

SUMMARY
The folate biosynthetic pathway offers many druggable targets that have yet to be exploited in TB therapy. Herein, we have identified a series of small molecules that interrupt Mycobacterium tuberculosis folate metabolism by dual targeting of dihydrofolate reductase (DHFR), a key enzyme in the folate pathway, and its functional analog, Rv2671. We have also compared the antifolate activity of these compounds to that of para-aminosalicylic acid (PAS). We found the bioactive metabolite of PAS, in addition to previously reported activity against DHFR, inhibits flavin-dependent thymidylate synthase (FDTS) in Mtb, suggesting a multi-targeted mechanism of action for this drug. Finally, we have shown that antifolate treatment in Mtb decreases the production of mycolic acids, most likely due to perturbation of the activated methyl cycle. We conclude that multi-targeting of the folate pathway in Mtb is associated with highly potent anti-mycobacterial activity.
Graphical Abstract
eTOC blurb
Hajian et al. have investigated the inhibitory activity of a series of small molecule antifolates and para-aminosalicylic acid against Mycobacterium tuberculosis, the causative agent of tuberculosis, and show that these compounds exert their activity by acting on multiple targets within the folate biosynthetic pathway.
INTRODUCTION
Tuberculosis (TB) has travelled with humans since the dawn of our species and has claimed millions of lives throughout human history. TB, caused by Mycobacterium tuberculosis (Mtb), is the leading cause of death from a single infectious agent. Despite more than half a century of anti-TB chemotherapy, the onerous drug regimens have led to poor adherence, treatment failure, and the growing frequency of drug- resistant TB. After decades of relative inactivity in TB drug discovery, the pressing need for novel therapeutics that can tackle these issues has renewed research interest in this area (Koul et al., 2011).
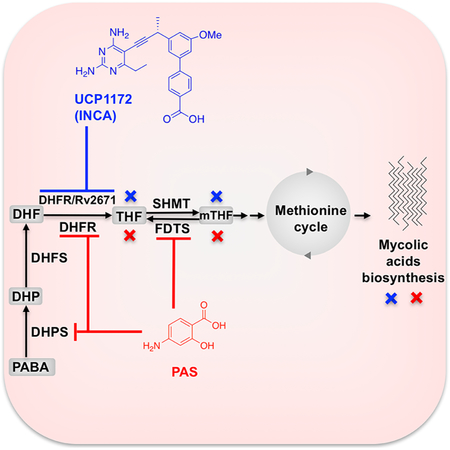
One class of therapeutics that have recently gained interest for their anti-tubercular activity are the folate antimetabolites, or antifolates (Kumar et al., 2015; Nixon et al., 2014). Antifolates interrupt the production of reduced folate cofactors by inhibiting key enzymes in the folate metabolic pathway (Figure 1) (Scocchera and Wright, 2017). Synthesis and recycling of reduced folates is essential for the de novo synthesis of methionine, purines, and deoxythymidine monophosphate (dTMP). A lynchpin in the folate pathway, dihydrofolate reductase (DHFR, encoded by folA), catalyzes the reduction of dihydrofolate (DHF) to tetrahydrofolate (THF) using NADPH as an electron donor. Due to its pivotal role in nucleic acid synthesis, and high conservation through evolution, DHFR has served as an ideal antiproliferative drug target for cancer and infectious diseases (Visentin et al., 2012). Despite the success of antifolates as anticancer and antimicrobial agents, they are an underutilized drug class in TB therapy, particularly those targeting DHFR. Current DHFR inhibitors either show poor potency against Mtb DHFR (e.g. trimethoprim) or fail to inhibit the growth of live Mtb despite good enzyme potency, most likely due to lack of permeability (e.g. methotrexate, pyrimethamine, and trimetrexate) (Nixon et al., 2014).
Figure 1.
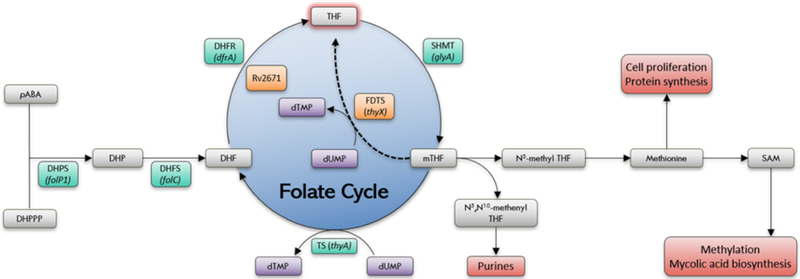
Mycobacterium tuberculosis folate pathway. Canonical pathway enzyme (green): DHPS: dihydropteroate synthase, DHFS: dihydrofolate synthase, DHFR: dihydrofolate reductase, TS: thymidylate synthase, SHMT: serine hydroxyl methyltransferase; Alternate pathway enzymes (orange) FDTS: flavin-dependent thymidylate synthase, Rv2671
Previously, we have reported the antimycobacterial activity of a series of small molecule antifolates that are designed to inhibit DHFR enzymes of various microorganism (Hajian et al., 2016). Screening of our compound library against wild-type and multidrug resistant Mtb strains identified a chemically distinct series that we have termed ionized non-classical antifolates (INCAs) (Figure 2) with remarkable activity at both the target and organism levels. INCAs enter cells through passive diffusion and are characterized by the presence of a single acidic functionality which affords ionic character at all pHs, either as a zwitterion or with net single negative charge (−1). This physicochemical profile stands in contrast to both lipophilic antifolates that are neutral or positively charged, and classical antifolates – like methotrexate – that carry a double negative charge and therefore, must be actively transported into cells.
Figure 2.
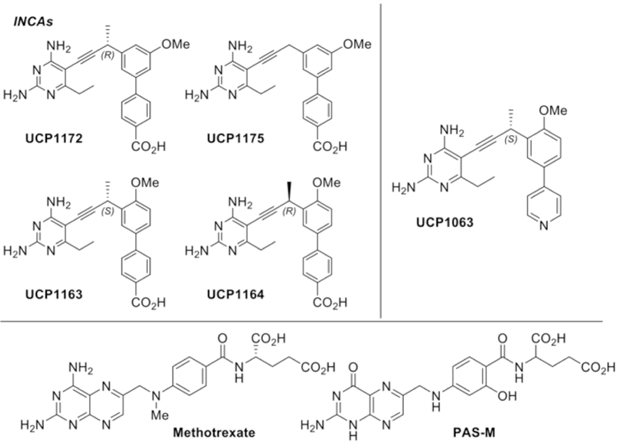
Structure of INCAs, UCP1063, methotrexate (MTX), and PAS-M
In this work, we describe in-depth mechanistic studies of select INCA inhibitors and compare their anti-DHFR activity to that of para-aminosalicylic acid (PAS), the only antifolate currently used in TB therapy. PAS, one of the few drugs available for treatment of drug resistant-TB, has been recently shown to act as an alternative substrate for folate biosynthesis in Mtb and the product of its metabolism has demonstrated inhibitory activity against Mtb DHFR (Chakraborty et al., 2013; Dawadi et al., 2017; Zheng et al., 2013).
We have also investigated the effect of these compounds on an alternate folate pathway present in Mtb. This alternate mechanism utilizes recently-discovered enzymes Rv2671 (a second DHFR found in Mtb) and flavin- dependent thymidylate synthase (FDTS, encoded by thyX) to generate THF and dTMP (Figure 1) (Cheng and Sacchettini, 2016; Mishanina et al., 2016; Zheng et al., 2013). The discovery of these enzymes has profound implications for antifolate drug discovery as they provide the organism with a source of facile resistance to antifolates. Overexpression of Rv2671 or thyX has been shown to be associated with resistance to DHFR inhibitors and PAS (Moradigaravand et al., 2016a; Zhang et al., 2015a; Zheng et al., 2013). However, it is not clear if the inhibition of these compensatory enzymes or multiple inhibition of both pathways improves the potency of antimycobacterial compounds against the live bacilli.
Here, we show that select INCAs are potent dual inhibitors of both Mtb DHFR and Rv2671 and this dual inhibition contributes to improved potency against Mtb cells. The INCA inhibitors exhibit a slow dissociation rate from DHFR, resulting in a long target occupancy, a desirable feature for antibacterials. PAS metabolite (PAS-M) was found to be a moderate Mtb DHFR when compared to INCAs with no significant inhibitory activity against Mtb Rv2671. Using X-ray crystallography, we have elucidated drug-target interactions that impart the potency of INCAs and PAS-M. Further investigations led us to identify FDTS as a critical secondary target for PAS-M suggesting a multi-targeted mechanism of action for PAS. Finally, we show that treatment of Mtb cells with INCAs or PAS leads to critical disruption of mycolic acids synthesis. Overall, our findings suggest that multi-targeted antifolates are promising candidates for development of new antitubercular agents.
RESULTS
Kinetic profiling of INCAs and PAS-M as DHFR inhibitors
INCAs feature a 2,4-diaminopyrimdine ring linked to a biaryl system through a propargyl bridge. The compounds chosen for this study included: UCP1172 and UCP1175, representing the INCA class, UCP1063 (a previous generation non-INCA), and PAS-M, which was synthesized through modification of a previously reported procedure (Scheme S1) (Dawadi et al., 2017).
To determine the relative affinity of these compounds for Mtb DHFR, the half maximal inhibitory concentrations (IC50) were measured using standard procedures that spectroscopically follow the oxidation of NADPH at 340 nm. The inhibitory constant, Ki, was determined from IC50 values using the Cheng-Prussof equation and KM values (Table S1). Methotrexate (MTX), a potent DHFR inhibitor among various species, was used as the positive control. INCAs and MTX showed potent inhibition of DHFR with Ki values below 10 nM (Table 1). However, PAS-M activity against Mtb DHFR was moderate (Ki = 750 nM) compared to INCAs and MTX. We also tested the inhibitory effect of PAS-M against Mtb DHFR under various incubation times, protein concentrations, and temperatures with no significant improvement detected (data not shown).
Table 1.
Microbiological and biochemical characteristics of INCAs, PAS-M, and MTX
| Mtb DHFR |
|||||||
|---|---|---|---|---|---|---|---|
| Compounds | MIC (μg/mL) | Ki(nM)a | koff(min-1)b | tR (min)c | Tm (°C)d | Hu DHFR Ki (nM) | Mtb Rv2671 Ki (nM) |
| MTX | >100 | 1.6 ± 0.16 | 0.056 ± 0.006 | 18 ± 1.5 | 76 ± 3.5 | 0.7 ±0.11 | ND |
| UCP1172 | 0.03 | 0.91 ±0.15 | 0.024 ± 0.003 | 42 ± 5.3 | 81 ± 3 | 60 ± 15 | 80 ± 6.5 |
| UCP1175 | 0.125 | 6.55 ± 0.26 | 0.072 ± 0.008 | 14 ± 1.5 | 73 ± 3.2 | 51 ±3 | 1170 ±140 |
| UCP1063 | 2 | 8 ± 1.3 | 0.096 ± 0.015 | 10.5 ± 1.6 | 71 ± 2.7 | 11.1 ± 1.3 | 52 ± 6.2 |
| PAS-M | 0.5e | 750 ± 120 | 0.643 ± 0.125 | 1.5 ± 0.3 | 68 ± 1.8 | ND | 27,500 ± 3500 |
| UCP1163 | 4 | 9.1 ± 0.25 | ND | ND | ND | 120 ± 7.5 | 350 ± 12.5 |
| UCP1164 | 0.5 | 9.9 ± 0.62 | ND | ND | ND | 144 ± 3.5 | 120 ± 8.5 |
Calculated as Ki=IC50/(1+[S]/KM),
koff is assumed to be equal to the activity recovery rate constant from jump-dilution assay,
Calculated as the reciprocal of the rate constant (kobs)
The Tm of the control (Mtb DHFR+NADPH) was determined to be 65 °C,
This value is the MIC of PAS. The MIC of PAS-M was determined to be 8 μg/ml (The difference is most likely due to permeability issues). All the experiments are performed in triplicates.
Puzzled by PAS-M’s low affinity for Mtb DHFR, and to obtain a better measurement of the mode of action of the inhibitors, we determined the dissociation rate of PAS-M and INCAs from Mtb DHFR. It has been shown that drug dissociation from its target is a critical factor for sustained drug efficacy in vitro and in vivo (Copeland, 2010; Walkup et al., 2015). Slow dissociation of a drug-target complex extends the duration of drug-target occupancy, or drug-target residence time (tR), which can be used as a parameter to quantify and compare the lifetime of drug-target interactions. Such parameters provide additional measurements of drug potency that may not be evident by thermodynamic parameters such as IC50 and Ki (Tonge, 2017).
The dissociation rates of the inhibitors were determined using a jump-dilution assay to determine whether PAS-M or INCAs binding is rapidly or slowly reversible. Briefly, Mtb DHFR was incubated with a stoichiometric equivalent of inhibitor at concentrations above Ki for three hours to allow the formation of the EI complex. The assay mixture was then diluted 100-fold into assay buffer containing a saturating concentration of DHF to disturb the equilibrium. The rapid dilution favors ligand dissociation while disfavoring ligand rebinding. The recovery of enzymatic activity was followed as a function of time after dilution. Under the condition of minimal inhibitor rebinding, the observed rate constant, kobs, can be considered as a reasonable estimate of the dissociation rate constant koff. The off-rate constant was then used to estimate the target-inhibitor residence time (tR) (Table 1).
After dilution, enzymatic activity was recovered for all compounds over the assay timeframe (Figure S1). The resulting progress curves for INCAs, UCP1063, and MTX were curvilinear (a lag phase followed by a linear phase) suggesting slowly reversible behavior. UCP1172 showed the longest residence time on Mtb DHFR (42 minutes) and as a result, the enzymatic activity recovery in the presence of UCP1172 was slower than the other compounds. The residence time of UCP1175 and UCP1063 were almost similar (14 and 10 minutes, respectively). The progress curve for PAS-M was linear with a slope nearly equal to the slope of the uninhibited control sample, suggesting fast dissociation. The tR for PAS-M was determined to be 1.5 minutes. Overall, a correlation between Ki and tR was observed, indicating that the dissociation rate is influenced by the affinity of the inhibitor for the target. The data suggest that unlike MTX and INCAs, PAS-M has a weak affinity for Mtb DHFR. However, given the nature of PAS’s bioactivation, in vitro enzyme assay is a poor metric for PAS-M’s Mtb DHFR activity in the cell. PAS-M biosynthesis requires a stoichiometric reduction in DHF, thereby reducing the natural substrate that PAS-M competes with for Mtb DHFR. Therefore, a Ki value measured in an in vitro assay may drastically underestimate PAS’s effect on the organism.
Thermal stability analysis of Mtb DHFR bound to the inhibitors
We evaluated the thermal stability of Mtb DHFR upon ligand binding using differential scanning calorimetry (DSC). It is known that ligand binding to a protein yields a complex with increased thermodynamic stability compared to the unbound protein. This difference in stability leads to a change in the protein’s apparent melting temperature (Tm) which can be measured using DSC. An increase in Tm upon binding typically correlates with better Ki values and thus increased ligand affinity (Celej et al., 2006; Hellman et al., 2016). A complex of Mtb DHFR and NADPH was used as the control sample to provide a comparison between the Tm of the uninhibited and inhibited protein. Overall, compound binding resulted in an increase in Tm (Table 1). UCP1172 and MTX showed the largest stabilizing effects on the protein (81°C and 75.5°C, respectively) compared to the control (65°C). UCP1175 and UCP1063 also showed an increased thermal stability (72.9°C and 71°C, respectively), although to a lesser extent compared to UCP1172 and MTX. Binding of PAS-M caused the smallest shift in Tm, again suggesting its weak affinity for Mtb DHFR. In sum, the changes in thermal stability of Mtb DHFR upon binding correlates well with Ki values.
Structural studies of Mtb DHFR in complex with INCAs and PAS-M
To investigate the ligand-target interactions that drive affinity and potency, we co-crystallized UCP1172, UCP1175, and PAS-M with Mtb DHFR and solved the protein ternary complex bound to the inhibitors and NADPH. UCP1172 and UCP1175 bind in a similar mode to that reported for MTX (Li et al., 2000). The 2,4-diaminopyrimidine ring of INCAs is deeply positioned into the hydrophobic pocket making two hydrogen bonds with the side chain of Asp27 and two hydrogen bonds with the main chain of Ile5 and Ile94 (Figure 3, top left and right). The hydroxyl of Tyr100 is also 3.4 Å away from the 4-amino group. Water-mediated interactions between the 2,4-diaminopyrimidine ring and Trp22, His30, and Ile113 and π-π stacking interactions with Phe31 are present. The biaryl system of INCAs makes hydrophobic interactions with Gln28, Leu50, Pro51, Ile54, and Leu57. The propargylic methyl of UCP1172 (Figure 3, top left) improves its hydrophobic interactions compared to UCP1175 which lacks propargylic functionality. The carboxylate of INCAs forms a hydrogen bond with a highly conserved Arg60 which is likely the interaction that defines the increased affinity of INCAs. Although our efforts to co-crystallize UCP1063 with Mtb DHFR were not successful, we believe the replacement of benzoic acid with pyridine in UCP1063 is responsible for its decreased affinity for the enzyme compared to UCP1172. The co-crystal structure of Mtb DHFR bound to PAS-M has revealed a likely reason for its poor affinity. Compared to MTX (Li et al., 2000), the pteridine ring of PAS-M is in an inverse orientation, with N1 and N8 too far for direct or water-mediated hydrogen bonds (Figure 3, middle left). The N1 and 2-amino group of the pteridine ring retain two hydrogen bonds with Asp27. The pteridine carbonyl and N5 make water-mediated interactions with Trp22, His30, and Thr113. The glutamate tail forms hydrogen bonds with Arg60 and Arg32. A large water network was also observed between the hydroxyl group and the residues of the binding pocket. Despite these interactions, the inability of the pteridine ring to interact with the highly conserved Ile5, Ile94, and Tyr100 is likely responsible for PAS-M’s weak affinity for Mtb DHFR. The flipped pteridine ring has also been observed in co-crystal structures of human DHFR bound to folate and is likely responsible for the difference in affinity between folate and MTX for Hu DHFR (Oefner et al., 1988).
Figure 3.
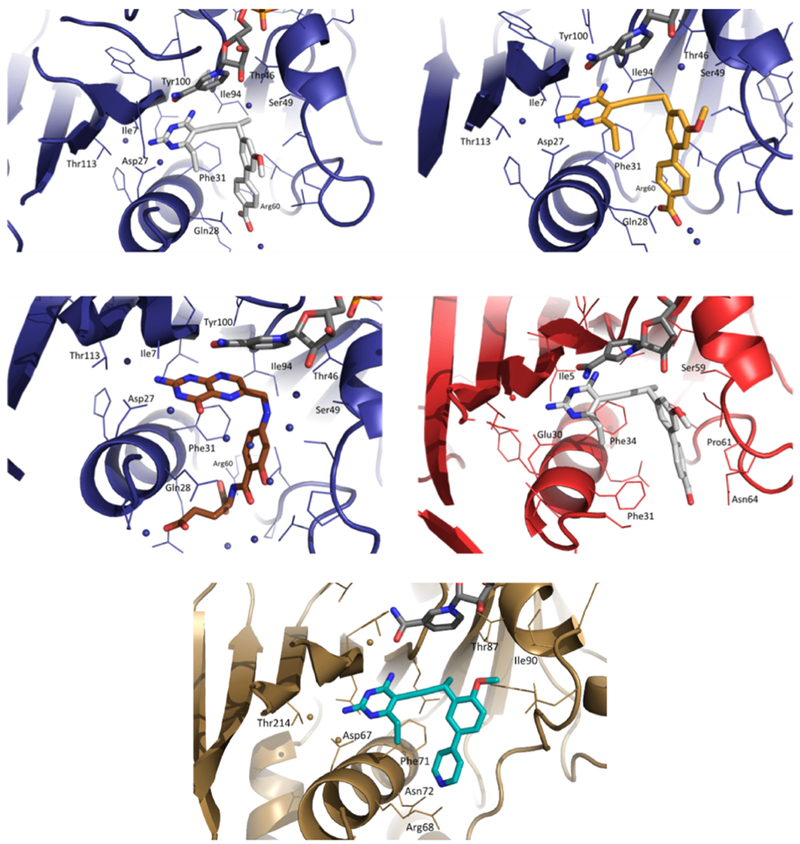
Top Left: Mtb DHFR in complex with NADPH (dark grey) and UCP1172 (light grey) at 1.4 Å (PDB ID: 6DDP); Top Right: Mtb DHFR in complex with NADPH and UCP1175 (yellow) at 1.7 Å (PDB ID: 6DDS); Middle Left: Mtb DHFR in complex with NADPH and PAS-M (brown) at 1.4 Å (PDB ID: 6DDW); Middle Right: Hu DHFR in complex with NADPH and UCP1172 (light grey) at 2.4 α (PDB ID: 6DE4); Bottom: Mtb Rv2671 in complex with NADPH and UCP1063 (cyan)
Selectivity of INCAs toward the microbial DHFR
Previously, we have shown that several INCAs, including UCP1172, have no measurable cytotoxicity toward mammalian cells at concentration up to 100 μM (Scocchera et al., 2016). The low toxicity of moderately potent DHFR inhibitors like the INCAs, may be attributed to the unique way in which human DHFR is regulated. In humans, DHFR is known to bind its cognate RNA and presence of a DHFR inhibitor leads to dissociation, relieving translational arrest, and rapid translation of DHFR. This provides a rapid, intrinsic protection to human cells that is unavailable to prokaryotic pathogens (Zhang and Rathod, 2002). Here, to understand the affinity differences between Mtb DHFR and Hu DHFR we co-crystallized Hu DHFR with UCP1172 (Figure 3, middle right). The interactions of the 2,4-diaminopyrimidine ring that anchors UCP1172 in the active site are very similar to what was observed in the Mtb DHFR and UCP1172 complex. The 4-amino moiety forms a hydrogen bond with the backbone carbonyl of Ile7 (3.0 Å) and another hydrogen bond with the backbone carbonyl oxygen of Val115 (3.3 Å). Atom N1 and the 2-amino group form two strong hydrogen bonds with Glu30 (2.8 and 2.6 Å, respectively). Glu30 corresponds to the conserved Asp27 among bacterial DHFRs. In Hu DHFR, the corresponding Arg70 is too far from the carboxylate of UCP1172 to form a hydrogen bond (6.0 Å). Although the carboxylate is near Asn64 (3.2-3.6 Å), the angle does not seem to favor hydrogen bonding. The lack of optimized interactions between the carboxylate group of UCP1172 and the Hu DHFR active site residues is likely the reason for its weaker affinity for Hu DHFR compared to Mtb DHFR (Table 1).
Inhibition of Mtb Rv2671
To further evaluate the effect of INCAs and PAS-M on Mtb folate metabolism, we tested the potency of INCAs and PAS-M against Mtb RV2671, an enzyme in Mtb recently shown to possess DHFR activity (Cheng and Sacchettini, 2016). Overexpression of Rv2671 has been shown to be associated with resistance to PAS and antifolates (Zhang et al., 2015b; Zheng et al., 2013).
The enzyme activity of Mtb Rv2671 was measured by following NADPH oxidation at 340 nm and its kinetic parameters were determined (Table S1). The KM of Mtb Rv2671 for DHF was determined to be 39 μM, about four times higher than the KM for Mtb DHFR. The kcat of Mtb Rv2671 is 0.69 s−1 which is almost 2.5 times less than Mtb DHFR. These numbers agree with what is known, that Mtb Rv2671 is less efficient as a reductase than DHFR (Cheng and Sacchettini, 2016).
We then evaluated the inhibitory activity of INCAs and PAS-M against Mtb Rv2671 (Table 1). Compounds UCP1172 and UCP1063 showed potent inhibition of Mtb Rv2671, with Ki values of 82 and 53 nM, respectively. UCP1175 showed moderate inhibition of the enzyme with a Ki of 1170 nM. PAS-M did not exhibit a significant inhibitory effect against Mtb Rv2671 (Ki= 27500 nM). To assess the impact of Rv2671 inhibition on the antimycobacterial activity of the compounds, we used an enantiomerically pure INCA couple, UCP1164 and UCP1163 (Figure 2), that possess similar DHFR inhibitory activity but different inhibition against Rv2671. UCP1164 and UCP1163 Ki values against Rv2671 were determined to be 120 nM and 350 nM, respectively (Table 1).
Interestingly, when tested against the organism, UCP1164 showed more potent inhibition of cell growth with MIC value of 0.5 μg/ml compared to UCP1163 (MIC= 4 μg/ml) (Table 1). Due to high structural similarity between these two compounds, we do not expect them to have significant permeability differences. Therefore, the observed difference in MIC values is most likely caused by different enzyme inhibitory activity against Mtb Rv2671. Taken together, the data suggest that dual inhibition of Mtb DHFR and Rv2671 contributes to potent antimycobacterial activity of INCAs.
Structural studies of Mtb Rv2671 bound to UCP1063
To investigate drug-target interactions, We co-crystallized Mtb Rv2671 in complex with NADPH and UCP1063 (Figure 3, bottom). The protein is composed of eight parallel β-strands, one anti-parallel β-strand, seven α-helices, and one 310-helix (η1). As previously reported (Cheng and Sacchettini, 2016), a weak density for the nicotinamide ribose moiety of NADPH and residues 90-95 was observed. This disorder is likely due to the lack of stabilized interactions in these regions and explains the high KM for DHF and NADPH. The 2,4-diaminopyrimidine ring of INCAs makes direct hydrogen bonds with Asn44, Asp67 (corresponding to Asp27 in Mtb DHFR), and Glu193. In addition, there is a water-mediated interaction with Thr214 and π-π stacking with Phe71 (corresponding to Phe31 in Mtb DHFR). The pyridine nitrogen of UCP1063 is too far from Asn72 and Arg68 to form direct hydrogen bonds (5.2 and 4 Å, respectively).
In vitro selection of PAS-resistant Mtb mutants
The study of PAS-resistant clinical isolates and spontaneous mutants has revealed no mutations in the dfrA gene; instead, mutations were identified in thyA, folC, thyX, and Rv2671 (Minato et al., 2015; Moradigaravand et al., 2016b; Zhang et al., 2015a). This seems to suggest other mechanisms of actions for PAS-M rather than DHFR inhibition. To improve our understanding of the underlying mechanisms of action of PAS, and its resistance, we performed in vitro generation of PAS-resistant isolates and investigated the mutations associated with PAS resistance (Table 2). Genome sequencing revealed folC (encoding for dihydrofolate synthase, DHFS) mutations (E153G and I43T) in two of the mutants. Both mutations have been previously shown to confer resistance to PAS in clinical isolates of Mtb (Zhao et al., 2014). It has been suggested PAS converts to its metabolite by acting as a competing substrate for dihydropteroate synthase (DHPS, encoded by folP1) and DHFS, sequentially (Chakraborty et al., 2013). Residues E153 and I43 in DHFS are important for dihydropteroate (H2Pte) binding. These mutants were shown to be much less efficient at catalyzing the glutamination of both the natural substrate (H2Pte), and the PAS derivative (hydroxyl-H2Pte). A mutation here would lead to a reduction in intracellular [PAS-M], and the organism would be spared of its antibacterial effects. We also identified a PAS-resistant isolate with a C to T upstream mutation at position −16 that has been previously found to result in thyX overexpression and PAS resistance (Moradigaravand et al., 2016a; Zhang et al., 2013, 2015b). The PAS-resistant isolates remained susceptible to INCAs although at slightly higher MIC values (especially for UCP1175). The largest increase in the MIC of INCAs was observed against the thyX overexpressor strain. The fact that thyX overexpression is a PAS resistance mechanism suggests that either, the organism is compensating for DHFR inhibition by PAS-M, or PAS/PAS-M directly interacts with FDTS.
Table 2.
Genetic determinants of PAS-resistance in Mtb spontaneous mutants and their susceptibility to PAS and INCAs (MIC in (μg/mL)
| Mutation |
||||
|---|---|---|---|---|
| WT Mtb |
folC (E153G) |
folC (I43T) |
thyX (−16 C->T) |
|
| PAS | 0.5 | 32 | 32 | 16 |
| UCP1172 | 0.03 | 0.125 | 0.015 | 1 |
| UCP1175 | 0.125 | 4 | 1 | 16 |
Inhibition of flavin-dependent thymidylate synthase (FDTS)
In human cells and almost 70% of microbial species, thymidylate synthase (TS, encoded by thyA) catalyzes the reductive methylation of dUMP using methylene-THF (mTHF) as both a one-carbon donor, and a reductant, to generate dTMP, an essential building block of DNA. However, in approximately 30% of microbial species, de novo synthesis of dTMP relies on flavin-dependent thymidylate synthase (FDTS, encoded by thyX). FDTS uses NADPH as the source of hydride, mTHF as the one-carbon donor and FAD as a relay system for hydride and methylene transfer, to catalyze the formation of thymidylate (Mishanina et al., 2016; Myllykallio et al., 2003). Unlike TS, the folate product of the FDTS reaction is the reduced folate THF, not DHF. Approximately 5% of microbial species, including Mtb, express all three folA, thyA, and thyX. Although it is unclear why both thyA and thyX genes are maintained in such organisms, studies have shown that thyX is essential for Mtb growth and survival, and its overexpression is associated with PAS-resistance in clinical isolates (Fivian-Hughes et al., 2012; Mathys et al., 2009; Moradigaravand et al., 2016a). Considering poor PAS-M affinity for Mtb DHFR and Rv2671, and PAS-M resistance due to thyX overexpression, we tested the inhibitory potential of PAS-M against Mtb FDTS. Due to the structural similarities between PAS-M and mTHF its ability to bind FDTS is likely.
We performed DSC experiments to determine PAS-M binding to Mtb FDTS via its effect on protein stability. A mixture of FDTS, dUMP, and FAD was used as the control. DSC showed the Tm of 66.6 °C for the control sample and 71.9 °C for the sampl e incubated with PAS-M (Figure 4, top left). This increase in Tm suggests the stabilizing effect of PAS-M on Mtb FDTS due to binding.
Figure 4.
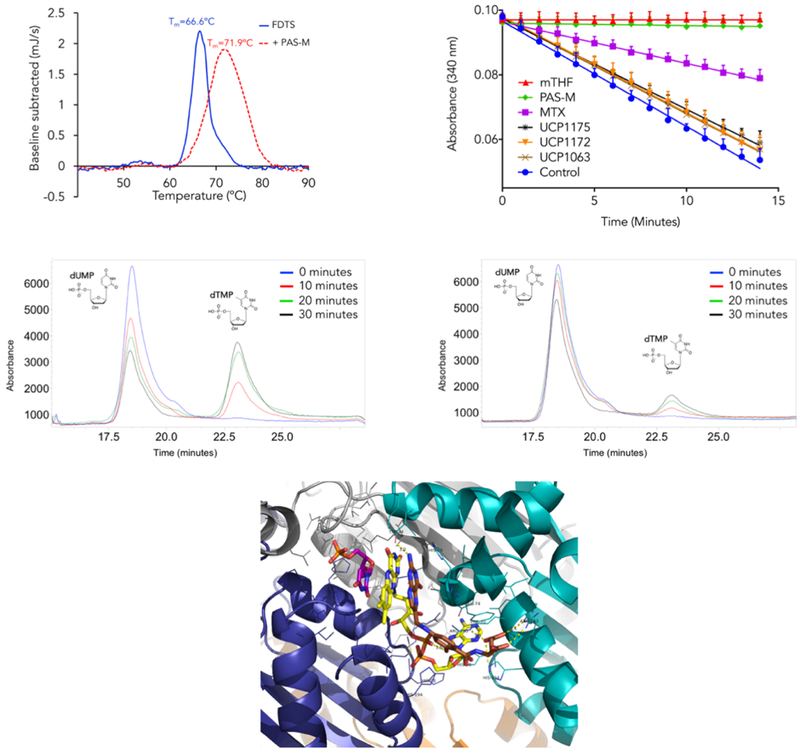
Top Left: Differential scanning calorimetry analysis of PAS-M binding to Mtb FDTS; Top right: Effect of INCAs, MTX, and PAS-M on the NADPH oxidation activity of Mtb FDTS. Data are presented as means ± SD, in three independent experiments; Middle left: HPLC analysis of Mtb FDTS reaction (uninhibited); Middle right: HPLC analysis of MtbFDTS reaction inhibited by PAS-M; Bottom: Modeling of PAS-M (brown) into the binding pocket of Mtb FDTS in complex with FAD (yellow) and dUMP (magenta)
It is known that mTHF and NADPH binding sites in FDTS partially overlap and high concentrations of mTHF inhibits NADPH oxidation due to competition for the binding site (Hunter et al., 2008; Koehn et al., 2012). A ligand that competes for the mTHF binding site should interrupt NADPH binding and ultimately oxidation event. The inhibitory effect of PAS-M, MTX, and INCAs on the NADPH oxidation activity of Mtb FDTS was tested by monitoring absorbance at 340 nM (Figure 4, top right). All the compounds were tested at stoichiometric equivalents of NADPH (200 μM). mTHF was used as the positive control with 100% inhibition of NADPH oxidation. Excitingly, PAS-M showed an inhibitory effect similar to mTHF, fully inhibiting NADPH oxidation. At the same concentration, MTX showed ~50% inhibition and no significant inhibition was observed with INCAs (less than 5%). We performed a dose-response NADPH oxidation inhibition assay to estimate the IC50 of PAS-M for the NADPH oxidation reaction (Figure S2). The IC50 was determined to be 14.4 ± 1.5 μM.
Although the NADPH oxidation assay is considered a valid method to identify FDTS inhibitors, it is not a direct measurement of dTMP synthesis. To evaluate the inhibitory effect of PAS-M on the production of dTMP, we monitored the formation of dTMP in FDTS reaction at 25 °C by employing high-pressure liquid chromatography (HPLC). The uninhibited reaction was used as the control and samples were taken at various times (0, 10, 20, and 30 minutes). Compared to the control, there is a significant decrease (~2.5 fold) in dTMP levels in the reaction incubated with PAS-M at stoichiometric equivalents with NADPH (100 μM) (Figure 4, middle left and right). This data suggests that PAS-M inhibits the formation of dTMP by the Mtb FDTS. To the best of our knowledge, this is the first report of PAS-M’s inhibitory effect on the Mtb FDTS.
Modelling PAS-M into FDTS
Our efforts to co-crystallize PAS-M with Mtb FDTS were not successful, even when attempting to soak PAS-M into a FDTS:FAD:dUMP complex. However, we solved a Mtb FDTS structure bound to FAD and dUMP at 3.5 Å resolution. We used this crystal structure for our structural analysis and modeled PAS-M into the Mtb FDTS binding site. Mtb FDTS is a tetramer with each monomer composed of a central α/β domain flanked by two helical top and bottom domains (Figure S3). The four monomers (a, b, c, d) come together to form a tetrameric molecule with four active sites. The adenine ring of FAD is buried in a deep binding pocket in the enzyme. The adenosine monophosphate (AMP) and flavin mononucleotide (FMN) moieties of FAD interact with conserved histidine and arginine residues in the binding pocket. The isoalloxazine ring forms hydrogen bonds with Arg95 of subunit a, Gln103 of subunit d, and Tyr144 of subunit c and stacks against the pyrimidine ring of dUMP. We then modelled PAS-M into the Mtb FDTS:FAD:dUMP complex using molecular modelling software (Schrädinger). In this model, the pterin moiety of PAS-M is stacked between the isoalloxazine ring of FAD (which itself is stacked against the pyrimidine ring of dUMP) and the imidazole of His69 of subunit c (Figure 4, bottom). This histidine residue is highly conserved among different species and its mutation has been shown to decrease the catalytic activity of FDTS from Thermotoga maritima (Koehn et al., 2012). The 2-amino group of the pteridine ring is 3.0 Å from Tyr44 of subunit c. The characteristic hydroxyl group of PAS-M is 3.1 Å from the conserved Arg190 and 3.5 Å from the phosphate groups of the FMN moiety of FAD. The carboxylates in the glutamate tail of PAS-M are within hydrogen bonding distance from Arg193, Arg190, and His194. We also assume that water-mediated interactions would further improve PAS-M affinity for Mtb FDTS.
Mutation rate and emergence of resistance to INCAs
To evaluate the development of resistance to INCAs and estimate mutation rates in vitro we performed fluctuation analysis to select spontaneously resistant mutants. Mutations conferring resistance to INCAs were measured to occur at the rate of 2.4 × 10−8 and 8.5 × 10−8 for UCP1172 and UCP1175, respectively (Table S2). This is similar to the rate reported for isoniazid, streptomycin, and bedaquiline (Andries et al., 2005; Huitric et al., 2010). The MIC values of the INAC-R strains were elevated by 32-fold, however owing to the high potency of UCP1172 against the wild-type organism, the MIC of resistant strains only rises to the level of 1 μg/mL. The mutation rate reported for PAS is 2 × 10−7 with MIC values rising up to 8 μg/mL(Zhao et al., 2014). The mutation rate of INCAs against methicillin-resistant Staphylococcus aureus (MRSA) is lower than what we observe for Mtb (Reeve et al., 2016). This is most likely due to lack of compensatory enzymes such as Rv2671 and FDTS in MRSA, an important distinction between Mtb and other common bacterial pathogens. Interestingly, resistance to INCAs also confers cross-resistance to PAS, showing a similar 32-fold loss of potency but with an MIC of 16 μg/mL. This also provides additional evidence that the major mechanism of antimycobacterial action of the INCAs is through blockade of the folate pathway.
The inhibitory effect of PAS and UCP1172 on mycolic acid production
It has been shown that antifolate treatment in Mtb leads to a critical disruption of the activated methyl cycle and decreases pools of methionine and S-adenosylmethionine (SAM) (Nixon et al., 2014). SAM is responsible for the supply of reactive methyl groups for a myriad of important cellular processes, including the formation of a variety of functional groups in meromycolic chain of mycolic acids that define several mycolate types in mycobacteria (Boissier et al., 2006). Given the link between the folate pathway and the activated methyl cycle, we hypothesized that treatment with INCAs or PAS may lead to changes in the lipid composition of the Mtb cell wall, particularly related to mycolic acids. We performed 14C-acetate labeling of model strain Mtb Erdman grown in the presence of UCP1172, which was added in early mid-log phase of growth at 0.36, 0.72, and 1.44 μg/ml final concentrations. As a control, the bacteria were treated with PAS at 0.36 and 0.72 μg/ml final concentrations in the same conditions. The treatment with UCP1172 or PAS did not lead to any dramatic alterations in the synthesis of major phospholipids, trehalose monomycolates, or trehalose dimycolates (Figure 5, left). However, total incorporation of 14C into the fractions of extracted lipids was reduced compared to the non-treated control (83 % and 66 % by 0.72 and 1.44 μg/ml UCP1172, respectively, and 82 % and 69 % by 0.36 and 0.72 μg/ml PAS, respectively). Analysis of fatty acid and mycolic acid methyl esters (FAME/MAME) extracted from radiolabeled cells revealed important changes in both UCP1172- and PAS-treated mycobacteria. An overall reduction of the 14C incorporation to the FAME/MAME fraction was observed (14, 36, and 56% reduction by 0.36, 0.72, and 1.44 μg/ml UCP1172, respectively, and 52% reduction by 0.36 and 0.72 μg/ml PAS). Interestingly, both PAS and UCP1172 treatment lead to a significant reduction of methoxymycolic acids (Figure 5, middle). For both PAS and UCP1172, further analysis of mycolic acids by argentation TLC revealed the appearance of other forms of mycolates which were not present in non-treated Mtb (Figure 5, right). Slower migration of these forms indicates the presence of mono- or di-unsaturation in their carbon chains. Since these forms of mycolic acids are precursors for further modifications with SAM-dependent methyltransferases we conclude that both UCP1172 and PAS interfere with these methylation processes. It has been shown that disruption of the folate pathway leads to perturbation of the activated methyl cycle and altered gene regulation involved in the biosynthesis and utilization of methionine and S-adenosyl methionine, including methyltransferases dependent on SAM (Nixon et al., 2014). We thus conclude that the observed effects (decrease in all forms of standard mycolates and accumulation of unsaturated mycolates) is most likely due to a lack of SAM for SAM-dependent methyltransferases involved in the modifications of mycolic acids.
Figure 5.
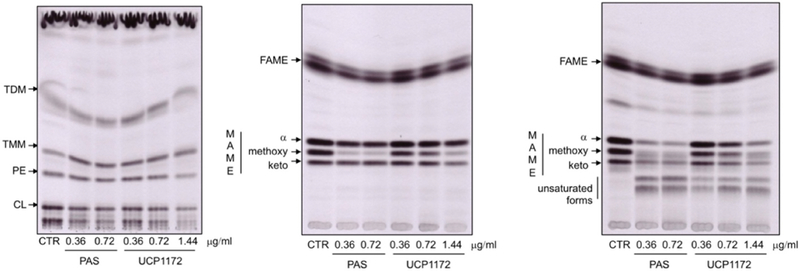
Left: Autoradiograph of TLC analysis of lipids extracted from 14C acetate labeled Mtb Erdman cells treated with UPC1172, PAS or DMSO as a control (CTR); Autoradiograph of standard (middle) and argentation (right) TLC analysis of methyl esters of fatty and mycolic acids isolated from 14C acetate labeled Mtb Erdman cells treated with UPC1172, PAS, or DMSO as a control (CTR)
DISCUSSION
Functional redundancy is commonly observed in Mtb and poses a challenge for drug discovery against this resilient pathogen (Barkan et al., 2010; Ganapathy et al., 2015; Johnston et al., 2010; van der Veen and Tang, 2015). One way to address this problem is multiple targeting of these functional analogs by use of a single pharmacophore. In fact, many successful monotherapeutic agents with low endogenous resistance rate have multiple targets (Silver, 2007, 2011). Although TB monotherapy is likely an unrealistic goal, anti-TB agents with multiple targets can simplify the regimen and improve treatment outcomes.
In this report, we have shown that both INCAs and PAS have multiple targets within the Mtb folate pathway. Biochemical studies indicate that INCA antimycobacterial activity is due to potent and long-lasting inhibition of canonical and non-canonical DHFR enzymes in Mtb. The importance of dual inhibition of Mtb DHFR and Mtb Rv2671, along with slow dissociation of the ligand from Mtb DHFR in improving the antimycobacterial activity of INCAs is reflected in the excellent MIC value of UCP1172 against Mtb (MIC= 0.03 μg/mL) (Table 1).
The elegant work by Chakraborty et al. (Chakraborty et al., 2013) and Zheng et al. (Zheng et al., 2013) showed that PAS is a competing substrate for DHPS and acts as a prodrug for DHFR. Our data elucidate the kinetics of PAS-M action against DHFR and reveals FDTS as a likely second target. To the best of our knowledge, inhibition of FDTS as part of the mechanism of action of PAS has not been reported before. This suggests that the downstream effects of PAS mis-incorporation into the folate pathway is more complicated than previously thought. We propose that PAS exerts its antimycobacterial activity by a multi targeted effect on the folate metabolism rather than acting on a single target within the pathway.
Finally, we have shown that antifolate treatment in Mtb results in a dramatic decrease in the level of all forms of standard mycolic acids, essential components of the mycobacterial cell wall. This adds to the previous report by Nixon et al. (2014) which indicated that antifolate treatment in Mtb disrupts the activated methyl cycle. They showed that treatment of Mtb cells with WR99210, a DHFR inhibitor, caused a 20-fold decrease in the concentration of SAM. SAM serves as the donor of reactive methyl groups for methylation of DNA, RNA, proteins, and lipids in reactions catalyzed by SAM-dependent methyltransferases. In Mtb, at least six SAM-dependent methyltransferases (encoded by mmaA1– mmaA4, cmaA2, and pcaA genes) were identified to catalyze the formation of mycolic acid chain (Barkan et al., 2009). These studies support the notion that inhibiting the production of reduced folates interrupts the biosynthesis of mycolic acids, most likely due to the interruption of the activated methyl cycle.
In the Mtb folate pathway, functional redundancy is used to ensure high levels of THF, considering DHFR, Rv2671, and FDTS all produce THF. We have identified compounds that target different pairs of these enzymes, but it is too enticing to not consider a compound capable of targeting all three enzymes. Such a compound would shut down all viable THF-generating enzymes and should prove highly potent against the organism. The work presented here can be further used in structure-based design of such inhibitors. It is important to note that the rate of mutation to INCAs suggest that compounds that inhibit multiple targets within the same pathway (series inhibition) are still prone to resistance as a single mutation can relieve inhibition of the sole essential pathway being targeted. This effect differs from classical combination chemotherapy approaches where multiple essential pathways are being simultaneously inhibited. Excitingly, the high initial potency of INCAs (UCP1172), means that the elevated MIC in the INCA-R strains only rises to 1 μg/mL, suggesting it may be possible to suppress these mutants at an achievable in vivo concentration.
New anti-TB agents and targets are required to address the issues associated with current drug regimens and to improve TB therapy outcomes. Our work on targeting the folate pathway in TB highlights the unique nature of this pathway relative to other pathogenic bacterial and suggests that it is possible to achieve highly potent agents by targeting multiple enzymes along the Mtb folate pathway. Downstream effects of the interrupted folate pathway in Mtb are manifold and include changes in many essential cell functions such as DNA and protein biosynthesis and cell wall metabolism. Continued efforts will have to consider the enzymes of the alternate pathway when designing antifolates as effective anti-TB agents.
STAR METHODS
CONTACT FOR REAGENTS AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dennis L. Wright (dennis.wright@uconn.edu).
BACTERIAL STRAINS AND CULTURE CONDITIONS
Mtb Erdman (ATCC 35801) was purchased from the American Type Culture Collection (Manassas, VA). The organism was grown in modified 7H10 Broth (pH 6.6; 7H10 agar formulation with agar and malachite green omitted) supplemented with 10% Middlebrook oleic acid-albumin-dextrose-catalase (OADC) enrichment and 0.05% Tween 80 on a rotary shaker at 37 °C for 7-10 days.
METHOD DETAILS
Synthesis of PAS-M and compound characterization
The synthesis of PAS-M was derived from a recent report by Dawadi et al (Dawadi et al., 2017). para-Nitrosalicylic acid was converted to its N-hydroxysuccinimidyl ester to activate the carboxylic acid to amidation with diethylglutamate. Subsequent nitro reduction by hydrogenation afforded the desired amine 1 (Scheme S1). Reductive amination with N-acetylformylpterin yielded the protected hydroxylated folate 2. 6-Formylpterin was converted to N-acetylformylpterin by refluxing in acetic anhydride. Global deprotection with sodium hydroxide afforded the desired hydroxylated folate adduct 3.
Compound characterization: 1,5-diethyl 2-[(4-amino-2-hydroxyphenyl) formamido]pentanedioate 1. para-Nitrosalicylic acid (2.10 g, 11.47 mmol), N-hydroxysuccinimide (1.45 g, 12.62 mmol), and DCC (2.60 g, 12.62 mmol) were added to a 100 mL round-bottomed flask fitted with a stir bar. DMF (50 mL) was added and stirred overnight. Overnight, a white precipitate had formed. The reaction was filtered through cotton into a separate 200 mL round-bottomed flask that contained a stirred solution of diethylglutamate HCl (3.03 g, 12.62 mmol), triethylamine (1.28 g, 12.62 mmol), and DMF (20 mL). The solution turned orange upon addition. After one hour, the DMF was removed in vacuo. The residue was brought up in ethanol (75 mL) and Pd/C (200 mg, 10% Pd) was added along with a stir bar. The flask was sealed, evacuated, and back-filled with a H2 balloon three times. The reaction stirred overnight with a H2 balloon to maintain positive pressure. The reaction was diluted with DCM, filtered through a celite plug, and the solvent was removed in vacuo. A column was run on the residue to afford the title compound as a yellow oil (1.61 g, 41 % three-step yield). 1H NMR (500 MHz, CDCl3) δ 12.40 (s, 1H), 7.27 (d, J = 8.4 Hz, 1H), 7.10 (d, J = 7.1 Hz, 1H), 6.21 – 6.10 (m, 2H), 4.73 (q, J = 7.4 Hz, 1H), 4.26 (q, J = 7.1 Hz, 2H), 4.21 – 4.05 (m, 4H), 2.53 (dt, J = 15.6, 7.1 Hz, 1H), 2.49 – 2.41 (m, 1H), 2.30 (dq, J = 13.2, 6.6, 6.1 Hz, 1H), 2.15 (dt, J = 14.4, 7.2 Hz, 1H), 1.32 (t, J = 7.1 Hz, 3H), 1.25 (t, J = 7.1 Hz, 3H). 13CNMR (126 MHz, CDCl3) δ 173.50, 172.04, 170.03, 163.65, 152.51, 127.60, 127.57, 106.47, 104.41, 101.69, 61.82, 60.96, 51.95, 30.54, 27.02, 14.16, 14.13. HRMS (DART, [M+H]+) m/z 339.1555, calculated for [C16H23N2O6+], 339.1551 (Data S1).
2-[(4-{[(2-amino-4-oxo-1,4-dihydropteridin-6-yl)methyl]amino}-2-hydroxyphenyl)formamido] pentanedioic acid 3. Aryl amine 1 (0.35 g, 1.03 mmol) was dissolved in AcOH (4 mL) and stirred. N-Acetylformyl pterin (0.22 g, 0.94 mmol) was suspended in AcOH (4 mL) and added slowly to the stirred solution of 1. The solution turned from cloudy to homogeneous within a few minutes. After 15 minutes, a solution of dimethylaminoborane (0.09 g, 1.50 mmol) in AcOH (1 mL) was added and stirred for 10 minutes. The reaction was heated to 65 °C for 20 minutes. The reaction volume was reduced in vacuo to 1 mL and slowly added to a stirred solution of Et2O (30 mL). A yellow solid crashed out immediately and was collected via filtration. The dried yellow solid was dissolved in NaOH (1.5 mL, 0.5 M) and heated to 70 °C for 1.5 h. Once cooled, the solution was acidified to pH 2 by addition of 1 M HCl and stored at 4 °C overnight. The material was isolated via centrifugation. Once the solution was decanted, the solid was washed three times each with water, EtOH, and Et2O. The material was dried in vacuo overnight to afford the title compound 3 (0.05 g, 25% two-step yield). 1H NMR (500 MHz, DMSO-d6) δ 12.60 (s, 1H), 12.40 (s, 2H), 11.44 (s, 1H), 8.66 (s, 1H), 8.43 (d, J = 7.6 Hz, 1H), 7.67 (d, J = 8.9 Hz, 1H), 7.06 (t, J = 5.9 Hz, 1H), 6.91 (s, 1H), 6.23 (dd, J = 8.8, 2.0 Hz, 1H), 6.00 (d, J = 2.0 Hz, 1H), 4.47 (d, J = 5.8 Hz, 2H), 4.38 (ddd, J = 9.6, 7.7, 5.0 Hz, 1H), 2.33 (t, J = 7.4 Hz, 2H), 2.09 (dq, J = 13.0, 7.7 Hz, 1H), 1.94 (ddd, J = 16.7, 14.1, 7.1 Hz, 1H). 13C NMR (126 MHz, DMSO-d6) δ 174.26, 173.76, 170.24, 162.95, 161.32, 157.09, 154.24, 153.54, 149.14, 148.81, 129.41, 128.46, 105.11, 103.64, 98.31, 51.83, 46.24, 30.78, 26.40. HRMS (DART, [M+H]+) m/z 458.1432, calculated for [C19H20N7O7+], 458.1419 (Data S2).
In vitro susceptibility testing
On the day of in vitro testing the drugs were thawed and diluted in modified 7H10 broth to 4-times the maximum concentration to be tested. The test range for INH was 8 μg/ml to 0.008 μg/ml and the test range for PAS was 64 μg/ml to 0.06 μg/ml. On the day of in vitro testing the organism was diluted in 7H10 broth to a final concentration of approximately 1 × 105 CFU/ml (in vitro inoculum). Polystyrene 96-well round-bottom plates (Corning Inc., Corning, NY) were prepared with 50 μl of 7H10 broth per well. The compounds were added to the first well prior to being serially 2-fold diluted throughout the row, leaving the last well with 7H10 broth only (growth control). Fifty μl of the in vitro inoculum was added to each well. Plates were sealed and incubated at 37 °C in ambient air for 14-21 days prior to reading. The minimal inhibitory concentration (MIC) was defined as the lowest concentration of drug required to inhibit growth of M. tuberculosis observed visually. The MIC assays were run in duplicate. The actual inoculum used was measured by titration in saline with Tween 80 and plating on 7H10 agar plates (Becton Dickinson, Sparks, MD). The plates were incubated in ambient air at 37°C for 4 weeks.
Transformation, expression and purification of Mtb DHFR, Hu DHFR, Mtb Rv2671, and Mtb FDTS
Recombinant plasmids harboring genes encoding Mtb DHFR (in pET-41a(+)),Hu DHFR (in pET-41a(+)) MtbRv2671 (in pET-28a(+)), and MtbFDTS (in pET-24d) were constructed by GenScript, separately. BL21(DE3) competent E. Coli cells (New England BioLabs) were transformed with the recombinant plasmids, separately. Transformed cells were grown in LB medium supplemented with 30 μg/mL kanamycin at 37°C until OD600 reached 0.6-0.7. The cells were induced with 1 mM IPTG for 20 hours at 20°C and spun down at 8000 rpm for 15 minutes. Each gram of wet cell pellet was resuspended in 5 ml of lysis buffer (25 mM Tris pH 8.0, 0.4 M KCl, 5 mM imidazole, 5 mM BME, 5% glycerol, 200 μg/ml lysozyme, 1 mM DNase I). The cell suspension was incubated for 30-60 minutes at 4°C with gentle rotation followed by sonication until a homogenous lysate was obtained. The lysate was centrifuged at 18,000 rpm for 30 minutes and supernatant was collected and filtered through 0.22 μm filter. The Mtb DHFR and Hu DHFR constructs did not contain histidine tag and were purified over methotrexate-agarose column pre-equilibrated with 4 CV of equilibration buffer (20mM Tris-HCl pH 7.5, 50 mM KCl, 2 mM DTT, 0.1 mM EDTA and 15% glycerol). The column was washed with 3 CV of wash buffer (20 mM Tris-HCl pH 7.5, 500 mM KCl, 2 mM DTT, 0.1 mM EDTA and 15% glycerol). The protein was eluted with 3 CV of elution buffer (equilibration buffer pH 8.5 + 2mM DHF). Mtb Rv2671 and Mtb FDTS constructs contained a histidine tag and were purified using Ni-NTA purification system (Thermo Scientific). The cell lysate supernatant was loaded over the column packed with HisPur Ni-NTA Superflow Agarose beads and washed with 5 CV of buffer A (25 mM Tris pH 8.0, 0.4 M KCl, 5 mM imidazole, 5 mM BME and 5% glycerol). The protein was eluted with a gradient of buffer B (25 mM Tris pH 8.0, 0.3 M KCl, 250 mM imidazole, 5 mM BME, 0.1 mM EDTA and 20% glycerol). For all the purifications, fractions containing DHFR protein were collected, concentrated and loaded onto a Hi-Prep 26/60 Sephacryl s-200 HR prepacked gel filtration/size exclusion column pre-equilibrated with 1 CV of final buffer (25 mM Tris pH 8.0, 50 mM KCl, 0.1 mM EDTA, 2 mM DTT and 15% glycerol). The column was washed with another 1 CV of final buffer and protein elution was monitored with AKTA UV/vis diode array spectrophotometer at 280 nm. Fractions containing pure enzyme were pooled, concentrated at 10 mg/ml and flash frozen in liquid nitrogen and stored at −80°C.
Enzyme inhibition assay for Mtb DHFR, Hu DHFR, and Mtb Rv2671
The DHFR activity of Mtb DHFR, Hu DHFR, and Mtb Rv2671 was measured in 500 μl of assay buffer containing 20 mM TES, 50 mM KCl, 0.5 mM EDTA, 10 mM 2-mercaptoethanol (BME) and 1 mg/ml bovine serum albumin (BSA) with various concentrations of NADPH and DHF ranging from 0 to 100 μM. The enzyme final concentration of 5-10 nM for MtbDHFR and 10-20 nM for other enzymes was used in the assay. Assay was started by adding DHF and monitoring NADPH oxidation at 340 nm. All measurements were performed at room temperature and in triplicate. Initial velocity data were fit with the Michaelis-Menten equation using Graphpad Prism 7.0 software. The DHFR activity inhibition assays and IC50 determination were performed in the same assay buffer with 100 μM NADPH and 100 μM DHF. For Mtb Rv2671 50 μM NADPH was used instead of 100 μM. Inhibitors, dissolved in 100% DMSO, were added to the mixture and incubated for 5 minutes prior to adding DHF.
Jump-Dilution Recovery Assay
Enzyme was incubated with the inhibitor (at concentration 10 times higher than IC50) for 16 hours at 4 °C in the absence of substrate to achieve nearly complete binding of the inhibitor to the DHFR enzyme. The mixture was then diluted 100-fold to stablish the final inhibitor concentration well below the IC50 into an assay mixture containing 100 mM DHF and 100 mM NADPH to disturb the equilibrium condition. Under this condition, minimum residual binding should occur and the observed rate constant, kobs, can be considered as a reasonable estimate of the dissociation rate constant koff. Enzymatic activity recovery was followed by monitoring NADPH oxidation at 340 nM for 60 minutes. Progress curves were fit into integrated rate Equation 1 using GraphPad Prism 7.0 using the initial velocity (the velocity observed in the presence of the inhibitor) as Vi the steady-state velocity (the final velocity following the dissociation of the inhibitor) as Vs to yield the observed activity rate constant (kobs). The residence time was estimated as the reciprocal of the observed activity recovery rate constant, kobs, as described in Equation (2) (Copeland et al., 2011; Walkup et al., 2015). The assay was performed in triplicate.
| Equation (1) |
| Equation (2) |
Mtb FDTS NADPH Oxidation Assay
Mtb FDTS NADPH oxidation assay was performed by monitoring NADPH oxidation at 340 nM using Genesys 150 UV-Vis spectrophotometer from Thermo Scientific. The assay buffer contained 100 μM dUMP, 50 μM FAD, 200 m=μM NADPH, 10 μM mTHF, and various concentrations of inhibitors (for inhibition assay). The assay was initiated by adding the enzyme at 10 nM final concentration. The assay was done in triplicate and each reaction was followed for 15 minutes (Hunter et al., 2008). The IC50 was determined by curve fitting using nonlinear regression and built-in equation ((log) inhibitor vs. response-Standard slope) in GraphPad Prism 7.0
Crystallization of Mtb DHFR, data collection, and structure determination
All the crystallization trials were performed by hanging drop vapor diffusion method and using EasyXtal 15-well plates (Qiagen). MtbDHFR was crystallized using slight modifications of conditions previously reported by Li et al (4). Pure protein was exchanged into the same final buffer containing 5% glycerol instead of 15%, incubated with 5 mM NADPH and 2mM ligand for two hours on ice. An equal volume of this mixture (final protein concentration 5 mg/ml) was mixed with crystallization solution (2 μl + 2 μl) and let to equilibrate against 500 μl of well solution at 4°C. Large pyramidal crystals grew within 8-12 weeks from solutions comprising 2.1-2.3 M (NH4)2SO4 and 0.1 M sodium acetate pH 4.5.
Crystals were flash frozen in the mother liquor supplemented with 20% glycerol. X-ray data were collected at Stanford Synchrotron Radiation Light Source (SSRL). Data were indexed and scaled using HKL2000. Structures were solved by molecular replacement using the Phaser (McCoy et al., 2007) and a previously reported complex (PDB ID: 5JA3) (Hajian et al., 2016). The programs Coot (Emsley and Cowtan, 2004) and Phenix (Adams et al., 2010) were used for structure refinement. Data collection and refinement statistics are reported in Table S3.
Crystallization of Hu DHFR, data collection, and structure determination
Hu DHFR was mixed with 10 mM NADPH and 2 mM ligand, incubated on ice for two hours and concentrated to 12 mg/ml. 2 μl of this solution was mixed with 2 μl of crystallization solutions containing 0.1 M Tris pH 7-7.5, 0.2-0.4 M Li2SO4, 25-35% PEG 4K and 7% EtOH. Large hexagonal crystals grew within 1-2 weeks at 4°C. Data collection and structure determination was performed similar to Mtb DHFR. A previously reported Hu DHFR (PDB ID: 4KAK) was used for molecular replacement (Lamb et al., 2013). Data collection and refinement statistics are reported in Table S3.
Crystallization of Mtb Rv2671, data collection, and structure determination
Mtb Rv2671 was mixed with 5 mM NADPH and 2 mM ligand, incubated at 4 °C overnight and concentrated to 5 mg/ml. 2 μl of crystallization solutions containing 0.1-0.3 M NaCl, 0.1 M Bis-Tris pH 5.5-7.5 and 20-30% PEG 3350 was mixed with 2 μl of protein solution and equilibrated against the well solution at 18 °C. Needle crystals grew within 2 weeks. Crystals were flash frozen in the mother liquor supplemented with 20% glycerol. X-ray data were collected at National Synchrotron Light Source II (NSLS II) at Brookhaven National Laboratory. Structures were solved by molecular replacement using the Phaser and previously reported complexes (Cheng and Sacchettini, 2016). The programs Coot and Phenix were used for structure refinement. Data collection and refinement statistics are reported in Table S3.
Crystallization of Mtb FDTS, data collection, and structure determination
The ternary complex of FDTS with FAD, dUMP was prepared by mixing the enzyme with a 2-fold molar excess of FAD and a 10-fold molar excess of dUMP. Prior to crystallization the complex was incubated at 4 °C for 18 hours and then concentrated to 12 mg/ml by ultrafiltration using Amicon Ultracel-10K filters (Millipore). Crystals were obtained by the hanging-drop vapor diffusion technique. An equal volume of the protein solution was mixed with a reservoir solution containing 18% PEG 8K, 0.15 M MgCl2, 0.1M Na Acetate pH 5.0 and 2 mM DTT, at 4 °C Initially, a shower of small crystals appeared in drops after 1 to 2 days. To delay the onset of crystallization and to reduce the number of crystals, an oil barrier (100 μl of 1:1 Paraffin: Silicon Oil mix) was applied over the reservoir solution in the standard hanging drops plates. Because of the favorable kinetics this process provided, larger and well-defined crystals were obtained after a week. Prior to data collection, crystals were soaked in the artificial mother liquor solution supplemented with 20% glycerol (v/v) and cryo-cooled in liquid nitrogen. X-ray data were collected at Stanford Synchrotron Radiation Light Source (SSRL). All data were integrated and reduced using iMOSFLM (Battye et al., 2011) and scaled using SCALA from the CCP4 suite (Winn et al., 2011). Data collection and refinement statistics are reported in Table S3.
Selection of PAS-resistant mutants
The resistant mutants were generated by growing M. tuberculosis Erdman (ATCC 35801) in modified 7H10 broth (pH 6.6; 7H10 agar formulation with agar and malachite green omitted) with 10% OADC (oleic acid, albumin, dextrose, catalase) enrichment (BBL Microbiology Systems, Cockeysville, MD) and 0.05% Tween 80 with 64 μg/ml of PAS added on a rotary shaker at 37°C for 14 days. After 14 days of incubation 1 ml of the broth was placed onto ten 7H10 agar plates with 10% OADC and 64 μg/ml of PAS. After 4 weeks of incubation at 37°C several colonies were selected, grown in 7H10 broth with 10% OADC, and tested for PAS resistance in a microtiter broth dilution assay.
Determination of mutation rates
The mutation rates were determined using fluctuation analysis and previously reported methods (Ford et al., 2011, 2013; Huitric et al., 2010; Pope et al., 2008). Briefly, Mtb cultures were grown to optical density (OD) of 0.7 to 1.0. Approximately 300,000 cells were used to inoculate 120mL of 7H10 media supplemented with 10% OADC and 0.05% tween 80, giving a total cell count of 10,000 cells per 4mL culture. This volume was immediately divided to 24 starter cultures of 4mL each in 15mL centrifugation tubes. 20 cultures were grown at 37°C with shaking for 14 days, until reaching an OD of 1.0. Cultures were spun at 4000 RPM for 10 minutes at 4°C and were resuspended in 250-500μL of the same growth media and spotted onto plates 7H10 agar plates supplemented with 10% OADC and 0.05% tween 80 and the inhibitor concentration at 10× times MIC. Plates were incubated at 37°C for 28 days. Cell counts were determined by serial dilution of the remaining four cultures. The experiment was performed in triplicate. The mutation rate (μ) was calculated using Equations explained in Quantification and Statistical analysis section.
DNA Extraction from PAS-Resistant isolates
DNA from the PAS-resistant strains were isolated using TRI Reagent purchased from Sigma Aldrich (St. Louis, MO) and following the TRI Reagent Protocol. Briefly, the PAS-resistant M. tuberculosis strains were grown in modified 7H10 broth with 10% OADC and 0.05% Tween 80 for 7-10 days. The isolates were diluted to 1 × 107 CFU/ml and 1ml of the isolates was placed into a sterile microfuge tube. The isolates were centrifuged for 10 minutes at 12,000 rpm. The supernatant was removed, and 1 ml of TRI Reagent was added to the pellet and vortexed. After standing at room temperature for 10 minutes 0.2 ml of chloroform was added to the tubes. The tubes were shaken for 15 seconds and allowed to stand for 10-15 minutes. The tubes were centrifuged for 15 minutes at 12,000 rpm at 4°C. The aqueous phase (containing RNA) was removed and discarded. To the interphase and organic phase (DNA and protein) 0.3 ml of 100% ethanol was added, mixed by inversion and allowed to stand for 2-5 minutes. The tubes were centrifuged in a cold room for 5 minutes at 6,000 rpm. The supernatant was discarded and the pellet containing the DNA was washed twice with a 0.1 M trisodium citrate: 10% ethanol solution. Between washing steps, the pellets were allowed to sit at room temperature for 30 minutes and then centrifuged at 6,000 rpm for 5 minutes. After the final wash the pellet was resuspended in 1 ml of 75% ethanol. Samples were plated on 7H10 agar to ensure that no viable Mtb remained.
Whole Genome Sequencing
One nanogram of genomic DNA (gDNA) was used for preparing the DNA library using Nextera XT DNA Library Prep Kit (Illumina), following the manufacturer’s recommended protocol. First, in a skirted PCR plate, gDNA was mixed with 10 μl of tagment DNA buffer and 5 μl of amplicon tagment mix. The plate was centrifuged at 280×g at 20°C for 1 minute followed by tagmentation program using thermal cycler setting of 55°C for 5 minutes and hold at 10 °C. After the sample reached 10 °C, 5 μl of neutralize tagment buffer was immediately added to neutralize the transposome. The sample was centrifuged at 280×g at 20°C for 1 minute followed by incubation at room temperature. The tagmented DNA was amplified using a limited-cycle PCR program. Nextera Index 1 (i7) and Index 2 (i5) adapters and Nextera PCR master mix were added to the tagmented gDNA and the samples were amplified using the following program on the thermal cycler:
72°C for 3 minutes
95 °C for 30 seconds
12 cycles of 95°C for 10 seconds, 55°C for 30 seconds, 72°C for 30 seconds
72°C for 5 minutes
Hold at 10°C
Library cleanup was performed using AMPure XP beads to remove short library fragments. The PCR products were centrifuged at 280×g at 20°C for 1 minute and the beads were added to each well. The ratio of PCR product to volume of beads was 3:2. The mixture was shaken at 1800 rpm for 2 minutes followed by incubation at room temperature for 5 minutes. The samples were places on magnetic stand and allowed for the liquid to become clear (2-3 minutes). Supernatant was removed and discarded from each well and the samples were washed with 200 μl of fresh 80% EtOH twice. The supernatant was again removed and discarded by gentle pipetting. The samples were air-dried on the magnetic stand for 15 minutes and then removed from the stand. The DNA was released from the beads by adding resuspension buffer followed by shaking at 1800 rpm for 2 minutes. The sample was placed on magnetic stand and allowed for the liquid to become clear. The supernatant containing clean DNA was transferred to a new plate. The size distribution of the fragments within the library was checked on an Agilent Technology 2100 Bioanalyzer using a high sensitivity DNA chip. The libraries were then normalized and pooled for sequencing step. The libraries were sequenced on the MiSeq using v2 2×250 base pair kit (Illumina). The genome assembly was performed using CLC Genomic Workbench and annotated on the RAST server.
Modelling of PAS-M into Mtb FDTS
Docking procedure was performed using version 2018-1 of the Schrädinger Biologics and Small- Molecule suites (for more details, see SI materials and methods). All calculations were performed using version 2018-1 of the Schrädinger Biologics and Small-Molecule suites. Solved FTDS in complex with FAD and dUMP was imported into Maestro and prepared with the Protein Preparation Wizard, which assigns tautomer and ionization states, water orientations, flips asparagine, glutamine, and histidine residues. All co-crystallization artifacts were removed from the structure, and waters only within 5Å of FAD and dUMP were kept (12). All other parameters were left unchanged on their default setting. Induced fit docking was performed with using the Induced Fit Docking protocol in Maestro (13). The centroid of the docking grid was generated by identifying the binding pocket residues: (Chain A) Tyr44, Tyr60, Val67, His69, Ser71; (Chain C) Met198 and His203. Extended sampling was selected, and all other parameters were left unchanged on their default setting. The ligand used for IFD was PAS-M. The structure was imported into Maestro and prepared for docking using the LigPrep protocol at pH 7.0 ± 2.0 (14).
Analysis of lipids and mycolic acids
The Mtb Erdman strain was grown by shaking at 37 °C in Midd lebrook 7H9 broth (Difco) supplemented with albumin-dextrose-catalase and 0.05% Tween 80. At O.D.600 nm of ~ 0.32 the culture was divided into 100 μl aliquots and tested compounds dissolved in DMSO were added in 0.36, 0.72 and 1.44 μg/ml final concentrations for UCP1172 and 0.36 and 0.72 μg/ml final concentrations for PAS. The final concentration of DMSO in each culture was kept 2 %. The cells grew 24 h statically in the presence of the tested compounds. Then 14C acetate (specific activity 106 mCi/mmol, ARC) was added in the final concentration 1 μCi/ml and the cultures continued growing for next 24 h. The lipids were extracted from 100 μl culture aliquots as described earlier with minor modifications (Stadthagen et al., 2005). Briefly, the cultures were transferred to 3 ml chloroform: methanol (2:1) and the mixtures were incubated at 65 °C for 2 h. After cooling down, 400 μl of water was added, the mixtures were mixed and centrifuged 15 min at 1 000 × g at room temperature. The organic phases were removed to the clean tube, dried under N2 and dissolved in 30 μl chloroform: methanol (2:1). 5 μl of each lipid sample were quantified by scintillation counting and 5 μl were loaded on the thin-layer chromatography (TLC) silica gel plates F254 (Merck). Lipids were separated in chloroform: methanol: water [20:4:0.5] and the plates were exposed to autoradiography film Biomax MR (Kodak) at −80 °C fo r 7 days. Methyl esters of fatty acids (FAME) and mycolic acids (MAME) were prepared from another 100 μl culture aliquots as previously described (Phetsuksiri et al., 1999). Dried extracts were dissolved in 30 μl chloroform: methanol (2:1), 5 μl of each sample were quantified by scintillation counting and 5 μl were loaded on TLC plates and different forms of methyl esters were separated in n-hexane/ethyl acetate [95:5], 3 runs and detected by autoradiography. Alternatively, methyl ester derivatives were loaded on TLC plates impregnated in 5% AgNO3 and activated at 100 °C for 1h and separated in the same solvent. The plates were exposed to autoradiography film Biomax MR (Kodak) at −80 °C for 7 days. The assay was performed in triplicate.
Differential Scanning Calorimetry (DSC)
To determine whether ligand binding to Mtb DHFR is stabilizing or destabilizing, we performed Differential Scanning Calorimetry (DSC) experiments on 1:1 molar ratio of protein:ligand incubated samples using a Nano-DSC instrument (TA instruments). 600 μl of Mtb DHFR at a concentration of 12 μM including NADPH and ligands or just NADPH (control) were heated 1 °C/min starting at 0 °C and up to 120 °C. For Mtb FDTS, the protein mixed with FAD and dUMP was used as the control. All samples were degassed extensively prior to injection into the calorimeter cell in order to prevent formation of air bubbles. The reference cell was filled with buffer for all runs. A pressure of 3 atm was applied to both cells during the run. The assay was performed in triplicate and the excess heat capacity scans for the protein transitions were obtained by subtracting a control scan of buffer versus buffer. The data were corrected for the difference in heat capacity between the initial and the final state by using a sigmoid baseline in the NanoAnalyze software (TA instruments) and fitted to a two-state transition model to determine the Tm values.
HPLC analysis of Mtb FDTS reaction
The reaction mixture was analyzed at different time points on a Shimadzu HPLC system using a reversed phase Phenomenex C 18 column (4.6 × 250 mm, 5 μm), with mobile phase A (5 mM potassium phosphate (pH 7.0), 5 mM tetrabutylammonium dihydrogen phosphate, and 5% acetonitrile) and mobile phase B (acetonitrile). The flow rate was 1 ml/min and the gradient was as follows: 100% A, 0-25 min; 100-10% A, 25-29 min; 10-100% A, 29-31 min; 100% A, 31-40 min. The injection volume for each sample was 50 μl and the autosampler was set at 4 °C. The standard solution of dUMP and dTMP were run to assign the retention times and the elution was monitored at 260nm. The experiment was performed in duplicate.
QUANTIFICATION AND STATISTICAL ANALYSIS
The signal intensity for spectrophotometric assay was quantified using Genesys 150 UV-Vis spectrophotometer from Thermo Scientific and the data were analyzed using GraphPad Prism 7.0. All the measurements were performed in triplicate and data shown in bar graphs are mean ± STDEV. Thermal stability data was analyzed using the NanoAnalyze software that is associated with the Nano-DSC instrument (TA instrument) and the data were corrected for the difference in heat capacity between the initial and the final state by using a sigmoid baseline and fitted to a two-state transition model to determine the Tm values.
The mutation rate (μ) was calculated using Equations 3 and 4
| Equation (3) |
| Equation (4) |
Nt= final number of cells (total CFU)
P0=number of independent cultures with zero mutant colonies
Ptot= total number of independent cultures
DATA AND SOFTWARE AVAILABILITY
All the macromolecular crystallography data including the protein sequences, coordinates, and the electron density map are publically available at the Protein Data Bank (https://www.rcsb.org/) using PDM IDs 6DDP, 6DDS, 6DDW, 6DE4, and 6DE5. PDB IDs are also provided in Key Resources Table. The genome sequencing data are publically available at NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra/) with the BioProject ID PRJNA523029. The accession numbers are provided in Key Resources Table.
KEY RESOURCE TABLE
| Reagents or Resources | Source | Identifier |
|---|---|---|
| Chemicals | ||
| Beta-nicotinamide adenine dinucleotide phosphate (NADPH) | Alfa-Aesar | Cat#J60387 |
| Dihydrofolic acid (DHF) | Sigma-Aldrich | Cat#D7006 |
| Tetrahydrofolic acid (THF) | Sigma-Aldrich | Cat#T3125 |
| Methotrexate | Sigma-Aldrich | Cat#A6770 |
| Isoniazid | Sigma-Aldrich | Cat#I3377 |
| Flavin adenine dinucleotide disodium salt hydrate (FAD) | Sigma-Aldrich | Cat#F6625 |
| 2’-deoxyuridine-5’-monophosphate disodium salt (dUMP) | Alfa-Aesar | Cat#J64627 |
| Thymidine 5’-monophosphate disodium salt (dTMP) | Sigma-Aldrich | Cat#T7004 |
| 5,10-Methylenetetrahydrofolate (mTHF) | Toronto Research Chemicals | Cat#F680350 |
| Isopropyl-1-thio-b-D-galacto-pyranoside (IPTG) | Thermo Scientific | Cat#R0392 |
| para-Aminosalicylic acid | Sigma-Aldrich | Cat#A79604 |
| Nextera XT DNA library preparation kit | Illumina | Cat#FC-131 |
| AMPure XP beads | Beckman-Coulter | Cat#A63880 |
| Microbial Strains | ||
| Mycobacterium tuberculosis Erdman Strain | ATCC | Cat#35801 |
| BL21 (DE3) competent E. Coli | New England BioLabs | Cat#C2527I |
| Recombinant DNA | ||
| Plasmid pET-28a(+)-MtbRv271 | GenScript | N/A |
| Plasmid pET-24d-MtbFDTS | GenScript | N/A |
| Plasmid pET-41a(+)-MtbDHFR | GenScript | N/A |
| Plasmid pET-41a(+)-HuDHFR | GenScript | N/A |
| Deposited Data | ||
| Macromolecular Crystallography | ||
| Structure of Mtb DHFR bound to UCP1172 | This study | PDB ID: 6DDP |
| Structure of Mtb DHFR bound to UCP1175 | This study | PDB ID: 6DDS |
| Structure of Mtb DHFR bound to PAS-M | This study | PDB ID: 6DDW |
| Structure of Hu DHFR bound to UCP1172 | This study | PDB ID: 6DE4 |
| Structure of Mtb Rv2671 bound to UCP1063 | This study | PDB ID: 6DE5 |
| Genome Sequences (BioProject ID: PRJNA523029) | ||
| Whole Genome Sequence of Wild-type Mtb | This paper | BioSample accession: SAMN10962817 |
| Whole Genome Sequence of PAS-R folC (E153G) mutant | This paper | BioSample accession: SAMN10962818 |
| Whole Genome Sequence of PAS-R folC (I43T) mutant | This paper | BioSample accession: SAMN10962820 |
| Whole Genome Sequence of PAS-R thyX mutant | This paper | BioSample accession: SAMN10962819 |
Supplementary Material
Data S1. NMR spectra of compound 1, Related to STAR methods. (top) 1H NMR of compound 1, (bottom) 13C NMR spectrum of compound 1.
Data S2. NMR spectra of 2compound 3, Related to STAR methods. (top) 1H NMR of compound 3, (bottom) 13C NMR spectrum of compound 3.
SIGNIFICANCE.
Tuberculosis therapy can specifically benefit from multi-targeting agents as Mycobacterium tuberculosis, the pathogen responsible for human tuberculosis, is treated by a long-term combination of drugs. We show that compounds with multiple targets within the M. tuberculosis folate pathway effectively inhibit the growth of live bacteria by disruption of essential cellular functions including DNA biosynthesis and cell wall metabolism. We also show that para-aminosalicylic acid (PAS), a TB drug used for years, has multiple targets and thus has provided a good example of a successful multi-targeted agent against tuberculosis.
Highlights.
Inhibitors of the folate pathway in Mycobacterium tuberculosis has been identified.
These molecules are dual inhibitors of dihydrofolate reductase and Rv2671.
Para-Aminosalicylic acid metabolite inhibits flavin-dependent thymidylate synthase.
Antifolates decrease the level of precursors of mycolic acids in M. tuberculosis.
ACKNOWLEDGEMENTS
We acknowledge that this research was funded by the US National Institute of Health (AI104841 and AI111957). We thank Stanford Synchrotron Radiation Lightsource (SSRL) and National Synchrotron Light Source II (NSLS-II) at Brookhaven National Lab for the beamline access and their staff members for their assistance. We also like to thank University of Connecticut Center for Open Research Sources and Equipment (COR2E) and Microbial Analysis, Resources, and Services (MARS). K.M. and J.K. also acknowledge support by Ministry of Education, Science, Research and Sport of the Slovak Republic (grant VEGA 1/0301/18 and 0395/2016 for Slovak/Russian cooperation in science 2015-15075/33841:1-15E0).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTEREST
D.W. is a co-founder of Quercus Molecular Design, LLC.
REFERENCES
- Adams PD, Afonine PV, Bunkóczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung L-W, Kapral GJ, Grosse-Kunstleve RW, et al. (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. Sect. D Biol. Crystallogr. 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andries K, Verhasselt P, Guillemont J, Gohlmann HWH, Neefs J-M, Winkler H, Van Gestel J, Timmerman P, Zhu M, Lee E, et al. (2005). A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science 307, 223–227. [DOI] [PubMed] [Google Scholar]
- Barkan D, Liu Z, Sacchettini JC, and Glickman MS (2009). Mycolic Acid Cyclopropanation is Essential for Viability, Drug Resistance, and Cell Wall Integrity of Mycobacterium tuberculosis. Chem. Biol. 16, 499–509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkan D, Rao V, Sukenick GD, and Glickman MS (2010). Redundant function of cmaA2 and mmaA2 in Mycobacterium tuberculosis cis cyclopropanation of oxygenated mycolates. J. Bacteriol. 192, 3661–3668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battye TGG, Kontogiannis L, Johnson O, Powell HR, and Leslie AGW (2011). iMOSFLM: a new graphical interface for diffraction-image processing with MOSFLM. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boissier F, Bardou F, Guillet V, Uttenweiler-Joseph S, Daffé M, Quémard A, and Mourey L (2006). Further insight into S-adenosylmethionine-dependent methyltransferases: structural characterization of Hma, an enzyme essential for the biosynthesis of oxygenated mycolic acids in Mycobacterium tuberculosis. J. Biol. Chem. 281, 4434–4445. [DOI] [PubMed] [Google Scholar]
- Celej MS, Dassie SA, González M, Bianconi ML, and Fidelio GD (2006). Differential scanning calorimetry as a tool to estimate binding parameters in multiligand binding proteins. Anal. Biochem. 350, 277–284. [DOI] [PubMed] [Google Scholar]
- Chakraborty S, Gruber T, Barry CE 3rd, Boshoff HI, and Rhee KY (2013). Para-aminosalicylic acid acts as an alternative substrate of folate metabolism in Mycobacterium tuberculosis. Science 339, 88–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Y, and Sacchettini JC (2016). Structural Insights into Mycobacterium tuberculosis Rv2671 Protein as a Dihydrofolate Reductase Functional Analogue Contributing to para -Aminosalicylic Acid Resistance. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Copeland RA (2010). The dynamics of drug-target interactions: drug-target residence time and its impact on efficacy and safety. Expert Opin. Drug Discov. 5, 305–310. [DOI] [PubMed] [Google Scholar]
- Copeland RA, Basavapathruni A, Moyer M, and Scott MP (2011). Impact of enzyme concentration and residence time on apparent activity recovery in jump dilution analysis. Anal. Biochem. 416, 206–210. [DOI] [PubMed] [Google Scholar]
- Dawadi S, Kordus SL, Baughn AD, and Aldrich CC (2017). Synthesis and Analysis of Bacterial Folate Metabolism Intermediates and Antifolates. Org. Lett. 19, 5220–5223. [DOI] [PubMed] [Google Scholar]
- Emsley P, and Cowtan K (2004). Coot : model-building tools for molecular graphics. Acta Crystallogr. Sect. D Biol. Crystallogr. 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- Fivian-Hughes AS, Houghton J, and Davis EO (2012). Mycobacterium tuberculosis thymidylate synthase gene thyX is essential and potentially bifunctional, while thyA deletion confers resistance to p-aminosalicylic acid. Microbiology 158, 308–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CB, Lin PL, Chase MR, Shah RR, Iartchouk O, Galagan J, Mohaideen N, Ioerger TR, Sacchettini JC, Lipsitch M, et al. (2011). Use of whole genome sequencing to estimate the mutation rate of Mycobacterium tuberculosis during latent infection. Nat. Genet. 43, 482–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ford CB, Shah RR, Maeda MK, Gagneux S, Murray MB, Cohen T, Johnston JC, Gardy J, Lipsitch M, and Fortune SM (2013). Mycobacterium tuberculosis mutation rate estimates from different lineages predict substantial differences in the emergence of drug-resistant tuberculosis. Nat. Genet. 45, 784–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganapathy U, Marrero J, Calhoun S, Eoh H, de Carvalho LPS, Rhee K, and Ehrt S (2015). Two enzymes with redundant fructose bisphosphatase activity sustain gluconeogenesis and virulence in Mycobacterium tuberculosis. Nat. Commun. 6, 7912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajian B, Scocchera E, Keshipeddy S, G-Dayanandan N, Shoen C, Krucinska J, Reeve S, Cynamon M, Anderson AC, and Wright DL (2016). Propargyl-linked antifolates are potent inhibitors of drug-sensitive and drug-resistant mycobacterium tuberculosis. PLoS One 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellman LM, Yin L, Wang Y, Blevins SJ, Riley TP, Belden OS, Spear TT, Nishimura MI, Stern LJ, and Baker BM (2016). Differential scanning fluorimetry based assessments of the thermal and kinetic stability of peptide-MHC complexes. J. Immunol. Methods 432, 95–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huitric E, Verhasselt P, Koul A, Andries K, Hoffner S, and Andersson DI (2010). Rates and Mechanisms of Resistance Development in Mycobacterium tuberculosis to a Novel Diarylquinoline ATP Synthase Inhibitor. Antimicrob. Agents Chemother. 54, 1022–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunter JH, Gujjar R, Pang CKT, and Rathod PK (2008). Kinetics and ligand-binding preferences of Mycobacterium tuberculosis thymidylate synthases, ThyA and ThyX. PLoS One 3, e2237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnston JB, Ouellet H, and Ortiz De Montellano PR (2010). Functional Redundancy of Steroid C 26-monooxygenase Activity in Mycobacterium tuberculosis Revealed by Biochemical and Genetic Analyses. J. Biol. Chem 285, 36352–36360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koehn EM, Perissinotti LL, Moghram S, Prabhakar A, Lesley S. a., Mathews II, and Kohen A (2012). Folate binding site of flavin-dependent thymidylate synthase. Proc. Natl. Acad. Sci. 109, 15722–15727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koul A, Arnoult E, Lounis N, Guillemont J, and Andries K (2011). The challenge of new drug discovery for tuberculosis. Nature 469, 483–490. [DOI] [PubMed] [Google Scholar]
- Kumar A, Guardia A, Colmenarejo G, Perez E, Gonzalez RR, Torres P, Calvo D, Gomez RM, Ortega F, Jimenez E, et al. (2015). A Focused Screen Identifies Antifolates with Activity on Mycobacterium tuberculosis. ACS Infect. Dis. 1, 604–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamb KM, G-Dayanandan N, Wright DL, and Anderson AC (2013). Elucidating Features That Drive the Design of Selective Antifolates Using Crystal Structures of Human Dihydrofolate Reductase. Biochemistry 52, 7318–7326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R, Sirawaraporn R, Chitnumsub P, Sirawaraporn W, Wooden J, Athappilly F, Turley S, and Hol WG. (2000). Three-dimensional structure of M. tuberculosis dihydrofolate reductase reveals opportunities for the design of novel tuberculosis drugs. J. Mol. Biol. 295, 307–323. [DOI] [PubMed] [Google Scholar]
- Mathys V, Wintjens R, Lefevre P, Bertout J, Singhal A, Kiass M, Kurepina N, Wang X-M, Mathema B, Baulard A, et al. (2009). Molecular genetics of para-aminosalicylic acid resistance in clinical isolates and spontaneous mutants of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 53, 2100–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy AJ, Grosse-Kunstleve RW, Adams PD, Winn MD, Storoni LC, Read RJ, and IUCr (2007). Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minato Y, Thiede JM, Kordus SL, McKlveen EJ, Turman BJ, and Baughn AD (2015). Mycobacterium tuberculosis folate metabolism and the mechanistic basis for para-aminosalicylic acid susceptibility and resistance. Antimicrob. Agents Chemother. 59, 5097–5106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mishanina TV, Yu L, Karunaratne K, Mondal D, Corcoran JM, Choi MA, and Kohen A (2016). An unprecedented mechanism of nucleotide methylation in organisms containing thyX. Science (80-. ). 351, 507–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradigaravand D, Grandjean L, Martinez E, Li H, Zheng J, Coronel J, Moore D, Torok ME, Sintchenko V, Huang H, et al. (2016a). dfrA thyA Double Deletion in para-Aminosalicylic Acid-Resistant Mycobacterium tuberculosis Beijing Strains. Antimicrob. Agents Chemother. 60, 3864–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moradigaravand D, Grandjean L, Martinez E, Li H, Zheng J, Coronel J, Moore D, Täräk ME, Sintchenko V, Huang H, et al. (2016b). dfrA thyA Double Deletion in para -Aminosalicylic Acid-Resistant Mycobacterium tuberculosis Beijing Strains. Antimicrob. Agents Chemother. 60, 3864–3867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myllykallio H, Leduc D, Filee J, and Liebl U (2003). Life without dihydrofolate reductase FolA. Trends Microbiol. 11, 220–223. [DOI] [PubMed] [Google Scholar]
- Nixon MR, Saionz KW, Koo MS, Szymonifka MJ, Jung H, Roberts JP, Nandakumar M, Kumar A, Liao R, Rustad T, et al. (2014). Folate pathway disruption leads to critical disruption of methionine derivatives in mycobacterium tuberculosis. Chem. Biol. 21, 819–830. [DOI] [PubMed] [Google Scholar]
- Oefner C, Winkler FK, to K Winkler CF, Zfe A, and Hoffmann-La Roche F (1988). Crystal structure of human dihydrofolate reductase complexed with folate. Eur. J. Biochem 174, 377–385. [DOI] [PubMed] [Google Scholar]
- Phetsuksiri B, Baulard AR, Cooper AM, Minnikin DE, Douglas JD, Besra GS, and Brennan PJ (1999). Antimycobacterial activities of isoxyl and new derivatives through the inhibition of mycolic acid synthesis. Antimicrob. Agents Chemother. 43, 1042–1051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pope CF, O’sullivan DM, Mchugh TD, and Gillespie SH (2008). MINIREVIEW A Practical Guide to Measuring Mutation Rates in Antibiotic Resistance. Antimicrob. Agents Chemother. 52, 1209–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeve SM, Scocchera E, Ferreira JJ, G-Dayanandan N, Keshipeddy S, Wright DL, and Anderson AC (2016). Charged Propargyl-Linked Antifolates Reveal Mechanisms of Antifolate Resistance and Inhibit Trimethoprim-Resistant MRSA Strains Possessing Clinically Relevant Mutations. J. Med. Chem. 59, 6493–6500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scocchera E, and Wright DL (2017). The Antifolates. (Springer, Berlin, Heidelberg: ), pp. 1–27. [Google Scholar]
- Scocchera E, Reeve SM, Keshipeddy S, Lombardo MN, Hajian B, Sochia AE, Alverson JB, Priestley ND, Anderson AC, and Wright DL (2016). Charged Nonclassical Antifolates with Activity Against Gram-Positive and Gram-Negative Pathogens. ACS Med. Chem. Lett. 7, 692–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silver LL (2007). Multi-targeting by monotherapeutic antibacterials. Nat. Rev. Drug Discov. 6, 41–55. [DOI] [PubMed] [Google Scholar]
- Silver LL (2011). Challenges of antibacterial discovery. Clin. Microbiol. Rev. 24, 71–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadthagen G, Korduláková J, Griffin R, Constant P, Bottová I, Barilone N, Gicquel B, Daffé M, and Jackson M (2005). p-Hydroxybenzoic acid synthesis in Mycobacterium tuberculosis. J. Biol. Chem. 280, 40699–40706. [DOI] [PubMed] [Google Scholar]
- Tonge PJ (2017). Drug-Target Kinetics in Drug Discovery. ACS Chem. Neurosci. 9, 29–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Veen S, and Tang CM (2015). The BER necessities: the repair of DNA damage in human-adapted bacterial pathogens. Nat. Rev. Microbiol. 13, 83–94. [DOI] [PubMed] [Google Scholar]
- Visentin M, Zhao R, and Goldman ID (2012). The antifolates. Hematol. Oncol. Clin. North Am. 26, 629–48, ix. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walkup GK, You Z, Ross PL, Allen EKH, Daryaee F, Hale MR, O’Donnell J, Ehmann DE, Schuck VJA, Buurman ET, et al. (2015). Translating slow-binding inhibition kinetics into cellular and in vivo effects. Nat. Chem. Biol. 11, 416–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AGW, McCoy A, et al. (2011). Overview of the CCP 4 suite and current developments. Acta Crystallogr. Sect. D Biol. Crystallogr. 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang K, and Rathod PK (2002). Divergent regulation of dihydrofolate reductase between malaria parasite and human host. Science (80-. ). 296, 545–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Li D, Zhao L, Fleming J, Lin N, Wang T, Liu Z, Li C, Galwey N, Deng J, et al. (2013). Genome sequencing of 161 Mycobacterium tuberculosis isolates from China identifies genes and intergenic regions associated with drug resistance. Nat. Genet. 45, 1255–1260. [DOI] [PubMed] [Google Scholar]
- Zhang X, Liu L, Zhang Y, Dai G, Huang H, and Jina Q (2015a). Genetic determinants involved in p-aminosalicylic acid resistance in clinical isolates from tuberculosis patients in northern China from 2006 to 2012. Antimicrob. Agents Chemother. 59, 1320–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Liu L, Zhang Y, Dai G, Huang H, and Jin Q (2015b). Genetic determinants involved in p-aminosalicylic acid resistance in clinical isolates from tuberculosis patients in northern China from 2006 to 2012. Antimicrob. Agents Chemother. 59, 1320–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao F, Wang X-D, Erber LN, Luo M, Guo A, Yang S, Gu J, Turman BJ, Gao Y, Li D, et al. (2014). Binding pocket alterations in dihydrofolate synthase confer resistance to para-aminosalicylic acid in clinical isolates of Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 58, 1479–1487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, Rubin EJ, Bifani P, Mathys V, Lim V, Au M, Jang J, Nam J, Dick T, Walker JR, et al. (2013). Para-aminosalicylic acid is a prodrug targeting dihydrofolate reductase in mycobacterium tuberculosis. J. Biol. Chem. 288, 23447–23456. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data S1. NMR spectra of compound 1, Related to STAR methods. (top) 1H NMR of compound 1, (bottom) 13C NMR spectrum of compound 1.
Data S2. NMR spectra of 2compound 3, Related to STAR methods. (top) 1H NMR of compound 3, (bottom) 13C NMR spectrum of compound 3.
Data Availability Statement
All the macromolecular crystallography data including the protein sequences, coordinates, and the electron density map are publically available at the Protein Data Bank (https://www.rcsb.org/) using PDM IDs 6DDP, 6DDS, 6DDW, 6DE4, and 6DE5. PDB IDs are also provided in Key Resources Table. The genome sequencing data are publically available at NCBI Sequence Read Archive (https://www.ncbi.nlm.nih.gov/sra/) with the BioProject ID PRJNA523029. The accession numbers are provided in Key Resources Table.
KEY RESOURCE TABLE
| Reagents or Resources | Source | Identifier |
|---|---|---|
| Chemicals | ||
| Beta-nicotinamide adenine dinucleotide phosphate (NADPH) | Alfa-Aesar | Cat#J60387 |
| Dihydrofolic acid (DHF) | Sigma-Aldrich | Cat#D7006 |
| Tetrahydrofolic acid (THF) | Sigma-Aldrich | Cat#T3125 |
| Methotrexate | Sigma-Aldrich | Cat#A6770 |
| Isoniazid | Sigma-Aldrich | Cat#I3377 |
| Flavin adenine dinucleotide disodium salt hydrate (FAD) | Sigma-Aldrich | Cat#F6625 |
| 2’-deoxyuridine-5’-monophosphate disodium salt (dUMP) | Alfa-Aesar | Cat#J64627 |
| Thymidine 5’-monophosphate disodium salt (dTMP) | Sigma-Aldrich | Cat#T7004 |
| 5,10-Methylenetetrahydrofolate (mTHF) | Toronto Research Chemicals | Cat#F680350 |
| Isopropyl-1-thio-b-D-galacto-pyranoside (IPTG) | Thermo Scientific | Cat#R0392 |
| para-Aminosalicylic acid | Sigma-Aldrich | Cat#A79604 |
| Nextera XT DNA library preparation kit | Illumina | Cat#FC-131 |
| AMPure XP beads | Beckman-Coulter | Cat#A63880 |
| Microbial Strains | ||
| Mycobacterium tuberculosis Erdman Strain | ATCC | Cat#35801 |
| BL21 (DE3) competent E. Coli | New England BioLabs | Cat#C2527I |
| Recombinant DNA | ||
| Plasmid pET-28a(+)-MtbRv271 | GenScript | N/A |
| Plasmid pET-24d-MtbFDTS | GenScript | N/A |
| Plasmid pET-41a(+)-MtbDHFR | GenScript | N/A |
| Plasmid pET-41a(+)-HuDHFR | GenScript | N/A |
| Deposited Data | ||
| Macromolecular Crystallography | ||
| Structure of Mtb DHFR bound to UCP1172 | This study | PDB ID: 6DDP |
| Structure of Mtb DHFR bound to UCP1175 | This study | PDB ID: 6DDS |
| Structure of Mtb DHFR bound to PAS-M | This study | PDB ID: 6DDW |
| Structure of Hu DHFR bound to UCP1172 | This study | PDB ID: 6DE4 |
| Structure of Mtb Rv2671 bound to UCP1063 | This study | PDB ID: 6DE5 |
| Genome Sequences (BioProject ID: PRJNA523029) | ||
| Whole Genome Sequence of Wild-type Mtb | This paper | BioSample accession: SAMN10962817 |
| Whole Genome Sequence of PAS-R folC (E153G) mutant | This paper | BioSample accession: SAMN10962818 |
| Whole Genome Sequence of PAS-R folC (I43T) mutant | This paper | BioSample accession: SAMN10962820 |
| Whole Genome Sequence of PAS-R thyX mutant | This paper | BioSample accession: SAMN10962819 |