

Abstract
Chronic lymphocytic leukemia (CLL) remains incurable with current standard therapy. We have previously reported that an increased expression of interleukin‐6 (IL‐6) receptor CD126 leads to resistance of CLL cells to chemotherapy and worse prognosis for patients with CLL. In this study, we determine whether autocrine IL‐6 production by CLL B cells is associated with poor clinical outcome and explore IL‐6‐mediated survival mechanism in primary CLL cells. Our results demonstrate that higher levels of autocrine IL‐6 are significantly associated with shorter absolute lymphocyte doubling time, patients received treatment, without complete remission, advanced Binet stages, 17p/11q deletion, and shorter time to first time treatment and progression‐free survival. IL‐6 activated both STAT3 and nuclear factor kappa B (NF‐κB) in primary CLL cells. Blocking IL‐6 receptor and JAK2 inhibited IL‐6‐mediated activation of STAT3 and NF‐κB. Our study demonstrates that an increased autocrine IL‐6 production by CLL B‐cells are associated with worse clinical outcome for patients with CLL. IL‐6 promotes CLL cell survival by activating both STAT3 and NF‐κB through diverse signaling cascades. Neutralizing IL‐6 or blocking IL‐6 receptor might contribute overcoming the resistance of CLL cells to chemotherapy. We propose that the measurement of autocrine IL‐6 could be a useful approach to predict clinical outcome.
Keywords: autocrine IL‐6, chronic lymphocytic leukemia, clinical outcome, tumor microenvironment
1. INTRODUCTION
Chronic lymphocytic leukemia (CLL), the most common adult leukemia, is characterized by the progressive accumulation of phenotypically mature monoclonal CD5+/CD23+malignant B cells in the peripheral blood, bone marrow, and lymphoid organs. CLL is heterogeneous disease with a highly variable clinical course and prognostication at time of diagnosis is mainly relying on clinicobiological parameters, such as deletions at chromosome 17p or 11q, absence of mutations in the immunoglobulin heavy‐chain gene (IgVH), expression of protein tyrosine kinase zeta‐associated protein‐70 (ZAP‐70), the cell surface glycoprotein CD38, and advanced clinical Binet stages (Caligaris‐Cappio & Ghia, 2008; Chiorazzi, Rai, & Ferrarini, 2005). Despite improved clinical outcomes using current chemoimmunotherapy regimens, CLL remains incurable.
Circulating CLL cells are typically long‐lived, however, they rapidly undergo spontaneous apoptosis in vitro (Liu et al., 2010; Liu, Jia, Wang, Farren, et al., 2016), indicating that tumor microenvironmental prosurvival signals in both lymphoid tissue and the circulation play a major role in preventing CLL cells from apoptotic cell death (Balakrishnan, Burger, Wierda, & Gandhi, 2009; Liu et al., 2010; Luqman et al., 2008). Many soluble factors including interleukin (IL) ‐4, ‐6, ‐10, and HMGB1, have been found in the plasma and tissue microenvironment in patients with CLL and play important roles in supporting CLL cell survival (Aguilar‐Hernandez et al., 2016; Antosz et al., 2015; Drennan et al., 2017; Jia et al., 2014). Inspite of the emergence of new treatment strategies with more specific targets, there is an increased need for personalized treatment strategies for patients with CLL.
IL‐6 belongs to a larger family of cytokines and is mainly secreted by T cells and macrophages to stimulate immune response during infection (Grivennikov et al., 2009). It has been demonstrated that higher levels of plasma IL‐6 are positively correlated with worse clinical outcome in patients with CLL (Fayad et al., 2001) and other cancers (Ebrahimi, Tucker, Li, Abbruzzese, & Kurzrock, 2004; Meyer et al., 2010). Although IL‐6 is elevated in the plasma of humans suffering from breast, prostate, lung, liver, and colon cancer, raised plasma IL‐6 is also seen in a wide variety of nonmalignant diseases (Heikkila, Ebrahim, & Lawlor, 2008). It was recently reported that constitutively activated nuclear factor kappa B (NF‐κB) in CLL cells induces the production of IL‐6 which further enhances B‐cell receptor and JAK2/STAT3 activation (Rozovski et al., 2017). IL‐6/JAK/STAT3 form a feed‐forward loop which drives tumorigenesis and metastasis (Chang et al., 2013). Many types of cancer cells secrete IL‐6 due to constitutive activation of STAT3 or NF‐κB (Rozovski et al., 2017; Yu, Pardoll, & Jove, 2009). Autocrine and paracrine IL‐6 further maintains STAT3 and NF‐κB activation through direct or indirect signal pathways (Iliopoulos, Hirsch, & Struhl, 2009; Lesina et al., 2011; Yang et al., 2007). Once activated, both transcription factors regulate gene expression with significant upregulation of genes involved in survival, proliferation, and immunosuppression in cancer cells. However, the precise mechanism by which IL‐6 activates both STAT3 and NF‐κB are elusive.
Autocrine IL‐6 plays a crucial role in carcinogenesis and cancer progression in different types of cancer cells (Fukuda et al., 2011; Gao et al., 2007). We previously reported that IL‐6 membrane‐bound receptor CD126 surface expression was found in all CLL cases and positively correlated with the levels of in vivo constitutive STAT3 activity and resistance of CLL cells to chemotherapy. Blocking CD126 using tocilizumab sensitized CLL cells to chemotherapy (Liu, Jia, Wang, Farren, et al., 2016). In the current study, we aimed to determine whether the levels of autocrine IL‐6 have prognostic value in patients with CLL. Using both STRING database prediction and experimental approaches, we explored the possible link between IL‐6‐mediated activation of STAT3 with NF‐κB in CLL cells.
2. MATERIALS AND METHODS
2.1. Patients, CLL cell separation, culture
The National Research Ethics Service, East London and the City HA Local Research Ethics Committee approved nondiagnostic analyses, and written informed consent was obtained (REC reference 07/Q0604/34; Farren et al., 2015; Liu, Jia, Wang, Farren, et al., 2016). Patients with CLL (n = 71) with a lymphocyte count greater than 50 × 109/L were enrolled in this study and all these cases were either untreated (n = 25) or had not received chemotherapy or steroids for over 6 months. Peripheral blood mononuclear cells (PBMC) were isolated by density‐gradient centrifugation over Ficoll‐Paque (GE Healthcare, Hatfield, UK). In all samples, the dual CD19+/CD5+ B‐CLL cells were confirmed to represent more than 95% of the PBMCs. Freshly isolated CLL cells were suspended in RPMI‐1640 medium containing 10% fetal bovine serum and 2 mM l‐glutamine (Invitrogen, Carlsbad, CA) at a density of 5 × 106 cells/ml and cultured at 37°C in a humidified incubator with 5% CO2. The datasets used or analyzed during the present study are available from the corresponding author on reasonable request.
2.2. Treatment of CLL cells
To activate STAT3 or RelA, CLL cells were incubated with 10 ng/ml of recombinant human IL‐6 (Novartis Pharma AG, Basel, Switzerland). To inhibit STAT3 or RelA activity, CLL cells were treated with 10 µg/ml anti‐IL‐6R antibody (MRA; Chugai Pharmaceutical Co. Ltd., Tokyo, Japan), 10 µM AG490 (Sigma‐Aldrich, Dorset, UK), or 10 µM Wortmannin (Sigma‐Aldrich, Dorset, UK), respectively, for the indicated time periods. Supernatants were collected for autocrine IL‐6 production assays and the cells were used for flow cytometry and western blot analysis.
2.3. Measurement of autocrine production IL‐6
Fresh CLL cells were isolated immediately after phlebotomy. Percentages of CLL B‐cells were determined by flow cytometry after stained with PerCP‐CY5.5‐conjugated anti‐CD19 antibody. If CLL samples contain >95% CD19 positive cells, cells will be directly used for IL‐6 autocrine assay. For samples with <95% CD19 positive cells, CLL B cells will be positively selected by B‐cell isolation kit (Miltenyi Biotec, Woking, UK.; Supporting Information Figure 1 a,b). A total of 5 × 106/ml cells were cultured for 24 hr (or for the indicated times) with or without treatment, the supernatants were collected and stored at −80°C until assayed. Autocrine production of IL‐6 in the supernatants was measured by the enzyme‐linked immunosorbent assay according to the manufacturer's instructions (R&D Systems, Minneapolis, MN) with medium alone as the baseline control. The wells with medium alone did not have measurable levels of either IL‐6. The concentration of IL‐6 in the patient samples was calculated against a standard curve. For the samples treated with IL‐6, the concentrations of added IL‐6 in control wells were subtracted from the values in test conditions (Liu, Jia, Wang, Wang, et al., 2016).
2.4. Caspase‐3 activity assay
Cytosolic proteins (50 µg) were diluted to 90 µl with fluorogenic assay buffer (20 mM HEPES‐KOH; pH 7.4; 10 mM dithiothreitol, 10% sucrose; 1.0 mM ethylenediaminetetraacetic acid A; 0.1% CHAPS). The reaction was initiated by addition of 10 µl of 400 µM (final concentration was 40 µM) fluorescent substrate Ac‐DEVE‐AFC (Merck‐Calbiochem, Nottingham, UK) for the caspase‐3 activity. The cleavage reaction was carried out at 37°C for 15 min. The fluorescence at 400/505 nm was measured with a BMG LABTECH POLARstar OPTIMA Microplate Reader (Offenburg, Germany; Liu, Agarwal, Gribben, et al., 2008).
2.5. Differential detergent fractionation for cellular proteins
Differential detergent fractionation involves sequential extraction of cells using detergent‐containing buffer separating cellular proteins into distinct compartments. To extract the cytosolic fraction (fraction 1), CLL cells were suspended into buffer 1 containing 0.02% digitonin and incubated on ice for 10 min. After centrifugation at 8,000 rpm for 2 min, the cytosolic extracts were transferred into eppendorf tubes. The pellets were washed with buffer 1 to reduce cytosolic protein contamination. Fraction 2 proteins (mitochondria and membranous organelles) were extracted with buffer 2 containing 0.5% Triton X‐100. After washing with buffer 2 twice, fraction 3 nuclear proteins were directly extracted with NuPAGE® LDS Sample Buffer (Invitrogen, Carlsbad, CA) and heated to 100°C for 5 min. Identification of cellular protein fractions was performed by western blot analysis using specific antibodies: LDH for cytosolic, COX IV for mitochondrial, and Rb for nuclear proteins (Jia et al., 2014).
2.6. Western blot analysis
Proteins were extracted with CelLyticTM M Cell Lysis Reagent (Sigma‐Aldrich, Dorset, UK) supplied with protease and phosphatase inhibitor cocktails (Sigma‐Aldrich, Dorset, UK). Proteins were mixed with NuPAGE LDS Sample Buffer (Invitrogen, Carlsbad, CA) and boiled for 5 min before analysis by western blot analysis. Proteins were subjected to 4–20% NuPAGE gels (Invitrogen) and transferred onto polyvinylidene fluoride (PVDF) membrane (Sigma‐Aldrich, Dorset, UK) at 20 V for 1 hr by semidry transfer. PVDF membrane was blocked with 5% nonfat milk in TBST for 1 hr and then incubated with primary antibodies overnight at 4°C against the following targets: Bcl‐2 (100), RelA (NF‐κB p65, F‐6), Bcl‐XL (H‐5), Bax (2D2), Survivin (D‐8), and LDH (H‐10) (Santa Cruz Biotechnology, Inc., Dallas, TX); β‐actin (AC‐74) (Sigma‐Aldrich, Dorset, UK); p‐RelA (phospho‐NF‐κB p65‐Ser536), p‐STAT3 (phosphor‐STAT3‐Tyr705), p‐AKT, AKT, Mcl‐1, and Rb antibodies (Cell Signaling Technology‐New England Biolabs, Hitchin, UK). Bound antibodies were detected using appropriate horseshoeradish peroxidase‐conjugated secondary antibodies and visualized by GeneSnap (SynGene, Cambridge, UK) after adding ECL plus (GE Healthcare Life Science, Hatfield, UK).
2.7. Immunofluorescence staining
CLL cells were fixed and permeabilized on slides with Cytofix/Cytoperm reagents (BD Biosciences, Franklin Lakes, NJ) and blocked with a buffer consisting of 0.1% saponin and 5% donkey serum. Cells were stained with either monoclonal anti‐p‐RelA antibody (at 1:100 dilutions; Cell Signaling Technology, Danvers, MA) or monoclonal anti‐RelA antibody (at 1:20; Santa Cruz Biotechnology, Inc., Dallas, TX) for 1 hr at room temperature. After washing with Tris‐buffered saline Tween20 (TBST), cells were incubated with Alexa‐Fluor 488 conjugated antimouse secondary antibody at a 1:100 dilution (Invitrogen, Carlsbad, CA). Slides were washed for three times with TBST, stained with 50 ng/ml 4′,6‐diamidino‐2‐phenylindole, air‐dried at 4°C in the dark, and mounted in ProLong® Gold antifade reagent (Invitrogen, Carlsbad, CA) before being viewed under an Olympus BX40 fluorescence microscope (Artisan Scientific Corporation, Champaign, IL; Jia et al., 2014).
2.8. Determining NF‐κB DNA binding activity by electrophoretic mobility shift assay
NF‐κB DNA binding activity was assessed using the electrophoretic mobility shift assay (EMSA). This assay is based on the fact that DNA‐protein complexes migrate slower than nonbound DNA in native polyacrylamide, resulting in a “shift” in the migration of the labeled DNA band. Nuclear protein–DNA complexes were extracted by NE‐PER® Nuclear and Cytoplasmic Extraction Reagents (Pierce‐Thermo Scientific, Waltham, MA). Double‐stranded NF‐κB consensus oligonucleotides 5′‐AGT TGA GGG GAC TTT CCC AGG C‐3 and 3′‐TCA ACT CCC CTG AAA GGG TCC G‐5′ (Santa Cruz Biotechnology, Inc., Dallas, TX) containing a putative binding site for NF‐κB were end‐labeled with biotin using Biotin 3′ End DNA Labeling Kit (Pierce‐Thermo Scientific, Waltham, MA). Bands were detected by “The Light ShiftTM Chemiluminescent EMSA kit” (Pierce‐Thermo Scientific, Waltham, MA) using a nonisotopic method to detect DNA‐protein interactions. Biotin end‐labeled DNA duplex of NF‐κB was incubated with the nuclear extracts (10 µg nuclear proteins). After the reaction, the DNA–protein complexes were subjected to a 6% DNA retardation gel (Invitrogen, Carlsbad, CA) and transferred to a positively charged nylon membrane (Pierce‐Thermo Scientific). After transfer, the membrane was immediately cross‐linked for 15 min on a Gene Flash UV transilluminator (Syngene, Cambridge, UK). A chemiluminescent detection method utilizing a luminol/enhancer solution and a stable peroxide solution (Pierce‐Thermo Scientific, Waltham, MA) was used as described by the manufacturer, and membranes were exposed under using the Luminescent Image Analyzer (FujiFilm, Tokyo, Japan) for visualization (Jia, Gopinathan, Sukumar, & Gribben, 2012).
2.9. Knockdown specific mRNAs by transfection of siRNAs
Human STAT3 (sc‐29493), RelA, JAK2, and AKT siRNA, (Santa Cruz Biotechnology, Inc., Dallas, TX) consisted of a pool of 3 target‐specific 19–25 nt siRNAs designed to knock down gene expression. Fresh primary CLL cells (5 × 107/ml) were suspended in 100 µl of Human B Cell Nucleofector (R) reagent (Lonza, Basel, Switzerland) and 2 µg of STAT3 siRNA or control siRNA A was added into the mixture. The transfection was performed using NucleofectorTM II apparatus with the program U‐015 (Lonza, Basel, Switzerland). Transfection efficiency and cell death were determined by flow cytometry after transfection with PMAX‐GFP plasmid for 24 hr (Liu, Jia, Wang, Wang, et al., 2016). Protein expression was determined at indicated times.
2.10. Flow cytometry assays for cell death and mitochondrial function
Cell viability was determined by propidium iodide (PI) dye exclusion. After treatment, cells were incubated with 10 µg/ml PI (Sigma‐Aldrich, Dorset, UK) and the integrity of cell membrane was measured by flow cytometry using a FACS Canto (Becton Dickinson, Oxford, UK). To determine the mitochondrial membrane potential (ΔΨm), cells were stained with 40 nM tetramethylrhodamine methyl ester (TMRM; Invitrogen) at 37°C for 15 min and changes in ΔΨm were measured on the FH2‐H channel (Liu, Agarwal, Movasaghi, et al., 2008). To measure the production of reactive oxidative species (ROS), cells were stained with 40 µM dihydroethidium (HE; Polyscience‐Park Scientific, Northampton, UK) at 37°C for 15 min and increases in ROS production were measured on the FH3‐H channel (Liu, Agarwal, Movasaghi, et al., 2008). For mitochondrial cytochrome c release, cells were permeabilized or fixed as previously reported (Liu et al., 2010) and washed three times with phosphate buffer saline (PBS). Cells were incubated with 20 µl of PE‐conjugated anti‐cytochrome c mAb (Santa Cruz Biotechnology, Inc., Dallas, TX) for 1 hr at room temperature in the dark. Before analysis by flow cytometry, cells were washed once in PBS.
2.11. Analysis of protein−protein associations by STRING
Search Tool for the Retrieval of Interacting Genes/Proteins (STRING; http://string‐db.org/) is a precomputed database for the exploration and analysis of protein–protein associations. The associations are derived from high‐throughput experimental data, mining of databases and literature, analyses of coexpressed genes and also from computational predictions, including those based on genomic context analysis. STRING employs a unique scoring framework based on benchmarks of the different types of associations against a common reference set, to produce a single confidence score per prediction (Lee et al., 2011; Rakshit, Rathi, & Roy, 2014).
2.12. Statistical analysis
Data are shown as either mean ± standard deviation or median with interquartile range when variation was high. Significant differences between groups were analyzed using Student's t test. Pearson's correlation coefficient method was used to analyze the linear correlation between two groups. Outcomes, measured from date of diagnosis to event occurrence or date of the last follow‐up, were progression‐free survival, the event being a relapse after first treatment, and time to first time treatment (TTFT), the event of patients with CLL receive first treatment after diagnosis. All p < 0.05 were considered statistically significant. *, **, and *** indicate p < 0.05, 0.01, and 0.001, respectively (Jia et al., 2014). Statistical analyses were performed using IBM SPSS (London, UK) version 22 for Windows and GraphPad Prism version 5.03.
The levels of plasma IL‐6 were analyzed using the statistical software package X‐tile (Yale University; Camp, Dolled‐Filhart, & Rimm, 2004) enabling cut points to be determined for markers without validated normal ranges. The X‐tile divides patients into two independent data sets (test and validation), in a 1:2 ratio, determines optimal cut point(s) for each marker in the test set and applies them to the validation set (Camp et al., 2004). In univariate analysis, Kaplan–Meier survival curves were generated using these cut points and p‐values, HR and 95% CIs generated using a log‐rank test. The Cox proportional hazards model was used to identify the independent prognostic factors (Greaves et al., 2013; Yu et al., 2014).
3. RESULTS
3.1. The significant impact of autocrine IL‐6 production on clinical outcomes in patients with CLL
Autocrine IL‐6 is intrinsic to CLL cells themselves and may better reflect the underlying biology of different prognostic CLL subgroups. The association between levels of autocrine IL‐6 with each prognostic subgroup was analyzed in 71 CLL samples. In vivo, CLL cells are typically long‐lived which can be evaluated clinically by the lymphocyte doubling time (LDT). The patients with a longer LDT (>12 months) will have a better response to the treatment and a less aggressive clinical course (Liu, Jia,Wang, Farren, et al., 2016). Patients with CLL with a longer LDT (>12 months) have significantly lower levels of autocrine IL‐6 compared to the cases with a shorter LDT (<12 months; Figure 1a). Similarly, patients with CLL with not yet required treatment have significantly lower average autocrine IL‐6 production than previously been treated patients (Figure 1b). In vitro studies showed that the level of autocrine IL‐6 positively correlated with cell viability and negatively correlated with chemo‐sensitivity. This suggests that the higher levels of IL‐6 protect CLL from spontaneous and chemotherapy‐induced cell death (Supporting Information Figure 2). To evaluate whether the levels of autocrine IL‐6 associated with patient's response to clinical treatment, patients were divided into two groups: achieving a complete response (CR) versus not achieving a CR (ie, a partial response, stable disease, and progressive disease). The levels of autocrine IL‐6 in the non‐CR group were significantly higher than in the CR group (Figure 1c), IL‐6 was significantly higher in Binet stage B/C versus stage A, and 17p‐/11q‐ versus non 17p‐/11q‐cytogenetics. There were no statistically significant differences of levels of IL‐6 in CD38+ versus CD38‐ and ZAP70 + versus ZAP70‐subgroups (Figure 1d).
Figure 1.
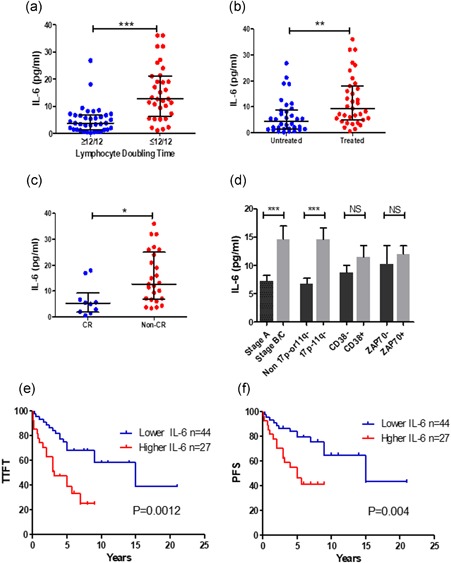
The association of autocrine IL‐6 with clinical outcome in patients with CLL. Autocrine IL‐6 production was determined by IL‐6 ELISA kit after 5 × 106/ml cells were cultured for 24 hr. Levels of autocrine IL‐6 production were compared in (a) LDT ≥ 12/12 (n = 38) vs. LDT ≤ 12⁄12 (n = 34), (b) treated (n = 35) vs. untreated (n = 34) groups, (c) patients with CR (n = 10) vs. non‐CR (n = 25), (d) other prognostic markers. Data were analyzed by the Mann–Whitney test. *p < 0.05, **p < 0.01, and ***p < 0.001. NS means not significant. (e) Time to the first treatment (TTFT) and (f) Progress‐free survival (PFS) of patients with CLL based on of patients with low autocrine IL‐6 (<9.35 pg/ml) and high IL‐6 (>9.35 pg/ml). Cut‐off point was obtained by the X‐tile software. CLL: chronic lymphocytic leukemia; CR: complete remission; ELISA: enzyme‐linked immunosorbent assay; IL‐6: interleukin 6; LDT: lymphocyte doubling time [Color figure can be viewed at wileyonlinelibrary.com]
To further explore the correlation between autocrine IL‐6 production and clinical prognosis, we determined the association between autocrine IL‐6 levels with the TTFT using categorical log‐rank analysis. The cut‐off point for IL‐6 (≥9.35 pg/ml) was obtained using the X‐tile software. Patients with CLL with lower levels of autocrine IL‐6 showed a significantly longer TTFT than patients with the higher levels of IL‐6 subset. The median TTFT in the higher IL‐6 group was 15 years versus 3.2 years in the lower IL‐6 group (Figure 1e). The TTFT is also significantly associated with other risk factors such as Binet stages and ZAP‐70 positivity, except CD38 (Supporting Information Figure 3). The median PFS in patients with lower IL‐6 group was 15 years versus 5 years in the higher IL‐6 group (Figure 1f). Median PFS in the stage A group was 15 years compared to 5 years in stage B/C group (Supporting Information Figure 3), and in the ZAP70‐ group was 15 years compared to 5 years in the ZAP70 + group, respectively (Supporting Information Figure 3). Multivariate analysis showed that the significance of autocrine IL‐6 levels remains after IL‐6 was stratified with all risk factors (Table 1) or a single risk factor (Table 2). This indicates that levels of autocrine IL‐6 on the prediction of clinical prognosis is independent of other prognostic markers. These findings suggest that the levels of autocrine IL‐6 have clinical utility as an independent prognostic marker for patients with CLL. However, this needs to be verified in other independent cohorts.
Table 1.
Uni‐ and multi‐variate analysis of categorical covariates of TTFT and PFS
| Outcome | Covariates (cut point) | Univariate analysis | Multivariate analysis | ||
|---|---|---|---|---|---|
| HR (95%CI) | p | HR (95% CI) | p | ||
| TTFT | IL‐6 a (>9.35vs.≤9.35) | 2.910 (1.461–5.796) | 0.002 | 3.597 (1.27–10.12) | 0.015 |
| Stage (B/C vs. A) | 3.770 (1.865–7.619) | <0.0001 | 0.751 (0.215–2.620) | 0.654 | |
| 17p11q (+vs.−) | 2.072 (1.033–4.155) | 0.040 | 1.711 (0.508–5.763) | 0.386 | |
| ZAP‐70 (+vs.−) | 3.033 (1.270–7.245) | 0.012 | 2.652 (0.843–8.349) | 0.095 | |
| CD38 (+vs.−) | 1.832 (0.908–3.693) | 0.091 | – | – | |
| PFS | IL‐6 a (>9.35vs.≤9.35) | 2.910 (1.345–6.298) | 0.007 | 3.581 (1.017–12.61) | 0.047 |
| Stage (B/C vs. A) | 4.245 (1.933–9.324) | <0.0001 | 1.246 (0.282–5.499) | 0.771 | |
| 17p11q (+vs.−) | 1.992 (0.908–4.371) | 0.086 | 1.571 (0.343–7.201) | 0.561 | |
| ZAP‐70 (+vs.−) | 2.854 (1.009–8.069) | 0.048 | 2.006 (0.528–7.614) | 0.307 | |
| CD38 (+vs. −) | 2.088 (0.957–4.558) | 0.065 | – | – | |
Note. Covariates that were included in the multivariate analysis were first selected using the Enter and then the backward/forward stepwise.
CI: confidence interval; HR: hazard ratio; PFS: progression‐free survival; TTFT: shorter time to first time treatment.
Categorical cut‐off points were defined by the X‐tile software, N = 71.
Table 2.
Multivariate analysis for IL‐6 on clinical outcome
| Covariate | TTFT a | PFS a | ||
|---|---|---|---|---|
| HR; 95% CI | p Value | HR; 95% CI | p Value | |
| Stage | 2.706 (1.257–5.823) | 0.011 | 3.231 (1.386–7.536) | 0.007 |
| IL‐6 | 2.361 (1.081–5.160) | 0.031 | 2.171 (0.922–5.109) | 0.076 |
| 17p11q | 1.409 (0.661–3.001) | 0.374 | 1.382 (0.597–3.199) | 0.450 |
| IL‐6 | 2.543 (1.198–5.398) | 0.015 | 2.599 (1.134–5.957) | 0.024 |
| ZAP‐70 | 2.394 (0.977–5.868) | 0.056 | 2.134 (0.720–6.326) | 0.172 |
| IL‐6 | 2.885 (1.141–7.296) | 0.025 | 2.957 (0.932–9.384) | 0.066 |
| CD38 | 1.517 (0.742–3.101) | 0.254 | 1.788 (0.806–3.969) | 0.153 |
| IL‐6 | 3.091 (1.488–6.421) | 0.002 | 2.886 (1.288–6.466) | 0.010 |
Note. Effect of IL‐6 on clinical outcome was adjusted individually by each risk factor.
CI: confidence interval; HR: hazard ratio; IL‐6: interleukin 6; PFS: progression‐free survival; TTFT: shorter time to first time treatment; ZAP‐70: zeta‐associated protein‐70.
Number of cases = 71.
3.2. IL‐6 induces NF‐κB and STAT3 activation in primary CLL cells in vitro
The signaling pathway by which IL‐6 induces STAT3 activation has been well documented (Akira et al., 1994; Antosz et al., 2015; Zhong, Wen, & Darnell, 1994). However, the mechanisms by which IL‐6‐mediated activation of NF‐κB is still elusive. We found IL‐6 induces time‐dependent upregulation of both p‐RelA (NF‐κB‐p65) and p‐STAT3 in primary CLL cells as detected by western blot analysis (Figure 2a). Immunofluorescent staining showed that the p‐RelA is weakly expressed in the cytosol of CLL cells in the resting stage. After stimulation with IL‐6, increased p‐RelA mainly localized in the nucleus of CLL cells. In contrast, non‐phosphorylated RelA was localized in the nucleus in both the control and IL‐6 treated CLL cells (Figure 2b). To further confirm whether IL‐6 induces NF‐κB activation, NF‐κB DNA binding activity was determined by EMSA. IL‐6 stimulation significantly increased NF‐κB DNA binding activity, as detected in three cases of primary CLL cells (Figure 2c,d). These results indicate that IL‐6 not only activates STAT3 but also stimulates NF‐κB, suggesting that the NF‐κB is involved in IL‐6‐mediated signaling network.
Figure 2.
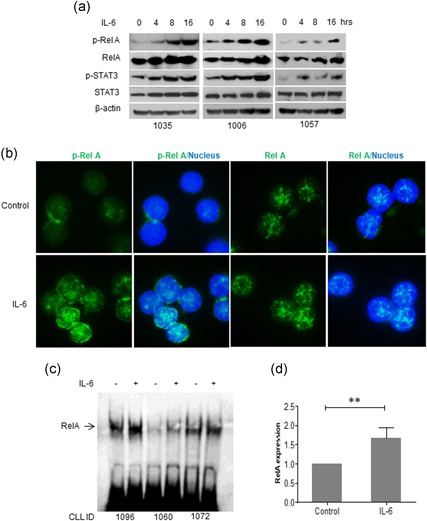
IL‐6‐mediated activation of NF‐κB and STAT3. (a) IL‐6‐induced phosphorylation of RelA and STAT3. Primary CLL cells were incubated with 10 ng/ml IL‐6 and cells were collected for western blot analysis at indicated times. Numbers on the bottom are patients ID. (b) Localization of P‐RelA and RelA. Primary CLL cells were incubated with IL‐6 for 4 hr. Cells on slides were stained with either P‐RelA or RelA antibodies, respectively. DAPI indicates nucleus location. (c) IL‐6‐mediated RelA binding to DNA as detected by EMSA. (d) Statistical analysis of IL‐6‐induced binding between RelA and DNA. CLL: chronic lymphocytic leukemia; DAPI: 4′,6‐diamidino‐2‐phenylindole; EMSA: electrophoretic mobility shift assay [Color figure can be viewed at wileyonlinelibrary.com]
3.3. Database prediction of functional associations between proteins in IL‐6 signaling pathway
The causal relationship between IL‐6‐mediated activation of STAT3 and NF‐κB is unclear. We established a theoretical IL‐6 signaling network using the online STRING program. First, five well‐known tumor survival signaling proteins, IL‐6, JAK2, AKT1, STAT3, and RelA were chosen to predict their functional associations (Rakshit et al., 2014). All of these proteins are involved in the cellular response to cytokine stimulus (showing in the yellow color) and four of them (except RelA) are responsible for regulation of reactive oxygen species (ROS) biosynthetic process (in the red color; Figure 3a). After randomly expanded this network, PIK3CA, PIK3CB, and PI3KCD, which activate AKT, bridge the link between JAK2 and AKT, are shown in the network. MTOR, which is a molecule in PI3K/AKT/mTOR pathway, has a strong association with JAK2 and STAT3 but a weak link with IL‐6. Similar to RelA, NFKB1A (p105/50) can also be activated by IL‐6 (Figure 3b).
Figure 3.
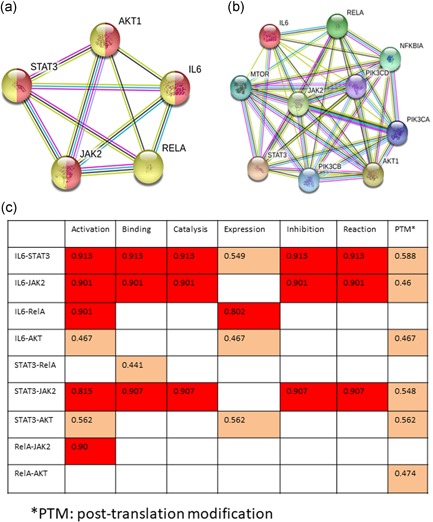
STRING prediction of functional associations of proteins in IL‐6 signaling pathway. (a) Functional network of 5 IL‐6 associated signaling molecules; (b) Expanded network. (c) Prediction of specific action scores between proteins. The red color indicates score >0.80 and orange color shows action scores are at medium levels. IL‐6: interleukin 6; STRING: search tool for the retrieval of interacting genes/proteins [Color figure can be viewed at wileyonlinelibrary.com]
The specific actions between proteins were predicted by STRING (Figure 3c). The stronger specific action was marked as red color and weaker action as orange color. IL‐6 has the strongest functional links with both STAT3 and JAK2, including activation, binding, catalysis, inhibition and posttranslation modification (PTM). The specific actions between STAT3 and JAK2 are similar to their own functional links to IL‐6. The specific action of IL‐6 to RelA includes activation and expression. Although IL‐6‐mediated activation of STAT3 and AKT also induces gene or protein expression, their scores of actions on expression are far lower than RelA. The association between STAT3 and RelA is only “binding,” suggesting that these two transcriptional factors may not direct activate each other.
3.4. JAK2 and PI3‐K/AKT are required for IL‐6‐induced RelA activation in CLL
To determine the experimental and biological association between IL‐6‐mediated NF‐κB activation, the anti‐IL‐6 receptor antibody was used to block IL‐6 binding to its surface receptor and the JAK2 inhibitor AG490 was used to inhibit JAK2‐mediated signaling. IL‐6‐mediated phosphorylation of STAT3 and RelA were significantly decreased after treatment with anti‐IL‐6 receptor antibody or AG490 (Figure 4a,b). To determine whether STAT3 is involved in IL‐6‐induced RelA phosphorylation in CLL, the levels of phosphorylated STAT3 and RelA were determined by western blot analysis after knockdown either STAT3 or RelA. Surprisingly, knockdown of STAT3 did not inhibit p‐RelA expression and similarly knockdown of RelA had no effect on p‐STAT3 expression (Figure 4c,d). In a good agreement with STRING prediction on protein–protein association, our results indicate that STAT3 and RelA do not activate each other in CLL cells, at least at the early time points after stimulation with exogenous IL‐6.
Figure 4.
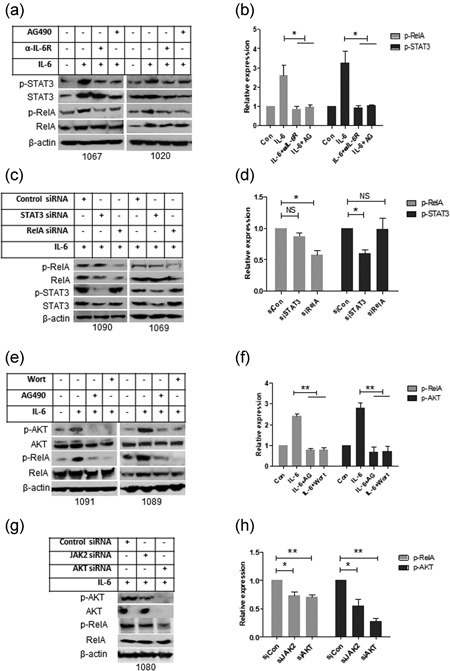
Functional analysis of the IL‐6‐mediated signaling pathway. (a,b) After IL‐6 receptor was blocked by anti‐IL‐6 receptor antibody and JAK2 was blocked by AG490; primary CLL cells were incubated with IL‐6 for 16 hr. (c,d) STAT3 or RelA was knocked down by transfection of STAT3 or RelA siRNA, and cells were incubated with IL‐6. The levels of P‐STAT3 and P‐RelA were determined by western blot analysis. (e,f) CLL cells were treated with PI3K inhibitor Wortmannin or AG490 and then incubated with IL‐6 for 16 hr. (g,h) JAK2 or AKT was knocked down by transfection of JAK2 or AKT siRNA and after 24 hr cells were incubated with IL‐6 for 16 hr. Phosphorylation of AKT and RelA was determined by western blot analysis. A, C, E, and G are representative western blot analysis and B, D, F, and H are statistical analysis of at least three cases data. Relative expression of phosphorylated protein was analyzed by densitometry using actin as control. Data shown are mean ± SD from at least three independent experiments. CLL: chronic lymphocytic leukemia; IL‐6:interleukin 6; SD: standard deviation
It was previously reported that PI3K/AKT are downstream effectors in IL‐6‐mediated survival signaling pathways (Hideshima, Nakamura, Chauhan, & Anderson, 2001). Treatment with wortmannin (PI3K inhibitor) or AG490 significantly inhibited IL‐6‐mediated phosphorylation of AKT and RelA in CLL cells (Figure 4e,f). Knockdown of JAK2 inhibited IL‐6‐mediated phosphorylation of AKT and RelA and silencing AKT significantly decreased IL‐6‐induced RelA phosphorylation (Figure 4g,h). Our results, in a good agreement with the STRING prediction, demonstrate that IL‐6 mediates NF‐κB activation is dependent, a least partly, on JAK2‐associated PI3K/AKT signaling pathway.
3.5. IL‐6 increases mitochondrial STAT3 expression and preserves mitochondrial function
Recent studies have revealed a novel role of p‐STAT3S727 in sustaining mitochondrial electron transport chain (ETC) activity, which regulates mitochondrial metabolic function and cell survival in normal and malignant cells (Gough et al., 2009; Wegrzyn et al., 2009). We determined whether mitochondrial STAT3 is involved in the improved CLL cell survival mediated by IL‐6. STAT3 protein was detected in cytoplasmic, mitochondrial and nuclear fractions (Figure 5a). Stimulation by IL‐6 increased expression of STAT3 in the mitochondrial fraction (Figure 5b). We previously reported that spontaneous apoptosis of CLL cells in vitro is via a mitochondria/caspase‐dependent pathway which involves ROS generation, ΔΨm depolarization, and cytochrome c release from mitochondria, resulting in activation of caspase‐3 (Liu, Jia, Wang, Wang, et al., 2016). CLL cells treated with IL‐6 showed reduced ROS generation, ΔΨm depolarization, cytochrome c release, and caspase‐3 activity (Figures 5c–f). Inhibition of JAK2 by AG490 significantly diminished the protective roles of IL‐6 on the mitochondria/caspase‐dependent apoptotic pathway (Figures 4c–f). Expression of Mcl‐1 and Bcl‐xL is coregulated by both STAT3 and NF‐κB. Inhibition of either STAT3 or NF‐κB decreases the expression of Mcl‐1 and Bcl‐xL in CLL cells (Liu, Jia, Wang, Wang, et al., 2016). It was shown above that IL‐6 increased phosphorylation of both STAT3 and RelA (Figure 2a). In turn, the activation of STAT3 and RelA by IL‐6 also increased expression of antiapoptotic protein Bcl‐xL and Mcl‐1, but not Bcl‐2 and Survivin (Figure 5g). In keeping with increased expression of antiapoptotic Bcl‐xL and Mcl‐1, stimulation with exogenous IL‐6 increased in vitro survival rate of CLL cells (Figure 5h), possibly in vivo as well. These results demonstrate that IL‐6 prevents apoptotic cell death by multiple mechanisms (Figure 6); (a) activating both STAT3 and NF‐κB via JAK2/PI3K/AKT axis and therefore upregulation of Bcl‐xL and Mcl‐1; and (b) promoting STAT3 mitochondrial translocation.
Figure 5.
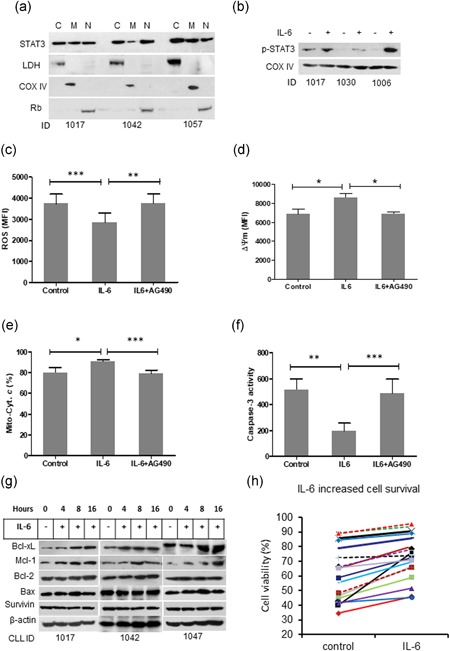
IL‐6 prevents mitochondria‐dependent apoptosis in CLL cells. (a) STAT3 intracellular distribution. Fresh primary CLL cells were fractionalized into cytoplasm (c), mitochondria (M) and nucleus (N). STAT3 expression was determined by western blot analysis. LDH, COX IV, and Rb are markers for cytoplasm, mitochondria and nucleus, respectively. (b) IL‐6 induced phosphorylation of STAT3. (c) ROS production, (d) Mitochondrial depolarization, and (e) cytochrome c release were measured by flow cytometry. (f) Caspase‐3 activity was measured by a fluorogenic assay. Data from A to F are mean ± SD from three independent experiments. Significant difference was analyzed by the Student t‐test. (g) Effect of IL‐6 on expression of Bcl‐xL, Mcl‐1, Bcl‐2, Bax, and Survivin was determined by western blot analysis. (h) CLL cell viability. CLL cells were incubated with or without IL‐6 for 24 hr. Cell viability was determined by flow cytometry after cells were stained with PI. CLL: chronic lymphocytic leukemia; IL‐6: interleukin 6; PI: propidium iodidie; ROS; reactive oxidative species; SD: standard deviation [Color figure can be viewed at wileyonlinelibrary.com]
Figure 6.
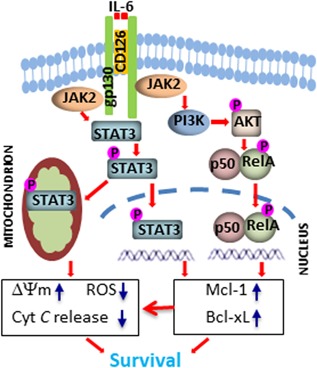
Schematic illustration of IL‐6‐mediated activation of STAT3 and NF‐κB. After engagement with receptor CD126, IL‐6 activates JAK2, which induces diverse signaling pathways: (1) activates STAT3 by inducing STAT3 phosphorylation, mitochondrial and nuclear translocation; (2) activates PI3K/AKT axis, which induces NF‐κB activation. Both activated STAT3 and NF‐κB induce expression of Bcl‐xL and Mcl‐1. Increased expression and Bcl‐xL and Mcl‐1 in the mitochondria, together with mitochondrial STAT3, prevent mitochondria‐dependent apoptosis and promote CLL cell survival. CLL: chronic lymphocytic leukemia [Color figure can be viewed at wileyonlinelibrary.com]
4. DISCUSSION
In the current study, we report for the first time that higher levels of autocrine IL‐6 are significantly associated with poor clinical outcomes in patients with CLL. Levels of autocrine IL‐6 could be used clinically to predict TTFT and PFS for patients with CLL. Moreover, using both STRING prediction and experimental approaches, we found that IL‐6 mediates activation of both STAT3 and NF‐κB via JAK2/PI3K/AKT axis in primary CLL cells.
It has been reported that IL‐6 levels in the plasma are associated with advanced stage and poor clinical outcome in patients with CLL (Fayad et al., 2001; Lai et al., 2002). However, the source of IL‐6 production was not specified. In this study, we determined autocrine IL‐6 production by CLL B cells. In tumor microenvironment, autocrine or paracrine IL‐6 mediates cell growth, differentiation and cytokine production through activation of the transcriptional factor STAT3 (Fukuda et al., 2011; Grivennikov et al., 2009; Iliopoulos et al., 2009). IL‐6 is a critical factor for tumorigenesis, cancer cell survival and proliferation (Bollrath et al., 2009; Lesina et al., 2011). In the dogma of inflammation‐associated cancer, the initial source of IL‐6 is from nonmalignant immune cells (Grivennikov et al., 2009), whereas most cancer cells can produce IL‐6, so both autocrine and paracrine IL‐6 in the cancer microenvironment maintain STAT3 and NF‐κB activation via an IL‐6 positive feedback loop (Iliopoulos et al., 2009; Lee et al., 2009). Autocrine production of IL‐6 by cancer cells promotes resistance to cell death: apoptosis‐sensitive cells do not express IL‐6, whereas high levels of IL‐6 are produced by apoptosis‐resistant cells in breast cancer (Conze et al., 2001).
We found a strong correlation between autocrine IL‐6 production, resistance to apoptosis and clinical prognosis: autocrine IL‐6 levels are variable in CLL, however, high IL‐6 levels are associated with resistance to spontaneous apoptosis in vitro and a poorer clinical outcome in patients with CLL. The presence of 17p and/or 11q deletion and the expression of ZAP‐70 or CD38 are all associated with poorer outcomes (Agrawal et al., 2008; Del Principe et al., 2006; Dohner et al., 2000; Wierda et al., 2011). Due to data unavailability of IgVH mutation status of patients with CLL, we were unable to analyze the association between autocrine IL‐6 and IgVH mutation. We found that regardless of the biological parameter studied, all risk‐marker positive patients have higher levels of autocrine IL‐6 production; furthermore, patients with higher autocrine IL‐6 levels have a shorter TTFT and PFS compared to patients with lower autocrine IL‐6. Only autocrine IL‐6 showed a significant correlation with TTFT; whereas in univariate analysis for PFS, autocrine IL‐6 and cytogenetic status showed statistically significant correlations, which were maintained in multivariate analysis.
Transcriptional factors STAT3 and NF‐κB play crucial roles in carcinogenesis, cancer cell proliferation and survival (Bollrath et al., 2009; Espinosa et al., 2010; Iliopoulos et al., 2009; Lesina et al., 2011). STAT3 and NF‐κB are constitutively active in most cancer cells in response to autocrine and paracrine signals that are produced within the tumor microenvironment (Grivennikov & Karin, 2010). We have previously reported there is marked heterogeneity of in vivo expression of constitutively activated p‐STAT3 and p‐RelA in CLL cells, which control autocrine IL‐6 and TNF‐α production, whereas inhibition of the constitutive activation of STAT3 or RelA results in decreased production of both IL‐6 and TNF‐α (Liu, Jia, Wang, Wang, et al., 2016). The functional relationship between STAT3 and NF‐κB is complex. Knocking down STAT3 decreases nuclear but increases the cytoplasmic distribution of p‐RelA in prostate cancer cells (Lee et al., 2009). Whereas NF‐κB can indirectly mediate STAT3 activation via IL‐6 production (Iliopoulos et al., 2009).
IL‐6 plays an important role in the crosstalk between STAT3 and NF‐κB in cancer cells, with IL‐6 involved in both STAT3 and NF‐κB activation, although diverse mechanisms of action have been reported in different types of cells. Iliopoulos et al reported that NF‐κB activation in tamoxifen‐treated MCF10 cells increases IL‐6 secretion and thereby enhances STAT3 activation (Iliopoulos et al., 2009). The activated STAT3 then directly activates miR‐21 and miR181b‐1, which leads to a positive feedback loop for NF‐κB activation by inhibiting PTEN and CYLD (Iliopoulos, Jaeger, Hirsch, Bulyk, & Struhl, 2010). In human mammary epithelial cells, IL‐6 enhances STAT3 transcription and expression, with the unphosphorylated STAT3 binding to unphosphorylated NF‐κB. The complex of unphosphorylated STAT3/NF‐κB can translocate into the nucleus and regulate target gene expression (Yang et al., 2007). In CLL, NOTCH1 mutations induce NF‐κB nuclear translocation and activation (Benedetti et al., 2018). Our data shows IL‐6 can induce NF‐κB activation in primary CLL cells, with enhanced RelA phosphorylation, nuclear translocation, DNA binding, and NF‐κB‐mediated IL‐6 production. Predicted by STRING database, STAT3 and NF‐κB bind but don't activate each other.
We dissected the pathway from IL‐6 to NF‐κB activation by blockade of the IL‐6 pathway (preventing IL‐6 binding to its receptor or inhibition of JAK2) and blockade of NF‐κB activation by inhibition of PI3K/AKT signaling. As expected, either approach leads to a decrease in IL‐6‐induced p‐AKT and p‐RelA expression. Surprisingly, knockdown of STAT3 did not affect IL‐6‐induced RelA or AKT phosphorylation. These results reveal a direct pathway of IL‐6‐induced NF‐κB activation in CLL cells involving JAK2, PI3K/AKT, and RelA, but independent of STAT3 (Figure 6).
The inhibition of apoptosis by either STAT3 or NF‐κB activation is mediated by transcription of several genes that are known to block the induction of apoptosis, such as Bcl‐2, Bcl‐xL and Mcl‐1 (Dai et al., 2004; Lesina et al., 2011; Oh et al., 2011; Yu et al., 2009), as well as upregulation of cytokines, such as IL‐6 (Bollrath et al., 2009; Iliopoulos et al., 2009). Expression of antiapoptotic proteins Bcl‐xL and Mcl‐1 in CLL cells is consistent with STAT3 and NF‐κB activation and the survival of primary CLL cells correlates with their STAT3 and/or NF‐κB activation status. Survival of CLL cells in vitro also showed a highly significant correlation with autocrine IL‐6 production.
Targeting IL‐6 has been used therapeutically in a variety of cancers and nonmalignant conditions (Jones, Scheller, & Rose‐John, 2011; Stone et al., 2012) but it has not been clinical investigated in CLL. As we previously reported, blocking IL‐6 membrane‐receptor CD126 using tocilizumab sensitizes CLL cells to chemotherapy (Liu, Jia, Wang, Farren et al., 2016). We propose that neutralizing IL‐6 could have great therapeutic potential in the improvement of life expectancy of patients with CLL.
In conclusion, our study demonstrates that higher capacity of autocrine IL‐6 production of CLL B‐cells is associated with shorter lymphocyte doubling time, shorter time to the time to first treatment and shorter progress‐free survival in patients with CLL. IL‐6 protects CLL cells from apoptosis by activating both STAT3 and NF‐κB via multiple signaling cascades. In CLL cells, autocrine IL‐6 provides a measure of the global activity of STAT3 and NF‐κB. As a simple laboratory assay, autocrine IL‐6 measurement may have clinical utility beyond CLL. Furthermore, our data also indicate a potential therapeutic role for interrupting IL‐6 signaling in CLL.
CONFLICTS OF INTEREST
The authors declare that there are no conflicts of interest.
AUTHOR CONTRIBUTIONS
F. T. L, S. G. A, and L. J conceived and designed this study. H. Q. W, F. T. L, T. F, and L. J performed experiments. H. Q. W and T. F collect clinical information, F. T. L and L. J analyzed data. All authors contributed to paper writing and consented for publication.
Supporting information
Supporting information
ACKNOWLEDGMENT
The authors thank patients with CLL who kindly donated samples for this research work. This work was supported by the National Natural Science Foundation of China (NNSF; 81172109) and (81570191) to F. T. L, and (81570194) to L. J.
Wang H-Q, Jia L, Li Y-T, Farren T, Agrawal SG, Liu F-T. Increased autocrine interleukin‐6 production is significantly associated with worse clinical outcome in patients with chronic lymphocytic leukemia. J Cell Physiol. 2018;234:13994–14006. 10.1002/jcp.28086
References
REFERENCES
- Agrawal, S. G. , Liu, F. T. , Wiseman, C. , Shirali, S. , Liu, H. , Lillington, D. , … Jia, L. (2008). Increased proteasomal degradation of Bax is a common feature of poor prognosis chronic lymphocytic leukemia. Blood, 111(5), 2790–2796. [DOI] [PubMed] [Google Scholar]
- Aguilar‐Hernandez, M. M. , Blunt, M. D. , Dobson, R. , Yeomans, A. , Thirdborough, S. , Larrayoz, M. , … Steele, A. J. (2016). IL‐4 enhances expression and function of surface IgM in CLL cells. Blood, 127(24), 3015–3025. [DOI] [PubMed] [Google Scholar]
- Akira, S. , Nishio, Y. , Inoue, M. , Wang, X. J. , We, S. , Matsusaka, T. , … Kishimoto, T. (1994). Molecular cloning of APRF, a novel IFN‐stimulated gene factor 3 p91‐related transcription factor involved in the gp130‐mediated signaling pathway. Cell, 77(1), 63–71. [DOI] [PubMed] [Google Scholar]
- Antosz, H. , Wojciechowska, K. , Sajewicz, J. , Choroszyńska, D. , Marzec‐Kotarska, B. , Osiak, M. , … Baszak, J. (2015). IL‐6, IL‐10, c‐Jun and STAT3 expression in B‐CLL. Blood Cells, Molecules, and Diseases, 54(3), 258–265. [DOI] [PubMed] [Google Scholar]
- Balakrishnan, K. , Burger, J. A. , Wierda, W. G. , & Gandhi, V. (2009). AT‐101 induces apoptosis in CLL B cells and overcomes stromal cell‐mediated Mcl‐1 induction and drug resistance. Blood, 113(1), 149–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benedetti, D. , Tissino, E. , Pozzo, F. , Bittolo, T. , Caldana, C. , Perini, C. , … Zucchetto, A. (2018). NOTCH1 mutations are associated with high CD49d expression in chronic lymphocytic leukemia: Link between the NOTCH1 and the NF‐kappaB pathways. Leukemia, 32(3), 654–662. [DOI] [PubMed] [Google Scholar]
- Bollrath, J. , Phesse, T. J. , von Burstin, V. A. , Putoczki, T. , Bennecke, M. , Bateman, T. , … Greten, F. R. (2009). gp130‐mediated Stat3 activation in enterocytes regulates cell survival and cell‐cycle progression during colitis‐associated tumorigenesis. Cancer Cell, 15(2), 91–102. [DOI] [PubMed] [Google Scholar]
- Caligaris‐Cappio, F. , & Ghia, P. (2008). Novel insights in chronic lymphocytic leukemia: Are we getting closer to understanding the pathogenesis of the disease? Journal of Clinical Oncology, 26(27), 4497–4503. [DOI] [PubMed] [Google Scholar]
- Camp, R. L. , Dolled‐Filhart, M. , & Rimm, D. L. (2004). X‐tile: A new bio‐informatics tool for biomarker assessment and outcome‐based cut‐point optimization. Clinical Cancer Research, 10(21), 7252–7259. [DOI] [PubMed] [Google Scholar]
- Chang, Q. , Bournazou, E. , Sansone, P. , Berishaj, M. , Gao, S. P. , Daly, L. , … Bromberg, J. (2013). The IL‐6/JAK/Stat3 feed‐forward loop drives tumorigenesis and metastasis. Neoplasia, 15(7), 848–862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiorazzi, N. , Rai, K. R. , & Ferrarini, M. (2005). Chronic lymphocytic leukemia. New England Journal of Medicine, 352(8), 804–815. [DOI] [PubMed] [Google Scholar]
- Conze, D. , Weiss, L. , Regen, P. S. , Bhushan, A. , Weaver, D. , Johnson, P. , & Rincon, M. (2001). Autocrine production of interleukin 6 causes multidrug resistance in breast cancer cells. Cancer Research, 61(24), 8851–8858. [PubMed] [Google Scholar]
- Dai, Y. , Pei, X. Y. , Rahmani, M. , Conrad, D. H. , Dent, P. , & Grant, S. (2004). Interruption of the NF‐kB pathway by bay 11‐7082 promotes UCN‐01‐mediated mitochondrial dysfunction and apoptosis in human multiple myeloma cells. Blood, 103(7), 2761–2770. [DOI] [PubMed] [Google Scholar]
- Del Principe, M. I. , Del Poeta, G. , Buccisano, F. , Maurillo, L. , Venditti, A. , Zucchetto, A. , … Amadori, S. (2006). Clinical significance of ZAP‐70 protein expression in B‐cell chronic lymphocytic leukemia. Blood, 108(3), 853–861. [DOI] [PubMed] [Google Scholar]
- Döhner, H. , Stilgenbauer, S. , Benner, A. , Leupolt, E. , Kröber, A. , Bullinger, L. , … Lichter, P. (2000). Genomic aberrations and survival in chronic lymphocytic leukemia. New England Journal of Medicine, 343(26), 1910–1916. [DOI] [PubMed] [Google Scholar]
- Drennan, S. , D'Avola, A. , Gao, Y. , Weigel, C. , Chrysostomou, E. , Steele, A. J. , … Forconi, F. (2017). IL‐10 production by CLL cells is enhanced in the anergic IGHV mutated subset and associates with reduced DNA methylation of the IL10 locus. Leukemia, 31(8), 1686–1694. [DOI] [PubMed] [Google Scholar]
- Ebrahimi, B. , Tucker, S. L. , Li, D. , Abbruzzese, J. L. , & Kurzrock, R. (2004). Cytokines in pancreatic carcinoma: Correlation with phenotypic characteristics and prognosis. Cancer, 101(12), 2727–2736. [DOI] [PubMed] [Google Scholar]
- Espinosa, L. , Cathelin, S. , D'Altri, T. , Trimarchi, T. , Statnikov, A. , Guiu, J. , … Bigas, A. (2010). The Notch/Hes1 pathway sustains NF‐kappaB activation through CYLD repression in T cell leukemia. Cancer Cell, 18(3), 268–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farren, T. W. , Giustiniani, J. , Fanous, M. , Liu, F. , Macey, M. G. , Wright, F. , … Agrawal, S. G. (2015). Minimal residual disease detection with tumor‐specific CD160 correlates with event‐free survival in chronic lymphocytic leukemia. Blood Cancer Journal, 5, e273–e273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fayad, L. , Keating, M. J. , Reuben, J. M. , O'Brien, S. , Lee, B. N. , Lerner, S. , & Kurzrock, R. (2001). Interleukin‐6 and interleukin‐10 levels in chronic lymphocytic leukemia: Correlation with phenotypic characteristics and outcome. Blood, 97(1), 256–263. [DOI] [PubMed] [Google Scholar]
- Fukuda, A. , Wang, S. C. , Morris, J. P. , Folias, A. E. , Liou, A. , Kim, G. E. , … Hebrok, M. (2011). Stat3 and MMP7 contribute to pancreatic ductal adenocarcinoma initiation and progression. Cancer Cell, 19(4), 441–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao, S. P. , Mark, K. G. , Leslie, K. , Pao, W. , Motoi, N. , Gerald, W. L. , … Bromberg, J. F. (2007). Mutations in the EGFR kinase domain mediate STAT3 activation via IL‐6 production in human lung adenocarcinomas. Journal of Clinical Investigation, 117(12), 3846–3856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough, D. J. , Corlett, A. , Schlessinger, K. , Wegrzyn, J. , Larner, A. C. , & Levy, D. E. (2009). Mitochondrial STAT3 supports Ras‐dependent oncogenic transformation. Science, 324(5935), 1713–1716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greaves, P. , Clear, A. , Coutinho, R. , Wilson, A. , Matthews, J. , Owen, A. , … Gribben, J. G. (2013). Expression of FOXP3, CD68, and CD20 at diagnosis in the microenvironment of classical Hodgkin lymphoma is predictive of outcome. Journal of clinical oncology: official journal of the American Society of Clinical Oncology, 31(2), 256–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov, S. , Karin, E. , Terzic, J. , Mucida, D. , Yu, G. Y. , Vallabhapurapu, S. , … Karin, M. (2009). IL‐6 and Stat3 are required for survival of intestinal epithelial cells and development of colitis‐associated cancer. Cancer Cell, 15(2), 103–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grivennikov, S. I. , & Karin, M. (2010). Dangerous liaisons: STAT3 and NF‐kappaB collaboration and crosstalk in cancer. Cytokine & growth factor reviews, 21(1), 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heikkilä, K. , Ebrahim, S. , & Lawlor, D. A. (2008). Systematic review of the association between circulating interleukin‐6 (IL‐6) and cancer. European Journal of Cancer, 44(7), 937–945. [DOI] [PubMed] [Google Scholar]
- Hideshima, T. , Nakamura, N. , Chauhan, D. , & Anderson, K. C. (2001). Biologic sequelae of interleukin‐6 induced PI3‐K/Akt signaling in multiple myeloma. Oncogene, 20(42), 5991–6000. [DOI] [PubMed] [Google Scholar]
- Iliopoulos, D. , Hirsch, H. A. , & Struhl, K. (2009). An epigenetic switch involving NF‐kappaB, Lin28, Let‐7 MicroRNA, and IL6 links inflammation to cell transformation. Cell, 139(4), 693–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iliopoulos, D. , Jaeger, S. A. , Hirsch, H. A. , Bulyk, M. L. , & Struhl, K. (2010). STAT3 activation of miR‐21 and miR‐181b‐1 via PTEN and CYLD are part of the epigenetic switch linking inflammation to cancer. Molecular Cell, 39(4), 493–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, L. , Gopinathan, G. , Sukumar, J. T. , & Gribben, J. G. (2012). Blocking autophagy prevents bortezomib‐induced NF‐kappaB activation by reducing I‐kappaBalpha degradation in lymphoma cells. PLOS One, 7(2), e32584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia, L. , Clear, A. , Liu, F. T. , Matthews, J. , Uddin, N. , McCarthy, A. , … Gribben, J. G. (2014). Extracellular HMGB1 promotes differentiation of nurse‐like cells in chronic lymphocytic leukemia. Blood, 123(11), 1709–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, S. A. , Scheller, J. , & Rose‐John, S. (2011). Therapeutic strategies for the clinical blockade of IL‐6/gp130 signaling. Journal of Clinical Investigation, 121(9), 3375–3383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai, R. , O'Brien, S. , Maushouri, T. , Rogers, A. , Kantarjian, H. , Keating, M. , & Albitar, M. (2002). Prognostic value of plasma interleukin‐6 levels in patients with chronic lymphocytic leukemia. Cancer, 95(5), 1071–1075. [DOI] [PubMed] [Google Scholar]
- Lee, H. , Herrmann, A. , Deng, J. H. , Kujawski, M. , Niu, G. , Li, Z. , … Yu, H. (2009). Persistently activated Stat3 maintains constitutive NF‐kappaB activity in tumors. Cancer Cell, 15(4), 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, S. A. , Tsao, T. , Yang, K. C. , Lin, H. , Kuo, Y. L. , Hsu, C. H. , … Kao, C. Y. (2011). Construction and analysis of the protein‐protein interaction networks for schizophrenia, bipolar disorder, and major depression. BMC Bioinformatics, 12(Suppl 13), S20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesina, M. , Kurkowski, M. U. , Ludes, K. , Rose‐John, S. , Treiber, M. , Klöppel, G. , … Algül, H. (2011). Stat3/Socs3 activation by IL‐6 transsignaling promotes progression of pancreatic intraepithelial neoplasia and development of pancreatic cancer. Cancer Cell, 19(4), 456–469. [DOI] [PubMed] [Google Scholar]
- Liu, F. T. , Jia, L. , Wang, P. , Wang, H. , Farren, T. W. , & Agrawal, S. G. (2016). STAT3 and NF‐kappaB cooperatively control in vitro spontaneous apoptosis and poor chemo‐responsiveness in patients with chronic lymphocytic leukemia. Oncotarget, 7(22), 32031–32045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, F. T. , Agrawal, S. G. , Movasaghi, Z. , Wyatt, P. B. , Rehman, I. U. , Gribben, J. G. , … Jia, L. (2008). Dietary flavonoids inhibit the anticancer effects of the proteasome inhibitor bortezomib. Blood, 112(9), 3835–3846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, F. T. , Agrawal, S. G. , Gribben, J. G. , Ye, H. , Du, M. Q. , Newland, A. C. , & Jia, L. (2008). Bortezomib blocks Bax degradation in malignant B cells during treatment with TRAIL. Blood, 111(5), 2797–2805. [DOI] [PubMed] [Google Scholar]
- Liu, F. T. , Giustiniani, J. , Farren, T. , Jia, L. , Bensussan, A. , Gribben, J. G. , & Agrawal, S. G. (2010). CD160 signaling mediates PI3K‐dependent survival and growth signals in chronic lymphocytic leukemia. Blood, 115(15), 3079–3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, F. T. , Jia, L. , Wang, P. , Farren, T. , Li, H. , Hao, X. , & Agrawal, S. G. (2016). CD126 and targeted therapy with tocilizumab in chronic lymphocytic leukemia. Clinical Cancer Research, 22(10), 2462–2469. [DOI] [PubMed] [Google Scholar]
- Luqman, M. , Klabunde, S. , Lin, K. , Georgakis, G. V. , Cherukuri, A. , Holash, J. , … Younes, A. (2008). The antileukemia activity of a human anti‐CD40 antagonist antibody, HCD122, on human chronic lymphocytic leukemia cells. Blood, 112(3), 711–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer, F. , Samson, E. , Douville, P. , Duchesne, T. , Liu, G. , & Bairati, I. (2010). Serum prognostic markers in head and neck cancer. Clinical Cancer Research, 16(3), 1008–1015. [DOI] [PubMed] [Google Scholar]
- Oh, H. M. , Yu, C. R. , Golestaneh, N. , Amadi‐Obi, A. , Lee, Y. S. , Eseonu, A. , … Egwuagu, C. E. (2011). STAT3 protein promotes T‐cell survival and inhibits interleukin‐2 production through up‐regulation of class O forkhead transcription factors. Journal of Biological Chemistry, 286(35), 30888–30897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakshit, H. , Rathi, N. , & Roy, D. (2014). Construction and analysis of the protein‐protein interaction networks based on gene expression profiles of Parkinson's disease. PLOS One, 9(8), e103047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozovski, U. , Harris, D. M. , Li, P. , Liu, Z. , Jain, P. , Veletic, I. , … Estrov, Z. (2017). Activation of the B‐cell receptor successively activates NF‐kappaB and STAT3 in chronic lymphocytic leukemia cells. International Journal of Cancer, 141(10), 2076–2081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone, R. L. , Nick, A. M. , McNeish, I. A. , Balkwill, F. , Han, H. D. , Bottsford‐Miller, J. , … Sood, A. K. (2012). Paraneoplastic thrombocytosis in ovarian cancer. New England Journal of Medicine, 366(7), 610–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn, J. , Potla, R. , Chwae, Y. J. , Sepuri, N. B. V. , Zhang, Q. , Koeck, T. , … Larner, A. C. (2009). Function of mitochondrial Stat3 in cellular respiration. Science, 323(5915), 793–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wierda, W. G. , O'Brien, S. , Wang, X. , Faderl, S. , Ferrajoli, A. , Do, K. A. , … Keating, M. J. (2011). Multivariable model for time to first treatment in patients with chronic lymphocytic leukemia. Journal of Clinical Oncology, 29(31), 4088–4095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, J. , Liao, X. , Agarwal, M. K. , Barnes, L. , Auron, P. E. , & Stark, G. R. (2007). Unphosphorylated STAT3 accumulates in response to IL‐6 and activates transcription by binding to NFkappaB. Genes and Development, 21(11), 1396–1408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, H. , Pardoll, D. , & Jove, R. (2009). STATs in cancer inflammation and immunity: A leading role for STAT3. Nature Reviews Cancer, 9(11), 798–809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu, L. X. , Yan, L. , Yang, W. , Wu, F. Q. , Ling, Y. , Chen, S. Z. , … Wang, H. Y. (2014). Platelets promote tumour metastasis via interaction between TLR4 and tumour cell‐released high‐mobility group box 1 protein. Nature Communications, 5, 5256. [DOI] [PubMed] [Google Scholar]
- Zhong, Z. , Wen, Z. , & Darnell, J. E., Jr. (1994). Stat3 and Stat4: Members of the family of signal transducers and activators of transcription. Proceedings of the National Academy of Sciences of the United States of America, 91(11), 4806–4810. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information