle, 2, 2–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.