

Abstract
Junctional adhesion molecule-A (JAM-A), an epithelial tight junction protein, plays an important role in regulating intestinal permeability through association with a scaffold signaling complex containing ZO-2, Afadin, and the small GTPase Rap2. Under inflammatory conditions, we report that the cytoplasmic tail of JAM-A is tyrosine phosphorylated (p-Y280) in association with loss of barrier function. While barely detectable Y280 phosphorylation was observed in confluent monolayers of human intestinal epithelial cells under basal conditions, exposure to cytokines TNFα, IFNγ, IL-22, or IL-17A, resulted in compromised barrier function in parallel with increased p-Y280. Phosphorylation was Src kinase dependent, and we identified Yes-1 and PTPN13 as a major kinase and phosphatase for p-JAM-A Y280, respectively. Moreover, cytokines IL-22 or IL-17A induced increased activity of Yes-1. Furthermore, the Src kinase inhibitor PP2 rescued cytokine-induced epithelial barrier defects and inhibited phosphorylation of JAM-A Y280 in vitro. Phosphorylation of JAM-A Y280 and increased permeability correlated with reduced JAM-A association with active Rap2. Finally, we observed increased phosphorylation of Y280 in colonic epithelium of individuals with ulcerative colitis and in mice with experimentally induced colitis. These findings support a novel mechanism by which tyrosine phosphorylation of JAM-A Y280 regulates epithelial barrier function during inflammation.
INTRODUCTION
A single layer of epithelial cells lines the gastrointestinal tract to form a critical barrier that prevents access of tissues to luminal antigens and pathogens. The epithelial barrier is maintained by intercellular junctions, the most apical of which are the tight junctions (TJs). TJs control epithelial paracellular permeability to water and ions, and are comprised of a complex series of transmembrane proteins that associate with scaffold proteins, actin-binding proteins, and signaling molecules to dynamically regulate epithelial barrier function.
A transmembrane TJ-associated protein termed junctional adhesion molecule-A (JAM-A) has two extracellular immunoglobulin-like domains that mediate interactions in cis and trans to form dimers/oligomers, and these homophilic interactions have been shown to play an important role in regulation of the epithelial barrier, cell migration, and proliferation (Mandell et al., 2005; Laukoetter et al., 2007; Nava et al., 2011; Monteiro and Parkos, 2012; Monteiro et al., 2013, 2014; Luissint et al., 2014; Mitchell et al., 2015). JAM-A has a single transmembrane domain and a short cytoplasmic tail ending in a C-terminal PDZ-binding motif that has been shown to interact with scaffold proteins zonula occludens (ZO-1,ZO-2), afadin, and MUPP1 and guanine nucleotide exchange factors, which mediate signaling functions (Bazzoni et al., 2000; Hamazaki et al., 2002; Nomme et al., 2011; Monteiro et al., 2013). JAM-A knockout mice have compromised intestinal epithelial barrier function, highlighting the importance of JAM-A in regulating intestinal permeability (Laukoetter et al., 2007). Furthermore, the role of JAM-A in regulating epithelial barrier function is supported by in vitro studies after knockdown of JAM-A in polarized epithelial cells (Mandell et al., 2007). Disruption of tight junctions and loss of key TJ-associated proteins, such as JAM-A, is associated with compromised intestinal epithelial barrier and altered epithelial cell function in pathologic conditions such as inflammatory bowel disease (IBD; Kucharzik et al., 2001). In IBD and in vitro studies, a cytokine-rich inflammatory microenvironment results in the internalization, loss, and/or mislocalization of JAM-A and other TJ proteins (Bruewer et al., 2003; Utech et al., 2005; Den Beste et al., 2013; Capaldo et al., 2014; Wise et al., 2014; Luissint et al., 2016). Despite these observations, the molecular details of JAM-A–mediated regulation of epithelial permeability remains incompletely understood.
Signaling molecules, including protein kinases and phosphatases such as protein kinase Cζ (PKCζ; Caraballo et al., 2011), Src family kinases (Kale et al., 2003), mitogen-activated protein kinase (MAPK; Basuroy et al., 2006), protein phosphatase 1 (PP1; Sallee and Burridge, 2009), and protein phosphatase 2A (PP2A) have been reported to interact with TJ proteins (Seth et al., 2007). Studies utilizing kinase inhibitors or conditions that alter tyrosine phosphatase expression have highlighted the importance of protein phosphorylation in the regulation of TJ function (Basuroy et al., 2006; Sallee and Burridge, 2009; Murchie et al., 2014).
While serine phosphorylation of JAM-A (S284) has been linked to stabilizing tight junctions in Madin–Darby canine kidney (MDCK) cells and in contributing to dynamic TJ assembly (Iden et al., 2012), functional consequence(s) of JAM-A tyrosine phosphorylation have not been explored. Altered expression of key protein tyrosine phosphatases has been reported in the colonic mucosa of people with IBD, and excessive tyrosine phosphorylation within intestinal crypts in mice is associated with defective barrier function (Murchie et al., 2014; Spalinger et al., 2016). These and other reports support a model of dynamic regulation of TJ function that is dependent, in part, on protein phosphorylation and dephosphorylation. Furthermore, such observations suggest that signaling events within the intestinal epithelium result in altered tyrosine phosphorylation that may be linked to barrier compromise during mucosal inflammation.
In this study, we report that epithelial JAM-A is phosphorylated at Y280 and provide a link between this phosphorylation event, proinflammatory cytokine exposure, and barrier compromise. Under baseline/normal conditions associated with mature TJs, very low- to undetectable levels of JAM-A Y280 phosphorylation was observed. Exposure of epithelial cells to several functionally important cytokines, TNFα, IFNγ, IL-22, or IL-17A, resulted in an increased p-JAM-A Y280. We identified the Src kinase family member Yes-1 and PTPN13 as a major tyrosine kinase and phosphatase, respectively, that regulate tyrosine phosphorylation of Y280. IL-22 or IL-17A exposure resulted in enhanced Yes-1 kinase activity, phosphorylation of JAM-A Y280, and compromised epithelial barrier function. Tyrosine phosphorylation of JAM-A also paralleled decreased association with active Rap2 and compromised epithelial barrier function. Finally, there was minimal tyrosine phosphorylation of JAM-A observed in normal colonic epithelium, whereas robust phosphorylation was present in colonic epithelium from individuals with ulcerative colitis and in mice with experimentally induced colitis (DSS). These results highlight a link between Yes-1, PTPN13, phosphorylation of JAM-A Y280, and the regulation of epithelial barrier function.
RESULTS
JAM-A is tyrosine phosphorylated at Y280
Serine phosphorylation of the cytoplasmic tail of murine JAM-A at S285 (S284 in human JAM-A) has been reported to regulate its localization in mature intercellular contacts of epithelial cells (Iden et al., 2012). Given a report using mass spectrometry indicating that JAM-A is also tyrosine phosphorylated (Van Itallie and Anderson, 2018), and the presence of a candidate tyrosine phosphorylation site at Y280, we investigated whether JAM-A function may be regulated by tyrosine phosphorylation at Y280. Experiments were performed using a commercially available antibody raised against phosphorylated Y280 JAM-A (p-JAM-A Y280; Figure 1a). Interestingly, p-JAM-A Y280 was barely detectible under basal conditions in confluent intestinal epithelial cells (IECs), whereas p-JAM-A Y280 was identified in intercellular junctions of epithelial cells after global inhibition of tyrosine phosphatases with pervanadate (Figure 1b, left). Immunoblotting experiments revealed labeling of a 37-kDa protein band consistent with JAM-A in lysates from cells treated with pervanadate. The addition of calf intestinal phosphatase (CIP) reversed pervanadate-induced JAM-A Y280 phosphorylation (Figure 1b, right). Such findings suggested minimal p-JAM-A Y280 in confluent monolayers under basal, non-pervanadate-treated conditions. Indeed, immunoprecipitation of total JAM-A from confluent monolayers of SK CO-15 and T84 cells followed by immunoblot with anti–p-Y280 antibody confirmed minimal phosphorylation of JAM-A Y280 in control monolayers under basal conditions compared with robust phosphorylation in cells treated with pervanadate (unpublished data). Specificity of the anti–p-JAM-A Y280 antibody was confirmed in studies using HEK 293T cells transfected with HA-tagged wild-type JAM-A (HA-JAM-A wt) and a nonphosphorylatable JAM-A mutant containing a tyrosine 280 to phenylalanine substitution (Y280F) followed by immunoblotting. As shown in Supplemental Figure 1a, pervanadate treatment induced tyrosine phosphorylation in JAM-A wt cells. However, phosphorylation of JAM-A was reduced to baseline in JAM-A Y280F cells, indicating that the antibody specifically recognizes phosphorylation of JAM-A at Y280. We also observed that pervanadate treatment led to a time-dependent increase in phosphorylation of JAM-A Y280 that correlated with decreased localization of JAM-A at cell–cell contacts (Figure 1c). In parallel, transepithelial electrical resistance (TEER) declined dramatically in pervanadate-treated cells (Supplemental Figure 1b). Confocal immunofluorescence imaging indicated that p-JAM-A Y280 colocalized with total JAM-A in TJs of confluent IECs after pervanadate treatment, similar to that observed with p-JAM-A S284 in polarized epithelial cells (Figure 1d and Supplemental Figure 1c).
FIGURE 1:
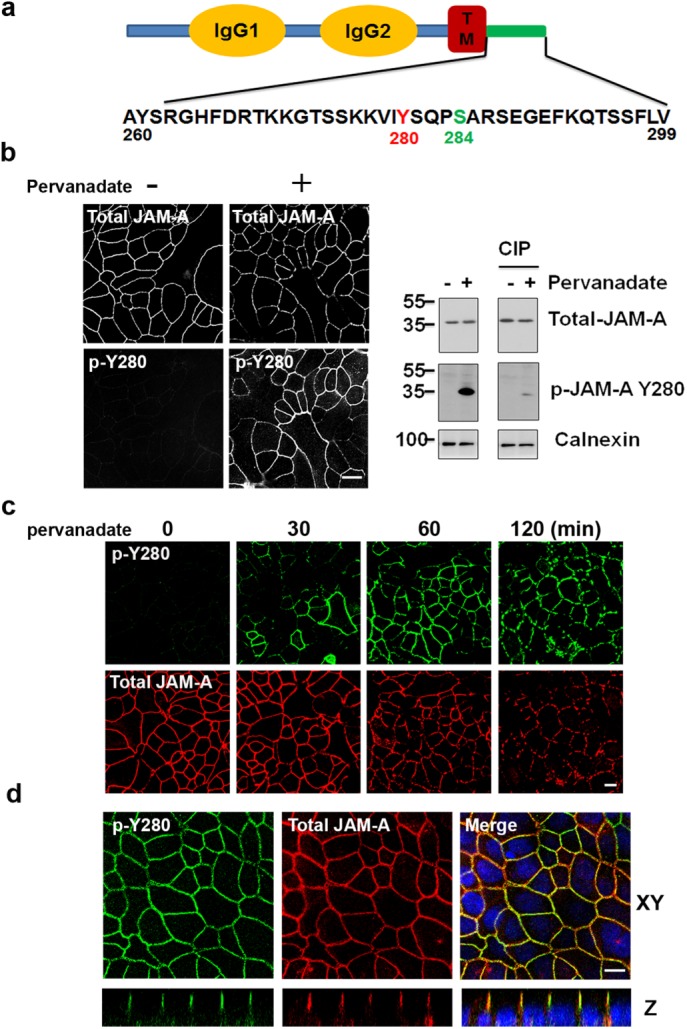
JAM-A is tyrosine phosphorylated at Y280. (a) Human JAM-A highlighting tyrosine 280 (murine JAM-A Y281) within the cytoplasmic tail in close proximity to S284. (b) Using a commercially available phosphospecific antibody, phosphorylation of JAM-A Y280 (p-Y280) is undetectable in confluent monolayers of SK CO-15 cells under basal conditions, but robust phosphorylation is observed after global inhibition of tyrosine phosphatase with pervanadate (25 μM) for 60 min (left). Detection of p-JAM-A Y280 by immunoblot after pervanadate treatment (60 min), compared with nontreated controls. Treatment with calf intestinal phosphatase (CIP) reversed pervanadate-induced phosphorylation of JAM-A Y280 (right). Similar results were observed in T84 cells. (c) Confluent SK CO-15 cells were treated with pervanadate as time points indicated, then cells were fixed and costained with anti–p-JAM-A Y280 and total JAM-A antibodies. (d) T84 cells were grown on Transwell filters until confluent. After 45 min of pervanadate treatment, cells were fixed with 4% PFA and permeabilized with 1% SDS followed by immunostaining with p-JAM-A Y280 and total JAM-A. Confocal Z-stacks indicate p-JAM-A Y280 localizes to the TJ. We also detected p-JAM-A Y280 localization to TJ in SK CO-15 cells (unpublished data). Scale bar: 10 μm.
JAM-A is tyrosine phosphorylated under cytokine treatment and impaired barrier function
Given the well-appreciated association of disrupted TJ function and impaired barrier upon exposure to certain cytokines, we performed experiments to assess whether JAM-A is tyrosine phosphorylated at Y280 under inflammatory conditions. For these experiments, confluent T84 monolayers were incubated with cytokines IL-17A, IL-22, TNFα, or IFNγ for 48 h followed by harvest of cells for immunoblotting. As shown in Figure 2a, left, increased phosphorylation of JAM-A Y280 was observed in T84 cells after exposure to all four cytokines. Similar results were obtained in SK CO-15 cells except TNFα treatment alone, but p-JAM-A Y280 was increased after IFNγ stimulated induction of TNFα receptor expression (Figure 2a, right; Pandita et al., 1992). In contrast to T84 cells, it is known that TEER in SK-CO15 cells is relatively insensitive to cytokine stimulation, likely due to altered signaling pathways in linking cytokine stimulation with regulation of TEER. Because paracellular permeability to solutes has been linked to the “leak” pathway (Anderson and Van Itallie, 1995; Anderson et al., 2004; Choi et al., 2017), we tested whether IFNγ induces changes in the leak pathway in SK CO-15 monolayers by measuring paracellular flux of fluorescein isothiocyanate (FITC) dextran. Indeed, we observed enhanced permeability to 10 kDa FITC dextran after exposure of SK CO-15 monolayers to 100 ng/ml IFNγ for 48 h (Supplemental Figure 2a). The relevance of these observations to natural human colonic epithelium was confirmed by demonstrating enhanced phosphorylation of JAM-A Y280 in monolayers derived from human colonoids after stimulation with IL-17A or IL-22 (Supplemental Figure 2b). Given the known effects of TNFα and/or IFNγ on epithelial barrier function under the conditions described here (Ma et al., 2004; Utech et al., 2005; Smyth et al., 2011; Al-Sadi et al., 2016), we performed experiments to determine whether IL-22 and IL-17A exposure would similarly result in altered epithelial barrier function. Indeed, both IL-22 and IL-17A enhanced transepithelial flux of 4 kDa FITC dextran (Figure 2, b and d). Similarly, TEER significantly declined in IL-22–treated T84 monolayers after 48 h. However, there was only a mild decrease in TEER observed after IL-17A treatment (Supplemental Figure 2c). Immunoblots revealed time-dependent increases in p-JAM-A Y280 in IECs incubated with IL-22 or IL-17A (Figure 2, c and e). These findings are consistent with a cytokine-induced increase in epithelial permeability that parallels tyrosine phosphorylation of JAM-A. However, the relatively small effect of IL-17A on TEER suggests that IL-22 and IL-17A may have differential effects on the leak and pore pathways (Anderson and Van Itallie, 1995; Anderson et al., 2004; Choi et al., 2017).
FIGURE 2:

Exposure of IECs to cytokines IL-17A, IL-22, TNFα, or IFNγ results in tyrosine phosphorylation of JAM-A at Y280 and a leaky barrier. (a) T84 was incubated with media containing cytokines IL-17A (100 ng/ml), IL-22 (40 ng/ml), TNFα (50 ng/ml), and IFN γ (100 ng/ml) for 48 h and then harvested and subjected for immunoblots as indicated (left). Similar experiments were performed in SK CO-15 except TNFα + INFγ combined treatment (right). (b, d) Confluent T84 monolayers were treated with 40 ng/ml IL-22 or 100 ng/ml IL-17A for 48 h and assessed for paracellular flux of 4 kDa FITC dextran for another hour compared with nontreated cells (CTL; N = 3 independent experiments. Bar graphs represent the mean ± SEM compared with control untreated cells. (c, e) Confluent T84 monolayers were incubated with media containing 40 ng/ml recombinant human IL-22 or 100 ng/ml recombinant human IL-17A for 0, 24, and 48 h, and then cells were harvested and lysed in RIPA buffer and immunoblotted for p-JAM-A Y280, total JAM-A, and calnexin (left). Densitometric analyses of p-JAM-A Y280 immunoblots normalized to calnexin bands in each condition. P-JAM-A Y280 bands’ density was relative to the band at time 0. Quantitation of p-JAM-A Y280 (N = 3 individual experiments, bar graphs represent mean ± SEM; right). For all experiments, *, p < 0.05; **, p < 0.01; and ***, p < 0.001 between the groups were determined by one-way ANOVA or two-tailed Student’s t tests.
Tyrosine phosphorylation of JAM-A is regulated by the Src family member Yes-1 and the phosphatase PTPN13
Experiments were performed to identify the kinase(s) and phosphatase(s) that regulate JAM-A Y280 tyrosine phosphorylation. We focused on members of the Src family of tyrosine kinases because they have previously been reported to modulate tyrosine phosphorylation of junctional proteins and influence epithelial barrier function (Meyer et al., 2001; Basuroy et al., 2003). Indeed, preincubation of IECs with the general Src kinase inhibitor PP2 (1 μM) completely inhibited the phosphorylation of JAM-A Y280 in response to pervanadate treatment (Figure 3a). Immunoblots of p-JAM-A Y280 revealed a PP2 dose-dependent decrease in tyrosine phosphorylation of JAM-A Y280 following pervanadate treatment (Figure 3b). To gain insight into which specific Src kinase associates with and phosphorylates JAM-A, coimmunoprecipitation experiments were performed with SK CO-15 cells that expressed HA-tagged JAM-A followed by immunoblotting to detect Src family members. As shown in the immunoblots in Figure 3c, HA-tagged JAM-A coprecipitated with Yes-1 and Lyn, but not Src. Parallel immunofluorescence labeling experiments revealed increased colocalization of JAM-A with Yes-1 compared with Lyn and Src (Supplemental Figure 3). These findings support Yes-1 as a tyrosine kinase that may play a role in tyrosine phosphorylation of JAM-A at Y280. To further investigate contributions of Yes-1 to phosphorylation of JAM-A Y280, small interfering RNA (siRNA)-mediated knockdown of Yes-1 was performed in SK CO-15 cells. As shown in Figure 3d, knockdown of Yes-1 resulted in a 65% reduction of p-JAM-A Y280 in pervanadate-treated SK CO-15 cells, as well as a 62% reduction of p-JAM-A Y280 in untreated cells (Supplemental Figure 4, a and b). Collectively, these results suggest that phosphorylation of JAM-A at Y280 is regulated by Yes-1.
FIGURE 3:

Yes-1 regulates phosphorylation of JAM-A Y280. (a) The Src family kinase inhibitor PP2 (1 μM) inhibits pervanadate-dependent phosphorylation of JAM-A Y280. Immunofluorescence staining of p-JAM-A Y280 and total JAM-A in T84 monolayers pretreated with/without PP2 followed by exposure to pervanadate. Similar results were observed in SK CO-15 cells (unpublished data). (b) Dose-dependent inhibition of p-JAM-A Y280 by PP2 after pervanadate treatment. T84 cells were lysed after treatment with increasing concentrations of PP2 followed by exposure to pervanadate. Lysates were then immunoblotted for p-JAM-A Y280, total JAM-A, and total Src. p-EGFR Y1068 was used as a positive control for PP2. Calnexin was used as a loading control. (c) Coimmunoprecipitation of Yes1/Lyn with JAM-A. SK CO-15 cells stably expressing HA-tagged full-length JAM-A were lysed with 1% Triton X-100 lysis buffer followed by immunoprecipitation with anti-HA antibody and probed with antibodies to Yes-1, Src, Lyn, and HA. Coimmunoprecipitation of HA-JAM-A with Yes-1 and Lyn was observed. HA-JAM-A did not coimmunoprecipitate with Src. ZO-2 served as a positive control for coimmunoprecipitation with JAM-A (Monteiro et al., 2013). (d) Knockdown of Yes-1 in IECs. SK CO-15 cells were transfected with siRNA against Yes-1. Posttransfection (48 h), the cells were incubated with pervanadate for 30 min before lysis of the cells for immunoblots. Both Yes-1 siRNAs suppressed Yes-1 efficiently. Yes-1 knockdown strongly inhibits p-JAM-A Y280 in response to pervanadate (left). Density of p-JAM-A Y280 immunoblots were normalized to total JAM-A and calnexin bands in each condition. p-JAM-A Y280 was compared with control siRNA (CTL siRNA). Quantification of immunoblots of p-JAM-A Y280 in Yes-1 knockdown and control cells (N = 3 individual experiments, bar graphs represent mean ± SEM; right). (e) T84 cells grown on Transwell filters and treated with IL-22 or IL-17A, or after pretreatment with 100 nM PP2 for 48 h. Cells were harvested, and then anti–Yes-1 immunoprecipitation followed by p-Src Y416 blot demonstrated the increased activity of Yes-1 after IL-22 or IL-17A treatment that was PP2 sensitive (top panels). Cells were treated with IL-22 or IL-17A as described above, and then lysed and Western blots probed for p-JAM-A Y280, total JAM-A, and calnexin as loading controls. IL-22 or IL-17A induced phosphorylation of JAM-A Y280 that was PP2 sensitive (bottom panels). Densities of p-JAM-A Y280 immunoblots were collected and normalized to total JAM-A and calnexin bands in each condition. p-JAM-A Y280 was compared with control untreated cells (CTL). Quantification of p-JAM-A Y280 in response to IL-22 or IL-17A treatment and PP2 rescue (N = 3 independent experiments; bar graph represents the mean ± SEM; right). (f) IL-22 or IL-17A induces barrier defect as assessed by increased paracellular flux of 4 kDa FITC dextran; PP2 rescues the barrier defect induced by IL-22 or IL-17A treatment compared with control untreated cells (CTL; N = 3 independent experiments; bar graph represents the mean ± SEM). For all experiments, *, p < 0.05; **, p < 0.01; and ***, p < 0.001 between the groups were determined by one-way ANOVA or two-tailed Student’s t tests. Scale bar: 10 μm.
As shown in Figure 2, exposure of intestinal epithelial monolayers to IL-22 and IL-17A resulted in a time-dependent increase in phosphorylation of JAM-A Y280 in parallel with increased permeability, suggesting that the barrier defect may be linked to phosphorylation of JAM-A Y280. Given the above results, experiments were performed to investigate whether IL-22 and IL-17A directly induce activation of Yes-1. Kinase activity of Yes-1 is regulated by phosphorylation of the Y426 residue, but there are no available antibodies that selectively target Y426. However, we were able to use another anti-Src kinase antibody, anti–p-Src Y416 that detects activation of Src family kinases including Yes-1. Yes-1 knockdown resulted in a 50–60% decrease in p-Src Y416, suggesting that anti–p-Src Y416 detects active Yes-1 (Supplemental Figure 5a). Immunoprecipitation of Yes-1 followed by Western blot for p-Src Y416 revealed increased phosphorylation activity of Yes-1 in IECs after exposure to IL-22 or IL-17A. Furthermore, PP2 effectively reversed the increase in activity of Yes-1 induced by IL-22 and IL-17A (Figure 3e, top panels). In Western blots of IL-22 and IL17A-treated IECs, increased p-JAM-A Y280 was detected after 48 h of IL-22 or IL-17A exposure. Inhibition of Src kinase activity with PP2 effectively reversed increases in phosphorylation observed after cytokine treatment (Figure 3e, bottom panels). These results lend support to the specificity of phosphorylation of JAM-A Y280 in response to IL-17A and IL-22. Finally, we observed that treatment of intestinal epithelial monolayers (T84 cells) with PP2 was effective in rescuing the barrier defects induced by IL-22 or IL-17A, as assessed by flux of 4 kDa FITC dextran (Figure 3f and Supplemental Figure 5b). Collectively, these results implicate cytokine-induced activation of Yes-1 in mediating tyrosine phosphorylation of JAM-A Y280 and subsequent increase in permeability.
To identify potential candidate phosphatase(s) that interact with and mediate dephosphorylation of p-JAM-A Y280, we screened for PDZ-dependent binding of the recombinant full-length cytoplasmic segment of JAM-A (amino acids 261–300) with a proteomic array of 96 PDZ domains derived from 48 distinct scaffold proteins (He et al., 2006; Monteiro et al., 2013). A positive binding signal was obtained for the first PDZ domain of the protein tyrosine phosphatase PTPN13. To confirm the interaction of PTPN13 with JAM-A, coimmunoprecipitation experiments were performed in polarized epithelial cells. As shown in Figure 4a, JAM-A and PTPN13 coimmunoprecipitated from lysates of confluent monolayers of SK CO-15 cells. To enhance potential interactions between the phosphatase and JAM-A, SK CO-15 cells were incubated with a buffer containing the global phosphatase inhibitor pervanadate, and JAM-A was immunoprecipitated followed by Western blotting for PTPN13 and p-JAM-A Y280. As shown in Figure 4b, coimmunoprecipitation of PTPN13 with JAM-A was significantly enhanced after pervanadate treatment suggesting preferential interactions of PTPN13 with tyrosine phosphorylated JAM-A. To directly test whether PTPN13 can dephosphorylate p-JAM-A Y280, lysates of pervanadate-treated IECs were incubated with increasing amounts of recombinant active or dead PTPN13 followed by Western blotting to detect p-JAM-A Y280 (Figure 4c). As can be seen, active PTPN13 significantly reduced tyrosine phosphorylation of Y280 in a dose-dependent manner. Moreover, we observed increased colocalization of PTPN13 with JAM-A in the cell–cell contacts after cytokine exposure (Supplemental Figure 6). These findings are consistent with recruitment of PTPN13 to TJ-associated JAM-A after cytokine exposure, presumably in response to tyrosine phosphorylation of JAM-A. Collectively, these observations support a role of PTPN13 as a JAM-A tyrosine phosphatase.
FIGURE 4:

PTPN13 acts as a phosphatase for p-JAM-A Y280. (a) Coimmunoprecipitation of PTPN13 and JAM-A. Confluent monolayers of SK CO-15 cells were lysed with buffer containing 1% Triton followed by immunoprecipitation with anti–JAM-A antibody and probed with antibodies to PTPN13 and JAM-A. (b) Pervanadate treatment resulted in increased binding of PTPN13 to JAM-A immunoprecipitated protein complexes. SK CO-15 cells were treated with pervanadate followed by immunoprecipitation with anti–JAM-A and probed with antibodies to PTPN13 and p-JAM-A Y280 (left). Quantification of PTPN13 coimmunoprecipitation with JAM-A with pervanadate treatment compared to untreated cells (CTL; N = 3 individual experiments; bar graphs represent mean ± SEM; right). (c) Active PTPN13 directly dephosphorylates p-JAM-A Y280. SK CO-15 cells were lysed in a modified lysis buffer containing 1% Triton X-100. Cell lysates (50 μl) were incubated with 1–3 μg of a recombinant active fragment of PTPN13 at 30oC for 10 min followed by SDS–PAGE, Western blot, and probing with antibodies to p-JAM-A Y280 and total JAM-A. Treatment of SK CO-15 cells with active PTPN13 results in dephosphorylation of p-JAM-A Y280 in a dose-dependent manner. Because PTPN13 does not affect phosphorylation of p-HER3 Y1289, it served as a negative control (left). An identical experiment performed using dead (boiled) PTPN13 instead of active phosphatase showed no dephosphorylation of p-JAM-A Y280 (middle). Densities of p-JAM-A Y280 immunoblots were normalized to total JAM-A and calnexin bands in each condition. P-JAM-A Y280 was compared with nontreated cells (CTL). Quantitation of p-JAM-A Y280 immunoblots after incubation of samples with active PTPN13 (N = 3 independent experiments; bar graphs represent mean ± SEM; right). For all experiments, *, p < 0.05; **, p < 0.01; and ***, p < 0.001 between the groups were determined by one-way ANOVA or two-tailed Student’s t tests.
Phosphorylation of JAM-A Y280 inhibits JAM-A association with TJ-associated scaffold signaling protein Rap2
It is well appreciated that transmembrane TJ proteins bind to scaffold proteins that associate with the actin cytoskeleton to regulate barrier function (Anderson and Van Itallie, 1995; Tsukita et al., 2001). Conversely, dissociation of transmembrane TJ proteins from scaffolding molecules has been shown to result in a compromised epithelial barrier (Utech et al., 2005). Given our findings linking inflammation-induced activation of Yes-1 to tyrosine phosphorylation of JAM-A Y280 and compromised barrier, we tested whether increased permeability after tyrosine phosphorylation of JAM-A Y280 coincides with dissociation of JAM-A and key signaling proteins known to play a role in regulating epithelial permeability. Specifically, we investigated whether tyrosine phosphorylation of JAM-A plays a role in regulating JAM-A association with the small GTPase Rap2c, which has previously been shown to play a key role in the maintenance of barrier function (Monteiro et al., 2013, 2014). We generated a GST-fused Rap2c full-length recombinant protein (GST-Rap2c wt), a dominant active mutation G12V, and a dominant inactive mutation S17N. In pull-down assays shown in Figure 5a, top panel, GST-Rap2c G12V pulled down HA-JAM-A wt, indicating preferential JAM-A association with active or GTP-bound Rap2c. As expected, the nonphosphorylatable mutant HA-JAM-A Y280F did not affect its binding to GST-Rap2 G12V (Figure 5a, bottom panel). Furthermore, assays with Rap2 G12V revealed very little association with HA-JAM-A wt after cells were treated with pervanadate to induce phosphorylation of JAM-A Y280. By contrast, GST-Rap2 G12V was able to pull down the nonphosphorylatable JAM-A mutant (HA-JAM-A Y280F) under the same conditions. These results are consistent with tyrosine phosphorylation-dependent dissociation of Rap2 with JAM-A (Figure 5b). In pull-down experiments with RalGDS that targeted active wild-type Rap2, we observed decreased active Rap2 association with JAM-A after stimulation of tyrosine phosphorylation with pervanadate. There was a striking absence of p-JAM-A Y280 in Rap2 pull downs (Figure 5c). Collectively, these findings suggest that association of JAM-A with active Rap2 is disrupted after Y280 phosphorylation. Consistent with RalGDS pull-down results, immunofluorescence localization experiments performed in confluent, pervanadate-treated IEC monolayers demonstrated reduced colocalization of Rap2 with JAM-A at TJs (Figure 5d).
FIGURE 5:

Phosphorylation of JAM-A Y280 results in decreased JAM-A association with active Rap2. (a) HA-JAM-A wt or Y280F were transfected into HEK 293T for 48 h, then cells were harvested and followed by GST-Rap2c wt, G12V, or S17N pull down and immunoblotting of anti-HA. (b) HA-JAM-A wt or Y280F were transfected to SK CO-15 for 48 h and then treated with pervanadate for 1 h followed by harvesting and GST-Rap2c G12V pull down and immunoblotting of anti-HA and p-JAM-A Y280. (c) Confluent monolayers of SK CO-15 cells were treated with/without pervanadate for 1 h, lysed with NP-40–containing buffer, and subjected to RalGDS pull down. As can be seen, RalGDS pulled down active Rap2 and JAM-A in nontreated cells. However, much less active Rap2 with JAM-A, and no p-JAM-A Y280 was pulled down in cells treated with pervanadate (left). Densitometric quantification of immunoblots of JAM-A and Rap2 by RalGDS pull down is shown in the middle and right panels (CTL is nontreated cells; N = 3 individual experiments; bar graphs represent mean ± SEM). (d) Monolayers of T84 cells were grown on Transwell filters until confluent. Cells were treated with pervanadate for 1 h (bottom panels) and fixed with 4% PFA and permeabilized with 1% SDS, followed by immunostaining to visualize Rap2, p-JAM-A Y280 (left), or JAM-A (right). (e) Confluent SK CO-15 cells were incubated with media containing IL-17A, IL-22, or TNFα + IFNγ for 48 h. Cells were lysed and pulled down with RalGDS and immunoblotted with anti-Rap2 and JAM-A. Densitometric quantification of immunoblots of Rap2 and JAM-A by RalGDS pull down is shown in the middle and right panels (CTL is untreated cells; N = 3 individual experiments; bar graphs represent mean ± SEM). For all experiments, *, p < 0.05; **, p < 0.01; and ***, p < 0.001 between the groups were determined by one-way ANOVA or two-tailed Student’s t tests. Scale bar: 10 μm.
Finally, we treated IECs with IL-17A, IL-22, or TNFα + IFNγ for 48 h followed by RalGDS pull down of active Rap2. As can be seen in Figure 5e, active Rap2 was significantly decreased in pull downs from cells treated with cytokines as compared with control nontreated cells. These results are consistent with cytokine-induced tyrosine phosphorylation of JAM-A at Y280 playing a role in disruption of the intestinal barrier under inflammatory conditions.
JAM-A Y280 is phosphorylated in dextran sulfa–induced colitis in mice and in humans with ulcerative colitis
Chronic intestinal inflammation, as observed in IBD, is associated with a leaky intestinal epithelial barrier and altered TJs (Luissint et al., 2016). Although mechanisms regulating barrier disruption in IBD are complex and poorly understood, there is a clear link between increased permeability and the cytokine-rich microenvironment. Given the results demonstrating increased tyrosine phosphorylation of JAM-A after cytokine exposure, we performed immunofluorescence labeling of p-JAM-A Y280 in the colons of mice with chronic dextran sulfate (DSS)–induced colitis. Colonic mucosa was fixed and stained with a pan-JAM-A antibody and compared with labeling with antibody against p-JAM-A Y280. Barely detectable levels of p-JAM-A Y280 were observed in colonic epithelium from control mice (Figure 6a, top panels), but robust tyrosine phosphorylation of JAM-A Y280 was detected in TJs of mucosal epithelial cells from colitic mice (Figure 6a, bottom panels). Similarly, we compared staining of p-JAM-A Y280 in colonic mucosa from individuals with ulcerative colitis (UC) to normal. Very low to undetectable staining of p-JAM-A Y280 was observed in the colonic epithelium from normal mucosa, which demonstrated characteristic TJ and basolateral localization of total JAM-A (Figure 6b, top panels). By contrast, strong immunolabeling of p-JAM-A Y280 was observed in TJs of crypt epithelial cells from colonic mucosa of individuals with UC (Figure 6b, bottom panels).
FIGURE 6:

p-JAM-A Y280 is increased in mouse models of experimental intestinal colitis and human UC. (a) Immunofluorescence staining of colonic mucosa from mice after chronic DSS treatment for p-JAM-A Y280 and total JAM-A as described in Material and Methods. Very low levels of p-JAM-A Y280 were detected in control (untreated) mucosa (top panels). By contrast, strong p-JAM-A Y280 staining is observed in the region of TJs in crypts of mice after DSS treatment (bottom panels). (b) Immunofluorescence staining of p-JAM-A Y280 and total JAM-A in frozen sections of human colonic mucosa obtained from normal and individuals with UC. Undetectable or low levels of p-JAM-A Y280 (top panels) in sections from normal individuals. In contrast, strong staining of p-JAM-A Y280 in crypt epithelium is observed in individuals with active UC (bottom panels). Scale bar: 100 μm.
DISCUSSION
Given that JAM-A has been shown to regulate intestinal epithelial barrier function (Liu et al., 2000; Laukoetter et al., 2007) and reports that link tyrosine phosphorylation of other TJ-associated proteins to regulation of barrier (Basuroy et al., 2003, 2006; Kale et al., 2003; Elias et al., 2009; Rahman et al., 2016), we investigated whether tyrosine phosphorylation plays a role in regulating JAM-A function. The cytoplasmic tail of JAM-A contains 40 amino acids with two tyrosine residues. Although tyrosine phosphorylation of JAM-A has not been studied in epithelial cells, a recent mass spectrometry study (Van Itallie and Anderson, 2018) reported tyrosine phosphorylation of JAM-A as well as another study that linked phosphorylation of JAM-A in platelets to protection from thrombosis (Naik et al., 2014). To explore the functional significance of tyrosine phosphorylation of JAM-A in epithelial cells, studies were performed with a rabbit polyclonal antibody raised against phospho-JAM-A Y280. Analyses of p-JAM-A Y280 in epithelial cells and tissue sections from intestinal mucosa revealed undetectable or very low levels of phosphorylation in confluent IEC and normal human/murine intestinal epithelial cells. Experiments were performed to define conditions and mechanisms by which JAM-A Y280 is phosphorylated.
Tyrosine phosphorylation of other TJ-associated proteins has been linked to impaired TJ function. For example, phosphorylation of Y398 and Y402 in occludin has been reported to impair interactions with ZO-1 and destabilize TJ assembly (Elias et al., 2009). In addition, tyrosine phosphorylation of TJ-associated proteins ZO-1 and occludin during DSS-induced colitis has been reported, suggesting tyrosine phosphorylation of TJ proteins occurs under conditions of inflammation. Unfortunately, there is a lack of specific antibodies to investigate the detailed cellular localization of individual tyrosine phosphorylated TJ proteins in mucosal specimens and cells.
With a new antibody raised against phospho-JAM-A Y280, we tested whether inflammatory conditions induce phosphorylation of JAM-A Y280 by exposing IEC monolayers to cytokines. Indeed, exposure of IECs (or human colonoids) to cytokines IFNγ, TNFα, IL-17A, or IL-22 increased phosphorylation of JAM-A Y280. Importantly, we observed that increased p-JAM-A Y280 paralleled the disruption of barrier function observed with IL-22 and IL-17A, suggesting that JAM-A Y280 phosphorylation may contribute to intestinal barrier defects during inflammation. It is well appreciated that IFNγ and TNFα induce epithelial barrier defects as many IFNγ- and TNFα-related signaling pathways have been shown to modulate barrier function (Ma et al., 2004; Utech et al., 2005; Scharl et al., 2009; Smyth et al., 2011; Feng and Teitelbaum, 2013; Al-Sadi et al., 2016). Levels of other key cytokines are also increased in the mucosa during intestinal inflammatory conditions. For example, the proinflammatory cytokine IL-17A is elevated in colitis (Jiang et al., 2014; Yang et al., 2014). In addition, IL-22 levels are also increased during colitis but, in contrast to IFNγ, TNFα, or IL-17A, this cytokine has been linked to promotion of mucosal repair (Sugimoto et al., 2008; Li et al., 2014). Consistent with the latter, we have observed that IL-22 promotes wound healing in cultured IECs in vitro (unpublished data). Intriguingly, we find that prorepair properties of IL-22 are associated with compromised barrier function in IECs as shown in this study (Figures 2 and 3) and recently reported by another group (Wang et al., 2017). Such observations are consistent with a hypothesis that links cytokine-induced phosphorylation of JAM-A Y280 and destabilization/disassembly of TJs with facilitated epithelial cell migration to promote wound healing during inflammation. Interestingly, we observed significantly enhanced permeability of FITC dextran in both T84 and SK CO-15 cells after exposure to IL-22 and IL-17A. However, there was only a mild decrease in TEER after IL-17A treatment (Supplemental Figure 2b), suggesting that disruption of barrier after cytokine-induced phosphorylation of JAM-A Y280 is preferentially mediated through changes in the leak pathway (Anderson and Van Itallie, 1995; Anderson et al., 2004; Choi et al., 2017). Such cytokine-induced epithelial barrier defects have also been strongly linked to disruption of intestinal homeostasis and to contribute to chronic inflammatory disease.
Coimmunoprecipitation and siRNA experiments defined a major role of the Src family tyrosine kinase Yes-1 in regulating phosphorylation of JAM-A at Y280. Indeed, we observed that Src family inhibitor PP2 inhibited pervanadate-induced phosphorylation of JAM-A Y280 in a dose-dependent manner. Furthermore, knockdown of Yes-1 resulted in significantly decreased p-JAM-A Y280, suggesting that Yes-1 is a positive regulator of JAM-A Y280 phosphorylation. Our findings linking Yes-1 to JAM-A phosphorylation were shown to be relevant under inflammatory conditions as exposure of cultured monolayers of human IECs to IL-22 and IL-17A resulted in increased activity of Yes-1, and p-JAM-A Y280 was readily detected after exposure of epithelial cells to these inflammatory mediators. Collectively, these findings suggest that IL-22– and IL-17A–induced activation of Yes-1 mediates phosphorylation of JAM-A Y280.
We also report that the tyrosine phosphatase PTPN13 negatively regulates phosphorylation of JAM-A at Y280. PTPN13 has been well studied as a tumor suppressor (Zhu et al., 2008) but there were no reports on PTPN13 regulation of epithelial barrier function via tyrosine phosphorylation of tight junction proteins. As a regulator of epithelial barrier function, PTPN13 provides new insights into tyrosine phosphatase contributions to barrier defense during inflammation. Given the delicate balance between kinase/phosphatase protein phosphorylation during homeostasis, it is not surprising that others have reported that tyrosine phosphorylation of junctional proteins is altered during colitis. For example, loss of the phosphatase PTPNσ has been reported to result in protein phosphorylation of ezrin and others in crypt epithelia and increased intestinal permeability in mice (Murchie et al., 2014). In humans, mutations in single nucleotide polymorphisms (SNPs) linked to the phosphatase PTPN2 are associated with increased susceptibility to IBD and other inflammatory disorders (Yu et al., 2012). Interestingly, PTPN2 has been shown to dephosphorylate many tyrosine kinases, including members of the Src family (Spalinger et al., 2016). Collectively, these observations suggest that increased activity of tyrosine kinases or reduced tyrosine phosphatase activity may contribute to the pathobiology of intestinal inflammation as observed in IBD.
We observed an epithelial barrier defect when JAM-A Y280 is phosphorylated in vitro. To mechanistically link tyrosine phosphorylation of JAM-A to regulation of permeability, we examined whether tyrosine phosphorylation affects JAM-A association with known signaling partners. Previous studies have shown that Rap2 colocalizes with JAM-A. Further studies indicated the active form of the GTPase Rap2c is associated with JAM-A and promotes barrier formation. Knockdown JAM-A or Rap2c was shown to result in increased paracellular permeability that paralleled decreases in Rap2 activity (Monteiro and Parkos, 2012; Monteiro et al., 2013, 2014). Results from this study suggest that phosphorylation of JAM-A at Y280 induces dissociation of JAM-A and active Rap2. Consistent with these findings, JAM-A association with active Rap2 was disrupted when IECs were either treated with pervanadate or cytokines to induce phosphorylation of JAM-A Y280. Collectively, these findings suggest that phosphorylation of JAM-A Y280-disrupts Rap2 association with JAM-A at TJs resulting in altered epithelial barrier function during inflammatory conditions.
Finally, we examined whether JAM-A Y280 is phosphorylated in normal and inflamed colonic mucosa in vivo. There was barely detectable staining of p-JAM-A Y280 in normal colonic mucosa from control mice, but a robust increase in staining was observed in colonic mucosa from colitic animals. Similar results were observed in normal human colonic mucosa when compared with p-JAM-A Y280 labeling in mucosa from individuals with active UC. These findings are consistent with p-JAM-A Y280 playing an important role in the leaky barrier associated with pathologic intestinal inflammation.
In conclusion, findings in this report are consistent with a model depicted in Figure 7. In this model, activation of Yes-1 by mucosal cytokines during inflammation promotes tyrosine phosphorylation of JAM-A at Y280, destabilizing TJ-associated signaling elements, which results in decreased Rap2 GTPase activity and compromised epithelial barrier. Collectively, these data provide new insights into how phosphorylation of JAM-A regulates epithelial permeability under inflammatory conditions.
FIGURE 7:
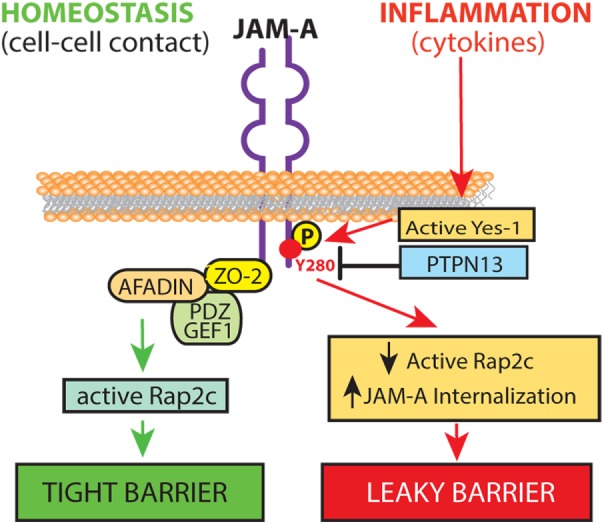
Model of tyrosine phosphorylation of JAM-A regulation of intestinal barrier during inflammation. During homeostasis, JAM-A forms cis- and trans-dimers, localizing mainly to the TJ with associated scaffolding and signaling molecules to form a tight barrier (left). During inflammation, cytokines (IL-22 and IL-17A) activate the Src family kinase Yes-1 resulting in phosphorylation of JAM-A at Y280. Increased phosphorylation of JAM-A Y280 results in dissociation of active Rap2 from TJ with JAM-A and internalization of JAM-A, resulting in a leaky barrier (right). A leaky barrier promotes increased microbial stimulation of mucosal immune cells to produce cytokines that perpetuate activation of Yes-1, tyrosine phosphorylation of JAM-A, and further deterioration of barrier function.
MATERIALS AND METHODS
Cells and human colonoid culture
Most experiments were performed in two intestinal epithelial cell lines: SK CO-15 and T84. SK CO-15 human IECs and HEK 293T were grown in high-glycose DMEM supplemented with 10% fetal bovine serum (FBS), 100 IU of penicillin, 100 μg/ml streptomycin, 2 mM l-glutamine, 15 mM HEPES, pH 7.4, and 1% nonessential amino acids. T84cells were grown in F12/MEM supplemented with 5% of FBS, 100 IU of penicillin, and 100 μg/ml streptomycin. HA-tagged JAM-A full-length plasmid (PcDNA3.1) was transfected with FuGENE 6 (Promega, Madison, WI) for HEK 293T cells and lipofectamine 3000 (Thermo Fisher Scientific, Waltham, MA) for SK CO-15 cells, according to manufacturer protocols.
Generation of human colonoid–derived monolayers: Three-dimensional (3D) colonoids were derived from human colonic resections and maintained in tissue culture as previously reported (Jung et al., 2011; Dame et al., 2014). To create colonoid monolayers, 3D colonoids were separated from matrigel and trypsinized to form a single cell suspension before being seeded on collagen-coated 0.33 cm2 Transwell filters as described previously (Zou et al., 2017). Following 5 d of culture, colonoids had formed two-dimensional polarized monolayers with TEER above 800 Ω cm2.
Antibodies
Murine monoclonal anti–human JAM-A (J10.4) was purified as described previously (Liu et al., 2000). Rabbit anti-p-JAM-A Y280 was from Rockland, Limerick, PA, catalogue: 600-401-GN5. Other antibodies were purchased: goat anti–JAM-A, mouse and goat anti-PTPN13, goat anti–E-cadherin (R & D Systems, Minneapolis, MN); polyclonal rabbit anti–JAM-A, ZO-2 and monoclonal mouse anti–ZO-2 (Thermo Fisher Scientific, Waltham, MA); monoclonal mouse anti–β catenin, mouse anti–Rap-2, Yes-1 (BD Transduction Laboratories, Lexington, KY); polyclonal rabbit anti-Src, Yes-1, Lyn, p-Src Y416, p-EGFR Y1068, p-HER3 Y1289 (Cell Signaling Technology, Beverly, MA); polyclonal rabbit anti-HA, calnexin, and monoclonal mouse anti-HA (Sigma-Aldrich, St. Louis, MO); polyclonal rabbit anti–Fap-1 (Santa Cruz, Dallas, Texas). For immunoblots, horseradish peroxidase–conjugated secondary antibodies were purchased (Jackson Immuno-Research Laboratories, West Grove, PA). For immunofluorescence, FITC and Alexa-conjugated secondary antibodies (Thermo Fisher Scientific, Waltham, MA) were used.
Yes-1 siRNA
Subconfluent SK CO-15 cells were transfected with Yes-1 siRNA (Sigma-Aldrich, St. Louis, MO) using lipofectamine RNAiMAX reagent (Thermo Fisher Scientific, Waltham, MA). Two days later, monolayers were harvested or fixed and knockdown verified by immunoblot and immunofluorescence. Because p-JAM-A Y280 levels were very low in normal IECs, some monolayers were treated with pervanadate (25 μM) to induce tyrosine phosphorylation.
DSS colitis
Cyclic exposure of 8-wk-old C57BL6 mice to drinking water with DSS followed by water alone was performed following standard protocols under guidelines approved by the University of Michigan Animal Care and Use Committee. Briefly, mice were allowed free access to food and drinking water containing 2.5% (wt/vol) DSS for 4 d, followed by a 5-d recovery without DSS for a total of three cycles of DSS and recovery before being killed. Daily disease activity was monitored as previously described (Laukoetter et al., 2007).
Immunoblots
After experimental treatments, confluent monolayers of IECs were homogenized in RIPA lysis buffer (10 mM Tris-HCl, pH 8.0, 140 mM NaCl, 1 mM EDTA, 1% Triton X-100, 0.1% sodium deoxycholate, 0.1% SDS, pH 7.4) or Triton lysis buffer (50 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 1 mM ethylene glycol-bis (b-aminoethyl ether)-N,N,N’,N’-tetraacetic acid [EGTA], 1% Triton X-100, 10% glycerol) supplemented with a cocktail of protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO). Lysates were cleared by centrifugation and protein concentrations determined by DC Protein Assay (Bio-Raid, Hercules, CA) using an Epoch microplate spectrophotometer (BioTek, Winooski, VT). Samples were boiled in sample buffer followed by SDS–PAGE and immunoblotting by standard methods. Calnexin was served as a protein loading control.
Immunoprecipitation
SK CO-15 or HEK 293T cells were harvested after 48 h of transfection in Triton lysis buffer (50 mM HEPES, 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 1% Triton X-100, 10% glycerol) supplemented with a cocktail of protease and phosphatase inhibitors (Sigma-Aldrich, St. Louis, MO). Lysates were incubated on ice for 20 min before centrifugation at 16,000 × g for 20 min. Collected supernatants were precleared with 50 μl of 50% protein A or G agarose beads for 1 h followed by incubation with rotation overnight in the presence of 5 μg/ml antibodies or control immunoglobulin G at 4°C. Immune complexes were precipitated by 50 μl of protein A or G for 2–4 h. Immunoprecipitated complexes were washed three times with Triton lysis buffer or HNTG buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 0.1% Triton X-100, and 10% glycerol) before being boiled in 2X LDS sample buffer. 2% lysate and immnunoprecipitates were then subjected to SDS–PAGE and immunoblotting.
Immunofluorescence and immunohistochemistry
IECs were grown on Transwell filters and fixed with 4% paraformaldehyde (PFA). For staining of most TJ proteins, fixed monolayers were permeabilized with 1% SDS for 10 min. In subsets of experiments, to visualize adhesion junction and intracellular components, fixed monolayers were permeabilized with 0.5% Triton X-100 instead of SDS for 10 min. Monolayers were blocked with 2–3% goat or donkey serum in PBS with 0.05% Tween 20 (TBST) for 30 min. Primary antibodies were diluted in block buffer, and cells were incubated overnight at 4°C. Cells were washed with PBST and fluorescently labeled secondary antibodies were diluted in blocking buffer followed by incubation for 1 h at room temperature. Washed monolayers were mounted in Prolong Gold anti-fade agent (Thermo Fisher Scientific). Samples of human colonic mucosa from individuals with UC or normal mucosa from colon cancer resections were collected anonymously under approved human subject guidelines at the University of Michigan. Human mucosal samples as well as colonic sections from mice with DSS colitis were fixed with 10% formaldehyde followed by paraffin embedding and sectioned (7–10 μm) onto glass slides. Paraffin sections were prepared for staining by deparaffinizing in xylene and rehydrating in ethanol. Antigen retrieval was performed in antigen unmasking buffer (Citra buffer; Vector Laboratories, Burlingame, CA). Tissue sections were prepared for immunofluorescence as described above for cellular staining. Fluorescence imaging was performed on a Nikon A1 confocal microscope (Nikon, Tokyo, Japan) in the Microscopy and Image Analysis Laboratory core of the University of Michigan.
Dephosphorylation of p-JAM-A Y280 by human PTPN13
Pervanadate-treated T84 cell lysates (50 μl) in modified Triton X-100 lysis buffer (50 mM HEPES, 2 mM EDTA, 3 mM dithiothreitol, 100 mM NaCl, and 1% Triton X-100) were incubated with 1–3 μg of active human PTPN13 fragment (Abcam, Cambridge, MA) for 10 min at 30°C, mixed with SDS sample buffer, and then subjected to SDS–PAGE and immunoblotting.
Rap2 activity assay
Rap2 activity assays were performed according to the manufacturer’s instructions (Cell BioLabs, San Diego, CA). Briefly, SK CO-15 were incubated with Hank’s balanced salt solution (HBSS) containing 25 μM pervanadate for 1 h before cells were lysed in kit-supplied buffer (25 mM HEPES, pH 7.5, 150 mM NaCl, 1% NP-40, 10 mM MaCl2, 1 mM EDTA, 2% glycerol). Nontreated SK CO-15 cells were lysed as controls. Cell lysates were incubated with 40 μl of RalGDS beads for 4 h at 4°C. The RalGDS beads were washed and boiled with 2X SDS sample buffer. Entire samples were then analyzed by SDS–PAGE and immunoblot.
Permeability assay
Cells were grown on 1.12 cm2, 3-μm-pore Transwell filters to confluence. TEER-to-passive ion flow was measured using an EVOMX voltmeter (World Precision Instruments, Sarasota, FL). Cytokines (TNFα, IFNγ, IL-22, IL-17A; R&D Systems, Minneapolis, MN) were added to cell culture media in both the top and bottom chambers of the Transwell filter once TEER reached ∼1000 Ω cm2 at specific time points per experimental design. TEER was recorded in response to cytokine treatments. For FITC dextran flux experiments, confluent monolayers of epithelial cells were grown on Transwell filters until TEER reached 1000 Ω cm2, and then cells were treated with cytokines at designated time periods. After cells were washed with HBSS+, 4 kD FITC-conjugated dextran (Sigma-Aldrich, St. Louis, MO) was placed on the top chamber of the Transwell filter and incubated for 1 h at 37°C. Samples from the bottom chamber of the Transwell filter were collected and the fluorescence intensity measured with a Cytation 5 imaging reader (BioTek, Winooski, VT).
Image preparation and statistical analyses
Image analyses were performed using ImageJ (National Institutes of Health, Bethesda, MD).
Statistical analyses were performed in Microsoft Excel (Microsoft, Redmond, WA) and GraphPad Prism (GraphPad, La Jolla, CA). Densitometric values obtained from immunoblots were normalized to band density of calnexin loading and nontreated controls. Data presented are from three individual experiments, and bar graphs represent mean ± SEM and statistical significance assessed by Student’s t test (independent samples, equal variance, two-tailed) and analysis of variance (ANOVA), where a P value of less than 0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
We thank Miguel Quiros Quesada, Monique O’Leary, and Mark Yulis for helpful comments and Robin Kunkel and Lynn McCain for assistance in manuscript and figures preparation. We acknowledge the Microscope and Image Analysis Laboratory of the University of Michigan for confocal image support, the Emory Integrated Genomics Core for cloning support, and Rockland Immunochemicals for design and production of anti–p-JAM-A Y280 antibodies. This work was supported by National Institutes of Health Grants no. DK-061379 and DK-079392 (C.A.P.) and DK-059888 and DK-055679 (A.N.) as well as a Joy Cappel Rockland Immunochemical grant for phospho-JAM-A antibody development (to C.W.).
Abbreviations used:
- CIP
calf intestinal phosphatase
- CTL
untreated cells
- DSS
dextran sulfate sodium
- EGTA
ethylene glycol-bis (b-aminoethyl ether)- N, N, N’, N’-tetraacetic acid
- FITC
fluorescein isothiocyanate
- GST-Rap2 wt
GST-fused Rap2c full-length recombinant protein
- HBSS
Hanks’ balanced salt solution
- HEK 293T
human embryonic kidney 293 cells with SV40 large T antigen mutant
- IBD
inflammatory bowel disease
- IECs
intestinal epithelial cells
- IFNg
interferon gamma
- IL-17A
interleukin-17A
- IL-22
interleukin-22
- JAM-A
junctional adhesion molecule-A
- MARK
mitogen-activated protein kinase
- MDCK
Madin–Darby canine kidney
- MUPP1
multi-PDZ domain protein 1
- PDZ
postsynaptic density protein (PSD95), Drosophila disc large tumor suppressor (Dlg1), and zonula occludens-1 protein (ZO-1)
- p-JAM-A Y280
phosphorylation of JAM-A at Y280
- PKCζ
protein kinase Cζ
- PP1
protein phosphatase 1
- PP2
Src family kinase inhibitor
- PP2A
protein phosphatase 2A
- PTPN13
tyrosine-protein phosphatase non-receptor type 13
- Rap2
Ras-related protein Rap2
- siRNA
small interfering RNA
- SNPs
single nucleotide polymorphisms
- Src
proto-oncogene tyrosine-protein kinase src
- TEER
transepithelial electrical resistance
- TJ
tight junction
- TNFα
tumor necrosis factor alpha
- UC
ulcerative colitis
- Yes-1
proto-oncogene tyrosine-protein kinase Yes
- ZO-1
zonula occludens-1
- ZO-2
zonula occludens-2
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E18-08-0531) on January 9, 2019.
REFERENCES
- Al-Sadi R, Guo S, Ye D, Rawat M, Ma TY. (2016). TNF-α modulation of intestinal tight junction permeability is mediated by NIK/IKK-α axis activation of the canonical NF-κB pathway. Am J Pathol , 1151–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson JM, Van Itallie CM. (1995). Tight junctions and the molecular basis for regulation of paracellular permeability. Am J Physiol , G467–G475. [DOI] [PubMed] [Google Scholar]
- Anderson JM, Van Itallie CM, Fanning AS. (2004). Setting up a selective barrier at the apical junction complex. Curr Opin Cell Biol , 140–145. [DOI] [PubMed] [Google Scholar]
- Basuroy S, Seth A, Elias B, Naren AP, Rao R. (2006). MAPK interacts with occludin and mediates EGF-induced prevention of tight junction disruption by hydrogen peroxide. Biochem J , 69–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basuroy S, Sheth P, Kuppuswamy D, Balasubramanian S, Ray RM, Rao RK. (2003). Expression of kinase-inactive c-Src delays oxidative stress-induced disassembly and accelerates calcium-mediated reassembly of tight junctions in the Caco-2 cell monolayer. J Biol Chem , 11916–11924. [DOI] [PubMed] [Google Scholar]
- Bazzoni G, Martinez-Estrada OM, Orsenigo F, Cordenonsi M, Citi S, Dejana E. (2000). Interaction of junctional adhesion molecule with the tight junction components ZO-1, cingulin, and occludin. J Biol Chem , 20520–20526. [DOI] [PubMed] [Google Scholar]
- Bruewer M, Luegering A, Kucharzik T, Parkos CA, Madara JL, Hopkins AM, Nusrat A. (2003). Proinflammatory cytokines disrupt epithelial barrier function by apoptosis-independent mechanisms. J Immunol , 6164–6172. [DOI] [PubMed] [Google Scholar]
- Capaldo CT, Farkas AE, Hilgarth RS, Krug SM, Wolf MF, Benedik JK, Fromm M, Koval M, Parkos C, Nusrat A. (2014). Proinflammatory cytokine-induced tight junction remodeling through dynamic self-assembly of claudins. Mol Biol Cell , 2710–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caraballo JC, Yshii C, Butti ML, Westphal W, Borcherding JA, Allamargot C, Comellas AP. (2011). Hypoxia increases transepithelial electrical conductance and reduces occludin at the plasma membrane in alveolar epithelial cells via PKC-ζ and PP2A pathway. Am J Physiol Lung Cell Mol Physiol , L569–L578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi W, Yeruva S, Turner JR. (2017). Contributions of intestinal epithelial barriers to health and disease. Exp Cell Res , 71–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dame MK, Jiang Y, Appelman HD, Copley KD, McClintock SD, Aslam MN, Attili D, Elmunzer BJ, Brenner DE, Varani J, et al. (2014). Human colonic crypts in culture: segregation of immunochemical markers in normal versus adenoma-derived. Lab Invest , 222–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Beste KA, Hoddeson EK, Parkos CA, Nusrat A, Wise SK. (2013). Epithelial permeability alterations in an in vitro air-liquid interface model of allergic fungal rhinosinusitis. Int Forum Allergy Rhinol , 19–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elias BC, Suzuki T, Seth A, Giorgianni F, Kale G, Shen L, Turner JR, Naren A, Desiderio DM, Rao R. (2009). Phosphorylation of Tyr-398 and Tyr-402 in occludin prevents its interaction with ZO-1 and destabilizes its assembly at the tight junctions. J Biol Chem , 1559–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng Y, Teitelbaum DH. (2013). Tumour necrosis factor–induced loss of intestinal barrier function requires TNFR1 and TNFR2 signalling in a mouse model of total parenteral nutrition. J Physiol , 3709–3723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamazaki Y, Itoh M, Sasaki H, Furuse M, Tsukita S. (2002). Multi-PDZ domain protein 1 (MUPP1) is concentrated at tight junctions through its possible interaction with claudin-1 and junctional adhesion molecule. J Biol Chem , 455–461. [DOI] [PubMed] [Google Scholar]
- He J, Bellini M, Inuzuka H, Xu J, Xiong Y, Yang X, Castleberry AM, Hall RA. (2006). Proteomic analysis of β1-adrenergic receptor interactions with PDZ scaffold proteins. J Biol Chem , 2820–2827. [DOI] [PubMed] [Google Scholar]
- Iden S, Misselwitz S, Peddibhotla SS, Tuncay H, Rehder D, Gerke V, Robenek H, Suzuki A, Ebnet K. (2012). aPKC phosphorylates JAM-A at Ser285 to promote cell contact maturation and tight junction formation. J Cell Biol , 623–639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang W, Su J, Zhang X, Cheng X, Zhou J, Shi R, Zhang H. (2014). Elevated levels of Th17 cells and Th17-related cytokines are associated with disease activity in patients with inflammatory bowel disease. Inflamm Res , 943–950. [DOI] [PubMed] [Google Scholar]
- Jung P, Sato T, Merlos-Suarez A, Barriga FM, Iglesias M, Rossell D, Auer H, Gallardo M, Blasco MA, Sancho E, et al. (2011). Isolation and in vitro expansion of human colonic stem cells. Nat Med , 1225–1227. [DOI] [PubMed] [Google Scholar]
- Kale G, Naren AP, Sheth P, Rao RK. (2003). Tyrosine phosphorylation of occludin attenuates its interactions with ZO-1, ZO-2, and ZO-3. Biochem Biophys Res Commun , 324–329. [DOI] [PubMed] [Google Scholar]
- Kucharzik T, Walsh SV, Chen J, Parkos CA, Nusrat A. (2001). Neutrophil transmigration in inflammatory bowel disease is associated with differential expression of epithelial intercellular junction proteins. Am J Pathol , 2001–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laukoetter MG, Nava P, Lee WY, Severson EA, Capaldo CT, Babbin BA, Williams IR, Koval M, Peatman E, Campbell JA, et al. (2007). JAM-A regulates permeability and inflammation in the intestine in vivo. J Exp Med , 3067–3076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li LJ, Gong C, Zhao MH, Feng BS. (2014). Role of interleukin-22 in inflammatory bowel disease. World J Gastroenterol , 18177–18188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Nusrat A, Schnell FJ, Reaves TA, Walsh S, Pochet M, Parkos CA. (2000). Human junction adhesion molecule regulates tight junction resealing in epithelia. J Cell Sci (Pt 13), 2363–2374. [DOI] [PubMed] [Google Scholar]
- Luissint AC, Nusrat A, Parkos CA. (2014). JAM-related proteins in mucosal homeostasis and inflammation. Semin Immunopathol , 211–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luissint AC, Parkos CA, Nusrat A. (2016). Inflammation and the intestinal barrier: leukocyte-epithelial cell interactions, cell junction remodeling, and mucosal repair. Gastroenterology , 616–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma TY, Iwamoto GK, Hoa NT, Akotia V, Pedram A, Boivin MA, Said HM. (2004). TNF-α-induced increase in intestinal epithelial tight junction permeability requires NF-κB activation. Am J Physiol Gastrointest Liver Physiol , G367–G376. [DOI] [PubMed] [Google Scholar]
- Mandell KJ, Babbin BA, Nusrat A, Parkos CA. (2005). Junctional adhesion molecule 1 regulates epithelial cell morphology through effects on β1 integrins and Rap1 activity. J Biol Chem , 11665–11674. [DOI] [PubMed] [Google Scholar]
- Mandell KJ, Berglin L, Severson EA, Edelhauser HF, Parkos CA. (2007). Expression of JAM-A in the human corneal endothelium and retinal pigment epithelium: localization and evidence for role in barrier function. Invest Ophthalmol Vis Sci , 3928–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer TN, Schwesinger C, Ye J, Denker BM, Nigam SK. (2001). Reassembly of the tight junction after oxidative stress depends on tyrosine kinase activity. J Biol Chem , 22048–22055. [DOI] [PubMed] [Google Scholar]
- Mitchell LA, Ward C, Kwon M, Mitchell PO, Quintero DA, Nusrat A, Parkos CA, Koval M. (2015). Junctional adhesion molecule A promotes epithelial tight junction assembly to augment lung barrier function. Am J Pathol , 372–386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro AC, Luissint AC, Sumagin R, Lai C, Vielmuth F, Wolf MF, Laur O, Reiss K, Spindler V, Stehle T, et al. (2014). Trans-dimerization of JAM-A regulates Rap2 and is mediated by a domain that is distinct from the cis-dimerization interface. Mol Biol Cell , 1574–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro AC, Parkos CA. (2012). Intracellular mediators of JAM-A-dependent epithelial barrier function. Ann NY Acad Sci , 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro AC, Sumagin R, Rankin CR, Leoni G, Mina MJ, Reiter DM, Stehle T, Dermody TS, Schaefer SA, Hall RA, et al. (2013). JAM-A associates with ZO-2, afadin, and PDZ-GEF1 to activate Rap2c and regulate epithelial barrier function. Mol Biol Cell , 2849–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murchie R, Guo CH, Persaud A, Muise A, Rotin D. (2014). Protein tyrosine phosphatase sigma targets apical junction complex proteins in the intestine and regulates epithelial permeability. Proc Natl Acad Sci USA , 693–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naik MU, Caplan JL, Naik UP. (2014). Junctional adhesion molecule-A suppresses platelet integrin αIIbβ3 signaling by recruiting Csk to the integrin-c-Src complex. Blood , 1393–1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nava P, Capaldo CT, Koch S, Kolegraff K, Rankin CR, Farkas AE, Feasel ME, Li L, Addis C, Parkos CA, et al. (2011). JAM-A regulates epithelial proliferation through Akt/β-catenin signalling. EMBO Rep , 314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nomme J, Fanning AS, Caffrey M, Lye MF, Anderson JM, Lavie A. (2011). The Src homology 3 domain is required for junctional adhesion molecule binding to the third PDZ domain of the scaffolding protein ZO-1. J Biol Chem , 43352–43360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pandita R, Pocsik E, Aggarwal BB. (1992). Interferon-γ induces cell surface expression for both types of tumor necrosis factor receptors. FEBS Lett , 87–90. [DOI] [PubMed] [Google Scholar]
- Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, Liu Y, Smith T, Neish AS, Li H, Tan S, et al. (2016). Loss of junctional adhesion molecule A promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology , 733–746. e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sallee JL, Burridge K. (2009). Density-enhanced phosphatase 1 regulates phosphorylation of tight junction proteins and enhances barrier function of epithelial cells. J Biol Chem , 14997–15006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharl M, Paul G, Barrett KE, McCole DF. (2009). AMP-activated protein kinase mediates the interferon-γ-induced decrease in intestinal epithelial barrier function. J Biol Chem , 27952–27963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seth A, Sheth P, Elias BC, Rao R. (2007). Protein phosphatases 2A and 1 interact with occludin and negatively regulate the assembly of tight junctions in the CACO-2 cell monolayer. J Biol Chem , 11487–11498. [DOI] [PubMed] [Google Scholar]
- Smyth D, Phan V, Wang A, McKay DM. (2011). Interferon-γ-induced increases in intestinal epithelial macromolecular permeability requires the Src kinase Fyn. Lab Invest , 764–777. [DOI] [PubMed] [Google Scholar]
- Spalinger MR, McCole DF, Rogler G, Scharl M. (2016). Protein tyrosine phosphatase non-receptor type 2 and inflammatory bowel disease. World J Gastroenterol , 1034–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K, Ogawa A, Mizoguchi E, Shimomura Y, Andoh A, Bhan AK, Blumberg RS, Xavier RJ, Mizoguchi A. (2008). IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J Clin Invest , 534–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukita S, Furuse M, Itoh M. (2001). Multifunctional strands in tight junctions. Nat Rev Mol Cell Biol , 285–293. [DOI] [PubMed] [Google Scholar]
- Utech M, Ivanov AI, Samarin SN, Bruewer M, Turner JR, Mrsny RJ, Parkos CA, Nusrat A. (2005). Mechanism of IFN-γ-induced endocytosis of tight junction proteins: myosin II-dependent vacuolarization of the apical plasma membrane. Mol Biol Cell , 5040–5052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Itallie CM, Anderson JM. (2018). Phosphorylation of tight junction transmembrane proteins: many sites, much to do. Tissue Barriers , e1382671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Mumm JB, Herbst R, Kolbeck R, Wang Y. (2017). IL-22 increases permeability of intestinal epithelial tight junctions by enhancing claudin-2 expression. J Immunol , 3316–3325. [DOI] [PubMed] [Google Scholar]
- Wise SK, Laury AM, Katz EH, Den Beste KA, Parkos CA, Nusrat A. (2014). Interleukin-4 and interleukin-13 compromise the sinonasal epithelial barrier and perturb intercellular junction protein expression. Int Forum Allergy Rhinol , 361–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Sundrud MS, Skepner J, Yamagata T. (2014). Targeting Th17 cells in autoimmune diseases. Trends Pharmacol Sci , 493–500. [DOI] [PubMed] [Google Scholar]
- Yu W, Hegarty JP, Berg A, Kelly AA, Wang Y, Poritz LS, Franke A, Schreiber S, Koltun WA, Lin Z. (2012). PTPN2 is associated with Crohn’s disease and its expression is regulated by NKX2–3. Dis Markers , 83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu JH, Chen R, Yi W, Cantin GT, Fearns C, Yang Y, Yates JR, 3rd, Lee JD. (2008). Protein tyrosine phosphatase PTPN13 negatively regulates Her2/ErbB2 malignant signaling. Oncogene , 2525–2531. [DOI] [PubMed] [Google Scholar]
- Zou WY, Blutt SE, Crawford SE, Ettayebi K, Zeng XL, Saxena K, Ramani S, Karandikar UC, Zachos NC, Estes MK. (2017). Human intestinal enteroids: new models to study gastrointestinal virus infections. Methods Mol Biol, doi: 10.1007/7651_2017_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.