

Abstract
Therapeutic options for patients with advanced‐stage hepatocellular carcinoma (HCC) are very limited. The only approved first‐line treatment is the multi‐tyrosine kinase inhibitor sorafenib, which shows low response rates and severe side effects. In particular, the compensatory activation of growth factor receptors leads to chemoresistance and limits the clinical impact of sorafenib. However, combination approaches to improve sorafenib have failed. Here we investigate the inhibition of cyclin‐dependent kinase 5 (Cdk5) as a promising combination strategy to improve sorafenib response in HCC. Combination of sorafenib with Cdk5 inhibition (genetic knockdown by short hairpin RNA or CRISPR/Cas9 and pharmacologic inhibition) synergistically impaired HCC progression in vitro and in vivo by inhibiting both tumor cell proliferation and migration. Importantly, these effects were mediated by a mechanism for Cdk5: A liquid chromatography–tandem mass spectrometry–based proteomic approach revealed that Cdk5 inhibition interferes with intracellular trafficking, a process crucial for cellular homeostasis and growth factor receptor signaling. Cdk5 inhibition resulted in an accumulation of enlarged vesicles and respective cargos in the perinuclear region, considerably impairing the extent and quality of growth factor receptor signaling. Thereby, Cdk5 inhibition offers a comprehensive approach to globally disturb growth factor receptor signaling that is superior to specific inhibition of individual growth factor receptors. Conclusion: Cdk5 inhibition represents an effective approach to improve sorafenib response and to prevent sorafenib treatment escape in HCC. Notably, Cdk5 is an addressable target frequently overexpressed in HCC, and with Dinaciclib, a clinically tested Cdk5 inhibitor is readily available. Thus, our study provides evidence for clinically evaluating the combination of sorafenib and Dinaciclib to improve the therapeutic situation for patients with advanced‐stage HCC.
Abbreviations
- BSA
bovine serum albumin
- Cdk5
cyclin‐dependent kinase 5
- EGFR
epidermal growth factor receptor
- EGF
epidermal growth factor
- ERK
extracellular signal‐regulated kinase
- GFP
green fluorescent protein
- HCC
hepatocellular carcinoma
- mTOR
mammalian target of rapamycin
- nt
nontargeting
- PBS
phosphate‐buffered saline
- PI3K
phosphoinositide 3 kinase
- shRNA
short hairpin RNA
Patients diagnosed with hepatocellular carcinoma (HCC) face a very poor prognosis.1 Treatment options are limited, and curative treatment strategies like surgical liver resection or liver transplantation are only suitable for patients with early‐stage HCC.2 For patients with advanced‐stage HCC, there are only few systemic therapies available, and the multi‐tyrosine kinase inhibitor sorafenib is the only approved first‐line therapy.
The small molecule sorafenib acts through the inhibition of various tyrosine kinases, such as vascular endothelial growth factor receptor, platelet‐derived growth factor receptor, and Raf family kinases.3 Unfortunately, only a small group of patients responds to sorafenib treatment and the survival benefit is limited to about 3 months, while severe side effects often lead to dose reduction or discontinuation of treatment.4 In particular, the compensatory activation of survival pathways like growth factor receptor signaling upon sorafenib treatment is responsible for chemoresistance and limited treatment efficacy.5 However, combinations of sorafenib with various chemotherapeutics to avoid chemoresistance have been unsuccessful so far.6 In the last decade, strong efforts were put into improving the therapeutic situation of advanced‐stage HCC patients, which led to significant advances in the recent past. For example, Regorafenib has recently been approved as second‐line treatment for patients with HCC progressing under sorafenib treatment.7 Further, Lenvatinib, as first‐line treatment, and Cabozantinib, as second‐line treatment, have shown promising results in clinical phase 3 trials and are expected to be approved for HCC treatment.8, 9 However, sorafenib is still the only approved first‐line treatment for patients with advanced‐stage HCC. Therefore, the search for new therapeutic approaches is urgent.
We recently elucidated an important role of cyclin‐dependent kinase 5 (Cdk5) in HCC.10 Cdk5 is an atypical member of the cyclin‐dependent kinase family, which has been extensively studied in the central nervous system, where it is involved in the development of various neurodegenerative diseases like Alzheimer’s and Parkinson’s disease.11 In recent years, information on the extraneuronal role of Cdk5 has been expanded and the involvement of Cdk5 in various types of cancer has been characterized (reviewed in12). We showed that Cdk5 is overexpressed in human HCC and promotes cancer progression by regulating DNA damage response. Consistently, inhibition of Cdk5 sensitized HCC cells to DNA damaging agents, which are only accepted for patients with intermediate‐stage HCC.
Here, we propose the inhibition of Cdk5 as an effective strategy to improve sorafenib therapy. We uncovered a mode of action for Cdk5 in HCC. By targeting the cellular trafficking equilibrium, rather than interfering with individual growth factor receptors, Cdk5 inhibition offers a global approach to prevent the compensatory activation of growth factor receptors and thus increases the therapeutic efficacy of sorafenib.
In summary, our study offers a reasonable preclinical basis for the combination of sorafenib with Cdk5 inhibitors for treatment of patients with advanced‐stage HCC.
Materials and Methods
IN VIVO EXPERIMENTS
All experiments were performed according to German legislation of animal protection and were approved by the local government authorities. As HUH7 cells are derived from human tumors, severe combined immunodeficient (SCID) mice had to be used to avoid tumor cell rejection. RIL175 cells originate from C57BL/6 mice, so the corresponding mice could be used. The albino phenotype was necessary for in vivo bioluminescence measurements.
Ectopic Tumor Model
Twenty female SCID “CB17/lcr‐PrkdcSCID/lcrlcocrl” mice, 6 weeks old, purchased from Charles River, were used. We subcutaneously injected 3.3 × 106 nontargeting (nt) or Cdk5 short hairpin RNA (shRNA) HUH7 cells into the flank of female SCID mice. Sorafenib was injected intraperitoneally (100 µL, solvent: 5% DMSO, 10% Solutol, 85% phosphate‐buffered saline [PBS]). Treatment with sorafenib was started 10 days after implantation with 10 mg/kg sorafenib injected daily for 7 days. Tumor volume was measured every second day with a caliper and calculated with the formula π/6 × L × W × H. Eighteen days after the implantation, all mice were sacrificed through cervical dislocation. For the statistical evaluation of tumor growth, an exponential growth model was used to model tumor volume, where the tumor volume at a given time t (N(t)) is a function of the starting volume N(0), the time of growth t and of the growth rate α: N(t) = N(0) × exp(α · t). Modelling was performed using nonlinear mixed effects modelling with the software NONMEM 7.3.
Dissemination Assay: Dinaciclib
Twenty female C57BL/6 albino “C57BL/6BrdCrHsd‐Tyrc” mice, 6 weeks old, purchased from Envigo, were used. The mice were pretreated intraperitoneally with 10 mg/kg Dinaciclib or solvent (5% DMSO, 10% Solutol, 85% PBS) three times (48, 24, and 0.5 hours) before cell injection. We intravenously injected 2 × 105 Ril175‐luc cells into the tail vein. Mice were imaged on day 3 after receiving an intraperitoneal injection of 6 mg/mL luciferin/mouse. Prior to imaging, mice were put under anesthesia with 3% isoflurane in oxygen. Mice were imaged in ventrodorsal position. During luminescence measurement using the IVIS Lumina system (PerkinElmer), mice were kept under narcosis with 2% isoflurane in oxygen, and hypothermia was prevented by a heating plate (37°C). The tumor signal per defined region of interest was calculated with the Living Image 4.4 software (Caliper Life Sciences) as photons/second/cm2 (total flux/area).
Dissemination Assay: Cdk5 KO
Twenty female C57BL/6 albino “C57BL/6BrdCrHsd‐Tyrc” mice, 6 weeks old, purchased from Envigo, were used. We intravenously injected 2 × 105 Ril175‐luc cells (either wild‐type or Cdk5 knockout [KO]) into the tail vein. Luminescence measurement was performed as described in Dissemination Assay: Dinaciclib.
IMMUNOSTAINING
Colocalization
For immunostaining experiments, nt and Cdk5 shRNA HUH7 cells were seeded into 8–well ibiTreat µ‐slides (Ibidi GmbH, Munich, Germany). Cells were washed with ice‐cold PBS+ Ca2+/Mg2+ once and fixed in 4% paraformaldehyde for 15 minutes, before being washed with PBS once. In order to permeabilize the cells, 0.2% Triton X‐100 (Merck, Darmstadt, Germany) was applied for 20 minutes. Unspecific antibody binding sites were blocked by incubation with 0.2 % bovine serum albumin (BSA; Sigma Aldrich, Taufkirchen, Germany) in PBS for 20 minutes. Afterward, cells were incubated with primary antibodies against epidermal growth factor receptor (EGFR; 1:150, Cell Signaling Technologies, 4267) and early endosome antigen 1 (1:150, Santa Cruz Biotechnology, sc‐6415) for 1 hour. Thereafter, cells were washed with PBS and incubated with Alexa Fluor 488 and 546 secondary antibodies (1:400, Molecular Probes/Invitrogen, A – 11008, A – 11056) together with 5 µg/µL Hoechst 33342 (1:200, Sigma Aldrich, Taufkirchen, Germany) in PBS containing 0.2% BSA for 30 minutes. Each well was then covered with FluorSave reagent mounting medium (Merck, Darmstadt, Germany) and glass coverslips. Images were taken with a Leica SP8 confocal laser scanning microscope (Leica Microsystems, Wetzlar, Germany).
EGFR Surface Localization
For the analysis of EGFR localized exclusively at the cell surface, nt and Cdk5 shRNA HUH7 or Hep3B cells were seeded into 8‐well ibiTreat µ‐slides and treated with sorafenib (0.5 µM, 5 µM, 24 hours). Afterward, incubation cells were immediately put on ice and incubated with a primary antibody targeting the extracellular domain of the EGFR (1:150, Calbiochem, GR01) for 1 hour at 4°C. After antibody staining, cells were washed twice with ice‐cold PBS and fixed in 4% paraformaldehyde for 8 minutes on ice. Thereafter, cells were washed with PBS and incubated with Alexa Fluor 488 secondary antibody together with 5 µg/mL Hoechst 33342 in PBS containing 0.2% BSA for 30 minutes. Each well was then covered with FluorSave reagent mounting medium and glass coverslips. Images were taken with a Leica SP8 confocal laser scanning microscope.
EGF Uptake and Elimination
nt and Cdk5 shRNA cells were seeded in 8‐well ibiTreat µ‐slides and treated with 100 ng/mL EGF Rhodamine (Thermo Fisher Scientific, E3481) for various time points. In the chase (elimination) experiments, EGF Rhodamine was removed after 30 minutes of incubation, and cells were washed twice with prewarmed PBS and incubated for various time points in medium without FCS. After incubation and chase, cells are immediately put on ice, washed twice with ice‐cold PBS, and incubated with acid wash solution (acetic acid 0.2 M, NaCl 0.5 M, pH 2.0) for 5 minutes to remove excess EGF. Cells are then washed with PBS twice and fixed in 4% paraformaldehyde. Each well was then covered with FluorSave reagent mounting medium and glass coverslips. Images were taken with a Leica SP8 confocal laser scanning microscope.
Live Cell Imaging/Time Lapse Microscopy
nt and Cdk5 shRNA HUH7 or Hep3B cells were seeded in 8‐well ibiTreat µ‐slides at a density of 5 × 104 and transfected with either EGFR‐green fluorescent protein (GFP; a gift from Alexander Sorkin, Addgene plasmid #32751), pLenti‐MetGFP (a gift from David Rimm, Addgene plasmid #37560), or Alpha 5 integrin‐GFP (a gift from Rick Horwitz, Addgene plasmid #15238) using DharmaFECT 1 transfection reagent (ThermoFisher Scientific, Waltham, MA). For the experiments on autophagic flux, the Premo Autophagy Tandem Sensor RFP‐GFP‐LC3B Kit with the RFP‐GFP‐LC3 plasmid (ThermoFisher Scientific, Waltham, MA) was used according to the manufacturer’s protocol. The use of this RFP‐GFP‐LC3 plasmid enables a detailed analysis of the autophagic flux, as GFP is quenched at the transition from autophagosome to lysosome, resulting in a shift from green and red fluorescent vesicles to only red fluorescent vesicles.
Cells were imaged using a Leica SP8 confocal laser scanning microscope. Frames were taken every 0.75 seconds for a total of 10 minutes. For the quantification of vesicle size, two types of objects have been considered: small vesicles (present in both conditions) and “ring shaped” vesicles (present only in Cdk5 shRNA HUH7 cells). The ParticleSizer Plugin of Fiji after background removal is used to recognize the small vesicles, and a Circular Hough Transform–based algorithm implemented by the Matlab imfindcircles function is used to recognize the “ring shaped” vesicles only in the Cdk5 knockdown condition after background removal. If the two kinds of vesicles are overlapping, only the donut‐shaped ones will be considered.
Results
Cdk5 INHIBITION IMPROVES SORAFENIB RESPONSE IN HCC IN VITRO AND IN VIVO
To test the functional effects of the combination of sorafenib and Cdk5 inhibition, we used the human HCC cell lines HUH7 and Hep3B, as well as RIL175, an HCC cell line derived from C57BL/6 mice. We silenced Cdk5 by shRNA, CRISPR/Cas9 knockout, or interfered with its kinase activity by using two well‐established pharmacologic inhibitors, Roscovitine and Dinaciclib, and the experimental Cdk5 inhibitor LGR1407 (Supporting Fig. S1A‐C). Cdk5 knockdown and inhibition synergistically enhanced the sorafenib‐mediated blockade of HCC cell proliferation and clonogenic survival (Fig. 1A‐D and Supporting Fig. S1D). For the evaluation of synergism, two separate models, namely Combination Subthresholding and Bliss Independence, were used. Both synergy models revealed a significant effect of the combination of sorafenib and Cdk5 inhibition compared with single treatments (respective Bliss values are indicated in Fig. 1A‐D). Importantly, similar and striking synergistic effects were observed when the combination of Cdk5 inhibition and sorafenib was administered in a murine HCC xenograft model (Fig. 1E,F). Tumor volume was observed over time and subjected to a nonlinear mixed effects modelling technique, which revealed a synergistic effect of the combination of sorafenib and Cdk5 inhibition resulting in a significantly reduced tumor growth rate.
Figure 1.
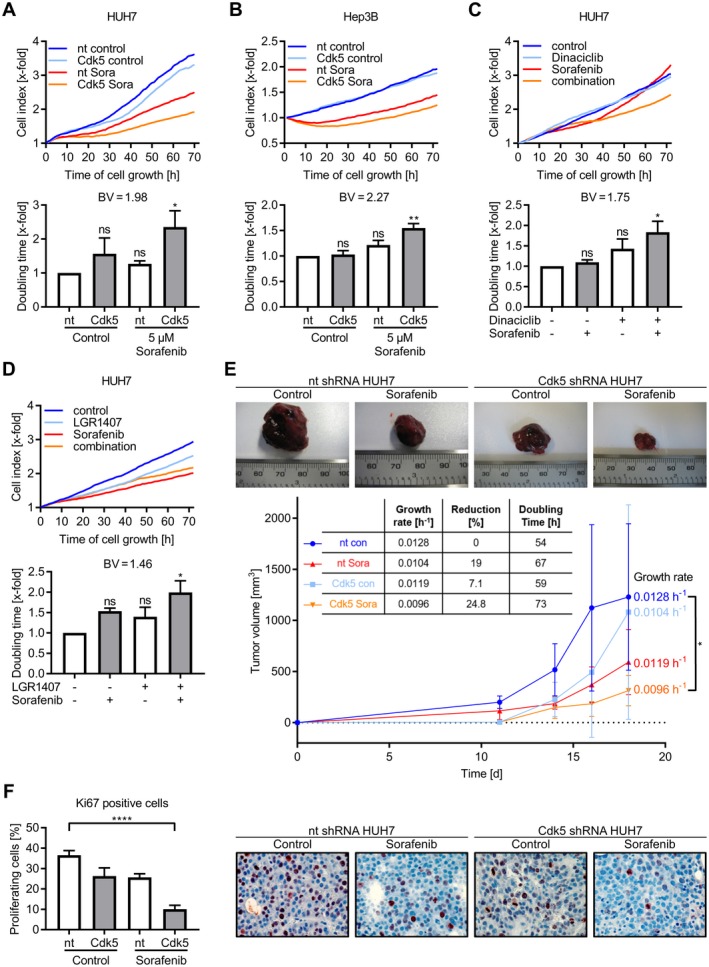
Combination of Cdk5 inhibition and sorafenib impairs HCC growth in vitro and in vivo. (A) Proliferation of nt and Cdk5 shRNA HUH7 cells after treatment with sorafenib (5 µM) is shown. Corresponding doubling time is shown. One‐way analysis of variance (ANOVA), Tukey *P < 0.05, n = 3. Bliss Value = 1.98. (B) Proliferation of nt and Cdk5 shRNA Hep3B cells treated with sorafenib (5 µM) is shown. Corresponding doubling time is shown. One‐way ANOVA, Tukey **P < 0.01, n = 3. Bliss Value = 2.27. (C) Proliferation of HUH7 cells treated with sorafenib (5 µM), dinaciclib (10 nM), or a combination of both is shown. Corresponding doubling time is shown. One‐way ANOVA, Tukey *P < 0.05, n = 3. Bliss Value = 1.75. (D) Proliferation of HUH7 cells treated with sorafenib (5 µM), LGR1407 (7.5 µM), or a combination of both is shown. Corresponding doubling time is shown. One‐way ANOVA, Tukey *P < 0.05, n = 3. Bliss Value = 1.46. (A‐D) Upper panel: one representative graph (out of three independent experiments) showing cell index over time. Lower panel: Bar diagram showing the statistical analysis of cell index (upper panel) expressed as doubling time. (E) Tumors of nt and Cdk5 shRNA HUH7 cells grown in SCID mice that were treated with either sorafenib or solvent are shown (n = 6). Tumor volume over the treatment period of 18 days is shown. The table shows the statistical evaluation of growth rates that were determined from tumor volumes by applying an exponential tumor growth model, which showed a significantly reduced tumor growth rate by combining Cdk5 inhibition with sorafenib (*P < 0.05). (F) Immunostaining of respective tumors from E for Ki67 (red) and hematoxylin (nuclei, blue) is shown. The bar graph indicates proliferating cells evaluated by counting Ki67‐positive cells. One‐way ANOVA, Tukey ****P < 0.0001, n = 6.
In addition to the antiproliferative effect, sorafenib and Cdk5 inhibition reduced HCC cell migration (Fig. 2A). Notably and somewhat surprisingly, low‐dose sorafenib treatment increased HCC migration and invasion (Fig. 2B‐F) in a proliferation‐independent fashion (Supporting Fig. S1E). Genetic knockdown and pharmacological inhibition of Cdk5, importantly, not only reduced the overall motility but also prevented the sorafenib‐induced increase of HCC cell migration and invasion (Fig. 2B‐F). Furthermore, Cdk5 knockout by CRISPR/Cas9 and pharmacologic inhibition impaired HCC cell dissemination in vivo (Fig. 2G,H).
Figure 2.
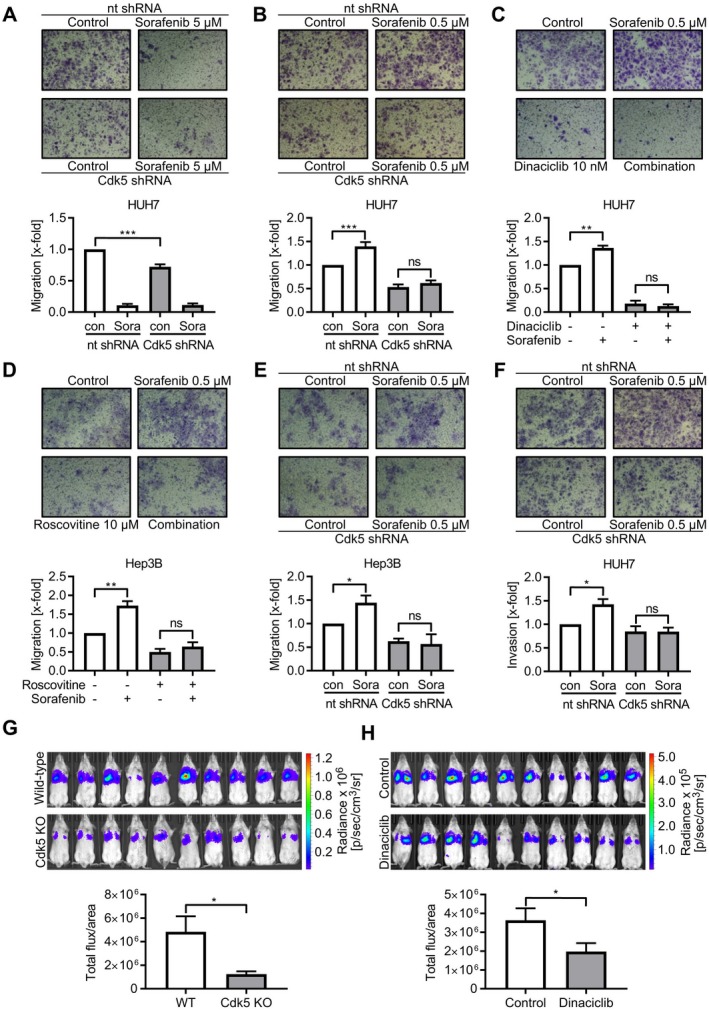
Cdk5 inhibition prevents sorafenib‐induced HCC cell migration. (A‐E) Transwell migration of nt and Cdk5 shRNA HUH7 (A,B), wild‐type HUH7 cells (C), wild‐type Hep3B cells (D) and nt and Cdk5 shRNA Hep3B cells (E) that were pretreated with the respective compounds in the indicated concentrations is shown. (F) Invasion of nt and Cdk5 shRNA HUH7 cells that were pretreated with sorafenib is shown. (A‐F) Bar graphs indicate the number of migrated cells normalized to control. One‐way analysis of variance (ANOVA), Tukey *P < 0.05, **P < 0.01, ***P < 0.001, n = 3. (G) Noninvasive images of tumor bearing mice injected with either RIL175 wild‐type cells or RIL175 Cdk5 knockout cells are shown. The bar graph shows corresponding signal intensities. t test, *P < 0.05, n = 10. (H) Noninvasive images of tumor‐bearing mice treated with either Dinaciclib or solvent are shown. The bar graph shows corresponding signal intensities. t test, *P < 0.05, n = 10. Abbreviations: Cdk5 KO, Cdk5 knockout; WT, wild‐type.
Cdk5 Inhibition Sensitizes to Sorafenib by Preventing the Compensatory Activation of Growth Factor Receptors
DNA damage is involved in the pathogenesis of liver disease and HCC progression.13 We have previously shown that Cdk5 inhibition sensitized HCC cells to the treatment with DNA damage‐inducing agents by regulating DNA repair mechanisms, ultimately resulting in apoptosis.10 However, DNA damage response and apoptosis were not modulated by the combination of sorafenib and Cdk5 inhibition (Supporting Fig. S2A‐C).
To obtain clues as to mechanisms through which Cdk5 inhibition sensitizes to sorafenib treatment, we took advantage of an LC‐MS/MS‐based proteomic approach and identified proteins whose levels were differentially controlled by Cdk5 knockdown alone and in combination with sorafenib (Fig. 3A,B; Supporting Fig. S3A,B and S4A‐C). The identified proteins were biologically related, as shown by a protein–protein interaction network analysis (Fig. 3C; Supporting Fig. S3C). In addition, functional enrichment analysis revealed a modulation of proteins associated with cellular metabolism (Fig. 3C). Moreover, interestingly, we observed an accumulation of proteins regulated by intracellular trafficking including proteins associated with autophagy like p62/Sequestosome1 and proteins trafficked via endocytosis like integrins or the EGFR (Fig. 3C,D). In order to elucidate the mode of action of Cdk5 inhibition to sensitize for sorafenib, we set out to test the importance of the identified pathways.
Figure 3.
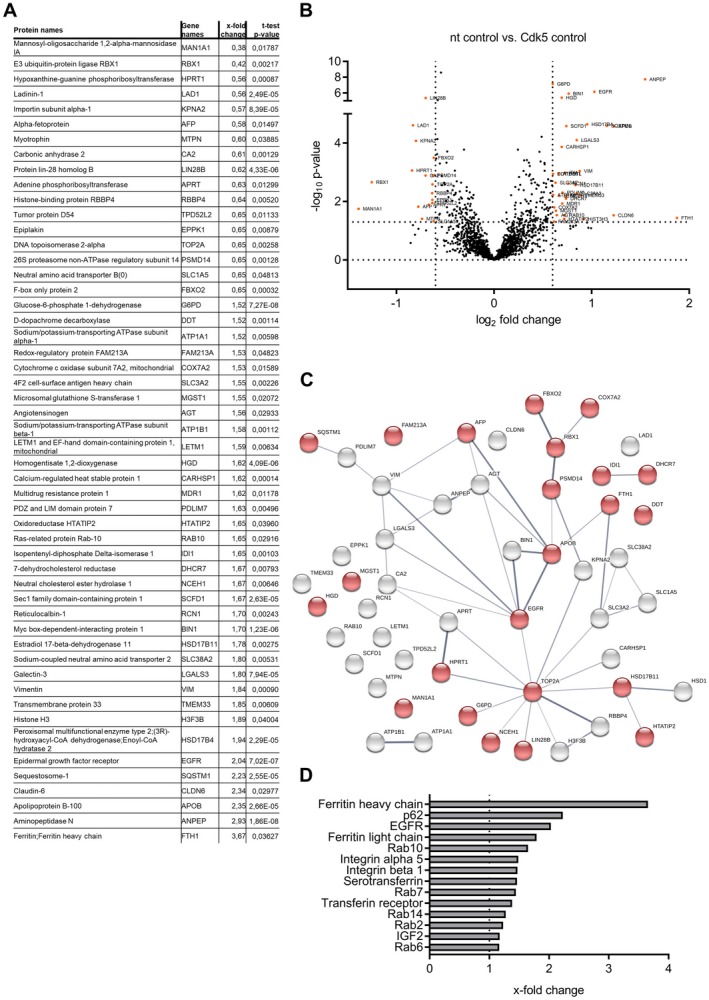
Proteomic analysis of Cdk5 knockdown cells. (A) Table of proteins showing alterations of protein abundance (P < 0.05; log2‐fold change > 0.6) between nt and Cdk5 shRNA HUH7 cells together with their respective gene names, x‐fold changes (nt shRNA HUH7 versus Cdk5 shRNA HUH7) and P Values. (B) Volcano Plot visualizing the protein hits given in table A. (C) Protein interaction map of protein hits given in table A created with string‐db.org (protein–protein interaction enrichment P Value: 0.0016). Proteins involved in metabolic processes, autophagy, and EGFR signaling are highlighted in red (false discovery rate: 0.0125). (D) The graph shows proteins associated with or regulated by endocytosis that were modulated by Cdk5 knockdown (x‐fold change compared with nt shRNA is displayed).
First, we analyzed HCC metabolism. Sorafenib reduced glycolysis and oxidative phosphorylation; however, Cdk5 inhibition had no additional effect (Supporting Fig. S5). Thus, modulation of cell metabolism is unlikely to mediate the sensitizing effect of Cdk5 inhibition and sorafenib.
Second, an accumulation of p62/Sequestosome1, a marker for proteins designated for autophagy, provided evidence that the autophagic flux was perturbed by Cdk5 inhibition (Fig. 4A,B). The relevance of this alteration was corroborated by an increased LC3‐II/I ratio upon Cdk5 knockdown (Fig. 4A,C). To assess whether Cdk5 knockdown induced a degradation block or increased autophagy, an artificial degradation block was applied by inhibiting vesicle fusion using Concanamycin A. Concanamycin A increased the LC3‐II/I ratio in nt shRNA cells but had no effect in Cdk5 shRNA cells, pointing to a degradation block caused by Cdk5 knockdown (Fig. 4D‐F). The disturbance in autophagic flux was further corroborated by using an RFP‐GFP‐LC3 sensor to visualize the progression from autophagosomes to autolysosomes. In fact, we observed strongly enlarged autophagosomes in Cdk5 shRNA HUH7 compared with nt shRNA HUH7 (Fig. 4G). However, sorafenib affected neither p62 levels nor LC3 conversion (Fig. 4A‐C). Thus, we concluded that inhibition of Cdk5 significantly disturbed autophagic flux, but this effect is not specifically responsible for the sensitizing effect of Cdk5 inhibition and sorafenib.
Figure 4.
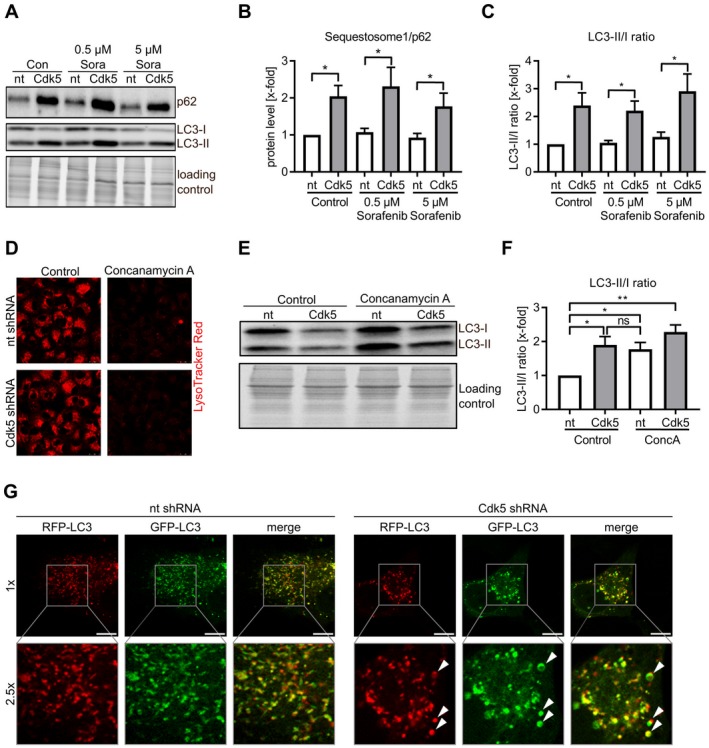
Cdk5 inhibition influences autophagic flux. (A) Immunoblots from nt and Cdk5 shRNA HUH7 cells treated with sorafenib probed with antibodies for p62/Sequestosome1 and LC3 are shown. (B) Quantitative evaluation of p62/Sequestosome1 from A is shown. One‐way analysis of variance (ANOVA), Holm‐Sidak *P < 0.05, n = 3. (C) Ratio of LC3‐II to LC3‐I after quantitative evaluation from A is shown. One‐way ANOVA, Holm‐Sidak *P < 0.05, n = 3. (D) LysoTracker Red staining of nt and Cdk5 shRNA HUH7 cells after treatment with concanamycin A (1 µM) is shown. (E) Immunoblot from nt and Cdk5 shRNA HUH7 cells treated with concanamycin A (1 µM) and probed with an antibody for LC3 is shown. (F) Ratio of LC3‐II/I is shown after quantitative evaluation of immunoblots from E. One‐way ANOVA, Newman‐Keuls *P < 0.05, **P < 0.01, n = 3. (G) Single frames from live cell imaging videos of nt and Cdk5 shRNA HUH7 cells expressing eGFP‐eRFP‐LC3. Scale bar 10 µm (1×).
Thirdly, interestingly, the proteomic screen demonstrated that Cdk5 inhibition induced an upregulation of proteins dependent on intracellular trafficking (Fig. 3D). This suggested that intracellular trafficking was interrupted by Cdk5 inhibition, resulting in accumulation of respective cargos. Among the cargos accumulated by Cdk5 inhibition, especially the EGFR attracted our attention, as the upregulation of growth factor receptors usually correlates with a more aggressive tumor progression.14 Therefore, in order to elucidate this apparent discrepancy between the elevation of the EGFR on the one hand, and inhibition of growth on the other hand, we used the EGFR as a good example to investigate the effects of sorafenib and Cdk5 inhibition on the compensatory activation of growth factor receptors.
Compensatory activation of growth factor signaling including EGF‐, insulin‐like growth factor‐, fibroblast growth factor‐, or hepatocyte growth factor signaling was described as a mechanism of HCC to adapt to sorafenib treatment.15, 16, 17 In line, our results corroborated compensatory activation of growth factor receptor signaling by sorafenib. Whereas the Ras/Raf/MEK/ERK pathway was impaired as shown by decreased ERK1/2 phosphorylation, phosphorylation of EGFR itself and its downstream target AKT was enhanced upon sorafenib (Fig. 5A,B and Supporting Fig. S6A‐C). Moreover, sorafenib treatment increased EGFR at the cell surface of nt shRNA HUH7 and Hep3B cells, likely accounting for the increased immediate response of EGFR to its ligand (Fig. 5C). Of note, Cdk5 inhibition prevented the compensatory activation of growth factor receptor signaling. In Cdk5 knockdown cells, AKT and EGFR phosphorylation were no longer induced upon sorafenib treatment (Fig. 5A,B and Supporting Fig. S6A‐C). In addition, in Cdk5 knockdown cells, EGFR surface levels remained unaffected by sorafenib (Fig. 5C).
Figure 5.
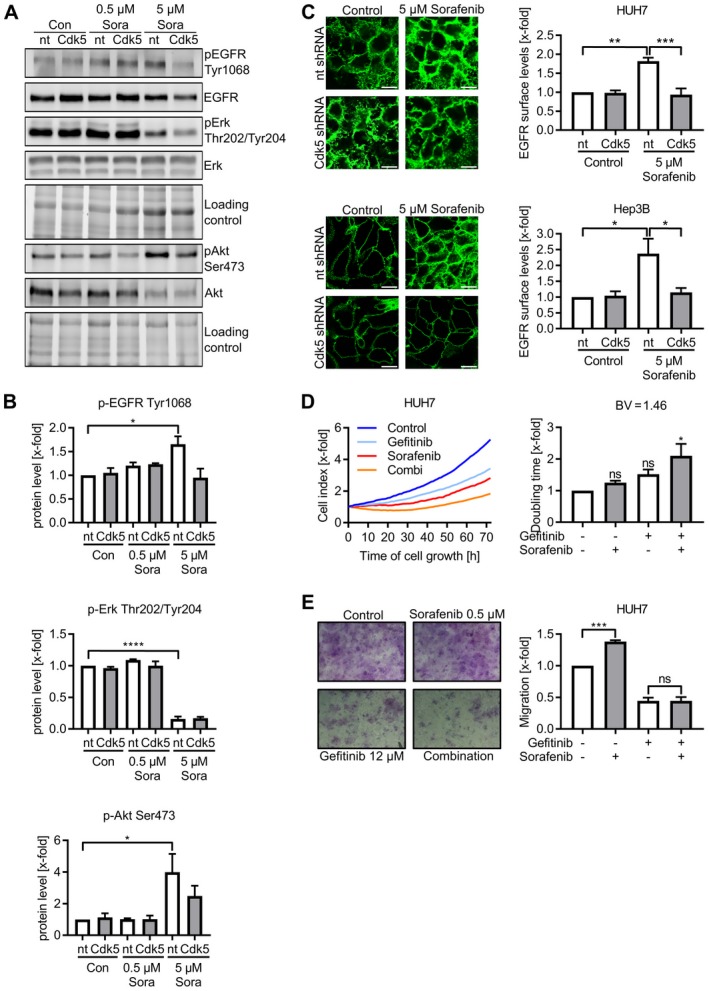
Cdk5 inhibition prevents compensatory activation of EGFR. (A) Immunoblots from nt and Cdk5 shRNA HUH7 cells treated with sorafenib probed with antibodies for pEGFR, EGFR, pErk, Erk, pAkt, and Akt are shown. (B) Quantitative evaluations of pEGFR, pErk and pAkt from A are shown. One‐way analysis of variance (ANOVA), Tukey *P < 0.05, ****P < 0.0001, n = 3. (C) Immunostaining for EGFR with an antibody specific to the extracellular domain in nt and Cdk5 shRNA HUH7 (upper panel) and Hep3B (lower panel) cells after sorafenib treatment is shown. Scale bar 20 µm. Relative evaluation of fluorescence intensity is shown. One‐way ANOVA, Tukey *P < 0.05, **P < 0.01, ***P < 0.001, n = 3. (D) Proliferation of HUH7 cells treated with sorafenib, gefitinib, or combination of both is shown. Corresponding doubling time is shown. One‐way ANOVA, Tukey *P < 0.05, n = 3. Left panel: One representative graph (out of three independent experiments) shows the cell index over time. Right panel: Bar diagram shows the statistical analysis of cell index expressed as doubling time. (E) Transwell migration of wild‐type HUH7 cells that were pretreated with the respective compounds in the indicated concentrations is shown. Representative pictures of migrated cells are shown together with bar diagrams showing the number of migrated cells normalized to the control. One‐way ANOVA, Tukey ***P < 0.001, n = 3. Abbreviations: pAKT, phosphorylated AKT; pEGFR, phosphorylated EGFR; pErk, phosphorylated Erk.
To more firmly establish the interference with growth factor receptor activation as a therapeutically relevant process that mediates the sensitizing effect of Cdk5 inhibition, we analyzed the effects of sorafenib in combination with the EGFR inhibitor gefitinib. Similar to Cdk5 inhibition, gefitinib in combination with sorafenib reduced HCC cell proliferation and migration (Fig. 5D,E). Further, the analysis of a human HCC tissue micro array revealed overall increased EGFR protein levels in HCC tissue compared with healthy liver tissue (Supporting Fig. 7A‐D). However, likely due to the small size and heterogeneity of the patient cohort, there was no correlation of EGFR positive staining, intensity, and immunoreactive score with tumor grading, r‐classification, tumor stage, frequency of recurrence, and cause of death (Supporting Table S1).
Collectively, this set of findings confirmed the compensatory activation of growth factor receptor pathways upon sorafenib treatment as a mechanism of HCC cells to sustain proliferative and migratory capacities and evade sorafenib treatment. Importantly, Cdk5 inhibition prevented the activation of the EGFR signaling cascade, despite not targeting the kinase activity of EGFR directly, suggesting a mode of action different from the classical inhibitors of growth factor receptors.
Cdk5 IS ESSENTIAL FOR INTRACELLULAR VESICLE TRAFFICKING
So far, our results demonstrated that Cdk5 inhibition interfered with the sorafenib‐induced compensatory activation of the EGFR. Moreover, our results indicated a mechanism different from the classical receptor tyrosine kinase inhibitors. Because the proteomic screen suggested that Cdk5 interfered with intracellular trafficking, in order to elucidate how Cdk5 perturbation interfered with EGFR activation, we focused on endocytosis of the receptor, which is crucial for EGFR signaling. After activation and dimerization, the EGFR is internalized and trafficked through early and late endosomes before the signaling is either terminated by degradation via lysosomes or maintained by recycling via endosomes.18 Cdk5 inhibition had no effect on the uptake of the EGF/EGFR complex (Fig. 6A). Conversely, it significantly delayed the clearance of internalized EGFR (Fig. 6B), suggesting that a late step of EGFR trafficking was affected. Consistently, we observed that upon the ablation of Cdk5, EGFR was perinuclearly enriched in aberrantly enlarged vesicle‐like structures (Fig. 6C). To gain detailed insights into how Cdk5 inhibition influences endosomal trafficking of EGFR, we performed live cell imaging using nt and Cdk5 shRNA HUH7 and Hep3B cells overexpressing eGFP‐tagged EGFR. Analysis of vesicle dynamics and size indicated, indeed, that vesicle trafficking was disturbed by Cdk5 inhibition. Control shRNA cells were characterized by small EGFR‐positive vesicles moving with high velocity and distinct directionality. Cdk5 shRNA cells, instead, had large ring‐shaped vesicles that showed impaired motility and accumulated in the perinuclear region (Fig. 6D; Supporting Video 1 and 2). Importantly, this effect was not limited to the EGFR as Cdk5 knockdown similarly affected the size and motility of vesicles carrying integrin α5, a model protein for endocytic trafficking, and c‐Met, the receptor for hepatocyte growth factor (Fig. 6E; Supporting Video [Link], [Link], [Link], [Link]). Especially the c‐Met receptor is of particular interest in this context as it represents one of the most prominent and frequently deregulated growth factor receptors in HCC,19, 20 indicating that several important receptors can be targeted by Cdk5 inhibition. These findings are in line with our observations of Cdk5 inhibition on the autophagic cascade, as autophagy is strongly interconnected with endocytic trafficking.
Figure 6.
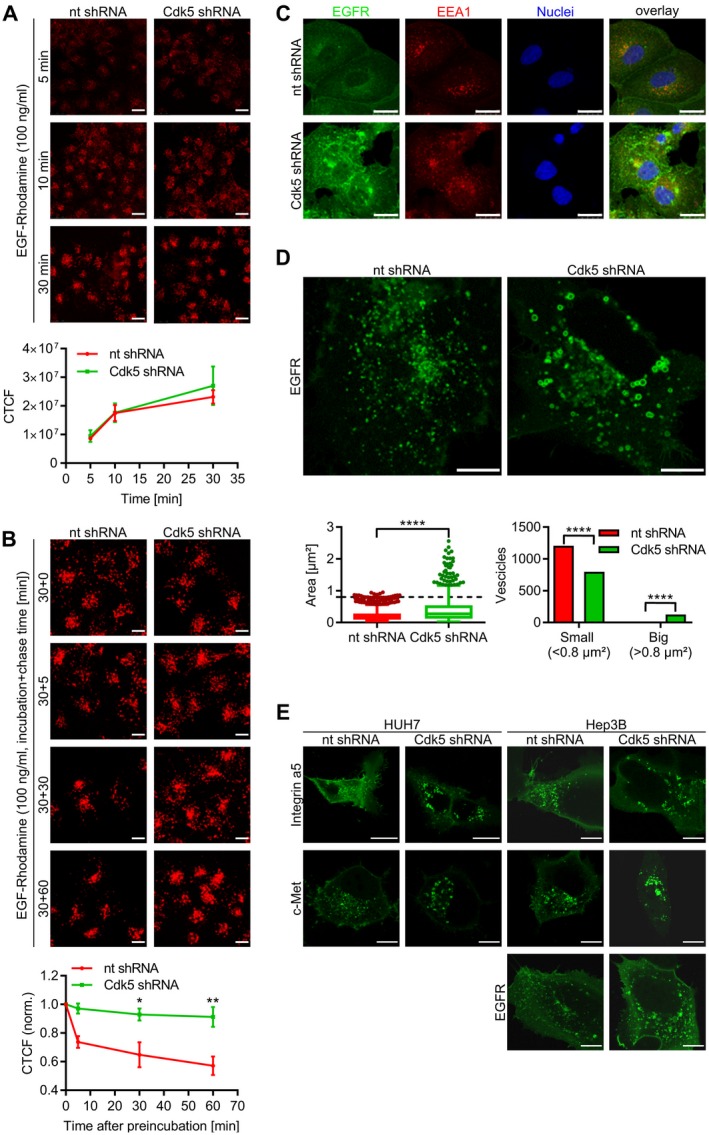
Cdk5 influences endosomal trafficking. (A) EGF‐uptake: Images display nt and Cdk5 shRNA HUH7 cells that were treated with EGF‐Rhodamine for various time points and analyzed by confocal microscopy. Scale bar 25 µm. Quantitative evaluation of CTCF is shown. For each condition, 30 cells were analyzed. (B) EGF‐elimination: Images show nt and Cdk5 shRNA HUH7 cells that were incubated with EGF‐Rhodamine (30 minutes) before EGF‐Rhodamine was removed and cells were chased for the given time points (0, 5, 30, and 60 minutes). Scale bar 25 µm. Quantitative evaluation of CTCF is indicated. One‐way analysis of variance, Tukey *P < 0.05, **P < 0.01, n = 3. (C) Immunostaining for EGFR (green), EEA1 (red), and Hoechst33342 (blue, nuclei) from nt and Cdk5 shRNA HUH7 cells is shown. Scale bar 25 µm. (D) Single frames from live cell imaging videos of nt and Cdk5 shRNA cells expressing eGFP‐EGFR are shown. Scale bar 10 µm. Box plot diagram and bar graph show the distribution of vesicle size comparing nt and Cdk5 shRNA. Mann‐Whitney, ****P < 0.0001, chi‐squared test, ****P < 0.0001. (E) Single frames from live cell imaging videos of nt and Cdk5 shRNA HUH7 and Hep3B cells expressing either eGFP‐Integrin‐α5, eGFP‐cMet, or eGFP‐EGFR are shown. Scale bar 25 µm (HUH7: integrin α5), 10 µm (HUH7: c‐Met, EGFR; Hep3B: integrin α5, EGFR, c‐Met). Abbreviation: CTCF, corrected total cell fluorescence.
In summary, our results demonstrated that Cdk5 inhibition affects intracellular trafficking, as shown by the effects on endocytosis and autophagy, leading to the intracellular accumulation of various cargos that likely affect the extent and quality of signal activation. As a consequence, Cdk5 inhibition offers a global strategy to block the compensatory activation of growth factor receptors usually observed upon sorafenib. An overview about the findings of our study is given in Fig. 7.
Figure 7.
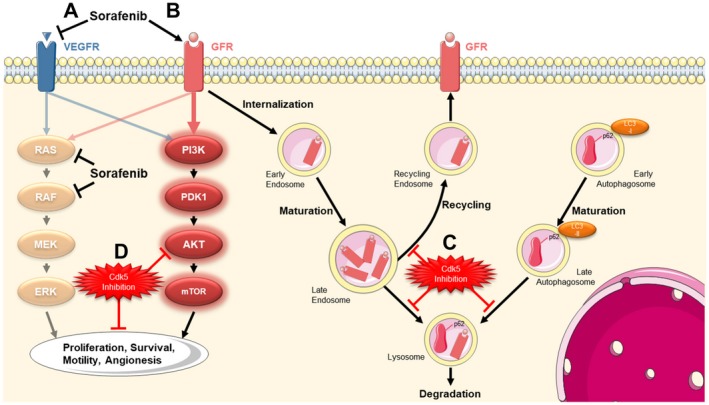
Summary. (A) The treatment of HCC cells with sorafenib causes an inhibition of VEGFR and its downstream targets RAS and RAF. (B) In turn, this leads to the compensatory activation of growth factor receptor signaling, which allows tumor cells to maintain proliferation and migration, mediated via the PI3K/AKT pathway. After activation, growth factor receptors have to be trafficked via the endosomal system and are either degraded via lysosomes or recycled via endosomes. (C) We uncovered that Cdk5 inhibition interferes with intracellular trafficking leading to an increase in vesicle size and an accumulation of respective cargos. (D) Thereby an inhibition of Cdk5 prevents the sorafenib‐induced compensatory activation of growth factor receptors and respective downstream targets and enhances the antitumor effects of sorafenib.
Discussion
In 2008, the approval of the multi‐tyrosine kinase inhibitor sorafenib fundamentally changed the treatment of patients with advanced‐stage HCC. The first approved systemic treatment increased both median overall survival and time to radiologic progression by about 3 months. However, the success was limited due to poor response rates and severe side effects.4 Since then, the efforts to improve sorafenib efficacy by combining it with conventional chemotherapeutics like doxorubicin or mammalian target of rapamycin (mTOR) inhibitors has remained unsuccessful (reviewed in6). Furthermore, clinical trials investigating new therapeutics, among others sunitinib,21 brivanib,22 and linifanib23 as HCC therapy options, failed to replace sorafenib as the first‐line treatment. Recently, palbociclib, an inhibitor of Cdk4/6, showed encouraging preclinical results in HCC treatment alone or in combination with sorafenib.24 By inhibiting cell cycle progression, palbociclib suppressed HCC progression in vitro and in vivo. As a loss of RB1 led to acquired resistance to palbociclib, treatment with palbociclib is suggested for patients with intact RB1. In addition, two new, promising multi‐tyrosine kinase inhibitors, regorafenib7 and cabozantinib,8 came into prospect as second‐line treatment options for patients with HCC and achieved encouraging results in clinical trials (reviewed in25). Along these lines, regorafenib was recently approved for the treatment of patients with advanced HCC progressing under sorafenib treatment, and cabozantinib is expected to be approved as a second‐line treatment in the foreseeable future. Further, lenvatinib, a multi‐tyrosine kinase inhibitor already used in the treatment of thyroid cancer, was shown to be noninferior to sorafenib in HCC treatment and is therefore considered for first‐line HCC therapy approval.9 However, sorafenib is thus far still the only first‐line treatment; therefore, it is of paramount importance to search for ways to improve the impact of sorafenib on HCC.25
Here, we propose Cdk5 inhibition as a promising combination approach to increase the efficacy of sorafenib in HCC. Our results provide direct evidence that Cdk5 inhibition improves sorafenib treatment by interfering with the compensatory activation of growth factor receptors. Activation of parallel pathways to avoid chemotherapeutic treatment is commonly observed in cancer.26 In HCC, in particular, activation of the Ras/Raf/MEK/ERK pathway, the PI3K/AKT/mTOR pathway, histone deacetylases (HDACs),27 and growth factor receptors account for escape from sorafenib treatment. Thus, during recent years, the effects of combinations of sorafenib with inhibition of these pathways have been evaluated. The Ras/Raf/MEK/ERK pathway is often activated by Ras mutations. Accordingly, the combination of sorafenib with MEK inhibitors has shown some efficacy in preclinical and clinical studies, especially if Ras was mutated.28 In addition, elevated MAPK14 was associated with poor response to sorafenib, and inhibition of MAPK14 was demonstrated to sensitize for sorafenib treatment.29 Activation of PI3K/AKT/mTOR signaling can render HCC less sensitive to sorafenib, and the combination of sorafenib and PI3K/AKT/mTOR inhibitors has shown encouraging results in a phase 1/2 study.30 However, other studies reported severe side effects and failure to improve overall survival.31, 32 HDAC inhibitors in combination with sorafenib have shown promising results in preclinical studies, but subsequent clinical trials had to be terminated due to severe toxicity (reviewed in33). Altogether, although there have been various approaches to improve the clinical effect of sorafenib, most of them failed to show significant clinical efficacy.34
Growth factor receptor pathways are characterized by high redundancy and are therefore able to overcome the malfunction of a pathway by over‐activating another, leading to treatment evasion and tumor progression, which is often observed in HCC.35 Studies showing increased metastasis upon sorafenib treatment36, 37 are in line with our results that demonstrate increased HCC cell migration and EGFR activation by sorafenib. However, up to now, there exist no detailed studies investigating a potential correlation between EGFR activation and sorafenib response in patients. Although EGFR is not an established target in HCC therapy, our results, which show high levels of EGFR in human HCC, are in line with previous studies that demonstrated frequent overexpression of the EGFR in human HCC. The altered EGFR protein levels correlated with metastasis, tumor aggressiveness, and poor patient survival.14, 38 However, a clinical benefit of the combination of EGFR inhibition with sorafenib has not been shown. According to the SEARCH trial, the combination of sorafenib with the EGFR inhibitor erlotinib failed to improve survival in patients with HCC.39 In summary, sorafenib combination therapies have shown some benefits, but exhibited severe toxicity and failed to improve overall survival. Thus, there is still no sorafenib combination approach that is clinically used in HCC therapy.
Importantly, our results reveal a mode of action of Cdk5 in HCC to interfere with growth factor receptor activation, different from the classical receptor tyrosine kinase inhibitors. By interfering with intracellular receptor trafficking, Cdk5 inhibition—in contrast to the unsuccessful inhibition of only EGFR by erlotinib—offers a global approach to block the compensatory activation of growth factor receptors. Thus, our study suggests Cdk5 inhibition as a potential option to improve sorafenib efficacy.
Like many growth factor receptor pathways, the EGFR pathway critically depends on endosomal trafficking to uphold or terminate the signaling.18 We showed that Cdk5 inhibition disturbs endosomal trafficking, which leads to an accumulation of respective cargo and an enlargement of endosomal vesicles, thereby inhibiting receptor activity. In the context of cancer, an involvement of Cdk5 in endocytic trafficking has not been described yet; however, there is evidence that Cdk5 plays an important role in regulating endocytosis in the neuronal system. In neurons, a role of Cdk5 in intracellular trafficking has been established.40 Cdk5 drives synaptic vesicle endocytosis by phosphorylating the dephosphins dynamin I, amphiphysin, and synaptojanin and regulates endocytosis of the N‐methyl‐D‐aspartate receptor at postsynaptic sites (reviewed in41). Moreover, during axon outgrowth, Cdk5 was shown to regulate Rab proteins, which coordinate vesicle trafficking, fusion, and maturation.42
During recent years, endocytosis has emerged as a central process in cancer. Endocytic circuitries are deeply interconnected with the activation and execution of various pathways that regulate cancer growth, progression, and metastasis, including small GTPases, integrin, epithelial‐mesenchymal transition, and growth factor receptor signaling.43 Thus, strategies to interfere with endocytosis have shown promising anticancer effects.44, 45, 46 Within this context, our results define Cdk5 as a central regulator of endocytic processes in cancer.
Along this line, our results show that Cdk5 inhibition influences and disturbs autophagic flux, an intracellular trafficking pathway highly interconnected with endocytosis. Autophagy has been associated with cancer progression and was recently shown to be involved in the development of resistance to sorafenib in HCC (reviewed in47). This underlines the global effect of Cdk5 inhibition on intracellular trafficking and establishes the possibility to target two separate mechanisms of resistance to sorafenib in HCC, autophagy and endocytosis.
Of note, as Cdk5 represents a clinically relevant target, Cdk5 inhibition is indicated as a promising option to prevent the compensatory activation of growth factor receptors. Cdk5 can be pharmacologically targeted by Dinaciclib, a small molecule Cdk5 inhibitor that has shown promising anticancer effects with a manageable side‐effect profile in clinical trials. Dinaciclib is a well‐tolerated compound and has been successfully evaluated as therapy of chronic lymphocytic leukemia in phase III clinical trials and is currently under clinical investigation for the treatment of other types of leukemia.48, 49 According to our study, Dinaciclib represents a promising therapeutic option for HCC that increases sorafenib efficiency and inhibits treatment escape.
In conclusion, our study presents Cdk5 inhibition as a promising approach to increase sorafenib efficacy in HCC that might contribute to improve the therapeutic situation for patients with advanced‐stage HCC.
Potential conflict of interest
Nothing to report.
Author names in bold designate shared co‐first authorship.
Supporting information
Acknowledgment
We thank Sara Sigismund (IFOM IEO Campus, Milan, Italy) for her help with the EGFR experiments. We thank F. Perocci (LMU gene center) for allowing us to use the seahorse device. We thank Jana Peliskova (Pharmaceutical Biology, LMU, Munich, Germany) for her help with the cell culture experiments. We thank Kerstin Loske (Pharmaceutical Biology, LMU, Munich, Germany) for her help with the animal experiments.
A.M.V. was funded by the DFG (VO376/17‐1).
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011;61:69‐90. [DOI] [PubMed] [Google Scholar]
- 2. Bruix J, Gores GJ, Mazzaferro V. Hepatocellular carcinoma: clinical frontiers and perspectives. Gut 2014;63:844‐855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Forner A, Gilabert M, Bruix J, Raoul JL. Treatment of intermediate‐stage hepatocellular carcinoma. Nat Rev Clin Oncol 2014;11:525‐535. [DOI] [PubMed] [Google Scholar]
- 4. Llovet JM, Ricci S, Mazzaferro V, Hilgard P, Gane E, Blanc JF, et al. Sorafenib in advanced hepatocellular carcinoma. N Engl J Med 2008;359:378‐390. [DOI] [PubMed] [Google Scholar]
- 5. Nishida N, Kitano M, Sakurai T, Kudo M. Molecular mechanism and prediction of sorafenib chemoresistance in human hepatocellular carcinoma. Dig Dis 2015;33:771‐779. [DOI] [PubMed] [Google Scholar]
- 6. Abdel‐Rahman O, Fouad M. Sorafenib‐based combination as a first line treatment for advanced hepatocellular carcinoma: a systematic review of the literature. Crit Rev Oncol Hematol 2014;91:1‐8. [DOI] [PubMed] [Google Scholar]
- 7. Bruix J, Qin S, Merle P, Granito A, Huang YH, Bodoky G, et al. Regorafenib for patients with hepatocellular carcinoma who progressed on sorafenib treatment (RESORCE): a randomised, double‐blind, placebo‐controlled, phase 3 trial. Lancet 2017;389:56‐66. [DOI] [PubMed] [Google Scholar]
- 8. Abou‐Alfa GK, Meyer T, Cheng A‐L, El‐Khoueiry AB, Rimassa L, Ryoo B‐Y, et al. Cabozantinib (C) versus placebo (P) in patients (pts) with advanced hepatocellular carcinoma (HCC) who have received prior sorafenib: Results from the randomized phase III CELESTIAL trial. J Clin Oncol 2018;36(4 Suppl):207‐207. [Google Scholar]
- 9. Kudo M, Finn RS, Qin S, Han KH, Ikeda K, Piscaglia F, et al. Lenvatinib versus sorafenib in first‐line treatment of patients with unresectable hepatocellular carcinoma: a randomised phase 3 non‐inferiority trial. Lancet 2018;391:1163‐1173. [DOI] [PubMed] [Google Scholar]
- 10. Ehrlich SM, Liebl J, Ardelt MA, Lehr T, De Toni EN, Mayr D, et al. Targeting cyclin dependent kinase 5 in hepatocellular carcinoma–A novel therapeutic approach. J Hepatol 2015;63:102‐113. [DOI] [PubMed] [Google Scholar]
- 11. Dhavan R, Tsai LH. A decade of CDK5. Nat Rev Mol Cell Biol 2001;2:749‐759. [DOI] [PubMed] [Google Scholar]
- 12. Pozo K, Bibb JA. The emerging role of Cdk5 in cancer. Trends Cancer 2016;2:606‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang SF, Chang CW, Wei RJ, Shiue YL, Wang SN, Yeh YT. Involvement of DNA damage response pathways in hepatocellular carcinoma. Biomed Res Int 2014;2014:153867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Ito Y, Takeda T, Sakon M, Tsujimoto M, Higashiyama S, Noda K, et al. Expression and clinical significance of erb‐B receptor family in hepatocellular carcinoma. Br J Cancer 2001;84:1377‐1383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Blivet‐Van Eggelpoel MJ, Chettouh H, Fartoux L, Aoudjehane L, Barbu V, Rey C, et al. Epidermal growth factor receptor and HER‐3 restrict cell response to sorafenib in hepatocellular carcinoma cells. J Hepatol 2012;57:108‐115. [DOI] [PubMed] [Google Scholar]
- 16. Firtina Karagonlar Z, Koc D, Iscan E, Erdal E, Atabey N. Elevated hepatocyte growth factor expression as an autocrine c‐Met activation mechanism in acquired resistance to sorafenib in hepatocellular carcinoma cells. Cancer Sci 2016;107:407‐416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tovar V, Cornella H, Moeini A, Vidal S, Hoshida Y, Sia D, et al. Tumour initiating cells and IGF/FGF signalling contribute to sorafenib resistance in hepatocellular carcinoma. Gut 2017;66:530‐540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sigismund S, Confalonieri S, Ciliberto A, Polo S, Scita G, DiFiore PP. Endocytosis and signaling: cell logistics shape the eukaryotic cell plan. Physiol Rev 2012;92:273‐366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bouattour M, Raymond E, Qin S, Cheng AL, Stammberger U, Locatelli G, et al. Recent developments of c‐Met as a therapeutic target in hepatocellular carcinoma. Hepatology 2018;67:1132‐1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. You H, Ding W, Dang H, Jiang Y, Rountree CB. c‐Met represents a potential therapeutic target for personalized treatment in hepatocellular carcinoma. Hepatology 2011;54:879‐889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cheng AL, Kang YK, Lin DY, Park JW, Kudo M, Qin S, et al. Sunitinib versus sorafenib in advanced hepatocellular cancer: results of a randomized phase III trial. J Clin Oncol 2013;31:4067‐4075. [DOI] [PubMed] [Google Scholar]
- 22. Johnson PJ, Qin S, Park JW, Poon RT, Raoul JL, Philip PA, et al. Brivanib versus sorafenib as first‐line therapy in patients with unresectable, advanced hepatocellular carcinoma: results from the randomized phase III BRISK‐FL study. J Clin Oncol 2013;31:3517‐3524. [DOI] [PubMed] [Google Scholar]
- 23. Cainap C, Qin S, Huang WT, Chung IJ, Pan H, Cheng Y, et al. Linifanib versus sorafenib in patients with advanced hepatocellular carcinoma: results of a randomized phase III trial. J Clin Oncol 2015;33:172‐179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Bollard J, Miguela V, Ruiz de Galarreta M, Venkatesh A, Bian CB, Roberto MP, et al. Palbociclib (PD‐0332991), a selective CDK4/6 inhibitor, restricts tumour growth in preclinical models of hepatocellular carcinoma. Gut 2017;66:1286‐1296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Kudo M. A new era of systemic therapy for hepatocellular carcinoma with regorafenib and lenvatinib. Liver Cancer 2017;6:177‐184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Engelman JA, Zejnullahu K, Mitsudomi T, Song Y, Hyland C, Park JO, et al. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007;316:1039‐1043. [DOI] [PubMed] [Google Scholar]
- 27. Chen MC, Chen CH, Chuang HC, Kulp SK, Teng CM, Chen CS. Novel mechanism by which histone deacetylase inhibitors facilitate topoisomerase IIalpha degradation in hepatocellular carcinoma cells. Hepatology 2011;53:148‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lim HY, Heo J, Choi HJ, Lin CY, Yoon JH, Hsu C, et al. A phase II study of the efficacy and safety of the combination therapy of the MEK inhibitor refametinib (BAY 86–9766) plus sorafenib for Asian patients with unresectable hepatocellular carcinoma. Clin Cancer Res 2014;20:5976‐5985. [DOI] [PubMed] [Google Scholar]
- 29. Rudalska R, Dauch D, Longerich T, McJunkin K, Wuestefeld T, Kang TW, et al. In vivo RNAi screening identifies a mechanism of sorafenib resistance in liver cancer. Nat Med 2014;20:1138‐1146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu AX, Abrams TA, Miksad R, Blaszkowsky LS, Meyerhardt JA, Zheng H, et al. Phase 1/2 study of everolimus in advanced hepatocellular carcinoma. Cancer 2011;117:5094‐5102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Koeberle D, Dufour JF, Demeter G, Li Q, Ribi K, Samaras P, et al. Sorafenib with or without everolimus in patients with advanced hepatocellular carcinoma (HCC): a randomized multicenter, multinational phase II trial (SAKK 77/08 and SASL 29). Ann Oncol 2016;27:856‐861. [DOI] [PubMed] [Google Scholar]
- 32. Finn RS, Poon RT, Yau T, Klumpen HJ, Chen LT, Kang YK, et al. Phase I study investigating everolimus combined with sorafenib in patients with advanced hepatocellular carcinoma. J Hepatol 2013;59:1271‐1277. [DOI] [PubMed] [Google Scholar]
- 33. Gao JJ, Shi ZY, Xia JF, Inagaki Y, Tang W. Sorafenib‐based combined molecule targeting in treatment of hepatocellular carcinoma. World J Gastroenterol 2015;21:12059‐12070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gerbes A, Zoulim F, Tilg H, Dufour JF, Bruix J, Paradis V, et al. Gut roundtable meeting paper: selected recent advances in hepatocellular carcinoma. Gut 2018;67:380‐388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Berasain C, Avila MA. The EGFR signalling system in the liver: from hepatoprotection to hepatocarcinogenesis. J Gastroenterol 2014;49:9‐23. [DOI] [PubMed] [Google Scholar]
- 36. Rose A, Grandoch M, vom Dorp F, Rubben H, Rosenkranz A, Fischer JW, et al. Stimulatory effects of the multi‐kinase inhibitor sorafenib on human bladder cancer cells. Br J Pharmacol 2010;160:1690‐1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Wang H, Xu L, Zhu X, Wang P, Chi H, Meng Z. Activation of phosphatidylinositol 3‐kinase/Akt signaling mediates sorafenib‐induced invasion and metastasis in hepatocellular carcinoma. Oncol Rep 2014;32:1465‐1472. [DOI] [PubMed] [Google Scholar]
- 38. Daveau M, Scotte M, Francois A, Coulouarn C, Ros G, Tallet Y, et al. Hepatocyte growth factor, transforming growth factor alpha, and their receptors as combined markers of prognosis in hepatocellular carcinoma. Mol Carcinog 2003;36:130‐141. [DOI] [PubMed] [Google Scholar]
- 39. Zhu AX, Rosmorduc O, Evans TR, Ross PJ, Santoro A, Carrilho FJ, et al. SEARCH: a phase III, randomized, double‐blind, placebo‐controlled trial of sorafenib plus erlotinib in patients with advanced hepatocellular carcinoma. J Clin Oncol 2015;33:559‐566. [DOI] [PubMed] [Google Scholar]
- 40. Smith DS, Tsai LH. Cdk5 behind the wheel: a role in trafficking and transport? Trends Cell Biol 2002;12:28‐36. [DOI] [PubMed] [Google Scholar]
- 41. Kawauchi T. Cdk5 regulates multiple cellular events in neural development, function and disease. Dev Growth Differ 2014;56:335‐348. [DOI] [PubMed] [Google Scholar]
- 42. Furusawa K, Asada A, Urrutia P, Gonzalez‐Billault C, Fukuda M, Hisanaga SI. Cdk5 regulation of the GRAB‐mediated Rab8‐Rab11 cascade in axon outgrowth. J Neurosci 2017;37:790‐806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Corallino S, Malabarba MG, Zobel M, Di Fiore PP, Scita G. Epithelial‐to‐mesenchymal plasticity harnesses endocytic circuitries. Front Oncol 2015;5:45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wiedmann RM, von Schwarzenberg K, Palamidessi A, Schreiner L, Kubisch R, Liebl J, et al. The V‐ATPase‐inhibitor archazolid abrogates tumor metastasis via inhibition of endocytic activation of the Rho‐GTPase Rac1. Cancer Res 2012;72:5976‐5987. [DOI] [PubMed] [Google Scholar]
- 45. Schneider LS, von Schwarzenberg K, Lehr T, Ulrich M, Kubisch‐Dohmen R, Liebl J, et al. Vacuolar‐ATPase inhibition blocks iron metabolism to mediate therapeutic effects in breast cancer. Cancer Res 2015;75:2863‐2874. [DOI] [PubMed] [Google Scholar]
- 46. Merk H, Messer P, Ardelt MA, Lamb DC, Zahler S, Muller R, et al. Inhibition of the V‐ATPase by Archazolid A: a new strategy to inhibit EMT. Mol Cancer Ther 2017;16:2329‐2339. [DOI] [PubMed] [Google Scholar]
- 47. Sun T, Liu H, Ming L. Multiple roles of autophagy in the sorafenib resistance of hepatocellular carcinoma. Cell Physiol Biochem 2017;44:716‐727. [DOI] [PubMed] [Google Scholar]
- 48. Kumar SK, LaPlant B, Chng WJ, Zonder J, Callander N, Fonseca R, et al. Dinaciclib, a novel CDK inhibitor, demonstrates encouraging single‐agent activity in patients with relapsed multiple myeloma. Blood 2015;125:443‐448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Ghia P, Scarfo L, Perez S, Pathiraja K, Derosier M, Small K, et al. Efficacy and safety of dinaciclib vs ofatumumab in patients with relapsed/refractory chronic lymphocytic leukemia. Blood 2017;129:1876‐1878. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials