

Abstract
Ribosomal frameshifting, a process whereby a translating ribosome is diverted from one reading frame to another on a contiguous mRNA, is an important regulatory mechanism in biology and an opportunity for therapeutic intervention in several human diseases. In HIV, ribosomal frameshifting controls the ratio of Gag and Gag-Pol, two polyproteins critical to the HIV life cycle. We have previously reported compounds able to selectively bind an RNA stemloop within the Gag-Pol mRNA; these compounds alter the production of Gag-Pol in a manner consistent with increased frameshifting. Importantly, they also display antiretroviral activity in human T-cells. Here, we describe new compounds with significantly reduced molecular weight, but with substantially maintained affinity and anti-HIV activity. These results suggest that development of more “ligand efficient” enhancers of ribosomal frameshifting is an achievable goal.
Graphical Abstract
Introduction:
The increasing recognition of RNA as an important player in human health and disease, and likewise its expanding status as a target for therapeutic intervention, stems from emerging discoveries of RNA’s role in regulating – or dysregulating – specific biological processes. Two examples of these processes include the direct action of micro RNAs (miRNA) on gene expression,1 and the ability of RNA repeat expansions to interfere with mRNA splicing in the myotonic dystrophies. 2 Both of these, along with other biomedically relevant RNA sequences, have been the subject of considerable effort by the RNA recognition community.3
Another, less-studied area of RNA influence with substantial potential importance is that of protein recoding, or “ribosomal frameshifting”.4 In this process, a ribosome progressing along mRNA is induced to shift forward or (most commonly) backward by one or two nucleotides out of its original reading frame. This generally occurs as it is transiting a pyrimidine-rich sequence of bases known as a “slippery sequence”. When the ribosome resumes translation, it is now in a new reading frame leading to production of either a defective protein (due to changes to the C-terminus), or an entirely new protein product.
Frameshifting is typically stimulated by a stable RNA secondary structure such as a stemloop or pseudoknot downstream of the slippery sequence. Other frameshift-inducing mechanisms are possible, however, as evidenced by a recent report indicating that −1 ribosomal frameshifting may be stimulated in trans by a protein in the life cycle of encephalomyocarditis virus.5 While the majority of frameshifting processes studied to date are in viruses and bacteria, emerging research indicates that this can also occur in humans. For example, a miRNA-regulated −1 ribosomal frameshift in the human CCR5 mRNA leads to production of non-functional CCR5, and degradation of the CCR5 mRNA via the nonsense-mediated decay (NMD) pathway.6 A human homolog of a bacterial gene producing both a copper transporter and copper chaperone from the same gene via −1 ribosomal frameshifting has also been reported.7
In the human immunodeficiency virus type 1 (HIV-1), −1 ribosomal frameshifting is used to determine the relative ratio of two polyproteins (Gag and Gag-Pol).8 Gag, a 55 kDa polyprotein, is produced with 90-95% frequency during translation of the Gag-Pol mRNA. Gag is subsequently proteolytically cleaved to yield HIV-1 structural proteins including Matrix, Capsid, and Nucleocapsid proteins. A −1 Programmed Ribosomal Frameshift (PRF) occurring 5-10% of the time allows read-through of the Gag stop codon, leading to production of Gag-Pol. This 160 kDa polyprotein is also processed posttranslation to yield structural proteins and essential HIV-1 enzymes (including Protease, Integrase, and Reverse Transcriptase). Early work using cotransfection of plasmids encoding Gag and Gag-Pol revealed that deviation from the normal ratio resulted in a drastic decrease in the infectivity of virus produced.9 The −1 PRF process in HIV-1 is believed to be regulated by a highly conserved, stable stemloop structure downstream of the UUUUUUA “slippery sequence” (Figure 1). This structure is known as the HIV-1 Frameshift Stimulatory Sequence, or HIV-1 FSS. While the precise mechanism by which the HIV-1 FSS exerts its influence is unknown, extensive mutagenesis experiments have confirmed its importance to the process.10
Figure 1:

The frameshift region of HIV-1 Gag-Pol mRNA. The “slippery sequence” (red) and frameshift-stimulating stem loop (green) are highlighted.
The possibility of using small molecules to influence frameshifting in HIV as a therapeutic strategy was recognized in 1998,11 although it was subsequently determined that the anti-HIV activity of the benzamidine derivative tested in that work was unrelated to frameshifting.12 Our group initially used a fragment-based in situ library synthesis and screening approach called Resin-Bound Dynamic Combinatorial Chemistry (RBDCC)13 to facilitate discovery of compounds able to bind the HIV-1 FSS. Disulfide 1 was identified as the primary hit in the screen, with low micromolar affinity for the FSS as measured by surface plasmon resonance (SPR).14 In subsequent efforts, we tested several structural hypotheses in an effort to enhance FSS binding affinity and selectivity, and to replace the labile disulfide moiety of 1 with bioisosteres suitable for assay in cells. Notable steps in this process included replacement of the disulfide with an olefin bioisostere,15 extension of the quinoline pi-surface to a benzo[g]quinoline (for example, structure 2),16 incorporation of amide N-methylation to enhance biostability and conformationally bias structures for RNA binding,17 and use of a triazole bioisostere (Compound 3).18 Compounds 2 and 3 were found to have affinities (Kd) of 102 and 16 nM, respectively, as measured by SPR. Both compounds inhibited both production and infectivity of pseudotyped HIV. Western blots of virus produced in the presence of either compound revealed concentration-dependent increases in p160 Gag-Pol consistent with frameshifting. Importantly, 3 inhibited both laboratory (HIV-1 IIIB) and patient-derived multidrug resistant (HIV-1 AD.MDR01)19 strains of HIV.
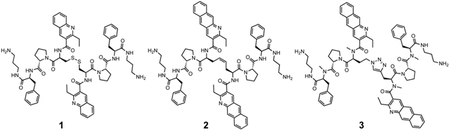
While these results provided strong validation of the HIV-1 FSS as an antiviral target, as well as our ability to develop compounds addressing this target, the disconnect between the nanomolar affinities and micromolar inhibitory potencies of compounds was a concern. Our work to date has involved a stepwise process of compound modification to improve RNA-binding affinity, selectivity, and anti-HIV activity. There is no guarantee that such a linear process produces the “best” compound. However, it is clear that structures such as 3 have some undesirable characteristics (including high molecular weight, potential intercalators, and several charged groups). As these might be contributors to the affinity/potency difference, we synthesized and tested a series of molecules retaining modifications that had the most favorable impacts (including replacement of the disulfide with a triazole bioisostere and amide N-methylation), while jettisoning “undesirable” functional groups (Figure 2). We also tested the hypothesis that increasing solubility without adding charge might also enhance affinity. The physical properties of compounds designed to address these goals represent a broad range of cLogP values20 (from −3.44 to +3.94), bracketing the 2.63 cLogP of compound 3. All have significantly reduced molecular weight vs. 2 and 3.
Figure 2:

Compounds synthesized for this work
Materials and Methods:
Synthesis of Compounds
Compounds were synthesized using standard solid-phase peptide protocols. Wang Resin (0.22 mmol/gram for alkyne containing monomers, 0.68 mmol/gram for azide containing monomers) was activated with carbonyl diimidazole (CDI, 10 eq.) in dimethylformamide (DMF) overnight followed by treatment with 1,3-diaminopropane (10 eq.) in DMF overnight. Amino acids were coupled using 3.1 eq. of Fmoc-AA-OH, where AA is Phe, Pro, Hyp(tBu) or N-Me-Phe; 2.9 eq. of HATU, and 5 eq. of DIPEA in DMF for 2 h. For coupling of Fmoc-AA-OH = Pra, Aha: 2 eq. of Fmoc-AA-OH, 1.93 eq. of HATU and 3.33 eq. of DIPEA were utilized. Deprotection of each Fmoc before each Fmoc-AA-OH coupling, was accomplished using 20% piperidine in DMF for 30 minutes. Where N-Me amino acids were needed that were not commercially available, Fmoc was first removed using standard methods followed by treatment of the resin with 4 eq. of 2-nitrobenzenesulfonyl chloride (NOSYL-Cl) and 10 eq. of collidine in DMF for 2 h. This activated resin was then treated with 20 eq. of TMS-diazomethane and 0.5 mL of methanol in DCM for 48 hours. Methylation was monitored by HPLC and MALDI-MS. To remove the NOSYL group after methylation completion, the resin was treated with 10 eq. of 2-mercaptoethanol and 5 eq. of DBU for 2 hrs. The end of each monomer was capped with quinoline or 2-ethyl quinoline. Quinoline-3-carboxylic acid or 2-ethyl quinoline-3-carboxylic acid, 3 eq., 2.9 eq. of HATU, 5 eq. of DIPEA were coupled in DMF overnight. To form the 1,4-substituted triazole ring, the azide-containing peptide was cleaved with 98% trifuoroacetic acid (TFA), 1% triethyl silane (TES), and 1% water. A crude solid was precipitated by cold ether and was used without further purification. Next, the resin-bound alkyne was dried under a vacuum and washed three times with dry THF. CuI (2 eq.) and DIPEA (50 eq.) were added to the vessel in anhydrous THF. After mixing for 15 min, azide (2 eq. based on resin loading) was added, and the resulting green/brown solution mixed overnight. The resin was then washed three times with THF, three times with DCM, three times with DMF, and three times with DCM. It was then cleaved with 98% TFA, 1% TES, and 1% water for 2 hrs. followed by rotary evaporation and then cold ether precipitation. Raw material was dissolved in water, and the final material was obtained using reverse phase preparative HPLC. The final products were characterized by high resolution mass spectrometry at the Chemistry Instrumentation Center of the University at Buffalo (Supplementary Information).
Surface Plasmon Resonance:
All SPR experiments were performed using a Biacore X (Biacore, Inc.) instrument. Gold sensor chips (CM5), coated with carboxymethylated dextran, were purchased from GE Life Sciences. A mixture of EDC and NHS was injected over both flow cells to make the surface amine-reactive. Streptavidin was then flowed through both flow cells, and ethanolamine was next used to deactivate the surface. Biotinylated HIV-1 FSS RNA (from Integrated DNA Technologies, Inc.) was then flowed through a single flow cell. Aliquots of RNA were refolded at 75 °C for 3 minutes in HBS buffer (10 mM HEPES, 150 mM NaCl, 3 mM EDTA, 0.005% Tween 20, pH 7.4) and allowed to cool before injection. Over these steps, 9000 RU of streptavidin per flow cell, and 2000 RU of RNA were immobilized. Compounds were injected at 80 μL/min for one minute in the HBS buffer described above. At least two injections were performed for each concentration. Each SPR curve was fit individually to a 1:1 Langmuir binding model. The two lowest compound concentrations with enough response to be fit reliably were utilized for final fitting and Kd determination. The average of the kinetic constants was reported. For neomycin, its on- and off-rates for the HIV-1 FSS RNA were found to be too rapid to permit kinetic analysis. Thus, its affinity was determined by steady-state methods. Injections of 6 concentrations ranging from 1-750 μM of neomycin were performed. The steady-state Kd was then determined by fitting the SPR curves with a 4-parameter logistic fit.
Competition Dialysis
Competition dialysis experiments were performed by analogy to methods reported in the literature.21 RNAs were refolded at 2.5 μM concentration in PBS buffer containing 1 mM EDTA in a thermocycler (3 minutes at 25 °C, 3 minutes at 90 °C, 5 minutes at 27°C). Dialysis tubes (MW cutoff = 3500) containing each RNA were prepared in duplicate. Each tube contained 200 μL of RNA (2.5 μM). The compound was dissolved in 180 mL of PBS buffer to yield a 2.0 μM solution of dialysate. The dialysis tubes containing RNA, and a control tube lacking RNA, were then dialyzed against this solution for 36 hours, a time determined in preliminary experiments to be sufficient for full equilibration.
After completing dialysis, 180 μL of solution from each dialysis tube was transferred into a 1 mL micro centrifuge tube. To each sample, 20 μL Triton X-100 (10% solution) was added to give a final volume of 200 μL (1%Triton X-100) to dissociate bound compound from RNA. The samples were then transferred into vials for quantitative analysis by HPLC, with comparison to a calibration curve. Data collected from HPLC was used to calculate the total concentration of compound in the dialysis tubes.
WST-1 Cytotoxicity Assay
HEK293FT cells were plated at 2.5 × 104 cells/well in triplicate in a 96-well plate in DMEM with 10% fetal bovine serum and 1% penicillin-streptomycin at 37 °C. After allowing cells to adhere to the plate for 4 hours, dilutions of compounds were added to wells. Cells were incubated with compound dilutions for 24 hours. A 10 μL solution of WST-1 premix (Clontech) was added and incubated for 1 h, followed by measurement using a PerkinElmer EnSpire plate reader according to the manufacturer’s protocol.
Effect of Compounds on Replication-Competent HIV IIIB
MT-2 cells were obtained from the NIH AIDS Reagent Program and grown in RPMI 1640 media supplemented with 10% fetal bovine serum. Cells were seeded in a T-25 cell culture flask at 6 × 106 cells/10 mL and were infected with HIVIIIB at a concentration of 32.41 ng p24/mL (as calibrated by p24 ELISA). Infection was allowed to proceed for 8 h in a humidified 37 °C incubator. Cells were then collected, centrifugated at 250 x g for 5 min, and were washed three times with PBS. Cells were then resuspended in fresh supplemented RPMI media and plated in a 48-well culture dish at 1 × 105 cells/well. Compounds were added to cells in triplicate to final concentrations of 25, 10, or 2.5 μM, such that the total volume was brought to 0.6 mL/well. On days 2 and 4 post-infection, 0.3 mL of supernatant was collected from each well and replaced with 0.3 mL of fresh media ± compound of the corresponding concentrations given above. On day 6 post-infection, 0.3 mL of supernatant was collected. All collected supernatants were frozen at −80 °C until subsequent analysis of viral load via p24 ELISA according to the manufacturer’s instructions (Advanced Bioscience Laboratories). ELISA plates were read on a DTX880 Mulitmode Detector (Beckman Coulter).
Results:
Compounds 4 – 11 were synthesized using methods previously described.17,18 Following purification, the affinity of each compound for the HIV-1 FSS RNA was tested by SPR (Table 2). We were gratified to observe that compound 6 was only 4-fold lower in affinity for the FSS than 3, despite its substantially reduced molecular weight. Differences in Kd between all four aminopropane-bearing compounds (4 – 7) were statistically insignificant. We had anticipated that compounds 5 and 7 would have higher affinity as their hydroxyproline residues could potentially hydrogen bond to the RNA. If that occurs, however, it may be counterbalanced by the known difference in hydroxyproline ring pucker relative to proline (a conformational effect shown to be important in the collagen triple helix) 22,23 The difference in Kd between compounds 4 – 7 and compounds lacking charged amines (8 – 11) is much more striking. These compounds were poorly soluble in buffer, and binding constants could only be measured for 8 and 10. Compound 8 was found to have an affinity of roughly 19 micromolar for the HIV-1 FSS, while compound 10 bound with a Kd of 1.47 micromolar. This 23-fold difference in Kd between 10 and 6 highlights the importance of charged groups in the binding process.
Binding selectivity was evaluated relative to bulk yeast tRNA in a competitive SPR assay. As an abundant cellular RNA, tRNA has commonly been employed as a “first pass” test for selectivity.24 We first validated this assay using neomycin, a well-studied, relatively nonselective RNA-binding natural product.25 Neomycin binds the FSS with rapid kinetics (kon, koff) and a Kd of 779 μM (Figure 3a, red and orange traces). When flowed over the SPR chip with 30 μM tRNA (a 3-fold excess relative to the amount of neomycin in solution), the SPR signal for neomycin – HIV-1 FSS binding is completely ablated (Figure 3a, blue and green traces). This is consistent with neomycin’s expected lack of binding selectivity. In contrast, SPR traces of compound 6 with and without excess tRNA produce binding constants that are within statistical error (Figure 3b). Small, statistically insignificant differences between the traces obtained with and without excess tRNA likely reflect aging of the SPR chip. Dissociation constants (Kd) measured for other new compounds likewise do not change appreciably in the presence of excess tRNA (Table 1). While these data do not constitute a comprehensive demonstration of specificity for the HIV-1 FSS, they nonetheless provide a strong initial indication of preferential binding.
Figure 3:

SPR competition assay. Competition with excess tRNA ablates signal from neomycin binding (a) but has little impact on binding of compound 6 (b). Two replicate injections are shown for each condition.
Table 1:
Binding constants measured by SPR for compounds binding the HIV-1 FSS. Binding constants represent the average of the constants obtained from individually fitting four SPR curves, at two different concentrations. Competition binding experiments with tRNA were conducted with HIV-1 FSS on-chip and a 5 fold excess (relative to compound) concentration of tRNA in solution. All dissociation constants KD are reported ± standard error.
| Compound | cLogP | ka (1/M·s) |
kd (1/s) | KD for FSS RNA (nM) |
KD with tRNA Competition (nM) |
|---|---|---|---|---|---|
| 3 | 2.63 | 1.9*105 | 3.0*10−3 | 16 ± 2 | 130 ± 10 |
| 4 | 0.31 | 6.49*104 | 2.37*10−2 | 497 ± 157 | 132 ± 34.5 |
| 5 | −1.52 | 1.49*104 | 6.80*10−3 | 583 ± 202 | 441 ± 121 |
| 6 | −1.6 | 5.66*104 | 2.26*10−3 | 62.2 ± 21.3 | 124 ± 37.4 |
| 7 | −3.44 | 6.74*104 | 5.42*10−3 | 110 ± 34.2 | 183 ± 32.5 |
| 8 | 3.94 | 1.69*103 | 2.07*10−2 | 19,300 ± 5,170 | 16,800 ± 7,020 |
| 9 | 2.11 | ND | ND | ND | ND |
| 10 | 0.19 | 4.09*103 | 5.41*10−3 | 1,470 ± 237 | ND |
| 11 | 2.02 | ND | ND | ND | ND |
As a further test of selectivity, we conducted competition dialysis experiments on compound 6. Eight human RNA sequences were chosen as competing RNAs based on their structural similarity to the HIV-1 FSS (Supplementary Figure 4). Among the 8 hairpins tested, compound 6 preferentially binds to HIV-1 FSS, consistent with recognition of specific structural features of the HIV-1 FSS RNA. Two other hairpins appreciably bound by compound 6 were those derived from IRF5 and BDNF mRNAs. Binding to IRF5 RNA is not surprising, as it is predicted to contain an ACAA tetraloop with closing C-G base pair, as does the HIV-1 FSS, and results with previous HIV-1 FSS binding compounds have suggested the tetraloop and closing base pair as the binding site.15 Binding to the hairpin derived from BDNF is somewhat more surprising, given its GAUG tetraloop. From the perspective of off-target activity in cells, in addition to the strong preferential binding of 6 to the HIV-1 FSS, the relative expression level of viral mRNA is as much as 300-fold that of cellular mRNAs.26 Therefore, minimal competitive binding to RNAs such as the IRF5 and BDNF hairpins would be expected in an infected cell.
Compounds 4 - 7 were chosen for further assessment based on their sub-micromolar affinity to the HIV-1 FSS. First, the impact of compounds on cellular metabolism was determined via WST-1 assay, a measure of mitochondrial activity. Only compound 4 showed significant effects below a concentration of 40 micromolar (Figure 5). Next, compounds were assayed for activity against live HIV IIIB (a reference laboratory strain of HIV-1) in human T-cells. Analysis of the results by HIV p24 ELISA showed that all four compounds decreased the amount of virus produced by up to 63% at the highest tested dose (25 μM), with a strong dose response. This is slightly less potent than compound 3 (approximately 5-fold, as the previously measured IC50 = 4.86 μM, consistent with the results of this assay), but nonetheless encouraging given the much smaller molecular weight of 4 - 7.
Figure 5:

Effect of HIV-FSS RNA-binding compounds on cellular metabolism as measured by a WST-1 assay. Error bars represent the standard deviation on measurements from triplicate wells (biological replicas) of each condition.
Conclusions:
We have demonstrated that the overall molecular weight of HIV-1 FSS RNA binding compounds may be reduced without dramatic reductions in affinity. This is encouraging from the standpoint of increasing the ligand efficiency of RNA-targeting compounds.27 Compound 6 in particular was found to have an affinity only 4-fold decreased from our previous “best” compound (3), indicating that both the quinoline ethyl groups and the extended pi surface provided by the benzo[g]quinoline are not essential as long as other features of the molecule are in place. The molecular weight and clogP for 6 are also reduced by 11.31% and 4.23 clogP, respectively, compared to compound 3. Compounds 4 – 7 also displayed moderate anti-HIV activity, although they were not able to completely ablate production of virus in the time course of the assay (in contrast to 3). We are continuing to study this phenomenon, as well as further structural modifications that may serve to enhance the affinity of reduced molecular weight FSS RNA targeted molecules.
Supplementary Material
Figure 4:

Competition dialysis results for compound 6.
Figure 6:

Reduced molecular-weight compounds inhibit HIVIIIB, albeit at lower potency than higher molecular-weight compound 3.
Acknowledgements:
We thank Professor Stephen Dewhurst and Mr. Jeffrey Chamberlain for assistance with HIV inhibition assays and for providing stocks of HIVIIIB. This work was supported by the National Institutes of Health via research grant GM100788 (to B.L.M.), and the Training in HIV Replication and Pathogenesis T32 grant (T32AI049815, to J.D.M.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References:
- 1 (a).Xiao W; Zhong Y; Wu L; Yang D; Ye S; Zhang M Mol. Clin. Oncol. 2019, 10, 67–77; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tesfaye AA; Azmi AS; Philip PA Am.J.Pathol. 2019, 189, 58–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2 (a).Osborne RJ, Thornton CA Hum. Mol. Genet. 2006, 15, 162–169; [DOI] [PubMed] [Google Scholar]; (b) LoRusso S; Weiner B; Arnold WD Neurotherapeutics 2018, 15, 872–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3 (a).Morgan BS; Forte JE; Hargrove AE Nucl. Acids Res. 2018, 19, 8025–8037; [DOI] [PMC free article] [PubMed] [Google Scholar]; Connelly CM; Moon MH; Schneekloth JS Jr. Cell Chem. Biol. 2016, 23, 1077–1090; [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Disney MD; Angelbello AJ Acc. Chem. Res. 2016, 49, 2698–2704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Atkins JF; Loughran G; Bhatt PR; Firth AE; Baranov PV Nucl. Acids Res. 2016, 44, 7007–7078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Napthine S; Ling R; Finch LK; Jones JD; Bell S; Brierley I; Firth AE Nature Commun. 2017, 8, 15582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Belew AT; Meskauskas A; Musalgaonkar S; Advani VM; Sulima SO; Kasprzak WK; Shapiro BA; Dinman JD Nature 2014, 512, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Meydan S; Klepacki D; Karthikeyan S; Margus T; Thomas P; Jones JE; Khan Y; Briggs J; Dinman JD; Vázquez-Laslop N; Mankin AS Mol. Cell 2017, 65, 207–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8 (a).Gareiss PC; Miller BL Curr. Opin. Invest. Drugs 2009, 10, 121–128; [PubMed] [Google Scholar]; (b) Brakier-Gingras L; Charbonneau J; Miller BL Frontiers in Clinical Drug Research: HIV 2014, 1, 3–18. [Google Scholar]
- 9.Shehu-Xhilaga M; Crowe SM; Mak JJ Virol. 2001, 75, 1834–1841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Garcia-Miranda P; Becker JT; Benner BE; Blume A; Sherer NM; Butcher SE J. Virol. 2016, 90, 6906–6917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hung M; Patel P; Davis S; Green SR J. Virol. 1998, 72, 4819–4824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Marcheschi RJ; Tonelli M; Kumar A; Butcher SA ACS Chem. Biol. 2011, 6, 857–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McNaughton BR; Miller BL Org. Lett. 2006, 8, 1803–1806. [DOI] [PubMed] [Google Scholar]
- 14.McNaughton BR; Gareiss PC; Miller BL J. Am. Chem. Soc. 2007, 129, 11306–11307. [DOI] [PubMed] [Google Scholar]
- 15.Palde PB; Ofori LO; Gareiss PC; Lerea J; Miller BL J. Med. Chem. 2010, 53, 6018–6027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ofori LO; Hilimire TA; Bennett RP; Brown NW; Smith HC; Miller BL J. Med. Chem. 2014, 57, 723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hilimire TA; Bennett RP; Stewart RA; Garcia-Miranda P; Blume A; Becker J; Sherer N; Helms ED; Butcher SE; Smith HC; Miller BL ACS Chem. Biol. 2016, 11, 88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hilimire TA; Chamberlain JM; Anokhina V; Bennett RP; Swart O; Myers JR; Ashton JM; Stewart RA; Featherston AL; Gates K; Helms ED; Smith HC; Dewhurst S; Miller BL ACS Chem. Biol. 2017, 12, 1674–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19 (a).Markowitz M; Mohri H; Mehandru S; Shet A; Berry L; Kalyanaraman R; Kim A; Chung C; Jean-Pierre P; Horowitz A; La Mar M; Wrin T; Parkin N; Poles M; Petropoulos C; Mullen M; Boden D; Ho DD Lancet 2005, 365, 1031–1038; [DOI] [PubMed] [Google Scholar]; (b) Mohri H; Markowitz MJ Acquir. Immune Defic. Syndr. 2008, 48, 511–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.cLogP values were calculated using Molinspiration (https://www.molinspiration.com) with primary amines drawn as +1 charged.
- 21.Satyanarayana S; Dabrowiak JC; Chaires JB Biochemistry 1993, 32, 2573–2584. [DOI] [PubMed] [Google Scholar]
- 22.Garbay-Jaureguiberry C; Arnoux B; Prangé T; Wehri-Altenburger S; Pascard C; Roques BP J. Am. Chem. Soc. 1980, 102, 1827–1837. [Google Scholar]
- 23.Chow WY; Bihan D; Forman CJ; Slatter DA; Reid DG; Wales DJ; Farndale RW; Duer MJ Sci. Rep. 2015, 5, 12556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Luedtke NW; Liu Q; Tor Y Biochemistry 2003, 42, 11391–11403. [DOI] [PubMed] [Google Scholar]
- 25.Thomas JR; Hergenrother PJ Chem. Rev. 2008, 108, 1171–1224. [DOI] [PubMed] [Google Scholar]
- 26.Martrus G; Niehrs A; Cornelis R; Rechtien A; García-Beltran W; Lütgehetmann M; Hoffmann C; Altfeld MJ Virol. 2016, 90, 9018–9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hopkins AL; Keserü GM; Leeson PD; Rees DC; Reynolds CH Nat. Rev. Drug. Disc. 2014, 13, 105–121 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.