

Abstract
We have addressed the role of bacterial co‐infection in viral oncogenesis using as model Epstein–Barr virus (EBV), a human herpesvirus that causes lymphoid malignancies and epithelial cancers. Infection of EBV carrying epithelial cells with the common oral pathogenic Gram‐negative bacterium Aggregatibacter actinomycetemcomitans (Aa) triggered reactivation of the productive virus cycle. Using isogenic Aa strains that differ in the production of the cytolethal distending toxin (CDT) and purified catalytically active or inactive toxin, we found that the CDT acts via induction of DNA double strand breaks and activation of the Ataxia Telangectasia Mutated (ATM) kinase. Exposure of EBV‐negative epithelial cells to the virus in the presence of sub‐lethal doses of CDT was accompanied by the accumulation of latently infected cells exhibiting multiple signs of genomic instability. These findings illustrate a scenario where co‐infection with certain bacterial species may favor the establishment of a microenvironment conducive to the EBV‐induced malignant transformation of epithelial cells.
Keywords: Epstein–Barr virus, virus reactivation, Gram‐negative bacteria, cytolethal distending toxin, DNA damage
Short abstract
What's new?
Little is known about the influence of coinfections, especially of bacteria, on viral oncogenesis. Here, the authors examined the effect of the cytolethal distending toxin (CDT) of Aggregatibacter actinomycetemcomitans, a common oral pathogen, on epithelial cells infected with Epstein–Barr virus (EBV). Exposure of EBV+ cells to CDT induced viral reactivation, while exposure of EBV‐ cells to low amounts of CDT led to the accumulation of latently infected cells upon infection, pointing to a multi‐layered role of bacterial co‐infection in viral oncogenesis.
Introduction
Infections are important contributors to the global cancer burden, particularly in less developed countries where genetic, socioeconomic, and geographic factors converge to promote specific cancer types1. Several infectious agents have been classified as human carcinogens.2 In addition, co‐infections with human immunodeficiency virus type 1 (HIV‐1) and Plasmodium falciparum are associated with a dramatic increase of cancers that are causally linked to other infectious agents, in particular Epstein–Barr virus (EBV) and Kaposi sarcoma herpes virus (KSHV).3 The mechanisms by which coinfection with components of the normal or pathogenic microbiome may contribute to viral oncogenesis are poorly understood.
EBV is a human herpes virus implicated in the pathogenesis of malignancies of lymphoid and epithelial cell origin, including endemic Burkitt's lymphoma, Hodgkin's lymphoma, posttransplant, and immunodeficiency‐associated lymphomas, midline granuloma, nasopharyngeal carcinoma (NPC), and gastric carcinoma.4 EBV oncogenicity is epitomized by the capacity of the virus to confer autonomous proliferation to B‐lymphocytes that become lymphoblastoid cell lines (LCLs) in vitro and can give rise to rapidly growing lymphomas in vivo. B‐cell immortalization is associated with the establishment of a nonproductive infection where only few latency‐associated viral genes are expressed.5 These encode for proteins, noncoding RNAs and microRNAs that, alone or in combination with alterations of cellular oncogenes and tumor suppressor genes, mediate the acquisition of several hallmarks of cancer, including autonomous growth, escape from apoptosis and replicative immortality.6
While EBV‐carrying B‐cell lines provide valuable tools for dissecting the key features of growth transformation and oncogenesis in this cell type, much less is known about the mechanisms by which the virus contributes to the pathogenesis of epithelial tumors. This is explained partly by the difficulty to infect primary epithelial cells in vitro, and partly by the rapid loss of the viral genome upon in vitro explant of biopsies from EBV‐positive NPC and gastric carcinoma.7, 8 The reasons for the poor susceptibility of epithelial cells to EBV infection are partially understood. Epithelial cells do not express the C3d receptor that serves as a high‐affinity EBV binding site in B‐lymphocytes.9 Furthermore, although alternative routes of entry, including polymeric IgA,8 integrins 10 or other surface moieties,11, 12 may be used, the establishment of persistent infection remains a rare event. The requirement for a particular cellular environment is substantiated by the findings that overexpression of cyclin D1 supports stable EBV infection in nasopharyngeal cells,13 while expression of interleukin 6 (IL6) and interleukin 6 receptor (IL6R) IL‐6/IL‐6R promotes the growth of EBV‐infected premalignant epithelial cells.14 Thus, both the efficiency and the outcome of infection appear to be influenced by environmental constraints that are not easily reproduced under in vitro culture conditions.
EBV carrying epithelial tumors arise in the nasopharynx and the stomach that are colonized by a highly diverse bacterial microflora. Oral bacterial biofilms are commonly linked to periodontitis.15 Epidemiologic studies implicate poor oral health in the pathogenesis for cancers of the head and neck, esophagus, stomach, and pancreas16 It is generally assumed that oncogenesis is linked to chronic inflammation via the local accumulation of genotoxic agents, such as reactive oxygen species and a variety of cytokines that sustain cell proliferation and inhibit apoptosis.17 Bacteria may contribute by triggering inflammation, or more directly via the release of toxins and metabolites that can influence the growth properties of epithelial cells.18
We have investigated the capacity of oral pathogenic bacteria to affect the outcome of EBV infection in epithelial cells. We found that bacteria commonly associated with periodontitis release effector molecules that induce EBV reactivation via different mechanisms. The cytolethal distending toxin (CDT) produced by Aggregatibacter actinomycetemcomitans (Aa) acted via induction of DNA damage and activation of the ataxia telangectasia mutated (ATM) kinase. Furthermore, EBV infection of epithelial cells in the presence of low amounts of CDT resulted in the accumulation of latently infected cells with signs of DNA damage and genomic instability, pointing to a role of co‐infection in viral oncogenesis.
Materials and Methods
Bacteria
The Aggregatibacter actinomycetemcomitans (Aa) strain HK1651 (ATCC 700685), the Aa D7SS‐smooth strain and its derivative D7SS‐smooth ΔcdtABC, with deletion of the CDT operon19 (gift of Dr. Casey Chen, Ostrow School of Dentistry, University of Southern California, California) were grown in Tryptic Soy Broth (BD, Franklin Lakes, New Jersey) at 37°C 5% CO2 for 48 hr. Corynebacterium kutscheri (Ck) (ATCC 15677) was grown in Brain Heart Infusion Broth (BD) at 37°C 5% CO2 for 24 hr. Klebsiella pneumonia subsp. pneumonia (Kp) (ATCC 13883) was grown in Nutrient Broth (BD) at 37°C in aerobic conditions. Porphyromonas gengivalis (Pg) (ATCC 33277) was grown in supplemented Tryptic Soy Broth (ATCC 2722) at 37°C in anaerobic conditions for 24 hr.
Cell lines
The AGS‐Bx1 gastric carcinoma cell line (gift of Alan K.S. Chiang, LKS Faculty of Medicine, The University of Hong Kong, Hong Kong) carries a recombinant EBV where transcription of GFP is driven by the CMV immediate early promoter.20 A stable subline of EBV‐negative AGS cells21 expressing the DsRed protein was generated by transfection with the pDsRed‐Express‐N1 plasmid (Clontech, Clontech, Mountain View, CA, Cat No. 632429) followed by selection in 500 μg/ml G418. The EBfaV‐GFP cell line (gift of Richard Longnecker, Northwestern University Medical School Chicago, Illinois) carries a recombinant EBV where the expression of GFP is driven by the LMP2 promoter.22 B95.8 virus transformed LCLs were generated as previously described.23
Treatments
AGS‐Bx1 cells were cultured in 6 wells plates in RPMI1640 supplemented with 10% FBS without antibiotics (complete medium). Cocultured with Aa or Pg was carried out at the indicated multiplicity of infection (MOI) in complete medium. As positive control for EBV reactivation, cells were treated with 30 ng/ml of 12‐o‐tetradecanoylphorbol‐13‐acetate (TPA) and 0.5 mM sodium butyrate (Bu) (Sigma‐Aldrich, Darmstadt, Germany). The bacteria were heat inactivated at 100°C for 15 min or fixed in 4% formaldehyde (MERK, Darmstadt, Germany) in PBS for 20 min. Supernatants from bacterial cultures grown for 24 hr (Pg, Ck, Kp) or 48 hr (Aa) were normalized at the optical density of 0.3 at 600 nm, cleared by centrifugation for 20 min at 3500 rpm and sterilized through a 0.2 μm filter. The supernatants were heat inactivated at 100°C for 15 min or fractionated by centrifugation for 30 min at 3500 rpm through an Amicon Ultra‐15 Centrifugal Filter Unit with Ultracel‐3 membrane (cutoff 3 kDa, Millipore, Bedford, Massachusetts).
DNA‐damaging agents
The wild type and the mutant CDTD273R from Heamophilus ducreyi were previously described.24 Cells were exposed for 6 hr or 24 hr to the following genotoxic agents: wild type or mutant CDT (1 μg/ml), Etoposide (40 μM, Sigma‐Aldrich), Camptothecin (5 μM, Selleckchem, Munich, Germany), UV irradiation by exposure to a UV transilluminator (Spectroline, Westbury, New York) for 20 sec. When indicated, cells were preincubated with the ATM inhibitor KU‐60019 (1 μM, Selleckchem) or SP1 inhibitor mithramycin A (Sigma‐Aldrich) for 1 hr prior to addition of the DNA damaging agents.
Western blot
Cell lysate was prepared in SDS lysis buffer (2% SDS, 10% glycerol, 65 mM TRIS–HCl pH 7.5), fractionated in NuPAGE 4–12% Bis‐Tris Gels (Invitrogen, Carlsbad, California), transferred to polyvinylidene difluoride (PVDF) membranes (Millipore) and probed with the indicated antibodies, followed by the appropriate horseradish peroxidase‐conjugated secondary antibody (GE Healthcare, Piscataway, New Jersey). The complexes were visualized by chemiluminescence (SuperSignal™, Thermo scientific, Waltham, Massachusetts). The following primary antibodies were used: mouse anti‐BZLF1 (sc‐53904; Santa Cruz Biotechnology, Dallas, Texas, USA), mouse anti‐BMRF1, rat anti‐BFRF3, rabbit anti‐BdRF1 (gifts of Jaap M. Middeldorp, VU University Medical Center, Amsterdam, Netherlands), mouse anti‐γH2AX and rabbit anti‐p53 (9718, 9282, Cell Signaling Technology, Danvers, Massachusetts), mouse anti‐phosphoATM‐Ser1981 (05–740, Millipore), rabbit anti‐phospho‐p53‐Ser15 (Ab1431, AbCam, Cambridge, UK), mouse anti‐p21 (610233, BD), mouse anti‐β‐actin (A5441, Sigma‐Aldrich).
Immunofluorescence
AGS‐Bx1 cells were grown on 13 mm diameter coverslips in 24 well plates. After treatment, cells were fixed for 20 min in 4% formaldehyde in PBS and permeabilized in PBS supplemented with 0.2% Triton X‐100 and 3% bovine serum albumin for 30 min at room temperature, following incubation with primary antibodies diluted 1:100, and the appropriate Alexa Fluor‐conjugated secondary antibodies. The slides were mounted in Vectashield‐containing DAPI mounting medium (Vector laboratories, Inc. Burlingame, California), and images were acquired with a fluorescence microscope (Leica DM RA2, Leica Microsystems, Wetzlar, Germany) equipped with a CCD camera (C4742–95, Hamamatsu, Japan). The following antibodies were used: rabbit anti‐phospho‐KAP1 (Nordic Biosite, Täby, Sweden), mouse‐anti 53BP1 (BD).
EBV infection of epithelial cells
EBfaV‐GFP cells were induced by treatment with 20 ng/ml TPA and 3 mM Bu in complete medium for 6 hr, followed by washing and culture for additional 18 hr in complete medium. For infection, 8 × 104 induced EBfaV‐GFP cells were mixed at 1:1 ratio with untreated AGS‐DsRed cells or AGS‐DsRed cells treated for 1 hr with 100 ng/ml purified CDT. The cells were seeded in wells of 12 well plates containing 13 mm diameter coverslips. After 48 hr, the adherent cells were fixed, permeabilized, and stained as described above. The following antibodies were used: mouse anti‐BZLF1 (sc‐53904, Santa Cruz Biotechnology) and mouse anti‐γH2AX (05‐636I, Merck‐Millipore, Darmstadt, Germany). Images were acquired with a confocal scanning microscope (Zeiss Confocal Microscope LSM510 META, Carl Zeiss Microscopy, Jena, Germany).
Results
Oral pathogenic bacteria promote EBV reactivation
To establish in vitro culture conditions that could mimic the long‐term exposure to bacteria and their products that may occur at mucosal sites, we turned to bacteria that cause chronic periodontitis and are implicated in the pathogenesis of systemic diseases including cancer. A list of the major microbial components of oral biofilms is shown in Supporting Information Table S1. Membrane‐associated virulence factors, including lipopolysaccharide (LPS), fimbriae, flagella, and various capsular components, and secreted virulence factors, including enzymes, toxins, and metabolites such as short‐chain fatty acids (SCFA), are differently expressed by these bacteria. Porphyromonas gingivalis (Pg) and Aggregatibacter actinomycetemcomitans (Aa) exhibit the largest difference in the secretion of SCFA and the cytolethal distending toxin (CDT), respectively, and were therefore chosen to probe the effect of coinfection.
To choose conditions of infection that allow eukaryotic cells survival while preventing bacteria overgrowth in the absence of antibiotics, the EBV‐positive epithelial cell line AGS‐Bx1 was infected with live bacteria at MOI between 12:1 and 100:1 (Supporting Information Fig. S1). Bacterial growth assessed by optical density (OD) of culture supernatants (Supporting Information Fig. S1A), and the recovery of viable AGS‐Bx1 cells (Supporting Information Fig. S1B), were monitored after 48 hr. Aa showed limited growth capacity not affected by the seeding dose or by the presence of AGS‐Bx1 cells. In contrast, the recovery of Pg increased in a dose‐dependent manner in the absence of cells but was strongly inhibited in the presence of AGS‐Bx1. Both bacteria decreased cell recovery (Supporting Information Fig. S1B). However, in line with the capacity of the secreted CDT to induce cell cycle arrest, the effect of Aa was independent of the initial MOI, while the effect of Pg was dose‐dependent, suggesting that the live bacterium is toxic to the cells.
We then asked whether infection with Aa or Pg may reactivate the productive virus cycle as assessed by expression of immediate early (BZLF1), early (BMFR1), and late (BdFR1 and BFRF3) viral proteins. Cells treated with the known inducers TPA/Bu were used as reference. Infection with both Aa and Pg induced the full spectrum of immediate early, early and late viral antigens (Fig. 1), suggesting that the productive virus cycle progresses to completion resulting in release of infectious virus. In Aa infected cells, BZLF1 and BMRF1 were expressed at levels compared to cells treated with TPA/Bu, while the expression of late antigens showed a clear dose dependent effect (Fig. 1 a). In line with the toxic effect of Pg, lower levels of all viral antigens were detected in infected cells compared to cells treated with TPA/Bu (Fig. 1 b). Based on bacterial recovery, cell viability and efficiency of EBV reactivation, subsequent experiments were performed at MOI 25:1.
Figure 1.
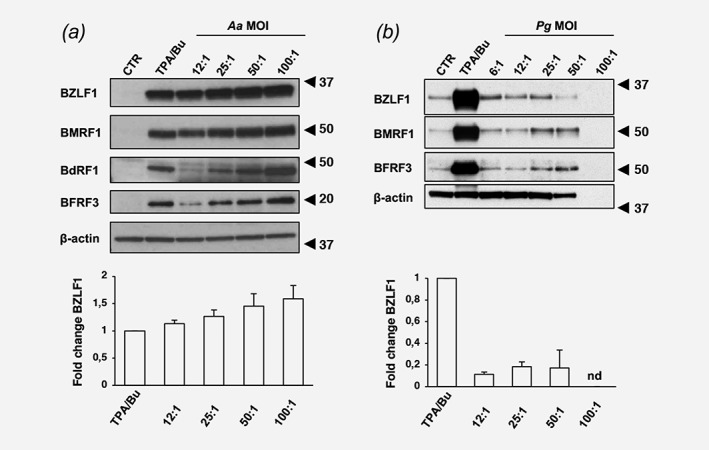
Exposure to live bacteria promotes EBV reactivation. AGS‐Bx1 cells were infected with Aa (a) or Pg (b) at the indicated MOI for 48 hr. EBV reactivation was assessed by probing Western blots with the indicated antibodies to lytic viral antigens. β‐actin was used as loading control. Representative Western blot illustrating the expression of viral proteins (upper panels) and the relative expression of BZLF1 calculated as the ratio between the intensity of the specific band in infected versus TPA/Bu treated cells in three independent experiments (lower panels) are shown. Nd, not done, due to low recovery of viable cells.
Different bacterial products induce EBV reactivation
In order to assess whether the inducing factors are secreted or associated with the bacterial cells, the effect of live bacteria was compared with that of bacteria killed by heat inactivation for 15 min at 100°C, or fixed in 4% PFA for 20 min. Both treatments abolished the capacity of Aa to promote EBV reactivation as assessed by expression of the immediate early antigen BZLF1, suggesting that the inducing factor(s) is secreted by the live bacterium (Fig. 2 a). In contrast, heat inactivation strongly enhanced the inducing capacity of Pg (Fig. 2 b), while the effect was abolished by PFA fixation, suggesting that the activity of a heat stable factor(s) may be masked by a heat‐sensitive toxin. The involvement of secreted factors was confirmed by the capacity of sterile bacterial culture supernatants from both Aa and Pg to induce EBV reactivation in a dose dependent manner (Fig. 2 c). Of note, the Pg culture supernatant did not exhibit the toxic effect of the live bacteria, indicating that the heat‐sensitive toxin may not be released (compare heat inactivation in Figs. 2 b and 2 d).
Figure 2.
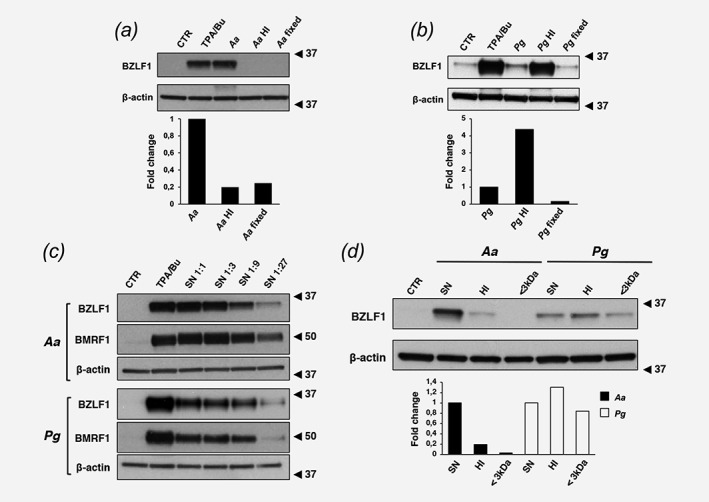
Factors secreted by Aa and Pg promote EBV reactivation. AGS‐Bx1 cells were cultured for 48 hr with live bacteria, heat inactivated bacteria (15 min at 100°C, HI) or bacteria fixed in 4% formaldehyde (fixed). The upper panels show representative Western blots of BZLF1 expression in cells infected with Aa (a) and Pg (b). The lower panels show the relative expression of BZLF1 calculated as ratio between the intensity of the specific bands in cells exposed to inactivated bacteria versus live bacteria. Mean of two independent experiments. (c) AGS‐Bx1 cells were exposed for 48 hr to serial dilutions of sterile culture supernatant from Aa (upper panel) or Pg (lower panel). TPA/Bu treated cells were used as positive control. Reactivation of the EBV lytic cycle was assessed by probing Western blot with the indicated antibodies. β‐actin was used as loading control. (d) AGS‐Bx1 cells were exposed for 48 hr to sterile bacterial culture supernatants from Aa or Pg that were either untreated (SN), heat inactivated for 15 min at 100°C (HI), or centrifuged through a 3 kDa cut‐off membrane (<3 kDa). Reactivation of the EBV productive cycle was assessed by Western blot as described in A. The graph shows the relative expression of BZLF1 in cells exposed to treated versus untreated bacterial supernatants. Mean of two independent experiments.
To further investigate the nature of the inducing factors, bacterial supernatants were heat inactivated or depleted of high molecular weight products by centrifugation through a 3 kDa cut‐off membrane. Heat inactivation and depletion of high molecular weight components strongly reduced the capacity of Aa supernatants to induce BZLF1 (Fig. 2 d), whereas neither treatment altered the activity of the Pg supernatant. Thus, while Aa secretes a heat‐sensitive factor, possibly a protein of molecular weight > 3 kDa, Pg produces a heat‐stable factor of small molecular weight. This bacterium secretes large amounts of the histone deacetylase (HDAC) inhibitor butyrate.25 The possibility that butyrate may be the active metabolite secreted by Pg was substantiated by the dose‐dependent induction of BZLF1 in butyrate treated AGS‐Bx1 (Supporting Information Fig. S2), whereas propionate and acetate (Supporting Information Fig S2), and supernatant from bacteria that produce propionate (Klebsiella pneumoniae) or acetate (Corynebacterium kutscheri) (Supporting Information Fig. S3) were inactive.
The properties of the factor produced by Aa point to the CDT. This DNAse‐like genotoxin induces single‐ and double‐strand (SSB, DSB) DNA breaks, and activates the DNA damage response (DDR).26 To test whether CDT promotes EBV reactivation, AGS‐Bx1 cells were infected with wild type Aa (Aawt) or an isogenic strain with deletion of the cdtABC operon (AaΔCDT). Production of the toxin was confirmed by the appearance of phosphorylated and ubiquitinated histone‐2AX (γH2AX and Ub‐H2AX). The induction of BZLF1 was drastically decreased in cells infected with AaΔCDT (Fig. 3 a). TPA/Bu, Aawt and to a minor extent AaΔCDT induced the accumulation of γH2AX, while Ub‐H2AX, signaling progression of the DNA damage response (DDR), was detected only in cells infected with Aawt. The capacity of the toxin to induce EBV reactivation was confirmed by comparing the expression of BZLF1 in cells treated with purified CDT (wt) or with a catalytically inactive toxin (mut) with mutation of the Mg2+ binding site of the enzyme24. Only the active toxin induced a dose‐dependent expression of BZLF1 (Fig. 3 b).
Figure 3.
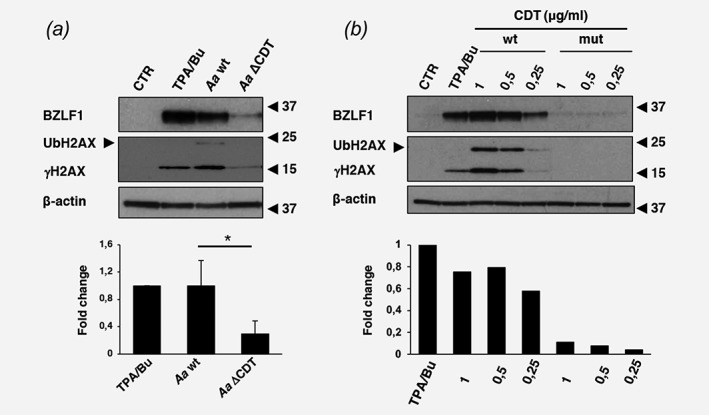
The CDT promotes EBV reactivation. (a) AGS‐Bx1 cells were infected for 48 hr at MOI of 25:1 with Aa isogenic strains encoding a functional CDT (Aa wt) or a deleted CdtABC operon (Aa ΔCDT). Reactivation of the EBV lytic cycle was assessed by Western blot using a BZLF1 specific antibody. CDT‐induced DNA damage was visualized using an antibody specific for phosphorylated H2AX (γH2AX). The arrow indicates ubiquitinated γH2AX (UbH2AX). β‐actin was used as loading control. The lower panel shows the relative BZLF1 expression calculated as the ratio between the intensity of the specific band in infected cells and cells treated with TPA/Bu. Mean±SD of three independent experiments. Statistical analysis was performed using the Student's t‐test, *p < 0,05. (b) AGS‐B× 1 cells were intoxicated for 48 hr with the indicated concentrations of the purified active CDT (wt) or catalytically inactive toxin (mut). Reactivation of the EBV lytic cycle and induction of the DNA damage response (DDR) were detected as described in A. The lower panel shows the relative BZLF1 expression calculated as the ratio between the intensity of the specific band in intoxicated cells versus cells treated with TPA/Bu. Mean of two independent experiments.
The bacterial CDT promotes EBV reactivation via induction of the DNA Damage response
CDT induces an ATM‐coordinated DDR that involves the phosphorylation of DNA damage sensors and response amplifiers, for example, KAP1 and H2AX, and subsequent recruitment of effectors of DNA repair, for example, 53BP1, and cell cycle arrest, for example, CHK2 and p53.27 Phosphorylation of H2AX is an early event of the DDR commonly used as a surrogate marker of DNA damage. Since a comparable accumulation of γH2AX was observed in AGS‐Bx1 cells treated for 48 hr with either CDT or TPA/Bu (Fig. 3 a), we asked whether both inducers acted via activation of the DDR. To this end, the phosphorylation of KAP1 and the formation of 53BP1 foci were monitored by immunofluorescence in AGS‐Bx1 cells treated for 6 hr or 24 hr with TPA/Bu, CDT, and etoposide (ETOP), a known inducer of DNA double strand breaks (Figs. 4 a, 4 b). As expected, both CDT and etoposide promoted phosphorylation of KAP1 and the formation of 53BP1 foci, which peaked already after 6 hr of treatment. In contrast, these early and late markers of DDR activation were not detected in cells exposed to TPA/Bu for 6 hr or 24 hr.
Figure 4.
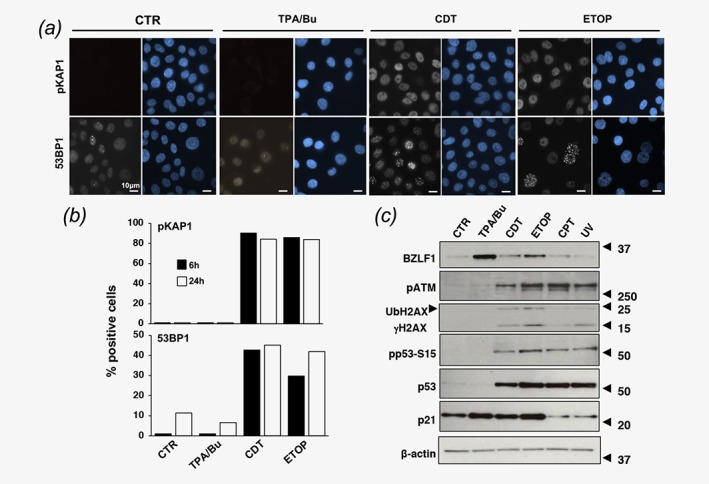
DNA double strand breaks promote EBV reactivation. (a). AGS‐Bx1 cells were exposed to TPA/Bu, CDT (1 μg/ml) or Etoposide (ETOP, 40 μM) for 6 hr or 24 hr. Activation of the DDR was monitored by detection of phosphorylated KAP1 (pKAP1, early marker) and 53BP1 foci (late marker). Nuclei were counterstained with DAPI (blue). Magnification 63 ×. (b). The percentage of positive cells assessed by scoring 90 cells per condition and time point. Cells with ≥5 53BP1 foci/nucleus were scored as positive. (c) AGS‐Bx1 cells were left untreated (CTR) or treated for 6 hr with TPA/Bu, CDT (1 μg/ml), Etoposide (ETOP, 40 μM), Camptothecin (CPT, 5 μM) or exposed to UV, and further incubated for 6 hr. Reactivation of the lytic cycle and induction of the DDR response were monitored by western blot using antibodies to BZLF1, phosphorylated ATM (pATM), γH2AX, p53, Ser15 phosphorylated p53 (pp53‐S15), and p21. The arrow indicates the ubiquitinated form of γH2AX (UbH2AX). β‐actin was used as loading control. One representative experiment out of three where all the antibodies were tested in parallel is shown.
To further explore the relationship between DNA damage and virus reactivation, AGS‐B × 1 cells were exposed to a panel of genotoxic agents that cause DNA DSBs (CDT and etoposide), DNA SSBs (Camptothecin, CPT) or pyrimidine dimers (UV irradiation, UV). In order to focus on the early triggering events, the expression of BZLF1 and markers of the DDR were monitored 6 hr after treatment (Fig. 4 c). BZLF1 was clearly detected in cells treated with CDT and etoposide, in parallel with phosphorylation of ATM, phosphorylation and ubiquitination of H2AX, phosphorylation of p53‐S15 and upregulation of p21. In contrast, treatment with CPT or UV had only marginal effects, suggesting that DNA double strand breaks are required for virus reactivation, which was confirmed by the failure to observe BZLF1 expression in cells treated with CPT for 24 hr (Supporting Information Fig. S4). In line with the data shown in Figures 3 and 4 a, phosphorylation of ATM, H2AX, and p53‐S15 were not detected in cells treated for with TPA/Bu (Fig. 4 c). Upregulation of p21 in the absence of p53 stabilization was observed in some of the experiments, probably due to the transient activation of p53 independent signaling pathways. Of note, ATM phosphorylation was observed in cells treated with TPA/Bu, CPT, or UV for 24 hr or longer (Supporting Information Fig. S4), suggesting that lytic viral products may activate some components of the DDR. The capacity of etoposide to trigger virus reactivation was reproduced in two freshly established EBV‐transformed LCLs (Supporting Information Fig. S5).
ATM plays a key role in the response to DNA double strand breaks through phosphorylation of a large number of substrates that regulate the cell cycle, DNA repair, gene transcription, and chromatin remodeling. Amongst the transcription factors targeted by ATM, Specific Protein‐1 (SP1) was previously shown to mediate the transcriptional activation of BZLF1 in response to butyrate.28 In order to assess whether the ATM‐SP1 signaling cascade is involved in the response to genotoxic agents, AGS‐Bx1 cells were pretreated with the ATM inhibitor KU‐60019 for 1 hr prior to exposure to CDT, etoposide or TPA/Bu in continuous presence of the inhibitor. The activity of KU‐60019 was confirmed by reduced phosphorylation of ATM and p53‐S15, and failure to upregulate p21 (Fig. 5 a). Inhibition of ATM abolished the induction of BZLF1 in cells treated with CDT and etoposide but had no effect in cells treated with TPA/Bu, confirming the involvement of different triggering pathways. Treatment with mithramycin A, a selective inhibitor of the binding of SP1 to target promoters,29 abolished the induction of BZLF1 in response to both TPA/Bu and etoposide (Fig. 5 b), indicating that, while differing in the upstream triggering events, the effects of HDAC inhibitors and DNA damage converge on activation of the SP1 transcription factor.
Figure 5.
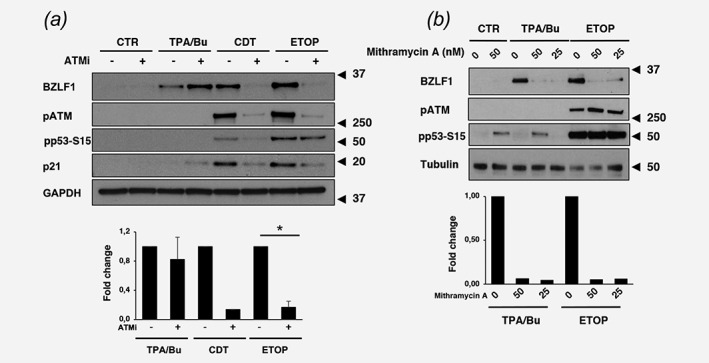
EBV reactivation by genotoxic agents is dependent on the ATM kinase and the SP1 transcription factor. (a) AGS‐Bx1 cells were pre‐incubated for 1 hr with the ATM inhibitor (ATMi) KU‐60019 (1 μM) prior to exposure to TPA/Bu, CDT, or Etoposide for 6 hr in the presence of the inhibitor. Reactivation of the lytic cycle and induction of the DDR was monitored by western blot using the indicated antibodies. GAPDH was used as loading control. The lower panel shows the quantification of BZLF1 expressed as the ratio between the intensity of the specific band in cells pretreated with the ATMi versus cells exposed to vehicle (DMSO). Mean ±SD of three independent experiments. Statistical analysis was performed using the Student's t‐test, *p < 0,05. (b) AGS‐BX1 cells were preincubated for 1 hr with the SP1 inhibitor mithramycin at the indicated concentrations (nM) prior to exposure to TPA/Bu or Etoposide for 6 hr in the presence of the inhibitor. Reactivation of the lytic cycle and induction of the DDR was monitored by western blot analysis. Tubulin was used as loading control. The lower panel shows the quantification of BZLF1 expressed as the ratio between the intensity of the specific band in cells pretreated with mithramycin versus cells exposed to vehicle (DMSO). Mean of two independent experiments.
Co‐exposure to EBV and CDT promotes genomic instability
In addition to inducing DNA damage and cell cycle arrest, the CDT triggers a cell survival response that leads to reorganization of the actin cytoskeleton and activation of the MAP kinases.30 To assess whether exposure to the toxin may influence the susceptibility of epithelial cells to EBV infection, EBV‐negative AGS cells were infected by cocultivation with EBfaV‐GFP cells that carry a recombinant EBV where the expression of GFP is driven by the LMP2 promoter that is expressed in latently infected cells. In order to distinguish between the two cell‐types, the AGS cells were stably transfected with a plasmid expressing the DsRed fluorescent protein. Latently EBV infected AGS cells were visualized by co‐localization of red and green fluorescence, while productive infection was monitored by BZLF1 specific immunofluorescence. Approximately 20% of AGS cells became infected upon cocultivation with induced EBfaV‐GFP for 48 hr and the majority of the cells were BZLF1 negative (Fig. 6 a). Exposure to CDT had a small enhancing effect on the activation of the productive virus cycle but induced a twofold to threefold increase in the number of latently infected cells with signs of genomic instability, including the accumulation of nuclear γH2AX foci (Fig. 6 b), and the formation of micronuclei (Fig. 6 c).
Figure 6.
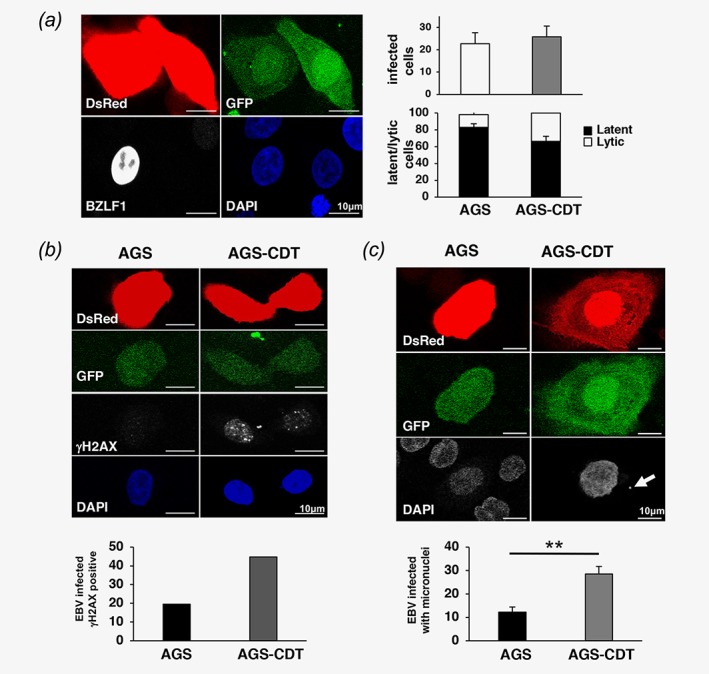
Co‐exposure to EBV and low doses of CDT induces signs of genomic instability in epithelial cells. EBV negative AGS cells stably expressing DsRed were infected with EBV by cocultivation with induced EBfaV cells. Where indicated, the cells were exposed to 0.1 μg/ml CDT for 1 hr (AGS‐CDT) prior to cocultivation with EBfaV for 48 hr. EBV infection was scored by colocalization of green and red fluorescence and productive infection was monitored by expression of BZLF1. (a) Left: representative scanning confocal micrograph of infected AGS‐CDT cells showing co‐expression of red and green fluorescence and expression of BZLF1 (white). The nuclei were counterstained with DAPI. Right: quantification of the percentage of infected cells and percentage of latently (green fluorescence only) versus productively infected cells (BZLF1 positive green cells). Mean±SEM of fours independent experiments. (b) Upper panels: representative scanning confocal micrograph of infected AGS illustrating the induction of DNA damage assessed by γH2AX immunofluorescence (white). Cells with ≥5 γH2AX foci/nucleus were scored as positive. Mean of two independent experiments. (c) Upper panels: representative scanning confocal micrograph of infected AGS cells illustrating the presence of micronuclei (white arrow) detected by DAPI staining. The lower panel shows the quantification of micronuclei positive infected cells. Mean±SD of three independent experiments. Statistical analysis was performed using the Student's t‐test, **p < 0,001.
Discussion
In this study, we have explored the role of bacterial coinfection in the pathogenesis of EBV‐associated epithelial cell malignancies and extended previous findings on the capacity of common oral pathogenic bacteria to modulate the fate of EBV infection in this cell type. We have found that a genotoxin produced by Aa and other Gram‐negative bacteria that colonize the gastrointestinal tract31 causes EBV reactivation via induction of DNA damage. Remarkably, we also found that CDT can synergize with EBV infection in the induction of genomic instability in epithelial cells, illustrating a new mechanism by which bacterial coinfection can contribute to the establishment of a cellular environment conducive to malignant transformation.
By comparing the effect of co‐infection with common oral pathogenic bacteria, we have found that factors released by Pg or Aa can trigger virus reactivation in EBV positive epithelial cells. Pg acted via the release of a low molecular weight metabolite, most likely butyrate that is known to promote EBV reactivation via inhibition of HDACs and epigenetic re‐modeling. Unexpectedly, we found that CDT produced by Aa triggers a different activation pathway that involves the induction of DNA damage and activation of the DDR. In this context, it is interesting to note that high loads of EBV and cytomegalovirus (CMV) DNA are frequently detected in the saliva and periodontal pockets of patients with periodontitis,36 suggesting that the reactivation of latent herpesvirus infections may be a common outcome of the bacterial colonization of this site.
Although the productive cycle of EBV is not permissive for cell growth, ample epidemiological evidence suggests the involvement of virus reactivation in malignant transformation, including the presence of elevated antibodies titers to lytic viral antigens and high EBV DNA load in the serum and nasopharynx of NPC patients,32 which often precedes by several years the onset of the disease.33 It has been speculated that the products of genes expressed during the productive cycle, including viral IL‐10 (vIL‐10) and cellular IL‐8, TGFβ, and VEGF, may establish a tumorigenic environment that sustains the proliferation of neighboring latently infected cells.34 In addition, viral proteins that cause DNA damage or interfere with the DDR may be expressed during abortive productive infection, as suggested by the finding that recurrent exposure to TPA/butyrate promoted the acquisition of chromosomal aberration and phenotypic properties of tumor progression in an EBV‐positive NPC cell line but not in the EBV‐negative parental cells.35 We have observed full reactivation of the productive virus cycle with expression immediate early, early, and late genes and presumably production of infectious virus following exposure of EBV positive epithelial cells to live Aa, bacterial culture supernatants or purified CDT. However, it seems possible that fine tuning of the in vitro culture conditions to resemble more closely the oropharyngeal microenvironment may favor abortive virus reactivation with expression of only a selection of immediate early/early genes, which would be compatible with cell survival while promoting the accumulation of genomic instability, due to the combined effect of viral gene products and the bacterial genotoxin.
The capacity of CDT to promote EBV reactivation via induction of DNA damage sheds new light on the regulation of viral latency. Our findings suggest that after DNA double strand breaks the activation of ATM is sufficient to initiate EBV reactivation. ATM is known to be activated following treatment with a variety of EBV inducers, including HDAC inhibitors, TGFβ, 5‐azacytidine, hydrogen peroxide, cisplatin, proteasome inhibitors, and B‐cell receptor crosslinking, but its contribution is unclear as blockade of the kinase had only partial effect on virus production.37 By focusing on the early events of productive infection, we have found that ATM is essential for BZLF1 expression in response to DNA damage but is not required in response to TPA/Bu, indicating that distinct signaling pathways trigger the lytic switch. Indeed, the phosphorylation of ATM and H2AX observed in cells exposed to TPA/Bu for 24 hr was not associated with activation of a canonical DDR, as assessed by failure to detect ubiquitination of H2AX, phosphorylation of KAP1 and p53, and expression of p21. Conceivably, lytic viral proteins, such as BZLF1 or BGLF4, may promote the late activation of ATM to enhance EBV replication through induction of cell cycle arrest and the assembly of replication compartments that are regulated by the kinase.38 In line with the notion that SP1 regulates the BZLF1 promoter, we found that the effects of TPA/Bu and DNA damage converge on activation of this cellular transcription factor. SP1 may serve both as transcriptional repressor and activator depending on the recruitment of HDAC‐containing repressor complexes.39, 40 This interaction is regulated by HDAC inhibitors such as butyrate and by phosphorylation of the interacting partners by various kinases including ATM. It is noteworthy that SP1 is differentially phosphorylated in response to distinct types of DNA damage.41 Thus, a different pattern of SP1 phosphorylation may explain the failure of UV irradiation to serve as an early trigger of BZLF1 expression.
Our findings delineate a scenario where, in addition to inducing virus reactivation, co‐infection with certain bacteria may directly contribute to the oncogenic process by causing genomic instability, establishing thereby a pre‐malignant phenotype that may, together with viral infection, progress to malignancy. We have previously reported that long‐term exposure to sub‐lethal doses of CDT increased mutation frequency and induced chromosomal aberrations in epithelial and mesenchymal cells.42 This tumor promoting effect of the toxin is at least partly dependent on the capacity to concomitantly trigger DNA damage and signaling pathways that favor cell survival.30 Conceivably, the pro‐survival effect of the toxin may be reinforced upon EBV infection by the expression of latent viral products that promote cell proliferation, inhibit apoptosis and support viral latency by inhibiting virus reactivation. Interestingly, infection of EBV negative epithelial cells in the presence of sublethal amounts of the toxin promoted the accumulation of cells expressing virus latency genes but negative for BZLF1, suggesting that the outcome of co‐infection may be influenced by environmental factors operating in vivo such as the relative abundance of different bacteria species or the formation of bacterial biofilms. It remains to be seen which molecular pathways activated by bacteria and their products regulate cellular properties that affect the susceptibility of epithelial cells to EBV infection in terms of enhanced virus entry and/or permissivity for different viral gene expression programs.
The observation that bacterial coinfection may affects in multiple ways the outcome of EBV infection in epithelial cells has interesting implications for the pathogenesis of NPC. NPC shows a typical geographic distribution with the majority of new cases reported in east and south Asia, northern Africa and in some ethnic groups, for example, the Inuits in the Artic.43 While EBV infection is a recognized etiological factor of NPC, genetic susceptibility loci have been identified in the MHC coding region and in genes involved in DNA repair, regulation of cell‐cycle checkpoints, cell adhesion and migration.44, 45 Several studies have identified poor oral health as a risk factor for NPC. In a hospital‐based case–control study, a significant positive association was found between the risk of NPC and infrequent tooth brushing or higher number of decayed teeth.46 In a large population‐based case control study performed in southern China where NPC is endemic, elevated risk of NPC was associated with indicators of poor oral health, such as a higher number of filled teeth.47 Interestingly, Aa is detected in the dental pockets of severe and chronic periodontitis in the majority of patients from southern China48 and north Africa,49 whereas the bacterium is less commonly associated with disease with in European populations. Collectively, these findings warrant a careful investigation of the composition of the oropharyngeal microbiome as a potential risk factor in the pathogenesis of NPC. It is noteworthy that bacteria‐induced DNA damage may also play a significant role in the pathogenesis of EBV positive gastric carcinoma, a distinct molecular subtype of the tumor characterized by recurrent PIK3CA mutations, and extreme DNA hypermethylation.50 Infection with H. pylori is a major risk factor for gastric carcinoma.51 Although there is no experimental evidence for the capacity of H. pylori to produce a CDT, this bacterium induces distinct patterns of DNA damage in epithelial cells 52, 53 Thus, it is conceivable that the interplay between H. pylori and EBV may promote the establishment of microenvironment required for the specific genomic and epigenetic modifications that characterize this tumor.
Supporting information
Figure S1. Recovery of bacteria and eukaryotic cell. Three hundred thousand AGS‐Bx1 cells were co‐cultured with Aa or Pg at the indicated MOI for 48 h and parallel aliquots of bacteria were maintained in complete medium without antibiotics in absence of the eukaryotic cells for the same time. A) Recovery of Aa (squares) or Pg (circles) measured as optical density at 600 nm after culture in the presence (white symbols) or absence (black symbols) of the AGS‐Bx1 cells. Mean ±SD of three independent experiments. B) Recovery of viable AGS‐BX1 cells after culture for 48 h with Aa (white squares) or Pg (black squares) at the indicated MOI. The data are presented as percentage of cells recovery relative to non‐infected cells (CTR). * indicates the initial seeding. Mean ±SD of three independent experiments.
Figure S2. Butyrate is the only SCFA promoting EBV reactivation in AGS‐Bx1 cells. AGS‐Bx1 cells were exposed to the indicated concentrations of butyrate, acetate or propionate for 48 h. The levels of BZLF1 expression was assessed by western blot analysis using an antibody specific for BZLF1. β‐actin was used as loading control.
Figure S3. Induction of EBV reactivation by oral pathogenic bacteria. AGS‐Bx1 cells were exposed for 24 h to sterile bacterial supernatant of Kp, Ck or Aa, normalized at optical density of 0.3 at 600 nm, and diluted 1:3 in complete medium. Cells exposed to TPA/Bu were used as positive control. The upper panel shows a representative western blot where reactivation of the EBV lytic cycle was monitored using an antibody specific for BZLF1. β‐actin was used as loading control. The lower panel shows the relative BZLF1 expression calculated as ratio between the intensity of the specific band in cells exposed to the bacterial supernatant versus TPA/Bu treated cells. Mean ±SD of three independent experiments.
Figure S4. Long‐term treatment with TPA/Bu induces ATM phosphorylation
AGS‐Bx1 cells were treated for 24 h with TPA/Bu, CDT (1 μg/ml), Etoposide (ETOP, 40 μM), Camptothecin (CPT, 5 μM) or exposed to UV, and further incubated for 24 h. Reactivation of the lytic cycle and induction of the DDR response was monitored by western blot analysis using the following antibodies: mouse anti‐BZLF1, mouse anti‐phospho‐ATM (pATM), mouse anti‐γH2AX, rabbit anti‐phospho‐p53‐S15 (pp53‐S15), mouse anti‐p53, mouse anti‐p21. The arrow indicates the ubiquitinated form of γH2AX (UbH2AX). β‐actin was used as loading control.
Figure S5. Effect of DNA‐DSB on EBV reactivation in epithelial and B cell lines. Two B95.8 virus transformed LCLs were exposed to increasing concentration of Etoposide (ETOP, 5 μM and 10 μM) for 24 h. Reactivation of the EBV lytic cycle and induction of the DDR was monitored by probing western blots with the indicated antibodies. β‐actin was used as loading control.
Appendix S1: Supplementary Material
Acknowledgements
We are grateful to Professor Anders Gustafsson, Department of Odontology, Karolinska Institutet for valuable comments and knowledge on oral pathogenic bacteria. This investigation was supported by grants from the Swedish Cancer Society, the Swedish Research Council and the Konung Gustaf V:s och Drottning Victoria Frimurarestiftelse to MGM, and the Swedish Cancer Society, the Swedish Research Council, the Lillian Sagens och Curt Ericssons forskningsstiftelse and Umeå University to TF.
The authors have no conflict of interest or financial disclosure to declare.
Contributor Information
Teresa Frisan, Email: teresa.frisan@ki.se, Email: teresa.frisan@umu.se.
Maria G. Masucci, Email: maria.masucci@ki.se.
REFERENCES
- 1. de Martel C, Ferlay J, Franceschi S, et al. Global burden of cancers attributable to infections in 2008: a review and synthetic analysis. Lancet Oncol 2012;13:607–15. [DOI] [PubMed] [Google Scholar]
- 2. Bouvard V, Baan R, Straif K, et al. A review of human carcinogens—Part B: biological agents. Lancet Oncol 2009;10:321–2. [DOI] [PubMed] [Google Scholar]
- 3. Rickinson AB. Co‐infections, inflammation and oncogenesis: future directions for EBV research. Semin Cancer Biol 2014;26:99–115. [DOI] [PubMed] [Google Scholar]
- 4. Young LS, Rickinson AB. Epstein‐Barr virus: 40 years on. Nat Rev. Cancer 2004;4:757–68. [DOI] [PubMed] [Google Scholar]
- 5. Young LS, Murray PG. Epstein‐Barr virus and oncogenesis: from latent genes to tumours. Oncogene 2003;22:5108–21. [DOI] [PubMed] [Google Scholar]
- 6. Kang MS, Kieff E. Epstein‐Barr virus latent genes. Exp Mol Med 2015;47:e131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Dittmer DP, Hilscher CJ, Gulley ML, et al. Multiple pathways for Epstein‐Barr virus episome loss from nasopharyngeal carcinoma. Int J. Cancer 2008;123:2105–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lin CT, Chan WY, Chen W, et al. Characterization of seven newly established nasopharyngeal carcinoma cell lines. Lab Invest 1993;68:716–27. [PubMed] [Google Scholar]
- 9. Tanner J, Weis J, Fearon D, et al. Epstein‐Barr virus gp350/220 binding to the B lymphocyte C3d receptor mediates adsorption, capping, and endocytosis. Cell 1987;50:203–13. [DOI] [PubMed] [Google Scholar]
- 10. Chesnokova LS, Nishimura SL, Hutt‐Fletcher LM. Fusion of epithelial cells by Epstein‐Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc Natl Acad Sci U S A 2009;106:20464–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chen J, Sathiyamoorthy K, Zhang X, et al. Ephrin receptor A2 is a functional entry receptor for Epstein‐Barr virus. Nat Microbiol 2018;3:172–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Zhang H, Li Y, Wang HB, et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein‐Barr virus entry. Nat Microbiol 2018;3:1–8. [DOI] [PubMed] [Google Scholar]
- 13. Tsang CM, Yip YL, Lo KW, et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc Natl Acad Sci U S A 2012;109:E3473–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Zhang G, Tsang CM, Deng W, et al. Enhanced IL‐6/IL‐6R signaling promotes growth and malignant properties in EBV‐infected premalignant and cancerous nasopharyngeal epithelial cells. PLoS One 2013;8:e62284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hall‐Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol 2004;2:95–108. [DOI] [PubMed] [Google Scholar]
- 16. Meyer MS, Joshipura K, Giovannucci E, et al. A review of the relationship between tooth loss, periodontal disease, and cancer. Cancer Causes Control 2008;19:895–907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Crusz SM, Balkwill FR. Inflammation and cancer: advances and new agents. Nat Rev Clin Oncol 2015;12:584–96. [DOI] [PubMed] [Google Scholar]
- 18. Gagnaire A, Nadel B, Raoult D, et al. Collateral damage: insights into bacterial mechanisms that predispose host cells to cancer. Nat Rev Microbiol 2017;15:109–28. [DOI] [PubMed] [Google Scholar]
- 19. Nalbant A, Chen C, Wang Y, et al. Induction of T‐cell apoptosis by Actinobacillus actinomycetemcomitans mutants with deletion of ltxA and cdt ABC genes: possible activity of GroEL‐like molecule. Oral Microbiol Immunol 2003;18:339–49. [DOI] [PubMed] [Google Scholar]
- 20. Borza CM, Hutt‐Fletcher LM. Alternate replication in B cells and epithelial cells switches tropism of Epstein‐Barr virus. Nat Med 2002;8:594–9. [DOI] [PubMed] [Google Scholar]
- 21. Barranco SC, Townsend CM Jr, Casartelli C, et al. Establishment and characterization of an in vitro model system for human adenocarcinoma of the stomach. Cancer Res 1983;43:1703–9. [PubMed] [Google Scholar]
- 22. Speck P, Kline KA, Cheresh P, et al. Epstein‐Barr virus lacking latent membrane protein 2 immortalizes B cells with efficiency indistinguishable from that of wild‐type virus. J Gen Virol 1999;80(Pt 8):2193–203. [DOI] [PubMed] [Google Scholar]
- 23. Miller G, Robinson J, Heston L, et al. Differences between laboratory strains of Epstein‐Barr virus based on immortalization, abortive infection, and interference. Proc Natl Acad Sci U S A 1974;71:4006–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Guerra L, Teter K, Lilley BN, et al. Cellular internalization of cytolethal distending toxin: a new end to a known pathway. Cell Microbiol 2005;7:921–34. [DOI] [PubMed] [Google Scholar]
- 25. Gorres KL, Daigle D, Mohanram S, et al. Activation and repression of Epstein‐Barr Virus and Kaposi's sarcoma‐associated herpesvirus lytic cycles by short‐ and medium‐chain fatty acids. J Virol 2014;88:8028–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Grasso F, Frisan T. Bacterial Genotoxins: Merging the DNA Damage Response into Infection Biology. Biomolecules 2015;5:1762–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010;40:179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Liu S, Borras AM, Liu P, et al. Binding of the ubiquitous cellular transcription factors Sp1 and Sp3 to the ZI domains in the Epstein‐Barr virus lytic switch BZLF1 gene promoter. Virology 1997;228:11–8. [DOI] [PubMed] [Google Scholar]
- 29. Blume SW, Snyder RC, Ray R, et al. Mithramycin inhibits SP1 binding and selectively inhibits transcriptional activity of the dihydrofolate reductase gene in vitro and in vivo. J Clin Invest 1991;88:1613–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Guerra L, Carr HS, Richter‐Dahlfors A, et al. A bacterial cytotoxin identifies the RhoA exchange factor Net1 as a key effector in the response to DNA damage. PLoS One 2008;3:e2254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Guerra L, Cortes‐Bratti X, Guidi R, et al. The biology of the cytolethal distending toxins. Toxins (Basel) 2011;3:172–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Chien YC, Chen JY, Liu MY, et al. Serologic markers of Epstein‐Barr virus infection and nasopharyngeal carcinoma in Taiwanese men. N Engl J Med 2001;345:1877–82. [DOI] [PubMed] [Google Scholar]
- 33. Henle W, Ho JH, Henle G, et al. Nasopharyngeal carcinoma: significance of changes in Epstein‐Barr virus‐related antibody patterns following therapy. Int J Cancer 1977;20:663–72. [DOI] [PubMed] [Google Scholar]
- 34. Murata T, Tsurumi T. Switching of EBV cycles between latent and lytic states. Rev Med Virol 2014;24:142–53. [DOI] [PubMed] [Google Scholar]
- 35. Fang CY, Huang SY, Wu CC, et al. The synergistic effect of chemical carcinogens enhances Epstein‐Barr virus reactivation and tumor progression of nasopharyngeal carcinoma cells. PLoS One 2012;7:e44810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Popovic J, Gasic J, Zivkovic S, et al. Prevalence of human cytomegalovirus and Epstein‐Barr virus in chronic periapical lesions. Intervirology 2015;58:271–7. [DOI] [PubMed] [Google Scholar]
- 37. Hagemeier SR, Barlow EA, Meng Q, et al. The cellular ataxia telangiectasia‐mutated kinase promotes epstein‐barr virus lytic reactivation in response to multiple different types of lytic reactivation‐inducing stimuli. J Virol 2012;86:13360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Hau PM, Deng W, Jia L, et al. Role of ATM in the formation of the replication compartment during lytic replication of Epstein‐Barr virus in nasopharyngeal epithelial cells. J Virol 2015;89:652–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Doetzlhofer A, Rotheneder H, Lagger G, et al. Histone deacetylase 1 can repress transcription by binding to Sp1. Mol Cell Biol 1999;19:5504–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Davie JR. Inhibition of histone deacetylase activity by butyrate. J Nutr 2003;133:2485S–93S. [DOI] [PubMed] [Google Scholar]
- 41. Iwahori S, Yasui Y, Kudoh A, et al. Identification of phosphorylation sites on transcription factor Sp1 in response to DNA damage and its accumulation at damaged sites. Cell Signal 2008;20:1795–803. [DOI] [PubMed] [Google Scholar]
- 42. Guidi R, Guerra L, Levi L, et al. Chronic exposure to the cytolethal distending toxins of Gram‐negative bacteria promotes genomic instability and altered DNA damage response. Cell Microbiol 2013;15:98–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Ferlay J, Soerjomataram I, Ervik M, Dirkshit R, Eser S, Mathers C, Rebelo M, Parkin DM, Formn D, Bray F. GLOBCAN 2012 v1.0, Cancer Incidence and Mortality Worldwide: IARC CancerBase N.11. International agency for cancer research, http://globocan.iarc.fr, 2013.
- 44. Bei JX, Li Y, Jia WH, et al. A genome‐wide association study of nasopharyngeal carcinoma identifies three new susceptibility loci. Nat Genet 2010;42:599–603. [DOI] [PubMed] [Google Scholar]
- 45. Yee Ko JM, Dai W, Wun Wong EH, et al. Multigene pathway‐based analyses identify nasopharyngeal carcinoma risk associations for cumulative adverse effects of TERT‐CLPTM1L and DNA double‐strand breaks repair. Int J Cancer 2014;135:1634–45. [DOI] [PubMed] [Google Scholar]
- 46. Turkoz FP, Celenkoglu G, Dogu GG, et al. Risk factors of nasopharyngeal carcinoma in Turkey‐an epidemiological survey of the Anatolian Society of Medical Oncology. Asian Pac J Cancer Prev 2011;12:3017–21. [PubMed] [Google Scholar]
- 47. Liu Z, Chang ET, Liu Q, et al. Oral hygiene and risk of nasopharyngeal carcinoma‐a population‐based case‐control study in China. Cancer Epidemiol Biomarkers Prev 2016;25:1201–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Wang X, Li L, Yang M, et al. Prevalence and distribution of Aggregatibacter actinomycetemcomitans and its cdtB gene in subgingival plaque of Chinese periodontitis patients. BMC Oral Health 2014;14:37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Haubek D, Johansson A. Pathogenicity of the highly leukotoxic JP2 clone of Aggregatibacter actinomycetemcomitans and its geographic dissemination and role in aggressive periodontitis. J Oral Microbiol 2014;6:1634–1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Cancer Genome Atlas Research N . Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Peek RM Jr, Crabtree JE. Helicobacter infection and gastric neoplasia. J Pathol 2006;208:233–48. [DOI] [PubMed] [Google Scholar]
- 52. Koeppel M, Garcia‐Alcalde F, Glowinski F, et al. Helicobacter pylori infection causes characteristic DNA damage patterns in human cells. Cell Rep 2015;11:1703–13. [DOI] [PubMed] [Google Scholar]
- 53. Toller IM, Neelsen KJ, Steger M, et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double‐strand breaks and a DNA damage response in its host cells. Proc Natl Acad Sci U S A 2011;108:14944–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Recovery of bacteria and eukaryotic cell. Three hundred thousand AGS‐Bx1 cells were co‐cultured with Aa or Pg at the indicated MOI for 48 h and parallel aliquots of bacteria were maintained in complete medium without antibiotics in absence of the eukaryotic cells for the same time. A) Recovery of Aa (squares) or Pg (circles) measured as optical density at 600 nm after culture in the presence (white symbols) or absence (black symbols) of the AGS‐Bx1 cells. Mean ±SD of three independent experiments. B) Recovery of viable AGS‐BX1 cells after culture for 48 h with Aa (white squares) or Pg (black squares) at the indicated MOI. The data are presented as percentage of cells recovery relative to non‐infected cells (CTR). * indicates the initial seeding. Mean ±SD of three independent experiments.
Figure S2. Butyrate is the only SCFA promoting EBV reactivation in AGS‐Bx1 cells. AGS‐Bx1 cells were exposed to the indicated concentrations of butyrate, acetate or propionate for 48 h. The levels of BZLF1 expression was assessed by western blot analysis using an antibody specific for BZLF1. β‐actin was used as loading control.
Figure S3. Induction of EBV reactivation by oral pathogenic bacteria. AGS‐Bx1 cells were exposed for 24 h to sterile bacterial supernatant of Kp, Ck or Aa, normalized at optical density of 0.3 at 600 nm, and diluted 1:3 in complete medium. Cells exposed to TPA/Bu were used as positive control. The upper panel shows a representative western blot where reactivation of the EBV lytic cycle was monitored using an antibody specific for BZLF1. β‐actin was used as loading control. The lower panel shows the relative BZLF1 expression calculated as ratio between the intensity of the specific band in cells exposed to the bacterial supernatant versus TPA/Bu treated cells. Mean ±SD of three independent experiments.
Figure S4. Long‐term treatment with TPA/Bu induces ATM phosphorylation
AGS‐Bx1 cells were treated for 24 h with TPA/Bu, CDT (1 μg/ml), Etoposide (ETOP, 40 μM), Camptothecin (CPT, 5 μM) or exposed to UV, and further incubated for 24 h. Reactivation of the lytic cycle and induction of the DDR response was monitored by western blot analysis using the following antibodies: mouse anti‐BZLF1, mouse anti‐phospho‐ATM (pATM), mouse anti‐γH2AX, rabbit anti‐phospho‐p53‐S15 (pp53‐S15), mouse anti‐p53, mouse anti‐p21. The arrow indicates the ubiquitinated form of γH2AX (UbH2AX). β‐actin was used as loading control.
Figure S5. Effect of DNA‐DSB on EBV reactivation in epithelial and B cell lines. Two B95.8 virus transformed LCLs were exposed to increasing concentration of Etoposide (ETOP, 5 μM and 10 μM) for 24 h. Reactivation of the EBV lytic cycle and induction of the DDR was monitored by probing western blots with the indicated antibodies. β‐actin was used as loading control.
Appendix S1: Supplementary Material