

Abstract
Pathogenic variants in BRCA1 and BRCA2 only explain the underlying genetic cause of about 10% of hereditary breast and ovarian cancer families. Because of cost‐effectiveness, multigene panel testing is often performed even if the clinical utility of testing most of the genes remains questionable. The purpose of our study was to assess the contribution of rare, deleterious‐predicted variants in DNA repair genes in familial breast cancer (BC) in a well‐characterized and homogeneous population. We analyzed 113 DNA repair genes selected from either an exome sequencing or a candidate gene approach in the GENESIS study, which includes familial BC cases with no BRCA1 or BRCA2 mutation and having a sister with BC (N = 1,207), and general population controls (N = 1,199). Sequencing data were filtered for rare loss‐of‐function variants (LoF) and likely deleterious missense variants (MV). We confirmed associations between LoF and MV in PALB2, ATM and CHEK2 and BC occurrence. We also identified for the first time associations between FANCI, MAST1, POLH and RTEL1 and BC susceptibility. Unlike other associated genes, carriers of an ATM LoF had a significantly higher risk of developing BC than carriers of an ATM MV (ORLoF = 17.4 vs. ORMV = 1.6; p Het = 0.002). Hence, our approach allowed us to specify BC relative risks associated with deleterious‐predicted variants in PALB2, ATM and CHEK2 and to add MAST1, POLH, RTEL1 and FANCI to the list of DNA repair genes possibly involved in BC susceptibility. We also highlight that different types of variants within the same gene can lead to different risk estimates.
Keywords: breast cancer, exome sequencing, multigene panel testing, variant, case–control study
Short abstract
What's new?
Pathogenic variants in BRCA1 and BRCA2 only explain the genetic cause of about 10% of hereditary breast and ovarian cancer families, and the clinical usefulness of testing other genes following the recent introduction of cost‐effective multigene panel sequencing in diagnostics laboratories remains questionable. This large case‐control study describes genetic variation in 113 DNA repair genes and specifies breast cancer relative risks associated with rare deleterious‐predicted variants in PALB2, ATM, and CHEK2. Importantly, different types of variants within the same gene can lead to different risk estimates. The results may help improve risk prediction models and define gene‐specific consensus management guidelines.
Abbreviations
- 95%CI
95% confidence interval
- BC
breast cancer
- ER
estrogen receptor
- HBOC
hereditary breast and ovarian cancer
- IGV
integrative genomics viewer
- LoF
loss‐of‐function variant
- MAF
minor allele frequency
- MV
missense variant
- OR
odds ratio
- P
probability value
- PR
progesterone receptor
- SNV
single nucleotide variant
- VUS
variant of uncertain clinical significance
- WES
whole‐exome sequencing.
Introduction
Women from families with multiple cases of breast or ovarian cancer have a higher risk of developing breast cancer (BC) during their lifetime than women in the general population.1 Since the identification of BRCA1 and BRCA2 in the late 1990s, genetic testing has become a routine clinical assessment for individuals with clinical features suggestive of a hereditary breast and ovarian cancer (HBOC) predisposition. However, a pathogenic variant in one of these two genes is found in only 10% of patients attending the cancer genetics clinics.2 HBOC families with no pathogenic variants in the known predisposition genes present challenges for molecular genetic diagnostics. In such families, predictive testing for healthy relatives, genetic counseling and preventive medical management are hampered.3
Paradoxically in recent years, the introduction of cost‐effective multigene panel sequencing in diagnostics laboratories has led to an explosion of genetic data in cancer‐prone families. Very often, 20–25 genes or more are being screened in parallel to BRCA1 and BRCA2 in HBOC families,4 but data attributing a causative role for potentially deleterious variants identified in many of these genes are usually lacking, and precise risk estimates per variant category (e.g. loss‐of‐function variants (LoF) or likely deleterious missense variants (MV)) have not been determined yet.
To date, the screened genes include known cancer susceptibility genes associated with a moderate (2‐ to 5‐fold) to high relative risk of BC (>5‐fold) because of their direct or indirect functional link with BRCA1 and BRCA2, such as PALB2,5, 6, 7, 8 ATM 9, 10 and CHEK2. 11 Other DNA repair genes, such as RAD51 paralogs,12, 13, 14 RINT1 15 and genes of the MRE11A‐RAD50‐NBN complex16 are also screened although replication studies conducted in independent at‐risk populations are lacking or failed to replicate initial findings.17, 18 The interpretation of the genetic data is also complicated. Most of the known pathogenic variants associated with BC are LoF (i.e. frameshift, stop‐gain and canonical splice variants predicted to result in a truncated protein)19 but a significant fraction of BC risk may be attributable to rare MV in some genes such as ATM, CHEK2 and TP53,9, 11, 20 and this fraction may be even bigger than that attributable to LoF.21
In the light of these issues, whether identifying a LoF or a MV in genes usually screened in parallel to BRCA1 and BRCA2 is clinically useful remains questionable. We therefore conducted a study to estimate BC relative risks associated with rare potentially pathogenic variants in DNA repair genes in a large well‐characterized and homogeneous population.
Participants, Material and Methods
Study participants
The study population consisted of women participating in GENESIS (GENE SISters), a French resource for familial BC research.22 In summary, 1,721 women affected with infiltrating mammary or ductal adenocarcinoma, not carrying a pathogenic variant in BRCA1 and BRCA2, and having a sister with BC were enrolled in GENESIS between 2007 and 2013 through the national network of cancer genetics clinics (http://www.unicancer.fr/en/unicancer-group). Affected sisters (N = 826) and 1,419 unrelated cancer‐free friends or colleagues of index cases (controls) were also included. These latter were aged‐matched (±3 years) to cases at interview. Blood samples, clinical and epidemiological data were collected for each participant. Written informed consent for our study was obtained from all participants. GENESIS study protocol and genetic subsequent studies were approved by the appropriate ethics committee (CCP Ile‐de‐France III) and by the French data protection authority (CNIL).
Selection of cases for the gene discovery stage
Women included in the WES stage consisted of 100 unrelated cases presenting with a strong personal or family history of BC. Among those, 53 cases belonged to a family with at least one case having developed bilateral BC, 43 cases had at least three sisters affected with BC, and 11 cases had a male first‐degree relative affected with BC. Seven of the selected cases fulfilled two of these criteria. The mean age at diagnosis of first BC was 48.4 (range: 28–71 years). Ninety‐eight of the cases were of European origin.
Selection of subjects for BC risk assessment
After quality control procedures, we analyzed DNA from 1,207 unrelated cases (none of whom had been included or were related to an individual who was included in the discovery stage), and 1,199 unrelated controls. The screened sample represented 77% of the GENESIS population. Distribution of cases and controls by age, ethnicity and number of cancers in the family is shown in Table 1.
Table 1.
Distribution of cases and controls by age, by population ancestry/ethnicity and by family history of cancer
| Controls (N = 1,199) | All cases (N = 1,207) | Pseudo‐incident1 cases (N = 663) | |
|---|---|---|---|
| Age 2 (years) | |||
| ≤30 | 9 | 0 | 0 |
| 31–40 | 61 | 20 | 13 |
| 41–50 | 265 | 269 | 171 |
| 51–60 | 429 | 465 | 250 |
| ≥61 | 435 | 453 | 229 |
| Mean age (SE) | 56.1 (0.3) | 57.5 (0.3) | 56.6 (0.4) |
| Range | 25–83 | 31–90 | 31–85 |
| Population ancestry/Ethnicity | |||
| Caucasian | 1,176 | 1,152 | 631 |
| Ashkenazi | 0 | 4 | 4 |
| African/Afro‐Caribbean | 1 | 3 | 1 |
| Arab/Berber/Asian/Arab+Berber | 4 | 13 | 6 |
| Mixed origin | 9 | 18 | 7 |
| Unknown | 9 | 17 | 14 |
| Number of cancers in family | |||
| Breast cancer (C50 + D05.1)3 | |||
| 0 | 851 | 0 | 0 |
| 1 | 261 | 0 | 0 |
| 2 | 68 | 294 | 167 |
| 3 | 14 | 467 | 252 |
| 4 | 3 | 280 | 161 |
| 5 | 1 | 104 | 46 |
| 6 | 0 | 46 | 28 |
| 7 | 1 | 11 | 6 |
| 8 | 0 | 5 | 3 |
| Ovarian cancer (C56) | |||
| 0 | 1,164 | 1,139 | 623 |
| 1 | 33 | 61 | 34 |
| 2 | 1 | 6 | 6 |
| 3 | 0 | 1 | 0 |
| 5 | 1 | 0 | 0 |
| Cancers from Lynch syndrome spectrum 4 | |||
| 0 | 781 | 678 | 396 |
| 1 | 306 | 335 | 158 |
| 2 | 81 | 130 | 74 |
| 3 | 20 | 44 | 23 |
| 4 | 5 | 14 | 8 |
| 5 | 5 | 5 | 4 |
| 6 | 1 | 0 | 0 |
| 7 | 0 | 1 | 0 |
Cases diagnosed with BC less than 5 years before enrollment in GENESIS.
Age at diagnosis for cases and age at inclusion for controls.
Number after excluding GENESIS index case and one affected sister.
Colon (C18), small bowel (C17), rectum (C19, C20), endometrium (C53, C54, C55), ovary (C56), stomach (C16), bile duct cancers (C24).
DNA preparation
Peripheral blood samples (10 mL) were collected in the presence of anticoagulant (sodium citrate), and genomic DNA (gDNA) extracted from peripheral blood leukocytes with a standard inorganic method23 using the DNA extractor Autopure‐LS (Qiagen). DNA handling (normalization and aliquoting) was done using a TECAN EVO instrument.
Sequencing, data processing and variant discovery
Library preparation, whole exome sequencing (WES) and gene panel sequencing were performed at the Centre National de Recherche en Génomique Humaine (Evry, France). Exomes were captured using the SureSelectXT Human All Exon V5 kit, and coding exons of candidate genes were captured using a custom SureSelect target Enrichment system (Agilent Technologies, Santa Clara, CA, USA). Final libraries were sequenced on an Illumina HiSeq 2500 instrument with 100‐bp paired end reads. Reads were mapped to the reference genome hg19 (1,000 Genomes GRCh37 build) using BWA aln v0.6.224 with the after parameters: two mismatches allowed in the 32 bp‐seed alignment and four mismatches allowed in the whole read, one alignment reported per read. For the exome‐sequenced samples, Picard Tools v.1.103 (http://picard.sourceforge.net) was used to remove duplicate reads in order to mitigate biases introduced by data generation steps such as PCR amplification. SNV and insertions/deletions (indels) were then called using three strategies: i. Freebayes (v0.9.9.9.2),25 ii. GATK’ Unified Genotyper (v2.2)26 on original alignment files and iii. on preprocessed alignment files using GATK modules for local realignment around indels and base quality score recalibration using databases of known polymorphisms, after best practice recommendations. All variant calls were applied using a minimum base quality of 17 and a minimum mapping quality of 10. A dedicated filtering process was applied per sample to prioritize variants of interest: variants covered by less than 5× and with phred quality of less than 30 were discarded, while only heterozygous variants (defined by an allelic ratio between 20 and 80%) and variants detected by at least two calling strategies were considered. For some complex events (indels or SNVs falling in repeated sequences) we also performed visual inspection of aligned reads using the Integrative Genomics Viewer (IGV)27, 28 to reduce the risk of false positives and help characterizing them.
The joint variant calling file was finally annotated with refGene regions, SNV effects, as well as 1,000 Genomes (1000G) minor allele frequency (MAF; august 2015 version) using Annovar (October 2015 version, http://annovar.openbioinformatics.org). Only exonic or splicing variants presenting a MAF below 0.5% in all represented ethnic groups of the 1000G database were kept in further analyses. In the gene discovery stage, variants carried by more than five cases and absent from 1000G were excluded as they were likely to represent a sequencing artifact or an unreported common polymorphism in our population. We then only considered LoF, inframe indels and conserved MV (PhyloP score > 0)29 predicted as deleterious by SIFT30 and/or PolyPhen231 in the analyses. In the case–control study, CADD32 (for all genes) and Align‐GVGD (for ATM, CHEK2 and PALB2)33 were also used to annotate and filter MV. In this latter stage, we applied a supplementary filter on MAF in order to exclude any variant with MAF > 0.5% in controls, as we observed that the MAF of some variants was underestimated in the public databases.
Statistical analyses
All LoF and MV with a phred CADD score equal or above 20 were considered in the analyses. Their frequencies among cases and controls were compared by unconditional logistic regressions, adjusting for ethnicity and age at inclusion in GENESIS. To address issues of multiple testing, by examining 113 genes and applying a Bonferroni correction, statistical significance was defined as p < 0.0004.
We used Likelihood ratio tests to test for heterogeneity according to variant type (LoF vs. MV), and polytomous logistic regressions to test for heterogeneity according to hormone status and according to time between inclusion and diagnosis.
All statistical tests were two‐sided and data was analyzed using STATA, version 11.2 (StataCorp, College Station, TX).
Results
Gene discovery stage
WES was performed on 100 unrelated high‐risk women to identify candidate genes. The average read depth achieved for target regions was 110X (range: 55X‐207X). At least 80% (average: 96.45%; range: 79.77%–99.35%) of the capture target regions were covered by 20 or more sequenced reads for all samples (Supporting Information Fig. S1). Whole exome sequencing performance and variants count for each sequenced DNA sample is shown in Supporting Information Table S1. After initial single nucleotide variants (SNV) filtering steps, an average of 18.2 (11–29) LoF, 6.8 (1–14) inframe indels and 133.7 (105–170) MV were identified per individual. In total, 2,584 genes were altered by such variants in at least two unrelated cases. Among those, 123 genes were known to be involved in DNA repair pathways. After having reviewed the potential involvement of each of them in cancer etiology or progression, we selected 77 genes for further investigation in the case–control study. This gene panel was completed with 36 genes currently included in commercial BC multigene panels. The workflow for gene prioritization and the final list of analyzed genes are shown on Figure 1.
Figure 1.
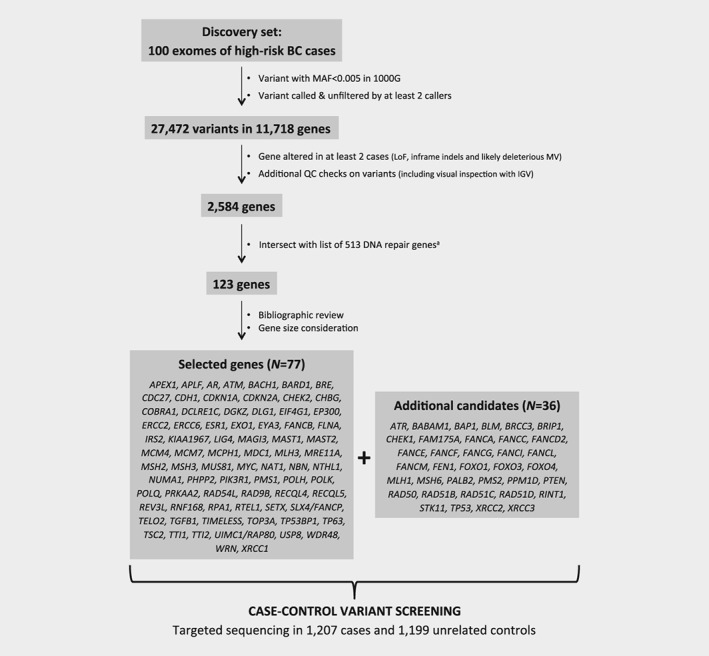
Strategy for genes prioritization. aThe DNA repair genes list was compiled from ACSN maps (https://acsn.curie.fr/), KEGG (http://www.genome.jp/kegg/pathway.html) and after review of literature.
Breast cancer risk assessment
Targeted sequencing of the 113 genes identified 3,930 distinct variants with MAF lower than 0.5% in GENESIS controls. These included 264 LoF variants (155 in cases and 132 in controls) and 1,994 MV bioinformatically predicted to alter protein function or stability (1,213 in cases and 1,103 in controls). The distribution of the number of altered genes per subject, according to their case/control status is shown on Supporting Information Figure S2. There was no overall difference in the distribution of number of altered genes between cases and controls (Chi2 (6df) = 10.0, p = 0.12) and only a modest increased risk of BC for subjects with at least two altered genes as compared to subjects with 0 or 1 altered gene (OR = 1.2; 95%CI, 1.0–1.4; p = 0.02). Nevertheless, we performed sensitivity analyses adjusted on the number of altered genes in the subsequent analyses. For each gene, we aggregated all rare LoF and MV, and also examined separately the two types of variants. The distribution of variants in cases and controls, per gene, and associated risk estimates are provided in Supporting Information Table S2, and results for all genes are summarized in Figure 2. Among those, PALB2, ATM, CHEK2, FANCI, MAST1, POLH and RTEL1 showed significant association with BC although only CHEK2 remained associated with BC after correcting for multiple testing.
Figure 2.
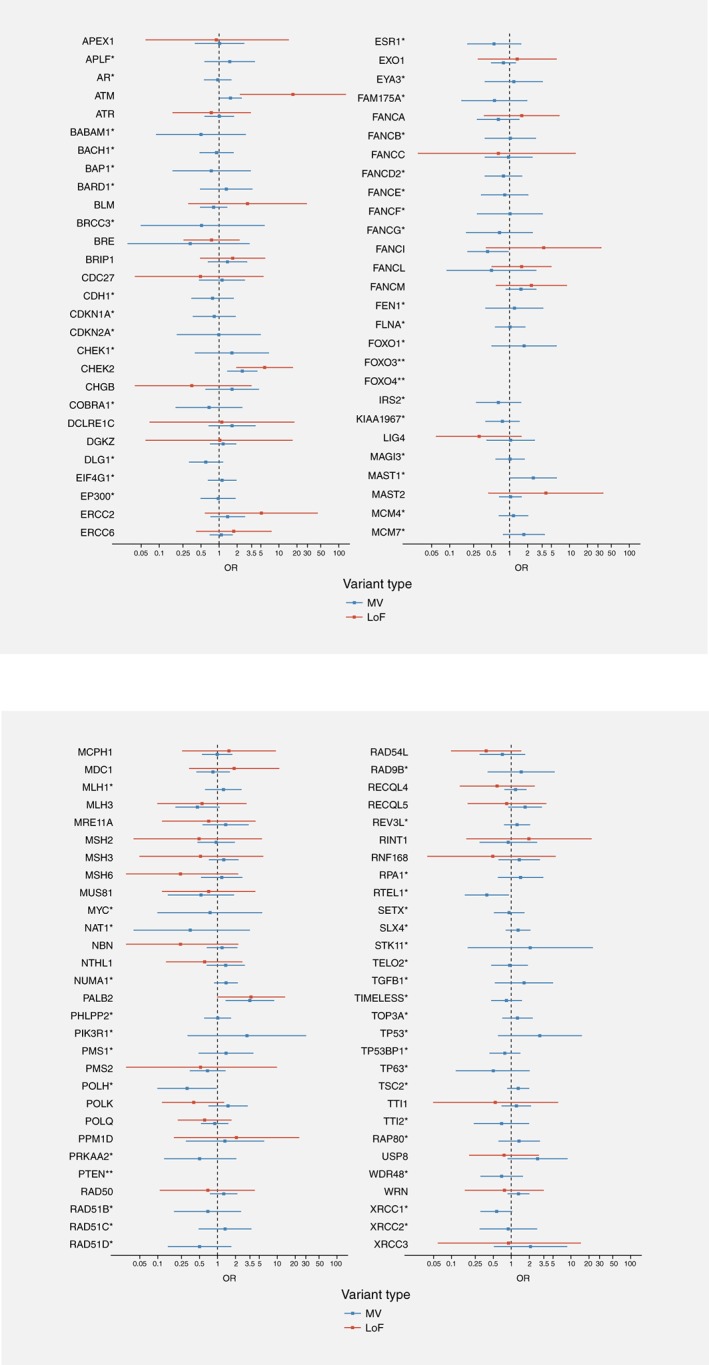
Result of the association tests per variant type for the 113 genes. Legend: *, no LoF identified in cases and/or in controls; **, no LoF and no likely deleterious MV identified in cases and/or in controls.
Both LoF and MV in PALB2, ATM and CHEK2 were associated with an increased BC risk, but carriers of an ATM LoF had a significantly higher risk of developing BC than carriers of an ATM MV (ORLoF = 17.4 vs. ORMV = 1.6; p Het = 0.002) (Table 2). In contrast for CHEK2 and PALB2 there was no significant heterogeneity in risk according to variant type (CHEK2: ORLoF + MV = 3.0; 95%CI 1.9–5.0; PALB2: ORLoF + MV = 3.5; 95%CI 1.7–7.5).
Table 2.
Distribution of rare variants and results of the association tests for the 7 genes showing association with BC (p ≤ 0.05)
| Any variant | LoF | Likely deleterious MV | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Control carriers | Case carriers | OR1 (95%CI) | p‐Value | OR2 (95%CI) | p‐Value | Control carriers | Case carriers | OR1 (95%CI) | p‐Value | Control carriers | Case carriers | OR1 (95%CI) | p‐Value | P het 3 |
| ATM | 40 | 77 | 1.9 (1.3, 2.9) | 0.001 | 1.8 (1.2, 2.7) | 0.003 | 1 | 16 | 17.4 (2.3, 132) | 0.006 | 39 | 61 | 1.6 (1.0, 2.3) | 0.04 | 0.002 |
| CHEK2 | 22 | 62 | 3.0 (1.9, 5.0) | 0.00001 | 2.9 (1.7, 4.7) | 0.0004 | 4 | 21 | 5.8 (2.0, 16.9) | 0.001 | 18 | 41 | 2.4 (1.4, 4.3) | 0.002 | 0.14 |
| FANCI | 22 | 13 | 0.6 (0.3, 1.2) | 0.13 | 0.5 (0.3, 1.1) | 0.08 | 1 | 4 | 3.7 (0.4, 33.6) | 0.24 | 21 | 9 | 0.4 (0.2, 1.0) | 0.04 | 0.04 |
| MAST1 | 8 | 17 | 2.2 (0.9, 5.1) | 0.07 | 2.0 (0.9, 4.7) | 0.11 | 1 | 0 | ‐ | ‐ | 7 | 17 | 2.5 (1.0, 6.0) | 0.04 | ‐ |
| PALB2 | 9 | 30 | 3.5 (1.7, 7.5) | 0.001 | 3.2 (1.5, 6.9) | 0.002 | 3 | 10 | 3.6 (1.0, 13.3) | 0.05 | 6 | 20 | 3.5 (1.4, 8.7) | 0.008 | 0.95 |
| POLH | 13 | 5 | 0.4 (0.1, 1.1) | 0.07 | 0.3 (0.1, 1.0) | 0.04 | 0 | 1 | ‐ | ‐ | 13 | 4 | 0.3 (0.1, 1.0) | 0.04 | ‐ |
| RTEL1 | 20 | 8 | 0.4 (0.2, 0.9) | 0.03 | 0.4 (0.2, 0.8) | 0.02 | 0 | 0 | ‐ | ‐ | 20 | 8 | 0.4 (0.2, 0.9) | 0.03 | ‐ |
Reference group: noncarriers of a variant in the tested gene; adjusting for ethnicity and age at inclusion.
Reference group: noncarriers of a variant in the tested gene; adjusting for ethnicity, age at inclusion and number of altered genes (continuous).
p‐Value of the likelihood ratio test to test for heterogeneity between the effect of LoF and MV.
The increased risk associated with variants in MAST1 was essentially driven by carrying a MV (ORMV = 2.5; 95%CI 1.0–6.0). Likewise, the decreased risk associated with variants in FANCI, POLH and RTEL1 was driven by carrying a MV (FANCI: ORMV = 0.4; 95%CI, 0.2–1.0; POLH: ORMV = 0.3; 95%CI 0.1–1.0; RTEL1: ORMV = 0.4; 95%CI 0.2–0.9). Intriguingly, four cases and one control carried a FANCI LoF, leading to a nonsignificant increase in BC risk (ORLoF = 3.7 vs. ORMV = 0.4; p Het = 0.04) (Table 2).
Furthermore, we found that carriers of a LoF in CHEK2 were diagnosed with BC at a younger age than noncarriers of a CHEK2 variant (46.7 years vs. 52.1 years, p T test = 0.007). Such a difference was not observed for carriers of a MV (50.1 years vs. 52.1 years, p T test = 0.16). For all other genes, no difference in age at diagnosis was observed (data not shown).
Breast cancer risk by hormone receptor status
Estrogen receptor (ER), progesterone receptor (PR) and HER2 gene amplification status of the breast tumor were available for 67%, 63% and 40% of the sequenced cases, respectively. For FANCI, POLH and RTEL1, there were too few observations in each stratum to draw any conclusion on whether carrying a likely deleterious variant was associated with a particular status. Information on HER2 status among carriers of a variant in any of the genes was too scarce to conclude. In the analyses comparing BC risk according to ER or PR status, none of the heterogeneity tests was significant. However, CHEK2 variants were associated with a higher BC risk point estimate for ER‐positive (OR = 3.5) compared to ER‐negative BC (OR = 1.3); a similar pattern was observed for PR status (Table 3). PALB2 variants were also associated with a higher BC risk point estimate for ER‐positive (OR = 4.6) compared to ER‐negative BC (OR = 1.1). MAST1 MV were associated with a higher point estimate for ER‐negative (OR = 4.3) compared to ER‐positive BC (OR = 2.1).
Table 3.
Analyses by hormone receptor status and restricted to pseudo‐incident cases, for the 7 genes associated with BC (P ≤ 0.05)
| Any variant | LoF | Likely deleterious MV | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene | Analysis1 | Case carriers | OR2 (95%CI) | p‐Value | Case carriers | OR2 (95%CI) | p‐Value | Case carriers | OR2 (95%CI) | p‐Value |
| ATM | ER positive | 47 | 2.1 (1.4, 3.3) | 0.001 | 9 | 17.7 (2.2, 140) | 0.006 | 38 | 1.7 (1.1, 2.8) | 0.02 |
| ER negative | 8 | 2.1 (0.9, 4.5) | 0.07 | 2 | 20.8 (1.9, 232) | 0.01 | 6 | 1.6 (0.7, 3.8) | 0.31 | |
| PR positive | 38 | 2.1 (1.3, 3.4) | 0.001 | 9 | 22.0 (2.8, 174) | 0.003 | 29 | 1.6 (1.0, 2.7) | 0.06 | |
| PR negative | 12 | 1.7 (0.9, 3.4) | 0.10 | 2 | 10.9 (1.0, 120) | 0.05 | 10 | 1.5 (0.7, 3.0) | 0.27 | |
| HER2 positive | 3 | 1.3 (0.4, 4.2) | 0.70 | 0 | ‐ | ‐ | 3 | 1.3 (0.4, 4.3) | 0.68 | |
| HER2 negative | 24 | 1.8 (1.1, 3.1) | 0.02 | 6 | 19.2 (2.3, 160) | 0.006 | 18 | 1.4 (0.8, 2.5) | 0.27 | |
| Pseudo‐incident cases3 | 41 | 1.9 (1.2, 2.9) | 0.006 | 8 | 15.6 (1.9, 125) | 0.01 | 33 | 1.5 (1.0, 2.5) | 0.08 | |
| CHEK2 | ER positive | 39 | 3.5 (2.1, 6.0) | 0.000004 | 16 | 7.9 (2.6, 23.9) | 0.0003 | 23 | 2.5 (1.3, 4.7) | 0.004 |
| ER negative | 3 | 1.3 (0.4, 4.5) | 0.64 | 1 | 2.5 (0.3, 22.2) | 0.42 | 2 | 1.1 (0.2, 4.8) | 0.91 | |
| PR positive | 35 | 3.9 (2.2, 6.7) | 0.000001 | 17 | 10.5 (3.5, 31.3) | 0.00003 | 18 | 2.4 (1.2, 4.7) | 0.01 | |
| PR negative | 6 | 1.6 (0.7, 4.1) | 0.29 | 0 | ‐ | ‐ | 6 | 2.0 (0.8, 5.2) | 0.14 | |
| HER2 positive | 5 | 3.8 (1.4, 10.4) | 0.009 | 1 | 4.3 (0.5, 39.1) | 0.2 | 4 | 3.7 (1.2, 11.3) | 0.02 | |
| HER2 negative | 27 | 4.1 (2.3, 7.3) | 0.000002 | 11 | 9.0 (2.8, 28.4) | 0.0002 | 16 | 3.0 (1.5, 6.0) | 0.002 | |
| Pseudo‐incident cases3 | 37 | 3.3 (1.9, 5.6) | 0.00001 | 12 | 5.9 (1.9, 18.4) | 0.002 | 25 | 2.7 (1.5, 5.0) | 0.001 | |
| FANCI | ER positive | 9 | 0.7 (0.3, 1.6) | 0.44 | 3 | 4.7 (0.5, 46.3) | 0.19 | 6 | 0.5 (0.2, 1.3) | 0.17 |
| ER negative | 0 | ‐ | ‐ | 0 | ‐ | ‐ | 0 | ‐ | ‐ | |
| PR positive | 8 | 0.9 (0.4, 1.9) | 0.70 | 2 | 4.7 (0.4, 52.0) | 0.21 | 6 | 0.7 (0.3, 1.7) | 0.39 | |
| PR negative | 1 | 0.2 (0.0, 1.8) | 0.17 | 1 | 4.3 (0.2, 79.5) | 0.33 | 0 | ‐ | ‐ | |
| HER2 positive | 1 | 0.7 (0.1, 5.4) | 0.75 | 1 | 15.1 (0.7, 319) | 0.09 | 0 | ‐ | ‐ | |
| HER2 negative | 3 | 0.4 (0.1, 1.4) | 0.20 | 0 | ‐ | ‐ | 3 | 0.4 (0.1, 1.5) | 0.19 | |
| Pseudo‐incident cases3 | 6 | 0.5 (0.2, 1.2) | 0.11 | 2 | 2.8 (0.2, 32.8) | 0.42 | 4 | 0.4 (0.1, 1.0) | 0.06 | |
| MAST1 | ER positive | 8 | 1.8 (0.7, 5.0) | 0.23 | 0 | ‐ | ‐ | 8 | 2.1 (0.8, 5.9) | 0.15 |
| ER negative | 3 | 3.8 (1.0, 14.4) | 0.05 | 0 | ‐ | ‐ | 3 | 4.3 (1.1, 16.8) | 0.04 | |
| PR positive | 5 | 1.2 (0.4, 3.9) | 0.79 | 0 | ‐ | ‐ | 5 | 1.7 (0.5, 5.3) | 0.38 | |
| PR negative | 4 | 3.0 (0.9, 10.0) | 0.08 | 0 | ‐ | ‐ | 4 | 3.4 (1.0, 11.9) | 0.05 | |
| HER2 positive | 2 | 4.2 (0.9, 20.0) | 0.08 | 0 | ‐ | ‐ | 2 | 4.7 (1.0, 23.1) | 0.06 | |
| HER2 negative | 2 | 0.8 (0.2, 3.7) | 0.75 | 0 | ‐ | ‐ | 2 | 0.9 (0.2, 4.3) | 0.89 | |
| Pseudo‐incident cases3 | 8 | 1.9 (0.7, 5.0) | 0.20 | 0 | ‐ | ‐ | 8 | 2.1 (0.8, 6.0) | 0.14 | |
| PALB2 | ER positive | 21 | 4.6 (2.1, 10.1) | 0.0002 | 8 | 5.4 (1.4, 20.4) | 0.01 | 13 | 4.2 (1.6, 11.1) | 0.004 |
| ER negative | 1 | 1.1 (0.1, 8.8) | 0.93 | 0 | ‐ | ‐ | 1 | 1.6 (0.2, 13.7) | 0.65 | |
| PR positive | 14 | 3.7 (1.6, 8.7) | 0.002 | 3 | 2.4 (0.5, 11.9) | 0.29 | 11 | 4.4 (1.6, 11.9) | 0.004 | |
| PR negative | 7 | 4.9 (1.8, 13.4) | 0.002 | 5 | 11.1 (2.6, 47.4) | 0.001 | 2 | 2.0 (0.4, 10.0) | 0.40 | |
| HER2 positive | 0 | ‐ | ‐ | 0 | ‐ | ‐ | 0 | ‐ | ‐ | |
| HER2 negative | 13 | 4.7 (2.0, 11.1) | 0.0004 | 3 | 3.4 (0.7, 16.7) | 0.14 | 10 | 5.3 (1.9, 14.8) | 0.001 | |
| Pseudo‐incident cases3 | 15 | 3.2 (1.4, 7.3) | 0.007 | 5 | 3.2 (0.8, 13.6) | 0.11 | 10 | 3.1 (1.1, 8.7) | 0.03 | |
| POLH | ER positive | 4 | 0.6 (0.2, 1.8) | 0.30 | 1 | ‐ | ‐ | 3 | 0.4 (0.1, 1.5) | 0.19 |
| ER negative | 0 | ‐ | ‐ | 0 | ‐ | ‐ | 0 | ‐ | ‐ | |
| PR positive | 4 | 0.7 (0.2, 2.2) | 0.60 | 1 | ‐ | ‐ | 3 | 0.5 (0.2, 1.9) | 0.34 | |
| PR negative | 0 | ‐ | ‐ | 0 | ‐ | ‐ | 0 | ‐ | ‐ | |
| HER2 positive | 0 | ‐ | ‐ | 0 | ‐ | ‐ | 0 | ‐ | ‐ | |
| HER2 negative | 1 | 0.2 (0.0, 1.8) | 0.20 | 1 | ‐ | ‐ | 0 | ‐ | ‐ | |
| Pseudo‐incident cases3 | 1 | 0.1 (0.0, 1.1) | 0.06 | 0 | ‐ | ‐ | 1 | 0.1 (0.0, 1.1) | 0.06 | |
| RTEL1 | ER positive | 5 | 0.5 (0.2, 1.2) | 0.12 | 0 | ‐ | ‐ | 5 | 0.5 (0.2, 1.2) | 0.12 |
| ER negative | 0 | ‐ | ‐ | 0 | ‐ | ‐ | 0 | ‐ | ‐ | |
| PR positive | 5 | 0.6 (0.2, 1.5) | 0.30 | 0 | ‐ | ‐ | 5 | 0.6 (0.2, 1.5) | 0.27 | |
| PR negative | 0 | ‐ | ‐ | 0 | ‐ | ‐ | 0 | ‐ | ‐ | |
| HER2 positive | 2 | 1.4 (0.3, 6.6) | 0.61 | 0 | ‐ | ‐ | 2 | 1.5 (0.3, 6.6) | 0.61 | |
| HER2 negative | 2 | 0.3 (0.1, 1.3) | 0.10 | 0 | ‐ | ‐ | 2 | 0.3 (0.1, 1.3) | 0.10 | |
| Pseudo‐incident cases3 | 7 | 0.6 (0.3, 1.5) | 0.30 | 0 | ‐ | ‐ | 7 | 0.6 (0.3, 1.5) | 0.31 | |
Abbreviations: ER, estrogen receptor; PR, progesterone receptor; HER2, HER2 gene amplification.
ER, PR and HER2 status were available for 788 (67%), 742 (63%), and 475 (40%) of sequenced GENESIS cases.
Reference group: noncarriers of a variant in the tested gene; adjusting for ethnicity and age at inclusion.
Cases diagnosed with BC less than 5 years before enrollment in GENESIS (N = 663).
Sensitivity analyses
Because GENESIS cases are prevalent cases, i.e. diagnosed with BC sometimes several years before being invited to participate in the study, cases carrying a pathogenic variant in a gene associated with a poor prognosis would have had less chance to be included and would be therefore under‐represented. To examine a survival bias, we excluded cases with a delay between diagnosis of BC and inclusion in GENESIS of greater than 5 years and performed analyses on pseudo‐incident cases (N = 663). There was no significant heterogeneity in BC risk according to time between diagnosis and inclusion for ATM, CHEK2, PALB2, FANCI, MAST1 and POLH (Table 3). The observed decreased BC risk associated with carrying a MV in RTEL1 seemed different, although not statistically significant, between cases diagnosed more than 5 years before inclusion and cases diagnosed less than 5 years before inclusion (OR>5years = 0.1 vs. OR≤5years = 0.6; p Het = 0.09). Therefore, we cannot conclude here that RTEL1 variant carriers have a poorer survival, but more studies are warranted to clarify this.
There was no change in BC risk estimates when adjusting for the number of self‐reported cancer cases in the family (BC, ovarian cancer or cancers from the Lynch syndrome spectrum; data not shown) or for the number of altered genes (Table 2). Finally, results by hormone receptor status were unchanged when restricting the analyses to pseudo‐incident cases (data not shown).
Discussion
By analyzing 113 DNA repair genes selected from either a WES or a candidate gene approach in familial BC cases and a well‐identified control group, we confirmed associations between variants predicted to impair protein function in PALB2, ATM and CHEK2 and occurrence of BC. We found that the risk associated with ATM variants can greatly vary according to variant type (ORATM‐LoF~ 10 x ORATM‐MV). The risk conferred by LoF would still significantly differ from the risk conferred by MV in this gene even if the number of ATM LoF carriers was four times higher in controls. There were no ATM LoF that were observed more than once in cases, and thus none could contribute disproportionately to the risk estimate (Supporting Information Table S3).
We also identified four genes (FANCI, MAST1, POLH and RTEL1) that had not been previously associated with BC susceptibility. Should the association with BC be confirmed in larger studies, the decreased BC risk associated with MV in FANCI, POLH and RTEL1 is puzzling. Because of a difference in BC risk between cases included more than 5 years after BC diagnosis and those included less than 5 years after BC diagnosis, we concluded that RTEL1 alterations might be associated with a poor prognosis. The same hypothesis was tested for carriers of a variant in FANCI or POLH and results were unchanged. Another possible explanation could be that FANCI and POLH are associated with another syndrome than the HBOC syndrome. Therefore due to recruitment criteria in GENESIS, carriers of a variant in one of these genes would be under‐represented among the cases of our study.
Apart from FANCI, which we included in our panel because it belongs to the same Fanconi Anemia gene family as BRCA1/FANCS, BRCA2/FANCD1 and PALB2/FANCN, we investigated MAST1, POLH and RTEL1 because they had been highlighted in the WES exploratory stage, as were the known BC genes ATM and CHEK2. This demonstrates the efficiency of our approach to prioritize new candidate genes. Another strength of our study is that the full coding sequence of the genes was screened in all subjects using the same sequencing techniques and platforms, thus avoiding biases that may arise when only the variants identified in cases are tested in controls. Moreover, unlike many other studies, controls were specifically recruited for the GENESIS study. They were cancer‐free friends or colleagues belonging to the same birth cohort as the cases, and were therefore naturally matched on many unmeasured potential confounders.
The literature on MAST1, POLH and RTEL1 has indicated relationships between these genes and carcinogenesis. Putative germline pathogenic variants in MAST1, encoding a microtubule‐associated serine–threonine kinase, were described in familial lung cancer cases.34 Transcriptome analysis of BC cell lines and tissue identified somatic recurrent rearrangements involving MAST1 and over‐expression of the resulting gene fusion.35 Inactivating variants of POLH, which encodes the translesion DNA polymerase η are responsible for Xeroderma Pigmentosum, a rare recessive syndrome associated with hypersensitivity to sunlight and a high frequency of skin cancers at an early age.36, 37 Of note, a germline POLH MV (c.2074A>G) was identified by WES in a Lebanese BC‐prone family38 but this variant, predicted to be benign by various tools, was not found in our population. RTEL1 encodes the regulator of telomere elongation helicase 1, which is essential for telomere maintenance and regulation of homologous recombination. Germline biallelic RTEL1 variants are responsible for dyskeratosis congenita and its severe variant Hoyeraal‐Hreidarsson syndrome,39 while heterozygous carriers with pulmonary fibrosis,40, 41, 42 myelodysplasia and liver disease have been described.43 Thus, our findings on FANCI, MAST1, POLH and RTEL1 would warrant further investigation in other large‐scale studies on familial BC and the setup of functional studies to assess the impact of the variants.
There was some discrepancy between our findings on ATM in GENESIS and previous findings from a pooled analysis of seven case–control studies that included the Breast Cancer Family Registry (BCFR) study9 in terms of risk estimates. In these latter analyses, we had found that carriers of a likely deleterious MV may be at higher risk of BC than LoF carriers.9 One explanation could be the difference in the tool predicting the impact of the MV. We therefore reanalyzed GENESIS data for MV in ATM, CHEK2 and PALB2 using Align‐GVGD33 which was used in the BCFR9, 44 and other studies.45, 46 Results obtained with Align‐GVGD were consistent with those obtained with CADD (Table 4). Another explanation could be the difference in the ages at diagnosis between the studies. In GENESIS, carriers of an ATM LoF or MV were diagnosed on average at age 50.0 (SD = 2.6 years) and 50.9 (SD = 1.2 years), respectively, while most of the cases in the pooled analysis were diagnosed before age 46. Even if in GENESIS carrying an ATM MV was associated with a higher BC risk point estimate in early‐onset cases (OR≤45years = 2.2 vs. OR>45years = 1.2, p Het = 0.06), the risk estimate associated with MV remained much lower than the risk estimate associated with LoF. No difference in BC risk was observed for LoF when stratifying by age (OR≤45years = 18.6 vs. OR>45years = 17.9, p Het = 0.95). Of note, the MV c.7271T>G (p.Val2424Gly), which had been associated with a substantial BC risk in several studies,47, 48, 49 and in particular in the Australian population, was not found in GENESIS cases and was found only once in controls. Moreover, there was no evidence that the observed association was driven by a specific variant (either a LoF or a MV) in our study set (Supporting Information Table S3). Conversely, the CHEK2 LoF c.1100delC was quite prevalent in the French population (Supporting Information Table S3). However, when excluding carriers of this variant in the analysis we found that other CHEK2 variants (both LoF and MV) contribute significantly to BC risk (OR = 2.5; 95%CI, 1.5–4.4; p = 0.0004).
Table 4.
Distribution of MV and results of the association tests for ATM, CHEK2 and PALB2 using Align‐GVGD classifiers (Tavtigian et al.33)
| Gene | Variant class | Controls | Cases | OR1 (95%CI) | p‐Value |
|---|---|---|---|---|---|
| ATM | Noncarriers | 1,129 | 1,102 | Ref. | |
| C0 | 43 | 47 | 1.1 (0.7, 1.6) | 0.75 | |
| C15‐C65 | 26 | 42 | 1.7 (1.0, 2.7) | 0.05 | |
| CHEK2 | Noncarriers | 1,174 | 1,137 | Ref. | |
| C0 | 7 | 23 | 3.4 (1.5, 8.0) | 0.005 | |
| C15‐C65 | 14 | 26 | 2.0 (1.1, 3.9) | 0.03 | |
| PALB2 | Noncarriers | 1,181 | 1,168 | Ref. | |
| C0 | 10 | 14 | 1.3 (0.6, 3.0) | 0.51 | |
| C15‐C65 | 5 | 15 | 3.3 (1.2, 9.0) | 0.02 |
Reference group: noncarriers of a variant in the tested gene; adjusting for ethnicity and age at inclusion.
There was also a noteworthy difference in PALB2 BC relative risk estimates when compared to those estimated by Antoniou et al.8 for carriers of a LoF. This may be explained by the difference in populations under study. Indeed, Antoniou et al. estimated BC risk from highly selected PALB2 families with uncertainty in modes of ascertainment. Although analyses were performed using modeling to reduce bias, their PALB2 families may have aggregated many other unmeasured genetic and nongenetic BC risk factors. Another notable finding in our study is the relatively high OR associated with MV in PALB2, which have not been systematically assessed in previous reports. Interestingly, the PALB2 MV c.2816T>G (p.Leu939Trp) was quite frequent in our study population: half of the PALB2 MV carriers carried this variant (12 out of 20 carriers in cases, and 3 out of 6 in controls; ORc.2816T>G = 4.3; 95%CI, 1.2–15.4; p = 0.02). To understand the origin of its high prevalence, which could explain our ability to detect its effect, we performed a haplotype analysis of the 16p12.2 locus containing PALB2 on GENESIS carriers of the c.2816T>G variant using MERLIN.50 In a companion study, genotypes of 38 SNPs localized +/−500 kb around the variant were obtained with the iCOGS array in the GENESIS population (authors’ unpublished data). This latter analysis suggests that the c.2816T>G variant could be a founder allele in the French BC cases (data not shown). A first study showed that this variant falls in the PALB2 WD40 domain and results in altered PALB2‐BRCA2 binding.51 However, it was then demonstrated that it does not disrupt the HR‐mediated DNA repair activity of PALB2.52 Two case–control studies conducted in the German population53 and in the British population6 on over 800 and 923 familial BC cases, respectively, showed that the c.2816T>G variant was not associated with BC. More recently, a multicenter case–control study conducted by the Breast Cancer Association Consortium (BCAC) on 42,671 invasive BC and 42,164 controls did not show any association with this variant (ORc.2816T>G = 1.05; 95%CI 0.83–1.32),49 leading to the conclusion that the PALB2 c.2816T>G variant should be classified as a likely neutral variant. However, even if we cannot rule out a false‐positive result in our study, one asset of the GENESIS population is to be homogeneous, in contrast with the countries heterogeneity in BCAC. Thus, further investigation on this variant may be worthwhile. Nonetheless, the risk estimates associated with PALB2 MVs was not driven by this peculiar variant (after excluding carriers of c.2816T>G, ORMV = 3.1; 95%CI, 1.2–7.9; p = 0.02).
With respect with the confirmed BC genes, deleterious variants in ATM, CHEK2 and PALB2 are present in 14% of GENESIS BC cases, of whom 1% carry a LoF in the actionable gene PALB2 and another 1% carry a PALB2 MV that would, for the time being, probably be considered as a variant of uncertain clinical significance in a cancer clinics setting. To further evaluate the potential clinical significance of variants considered in our case–control analysis, we determined how many of the 264 LoF and 1,994 likely deleterious MV identified in the 113 genes were present in ClinVar.54, 55 Only 78 (3.5%) were reported as “pathogenic” or “likely pathogenic” variants (Supporting Information Fig. S3). They were located in 30 genes including PALB2, TP53, CDH1, RAD51C, RAD51D, MLH1, MSH2 and PMS2 for which specific management guidelines have been elaborated in France in some specific cancer predisposition contexts (http://www.unicancer.fr/recherche/les‐groupes‐recherche/groupe‐genetique‐et‐cancer‐ggc). This illustrates that although ClinVar is a useful tool for variant interpretations, the majority of variants that will be identified by multigene panel testing will be unreported, and efforts to facilitate sharing of data, in particular observation in unaffected populations, together with methods for variant assessment and expertise in specific disease areas are needed.
We chose to investigate each gene independently in order to be able to compare our relative risk estimates with those of other published data. However, we also performed the analyses adjusting for the number of altered genes, and no significant change in relative risk estimates was observed (Table 2). Nevertheless, one cannot exclude that there is a residual quantitative effect shift of number of altered genes in cases as compared to controls (Supporting Information Fig. S2). A potential polygenic effect might be further investigated in larger studies where a larger number of genes can be tested.
In conclusion, this case–control study conducted in a well‐characterized population allowed us to describe genetic variation in 113 DNA repair genes and to specify BC relative risks associated with rare deleterious‐predicted variants in PALB2, ATM and CHEK2. More generally, this work highlights the importance of considering all types of variants in the analyses, as their contribution might differ from one gene to another. Further family‐based studies and gene‐specific prospective cohorts are needed to assess absolute risk for carriers of a deleterious variant by taking into account other factors such as family history of cancer, polygenic models or even environmental/lifestyle factors. They will be useful to improve risk prediction models and will help defining gene‐specific consensus management guidelines.
Authors’ contribution
Conceived and designed the experiments: RO, FD, AB, JFD, FL. Performed the experiments: LB, NM, ALR. Analyzed the data: EG, ALR, NA, FL. Contributed reagents/materials/analysis tools: MGD, DG, VM, CL, JC, NS. Coordinated the GENESIS study: SM, DSL, NA. Included GENESIS participants, collected data: SEM, DLG, JB, NM, MM, EC. Invited GENESIS index cases: OCH, DL, CP, SFF, CC, TF, FE, AC, CA, AF, LG, JT, AF, JC, SG, IM, FS, SAB, JML, CL, SLD, HD, YJB, ML, PP, LVB, VB, PB, EL, CM, CN, CD, JPF, PG, LF, AL, BB, OC, MGV, IC, DSL. Wrote the paper: EG, NA, FL. All authors read and approved the final version of the study.
Supporting information
Supplementary Table S1. Whole‐exome sequencing performance and variants count.
Supplementary Table S2. Distribution of rare variants and result of the association tests for the 113 genes when pooling LoF and MV variants together in the analysis, and per variant type.
Supplementary Table S3. Loss‐of‐function variants and likely deleterious missense variants identified in ATM, CHEK2, PALB2, FANCI, MAST1, POLH and RTEL1 in the case–control study.
Supplementary Figure S1. Coverage summary across the 100 exomes.
Supplementary Figure S2. Distribution of the number of altered genes per subject, according to case/control status.
Supplementary Figure S3. Frequency of pathogenic variants by gene, according to ClinVar.
Acknowledgements
We wish to thank all the families who so willingly participated in the GENESIS study.
This article is dedicated to the memory of Olga M. Sinilnikova who died prematurely on June 30, 2014. Olga participated decisively in structuring research around hereditary predisposition to BC and in leading the GENESIS study with Nadine Andrieu and Dominique Stoppa‐Lyonnet. She also contributed to the design of the GENESIS‐NGS project.
We thank all the GENESIS collaborating cancer clinics (Clinique Sainte Catherine, Avignon: H. Dreyfus; Hôpital Saint Jacques, Besançon: M‐A. Collonge‐Rame; Institut Bergonié, Bordeaux: M. Longy, A. Floquet, E. Barouk‐Simonet; CHU, Brest: S. Audebert; Centre François Baclesse, Caen: P. Berthet; Hôpital Dieu, Chambéry: S. Fert‐Ferrer; Centre Jean Perrin, Clermont‐Ferrand: Y‐J. Bignon; Hôpital Pasteur, Colmar: J‐M. Limacher; Hôpital d'Enfants CHU – Centre Georges François Leclerc, Dijon: L. Faivre; CHU, Fort de France: O. Bera; CHU Albert Michallon, Grenoble: D. Leroux; Hôpital Flaubert, Le Havre: V. Layet; Centre Oscar Lambret, Lille: P. Vennin (deceased prematurely), C. Adenis; Hôpital Jeanne de Flandre, Lille: S. Lejeune‐Dumoulin, S. Manouvier‐Hanu; CHRU Dupuytren, Limoges: L. Venat‐Bouvet; Centre Léon Bérard, Lyon: C. Lasset, V. Bonadona; Hôpital Edouard Herriot, Lyon: S. Giraud; Institut Paoli‐Calmettes, Marseille: F. Eisinger, L. Huiart; Centre Val d'Aurelle – Paul Lamarque, Montpellier: I. Coupier; CHU Arnaud de Villeneuve, Montpellier: I. Coupier, P. Pujol; Centre René Gauducheau, Nantes: C. Delnatte; Centre Catherine de Sienne, Nantes: A. Lortholary; Centre Antoine Lacassagne, Nice: M. Frénay, V. Mari; Hôpital Caremeau, Nîmes: J. Chiesa; Réseau Oncogénétique Poitou Charente, Niort: P. Gesta; Institut Curie, Paris: D. Stoppa‐Lyonnet, M. Gauthier‐Villars, B. Buecher, A. de Pauw, C. Abadie, M. Belotti; Hôpital Saint‐Louis, Paris: O. Cohen‐Haguenauer; Centre Viggo‐Petersen, Paris: F. Cornélis; Hôpital Tenon, Paris: A. Fajac; GH Pitié Salpétrière et Hôpital Beaujon, Paris: C. Colas, F. Soubrier, P. Hammel, A. Fajac; Institut Jean Godinot, Reims: C. Penet, T. D. Nguyen; Polyclinique Courlancy, Reims: L. Demange (deceased prematurely), C. Penet; Centre Eugène Marquis, Rennes: C. Dugast; Centre Henri Becquerel, Rouen: A. Chevrier, T. Frebourg, J. Tinat, I. Tennevet, A. Rossi; Hôpital René Huguenin/Institut Curie, Saint Cloud: C. Noguès, L. Demange (deceased prematurely), E. Mouret‐Fourme; CHU, Saint‐Etienne: F. Prieur; Centre Paul Strauss, Strasbourg: J‐P. Fricker, H. Schuster; Hôpital Civil, Strasbourg: O. Caron, C. Maugard; Institut Claudius Regaud, Toulouse: L. Gladieff, V. Feillel; Hôpital Bretonneau, Tours: I. Mortemousque; Centre Alexis Vautrin, Vandoeuvre‐les‐Nancy: E. Luporsi; Hôpital de Bravois, Vandoeuvre‐les‐Nancy: P. Jonveaux; Gustave Roussy, Villejuif: A. Chompret (deceased prematurely), O. Caron).
Conflict interest: All authors, except for DG, FE and OC declare no conflict of interest. DG is employed at Agilent Technologies Inc. FE is a consultant for ROCHE S.A., Genome Quebec and the International Biobank of Luxembourg. OC received honoraria for AstraZeneca and Ipsen.
References
- 1. Collaborative Group on Hormonal Factors in Breast C . Breast cancer and hormonal contraceptives: collaborative reanalysis of individual data on 53 297 women with breast cancer and 100 239 women without breast cancer from 54 epidemiological studies. Lancet 1996;347:1713–27. [DOI] [PubMed] [Google Scholar]
- 2. Antoniou AC, Cunningham AP, Peto J, et al. The BOADICEA model of genetic susceptibility to breast and ovarian cancers: updates and extensions. Br J Cancer 2008;98:1457–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Gadzicki D, Evans DG, Harris H, et al. Genetic testing for familial/hereditary breast cancer‐comparison of guidelines and recommendations from the UK, France, The Netherlands and Germany. J Community Genet 2011;2:53–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Thompson ER, Rowley SM, Li N, et al. Panel testing for familial breast cancer: calibrating the tension between research and clinical care. J Clin Oncol 2016;34:1455–9. [DOI] [PubMed] [Google Scholar]
- 5. Southey MC, Teo ZL, Dowty JG, et al. A PALB2 mutation associated with high risk of breast cancer. Breast Cancer Res 2010;12:R109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rahman N, Seal S, Thompson D, et al. PALB2, which encodes a BRCA2‐interacting protein, is a breast cancer susceptibility gene. Nat Genet 2007;39:165–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Casadei S, Norquist BM, Walsh T, et al. Contribution of inherited mutations in the BRCA2‐interacting protein PALB2 to familial breast cancer. Cancer Res 2011;71:2222–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Antoniou AC, Casadei S, Heikkinen T, et al. Breast‐cancer risk in families with mutations in PALB2. N Engl J Med 2014;371:497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Tavtigian SV, Oefner PJ, Babikyan D, et al. Rare, evolutionarily unlikely missense substitutions in ATM confer increased risk of breast cancer. Am J Hum Genet 2009;85:427–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Renwick A, Thompson D, Seal S, et al. ATM mutations that cause ataxia‐telangiectasia are breast cancer susceptibility alleles. Nat Genet 2006;38:873–5. [DOI] [PubMed] [Google Scholar]
- 11. Le Calvez‐Kelm F, Lesueur F, Damiola F, et al. Rare, evolutionarily unlikely missense substitutions in CHEK2 contribute to breast cancer susceptibility: results from a breast cancer family registry case‐control mutation‐screening study. Breast Cancer Res 2011;13:R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Meindl A, Hellebrand H, Wiek C, et al. Germline mutations in breast and ovarian cancer pedigrees establish RAD51C as a human cancer susceptibility gene. Nat Genet 2010;42:410–4. [DOI] [PubMed] [Google Scholar]
- 13. Golmard L, Castera L, Krieger S, et al. Contribution of germline deleterious variants in the RAD51 paralogs to breast and ovarian cancers. Eur J Hum Genet 2017;25:1345–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Park DJ, Lesueur F, Nguyen‐Dumont T, et al. Rare mutations in XRCC2 increase the risk of breast cancer. Am J Hum Genet 2012;90:734–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Park DJ, Tao K, Le Calvez‐Kelm F, et al. Rare mutations in RINT1 predispose carriers to breast and lynch syndrome‐spectrum cancers. Cancer Discov 2014;4:804–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Damiola F, Pertesi M, Oliver J, et al. Rare key functional domain missense substitutions in MRE11A, RAD50, and NBN contribute to breast cancer susceptibility: results from a breast cancer family registry case‐control mutation‐screening study. Breast Cancer Res 2014;16:R58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hilbers FS, Wijnen JT, Hoogerbrugge N, et al. Rare variants in XRCC2 as breast cancer susceptibility alleles. J Med Genet 2012;49:618–20. [DOI] [PubMed] [Google Scholar]
- 18. Li N, Thompson ER, Rowley SM, et al. Reevaluation of RINT1 as a breast cancer predisposition gene. Breast Cancer Res Treat 2016;159:385–92. [DOI] [PubMed] [Google Scholar]
- 19. Easton DF, Pharoah PD, Antoniou AC, et al. Gene‐panel sequencing and the prediction of breast‐cancer risk. N Engl J Med 2015;372:2243–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ognjanovic S, Olivier M, Bergemann TL, et al. Sarcomas in TP53 germline mutation carriers: a review of the IARC TP53 database. Cancer 2012;118:1387–96. [DOI] [PubMed] [Google Scholar]
- 21. Tavtigian SV, Chenevix‐Trench G. Growing recognition of the role for rare missense substitutions in breast cancer susceptibility. Biomark Med 2014;8:589–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sinilnikova OM, Dondon MG, Eon‐Marchais S, et al. GENESIS: a French national resource to study the missing heritability of breast cancer. BMC Cancer 2016;16:13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Grimberg J, Nawoschik S, Belluscio L, et al. A simple and efficient non‐organic procedure for the isolation of genomic DNA from blood. Nucleic Acids Res 1989;17:8390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Li H, Durbin R. Fast and accurate long‐read alignment with burrows‐wheeler transform. Bioinformatics 2010;26:589–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garrison EM. G. Haplotype‐based variant detection from short‐read sequencing. ar Xiv. preprint . ArXiv 2012;1207:3907. [Google Scholar]
- 26. McKenna A, Hanna M, Banks E, et al. The genome analysis toolkit: a map reduce framework for analyzing next‐generation DNA sequencing data. Genome Res 2010;20:1297–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Robinson JT, Thorvaldsdottir H, Winckler W, et al. Integrative genomics viewer. Nat Biotechnol 2011;29:24–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative genomics viewer (IGV): high‐performance genomics data visualization and exploration. Brief Bioinform 2013;14:178–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Pollard KS, Hubisz MJ, Rosenbloom KR, et al. Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res 2010;20:110–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ng PC, Henikoff S. Predicting deleterious amino acid substitutions. Genome Res 2001;11:863–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Adzhubei I, Jordan DM. Sunyaev SR. predicting functional effect of human missense mutations using poly Phen‐2. Curr Protoc Hum Genet . Chapter 7: Unit 2013;7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Kircher M, Witten DM, Jain P, et al. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet 2014;46:310–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Tavtigian SV, Byrnes GB, Goldgar DE, et al. Classification of rare missense substitutions, using risk surfaces, with genetic‐ and molecular‐epidemiology applications. Hum Mutat 2008;29:1342–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tomoshige K, Matsumoto K, Tsuchiya T, et al. Germline mutations causing familial lung cancer. J Hum Genet 2015;60:597–603. [DOI] [PubMed] [Google Scholar]
- 35. Robinson DR, Kalyana‐Sundaram S, Wu YM, et al. Functionally recurrent rearrangements of the MAST kinase and notch gene families in breast cancer. Nat Med 2011;17:1646–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gratchev A, Strein P, Utikal J, et al. Molecular genetics of Xeroderma pigmentosum variant. Exp Dermatol 2003;12:529–36. [DOI] [PubMed] [Google Scholar]
- 37. De Palma A, Morren MA, Ged C, et al. Diagnosis of Xeroderma pigmentosum variant in a young patient with two novel mutations in the POLH gene. Am J Med Genet A 2017;173:2511–6. [DOI] [PubMed] [Google Scholar]
- 38. Jalkh N, Chouery E, Haidar Z, et al. Next‐generation sequencing in familial breast cancer patients from Lebanon. BMC Med Genomics 2017;10:8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Speckmann C, Sahoo SS, Rizzi M, et al. Clinical and molecular heterogeneity of RTEL1 deficiency. Front Immunol 2017;8:449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kannengiesser C, Borie R, Menard C, et al. Heterozygous RTEL1 mutations are associated with familial pulmonary fibrosis. Eur Respir J 2015;46:474–85. [DOI] [PubMed] [Google Scholar]
- 41. Cogan JD, Kropski JA, Zhao M, et al. Rare variants in RTEL1 are associated with familial interstitial pneumonia. Am J Respir Crit Care Med 2015;191:646–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stuart BD, Choi J, Zaidi S, et al. Exome sequencing links mutations in PARN and RTEL1 with familial pulmonary fibrosis and telomere shortening. Nat Genet 2015;47:512–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Cardoso SR, Ellison ACM, Walne AJ, et al. Myelodysplasia and liver disease extend the spectrum of RTEL1 related telomeropathies. Haematologica 2017;102:e293–e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Le Calvez‐Kelm F, Lesueur F, Damiola F, et al. Rare, evolutionarily unlikely missense substitutions in CHEK2 contribute to breast cancer susceptibility: results from a breast cancer family registry (CFR) case‐control mutation screening study. Breast Cancer Res 2011;13:R6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tischkowitz M, Capanu M, Sabbaghian N, et al. Rare germline mutations in PALB2 and breast cancer risk: a population‐based study. Hum Mutat 2012;33:674–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Teo ZL, Park DJ, Provenzano E, et al. kConFab, Dowty JG, hopper JL, Winship I, Goldgar DE, Southey MC. Prevalence of PALB2 mutations in Australasian multiple‐case breast cancer families. Breast Cancer Res 2013;15:R17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bernstein JL, Teraoka S, Southey MC, et al. Population‐based estimates of breast cancer risks associated with ATM gene variants c.7271T>G and c. 1066‐6T>G (IVS10‐6T>G) from the breast cancer family registry. Hum Mutat 2006;27:1122–8. [DOI] [PubMed] [Google Scholar]
- 48. Goldgar DE, Healey S, Dowty JG, et al. Rare variants in the ATM gene and risk of breast cancer. Breast Cancer Res: BCR 2011;13:R73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Southey MC, Goldgar DE, Winqvist R, et al. PALB2, CHEK2 and ATM rare variants and cancer risk: data from COGS. J Med Genet 2016;53:800–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Abecasis GR, Cherny SS, Cookson WO, et al. Merlin‐‐rapid analysis of dense genetic maps using sparse gene flow trees. Nat Genet 2002;30:97–101. [DOI] [PubMed] [Google Scholar]
- 51. Park JY, Singh TR, Nassar N, et al. Breast cancer‐associated missense mutants of the PALB2 WD40 domain, which directly binds RAD51C, RAD51 and BRCA2, disrupt DNA repair. Oncogene 2014;33:4803–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Catucci I, Radice P, Milne RL, et al. The PALB2 p.Leu939Trp mutation is not associated with breast cancer risk. Breast Cancer Res 2016;18:111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Hellebrand H, Sutter C, Honisch E, et al. Germline mutations in the PALB2 gene are population specific and occur with low frequencies in familial breast cancer. Hum Mutat 2011;32:E2176–88. [DOI] [PubMed] [Google Scholar]
- 54. Landrum MJ, Lee JM, Riley GR, et al. ClinVar: public archive of relationships among sequence variation and human phenotype. Nucleic Acids Res 2014;42:D980–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Harrison SM, Riggs ER, Maglott DR, et al. Using ClinVar as a resource to support variant interpretation. Curr Protoc Hum Genet 2016;89:8.16. 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table S1. Whole‐exome sequencing performance and variants count.
Supplementary Table S2. Distribution of rare variants and result of the association tests for the 113 genes when pooling LoF and MV variants together in the analysis, and per variant type.
Supplementary Table S3. Loss‐of‐function variants and likely deleterious missense variants identified in ATM, CHEK2, PALB2, FANCI, MAST1, POLH and RTEL1 in the case–control study.
Supplementary Figure S1. Coverage summary across the 100 exomes.
Supplementary Figure S2. Distribution of the number of altered genes per subject, according to case/control status.
Supplementary Figure S3. Frequency of pathogenic variants by gene, according to ClinVar.