

Abstract
Traumatic Brain Injury (TBI) affects more than 1.7 million Americans each year and about 30% of TBI-patients having visual impairments. The loss of retinal ganglion cells (RGC) in the retina and axonal degeneration in the optic nerve have been attributed to vision impairment following TBI; however, the molecular mechanism has not been elucidated. Here we have shown that an increase in histone di-methylation at lysine 9 residue (H3K9Me2), synthesized by the catalytic activity of a histone methyltransferase, G9a is responsible for RGC loss and axonal degeneration in the optic nerve following TBI. To elucidate the molecular mechanism, we found that an increase in H3K9Me2 results in the induction of oxidative stress both in the RGC and optic nerve by decreasing the mRNA level of antioxidants such as Superoxide dismutase (sod) and catalase through impairing the transcriptional activity of Nuclear factor E2-related factor 2 (Nrf2) via direct interaction. The induction of oxidative stress is associated with death in RGC and oligodendrocyte precursor cells (OPCs). The death in OPCs is correlated with a reduction in myelination, and the expression of myelin binding protein (MBP) in association with degeneration of neurofilaments in the optic nerve. This event allied to an impairment of the retrograde transport of axons and loss of nerve fiber layer in the optic nerve following TBI. An administration of G9a inhibitor, UNC0638 attenuates the induction of H3K9Me2 both in RGC and optic nerve and subsequently activates Nrf2 to reduce oxidative stress. This event was concomitant with the rescue in the loss of retinal thickness, attenuation in optic nerve degeneration and improvement in the retrograde transport of axons following TBI.
1. Introduction
Traumatic brain injury (TBI) is a significant cause of death and disability, with an estimated worldwide incidence of about 10 million cases per year [1–3]. The ocular and vision damage has been reported previously as a consequence of TBI, and approximately 20–40% of people with brain injury experience related vision disorders [4], as part of the post-concussion syndrome [5–8]. The incidence of TBI and the symptoms of photo-sensitivity, blurred vision, double vision, decreased visual acuity, and visual field defects in the US has increased markedly in recent decades [1,9]. The loss of retinal ganglion cells (RGCs) and structural damage to the optic nerve [1,9,10] have shown to contribute to the TBI induced retinal dysfunction; however, the underlying mechanism has not been elucidated yet.
Even though the retina is composed of several layers, RGCs are the primary cell type in the innermost cellular layer of the retina, responsible for carrying visual information between the eye and the brain [11]. Considering that Brn3a exclusively expressed in the nucleus of RGC, Brn3a has been recognized as an exclusive and reliable marker for RGCs [12,13]. The optic nerve is comprised of axons from RGCs, whose somas reside within the retina. The oligodendrocyte progenitor cells (OPCs) persist in substantial numbers in the adult optic nerve in a quiescent state and provide a source of new oligodendrocytes after injury [14,15]. The proliferation and differentiation of OPCs into oligodendrocytes is critical for myelination of optic nerves, which is required to establish the proper communication between the retina and the brain [16–18]. Damage to the myelin sheath and oligodendrocytes of the optic nerve fibers directly affects the neurofilament composition and functions of axons following TBI [19].
Most of the biochemical cascades which occur in response to primary and secondary injury after TBI generate oxidative stress, due to an imbalance between oxidant and antioxidant agents. Several oxidative stress markers (carbonylated proteins, lipid peroxides, reactive oxygen species) are increased, while antioxidant defense enzymes such as GSH, superoxide dismutase (SOD), and catalase (CAT) were decreased in the brain after TBI. This imbalance results in cellular dysfunction and death and is directly related to the pathogenesis of TBI [20,21]. RGCs are very susceptible to oxidative stress, and it was shown that oxidative stress or reactive oxidants are the significant factors involved in retinal RGCs death in several ocular neurodegenerative diseases such as glaucoma, AMD and optic nerve degeneration [22]. Retinal ganglion cell axons have been considered essential for migration, proliferation, and survival of oligodendrocyte lineage cells in the optic nerve {Ueda, 1999 #6212}. Under normal condition, oligodendrocyte precursors cells (OPCs) migrated along the length of the nerve and subsequently multiplied and differentiated into myelin basic protein (MBP)–positive oligodendrocytes, which is followed by axonal ensheathment and myelination. The appearance of OPCs, oligodendrocytes, and myelin in the optic nerve follows a reproducible temporal and spatial pattern [14,23]. Under disease condition, oligodendroglia is particularly vulnerable to oxidative damage after neurotrauma in vivo [24] and affect myelination [14,25,26].
Oxidative damage is typically minimized by the presence of a range of antioxidant and efficient repair systems. A primary mechanism in the cellular defense against oxidative stress is the activation of the Nuclear factor E2-related factor 2 (Nrf2)-antioxidant response element signaling pathway, which controls the expression of genes whose protein products are involved in the detoxication and elimination of reactive oxidants agents mostly by enhancing cellular antioxidant capacity [27–29]. Nrf2 is a basic-region leucine zipper transcription factor that acts to synthesize genes for antioxidants such as superoxide dismutase (sod) and catalase [30–33]. In basal conditions, Nrf2 is retained in cytoplasm along with other complex proteins and undergoes ubiquitination and proteasomal degradation. Under stress condition, Nrf2 translocates and accumulates in the nucleus where it binds to cis-acting antioxidant response elements (ARE) in the promoter region of antioxidant genes [30–34]. The impairment of Nrf2 has been associated with diabetic retinopathy [35], glaucoma [36], ischemia-reperfusion [37], and age-related macular dystrophy [36].
Other than self-regulation, activation of transcription factors also depends on the status of chromatin and the modification of histone tails [38]. One of the most studied post-translational modifications of histone is histone methylation, which primarily occurs on the lysine residues of histones. Methylation of histone polypeptides marks a gene to be or not be transcribed depending on whether it is monomethylated or dimethylated [39,40]. The di-methylation of histone three at lysine 9 (H3K9Me2) has been identified as a chromatin silencer, and it is preferentially catalyzed by a histone methyltransferase, G9a [41–43]. In general, an increase in H3K9Me2 has been shown to restrict the binding of transcription factors to the promoters of their downstream genes and thereby reduces their mRNA and protein levels. G9a is substantially expressed in the mouse retina throughout development and in the adult mice [44]. The G9a inhibitor UNC0638 inhibits the catalytic activity of G9a with excellent potency and selectivity over a wide range of epigenetic and non-epigenetic targets [45]. Inhibition of G9a complex has shown to enhance memory in neural networks [46]. However, its role in visual functions after TBI has not been elucidated.
In the present study, we have shown that an increase in G9a/ H3K9Me2 contributes to RGC loss and optic nerve degeneration following TBI. As a part of the mechanism, we have demonstrated that an increase in H3K9Me2 blocks the transcriptional activity of Nrf2, which increases oxidative stress in RGC and optic nerve following TBI. These events, in turn, prevents the RGC loss and optic nerve degeneration after TBI.
2. Materials and methods
2.1. TBI procedure
The Committee on Animal Use for Research and Education at the University of Pittsburgh approved all animal studies, in compliance with National Institutes of Health guidelines. The procedure was performed based on our previously published protocol [47–49]. Briefly, 8–12-week old adult male C57BL/6 (Jackson Laboratory) mice were anesthetized with xylazine (8 mg/kg)/ketamine (60 mg/kg) and subjected to a sham injury or controlled cortical impact (CCI). Lubricant eye drop (Allergan Inc.) were applied frequently, with saline wash throughout the surgical procedure. Mice were placed in a stereotaxic frame (Ambient Instruments), and a 3.5 mm craniotomy was made in the right parietal bone targeting primary visual cortex (0–1 mm anterior to the lambda suture, 1.5–3 mm lateral to the midline). Mice were impacted at 4.5 m/s with a 20 ms dwell time and 1.2 mm depression using a 3-mm-diameter convex tip, mimicking a moderate TBI. Sham-operated mice underwent identical surgical procedures but were not impacted. The incision was closed with 3 M Vetbond tissue adhesive and mice were allowed to recover on the heat pad.
2.2. UNC0638 treatment to mice
UNC0638 was purchased from Sigma Aldrich and was dissolved in DMSO. UNC0638 (5 mg/kg) was delivered by intraperitoneal injection to sham and TBI groups at 30 min after TBI, followed by a daily dose for 7 days. Because UNC0638 was dissolved in DMSO, only sham and TBI mice received DMSO as controls.
2.3. Chromatin immunoprecipitation (ChIP) assays
ChIP assays were performed to study the transcription factor Nrf2 binding to sod promoter were performed following the manufacturer (Millipore) as published previously with minor modifications [47,48,50]. In each assay, six retinas or optic nerves were pooled from either sham or TBI and half of each sample was mixed with either Nrf2 antibody or normal rabbit serum for a control (Jackson ImmunoResearch Laboratories). The supernatant obtained from the normal serum samples following immunoprecipitation was regarded as the input. DNA from immunoprecipitates was unlinked from protein complexes and purified further by phenol/chloroform extraction. Samples were analyzed in triplicate using qPCR. The data represents as fold change between TBI and sham group of mice.
2.4. Oxidative stress measurement
The oxidative stress was measured using a novel fluorogenic probe CellROX® Green from Invitrogen. The freshly isolated retinas and optic nerves were washed with Dulbecco’s phosphate-buffered saline and stained with 5 μM CellRox Green Reagent (Invitrogen) at 4 °C for 6 h in the dark. After two washes with Dulbecco’s phosphate-buffered saline, the retinas were fixed with 4% PFA at 4 °C for 2 h and were then flat mounted onto glass slides. Imaging was performed with a confocal laser-scanning microscope as described previously [51].
2.5. TBARS assay
The TBARS assay (thiobarbituric acid reactive substance assay) was carried out according to previously reported methods with minor modifications [52–54]. TBARS assays measure the total level of oxidized lipids based on the reaction of malondialdehyde (MDA), one of the end products of lipid peroxidation, with thiobarbituric acid (TBA) [53]. Briefly, the retinal and optic nerve homogenates, in 1.15% KCl containing 1% protease inhibitor cocktail and 0.5 mM butylated hydroxytoluene (BHT), was added to a reaction mixture (0.81% SDS, 0.36% TBA, and 9% acetic acid) on ice. After heating the reaction mixture to 100 °C for 1 h, it was centrifuged at 20,000g for 10 min at 4 °C. The supernatant was collected, and its fluorescence was measured at 530 nm excitation and 550 nm emissions. The results were normalized to protein concentration, which was determined with the bicinchoninic acid (BCA) protein assay kit (Thermo Scientific).
2.6. 4-HNE staining to detect lipid peroxidation
The lipid peroxidation product 4 hydroxy-2-noneal or 4-HNE, a sensitive marker of oxidative damage and lipid peroxidation and was evaluated by immunofluorescence staining using tissue sections. For immunofluorescence staining, fixed frozen retina or optic nerve sections were washed three times with PBS and permeabilized for 30 min with 0.1% Triton-X-100 in PBS as described previously with minor modifications [55]. Unreacted aldehydes were quenched by 5 min wash with 1 mg/ml sodium borohydride in PBS. Sections were blocked with 5% normal goat serum containing 0.5% Triton X-100 for 1 h, followed by incubation with the primary antibody against 4-HNE (JaICA, Japan; 1:100), overnight at 4 °C. Sections were then washed and incubated with the appropriate Alexa, Cy5 Fluor-tagged secondary antibody. Sections were mounted with ProlongTM Gold antifade reagent with DAPI (Invitrogen).
2.7. Terminal-deoxynucleotidyltransferase mediated nick end labeling (TUNEL) assay
TUNEL reactions (DeadEnd™ fluorometric TUNEL System, Promega Corporation) were performed to detect apoptotic cell. To ensure the use of similar fields for comparison, all frozen retinal and optic nerve sections were prepared with retinas at 1–2 mm distance from the optic nerve head. The TUNEL positive cells in the RGC layer of each sample were counted in ten high powered fields, and three sections per eye were averaged [56,57].
2.8. Nuclear fraction isolation from both retina and optic nerve
Retina and optic nerve tissues were extracted and separated into nuclear and cytoplasmic fractions using a nuclear/cytoplasmic separation kit (Bio Vision), following the manufacturer’s instruction [58]. Briefly, 1 mg of the retina or optic nerve tissues were gently dissociated in 200 μl of Cytosol Extraction Buffer A (supplemented with DDT and protease inhibitor) using a Dounce homogenizer at 4 °C and cells were pelleted by centrifugation at 500×g for 2–3 min. Cytosolic fraction was collected after processing the tissue in Cytosol Extraction Buffer A and B according to the manufacturer’s instructions. Nuclear extract was prepared after processing the remaining nuclei pellet in 50 μl of ice-cold Nuclear Extraction Buffer Mix. Protein concentration was determined using the BCA method (Thermo Scientific), and extracts were stored at −80 °C until use.
2.9. Western blot and co-immunoprecipitation
Retina or Optic nerve lysates were prepared as described previously [47,57,59,60]. Tissue was placed in RIPA buffer (containing protease and phosphatase inhibitor, Invitrogen), sonicated, and centrifuged for 5 min at 12,000×g at 4 °C. Fifty micrograms of protein were resolved on a 4–20% SDS-polyacrylamide gel and transferred on to a nitrocellulose membrane. Blots were incubated overnight at 4 °C in primary antibody against Nrf2 (Cell Signaling, 1:500), H3KMe2 (Active Motif., 1:500), G9a (Perseus proteomics, Japan, 1:100), SOD (Sigma-Aldrich, 1:500), Catalase (Rockland Immunochemicals Inc, 1:500), and Actin (Sigma-Aldrich, 1:5000) followed by a 2 h incubation with a Licor IRDye secondary antibody at room temperature. Blots were visualized using a LiCor Odyssey near-infrared imaging system, and densitometry analysis for each band was performed using Image Studio Lite software, and the changes in the experimental band intensities were represented as the fold change as described previously [47,57,59,60]. Protein-protein interactions were measured by co-immunoprecipitation assay per our method [47,57,59,60]. Briefly, tissue was homogenized in lysis buffer containing 50 mmTris, pH 7.4, 150 mM NaCl, 0.5% (v/v) tween- 20, 50 mM Tris (pH 7.5), 1 mM EDTA with protease and phosphatase inhibitor by passing through a 26-gauge syringe needle and centrifuged at 12,000×g at 4 °C for 5 min. 400 μg of the total protein for each sample was incubated overnight with Nrf2 (Cell Signaling, 1:100). 30 μl of protein G agarose was added, and SDS-PAGE resolved co-immunoprecipitates and analyzed by western blotting with H3KMe2 antibody (Active Motif., 1:500).
2.10. Quantitative real-time PCR
Quantitative real-time PCR (qRT-PCR) was performed per our method [47,49]. Total RNA was isolated using the kit (QIAGEN), and the cDNA was subjected to quantitative real-time-PCR (qRT-PCR) analysis with fast-standard SYBR green dye (Applied Biosystems) using an AB7500 RT-PCR instrument (Applied Biosystems). Results were normalized using total input DNA and expressed as bound/Input (fold). Optic nerve and retina were taken after 7d of TBI induction on mouse brain. The primers used to carry out PCR analysis were purchased from realtimeprimers.com and Invitrogen are as follows.
catalase (NM_009804): TGAGAAGCCTAAGAACGCAATTC, CCCTTC GCAGCCATGTG;
sod (NM_011434): GTGATTGGGATTGCGCAGTA, TGGTTTGAGGG TAGCAGATGAGT; and
Actin: AAGAGCTATGAGCTGCCTGA, TACGGATGTCAACGTCACAC.
Product specificity was confirmed by melting curve analysis. Gene expression levels were quantified, and data were normalized to Actin, a housekeeping gene. Data are expressed as mean fold change versus vehicle placebo treatment.
2.11. Quantification of RGCs in mice retinal flat mounts using fluorogold staining
Retrograde staining of the RGCs of the eye was achieved by injecting a fluorescent dye into the superior colliculus bilaterally as described previously with some modifications [61,62]. The mice (n = 5/ group) were placed in a stereotactic apparatus (RWD Life Science Co. Ltd., Shenzhen, China), following intraperitoneal injection of ketamine and xylazine to ensure the animals remained immobile and the skin of the skull was incised. The brain surface was exposed by perforating the parietal bone with a drill to facilitate dye injection. Fluorogold [2-Hydroxystilbene-4,4′-dicarboxamidine bis(methanesulfonate), Sigma-Aldrich] was injected (3%; 5.0 μl each) at a point 2.92 mm caudal to the bregma and 0.50 mm lateral to the midline on the two sides, to a depth of 1.60 mm from the surface of the skull. Further, on 5th day TBI was induced on right visual cortex by CCI method as described above.
Subsequently, on the 11th day after the injection of Fluorogold (FG) into the visual cortex, the left eye was enucleated following the sacrifice of the animals with an overdose of intraperitoneal ketamine/xylazine cocktail. Immediately, cornea and lens were removed from the eye, and the remaining cup was fixed in 4% paraformaldehyde (PFA) in PBS for 1 h at room temperature. Eyecup was washed with PBS for 15 min in the same conditions. A total of four radial cuts were made in the periphery of the eyecup, and the retina was carefully separated from the retinal pigment epithelium.
The retinae were then flat mounted on a glass slide and preserved in the dark at 4 °C. The retinal flat mounts were viewed using a fluorescence microscope (Carl Zeiss, magnification, 20 × ). The FG labelled RGCs was manually counted in a blinded manner, by another investigator, in each quadrant at 0.5, 1 and 1.5 mm radial from the optic nerve. The total quantity of RGCs per mm2 in all four quadrants was calculated. Cells with an irregular shape, intense dye staining, or a smaller or larger size than typical RGCs were considered as non-RGC cells and excluded from counting.
2.12. IHC protocol for retina and optic nerve sections
The IHC was performed per our method as described previously with some modifications [63,64]. Immediately following euthanasia, mice were transcardially perfused with 0.1 M phosphate-buffered saline (PBS; pH 7.4) followed by 4% paraformaldehyde (over 10 min). After perfusion eyes were enucleated and immediately eye limbus was punctured. Subsequently, eyes were fixed overnight in 4% PFA at 4 °C, then cryoprotected overnight by incubation in 10% then 18% sucrose at 4 °C. Eyeballs were embedded in optimum cutting temperature (OCT) medium and frozen at −80 °C. Eyes were sectioned (12 μm) on a cryostat at −22 °C and mounted on Superfrost Plus slides. Optic nerve sections were sectioned at 12 μm and stained for selected antigens.
For immunofluorescence staining, fixed frozen eye or optic nerve sections were washed three times with PBS and permeabilized for 30 min with 0.1% Triton-X-100 in PBS. Unreacted aldehydes were quenched by 5 min wash with 1 mg/ml sodium borohydride in PBS. Sections were blocked with 5% normal goat serum containing 0.5% Triton X-100 for 1 h, followed by incubation with the primary antibody against Brn3a (Santa Cruz, 1:100; Brn3a is a marker of RGCs), G9a (Perseus proteomics, Japan, 1:100; G9a is a histone methyltransferase enzyme that catalyzes the mono- and di-methylated states of histone H3 at lysine residue), H3K9me2 (Active Motif, 1:100; H3K9me2 is a covalent histone modification that is catalyzed by G9a and used as an epigenetic mark associated with transcriptional repression), MBP (Santa Cruz, 1:100; MBP is an essential component of the myelin sheath, and it is considered to be a marker for oligodendrocyte differentiation and myelination of neurons), SMI-32 (BioLegend, 1:100; SMI-32 antibody is a marker of axonal damage, it recognizes a non-phosphorylated epitope of neurofilament proteins that maintenance of large neurons with highly myelinated processes), olig2 (Santa Cruz, 1:100; Oligodendrocyte transcription factor or olig2 is an oligodendroglial lineage marker), 8-OhDG (Antibody Online, 1:100; 8-OhDG is a marker for oxidatively modified DNA) and caspase 3 (GeneTex, 1:100; Caspase 3 is a marker of cellular damage and apoptosis) overnight at 4 °C. Sections were then washed and incubated with the appropriate Alexa, Cy3 or Cy5 Fluor-tagged secondary antibody (Invitrogen). Sections were mounted with ProlongTM Gold antifade reagent with DAPI (Invitrogen). For Fluorescent Myelin staining optic nerve sections were incubated with FluoroMyelin Red (Invitrogen, 1:300) for 30 min at room temperature, washed with PBS before mounting. All the histological analysis was carried out in a blinded manner.
2.13. Spectral domain optical coherence tomography (SD-OCT)
Spectral-domain optical coherence tomography (SD-OCT) scanning was adapted from the procedures described previously with modifications [65–67]. Mice were anesthetized with xylazine (8 mg/kg)/ketamine (60 mg/kg), and lubricant eye drop was applied frequently, with saline wash throughout to maintain corneal clarity. Mice were secured on a custom stage that allowed for free rotation to acquire images focusing on the retina. Images were obtained including averaged single B scan and volume intensity scans centered on the retinal layers using SD-OCT (Bioptigen, Inc). The post-imaging analysis included segmentation report and manual assessment of standard retinal nerve fiber layers (RNFL) thickness using InVivoVue Diver 3.0 software (Bioptigen). The RNFL thickness was measured in 16 points of a 5 × 5 grid using the caliper tool provided by the Bioptigen software where the white color reflects thicker RNFL, while the dark blue represents thinner RNFL. The ETDRS stands for the Early Treatment Diabetic Retinopathy Study (ETDRS), where the retinal thickness is compared to that of a normative database and classified as ‘within normal limits,’ ‘borderline’ or ‘normal outside limits.’ Data collected were represented as a heat map, and RNFL thickness was plotted.
2.14. Statistical analysis
The biochemical studies, confocal analysis, immunoblotting analysis, and SD-OCT assay were statistically analyzed using one-way analysis of variance (one-way ANOVA), and multiple comparisons were performed using the Tukey–Kramer posthoc test (p < 0.05) unless noted otherwise. All the measurements including the histological analysis were carried out in a blinded manner. For in vivo experiments we used at least 5 mice for each set of experiments. Mean values were calculated for each experiment (n = 5), and all the data were depicted as the mean ± SEM.
3. Results
3.1. TBI induces oxidative stress in RGC following TBI
Considering that induction of oxidative stress is the primary outcome after TBI [20,21], we monitored whether TBI can induce oxidative stress in the retina, which is formed of several layers of cells and out of them RGCs are a type of neuron located near the inner surface of the retina and are the last output neurons of the vertebrate retina to communicate with the brain [68] (Fig. S1). For that purpose, we stained the retina with a fluorogenic probe, known as CellROX Deep Green reagent, which is a cell-permeant dye, weakly fluorescents in a reduced state but exhibits bright green photostable fluorescence upon oxidation by reactive oxygen species (ROS) [69]. We found that a significant number of ROS positive cells (green) were mostly enriched in Retinal Ganglion cells (RGC) of the retina after 7 days after TBI compared to sham condition (Fig. 1A and B).
Fig. 1. TBI induces oxidative stress in RGC.

(A–B) Confocal microscopic analysis (A) and quantitative analysis (B) shows that CellROX positive cells were increased more than 4-fold significantly in RGC following TBI. (C–D) Confocal microscopic analysis (C) and quantitative analysis (D) show that the 8-OhDG (a marker for oxidatively modified DNA) positive cells were increased more than 3-fold significantly in RGC following TBI. (E–F) Confocal microscopic analysis (E) and quantitative analysis (F) show that 4-HNE (lipid peroxidation marker) positive cells were increased more than 3-fold significantly in RGC following TBI. (G) The TBARS analysis suggests that TBI leads to an increase in lipid peroxidation significantly. Statistical significance was measured by one-way ANOVA with a Tukey-Kramer post-hoc correction, n = 5, *p < 0.05. All data are expressed as mean ± S.E.M.
An increase in oxidative stress results in the formation of 8-hydroxy-2′-deoxyguanosine (8-OHdG) which serves as a well-known marker for oxidative stress-induced DNA damage. To assess cellular DNA damage, we performed IHC for the anti-8-OHdG antibody. The 8OHdG-positive cells were observed in RGC 7 days after TBI, while almost none were seen in retinas of sham mice (Fig. 1C and D). We also measured the total level of oxidized lipids by immunohistochemical staining using an anti-4-hydroxynonenal (4-HNE) monoclonal antibody to detect (4-HNE) (Fig. 1E and F), an aldehyde product of lipid peroxidation; and Thiobarbituric acid reactive substances (TBARS) assay where TBARS are formed as a byproduct of lipid peroxidation. Consistent with 4-HNE data, the level of TBARS in the retina increased with time after TBI (Fig. 1G); suggests that the level of oxidized lipids was increased along with oxidative stress significantly in the retina in comparison with the sham group.
3.2. TBI-induced oxidative stress causes cell death in RGC
Since an increase in oxidative stress affects cell survival, we monitored cell death in RGC by monitoring the expression level of Brn3a (RGC marker) and TUNEL assay. We found that the number of Brn3a positive cells was decreased significantly after TBI (Fig. 2A and B). To see whether the loss of RGC is due to apoptotic cell death, we performed the TUNEL assay, which detects DNA fragmentation by labeling the 3′-hydroxyl termini in the double-strand DNA breaks generated during apoptosis. We found that the TUNEL positive cells were increased in RGC following TBI; however, there is no TUNEL positive cells were observed in sham mice (Fig. 2C and D). This data suggests that TBI induces RGC death and as a result, the number of RGC was reduced after TBI.
Fig. 2. TBI induces cell death in RGC.

(A–B) Confocal microscopic data (A) and quantitative analysis (B) shows that the number of Brn3a positive cells was decreased in RGC following TBI. (C–D) Confocal microscopic data (C) and quantitative analysis (D) show that the number of TUNEL positive cells (green fluorescence) was increased in RGC following TBI. (E–F) The SD-OCT data shows that the RNFL thickness has been decreased in ETDRS measured after 100, 300 and 600 μM diameter. (F) The quantitative analysis of RNFL thickness measured by SD-OCT in both TBI and sham mice. (G–H) FG staining shows that TBI leads to a decrease FG in RGC compared to sham mice. Box 1, 2 and 3 are 0.5 mm, 1 mm and 1.5 mm from the center. Statistical significance was measured by one-way ANOVA with a Tukey-Kramer post-hoc correction, n = 5, *p < 0.05. All data are expressed as mean ± S.E.M. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
The loss of RGC ultimately results in the decrease in the retinal nerve fiber layer (RNFL) thickness [41–43], which can be measured by Spectral Domain Optical Coherence Tomography (SD-OCT). Thus, to study the objective measurements of anatomical structures related to RGCs, we measured RNFL by SD-OCT after TBI. We found that TBI causes a significant loss in RNFL thickness compared to sham mice (Fig. 2E and F). The reduction in RNFL level indicates that the communication between RGC and the brain could be affected.
In general, the axon of RGC allows the cell body to communicate with its terminal neuronal targets in the lateral geniculate nucleus and the superior colliculus located in the visual cortex of the brain [70,71]. This communication is achieved by the transport of molecules through axons of RGC in retrograde directions [71–73], which can be monitored by Fluorogold (FG) staining of RGC. In this procedure, FG can accumulate in the retinal RGC cells and make it fluorescently marked after stereotactic injection of FG into the superior colliculus of the mice following either sham or TBI. We found that TBI leads to a significant decrease in FG-labelled RGC density following TBI compared to sham mice (Fig. 2G–J). The total number of RGCs were counted in either 0.5 mm (box 1), 1 mm (box 2) or 1.5 mm (box 3) distance from the center. This data suggests that TBI affects the communication between the brain and retina. However, it is not clear whether the disruption in connection occurs only due to the cell death in RGC or also contributed by the damage in the optic nerve which connects the soma of the RGC to the brain.
3.3. TBI induced oxidative stress causes death in oligodendrocytes in association with loss of myelin and degeneration of neurofilaments
To explore the above possibility, we monitored whether TBI leads to cell death in the optic nerve by doing the TUNEL assay using optic nerve sections isolated from TBI and sham mice. We found that the number of TUNEL positive cells were increased in the optic nerve section isolated from TBI mice compared to sham mice (Fig. 3A and B). The bHLH transcription factor, olig2, is universally expressed in the optic nerve [16] and serves as a well-known marker of OPCs, which are highly abundant in the optic nerve and the proliferation and differentiation of OPCs into oligodendrocytes is critical for myelination of optic nerves [16–18]. The myelination of the optic nerve is required to establish the proper communication between the retina and the brain. We found that in optic nerve most of the TUNEL positive cells were colocalized with olig2 after TBI; indicates that TBI leads to death in OPCs in the optic nerve (Fig. 3C and D).
Fig. 3. TBI induces oxidative stress and optic nerve degeneration.

(A–B) Confocal microscopic data (A) and quantitative analysis (B) show that TUNEL positive cells were enriched in Olig2 positive cells following TBI. The quantification of TUNEL positive cells shows that TBI increased cell death in Olig2 positive cells significantly. (C–D) Confocal analysis of Fluoromyelin staining (C) and quantitative analysis (D) in optic nerve shows that its level was decreased significantly. The quantitation of Fluoromyelin staining was reduced in the optic nerve following TBI. (E–F) Confocal analysis of MBP staining (E) and quantitative analysis (F) in optic nerve shows that its level was decreased significantly. The quantitation of MBP staining was reduced in the optic nerve following TBI. (G–I) Confocal microscopic data (G) shows that SMI-32 positive axonal bulbs (G, H), axonal retraction (G, I) and holes (G, J) were increased after TBI. The white arrowhead indicates axonal bulbs; the yellow arrowhead indicates axonal retraction and asterisks indicates the axonal holes. (K–L) Confocal microscopic data (K) and quantitative analysis (L) shows that CellROX positive cells were increased in the optic nerve following TBI. The CellROX positive cells were quantitated in the optic nerve of both sham and TBI mice. (M–N) Confocal microscopic analysis (M) and quantitative analysis (N) show that 4-HNE positive cells were increased significantly in optic nerve following TBI. (O) The level of TBARS was increased in the optic nerve of TBI mice compared to sham mice. Statistical significance was measured by one-way ANOVA with a Tukey-Kramer post-hoc correction, n = 5, *p < 0.05. All data are expressed as mean ± S.E.M. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
To see whether TBI-induced loss of OPCs in optic nerve influence the overall myelination, we stained the myelin with a fluorescent dye, FluoroMyelin™ Red [74]. We found that red fluorescent intensity in the optic nerve isolated from TBI mice was significantly decreased compared to optic nerve isolated from sham mice (Fig. 3E and F). Myelin basic protein (MBP) is the major constituents of the myelin sheath of oligodendrocytes and essentially contributes in the process of myelination [75]. We monitored the level of MBP in the optic nerve of both TBI and sham mice. Similar to myelination data, we found that the level of MBP was decreased significantly in the optic nerve of TBI mice compared to sham mice (Fig. 3G and H); suggests that a loss of OPCs may contribute to the loss of myelin by decreasing the level of MBP in the optic nerve after TBI.
Myelination modulates phosphorylation of neurofilaments which is critical for axonal retrograde transport [76,77]. Phosphorylation of neurofilaments indicates the well-organized axons; however, the dephosphorylation of neurofilaments causes axonal swelling with short segments of axons and can be monitored by staining the neurofilaments with SMI-32 [77,78]. After TBI, SMI-32-positive large axonal swellings, axonal retraction, and holes were observed along with short segments of axons, reflecting early stages of axonal degeneration (Fig. 3G, I); although the control optic nerves had long nerve fibers, which were arranged in parallel. Semi-quantitative counting revealed that axonal degeneration was increased significantly after TBI compared to sham mice (Fig. 3G, J).
Considering that oxidative stress leads to impairment in myelination of axons [14,79], we stained both TBI and sham optic nerve sections with CellROX. Consistent with RGC data, we found that TBI leads to an increase in the CellROX positive cells in the optic nerve isolated from TBI mice compared to sham mice (Fig. 3K and L). We also monitored the total level of oxidized lipids by staining with 4-HNE and doing a TBARS assay in the optic nerve isolated from TBI and sham mice. We found that the level of 4-HNE positive cells (Fig. 3M and N) was increased along with an increase in TBARS in the optic nerve significantly in TBI samples compared to sham mice (Fig. 3O). Taken together, our data suggest that augmentation of oxidative stress and lipid peroxidation occurs in concomitant with an increase in cell death in both RGC and optic nerve.
Collectively, our data show that TBI leads to an increase in oxidative stress which is closely associated with structural and functional dysfunction of RGC and optic nerve. This event, in turn, can disrupt the communication between the brain and the retina; however, the underlying mechanism responsible for the induction of oxidative stress has not been identified yet.
3.4. An increase in H3K9Me2 impairs transcriptional activity of Nrf2 and induces oxidative stress in RGC and oligodendrocytes in the optic nerve following TBI
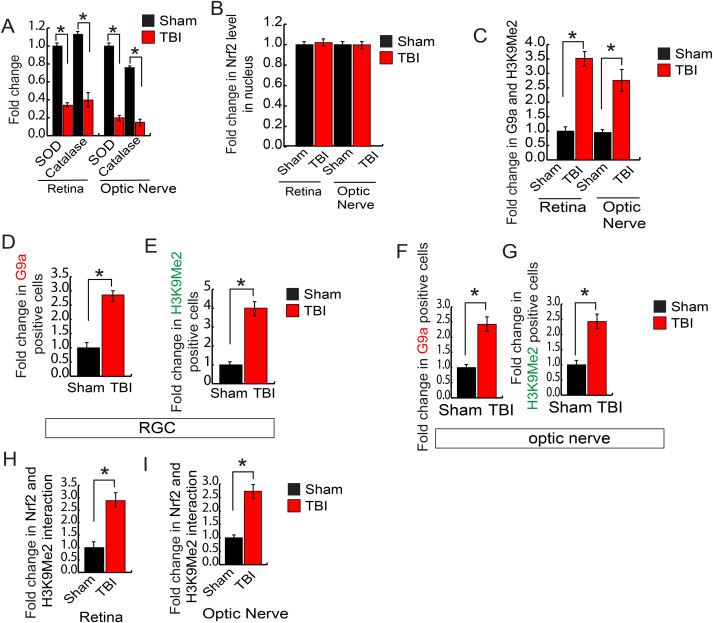
Given the fact that a decrease in antioxidants is mostly responsible for an increase in oxidative stress, we monitored the level of mRNA level of antioxidants by quantitative RT-PCR analysis using the total RNA isolated from the retina and optic nerve following sham and TBI. We found that the mRNA level of superoxide dismutase (sod) and catalase were decreased significantly both in the retina and optic nerve following TBI compared to sham mice (Fig. 4A). In another set of experiments, we monitored the expression level of antioxidants in both retinal and optic nerve-lysates isolated after TBI and sham. Consistent with quantitative RT-PCR data, our Western blot analysis showed that TBI leads to a decrease in the expression level of SOD and catalase both in the retinal lysates and lysates isolated from optic nerve (Fig. 4B, S2A).
Fig. 4. An induction of H3K9Me2 attenuated the transcriptional activity of Nrf2 and reduced the antioxidant levels.

(A) The quantitative RT-PCR analysis data shows that TBI causes a decrease in the mRNA level of sod and catalase both in the retina and optic nerve compared to sham mice. (B) Western blot analysis data suggests that TBI leads to a decrease in the expression level of SOD and Catalase in both retina and optic nerve compared to sham mice. (C) The expression level of Nrf2 in nucleus show that there is no difference either in the retina or optic nerve following TBI compared to sham. (D) The chromatin immunoprecipitation (ChIP) data shows that the binding of Nrf2 to the sod promoter was decreased in the retina and optic nerve after TBI compared to sham mice. (E) Western blot analysis show that the level of H3K9Me2 and G9a was increased in the retina and optic nerve after TBI compared to sham mice. (F) Confocal microscopic analysis shows that TBI leads to an increase in the expression level of G9a and H3K9Me2 in RGC after TBI. (G) Confocal microscopic data shows that TBI leads to an increase in G9a and H3K9Me2 in the optic nerve following TBI. (H) The co-immunoprecipitation (IP) analysis show that the interaction between Nrf2 and H3K9Me2 was increased significantly in both retina and optic nerve following TBI. Statistical significance was measured by one-way ANOVA with a Tukey-Kramer post-hoc correction, n = 5, *p < 0.05. All data are expressed as mean ± S.E.M.
Nrf2 is a redox-sensitive transcription factor that binds to antioxidant response elements located in the promoter region of genes encoding many antioxidant enzymes [30–33]. Thus, upon activation Nrf2 accumulates in the nucleus and synthesize the mRNA of antioxidants. To see whether TBI has any influence of the Nrf2 level, we monitored its level in the nuclear lysates isolated from the retina and optic nerve following either sham or TBI. Surprisingly, we found that there was no difference in the nuclear Nrf2 level following either TBI or sham (Fig. 4C, S2B). To see whether the DNA binding property of Nrf2 has been affected after TBI, we monitored Nrf2 binding to one of its downstream genes such as the sod promoter. We found that Nrf2 binding to the sod promoter was decreased significantly both in the retina and optic nerve after TBI compared to sham (Fig. 4D) as monitored by Chromatin immunoprecipitation (ChIP) assay. This data raised the possibility that the epigenetic modification of histones could be responsible for transcriptional inactivation of Nrf2.
Di-methylation of histones at lysine 9 residue (H3K9Me2) is known to silence transcription by blocking the access of transcription factor to its promoter [41–43]. A methyltransferase G9a is known to catalyze H3K9Me2 [41–43], and the expression level of G9a remains low under physiological condition. Thus, we monitored the level of H3K9Me2 and G9a in the retina and optic nerve following TBI or sham. The Western blot analysis suggests that TBI leads to a significant increase in the level of H3K9Me2 and G9a in the RGC and optic nerve compared to sham mice (Fig. 4E, S2C). We monitored the level of H3K9Me2 and G9a in the RGC and optic nerve through confocal microscopic analysis. Consistent with our Western blot data, we found that TBI leads to an increase in G9a (Fig. 4F, S2D) along with H3K9Me2 (Fig. 4F, S2E) level in RGC after TBI compared to sham. Similarly, the level of G9a (Fig. 4G, S2F) along with H3K9Me2 (Fig. 4G, S2G) were increased in the optic nerve following TBI.
To monitor whether H3K9Me2 has any direct influence on the transcriptional activity of Nrf2, we have performed the immunoprecipitation (IP) assay to monitor whether Nrf2 and H3K9Me2 can interact with each other. We found that TBI leads to an increase in Nrf2-H3K9Me2 interaction in the retina (Fig. 4H, S2H) and optic nerve lysates following TBI (Fig. 4H, S2I). Taken together, our data suggest that TBI leads to an increase in H3K9Me2 due to an induction in the level of G9a. An augmented H3K9Me2 interacts with Nrf2 and subsequently attenuates the binding of Nrf2 to its downstream antioxidant genes such as sod. As a result, the level of antioxidants was decreased, in association with an increase in oxidative stress and cell death both in RGC and optic nerve. Thus, we were interested in testing whether blocking the induction of H3K9Me2 by preventing the catalytic activity of G9a can reduce the oxidative stress, inhibit cell death and ultimately restores the communication between the brain and retina to prevent the loss of RNFL thickness.
3.5. Treatment with a catalytic inhibitor of G9a, UNC0638 reduces the induction in H3K9Me2 and the augments transcriptional activity of Nrf2 to synthesize antioxidants following TBI
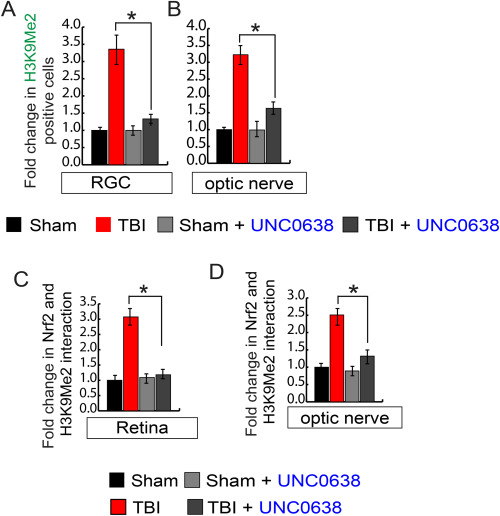
A chemical compound, UNC0638 has been identified as an inhibitor of G9a with excellent potency and selectivity over a wide range of epigenetic and non-epigenetic targets [45]. To understand whether treatment with a G9a inhibitor, UNC0638 can reduce the induction of H3K9Me2 in the retina or optic nerve, we monitored its level by confocal microscopy. We found that H3K9Me2 level was decreased significantly in RGC (Fig. 5A, S3A) and optic nerve (Fig. 5B, S3B) treated with UNC0638 following TBI compared to only TBI-mice. To monitor whether the reduction in H3K9Me2 after treatment with UNC0638 results in an improvement in mRNA level of sod and catalase in the retina and optic nerve following TBI, we performed quantitative RT-PCR analysis using total RNA isolated from the retina and optic nerve. We found that the loss of sod and catalase was rescued in the retina (Fig. 5C and D) and optic nerve (Fig. 5E and F) after treatment with UNC0638 following TBI. To monitor whether treatment with UNC0638 improves Nrf2 binding on the sod promoter, we performed the ChIP assay and found that treatment with UNC0638 recovers Nrf2 binding to the sod promoter in the retina (Fig. 5G) and optic nerve (Fig. 5H). To monitor whether inhibition of H3K9Me2 contributes an improvement in transcriptional activity of Nrf2, we monitored the interaction between Nrf2 and H3K9Me2 in the retinal and optic nerve lysates. We found that the decrease in the expression level of H3K9Me2 reduced its interaction with Nrf2 in both the retina (Fig. 5I, S3C) and optic nerve (Fig. 5J, S3D) in TBI-mice treated with UNC0638 compared to TBI mice only. These data suggest that TBI leads to an increase in G9a and H3K9Me2 both in RGC and optic nerve and the interaction between H3K9me2 and Nrf2 is responsible for the impairment in transcriptional activity of Nrf2 by preventing its binding to the promoters of antioxidant genes.
Fig. 5. Treatment with a G9a inhibitor, UNC0638 reduced the induction of H3K9Me2, and transcriptional activation of Nrf2 following TBI.

(A) Confocal microscopic analysis shows that the induction of H3K9Me2 in RGC was decreased after treatment with UNC0638 following TBI. (B) Confocal microscopic examination shows that the induction of H3K9Me2 in the optic nerve was reduced after treatment with UNC0638 following TBI. (C–F) the quantitative RT-PCR analysis shows that the decrease in mRNA level of sod (C, E) and catalase (D, F) either in the retina or optic nerve was rescued in TBI-mice treated with UNC0638. (G–H) The ChIP assay suggests that a decrease in Nrf2 binding to the sod promoter was attenuated in the retina (G) and optic nerve (H) in TBI-mice treated with UNC0638. (I–J) The co-immunoprecipitation (Co-IP) study shows that treatment with UNC0638 decreases the interaction between Nrf2 and H3K9Me2 after TBI in the retina (I) and optic nerve (J).
3.6. Treatment with UNC0638 reduced oxidative-stress by activating Nrf2 in association with a reduction in cell death in RGC and optic nerve, and optic nerve degeneration following TBI
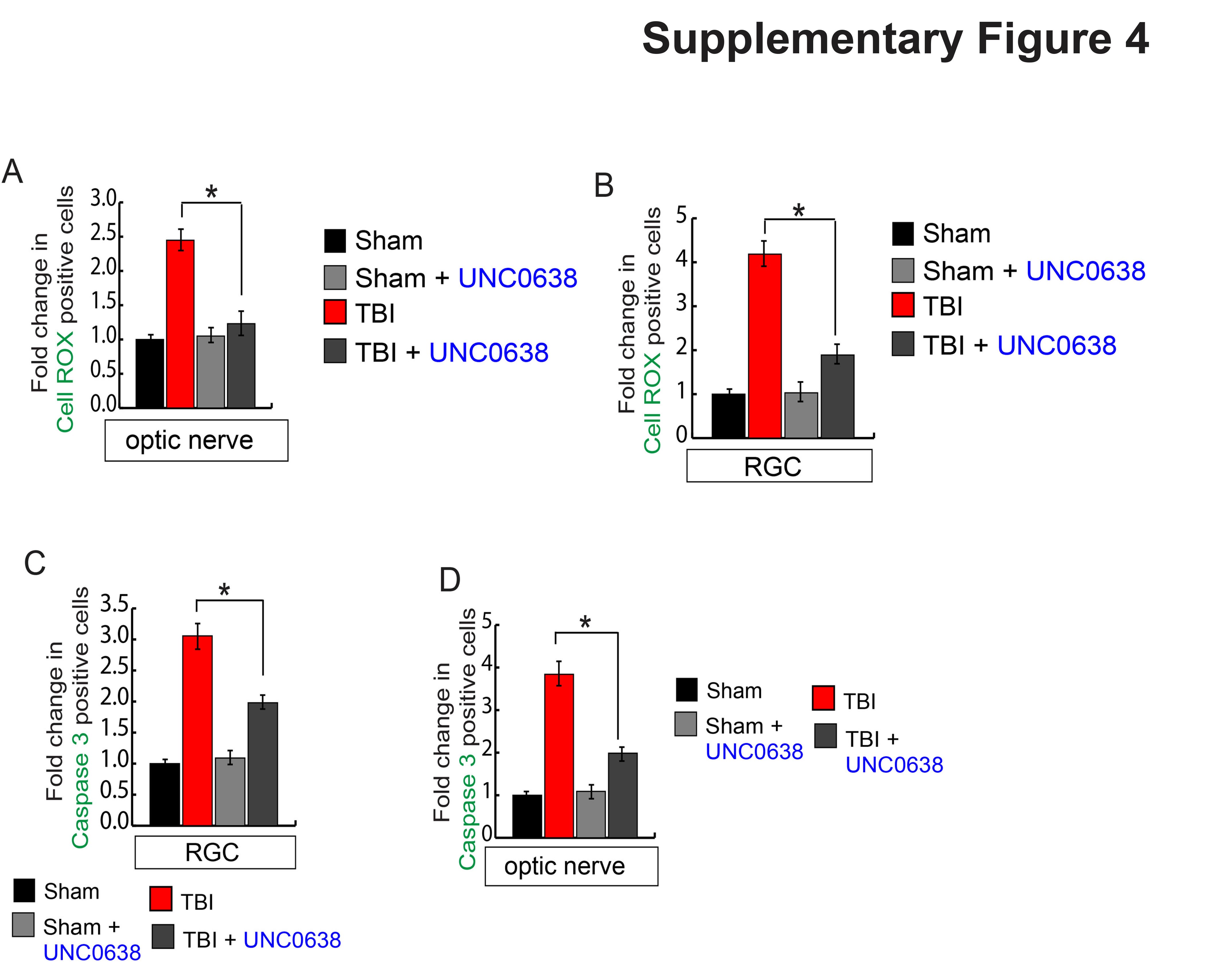
Since the DNA binding activity of Nrf2 has been restored after reducing H3K9Me2 by treating with UNC0638, we monitored the level of oxidative stress in RGC and optic nerve by staining with CellROX. We found that the green fluorescent intensity which indicates the oxidative stress was reduced in the optic nerve (Fig. 6A, S4A) and RGC (Fig. 6B, S4B) in TBI-mice treated with UNC0638. This data was further confirmed by the TBARS assay where we monitored the level of TBARS (Thiobarbituric acid reactive substances; oxidized lipids), and we found that the level of TBARS level was decreased significantly in the retina (Fig. 6C) and optic nerve (Fig. 6D) in TBI-mice treated with UNC0638 compared to TBI mice. These data suggest that UNC0638 treatment reduced oxidative stress and lipid peroxidation in association with induction of antioxidants in the RGC and optic nerve following TBI. To see whether reduction in oxidative stress is associated with the cell death, we analyzed the cell death by monitoring caspase 3 positive cells and we found that treatment with UNC0638 prevents cell death both in RGC (Fig. 6E, S4C) and optic nerve (Fig. 6F, S4D) following TBI significantly compared to only TBI mice.
Fig. 6. Treatment with UNC0638 reduced oxidative stress and cell death in the retina and optic nerve following TBI.

(A–B) The confocal analysis shows that CellROX positive cells were decreased in TBI-mice treated with UNC0638 compared to TBI-mice in both optic nerve (A) and RGC region (B). (C–D) The quantitative analysis shows that TBARS were decreased in TBI-mice treated with UNC0638 compared to TBI-mice in both retinas (C) and optic nerve (D) sections. (E–F) The confocal shows that caspase 3 positive cells were decreased in TBI-mice treated with UNC0638 compared to TBI-mice in both retina (E) and optic nerve (F) sections. Statistical significance was measured by one-way ANOVA with a Tukey-Kramer post-hoc correction, n = 5, *p < 0.05. All data are expressed as mean ± S.E.M.
3.7. Treatment with a catalytic inhibitor of G9a, UNC0638 rescues retinal thickness, and rescue retrograde transport after TBI
Since the death of oligodendrocytes contributes significantly to demyelination in the optic nerve, and treatment with UNC0638 prevents the cell death; we monitored myelination in optic nerve isolated from TBI and sham mice using red Fluoromyelin staining. We found that the loss of myelin after TBI was rescued in the optic nerve in TBI mice treated with UNC0638 (Fig. 7A and B). This data was further supported by analysis of MBP protein. Like red Fluoromyelin staining, the reduction in the level of MBP in the optic nerve was restored in TBI-mice treated with UNC0638 (Fig. 7C and D). To monitor whether treatment with UNC0638 affects neurofilament degeneration following TBI, we stained optic nerve isolated from TBI and TBI-mice treated with UNC0638 using SMI-32. We found that the number of axonal bulbs (Fig. 7E and F), axonal retraction (Fig. 7E, G) and holes (Fig. 7E, H) were reduced and neurofilaments stacked in parallel; indicates that abnormalities in neurofilaments were rescued significantly after UNC0638 treatment.
Fig. 7. Treatment with UNC0638 rescues demyelination, abnormalities in neurofilaments, and RNFL thickness following TBI.

(A, B) Confocal microscopic analysis (A) and the quantitative analysis (B) show that a loss of Fluoromyelin red staining after TBI was rescued in TBI + UNC0638 treated mice. (C–D) Confocal microscopic analysis (C) and the quantitative analysis (D) show that a loss of MBP staining after TBI was rescued in TBI + UNC0638 treated mice. (E–H) The confocal microscopy (E) and the quantitative analysis (F–H) show that an increase in SMI-32 positive axonal bulbs (F), the axonal retraction (G) and holes (H) was recovered in optic nerve isolated from TBI + UNC0638 mice. (I–J) The SD-OCT data shows that the RNFL thickness has been decreased in ETDRS measured after 100, 300 and 600 μM diameter in TBI; however, it was rescued in TBI-mice treated with UNC0638 (I). The quantitative analysis of RNFL thickness measured by SD-OCT in both TBI and sham mice treated with or without UNC0638 (J). (K–L) The Fluorogold (FG) staining suggests that TBI-induced loss of RGCs was rescued in TBI-mice treated with UNC0638. Box 1, 2 and 3 are 0.5 mm, 1 mm and 1.5 mm from the center. Statistical significance was measured by one-way ANOVA with a Tukey-Kramer post-hoc correction, n = 5, *p < 0.05. All data are expressed as mean ± S.E.M. (For interpretation of the references to color in this figure legend, the reader is referred to the Web version of this article.)
Given that treatment with UNC0638 restored RGC death and optic nerve degeneration, we analyzed whether UNC0638 can rescue the loss of RNFL thickness. Thus, we performed SD-OCT analysis using TBI mice, and TBI-mice treated with UNC0638. We found that the TBI-induced loss of RNFL thickness was rescued significantly in TBI-mice treated with UNC0638 (Fig. 7I and J). Lastly, we measured the retrograde transport which is critical to communicate between the retina and the brain. We found that treatment with UNC0638 rescues the reduction in the density of FG-positive cells significantly after TBI compared to only TBI mice (Fig. 6K and L). This data suggests that UNC0638 is instrumental in providing neuroprotection and improving retrograde transportation of axons following TBI.
4. Discussion
TBI frequently involves the loss of RGCs and damage to their axons which constitute the optic nerve. In the present study, we have shown that induction of H3K9Me2 was correlated with RGC loss and axonal degeneration and it manifests these processes mostly via inducing oxidative stress through impairing the transcriptional activity of Nrf2, which is essential to synthesize antioxidants. Attenuating the induction of H3K9Me2 by inhibiting its catalytic enzyme, G9a blocks both processes following TBI.
After TBI, an assembly of oxidative stress markers such as lipid peroxides and ROS are produced in the eye, while antioxidant defense enzymes such as SOD and Catalase are decreased. We analyzed lipid peroxidation in two methods; TBARS assay and 4-HNE. However, the limitation of TBAR assay is that it mainly measures malondialdehyde (MDA) which is not only generated by oxidative damage caused by lipid peroxidation but also during enzymatic lipid peroxidation (e.g., cycloxygenase or 5-lipoxygenase activity). Thus, data resulted from 4-HNE staining, which is specific for oxidative damage-induced lipid peroxidation has overcome the limitation. This imbalance in the oxidative stress and the level of antioxidants is directly related to the pathogenesis of TBI. Therefore, the development of antioxidant strategies is required to have as a primary interest to optimize brain injury treatment.
Several studies have shown that the Nrf2 is an established regulator of cellular resistance to oxidants [80–82]. The deficiency in Nrf2 substantially increased the susceptibility of mice to a broad range of disease conditions associated with oxidative pathology [80–82]. Moreover, augmentation of Nrf2 is known to pharmacologically protected animals from oxidative damage [83,84]. The current literature suggests that upon activation, Nrf2 mostly enters into the nucleus to carry out the transcriptional activation to synthesize genes for antioxidants. Thus, nuclear accumulation of Nrf2 could be the determining factor for its transcriptional activity [30–34]. However, our study suggests that the total level of Nrf2 remained unaltered in the nucleus following sham or TBI. Thus, the nuclear accumulation of Nrf2 may not be enough to conclude whether Nrf2 will be transcriptionally active, because the binding of Nrf2 to its promoter depends on the status of the histones. Our data shows that an increase in histone di-methylation (H3K9Me2) prevented the DNA binding activity of Nrf2 from synthesizing mRNA level of its downstream antioxidants and subsequently induced the oxidative stress in the RGC and optic nerve following TBI. Thus, our study unfolds a novel mechanism for TBI-induced vision impairment where the induction of an epigenetic silencer of transcription such as H3K9Me2 is responsible for impairment of transcriptional activation of Nrf2. However, we cannot rule out the possibility that an alteration in H3K9Me2 may affect other transcription factors including Nrf2, which may directly or indirectly affect the cellular redox status either in RGC or optic nerve. Further studies are required to give more insight regarding the interaction between Nrf2 and H3K9Me2 based on our current finding, where it was shown that the interaction between Nrf2 and H3K9Me2 leads to form an inactive transcriptional complex on the sod promoter.
Disruption in the retrograde transport process, either due to the damage to the optic nerve or RGC population, has been implicated in several retinal disorders such as glaucoma or optic nerve degeneration [73,85,86]. In this study, we provided further evidence that TBI also affects the retrograde transport in association to induction of oxidative stress. In general, axons are wrapped by the myelin which is mainly synthesized by oligodendrocytes in the optic nerve to process the retrograde transport efficiently. Our data shows that TBI induced cell death in OPCs is correlated with induction in the loss of myelin which was evidenced by a decrease in Fluoromyelin staining. Since the recovery of myelin loss is correlated with the reduction of oxidative stress, we consider that oxidative stress may be directly or indirectly responsible for the impairment of myelination following TBI. Indeed, previously another study showed that OPCs are particularly vulnerable to oxidative damage after neurotrauma in vivo [24] and the loss of OPCs results in loss of myelination [25]. Also, our study shows that, inhibiting G9a using UNC0638 causes reduction in oxidative stress by restoring the transcriptional activity of Nrf2 in concomitant with an improvement in retrograde transport; thus, we concluded that our study provides a previously unknown role of histone methylation in the regulation of myelination; although, the detailed underlying mechanism remains to be elucidated.
Demyelination of optic nerve in association with degeneration is a standard feature in ocular disorders (e.g., optic neuritis, optic nerve injury, etc.) or CNS disorders (e.g., ischemia, EAE, Alzheimer’s disease, etc.). Since preventing the induction of H3K9Me2 by treatment with UNC0638 restores the myelination, the results from our study can be applied to develop novel therapeutic strategies against these diseases. Moreover, identifying a common mechanism to address critical features of vision impairments such as RGC loss and optic nerve degeneration provide a novel opportunity to treat vision-related problems in TBI patients.
5. Conclusion
TBI-induced vision impairment is multifactorial; more specifically, the loss of RGC in the retina and optic nerve degeneration contribute significantly to vision impairment following TBI. Thus, targeting each event individually will not result in an effective therapeutic strategy to improve vision deficiency in TBI survivors. It is required to identify the common mechanism that is accountable for both events. Our study shows that an epigenetic alteration such as induction of H3K9Me2 is responsible for both RGC loss and axonal degeneration following TBI. This study provides a proof-of-concept that blocking G9a, an enzyme responsible for catalyzing H3K9Me2 will provide a neuroprotective effect against vision impairment following TBI.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgement
We would like to like to acknowledge funding from NIH/NINDS (RO1NS094516) and NIH/NEI (R01EY025622) to N.S. We want to thank Kira Lathrop from the Department of Opthalmology for her generous help to perform confocal analysis and SD-OCT analysis. This work was also supported by NIH CORE Grant P30 EY08098 to the Department of Ophthalmology, the Eye and Ear Foundation of Pittsburgh, and from an unrestricted grant from Research to Prevent Blindness, New York, NY
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.freeradbiomed.2019.02.015.
References
- [1].Gupta R, Sen N, Traumatic brain injury: a risk factor for neurodegenerative diseases, Rev. Neurosci 27 (1) (2016) 93–100. [DOI] [PubMed] [Google Scholar]
- [2].Hyder AA, Wunderlich CA, Puvanachandra P, Gururaj G, Kobusingye OC, The impact of traumatic brain injuries: a global perspective, NeuroRehabilitation 22 (5) (2007) 341–353. [PubMed] [Google Scholar]
- [3].Dewan MC, Rattani A, Gupta S, Baticulon RE, Hung YC, Punchak M, Agrawal A, Adeleye AO, Shrime MG, Rubiano AM, Rosenfeld JV, Park KB, Estimating the global incidence of traumatic brain injury, J. Neurosurg (2018) 1–18. [DOI] [PubMed]
- [4].Roberts PS, Rizzo JR, Hreha K, Wertheimer J, Kaldenberg J, Hironaka D, Riggs R, Colenbrander A, A conceptual model for vision rehabilitation, J. Rehabil. Res. Dev 53 (6) (2016) 693–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Greenwald BD, Kapoor N, Singh AD, Visual impairments in the first year after traumatic brain injury, Brain Inj 26 (11) (2012) 1338–1359. [DOI] [PubMed] [Google Scholar]
- [6].Cockerham GC, Goodrich GL, Weichel ED, Orcutt JC, Rizzo JF, Bower KS, Schuchard RA, Eye and visual function in traumatic brain injury, J. Rehabil. Res. Dev 46 (6) (2009) 811–818. [DOI] [PubMed] [Google Scholar]
- [7].Atkins EJ, Newman NJ, Biousse V, Post-traumatic visual loss, Rev. Neurol. Dis 5 (2) (2008) 73–81. [PMC free article] [PubMed] [Google Scholar]
- [8].Sen N, An insight into the vision impairment following traumatic brain injury, Neurochem. Int 111 (2017) 103–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Armstrong RA, Visual problems associated with traumatic brain injury, Clin. Exp. Optom 101 (6) (2018. November) 716–726, 10.1111/cxo.12670. [DOI] [PubMed] [Google Scholar]
- [10].Mohan K, Kecova H, Hernandez-Merino E, Kardon RH, Harper MM, Retinal ganglion cell damage in an experimental rodent model of blast-mediated traumatic brain injury, Invest. Ophthalmol. Vis. Sci 54 (5) (2013) 3440–3450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Yu DY, Cringle SJ, Retinal degeneration and local oxygen metabolism, Exp. Eye Res 80 (6) (2005) 745–751. [DOI] [PubMed] [Google Scholar]
- [12].Wang SW, Mu X, Bowers WJ, Kim DS, Plas DJ, Crair MC, Federoff HJ, Gan L, Klein WH, Brn3b/Brn3c double knockout mice reveal an unsuspected role for Brn3c in retinal ganglion cell axon outgrowth, Development 129 (2) (2002) 467–477. [DOI] [PubMed] [Google Scholar]
- [13].Nadal-Nicolas FM, Jimenez-Lopez M, Sobrado-Calvo P, Nieto-Lopez L, Canovas-Martinez I, Salinas-Navarro M, Vidal-Sanz M, Agudo M, Brn3a as a marker of retinal ganglion cells: qualitative and quantitative time course studies in naive and optic nerve-injured retinas, Invest. Ophthalmol. Vis. Sci 50 (8) (2009) 3860–3868. [DOI] [PubMed] [Google Scholar]
- [14].Bradl M, Lassmann H, Oligodendrocytes: biology and pathology, Acta Neuropathol 119 (1) (2010) 37–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Shi J, Marinovich A, Barres BA, Purification and characterization of adult oligodendrocyte precursor cells from the rat optic nerve, J. Neurosci 18 (12) (1998) 4627–4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zhou Q, Choi G, Anderson DJ, The bHLH transcription factor Olig2 promotes oligodendrocyte differentiation in collaboration with Nkx2.2, Neuron 31 (5) (2001) 791–807. [DOI] [PubMed] [Google Scholar]
- [17].Wegener A, Deboux C, Bachelin C, Frah M, Kerninon C, Seilhean D, Weider M, Wegner M, Nait-Oumesmar B, Gain of Olig2 function in oligodendrocyte progenitors promotes remyelination, Brain 138 (Pt 1) (2015) 120–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Juarez A. Lopez, He D, Richard Lu Q, Oligodendrocyte progenitor programming and reprogramming: toward myelin regeneration, Brain Res 1638 (Pt B) (2016) 209–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Maxwell WL, Damage to myelin and oligodendrocytes: a role in chronic outcomes following traumatic brain injury? Brain Sci 3 (3) (2013) 1374–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Rodriguez-Rodriguez A, Egea-Guerrero JJ, Murillo-Cabezas F, Carrillo-Vico A, Oxidative stress in traumatic brain injury, Curr. Med. Chem 21 (10) (2014) 1201–1211. [DOI] [PubMed] [Google Scholar]
- [21].Cornelius C, Crupi R, Calabrese V, Graziano A, Milone P, Pennisi G, Radak Z, Calabrese EJ, Cuzzocrea S, Traumatic brain injury: oxidative stress and neuroprotection, Antioxidants Redox Signal 19 (8) (2013) 836–853. [DOI] [PubMed] [Google Scholar]
- [22].Birben E, Sahiner UM, Sackesen C, Erzurum S, Kalayci O, Oxidative stress and antioxidant defense, World Allergy Organ J 5 (1) (2012) 9–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Ueda H, Levine JM, Miller RH, Trapp BD, Rat optic nerve oligodendrocytes develop in the absence of viable retinal ganglion cell axons, J. Cell Biol 146 (6) (1999) 1365–1374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Giacci MK, Bartlett CA, Smith NM, Iyer KS, Toomey LM, Jiang H, Guagliardo P, Kilburn MR, Fitzgerald M, Oligodendroglia are particularly vulnerable to oxidative damage after neurotrauma in vivo, J. Neurosci 38 (29) (2018) 6491–6504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Ravera S, Bartolucci M, Cuccarolo P, Litame E, Illarcio M, Calzia D, Degan P, Morelli A, Panfoli I, Oxidative stress in myelin sheath: the other face of the extramitochondrial oxidative phosphorylation ability, Free Radic. Res 49 (9) (2015) 1156–1164. [DOI] [PubMed] [Google Scholar]
- [26].Giacci M, Fitzgerald M, Oligodendroglia are particularly vulnerable to oxidative damage after neurotrauma in vivo, J. Exp. Neurosci 12 (2018) 1179069518810004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Nguyen T, Nioi P, Pickett CB, The Nrf2-antioxidant response element signaling pathway and its activation by oxidative stress, J. Biol. Chem 284 (20) (2009) 13291–13295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Moi P, Chan K, Asunis I, Cao A, Kan YW, Isolation of NF-E2-related factor 2 (Nrf2), a NF-E2-like basic leucine zipper transcriptional activator that binds to the tandem NF-E2/AP1 repeat of the beta-globin locus control region, Proc. Natl. Acad. Sci. U. S. A 91 (21) (1994) 9926–9930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Venugopal R, Jaiswal AK, Nrf1 and Nrf2 positively and c-Fos and Fra1 negatively regulate the human antioxidant response element-mediated expression of NAD(P) H:quinone oxidoreductase 1 gene, Proc. Natl. Acad. Sci. U. S. A 93 (25) (1996) 14960–14965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ma Q, Role of nrf2 in oxidative stress and toxicity, Annu. Rev. Pharmacol. Toxicol 53 (2013) 401–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Nioi P, McMahon M, Itoh K, Yamamoto M, Hayes JD, Identification of a novel Nrf2-regulated antioxidant response element (ARE) in the mouse NAD(P)H:quinone oxidoreductase 1 gene: reassessment of the ARE consensus sequence, Biochem. J 374 (Pt 2) (2003) 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Kwak MK, Wakabayashi N, Greenlaw JL, Yamamoto M, Kensler TW, Antioxidants enhance mammalian proteasome expression through the Keap1-Nrf2 signaling pathway, Mol. Cell Biol 23 (23) (2003) 8786–8794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Hu R, Xu C, Shen G, Jain MR, Khor TO, Gopalkrishnan A, Lin W, Reddy B, Chan JY, Kong AN, Identification of Nrf2-regulated genes induced by chemopreventive isothiocyanate PEITC by oligonucleotide microarray, Life Sci 79 (20) (2006) 1944–1955. [DOI] [PubMed] [Google Scholar]
- [34].Gazaryan IG, Thomas B, The status of Nrf2-based therapeutics: current perspectives and future prospects, Neural Regen Res 11 (11) (2016) 1708–1711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Xu Z, Wei Y, Gong J, Cho H, Park JK, Sung ER, Huang H, Wu L, Eberhart C, Handa JT, Du Y, Kern TS, Thimmulappa R, Barber AJ, Biswal S, Duh EJ, NRF2 plays a protective role in diabetic retinopathy in mice, Diabetologia 57 (1) (2014) 204–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Batliwala S, Xavier C, Liu Y, Wu H, Pang IH, Involvement of Nrf2 in ocular diseases, Oxid Med Cell Longev (2017) 1703810 2017. [DOI] [PMC free article] [PubMed]
- [37].Xu Z, Cho H, Hartsock MJ, Mitchell KL, Gong J, Wu L, Wei Y, Wang S, Thimmulappa RK, Sporn MB, Biswal S, Welsbie DS, Duh EJ, Neuroprotective role of Nrf2 for retinal ganglion cells in ischemia-reperfusion, J. Neurochem 133 (2) (2015) 233–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Voss TC, Hager GL, Dynamic regulation of transcriptional states by chromatin and transcription factors, Nat. Rev. Genet 15 (2) (2014) 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Zhang Y, Reinberg D, Transcription regulation by histone methylation: interplay between different covalent modifications of the core histone tails, Genes Dev 15 (18) (2001) 2343–2360. [DOI] [PubMed] [Google Scholar]
- [40].Greer EL, Shi Y, Histone methylation: a dynamic mark in health, disease and inheritance, Nat. Rev. Genet 13 (5) (2012) 343–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Tachibana M, Sugimoto K, Nozaki M, Ueda J, Ohta T, Ohki M, Fukuda M, Takeda N, Niida H, Kato H, Shinkai Y, G9a histone methyltransferase plays a dominant role in euchromatic histone H3 lysine 9 methylation and is essential for early embryogenesis, Genes Dev 16 (14) (2002) 1779–1791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Peters AH, Kubicek S, Mechtler K, O’Sullivan RJ, Derijck AA, Perez-Burgos L, Kohlmaier A, Opravil S, Tachibana M, Shinkai Y, Martens JH, Jenuwein T, Partitioning and plasticity of repressive histone methylation states in mammalian chromatin, Mol. Cell 12 (6) (2003) 1577–1589. [DOI] [PubMed] [Google Scholar]
- [43].Rice JC, Briggs SD, Ueberheide B, Barber CM, Shabanowitz J, Hunt DF, Shinkai Y, Allis CD, Histone methyltransferases direct different degrees of methylation to define distinct chromatin domains, Mol. Cell 12 (6) (2003) 1591–1598. [DOI] [PubMed] [Google Scholar]
- [44].Katoh K, Yamazaki R, Onishi A, Sanuki R, Furukawa T, G9a histone methyltransferase activity in retinal progenitors is essential for proper differentiation and survival of mouse retinal cells, J. Neurosci 32 (49) (2012) 17658–17670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Vedadi M, Barsyte-Lovejoy D, Liu F, Rival-Gervier S, Allali-Hassani A, Labrie V, Wigle TJ, Dimaggio PA, Wasney GA, Siarheyeva A, Dong A, Tempel W, Wang SC, Chen X, Chau I, Mangano TJ, Huang XP, Simpson CD, Pattenden SG, Norris JL, Kireev DB, Tripathy A, Edwards A, Roth BL, Janzen WP, Garcia BA, Petronis A, Ellis J, Brown PJ, Frye SV, Arrowsmith CH, Jin J, A chemical probe selectively inhibits G9a and GLP methyltransferase activity in cells, Nat. Chem. Biol 7 (8) (2011) 566–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Sharma M, Razali NB, Sajikumar S, Inhibition of G9a/GLP complex promotes long-term potentiation and synaptic tagging/capture in hippocampal CA1 pyramidal neurons, Cerebr. Cortex 27 (6) (2017) 3161–3171. [DOI] [PubMed] [Google Scholar]
- [47].Sen T, Gupta R, Kaiser H, Sen N, Activation of PERK elicits memory impairment through inactivation of CREB and downregulation of PSD95 after traumatic brain injury, J. Neurosci 37 (24) (2017) 5900–5911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Saha P, Gupta R, Sen T, Sen N, Activation of cyclin D1 affects mitochondrial mass following traumatic brain injury, Neurobiol. Dis 118 (2018) 108–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Sen T, Sen N, Treatment with an activator of hypoxia-inducible factor 1, DMOG provides neuroprotection after traumatic brain injury, Neuropharmacology 107 (2016) 79–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Pelzel HR, Schlamp CL, Nickells RW, Histone H4 deacetylation plays a critical role in early gene silencing during neuronal apoptosis, BMC Neurosci 11 (2010) 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Himori N, Yamamoto K, Maruyama K, Ryu M, Taguchi K, Yamamoto M, Nakazawa T, Critical role of Nrf2 in oxidative stress-induced retinal ganglion cell death, J. Neurochem 127 (5) (2013) 669–680. [DOI] [PubMed] [Google Scholar]
- [52].Takemura N, Takahashi K, Tanaka H, Ihara Y, Ikemoto A, Fujii Y, Okuyama H, Dietary, but not topical, alpha-linolenic acid suppresses UVB-induced skin injury in hairless mice when compared with linoleic acids, Photochem. Photobiol 76 (6) (2002) 657–663. [DOI] [PubMed] [Google Scholar]
- [53].Esterbauer H, Cheeseman KH, Determination of aldehydic lipid peroxidation products: malonaldehyde and 4-hydroxynonenal, Methods Enzymol 186 (1990) 407–421. [DOI] [PubMed] [Google Scholar]
- [54].Aitken RJ, Harkiss D, Buckingham DW, Analysis of lipid peroxidation mechanisms in human spermatozoa, Mol. Reprod. Dev 35 (3) (1993) 302–315. [DOI] [PubMed] [Google Scholar]
- [55].Tanito M, Elliott MH, Kotake Y, Anderson RE, Protein modifications by 4-hydroxynonenal and 4-hydroxyhexenal in light-exposed rat retina, Invest. Ophthalmol. Vis. Sci 46 (10) (2005) 3859–3868. [DOI] [PubMed] [Google Scholar]
- [56].Tsai RK, Chang CH, Wang HZ, Neuroprotective effects of recombinant human granulocyte colony-stimulating factor (G-CSF) in neurodegeneration after optic nerve crush in rats, Exp. Eye Res 87 (3) (2008) 242–250. [DOI] [PubMed] [Google Scholar]
- [57].Farook JM, Shields J, Tawfik A, Markand S, Sen T, Smith SB, Brann D, Dhandapani KM, Sen N, GADD34 induces cell death through inactivation of Akt following traumatic brain injury, Cell Death Dis 4 (2013) e754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Yamazaki F, Kim HH, Lau P, Hwang CK, Iuvone PM, Klein D, Clokie SJ, pY RNA1-s2: a highly retina-enriched small RNA that selectively binds to Matrin 3 (Matr3), PLoS One 9 (2) (2014) e88217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Kapoor S, Kim SM, Farook JM, Mir S, Saha R, Sen N, Foxo3a transcriptionally upregulates AQP4 and induces cerebral edema following traumatic brain injury, J. Neurosci 33 (44) (2013) 17398–17403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Mir S, Sen T, Sen N, Cytokine-induced GAPDH sulfhydration affects PSD95 degradation and memory, Mol. Cell 56 (6) (2014) 786–795. [DOI] [PubMed] [Google Scholar]
- [61].Qin Q, Patil KA, Gronert K, Sharma SC, Neuroprotectin D1 inhibits retinal ganglion cell death following axotomy, Prostaglandins Leukot. Essent. Fatty Acids 79 (6) (2008) 201–207. [DOI] [PubMed] [Google Scholar]
- [62].Sapieha PS, Duplan L, Uetani N, Joly S, Tremblay ML, Kennedy TE, Di Polo A, Receptor protein tyrosine phosphatase sigma inhibits axon regrowth in the adult injured CNS, Mol. Cell. Neurosci 28 (4) (2005) 625–635. [DOI] [PubMed] [Google Scholar]
- [63].Chintalapudi SR, Djenderedjian L, Stiemke AB, Steinle JJ, Jablonski MM, Morales-Tirado VM, Isolation and molecular profiling of primary mouse retinal ganglion cells: comparison of phenotypes from healthy and glaucomatous retinas, Front. Aging Neurosci 8 (2016) 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Stowell C, Wang L, Arbogast B, Lan JQ, Cioffi GA, Burgoyne CF, Zhou A, Retinal proteomic changes under different ischemic conditions - implication of an epigenetic regulatory mechanism, Int J Physiol Pathophysiol Pharmacol 2 (2) (2010) 148–160. [PMC free article] [PubMed] [Google Scholar]
- [65].Yun H, Lathrop KL, Yang E, Sun M, Kagemann L, Fu V, Stolz DB, Schuman JS, Du Y, A laser-induced mouse model with long-term intraocular pressure elevation, PLoS One 9 (9) (2014) e107446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Tzekov R, Quezada A, Gautier M, Biggins D, Frances C, Mouzon B, Jamison J, Mullan M, Crawford F, Repetitive mild traumatic brain injury causes optic nerve and retinal damage in a mouse model, J. Neuropathol. Exp. Neurol 73 (4) (2014) 345–361. [DOI] [PubMed] [Google Scholar]
- [67].Tao W, Dvoriantchikova G, Tse BC, Pappas S, Chou TH, Tapia M, Porciatti V, Ivanov D, Tse DT, Pelaez D, A novel mouse model of traumatic optic neuropathy using external ultrasound energy to achieve focal, indirect optic nerve injury, Sci. Rep 7 (1) (2017) 11779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Watson AB, A formula for human retinal ganglion cell receptive field density as a function of visual field location, J. Vis 14 (7) (2014). [DOI] [PubMed] [Google Scholar]
- [69].Kondadi AK, Wang S, Montagner S, Kladt N, Korwitz A, Martinelli P, Herholz D, Baker MJ, Schauss AC, Langer T, Rugarli EI, Loss of the m-AAA protease subunit AFG(3)L(2) causes mitochondrial transport defects and tau hyperphosphorylation, EMBO J 33 (9) (2014) 1011–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Callaway EM, Structure and function of parallel pathways in the primate early visual system, J. Physiol 566 (Pt 1) (2005) 13–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Erskine L, Herrera E, Connecting the retina to the brain, ASN neuro 6 (6) (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Morgan JE, Circulation and axonal transport in the optic nerve, Eye 18 (11) (2004) 1089–1095. [DOI] [PubMed] [Google Scholar]
- [73].Chidlow G, Ebneter A, Wood JP, Casson RJ, The optic nerve head is the site of axonal transport disruption, axonal cytoskeleton damage and putative axonal regeneration failure in a rat model of glaucoma, Acta Neuropathol 121 (6) (2011) 737–751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Monsma PC, Brown A, FluoroMyelin Red is a bright, photostable and non-toxic fluorescent stain for live imaging of myelin, J. Neurosci. Methods 209 (2) (2012) 344–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Marty MC, Alliot F, Rutin J, Fritz R, Trisler D, Pessac B, The myelin basic protein gene is expressed in differentiated blood cell lineages and in hemopoietic progenitors, Proc. Natl. Acad. Sci. U. S. A 99 (13) (2002) 8856–8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Louis ED, Ma K, Babij R, Cortes E, Liem RK, Vonsattel JP, Faust PL, Neurofilament protein levels: quantitative analysis in essential tremor cerebellar cortex, Neurosci. Lett 518 (1) (2012) 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Omary MB, Ku NO, Tao GZ, Toivola DM, Liao J, “Heads and tails” of intermediate filament phosphorylation: multiple sites and functional insights, Trends Biochem. Sci 31 (7) (2006) 383–394. [DOI] [PubMed] [Google Scholar]
- [78].Petzold A, Neurofilament phosphoforms: surrogate markers for axonal injury, degeneration and loss, J. Neurol. Sci 233 (1–2) (2005) 183–198. [DOI] [PubMed] [Google Scholar]
- [79].Chidlow G, Wood JPM, Casson RJ, Investigations into hypoxia and oxidative stress at the optic nerve head in a rat model of glaucoma, Front. Neurosci 11 (2017) 478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Kensler TW, Wakabayashi N, Biswal S, Cell survival responses to environmental stresses via the Keap1-Nrf2-ARE pathway, Annu. Rev. Pharmacol. Toxicol 47 (2007) 89–116. [DOI] [PubMed] [Google Scholar]
- [81].Motohashi H, Yamamoto M, Nrf2-Keap1 defines a physiologically important stress response mechanism, Trends Mol. Med 10 (11) (2004) 549–557. [DOI] [PubMed] [Google Scholar]
- [82].Chan K, Han XD, Kan YW, An important function of Nrf2 in combating oxidative stress: detoxification of acetaminophen, Proc. Natl. Acad. Sci. U. S. A 98 (8) (2001) 4611–4616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Jadeja RN, Upadhyay KK, Devkar RV, Khurana S, Naturally occurring Nrf2 activators: potential in treatment of liver injury, Oxid Med Cell Longev (2016) 3453926 2016. [DOI] [PMC free article] [PubMed]
- [84].David JA, Rifkin WJ, Rabbani PS, Ceradini DJ, The Nrf2/keap1/ARE pathway and oxidative stress as a therapeutic target in type II diabetes mellitus, J Diabetes Res 2017 (2017) 4826724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Pease ME, McKinnon SJ, Quigley HA, Kerrigan-Baumrind LA, Zack DJ, Obstructed axonal transport of BDNF and its receptor TrkB in experimental glaucoma, Invest. Ophthalmol. Vis. Sci 41 (3) (2000) 764–774. [PubMed] [Google Scholar]
- [86].Chen H, Weber AJ, BDNF enhances retinal ganglion cell survival in cats with optic nerve damage, Invest. Ophthalmol. Vis. Sci 42 (5) (2001) 966–974. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.