

Abstract
Caspases play a central role in apoptosis, inflammation, and fibrosis. They produce hemodynamically active, proinflammatory microparticles that cause intrahepatic inflammation, vasoconstriction, and extrahepatic splanchnic vasodilation. Emricasan is a pan‐caspase inhibitor that lowers portal hypertension (PH) and improves survival in murine models of cirrhosis. This exploratory study assessed whether emricasan lowers PH in patients with compensated cirrhosis. This multicenter, open‐label study enrolled 23 subjects with compensated cirrhosis and PH (hepatic vein pressure gradient [HVPG] >5 mm Hg). Emricasan 25 mg twice daily was given for 28 days. HVPG measurements were standardized and performed before and after emricasan. A single expert read all HVPG tracings. Median age was 59 (range 49‐80); 70% were male. Cirrhosis etiologies were nonalcoholic steatohepatitis and hepatitis C virus. Subjects were Child class A (87%) with a median Model for End‐Stage Liver Disease score of 8 (range 6‐15). Twelve had severe PH (HVPG ≥12 mm Hg). Overall, there was no significant change in HVPG after emricasan (mean [standard deviation, SD] –1.1 [4.57] mm Hg). HVPG decreased significantly (mean [SD] –3.7[4.05] mm Hg; P = 0.003) in those with severe PH: 4/12 had a ≥20% decrease, 8/12 had a ≥10% decrease, and 2/12 HVPG decreased below 12 mm Hg. There were no significant changes in blood pressure or heart rate. Aspartate aminotransferase (AST) and alanine aminotransferase (ALT) decreased significantly in the entire group and in those with severe PH. Serum cleaved cytokeratin 18 and caspase‐3/7 decreased significantly. Emricasan was well tolerated. One subject discontinued for nonserious adverse events. Conclusion: Emricasan administered for 28 days decreased HVPG in patients with compensated cirrhosis and severe PH; an effect upon portal venous inflow is likely, and concomitant decreases in AST/ALT suggest an intrahepatic anti‐inflammatory effect.
Abbreviations
- AE
adverse event
- ALT
alanine aminotransferase
- AST
aspartate aminotransferase
- cCK18
cleaved cytokeratin 18
- CSPH
clinically significant portal hypertension
- HCV
hepatitis C virus
- HVPG
hepatic vein pressure gradient
- INR
international normalized ratio
- MELD
Model for End‐Stage Liver Disease
- NASH
nonalcoholic steatohepatitis
- NSBB
nonselective beta‐blocker
- PH
portal hypertension
- RLU
relative light units
- SD
standard deviation
Portal hypertension (PH) is the main driver of decompensation and death in patients with compensated cirrhosis.1 The initial mechanism is increased intrahepatic resistance to blood flow, but PH is maintained and enhanced by extrahepatic hemodynamic disturbances, including splanchnic vasodilation and increased cardiac output, which lead to increased portal venous inflow.2
Emricasan (IDN‐6556) is an oral pan‐caspase inhibitor that decreased apoptosis, inflammation, fibrosis, cirrhosis, and death in animal models of acute hepatitis and chronic models of nonalcoholic steatohepatitis (NASH).3, 4, 5 In two NASH models, emricasan reduced caspase activity, apoptosis, inflammation, proinflammatory cytokines, and fibrogenesis. In a murine model of cirrhosis induced by bile duct ligation, emricasan decreased portal pressure and improved survival.3 More recently, emricasan was studied in a rat CCl4 model of cirrhosis and was shown to decrease portal pressure, hepatic vascular resistance, inflammation, and liver fibrosis.5 These preclinical studies suggested that emricasan may be effective at treating PH, improving hepatocellular function, and decreasing hepatic inflammation and fibrosis.
The aim of this exploratory study was to investigate the safety and efficacy of emricasan at reducing portal pressure, as determined by the hepatic venous pressure gradient (HVPG), in patients with compensated cirrhosis and PH.
Materials and Methods
Subjects
Patients had compensated cirrhosis and PH. The diagnosis of cirrhosis was established by clinical, biochemical, imaging, and/or histological criteria. Compensated cirrhosis was defined by absence of overt ascites or encephalopathy, history of variceal hemorrhage, and no jaundice. Patients were required to have HVPG >5 mm Hg and PH based on any one of the following: (1) splenomegaly on imaging and/or clinical evaluation with platelet count of <120,000, (2) small varices on screening endoscopy and/or collateral circulation on imaging, or (3) medium/large varices that had never bled and had been obliterated with endoscopic ligation.
Exclusion criteria were age <18, decompensated cirrhosis defined by clinically overt ascites (requiring diuretics), overt encephalopathy (grade II or higher and requiring therapy), or history of variceal hemorrhage. Subjects with a history of overt ascites or grade II or higher encephalopathy who were currently stable (no ascites on physical examination and encephalopathy grade ≤I, with or without specific medication) were eligible. Other exclusions included Child‐Pugh class C; other nonliver organ failure; total bilirubin >12 mg/dL; international normalized ratio (INR) >2.5; platelets <20 × 109/L; overt hepatic encephalopathy of grade III or higher; serum creatinine >2 mg/dL; use of nonselective beta‐blockers (NSBBs), carvedilol, or nitrates; known human immunodeficiency virus infection; pancreatitis; portal vein thrombosis; plan to receive anti–hepatitis C virus (HCV) therapy during the study; hepatitis B virus on stable therapy for less than 3 months; or hepatocellular carcinoma. An attempt was made to primarily enroll subjects with a history of NASH and/or HCV‐related cirrhosis and PH.
Concomitant use of fibrates, statins, angiotensin II receptor antagonists, or angiotensin converting enzyme inhibitors was allowed if there was no change in dose or regimen within 3 months of screening.
Male and female subjects of childbearing potential were required to use two reliable forms of contraception from screening to 1 month after the last dose of study drug.
The study was conducted following the principles of the Declaration of Helsinki and approved by the institutional review committee of each participating hospital. All patients gave written informed consent.
Study Protocol
Medical/surgical history, laboratory tests, medication record, and physical examination including vital signs were obtained for all subjects. Eligible subjects had the baseline HVPG measured prior to starting emricasan treatment.
This was an open‐label study where all subjects received emricasan 25 mg orally twice a day for 28 days. Clinical, laboratory, and safety assessments were performed on days 1, 7, and 28. The HVPG was repeated at the day 28 visit. Follow‐up assessment off emricasan was approximately 28 days after the day 28 visit (day 56 visit). Clinical, laboratory, and safety assessments were repeated at that visit.
HVPG Measurement
Portal pressure was measured indirectly by determining the HVPG as described.6 An HVPG study manual was provided to all sites, and the central HVPG reader (J.B.) instructed the sites on HVPG measurement procedures. Each center provided at least two satisfactory HVPG measurements before being qualified to screen subjects. The central reader reviewed all HVPG tracings and provided ongoing feedback on technical aspects of the procedure. If a tracing was not adequate, the investigator was contacted promptly and the measurement repeated. Several tracings not meeting the prespecified quality criteria were subsequently repeated in order to obtain adequate tracings. All 44 tracings included in the analysis were of at least “adequate” quality. The procedure was performed after fasting all night, and sites were to perform the procedure at roughly the same time of day due to circadian variation in HVPG measurements.
Using the transjugular approach, a balloon‐tipped catheter was advanced into a hepatic vein under fluoroscopic guidance. The free hepatic venous pressure (FHVP) was measured with the balloon deflated and floating freely in the hepatic vein close to its junction with the inferior vena cava. The wedged hepatic venous pressure (WHVP) was measured with the balloon inflated until the branch of the hepatic vein was completely occluded. HVPG was obtained by subtracting the FHVP from the WHVP. All measurements were performed in triplicate, and permanent tracings were obtained.
Study Endpoints
The objectives of this study were to assess whether emricasan would decrease markers of caspase activation and improve portal hypertension in patients with compensated cirrhosis and PH. The primary endpoints of the study were change in cleaved cytokeratin 18 (cCK18/M30), a marker of apoptosis and therefore of caspase activity, and change in HVPG from baseline to day 28.
Other biomarker measures of pharmacodynamic activity included caspase‐3/7 activity, serum aminotransferases (alanine aminotransferase [ALT] and aspartate aminotransferase [AST]) and liver function (total bilirubin, INR, and albumin). Changes in Model for End‐Stage Liver Disease (MELD) and Child‐Pugh scores were also analyzed.
Safety Assessments
Safety assessments included collection of adverse events (AE), clinical examination, vital signs, laboratory tests, and electrocardiograms (ECGs). Both the severity of AEs and their relation to study medication treatment were collected.
Statistical Analyses
Because this was an exploratory study, the sample size for HVPG change from baseline was not statistically based. In a previously completed study (A8491003) that was conducted in subjects without cirrhosis but with chronic hepatitis C infection who had failed previous HCV therapy and had elevated aminotransferases, the observed mean change from baseline in log‐transformed cCK18/M30 at week 10 for the 25 mg twice daily treatment group was –0.423 with a standard deviation (SD) of 0.468.7 If a similar effect size was observed in this study population, a sample size of n = 18 would provide in excess of 90% power to detect a change from baseline in cCK18/M30.
Baseline characteristics of patients were summarized as median SD for continuous variables and as percentages for nominal variables.
Analyses of the mean change from baseline in HVPG and log‐transformed cCK18/M30 at day 28 were conducted using a paired t test. For the HVPG subgroup analyses (<12 and ≥12 mm Hg), an analysis of variance (for HVPG) or analysis of covariance (for cCK18/M30) including baseline HVPG category and baseline value (only analysis of covariance) as factors were used. No adjustments for multiplicity were used in any of the analyses.
Additional post hoc sensitivity analyses were performed for the subgroups with and without clinically significant PH (<10 and ≥10 mm Hg). Responder analyses looking at 10% improvement from baseline (prespecified analysis) and 20% improvement from baseline (post hoc analysis) were also performed using descriptive statistics.
The mean change from baseline in caspase‐3/7 at day 28 was summarized with descriptive statistics for the absolute and relative changes. The relative change was calculated by back‐transforming the log‐transformed changes from baseline.
Changes in aminotransferases, total bilirubin, and INR were tested using paired t test.
All concomitant medications that might affect portal pressure, including diuretics, nitrates, antihypertensives, and statins, were reviewed prior to database lock. No patient had new medications added or changes in dosages of medications that may have confounded interpretation of the HVPG results.
Results
Subject Disposition
Subject disposition is shown in Fig. 1. Between October 2014 and April 2015, 30 patients were screened for the study at nine sites in the United States and had a baseline HVPG performed. Seven subjects were not eligible (5 did not meet all inclusion criteria, and 4 met exclusion criteria). Baseline characteristics of the 23 treated patients are shown in Table 1. Median age was 59 years, and 70% were male. The most common etiologies of cirrhosis were NASH (56.5%) and HCV (39.1%), with only one patient having alcoholic cirrhosis. The baseline median HVPG was 13.5 mm Hg (range 5.5‐32.0); 10/23 (43%) patients had HVPG <12 mm Hg with a median value of 7.8 mm Hg (range 5.5‐11 mm Hg), and 13/23 (57%) patients had severe PH (baseline HVPG ≥12 mm Hg) with a median value of 21.5 mm Hg (range 13‐32 mm Hg). One patient (baseline HVPG 13 mm Hg) did not complete treatment and withdrew on study day 1 because of AEs, none of which were serious or assessed as being related to study medication. The evaluable patient population consisted of 22 patients (12 with severe PH) who completed 28 days of treatment with emricasan and had day 28 HVPG measurements.
Figure 1.
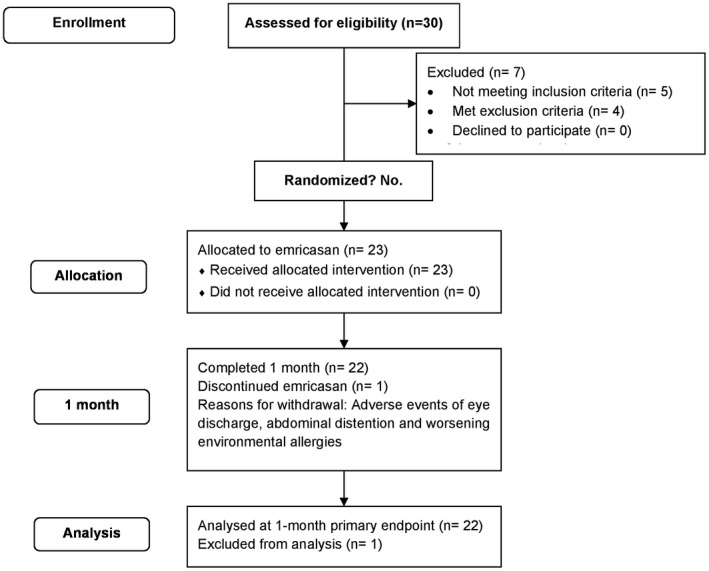
Subject flowchart and disposition. A total of 30 subjects were screened; 23 met the inclusion/exclusion criteria and were treated with emricasan. Of the 23 subjects, 22 had day 28 HVPG measurements. Data from all subjects were included in the primary efficacy analyses.
Table 1.
Baseline Demographics and Disease Characteristics in the Overall Patient Population, Baseline HVPG <12 mm Hg, and Baseline HVPG ≥12 mm Hg
| Median (minimum‐maximum) |
All Subjects (N = 23) |
HVPG <12 mm Hg (n = 10) |
HVPG ≥ 12 mm Hg (n = 13) |
|---|---|---|---|
| Age | 59 (48‐80) | 57.5 (49‐71) | 60 (49‐80) |
| Gender (% male) | 16 (70%) | 9 (90%) | 7 (54%) |
| Race (% Caucasian) | 21 (91%) | 9 (90%) | 12 (92%) |
| Etiology of cirrhosis | |||
| NASH | 13 (56.5%) | 7 (70%) | 6 (46.2%) |
|
HCV only HCV primary + alcohol |
5 (21.7%) 4 (17.4%) |
1 (10%) 2 (20%) |
4 (30.8%) 2 (15.4%) |
| Alcohol alone | 1 (4.3%) | 0 | 1 (7.7%) |
| Child‐Pugh class A | 20 (87%) | 10 (100%) | 10 (77%) |
| Child‐Pugh class B | 3 (13%) | 0 | 3 (23%) |
| Esophageal varices | |||
| None | 7 (30.4%) | 3 (30.0%) | 4 (30.8%) |
| Small | 12 (52.2%) | 7 (70.0%) | 5 (38.5%) |
| Medium or large | 4 (17.4%) | 0 | 4 (30.8%) |
| BMI (kg/m2) | 32.4 (17.9‐44.9) | 32.1 (25.4‐42.4) | 33.0 (17.9‐44.9) |
| Systolic BP | 126.0 (108‐171) | 127.0 (108‐145) | 126.0 (109‐171) |
| Heart rate | 76.0 (50‐99) | 78.5 (60‐92) | 76.0 (50‐99) |
| HVPG (mm Hg)* | 13.5 (5.5‐32.0) | 7.8 (5.5‐11.0) | 21.5 (13.0‐32.0) |
| cCK18 (U/L) | 191.0 (78‐994) | 189.0 (126, 442) | 221.0 (78‐994) |
| Caspase 3/7 (RLU) | 1,997 (750‐37,428) | 1,598 (750‐4,970) | 2,305 (1,457‐37,428) |
| MELD score | 8 (6‐15) | 7 (6‐10) | 8 (6‐15) |
| Total bilirubin (mg/dL) | 0.7 (0.31‐3.08) | 0.6 (0.31‐1.08) | 0.8 (0.31‐3.08) |
| INR | 1.1 (0.9‐1.6) | 1.0 (1.0‐1.2) | 1.1 (0.9‐1.6) |
| Albumin (g/dL) | 4.2 (2.7‐4.9) | 4.4 (3.6‐4.9) | 3.9 (2.7‐4.5) |
| Platelet count (K/mm3) | 104.0 (43‐199) | 127.0 (63‐199) | 77.0 (43‐191) |
| AST (U/L) | 35.0 (16‐83) | 27.0 (18‐71) | 39.0 (16‐83) |
| ALT (U/L) | 25.0 (10‐99) | 23.5 (16‐99) | 32.0 (10‐61) |
Abbreviations: BMI, body mass index; BP, blood pressure.
Values that are statistically different (P < 0.05) are indicated.
Changes In HVPG
In the evaluable population (n = 22 with baseline and day 28 HVPG values), there was a nonsignificant trend for the HVPG to decrease after 28 days of emricasan (mean [SD] change from baseline of –1.1 [4.6] mm Hg). Individual patient responses in the overall population are shown in Fig. 2. When patients were categorized by HVPG of <12 or ≥12 mm Hg, those with severe PH (HVPG ≥12 mm Hg, n = 12) demonstrated a clinically significant reduction in HVPG (mean [SD] –3.7 [4.0] mm Hg; P = 0.0025), while patients with HVPG <12 mm Hg (n = 10) did not (mean [SD] +1.9 [3.2] mm Hg; P = not significant). Individual patient responses in the <12 (black) and >12 groups (red) are shown in Fig. 2. In the severe PH group, HVPG was reduced to levels <12 mm Hg in 2 patients (from 13.5 to 8 mm Hg and 17.0 to 9.5 mm Hg). One third (4/12) of patients had a ≥20% decrease from baseline, and two thirds (8/12) had a ≥10% decrease from baseline. The effects of emricasan on HVPG in patients with severe PH were similar in patients with different etiologies for cirrhosis (data not shown).
Figure 2.
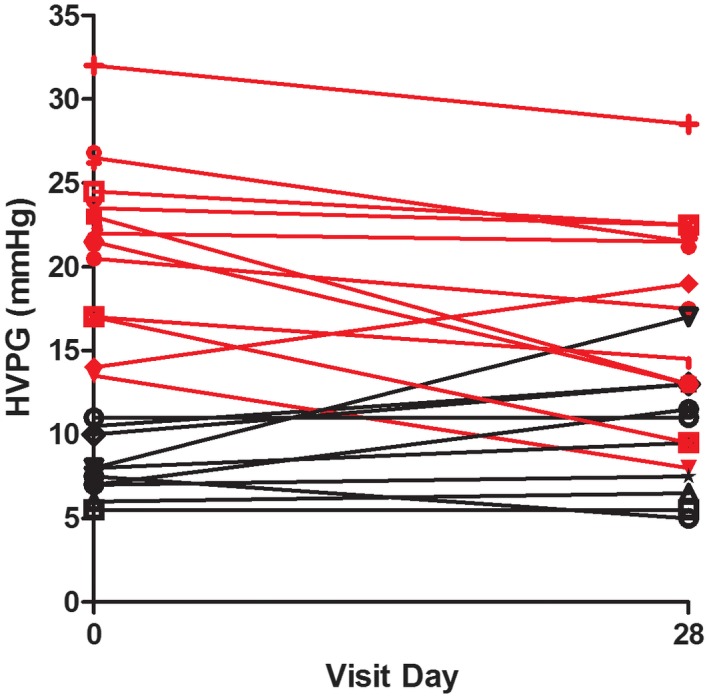
HVPG was measured at baseline and after 28 days of treatment with emricasan. Twenty‐two subjects had baseline and day 28 HVPG values. Ten subjects with a baseline HVPG of <12 mm Hg are shown in black. In them, emricasan did not significantly modify the HVPG (mean [SD] +1.9 [3.2] mm Hg, P = 0.12). Twelve subjects who had a baseline HVPG of >12 mm Hg and a follow‐up value at day 28 are shown in red. In those subjects, emricasan significantly decreased the HVPG (mean [SD] 3.7 [4.0] mm Hg; P = 0.003).
A sensitivity analysis using an HVPG cutoff of 10 mm Hg showed similar results. The baseline demographics of the subgroups with HVPG <10 mm Hg and ≥10 mm Hg are shown in Supporting Table S1. The subgroup of subjects with a baseline HVPG of ≥10 mm Hg (n = 15) had their HVPG decrease from 19.1 ± 6.5 to 16.5 ± 5.8 mm Hg. At day 28, 8/15 decreased the baseline value by ≥10%, 4/15 decreased the baseline value by ≥20%, and 2/15 decreased the baseline value to <10 mm Hg.
Although subjects using NSBBs were to be excluded from the study, 2 subjects taking stable doses of NSBBs for over 3 months were identified after treatment with emricasan had been initiated. These subjects remained in the study and were included in analyses of efficacy and safety. Both subjects had severe PH (baseline HVPG measurements of 23 and 21.5 mm Hg), and both had responses to emricasan treatment (HVPG decreases of 10 and 8.5 mm Hg, respectively).
A change in body weight between baseline and day 28 did not appear to confound interpretation of the results. Only 4 subjects gained or lost more than 5 lb between day 1 and day 28. One subject in the HVPG <12 mm Hg group gained 11 lb, and the HVPG increased from 8 to 17 mm Hg. Excluding this subject, the mean [SD] change from baseline at day 28 in the HVPG <12 mm Hg group was +1.1 [2.0] mm Hg. One subject in the HVPG ≥12 mm Hg group gained 8.9 lb, and 2 subjects lost 8 and 10.2 lb, respectively. Excluding them from the analysis yielded a mean [SD] change from baseline at day 28 of –3.5 [4.5] mm Hg. Thus, changes in HVPG followed the same pattern as that observed in the main analysis.
Five subjects in the study had a history of alcohol use disorder. Alcohol was the primary cause of cirrhosis in 1 subject, and contributory in 4 of the 9 subjects whose cirrhosis was deemed secondary to HCV. Four of the 5 subjects had stopped alcohol use in 1987, 1989, 2009, and 2012, respectively. To assess the possibility that surreptitious alcohol use may have affected the study results, a sensitivity analysis excluded those 5 subjects. The mean HVPG decrease from baseline at day 28 was slightly greater in this subgroup than in the overall population (mean [SD] change of –1.4 [4.7] mm Hg) but not significant. Two subjects were in the HVPG <12 mm Hg subgroup, and when excluding them, the mean [SD] change from baseline in HVPG at day 28 was +2.3 [3.1] mm Hg (P = not significant). The other 3 patients were in the HVPG ≥12 subgroup, and when excluding them, the mean [SD] change from baseline in HVPG at day 28 was –4.8 [3.2] mm Hg (P = 0.0004). Thus, changes in HVPG followed the same pattern observed in the main analysis.
Eight investigators at 9 sites enrolled the 23 subjects who were treated in this study. Of the sites that enrolled subjects with severe PH, no site had more than 2 subjects who had a decrease in HVPG. Thus, there was no center effect.
Changes in the Mechanism‐Specific Biomarker CASPASE‐3/7
Changes in caspase‐3/7 activity were also assessed. All 23 subjects had baseline and day 28 values for analysis. Values at baseline were clearly elevated in the overall patient population (median 1,997 relative light units [RLU], n = 23) compared to healthy subjects of previous studies (median of 919.5 RLU in study IDN‐6556‐05 and median of 963.5 RLU in study IDN‐6556‐08) and decreased significantly at day 28 with emricasan treatment (mean reduction of –29.2%, P = 0.002) (Fig. 3A). Baseline caspase‐3/7 activity was elevated in both HVPG subgroups but higher in the severe PH subgroup (median of 2,305 versus 1,598 RLU). Reductions from baseline in caspase‐3/7 activity at day 28 were qualitatively similar and statistically significant in both the low and high HVPG subgroups (mean reduction of –50.2%, P = 0.003, and –26.1%, P = 0.028, respectively). Caspase‐3/7 activity was reduced in both HVPG subgroups relative to baseline at the first postbaseline visit (mean –48.1% and –52.1%, respectively, at day 7; significance not tested). Excluding the 5 subjects with a history of alcohol use disorder yielded similar results, with a mean reduction of –57.1% at day 28 (P = 0.014). Two of the 5 subjects with a history of alcohol use disorder were in the HVPG <12 mm Hg subgroup, and when excluding them, the mean change from baseline in caspase‐3/7 activity at day 28 was –42.0% (P = 0.030). The other 3 patients were in the HVPG ≥12 subgroup, and when excluding them, the mean change from baseline in caspase‐3/7 activity at day 28 was –61.5% (P = 0.157). Thus, the directional changes in caspase‐3/7 activity were similar to the main analysis.
Figure 3.
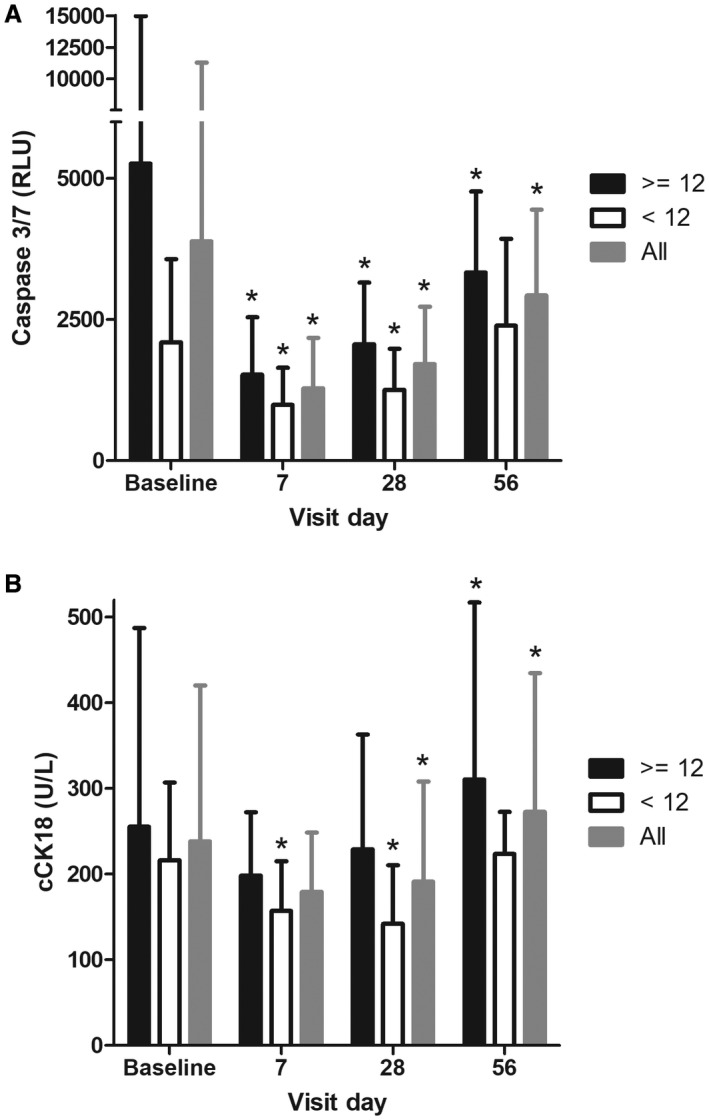
Caspase 3/7 (A) and cCK18 (B) were measured at baseline and on days 7, 28 (last day on treatment), and 56 (28 days following treatment with emricasan). Subjects were analyzed as follows: all subjects (gray columns), baseline HVPG <12 mm Hg (white columns), and baseline HVPG >12 mm Hg (black columns). Mean values with SD are shown for each visit. Values that were statistically different from baseline (P < 0.05, two‐sided Wilcoxon signed‐rank test) are indicated with an asterisk.
Changes in cCK18
Baseline values of cCK18 in the overall population (median 191.0 U/L) were relatively low and in the range of values previously observed in healthy subjects (median of 240 U/L in study IDN‐6556‐05 and median of 162 U/L in study IDN‐6556‐08) and subjects with mild hepatic impairment (median of 147.5 U/L in study IDN‐6556‐08). Nevertheless, emricasan treatment decreased cCK18 values by a mean of 20% between baseline and day 28 (P = 0.026) (all subjects had a day 28 value). Baseline cCK18 levels were also similar in each HVPG subgroup (median baseline values of 189.0 and 221.0 U/L in the <12 and ≥12 mm Hg HVPG subgroups, respectively). cCK18 values decreased more at day 28 in the patients with baseline HVPG <12 mm Hg (mean 37% decrease, P = 0.001) than in the patients with severe PH (4% decrease, P = 0.716) (Fig. 3B). There was no correlation between the change in HVPG and change in cCK18.
Excluding the 5 subjects with a history of alcohol use disorder yielded similar results, with a mean reduction of –19.7% at day 28 (P = 0.056). Two of the 5 subjects with a history of alcohol use disorder were in the HVPG <12 mm Hg subgroup, and when excluding them, the mean change from baseline in cCK18 at day 28 was –38.5% (P = 0.017). The other 3 patients were in the HVPG ≥12 subgroup, and when excluding them, the mean change from baseline in cCK18 at day 28 was –7.6% (P = 0.852). Thus, changes in cCK18 were qualitatively similar to the main analysis.
Changes in Markers of Hepatocellular Injury and Inflammation (ALT AND AST)
Baseline ALT values were low in the overall patient group (median 25.0 IU/L) and slightly lower in the low versus high HVPG subgroup (23.5 and 32.0 IU/L, respectively). Figure 4A shows the median ALT values by visit day. ALT decreased significantly in the overall population (n = 23) and in the severe PH subgroup (n = 13) at day 28 (both P < 0.05, two‐sided Wilcoxon signed‐rank test). ALT values decreased by day 7 in all groups and remained lower through day 28/end of treatment. Values tended to increase back toward baseline values by the day 56 visit.
Figure 4.
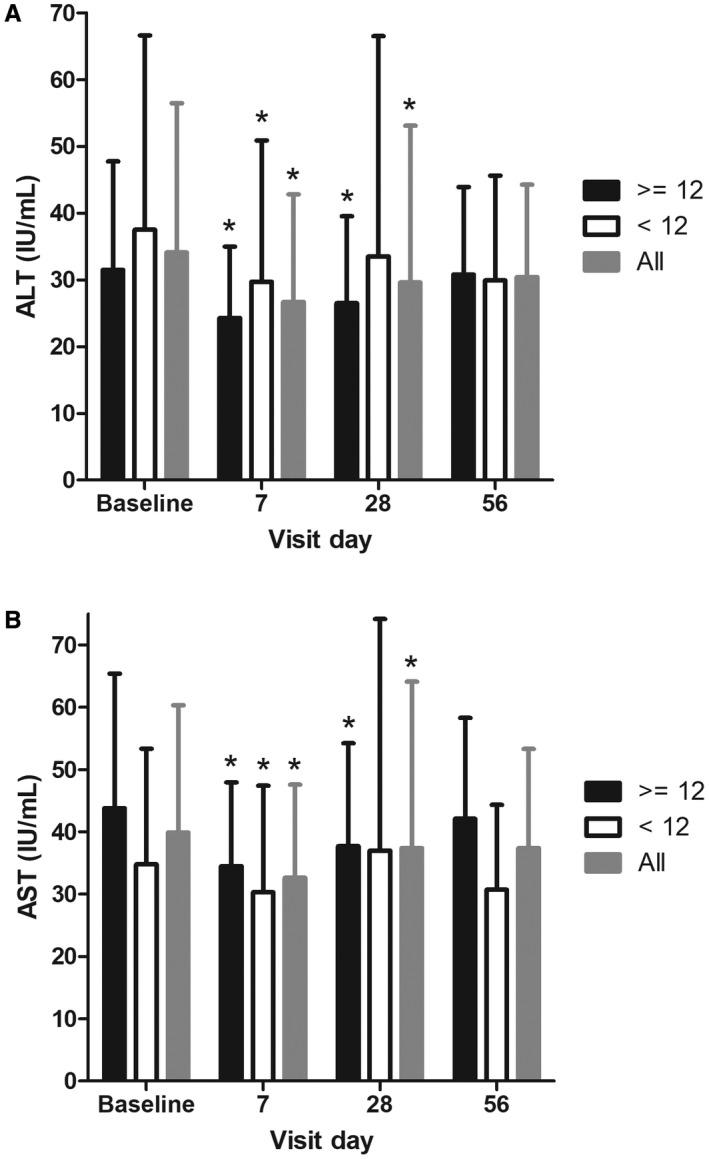
ALT (A) and AST (B) were measured at baseline and on days 7, 28 (last day on treatment), and 56 (28 days following treatment with emricasan). Subjects were analyzed as follows: all subjects, baseline HVPG <12 mm Hg, and baseline HVPG >12 mm Hg. Mean values with SD are shown for each visit. Values that were statistically different from baseline (P < 0.05, two‐sided Wilcoxon signed‐rank test) are indicated with an asterisk.
A similar pattern was observed for AST. Baseline values in the overall group were mildly abnormal (median 35.0 IU/L) and lower in the low versus high HVPG subgroup (27.0 and 39.0 IU/L, respectively) (Table 1). AST decreased in the overall population (n = 23) and in both subgroups (Fig. 4B). AST values decreased by day 7 and were statistically different in the overall and severe PH subgroup at day 28/end of treatment (P < 0.05). Like ALT, AST values tended to increase back toward baseline values by the day 56 visit.
Changes in Measures of Hepatic Function and Prognosis
There were no significant changes from baseline in MELD score (mean change from baseline in both subgroups 0.0), Child‐Pugh score, total bilirubin, INR, or serum albumin following 28 days of treatment with emricasan. Two subjects (one HVPG ≤12 mm Hg, one HVPG ≥12 mm Hg) who were Child‐Pugh class A at baseline became Child‐Pugh class B at day 28. One subject (with HVPG ≥12 mm Hg) had a shift from Child Pugh class B at baseline to Child Pugh class A at day 28. All other subjects had no change in Child‐Pugh class.
Safety
Treatment with emricasan was generally well tolerated. Overall, 60 AEs were reported by 15 subjects; most were nonserious, and the most common was fatigue (Table 2). Three AEs reported by 1 subject were assessed by the investigator as serious (systemic inflammatory response syndrome, acute respiratory failure, and shortness of breath, all 3 occurring approximately 10 days after the last dose of study drug). One subject reported four AEs, which led to discontinuation from the study (eye discharge, abdominal distension, hypersensitivity [due to environmental allergy], and bilateral conjunctivitis) 1 day after starting emricasan.
Table 2.
AEs in the Overall Patient Population, Baseline HVPG <12 mm Hg, and Baseline HVPG ≥12 mm Hg
|
All Subjects (N = 23) |
HVPG <12 mm Hg (n = 10) |
HVPG ≥12 mm Hg (n = 13) |
|
|---|---|---|---|
| Subjects with AEs | 15 (65%) | 6 (60%) | 9 (69%) |
| Subjects with serious AEs* | 1 (4%) | 0 | 1 (8%) |
| Subjects with severe AEs* | 1 (4%) | 0 | 1 (8%) |
| Subjects with AEs leading to discontinuation | 1 (4%) | 0 | 1 (8%) |
| Number of AEs | 60 | 11 | 49 |
| Number of AEs assessed as related | 15 | 7 | 8 |
| AEs occurring in >5% of subjects | |||
| Fatigue | 5 (22%) | 2 (20%) | 3 (23%) |
| Headache | 3 (13%) | 3 (30%) | 0 |
| Peripheral edema | 3 (13%) | 1 (10%) | 2 (15%) |
| Dehydration | 2 (9%) | 0 | 2 (15%) |
| Diarrhea | 2 (9%) | 0 | 2 (15%) |
| Constipation | 2 (9%) | 1 (10%) | 1 (8%) |
| Nausea | 2 (9%) | 0 | 2 (15%) |
*Subject with systemic inflammatory response syndrome, felt to be unrelated to emricasan.
The number of AEs reported was higher in subjects in the severe PH subgroup (n = 49) than in the HVPG <12 mm Hg subgroup (n = 11), which was mainly due to 1 subject who had 27 AEs, most of which occurred in association with a hospitalization and subsequent transfusion reaction that occurred approximately 10 days after the last dose of study drug. This subject had three serious AEs (the only serious AEs reported in the study) of systemic inflammatory response syndrome, acute respiratory failure, and dyspnea, considered not related to study drug. The percentage of subjects with AEs was similar in both subgroups (69% in the HVPG ≥12 mm Hg versus 60% in the HVPG <12 mm Hg subgroup). Most AEs were mild, and 10 subjects reported moderate AEs (6 with HVPG ≥12 mm Hg, and 4 with HVPG <12 mm Hg). No subject died, developed decompensating events (ascites, variceal hemorrhage, encephalopathy, or jaundice), or developed a malignancy. There were no apparent effects of emricasan upon non‐liver‐related laboratory tests or ECG parameters.
Systemic hemodynamics were analyzed for any potential effect of emricasan treatment (Table 3). There was no detectable effect of emricasan upon systolic blood pressure, diastolic blood pressure, or heart rate.
Table 3.
Change From Baseline in Hemodynamic Parameters
| Median | |||
|---|---|---|---|
|
HVPG <12 mm Hg (n = 10) |
HVPG ≥12 mm Hg (n = 12) |
Evaluable Patients (N = 22) |
|
| Systolic BP (mm Hg) | |||
| Baseline | 127.0 | 126.0 | 126.0 |
| Change to day 28 | –2.0 | 0 | –1.0 |
| Diastolic BP (mm Hg) | |||
| Baseline | 72.5 | 75.0 | 75.0 |
| Change to day 28 | –2.0 | –2.0 | –2.0 |
| Heart rate (beats/minute) | |||
| Baseline | 78.5 | 76.0 | 76.0 |
| Change to day 28 | –4.0 | 0 | –1.0 |
Abbreviations: BP, blood pressure.
Discussion
This study assessed whether the pan‐caspase inhibitor emricasan might decrease PH by inhibiting apoptosis and inflammation. Apoptosis is a hallmark of hepatocellular damage in many chronic liver diseases, including nonalcoholic fatty liver disease (NAFLD), hepatitis C, hepatitis B, and alcoholic liver disease.8, 9, 10, 11 Caspases are a family of at least 11 human intracellular cysteine proteases that mediate apoptosis and inflammation.12 Caspase activation and apoptosis result in the production of proinflammatory, hemodynamically active microvesicles that lead to important biological effects within the liver and systemically.13, 14 Caspases are an important link between hepatocyte steatosis, lipotoxic damage, apoptosis, inflammation, and fibrogenesis in NAFLD. Caspases 2, 3/7, and 9 are integral to the induction of cell death by lipotoxic stress and to the pathogenesis of NASH‐related cirrhosis.15, 16, 17, 18, 19, 20
Microvesicle release as a result of lipotoxicity or ethanol exposure is also dependent upon caspase.21, 22, 23 Cannito et al. showed that steatosis and lipotoxicity led to microvesicle release, and that following uptake by hepatocytes or macrophages, the microvesicles caused inflammasome activation and inflammation.24 These intrahepatic effects alone would provide a strong rationale for the use of a pan‐caspase inhibitor in NASH.
Increased apoptosis and inflammation are also hallmarks of active hepatitis C infection, and the resulting release of hepatic microvesicles/apoptotic bodies triggers inflammation and fibrosis upon phagocytosis by Kupffer cells and hepatic stellate cells.25, 26 HCV infection results in death receptor–mediated mitochondrial damage, leading to downstream caspase activation and apoptosis.27 Pan‐caspase inhibition with emricasan acutely decreased ALT levels in subjects with active HCV infection.7, 28 There is therefore a pathophysiologic basis for believing that caspase inhibition may be beneficial in HCV infection as well as NASH.29
Microvesicles may also worsen PH in cirrhosis by acting on the increased portal blood flow, a mechanism that contributes to worsening PH in advanced stages, when the systemic circulation becomes hyperdynamic.2 Microvesicles from patients with decompensated cirrhosis impair the vasoconstrictor response to phenylephrine and decrease mean arterial pressure,13 increasing portal blood inflow and worsening PH.
Microvesicles may also contribute to increased intrahepatic vascular resistance by increasing the hepatic vascular tone through changes in sinusoidal endothelial cell gene expression, sinusoidal remodeling, and reduced production of the endothelial vasodilator nitric oxide.30 Caspases have been shown to cleave endothelial nitric oxide synthase,31 which may partly contribute to the reduced nitric oxide availability in the liver microcirculation in cirrhosis.32
Emricasan treatment in the current study demonstrated an intrahepatic effect as shown by decreases in ALT and AST. Elevations in ALT and AST are indicative of hepatocellular damage and/or inflammation. This is consistent with previous studies with emricasan in patients with active hepatitis C and elevated aminotransferase levels.7, 28 Baseline levels of ALT and AST were low in this study, and decreases in ALT and AST were more apparent in the severe PH subgroup, likely due to the higher baseline values in that subgroup, making it easier to show a treatment effect.
Although emricasan did not decrease HVPG in the overall group of patients with compensated cirrhosis, there was a clinically significant reduction in HVPG in patients with an HVPG ≥12 mm Hg and in those with clinically significant PH (CSPH). This is relevant because these are the patients who are at the greatest risk of decompensation and in whom reductions in portal pressure will lead not only to a reduction in the development of variceal hemorrhage but also to a reduction in ascites, which is the most common cause of decompensation of cirrhosis.33 Sensitivity analyses assessing the potential effects of weight loss and alcohol use on HVPG did not suggest a confounding effect. The sample size of this study was not chosen to detect a difference in HVPG. However, if the true treatment effect of emricasan is to lower HVPG 3.7 mm Hg following 28 days of treatment, and the variability is also as observed in this study (SD, 4.0), 12 subjects would have provided 81% power to detect that treatment difference using a paired t test. Additional larger studies in more homogeneous patient populations will be necessary to better estimate the true treatment effect of emricasan upon HVPG.
Importantly, emricasan was generally safe and well tolerated, with the most common AE being fatigue (in 22% of the patients) and with only one patient developing a severe AE that was thought to be unrelated to the drug.
The results should be taken in the context of an exploratory study, with a short duration of treatment, a small sample size, lack of a control group or adjustment for multiple testing, and without additional hemodynamic investigations that could provide a more precise mechanistic explanation of how caspase inhibition induced the decrease in HVPG in the subgroup of patients with severe PH.
The acute hemodynamic effects of vasoactive drugs (propranolol, nadolol, vasopressin, terlipressin, somatostatin, etc.) on portal pressure have routinely been demonstrated 15‐20 minutes after intravenous administration.33, 34 For drugs acting on intrahepatic resistance, simvastatin significantly decreased HVPG after 28 days of oral administration in a double‐blind, phase II, randomized controlled trial in patients with cirrhosis.35 So, it is conceivable that changes in HVPG due to either intrahepatic or hemodynamic mechanisms were captured in the current study. Whether the portal pressure–reducing effect of emricasan might be potentiated on longer administration should be verified in longer, adequately designed studies.
Mechanisms of PH differ in patients with mild portal hypertension (HVPG >5 but <10 mm Hg) compared to those with CSPH.2 In mild PH, the main mechanism leading to portal hypertension is increased intrahepatic resistance, while in those with CSPH/varices, increased portal flow plays a major role in maintaining and aggravating the PH state. Patients with mild PH have a significantly lower response to NSBBs (which decrease portal flow) compared to those with CSPH/varices, who have a hyperkinetic circulation.33 This study suggests that the same may happen with emricasan, which, analogously to NSBBs, decreases portal pressure mostly in patients with severe PH.
It is unlikely that emricasan reduced intrahepatic resistance through an antifibrotic effect in 28 days, although the decreases in aminotransferases observed on emricasan are consistent with decreased inflammation and hepatocellular injury. Short‐term decreases in HVPG were observed in patclarify its mechanism of action, and disclose whetherients with HCV cirrhosis shortly after achieving viral elimination, before fibrosis would have regressed.36 Longer‐term emricasan administration could further decrease portal pressure due to an antifibrotic effect and architectural remodeling, as observed in rats with cirrhosis30 and after treatment of HCV‐related cirrhosis where there was a small decrease in HVPG at the time of sustained virologic response but a marked decline during longer‐term follow‐up.37
In summary, this exploratory study reveals that oral treatment with the caspase inhibitor emricasan for 28 days lowered portal pressure in patients with severe PH and was generally safe and well tolerated. The relatively short period of drug administration suggests that the decrease in portal pressure was likely due to a hemodynamic effect inducing a decrease in portal blood flow and/or in intrahepatic vascular resistance, although an intrahepatic effect upon hepatocellular damage and inflammation may also have contributed. Future randomized, placebo‐controlled studies will elucidate the potential for additional long‐term effects of the drug, clarify its mechanism of action, and disclose whether continued emricasan administration may represent an effective treatment for cirrhotic PH.
Potential conflict of interest
Dr. Borg received grants from Conatus. Dr. Bosch consults for, advises, and received grants from Conatus. He consults for and advises Actelion. He consults for and received grants from Gilead and Exalenz. He is director of, owns stock in, and holds intellectual property rights with Barcelona Liver Biosystems. Dr. Chan is employed by and owns stock in Conatus. Dr. Garcia‐Tsao consults for and advises Conatus. She advises and received grants from Intercept. She consults for Galectin. She advises Biovie. Dr. Hagerty is employed by Conatus. Dr. Pyrsopoulos received grants from Conatus. Dr. Reddy consults for and received grants from Merck, AbbVie, and Gilead. He received grants from Conatus and Intercept. Dr. Robinson is employed by and owns stock in Conatus. Dr. Spada is employed by and owns stock in Conatus. Dr. Fuchs received grants from Conatus. Dr. Shiffman advises, is on the speakers’ bureau of, and received grants from AbbVie, Bristol‐Myers Squibb, Gilead, Intercept, and Merck. He consults for Optum Rx. He advises Salix. He is on the speakers’ bureau for Bayer and Daiichi Sankyo. He received grants from Conatus, CymaBay, Exalenz, Galectin, Genfit, Immuron, NGMBio, Novartis, and Shire.
Supporting information
Contributor Information
Guadalupe Garcia‐Tsao, Email: guadalupe.garcia-tsao@yale.edu.
Jaime Bosch, Email: jbosch@clinic.cat.
References
- 1. Ripoll C, Groszmann R, Garcia‐Tsao G, Grace N, Burroughs A, Planas R, et al. Hepatic venous pressure gradient predicts clinical decompensation in patients with compensated cirrhosis. Gastroenterology 2007;133:481–488. [DOI] [PubMed] [Google Scholar]
- 2. Bosch J, Groszmann RJ, Shah VH. Evolution in the understanding of the pathophysiological basis of portal hypertension: how changes in paradigm are leading to successful new treatments. J Hepatol 2015;62:S121–S130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Canbay A, Feldstein A, Baskin‐Bey E, Bronk SF, Gores GJ. The caspase inhibitor IDN‐6556 attenuates hepatic injury and fibrosis in the bile duct ligated mouse. J Pharmacol Exp Ther 2004;308:1191–1196. [DOI] [PubMed] [Google Scholar]
- 4. Barreyro FJ, Holod S, Finocchietto PV, Camino AM, Aquino JB, Avagnina A, et al. The pan‐caspase inhibitor Emricasan (IDN‐6556) decreases liver injury and fibrosis in a murine model of non‐alcoholic steatohepatitis. Liver Int 2015;35:953–966. [DOI] [PubMed] [Google Scholar]
- 5. Gracia‐Sancho J, Contreras PC, Vila S, Garcia‐Caldero H, Spada AP, Bosch J. The pan‐caspase inhibitor emricasan improves the hepatic microcirculatory dysfunction of CCL4‐cirrhotic rats leading to portal hypertension amelioration and cirrhosis regression. Hepatology 2016;64:1043A. [Google Scholar]
- 6. Groszmann RJ, Wongcharatrawee S. The hepatic venous pressure gradient: anything worth doing should be done right. Hepatology 2004;39:280–282. [DOI] [PubMed] [Google Scholar]
- 7. Shiffman ML, Pockros P, McHutchison JG, Schiff ER, Morris M, Burgess G. Clinical trial: the efficacy and safety of oral PF‐03491390, a pan‐caspase inhibitor—a randomized placebo‐controlled study in patients with chronic hepatitis C. Aliment Pharmacol Ther 2010;31:969–978. [DOI] [PubMed] [Google Scholar]
- 8. Cao ZJ, Li J, Wang Y, Bao R, Liu YH, Xiang XG, et al. Serum hepatocyte apoptosis biomarker predicts the presence of significant histological lesion in chronic hepatitis B virus infection. Dig Liver Dis 2016;48:1463–1470. [DOI] [PubMed] [Google Scholar]
- 9. Brunt EM, Tiniakos DG. Alcoholic and nonalcoholic fatty liver disease In: Odze RD, Goldblum JR, eds. Surgical Pathology of the GI Tract, Liver, Biliary Tract, and Pancreas, 2nd ed Philadelphia: Elsevier; 2009:1007–1014. [Google Scholar]
- 10. Yilmaz Y, Dolar E, Ulukaya E, Akgoz S, Keskin M, Kiyici M, et al. Elevated serum levels of caspase‐cleaved cytokeratin 18 (CK18‐Asp396) in patients with nonalcoholic steatohepatitis and chronic hepatitis C. Med Sci Monit 2009;15:189–193. [PubMed] [Google Scholar]
- 11. Lavallard VJ, Bonnafous S, Patouraux S, Saint‐Paul MC, Rousseau D, Anty R, et al. Serum markers of hepatocyte death and apoptosis are non invasive biomarkers of severe fibrosis in patients with alcoholic liver disease. PLoS One 2011;6:e17599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Galluzzi L, Lopez‐Soto A, Kumar S, Kroemer G. Caspases connect cell‐death signaling to organismal homeostasis. Immunity 2016;44:221–231. [DOI] [PubMed] [Google Scholar]
- 13. Rautou PE, Bresson J, Sainte‐Marie Y, Vion AC, Paradis V, Renard JM, et al. Abnormal plasma microparticles impair vasoconstrictor responses in patients with cirrhosis. Gastroenterology 2012;143:166–176. [DOI] [PubMed] [Google Scholar]
- 14. Lemoinne S, Thabut D, Housset C, Moreau R, Valla D, Boulanger CM, et al. The emerging roles of microvesicles in liver diseases. Nat Rev Gastroenterol Hepatol 2014;11:350–361. [DOI] [PubMed] [Google Scholar]
- 15. Lupi R, Dotta F, Marselli L, Del Guerra S, Masini M, Santangelo C, et al. Prolonged exposure to free fatty acids has cytostatic and pro‐apoptotic effects on human pancreatic islets: evidence that beta‐cell death is caspase mediated, partially dependent on ceramide pathway, and Bcl‐2 regulated. Diabetes 2002;51:1437–1442. [DOI] [PubMed] [Google Scholar]
- 16. Hirota N, Otabe S, Nakayama H, Yuan X, Yamada K. Sequential activation of caspases and synergistic beta‐cell cytotoxicity by palmitate and anti‐Fas antibodies. Life Sci 2006;79:1312–1316. [DOI] [PubMed] [Google Scholar]
- 17. Wei Y, Wang D, Topczewski F, Pagliassotti MJ. Saturated fatty acids induce endoplasmic reticulum stress and apoptosis independently of ceramide in liver cells. Am J Physiol Endocrinol Metab 2006;291:E275–E281. [DOI] [PubMed] [Google Scholar]
- 18. Ricchi M, Odoardi MR, Carulli L, Anzivino C, Ballestri S, Pinetti A, et al. Differential effect of oleic and palmitic acid on lipid accumulation and apoptosis in cultured hepatocytes. J Gastroenterol Hepatol 2009;24:830–840. [DOI] [PubMed] [Google Scholar]
- 19. Johnson ES, Lindblom KR, Robeson A, Stevens RD, Ilkayeva OR, Newgard CB, et al. Metabolomic profiling reveals a role for caspase‐2 in lipoapoptosis. J Biol Chem 2013;288:14463–14475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Machado MV, Michelotti GA, de Almeida Pereira T, Boursier J, Kruger L, Swiderska‐Syn M, et al. Reduced lipoapoptosis, hedgehog pathway activation and fibrosis in caspase‐2 deficient mice with non‐alcoholic steatohepatitis. Gut 2015;64:1148–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Povero D, Eguchi A, Niesman IR, Andronikou N, de Mollerat du Jeu X, Mulya A, et al. Lipid‐induced toxicity stimulates hepatocytes to release angiogenic microparticles that require Vanin‐1 for uptake by endothelial cells. Sci Signal 2013;6:ra88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Verma VK, Li H, Wang R, Hirsova P, Mushref M, Liu Y, et al. Alcohol stimulates macrophage activation through caspase‐dependent hepatocyte derived release of CD40L containing extracellular vesicles. J Hepatol 2016;64:651–660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hirsova P, Ibrahim SH, Krishnan A, Verma VK, Bronk SF, Werneburg NW, et al. Lipid‐induced signaling causes release of inflammatory extracellular vesicles from hepatocytes. Gastroenterology 2016;150:956–967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cannito S, Morello E, Bocca C, Foglia B, Benetti E, Novo E, et al. Microvesicles released from fat‐laden cells promote activation of hepatocellular NLRP3 inflammasome: a pro‐inflammatory link between lipotoxicity and non‐alcoholic steatohepatitis. PLoS One 2017;12:e0172575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Canbay A, Taimr P, Torok N, Higuchi H, Friedman S, Gores GJ. Apoptotic body engulfment by a human stellate cell line is profibrogenic. Lab Invest 2003;83:655–663. [DOI] [PubMed] [Google Scholar]
- 26. Canbay A, Feldstein AE, Higuchi H, Werneburg N, Grambihler A, Bronk SF, et al. Kupffer cell engulfment of apoptotic bodies stimulates death ligand and cytokine expression. Hepatology 2003;38:1188–1198. [DOI] [PubMed] [Google Scholar]
- 27. Mengshol JA, Golden‐Mason L, Rosen HR. Mechanisms of disease: HCV‐induced liver injury. Nat Clin Pract Gastroenterol Hepatol 2007;4:622–634. [DOI] [PubMed] [Google Scholar]
- 28. Pockros PJ, Schiff ER, Shiffman ML, McHutchison JG, Gish RG, Afdhal NH, et al. Oral IDN‐6556, an antiapoptotic caspase inhibitor, may lower aminotransferase activity in patients with chronic hepatitis C. Hepatology 2007;46:324–329. [DOI] [PubMed] [Google Scholar]
- 29. Masuoka HC, Guicciardi ME, Gores GJ. Caspase inhibitors for the treatment of hepatitis C. Clin Liver Dis 2009;13:467–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Witek RP, Yang L, Liu R, Jung Y, Omenetti A, Syn WK, et al. Liver cell–derived microparticles activate hedgehog signaling and alter gene expression in hepatic endothelial cells. Gastroenterology 2009;136:320–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Tesauro M, Thompson WC, Moss J. Effect of staurosporine‐induced apoptosis on endothelial nitric oxide synthase in transfected COS‐7 cells and primary endothelial cells. Cell Death Differ 2006;13:597–606. [DOI] [PubMed] [Google Scholar]
- 32. Garcia‐Pagan JC, Gracia‐Sancho J, Bosch J. Functional aspects on the pathophysiology of portal hypertension in cirrhosis. J Hepatol 2012;57:458–461. [DOI] [PubMed] [Google Scholar]
- 33. Villanueva C, Albillos A, Genesca J, Abraldes JG, Calleja JL, Aracil C, et al. Development of hyperdynamic circulation and response to beta‐blockers in compensated cirrhosis with portal hypertension. Hepatology 2016;63:197–206. [DOI] [PubMed] [Google Scholar]
- 34. Villanueva C, Aracil C, Colomo A, Hernandez‐Gea V, Lopez‐Balaguer JM, Alvarez‐Urturi C, et al. Acute hemodynamic response to beta‐blockers and prediction of long‐term outcome in primary prophylaxis of variceal bleeding. Gastroenterology 2009;137:119–128. [DOI] [PubMed] [Google Scholar]
- 35. Abraldes JG, Albillos A, Banares R, Turnes J, Gonzalez R, Garcia‐Pagan JC, et al. Simvastatin lowers portal pressure in patients with cirrhosis and portal hypertension: a randomized controlled trial. Gastroenterology 2009;136:1651–1658. [DOI] [PubMed] [Google Scholar]
- 36. Mandorfer M. Sustained virologic response to interferon‐free therapies ameliorates HCV induced portal hypertension. J Hepatol 2016;65:692–699. [DOI] [PubMed] [Google Scholar]
- 37. Afdhal N, Everson GT, Calleja JL, McCaughan GW, Bosch J, Brainard DM, et al. Effect of viral suppression on hepatic venous pressure gradient in hepatitis C with cirrhosis and portal hypertension. J Viral Hepat 2017;24:823–831. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials