

Abstract
Cystic kidneys are common causes of end-stage renal disease, both in children and in adults. Autosomal dominant polycystic kidney disease (ADPKD) and autosomal recessive polycystic kidney disease (ARPKD) are cilia-related disorders and the two main forms of monogenic cystic kidney diseases. ADPKD is a common disease that mostly presents in adults, whereas ARPKD is a rarer and often more severe form of polycystic kidney disease (PKD) that usually presents perinatally or in early childhood. Cell biological and clinical research approaches have expanded our knowledge of the pathogenesis of ADPKD and ARPKD and revealed some mechanistic overlap between them. A reduced ‘dosage’ of PKD proteins is thought to disturb cell homeostasis and converging signalling pathways, such as Ca2+, cAMP, mechanistic target of rapamycin, WNT, vascular endothelial growth factor and Hippo signalling, and could explain the more severe clinical course in some patients with PKD. Genetic diagnosis might benefit families and improve the clinical management of patients, which might be enhanced even further with emerging therapeutic options. However, many important questions about the pathogenesis of PKD remain. In this Primer, we provide an overview of the current knowledge of PKD and its treatment.
In patients with polycystic kidney diseases (PKDs), the kidneys contain multiple fluid-filled cysts, although other organs may also be affected (FIG. 1). Although PKD is inherited monogenically, it is phenotypically, genically and allelically heterogeneous. Autosomal dominant PKD (ADPKD) is the most common form of PKD and is generally an adult-onset, multisystem disorder that is characterized by gradually growing renal cysts that start to develop in utero and can originate from all areas of the kidneys, although cysts usually form in the distal regions of the nephron and the collecting duct. Progressive fibrocystic renal disease in ADPKD is often accompanied by hepatobiliary changes or other extrarenal abnormalities, such as intracranial arterial aneurysms1,2. Mutations in PKD1 or PKD2, which encode polycystin 1 (PC1) and PC2, respectively, are the most common cause of ADPKD. PC1 and PC2 form a heterodimeric complex through the interaction of the coiled-coil motifs in the carboxy-terminal tails of each protein, which likely functions at primary cilia. PC2 is a cation channel that is a member of the transient receptor potential (TRP) family of ion channels, which are ubiquitously present across evolution and usually contain six membrane-spanning helices that are flanked by an intracellular amino terminus and carboxyl terminus (FIG. 2). By contrast, the roles of PC1 and the PC1–PC2 complex are only poorly understood. Autosomal recessive PKD (ARPKD) is rarer, and the clinical course is usually much more severe than with ADPKD. ARPKD typically manifests perinatally or in childhood, and patients often die perinatally or in infancy. Manifestations of the disease include hepatic fibrosis and greatly enlarged kidneys, and cysts typically affect the collecting ducts. ARPKD is primarily caused by mutations in polycystic kidney and hepatic disease 1 (PKHD1), which encodes fibrocystin (also known as polyductin), a protein that localizes to the primary cilium and basal body.
Fig. 1 |. Renal and extrarenal manifestations in polycystic kidney disease.
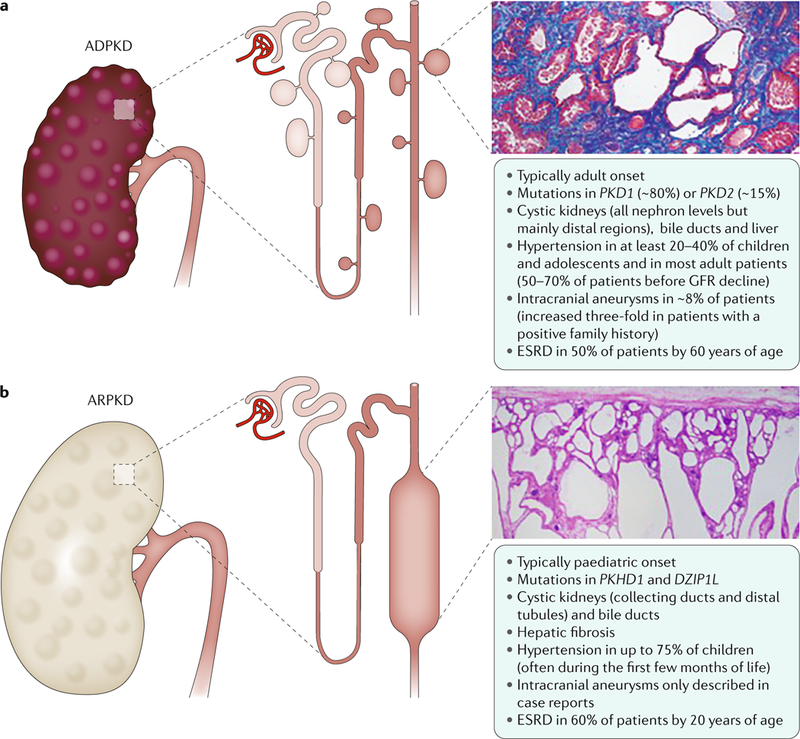
a | Autosomal dominant polycystic kidney disease (ADPKD) is the most common form of polycystic kidney disease (PKD) and is mainly caused by mutations in PKD1 and PKD2, which encode polycystin 1 and polycystin 2, respectively. ADPKD is usually an adult-onset disease that is characterized by the formation of fluid-filled cysts in various locations in the kidneys, but mostly in the distal regions. The histology image is of the kidney (stained with Malloy trichrome stain) of a 49-year-old patient with end-stage ADPKD. Small cysts and extensive fibrosis (blue) are visible. b | Autosomal recessive PKD (ARPKD) is rarer and more severe than ADPKD and is caused by mutations in polycystic kidney and hepatic disease 1 (PKHD1; which encodes fibrocystin) and DZIP1L (which encodes DAZ-interacting protein 1-like protein (DZIP1L)). ARPKD usually presents in utero, perinatally or in infancy and is characterized by the formation of cysts from the renal distal tubules and collecting ducts. Microscopically, cysts are fusiform dilatations of the aforementioned distal parts of the nephron, which are lined by a columnar or cuboidal epithelium. Respective dilated collecting ducts run perpendicular to the renal capsule (renal section is stained with haematoxylin and eosin). Both diseases often progress to end-stage renal disease (ESRD) that requires renal replacement therapy. GFR,glomerular filtration rate.
Fig. 2 |. Domain organization of proteins implicated in polycystic kidney disease.
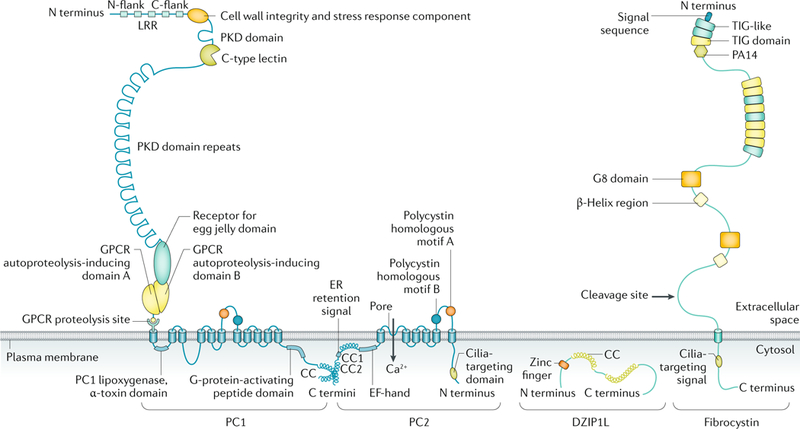
The structure of polycystin 1 (PC1), polycystin 2 (PC2), the membrane-bound form of fibrocystin and DAZ-interacting protein 1-like protein (DZIP1L) are depicted (not to scale). PC1 and PC2 are multispan membrane proteins that form a complex that is localized to multiple subcellular locations, including the primary cilium. Fibrocystin is also localized to the primary cilium and is subject to Notch-like proteolytic processing, resulting in release of the carboxy-terminal tail, which can translocate to the nucleus and may regulate gene expression. DZIP1L is a soluble zinc-finger protein that is localized to the centrioles and basal bodies at the ciliary transition zone. CC, coiled coil; C flank, carboxyl flank; C terminus, carboxyl terminus; ER, endoplasmic reticulum; GPCR, G protein-coupled receptor; LRR, leucine-rich repeat; N flank, amino flank; N terminus, amino terminus; PKD, polycystic kidney disease. Adapted from REF.293, Springer Nature Limited.
Although single renal cysts are common and usually benign in adults, bilateral cystic kidneys require careful clinical assessment to identify the underlying genetic disorder, especially in children, owing to major implications for the patient and family. Both ADPKD and ARPKD often result in chronic kidney disease (CKD; a gradual loss of kidney function) and end-stage renal disease (ESRD; severe kidney disease that requires dialysis or kidney transplantation for patient survival), which are associated with substantial associated morbidity and mortality.
In the past decade, progress has been made in unravelling the aetiology of PKD. Studies have shown that the proteins encoded by genes associated with PKD colocalize in multimeric complexes at distinct subcellular sites in epithelial cells, and compelling evidence exists to suggest that primary cilia have a central pathogenic role in PKD3–5. Cell biological and clinical research approaches have further extended our understanding of the pathophysiology of PKD and are starting to help in the identification of rational personalized therapies.
In this Primer, we provide a general overview of the current knowledge of ADPKD and ARPKD, including the epidemiology, pathophysiology, prevention, prognosis and quality of life (QOL). Furthermore, we discuss the diagnostic approaches and the clinical management of PKD and its comorbidities in greater detail. Finally, we provide a brief outlook and discuss potential future therapeutic options.
Epidemiology
ADPKD
ADPKD is the most common potentially lethal (that is, it results in renal failure and death in the absence of renal replacement therapy (RRT)) single-gene disorder and the most prevalent inherited progressive kidney disease2. The prevalence of ADPKD is reported to be between 1 in 400 and 1 in 1,000 live births, mainly on the basis of two early landmark clinical studies, in Denmark6 and in Minnesota, USA7. On the basis of this prevalence, ADPKD is predicted to affect >10 million people worldwide in all ethnic groups and therefore constitutes a major public health burden.
To ascertain the exact prevalence of a hereditary disease, several factors must be considered8 — the geographic and ethnic composition and the size of the population, the choice and mode of calculation of epidemiologic measurement, the screening policies and other characteristics of the health-care system, the disease definition and diagnostic criteria that are used, the sources of ascertainment and possible causes of under-ascertainment and the period of time during which events are counted (ascertainment period). Thus, variability in study design and ascertainment might partially explain the large differences in estimates of the prevalence of ADPKD that have been reported in some studies. For example, two clinical surveys in Europe (carried out in the United Kingdom9 and in France10), as well as a survey in the Seychelles11 measured the prevalence of symptomatic individuals and estimated the prevalence of asymptomatic individuals. The estimated prevalence of ADPKD was 1 in 2,459 in the UK study, 1 in 1,111 in the French study and 1 in 542 for the Seychelles. In the study of the Seychelles population, the researchers speculated that the high prevalence among white individuals might be due to a founder effect (that is, low genetic variation in a population because it was started by a small number of individuals).
Subsequent studies based on renal phenotypic features reported an ADPKD prevalence of 1 in 2,700 to 1 in 4,000 in different populations in Europe and Japan12–15. A recent study on ADPKD prevalence in the European Union, which reviewed the epidemiology literature from 1980 to 2015 and was based mainly on the large Registry of the European Renal Association (ERA)–European Dialysis and Transplant Association (EDTA), estimated that the prevalence of ADPKD in 19 countries was 1 in 2,525 (REF.16); that is, fewer than 5 cases in 10,000, which is the threshold for a rare disease in the European Union.Importantly, the prevalence rates estimated in all these studies were much lower than the ADPKD prevalence estimated from autopsy studies (≥1 in 500)6,7,17. These data suggest that marked under-ascertainment exists and that a considerable number of (probably only mildly or moderately) affected individuals remain undiagnosed18.
ADPKD is an important cause of ESRD, for which patients receive RRT, which includes haemodialysis, peritoneal dialysis, haemofiltration, haemodiafiltration and kidney transplantation. In a population-based registry in Germany, 32% of patients with ADPKD were receiving RRT14. ESRD occurs as a result of ADPKD in up to 75% of patients by 70 years of age14,19–21. Age-adjusted sex ratios suggest that ADPKD is more progressive in men than in women (1.2–1.3 men to women) on the basis of rates of ESRD. A Canadian study reported that 25% of patients with ADPKD had ESRD by 47 years of age, 50% by 59 years of age and 75% by 70 years of age22. In a French study, 22% of patients with ADPKD had ESRD by 50 years of age, 42% by 58 years of age and 72% by 73 years of age23. A European study of >300,000 patients starting RRT between 1991 and 2010 reported that the age-adjusted and sex-adjusted incidence of RRT in patients with ADPKD increased slightly over the study period from 7.6 per million to 8.3 per million population24. The researchers in this study noted that the incidence of RRT had markedly increased over time in patients >70 years of age, whereas no change was observed for the cohort of patients with ADPKD who were <50 years of age, suggesting that older patients are receiving RRT. In the UK Renal Registry, mortality in patients with ADPKD receiving RRT was 3.18–4.96 per 100 person-years, which was lower than that of patients receiving RRT who had other renal diseases. The ADPKD group had a lower hazard for all-cause mortality than the other renal diseases group (adjusted HR 0.45, 95% CI 0.38–0.53)25. Cardiac diseases and infections were the leading causes of death in patients with ADPKD who received RRT24.
ARPKD
As ARPKD is much rarer than ADPKD, the available epidemiological data for ARPKD are not as extensive as those for ADPKD. The reported incidence of ARPKD in North, Central and South America is 1 in 26,500 live births, corresponding to a carrier frequency of ~1 in 70 in non-isolated populations26. Isolated or inbred populations with many consanguineous marriages might have a much higher prevalence — an incidence of 1 in 8,000 births in Finland was reported27. Boys and girls seem to be equally affected by ARPKD. Of note, among children with PKD in paediatric nephrology departments, the total number of patients with early-onset ADPKD might be comparable to the number of children with ARPKD28. The overall prevalence of PKD in children is estimated to be ~1 in 10,000.
After delivery, the pulmonary status of neonatal infants usually dictates a requirement for early disease management of ARPKD. Mortality of 30–40% due to pulmonary hypoplasia has been reported for ARPKD29,30. However, for infants who survive the perinatal period, the long-term prognosis for patient survival is much better than generally perceived by many medical professionals. One-year and 10-year survival are ~85% and ~82%, respectively29–32.
Mechanisms/pathophysiology
The polycystin proteins are localized predominantly, although not exclusively, in the primary cilium, which is a hair-like structure that protrudes from the apical membrane of renal epithelial cells into the lumen of the nephron. The distribution of the polycystin proteins at different subcellular locations is required for them to orchestrate a network of signalling pathways that establish and maintain a differentiated renal epithelium33.
ADPKD
Genes and proteins.
Mutations in PKD1 (chromo-some 16p13.3) are responsible for almost 80% of cases of ADPKD, whereas ~15% of ADPKD cases are attributed to mutations in PKD2 (chromosome 4q22.1) and the remaining ~5–10% of ADPKD cases are genetically unresolved or are due to rare mutations in other loci18. These rare mutations that result in an ADPKD-like phenotype occur in hepatocyte nuclear factor 1β (HNF1B; which is a transcription factor that upregulates the expression of multiple PKD-associated genes, including PKHD1 and PKD2), neutral α-glucosidase AB (GANAB; which is involved in protein folding)34,35 and DNAJB11 (which encodes a chaperone protein that is associated with binding-immunoglobulin protein (BiP; also known as HSPA5))36. In addition, ADPKD can result from mutations in genes that are primarily associated with autosomal dominant polycystic liver disease (ADPLD), including SEC63 (which encodes a protein that is required for protein translocation across the endoplasmic reticulum (ER) membrane) and PRKCSH (which encodes the regulatory β-subunit of glycosidase 2 that is involved in protein folding)37–39. Patients with mutations in GANAB can manifest with an ADPKD or ADPLD phenotype, and other possible ADPLD-causing genes include LRP5 (which encodes a WNT co-receptor), ALG8 (which encodes an α−1,3-glucosyltransferase) and SEC61B (which encodes the β-subunit of the SEC61 trans-locon)35,40. Carriers with a mutation in one PKHD1 allele may also sometimes manifest with a few liver or kidney cysts35,41. In addition, autosomal dominant tubulointer-stitial kidney disease (ADTKD) caused by mutations in uromodulin (UMOD), mucin 1 (MUC1), renin (REN) and SEC61A (which encodes the α-subunit of the SEC61 translocon) can also sometimes be mistaken for ADPKD, although ADTKD is usually associated with renal impairment without renal enlargement and with only a few cysts42–45. Mutations in COL4A1 (which encodes a collagen subunit), including those associated with hereditary angiopathy with nephropathy, aneurysms and muscle cramps (HANAC) syndrome46, can sometimes cause an ADPKD-like phenotype18. The X-linked ciliopathy caused by mutations in OFD1 (which encodes a centriole and centriolar satellite protein and is associated with oral-facial-digital syndrome type I) can also sometimes be confused with ADPKD, although additional facial and digital phenotypes are usually present. In summary, mutations in PKD1 and PKD2 are the main cause of ADPKD, but mutations that interfere with the biogenesis of PC1, reduce the expression of PKD1 or PKD2 or ciliopathy and other mutations can sometimes mimic ADPKD18. ADPKD can sometimes also be mimicked by the hereditary cancer syndromes tuberous sclerosis and von Hippel–Lindau syndrome in cases in which extrarenal features are either absent or mild47.
PC1 is a large, integral membrane protein of 4,303 amino acids with 11 transmembrane domains, an extensive extracellular domain containing multiple predicted motifs and a small, ~200-amino acid intracellular carboxy-terminal tail that regulates multiple signalling cascades48 (FIG. 2). PC1 is cleaved at the G protein-coupled receptor proteolytic site to generate a large amino-terminal fragment that remains non-covalently tethered to the carboxy-terminal fragment at the plasma membrane49. This cleavage seems to regulate the biogenesis and trafficking of PC1 (REF.50). In addition, regulated cleavage of the intracellular C-terminal tail releases PC1 fragments into the cytoplasm, which translocate to the nucleus and interact with transcription activators, repressors and co-activators to modulate various signalling pathways51,52.
Although many of the functions of PC1 remain unclear, the C-terminal tail of PC1 is known to mediate the interaction of PC1 with PC2 (REF.53), which is involved in the regulation of ion transport and indirectly affects Ca2+ signalling. The cryo-electron microscopy structure of PC2 establishes it as a homotetrameric ion channel, although it is possible that PC2 heteromultimerizes with other TRP channel subunits or with PC1 (REFS54,55).
Patients with ADPKD usually carry a germline mutation in one allele of either PKD1 or PKD2 (see Diagnosis section, below), although at least one second event, such as somatic inactivation of the remaining wild-type PKD1 or PKD2 allele or loss of heterozygosity, is required to initiate cyst formation56–58. In addition, variants in other genes linked to PKD59 or unidentified modifying genes, as well as environmental factors such as acute kidney injury, can modulate cyst formation and disease progression60,61. The likelihood of cyst formation substantially increases when the level of functional PC1 or PC2 drops below a critical threshold56–58,62,63; accordingly, the amount of functional PC1 or PC2 and, thereby, the type of mutation (for example, inactivating mutations or mutations that reduce the function of the gene product (hypomorphic mutations)) affect the likelihood of cyst formation and the disease progression62–64 (FIG. 3). In mice, disruption of Pkd1 in a small number (10%) of cells leads to an ~6-month dormant period in which there is no renal tubular dilatation, followed by a period with rapid cyst formation, suggesting that additional triggers are required to initiate cyst formation60,65.
Fig. 3 |. The dosage model of cystogenesis in autosomal dominant polycystic kidney disease.
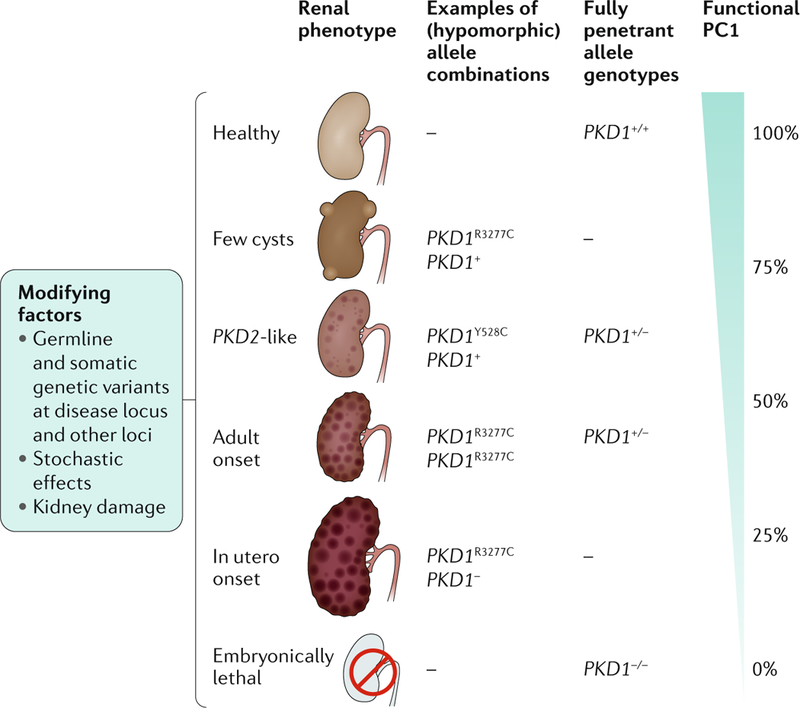
The level of functional polycystin 1 (PC1; encoded by PKD1) directly influences the renal phenotype in patients with autosomal dominant polycystic kidney disease (ADPKD) — an ~50% reduction in PC1 levels (for example, from haploinsufficiency due to a single inactivating allele) is associated with adult-onset PKD, whereas the complete absence of PC1 is lethal. Furthermore, incompletely penetrant (hypomorphic) PKD1 alleles of different strengths and combinations also influence the renal phenotype. For example, the PKD1Y528C allele results in a phenotype similar to that in patients with mutations in PKD2, whereas the PKD1R3277C allele can result in a phenotype that ranges in severity from just a few cysts to adult-onset disease to early-onset disease, depending on which PKD1 allele is present in trans. Additional mutations and/or variants of the disease-causing locus and somatic and germline mutations at other loci, as well as chance and environmental factors, influence the disease course by determining the frequency of cyst development and their progression. Adapted with permission from REF.294, Elsevier.
Determinants of disease severity.
The renal phenotype in patients with ADPKD ranges from patients in old age without renal failure to rare cases of enlarged kidneys that are detected in utero18. The identity of the gene that is mutated in patients with ADPKD explains part of this phenotypic variability, so that patients with a mutation in PKD1 have earlier-onset ESRD, lower glomerular filtration rate (GFR) and larger kidney volumes than patients with a mutation in PKD2 (REFS64,66). Sex is increasingly recognized as important to the outcome of renal disease in patients with ADPKD — in general, renal disease is substantially more severe in men than in women66,67. However, >80% of patients with ADPKD and severe polycystic liver disease (PLD) are female, suggesting that hormonal differences might influence disease severity68. The role of PKD1 allelic effects is becoming clearer, as a considerable proportion of in-frame PKD1 mutations are incompletely penetrant (hypomorphic) and result in ESRD later in life, lower GFR and smaller kidneys than with more penetrant alleles64,66. However, genic or allelic effects do not seem to be important for the development of severe PLD in patients with ADPKD68.
Mutational inactivation of both alleles of PKD1 or PKD2 is thought to cause embryonic lethality in humans (as in mice); however, patients with biallelic mutations who are homozygous or compound heterozygous, with at least one weak PKD1 or PKD2 hypomorphic allele (that is, with a reduced level of activity or expression), do survive63. Biallelic mutations cause rare severe cases of onset of ADPKD in utero59,69–72. A mouse model mimicking the hypomorphic PKD1R3277C allele shows the association of the dosage of functional PC1 to disease severity63. Digenic patients with ADPKD (that is, those with mutations in both PKD1 and PKD2) have more severe disease than monogenic family members, but digenic mutations are not lethal in humans73. The combination of an ADPKD allele with an allele of another cystogene, such as HNF1B, may also exacerbate the renal disease phenotype59.
At least 10–15% of families with ADPKD have no family history of the disease, suggesting that de novo mutations occur at a considerable rate, and intrafamilial variability in this case can be explained by mosaicism, in which the proportion of cells that carry the mutation and those that contain the wild-type allele can differ substantially74,75. Mosaicism can be more reliably detected by next-generation sequencing (NGS)-based approaches than by conventional sequencing owing mainly to the superior sequencing depth in NGS47.
Finally, studies of twins and those that analysed inter-familial and intrafamilial variability in ADPKD severity suggest that other genetic factors contribute substantially to the variability in the severity of renal disease66,76–78, although, to date, the identity of these other genetic factors is unknown79.
Arterial hypertension.
Early in the course of ADPKD when renal function is still normal, most patients already develop arterial hypertension that contributes considerably to the increased cardiovascular morbidity and mortality observed in patients with ADPKD. Although various pathogenic mechanisms are known and activation of the renin–angiotensin–aldosterone system (RAAS; a hormone system that regulates blood pressure and fluid balance) is clearly the most prominent, other mechanisms have also been described, including increased activity of the sympathetic nervous system and disturbances in the fine-tuning of vascular tone through the action of endothelin, nitric oxide (NO) and intracellular calcium (reviewed elsewhere33).
Cyst formation.
The cysts in patients with ADPKD are fluid-filled focal outgrowths from the renal epithelium, which arise in a minority (~1%) of nephrons and eventually detach when the amount of functional PC1 or PC2 drops below a critical threshold level. The polycystin proteins localize to the apical and basolateral plasma membrane, adherens junctions, desmosomes, focal adhesions and the primary cilium and are also secreted in microvesicles80. In addition, PC2 is localized to the ER and mediates Ca2+ release from this organelle. The polycystin proteins form multimeric protein complexes that modulate several signalling pathways, such as Ca2+, cAMP, mechanistic target of rapamycin (mTOR), WNT, vascular endothelial growth factor (VEGF) and Hippo signalling. Consequently, numerous cellular changes have been observed in cyst-lining cells, including alterations in apical–basal polarity, planar cell polarity (PCP), increased extracellular matrix production and cellular metabolism, involving essential cellular functions such as fluid transport, proliferation, apoptosis, cell adhesion and differentiation81–85 (FIG. 4).
Fig. 4 |. Mechanisms of cyst formation and expansion.
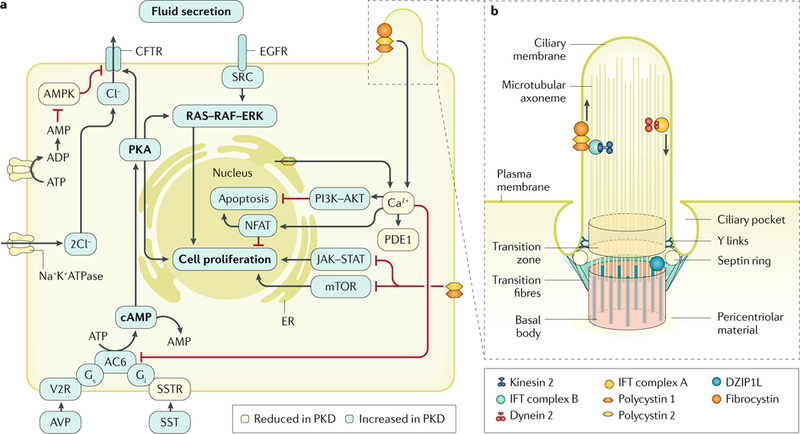
Polycystin 1 (PC1) and PC2 form a multimeric protein complex that is localized to several cellular compartments, including cell–cell junctions, cell–matrix interfaces and in the primary cilium (part a; the ciliary localization of the polycystins is shown in part b). The polycystin proteins are also post-translationally modified, which regulates their transport, localization and/or function52,121,288,295. In addition, many proteins have been reported to bind directly to polycystin proteins; for example, PC1 and fibrocystin bind to PC2 and modulate its channel activity53,296,297. Furthermore, DAZ-interacting protein 1-like protein (DZIP1L) interacts with septin 2 (SEPT2; in the septin ring), a protein implicated in maintenance of the periciliary diffusion barrier at the ciliary transition zone. Consistent with a defect in the diffusion barrier, the localization of PC1 and PC2 to the ciliary membrane is compromised in DZIP1L-mutant cells, suggesting that DZIP1L is required for regulating the integrity of the transition zone. How PC1, PC2, fibrocystin and DZIP1L directly affect cellular signalling is not known with certainty. However, these proteins modulate several signalling pathways, which in turn control essential cellular functions, such as proliferation, apoptosis, cell adhesion and differentiation54. Reduced Ca2+ influx, increased cAMP levels and aberrant activation of RAS–RAF–ERK signalling in renal epithelial cells are important mediators of cyst growth. Other proposed mechanisms that regulate cystogenesis and/or cyst growth include altered signalling mediated by G proteins, mechanistic target of rapamycin (mTOR), phosphoinositide 3-kinase (PI3K)–AKT, AMP-activated protein kinase (AMPK), Janus kinase 2 (JAK2)–signal transducer and activator of transcription 1 (STAT1; or STAT3 or STAT6), nuclear factor of activated T cells (NFAT) and nuclear factor-κB. In addition, cyst expansion is accompanied by changes in cellular metabolism, such as a switch to aerobic glycolysis as well as impaired fatty acid oxidation. AC6, adenylyl cyclase 6; AVP, arginine vasopressin; CFTR, cystic fibrosis transmembrane conductance regulator; EGFR, epidermal growth factor receptor; ER, endoplasmic reticulum; IFT, intraflagellar transport; PDE1, phosphodiesterase 1; PKA, protein kinase A; PKD, polycystic kidney disease; SST, somatostatin; SSTR, somatostatin receptor; V2R, vasopressin V2 receptor.Part a is adapted from REF.293, Springer Nature Limited. Part b is adapted from REF.298, Springer Nature Limited.
However, as PKD progresses, the molecular alterations in the kidneys are increasingly the consequence of secondary events that disrupt the network of signalling pathways. Reduced Ca2+ influx, increased cAMP levels and aberrant RAS–RAF–ERK activation in renal epithelial cells are important mediators of cyst growth. which is further sustained by increased growth factor signalling86,87. For example, increased levels of epidermal growth factor receptor (EGFR) and its mislocalization to the apical surface of cystic epithelial cells, together with the presence of the ligands EGF and transforming growth factor-α (TGFα) in cyst fluid, probably promote tubular epithelial cell proliferation88,89. Other mechanisms that might be involved in cyst growth include altered signalling through G proteins, mTOR, phosphoinositide 3-kinase (PI3K)–AKT, AMP-activated protein kinase (AMPK), Janus kinase 2 (JAK2)–signal transducer and activator of transcription 1 (STAT1; or STAT3 or STAT6), nuclear factor of activated T cells (NFAT) and nuclear factor-κB (NF-κB)90–97. In addition, cyst expansion is accompanied by changes in cellular metabolism — increased glucose consumption that is indicative of ATP synthesis through aerobic glycolysis, and impaired fatty acid oxidation, seem to be additional features of ADPKD83,98–102. Cysts can induce stress (presumably mechanical) on neighbouring nephrons, resulting in apoptosis of renal epithelial cells and thereby locally increasing the likelihood of cyst formation65.
The polycystin complex localized in the primary cilium was initially thought to transduce extracellular fluid flow-generated shear stress into a cellular Ca2+ signal86, but subsequent studies suggested that although polycystin proteins induce local changes in ciliary calcium concentration through their channel function, global cytoplasmic calcium levels are not substantially altered87. Intriguingly, the engineered loss of cilia ameliorates the cystic phenotype in Pkd1-mutant mice or Pkd2-mutant mice, suggesting that, in healthy kidneys, the polycystin proteins inhibit a cilia-regulated cystogenic signalling pathway that controls renal epithelial cell proliferation103.
However, the function of the polycystin proteins at other subcellular locations should not be underestimated. Simultaneous ablation of Pkd1 and Itgb1 (which encodes β1 integrin) in mice resulted in inhibition of Pkd1 ablation-dependent cystogenesis and suppression of fibrosis, suggesting that PC1 represses signalling that regulates the crosstalk between the extracellular matrix and integrins104. Although a direct association between PC1 and β1 integrin has not been detected, PC1 interacts with the cytoskeletal adaptor proteins vinculin and paxillin, which are essential for integrin signalling through focal adhesion complex proteins, such as focal adhesion kinase (FAK) and SRC105. The accumulation of the extracellular matrix and the recruitment of inflammatory cells can already be observed at early stages of the disease. In particular, M1 and M2 macrophages are important contributors to disease progression. Macrophages and interstitial fibroblasts also produce cytokines, and abnormal cytokine-mediated crosstalk between renal epithelial cells and inflammatory cells results in a positive feedback loop of increasing fibrosis (FIG. 5).
Fig. 5 |. Renal fibrosis in autosomal dominant polycystic kidney disease.
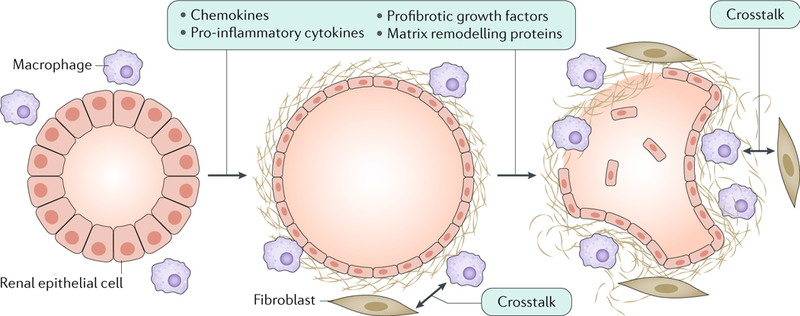
Altered expression of cystic proteins and dysregulated signalling in renal epithelial cells affect cell–extracellular matrix interactions and induce the production of chemokines (such as monocyte chemotactic protein 1 (MCP1), CC-chemokine ligand 6 (CCL6), CCL28, CXC-chemokine ligand 1 (CXCL1), CXCL8 and CX3C-chemokine ligand 1 (CX3CL1)), pro-inflammatory cytokines (such as tumour necrosis factor (TNF), IL-1, IL-2, IL-6, IL-8, macrophage colony-stimulating factor 1 (CSF1) and macrophage migration inhibitory factor (MIF)), profibrotic growth factors (such as transforming growth factor-β (TGFβ), TGFα, epidermal growth factor (EGF), fibroblast growth factors (FGFs) and platelet-derived growth factor (PDGF)) and proteins involved in matrix remodelling (such as matrix metalloproteinases (MMPs) and tissue inhibitors of metalloproteinases (TIMPs))299,300. These changes result in the accumulation of extracellular matrix and the recruitment of inflammatory cells, which is already observed in the early stages of the disease. In particular, M1 and M2 macrophages are important contributors to disease progression. Macrophages and interstitial fibroblasts also produce cytokines, and abnormal cytokine-mediated crosstalk between renal epithelial cells and inflammatory cells results in a positive feedback loop of increasing fibrosis.
In addition to altered cell–matrix interactions, the cell–cell junctions in cyst-lining epithelia are also abnormal106. The abnormal composition and localization of adherens junctions and desmosomes in ADPKD suggest that the polycystin proteins regulate the dynamics of cell–cell adhesions to reduce their mechanical strength and allow cytoskeletal remodelling in ADPKD106,107.
As a consequence, the integrity of apical–basal polarity and PCP is compromised in cystic epithelial cells108. However, although disruption of PCP signalling results in inaccurate control of tubule diameter, it is not cystogenic109. These data suggest that PKD mutations do not disrupt PCP signalling but instead act independently of and in parallel with PCP signalling to affect oriented epithelial cell division107.
Overall, cystic epithelia often have a partially de-differentiated phenotype, suggesting that mutations in the PKD genes result in cells reverting to a developmental pattern of gene expression. Although the molecular mechanisms of polycystin regulation of various signalling pathways at different subcellular locations are not fully understood, this network of signalling pathways seems to be crucial for establishing and maintaining a differentiated renal epithelium60.
Histopathological changes.
Epithelial cell proliferation, abnormal fluid secretion and excessive extracellular matrix deposition are the major characteristics of cystic epithelial cells. These changes are accompanied by alterations in the pericystic blood and lymphatic microvas-culature110–112. Most cysts detach from the tubules from which they form and fill with fluid by transepithelial secretion113. Cyst enlargement also compresses the surrounding nephrons, interstitium and vasculature. The obstructed nephrons eventually form atubular glomeruli and apoptotic proximal tubules114.
These events are accompanied by the production of chemokines, cytokines and growth factors by epithelial cells, interstitial fibroblasts and inflammatory cells, such as macrophages115,116. Abnormal cytokine-mediated crosstalk between epithelial and inflammatory cells promotes more inflammation and fibrosis, new cyst formation and disease progression, which result in massively fibrotic kidneys at end-stage disease117 (FIG. 5). Increased inflammation is probably an early event in disease progression, and the selective depletion of macrophages in the kidneys of a Pkd1 mouse model resulted in considerable amelioration of the cystic phenotype and improvement in renal function118. Macrophage migration inhibitory factor (MIF) is an important pro-inflammatory cytokine in PKD, as deletion of Mif delayed cyst growth and improved renal function in Pkd1 -knockout mice119.
Extrarenal pathology.
ADPKD is a systemic disease and, in addition to the renal pathology, patients develop liver cysts and cardiovascular abnormalities. PLD is characterized by the presence of multiple cysts scattered throughout the liver parenchyma, which form owing to overgrowth of the biliary epithelium. The severity of PLD can range from a few hepatic cysts to a massively enlarged cystic liver. As in renal epithelia, a substantial reduction or complete loss of function of the polycystin proteins underlies liver cyst formation120. PLD can also occur as a distinct disease, without the involvement of renal cysts, as ADPLD. Although ADPLD is caused by mutations in a set of genes that are distinct from those involved in ADPKD, studies in mice suggest that mutations in these PLD genes affect the dosage of PC1, which seems to be the rate-limiting determinant of liver cyst formation121.
Patients with ADPKD also have cardiovascular abnormalities, including hypertension, left ventricular hypertrophy, aortic root dilatation, arterial aneurysms, heart valve abnormalities and intracranial aneurysms (ICAs) (reviewed elsewhere122). Numerous lines of evidence suggest that the vascular and cardiovascular defects are directly related to reduced expression of the polycystin proteins (haploinsufficiency) in the endothelial cells and vascular smooth muscle cells (VSMCs) of most blood vessels, including the aorta and cerebral arteries110,114. Indeed, the risk of cerebral aneurysms is higher in families with a positive family history of ADPKD than those without, probably because of modifying genes123. The functions of PC1 and PC2 in the vasculature indicate that they have a crucial role in mechanosensation. In endothelial cells, the proteins are involved in fluid-shear stress sensing, thereby regulating calcium signalling and NO release124. NO released by endothelial cells affects vasodilatation in response to increased blood flow. In VSMCs, the polycystins regulate pressure sensing by modulating the activity of the stretch-activated cation channels and myogenic contraction125. A loss of myogenic tone may contribute to aneurysm formation owing to an increase in arterial wall stress125. Finally, studies of haploinsufficient and hypomorphic models of ADPKD have shown that vascular changes are associated with a reduction of the levels of polycystin proteins126–128.
The PKD genes are expressed in a wide range of tissues beyond the kidney, and their expression is developmentally regulated in most of these tissues. Accordingly, other extrarenal manifestations can occur, including cysts in the pancreas, seminal vesicles and the arachnoid membrane, in patients with ADPKD115. Although patients with a single or those with more than one mutant PKD allele usually have relatively restricted disease phenotypes (that is, cysts or manifestations in only a few organs), complete knockout of Pkd1 or Pkd2 in mice results in embryonic lethality that is associated with a wide range of abnormalities, including: severely cystic pancreas, kidneys and liver; cardiac abnormalities; vascular defects (such as oedema and vascular leakage); craniofacial defects; bone defects; and, rarely, polyhydramnios (excess amniotic fluid)129–133. Tissue-specific Pkd1 or Pkd2 disruption results in hydrocephalus and lymphatic abnormalities in mice112,134. The species-specific differences in ADPKD phenotypic severity suggest that the levels of polycystin proteins in most human tissues do not drop below a critical threshold or that when this happens, the affected cells might undergo apoptosis.
Experimental models.
To gain insight into the disease mechanisms and pathogenesis of ADPKD, various mouse models have been genetically engineered33, including inducible, conditional knockout mice and mouse models with hypomorphic or hypermutable alleles of Pkd1 or Pkd2, which are extremely useful as ablation of Pkd1 or Pkd2 results in embryonic lethality and heterozygous mice develop only very mild disease, even in old age62,63,135–139. These models are characterized by the age of disease onset, the progressiveness of the disease, the nephron segment that is affected, the number of affected nephrons, whether cyst formation is synchronized or unsynchronized and the extent of renal fibrosis and inflammation. These models enable step-by-step analysis of the pathogenesis of ADPKD and have provided valuable mechanistic insights into the early stages and the later, progressive stages of the disease. Findings in tissues from mouse models of early disease are complementary to those in renal tissues from patients with ADPKD, as the kidneys are usually removed from patients with the progressive stages of ADPKD or ESRD. Examples of the value of animal models include their role in highlighting the difference in the severity of renal disease observed when Pkd1 is inactivated during renal development compared with after the kidneys have formed137,138. In addition, renal damage was shown to increase the severity of PKD in haploinsufficient mice60,140. Finally, hypomorphic models have shown that polycystic kidneys can develop when polycystin function is reduced but not eliminated62,63.
ARPKD
Genes and proteins.
The polycystic kidneys and congenital hepatic fibrosis (CHF) observed in ARPKD suggest that terminal differentiation of the renal collecting ducts and intrahepatic biliary ducts is disordered in this disease. Histological analysis has revealed that the changes in the liver that are invariably present from early embryonic development in patients with ARPKD are characterized by defective remodelling of the ductal plate with CHF and biliary duct ectasia, defined as ductal plate malformation (DPM; FIG. 6), which also occurs frequently in other cilia-related disorders (ciliopathies), such as nephronophthisis, Joubert syndrome and Bardet–Biedl syndrome, some ofwhich may have an ARPKD-like phenotype1,141. Different types of variants in PKHD1 (6p12.3–12.2), such as missense and truncating mutations, are responsible for most cases of ARPKD (discussed further in the Diagnosis section).
Fig. 6 |. Hepatobiliary lesions in hepatorenal disease.
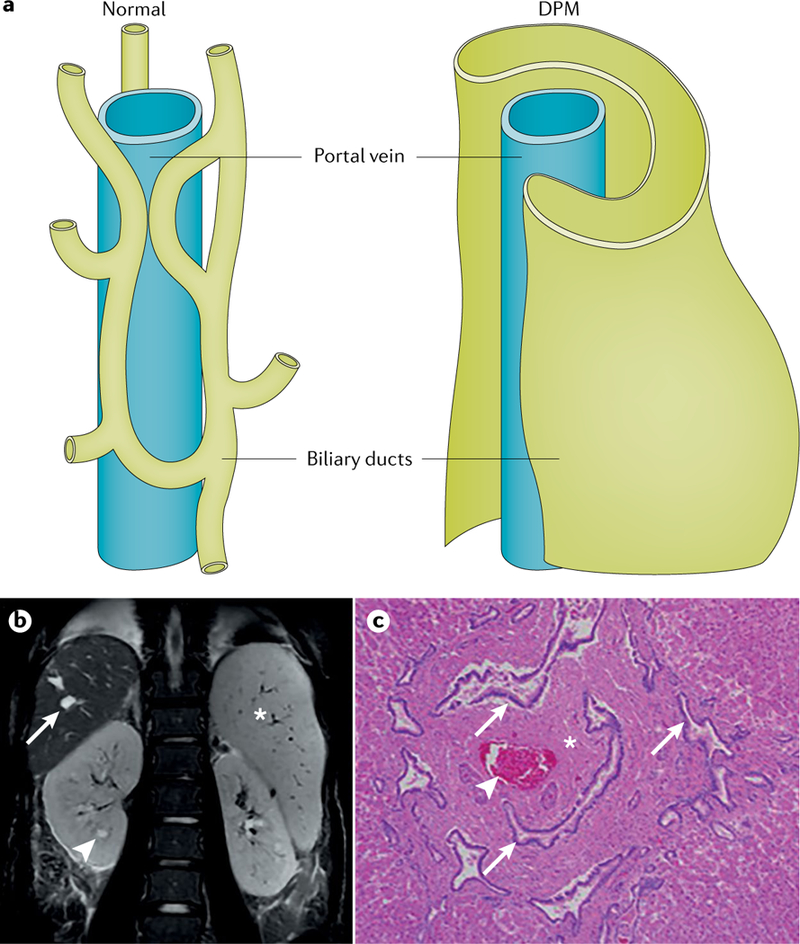
a | Hepatobiliary lesions result from an architectural defect in the developing biliary tree. The normal ramifications of the portal venous system and the lattice-like network of associated biliary ducts (left) are disrupted owing to ductal plate malformation (DPM) (right), likely owing to a defect in terminal differentiation of cholangiocytes. b | The DPM results in marked cystic and fusiform dilatation of the intrahepatic biliary system (coronal T2-weighted image of the abdomen), nephromegaly with small cysts (arrowhead), cystic biliary disease (arrow) and marked splenomegaly (asterisk). c | The histopathological manifestation of the DPM is congenital hepatic fibrosis (section stained with haematoxylin and eosin), which is characterized by extensive fibrosis of the portal area (asterisk), ectatic, tortuous bile ducts (arrows) and hypoplasia of the portal vein (arrowhead). Magnification is 40×. Part a is reprinted, with permission, from Marchal G J, Desmet V J, Proesmans W C, et al. Caroli disease: high-frequency US and pathologic findings.
The mRNA of PKHD1 is alternatively spliced to produce multiple transcripts142,143. The longest transcript encodes a 4,074-residue protein comprising a single transmembrane domain, an extensive extracellular N-terminal domain and a short C-terminal cytoplasmic tail (FIG. 2). Fibrocystin localizes to the primary cilium and to other cellular compartments142,144,145 and is targeted to the ciliary membrane by a specific motif in its C terminus146. In addition, similar to Notch, fibrocystin is proteolytically cleaved to release the cytoplasmic tail, which translocates to the nucleus and presumably regulates downstream target genes. Although this remains to be conclusively established, it is possible that the C-terminal tail of fibrocystin regulates the expression of genes that encode proteins that drive cyst formation147,148.
In addition, mutations in the gene encoding the ciliary transition zone protein DAZ-interacting protein 1-like protein (DZIP1L) have been described in some pedigrees in which the affected children had clinically moderate ARPKD and no evidence of mutations in PKHD1 (REF.132; C.B., unpublished data). DZIP1L is a 767-residue soluble zinc-finger protein that, similar to PC1, PC2 and fibrocystin, localizes to centrioles and the distal end of basal bodies. DZIP1L interacts with septin 2 (SEPT2; in the septin ring), a protein that is implicated in the maintenance of the periciliary diffusion barrier at the ciliary transition zone. Consistent with a defect in the diffusion barrier, the localization of PC1 and PC2 to the ciliary membrane is compromised in DZIP1L-mutant cells132. Diseases with symptoms similar to those in ADPKD and ARPKD might also be caused by mutations in other genes (reviewed in detail elsewhere1). Of note, mutations in PKD1 and PKD2 can also be inherited in a recessive manner (homozygous or compound heterozygous) with at least one weak PKD1 or PKD2 hypomorphic allele; these patients can present with an ARPKD-like phenotype141.
Finally, PKD (mimicking ARPKD or ADPKD) with hyperinsulinie hypoglycaemia was described in 11 pedigrees and was linked to biallelic mutations (including a specific promoter mutation) in phosphomannomutase 2 (PMM2)149. PMM2 is a key enzyme in N-linked glycosylation, and recessive mutations in PMM2 are usually associated with congenital disorder of glycosylation type 1a, which has a severe, pleiotropic phenotype.
Determinants of disease severity.
Efforts to assess geno-type–phenotype correlations for PKHD1-linked ARPKD have been hampered by multiple allelism (that is, the presence of many different mutations in the same gene) and the high incidence of different compound heterozygotes (that is, the presence of two different mutant alleles at a single locus). However, genotype–phenotype correlations have been made for the type of mutation instead of the site of individual mutations150. Allelic effects are known; for example, patients with two truncating mutations in PKHD1 often have severe disease that is usually lethal (although a few exceptions have been described) and surviving patients typically have at least one inframe mutation30,150. These data indicate that many missense mutations do not completely abrogate PKHD1 function. Notwithstanding, some missense mutations in PKHD1 result in disease that is as severe as that resulting from truncating mutations. No substantial clinical differences have been observed between patients with two missense mutations and patients with a truncating mutation in trans; thus, the milder mutation seems to define the disease severity. Substantial intrafamilial variability among a considerable number of siblings indicates that other genetic and environmental factors also influence the disease phenotype32.
The clinical course in patients and siblings with DZIP1L mutations described above was invariably moderate. Although the clinical manifestations in all of these patients were initially detected prenatally or during early childhood, none of the patients died perinatally. To date, the data are consistent with the idea that the type or location of the mutation in DZIP1L does not determine the severity of the clinical course. Patients who have missense mutations in both parental DZIP1L alleles have a phenotype comparable to that of patients who have two truncating DZIP1L alleles. Although genetic screening of a large number of patients who died perinatally has not been carried out, in view of homozygous truncating mutations in two patients132, we hypothesize that it is unlikely that DZIP1L mutations play a major role in this cohort of mostly severely affected patients. This hypothesis is consistent with the birth of homozygous Dzip1l-mutant outbred mice at the expected Mendelian frequencies132. However, the identification of additional families with DZIP1L mutations is required to draw firm conclusions.
Cyst formation.
ARPKD cysts arise predominantly from the distal tubules and collecting ducts, but the detailed mechanisms of cyst formation are only poorly understood. Fibrocystin is expressed postnatally predominantly in the kidneys, liver and pancreas, and as mentioned earlier, fibrocystin localizes to the apical membranes as well as to primary cilia, especially at the basal body and the mitotic spindle in renal tubular cells and biliary epithelial cells145,151,152. DZIP1L is widely expressed and is localized primarily to centrioles and the distal end of basal bodies. Although little is known about the molecular functions of fibrocystin and DZIP1L, fibrocystin has been shown to interact with PC2 within the PC1–PC2 complex153. In addition, as in ADPKD, the cAMP, MYC and mTOR signalling pathways are activated in ARPKD154 and thus represent potential targets for directed therapeutic intervention.
Experimental models of ARPKD.
To date, multiple Pkhd1-based models of ARPKD have been described, which were primarily generated by gene-targeting methods in mice. Although an ARPKD-like liver phenotype is invariably present in all of these mouse models, a renal phenotype is often absent, and when present, it is a mild, slowly progressive lesion that typically involves the proximal tubules rather than the collecting ducts. Interestingly, several models also have a severe pancreatic ductal phenotype, whereas clinically significant pancreatic disease in patients with ARPKD is quite rare. The mechanism that underlies the mild renal phenotype in these animal models remains unclear. However, as genetic background seems to have a major effect on renal pathology, genetic modifiers likely modulate renal disease155. In the PCK rat, which arose owing to a spontaneous mutation in Pkhd1 (REF.144), the hepatic phenotype is very similar to that in ARPKD, whereas the slowly progressive renal cystic disease in this model more closely resembles the renal cystic lesions in ADPKD than in ARPKD156. Introgression of the Pkhd1 mutation in the PCK rat into a different background ameliorated the renal but not the hepatic phenotype, again suggesting that genetic factors modulate the severity of the renal cystic phenotype157.
Consistent with the extensive phenotypic variability observed in experimental models of ARPKD, Dzip1lwpy/wpy mice (which were identified in an N-ethyl-N-nitrosourea-induced mutagenesis screen and contain a nonsense mutation in Dzip1l) display a number of embryonic morphological defects, including limb and craniofacial defects, in addition to cystic kidney disease and hepatobiliary DPM132. The amelioration of the craniofacial phenotypes by crossing Dzip1lwpy/wpy mice with an outbred strain suggests that at least some of the phenotypic differences with the human ARPKD may be due to genetic background effects. Tissue-specific changes in the expression of genes involved in Hedgehog signalling, such as gene expression changes in the limb that are consistent with polydactyly, were detected in Dzip1lwpy/wpy embryos132.
Diagnosis, screening and prevention
ADPKD
Diagnosis.
ADPKD is a progressive disease in which renal cyst development begins in utero, although cyst initiation and enlargement continue throughout the patient’s life158. Cyst enlargement results in an exponential increase in total kidney volume (TKV; expressed relative to height (htTKV)) and often results in ESRD159. Clinical symptoms, including early-onset hypertension, abdominal fullness and pain, haematuria and urinary tract infections (UTIs), are usually first observed decades (sometimes even in childhood) before the onset of renal insufficiency2 (the numerous manifestations of the disease are described in TABLE 1).
Table 1 |.
Renal and extrarenal manifestations in ADPKD
| Manifestation | Prevalence | Comments | Refs |
|---|---|---|---|
| Renal | |||
| Urinary concentration defecta | Up to 60% of children | Earliest manifestation of mild polyuria is often undetected | 2,302 |
| Hypertensiona | • 50–70% of patients prior to GFR decline • Average age of onset is 30 years • At least 20–40% of children |
Screen children with family history of ADPKD from 5 years of age, then at 3-year intervals if negative for hypertension | 122,176 |
| ESRDa | 50% of patients by 60 years of age | Mean age of onset of 56 years (truncating PKD1 mutations), 68 years (non-truncating PKD1 mutations) or 78 years (PKD2 mutations) | 64 |
| Proteinuria (>300 mg/day) | Associated with GFR decline | Prognostic marker of ADPKD | 2 |
| Abdominal or flank pain | >60% of adult patients | • Acute or chronic • Multiple causes |
115 |
| Nephrolithiasis | 20–35% of adult patients | Uric acid and/or calcium oxalate stones | 2 |
| Cyst haemorrhage and/or gross haematuria | Up to 60% of adult patients | Most haemorrhages resolve within 2–7 days without intervention | 115 |
| Urinary tract infectiona | 30–50% of adult patients | More common in women than in men | 115 |
| Renal cell carcinoma | <1% of adult patients | Risk not increased compared with the general population, but patients can present with systemic symptoms of cancer | 68,219 |
| Extrarenal | |||
| Polycystic liver disease | >80% of patients by 30 years of age | Include liver imaging in initial visit; further follow-up dependent on result of imaging | 68 |
| ICA | • 8% of all adult patients • 21% of adult patients with a family history of ICA |
Screen if family history of subarachnoid haemorrhage or ICA, personal history of intracranial haemorrhage, individuals working in high-risk professions and before major elective surgery (including before transplantation) | 238 |
| Arachnoid cysts | 8% of adult patients | Possible increased risk of spontaneous subdural haematoma | 115 |
| Mitral valve prolapse or bicuspid aortic valve | Up to 25% of adult patients | Screen when there is a heart murmur or symptoms | 122 |
| Idiopathic dilated cardiomyopathy or left ventricular non-compaction | Rare | Screen when there is a family history of these conditions | 122 |
| Pericardial effusion | Up to 35% of adult patients | Screen if symptoms of pericardial effusion are present | 122 |
| Pancreatic cysts | 10% of adult patients | No screening needed | 115 |
| Diverticulosis | Up to 50% of patients with ESRD | Increased risk of diverticulum perforation following renal transplantation | 115 |
| Bronchiectasis | Up to 35–40% of adult patients | Mild; no screening needed | 115 |
| Congenital hepatic fibrosisa | Rare (on the basis of case reports) | No screening needed | 141,303 |
| Seminal vesicle cysts | Up to 40% of men | No correlation to semen abnormalities | 304 |
| Male infertility | Associated with ADPKD | Abnormal semen parameters reported | 304 |
ADPKD, autosomal dominant PKD; ESRD, end-stage renal disease; GFR, glomerular filtration rate; ICA, intracranial aneurysm.
Manifestations that are also present in patients with autosomal recessive polycystic kidney disease.
Presymptomatic diagnosis can usually be made in at-risk family members (such as children or siblings of affected individuals) by abdominal imaging, and the presence of multiple bilateral cysts identifies affected individuals. Ultrasonography is the most common and least expensive imaging method, but CT or MRI can provide more quantitative data (that is, htTKV) of prognostic value (FIG. 7). As cyst development in ADPKD is related to age, and even genetically unaffected individuals can develop a few simple cysts as they age160, criteria have been developed to define whether at-risk individuals are affected on the basis of number of cysts by age (TABLE 2).
Fig. 7 |. Diagnosis of autosomal dominant polycystic kidney disease using different imaging techniques.
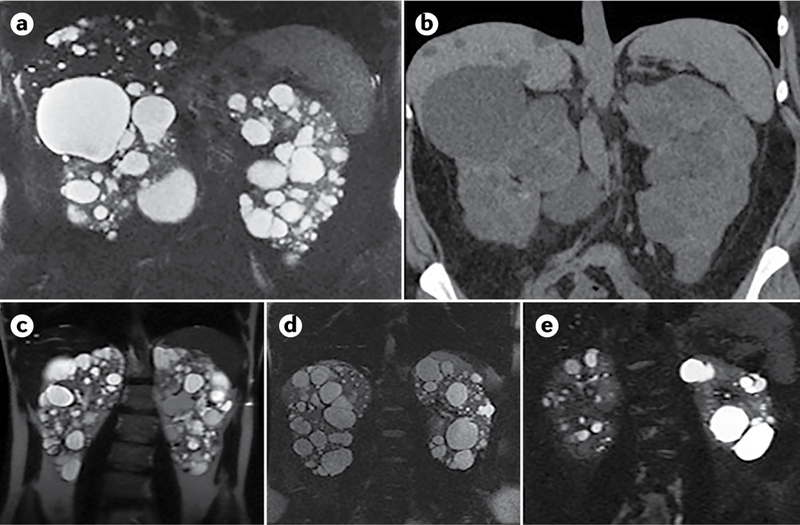
a–b | Comparison of MRI (T2; part a) and CT (without contrast; part b) scans of a 35-year-old woman with autosomal dominant polycystic kidney disease (ADPKD), showing widespread kidney cysts and a few liver cysts. c–e | T2 MRI images of patients with ADPKD who have a truncating mutation in PKD1 (part c; 41-year-old man), a non-truncating mutation in PKD1 (part d, 40-year-old man) or a splicing mutation in PKD2 (part e; 41-year-old man). Patients with truncating mutations in PKD1 typically have more cysts, whereas patients with non-truncating mutations in PKD1 have an intermediate number of cysts and patients with splicing mutations in PKD2 have the fewest cysts.
Table 2 |.
Ultrasonography criteria for an ADPKD diagnosis in at-risk individuals
| Age | Genotype | ||
|---|---|---|---|
| PKD1 mutation | PKD2 mutation | Unknown ADPKD genotype |
|
| 15–30 years | • ≥3 cystsa • PPV = 100% • SEN = 94.3% |
• ≥3 cystsa • PPV = 100% • SEN = 69.5% |
• ≥3 cystsa • PPV = 100% • SEN = 81.7% |
| 30–39 years | • ≥3 cystsa • PPV = 100% • SEN = 96.6% |
• ≥3 cystsa • PPV=100% • SEN = 94.9% |
• ≥3 cystsa • PPV=100% • SEN = 95.5% |
| 40–59 years | • ≥2 cysts in each kidney • PPV = 100% • SEN = 92.6% |
• ≥2 cysts in each kidney • PPV = 100% • SEN = 88.8% |
• ≥2 cysts in each kidney • PPV = 100% • SEN = 90% |
Compiled with data from Ref.161. ADPKD, autosomal dominant polycystic kidney disease; PPV, positive predictive value; SEN, sensitivity.
Unilateral or bilateral. All values are mean estimates.
The corresponding number of MRI-detected cysts that are considered to be diagnostic has been defined as more than ten in those aged 16–40 years; fewer than ten cysts does not classify as PKD161. In patients without an established family history, the detection of more than ten cysts per kidney by ultrasonography is usually considered diagnostic162, but genetic diagnostic testing would be valuable.
Imaging provides a less certain diagnosis in young adults owing to the late and only mild disease manifestation in some patients; therefore, in these cases and others, genetic analysis may be helpful to obtain a definite diagnosis (BOX 1).
Box 1 |. Indications for genetic testing in ADPKD.
In a young relative who is a potential kidney donor for a patient with autosomal dominant polycystic kidney disease (ADPKD), especially if one or two cysts are detected by imaging methods — to exclude genetically affected relatives291.
In individuals with a negative family history — to establish a firm diagnosis or to test for mosaicism74 (note that at least 10–15% of families with ADPKD can be traced to a new (de novo) mutation).
In cases of polycystic kidney disease of unknown origin and a negative family history — to assess recurrence risk and for family planning47.
In patients with very early-onset disease — to identify biallelic disease or other complex genetics59,248,292.
In patients with mild disease — to establish whether the disease is genetic and its cause292.
In patients who are starting treatment or are being considered for a clinical trial — to establish a firm diagnosis and help determine whether the disease is rapidly progressive.
In all patients with ADPKD — if treatment is fairly inexpensive and reliable, most patients can obtain a firm diagnosis and prognostic information.
Obtaining a genetic diagnosis of ADPKD is complex because of historical segmental intrachromosomal duplication of three-quarters of the PKD1 gene (at the 5′ end), which produced six pseudogenes that have up to 99% sequence homology with PKD1. Consequently, specialized screening of PKD1 is required because mutations are not always detected by whole-exome sequencing (WES). Traditionally, genetic diagnostic analysis has involved using PKD1 -specific long-range PCR fragments that exploit the rare sequence differences between PKD1 and the pseudogenes, followed by Sanger sequencing or NGS of the long-range products to identify mutations163–165. However, newer sophisticated NGS methods that use specific capture workflows and in silico analyses166,167 or whole-genome sequencing (WGS)168 have been reported, and capture-panel-based approaches are now favoured for genetic diagnosis of ADPKD.
Furthermore, interpretation of the results of genetic diagnostic analysis is complicated not only by the PKD1 pseudogenes but also by the high level of allelic heterogeneity in PKD1 and PKD2, in which >1,900 different mutations have been described, and no single mutation accounts for >2% of the total patient population (ADPKD Mutation Database). A substantial proportion of PKD1 mutations are in-frame (~30%), and because this gene has >300 described neutral non-synonymous variants, determining whether a missense variant is pathogenic is not always straightforward. Inquiry of the ADPKD Mutation Database and the use of scoring algorithms together with contextual information from population databases (for example, the Genome Aggregation Database (gnomAD) and family studies can help determine the pathogenicity of these variants66,163,169,170. In ~7% of ADPKD families, no mutation is detected64,66,163,164; some of these families probably have mutations at the known PKD loci that were not detected (for example, deep intronic or regulatory mutations), including mosaic cases that are probably more common in patients with de novo mutations than is currently reported75,171,172. In addition, further genetic heterogeneity is likely, as mutations in GANAB were identified as a cause of mild ADPKD, which represents ~0.3% of all cases34, and DNAJB11 mutations cause a hybrid of ADPKD and ADTKD36. Phenocopies of the ADPKD phenotype can also be due to mutations in HNF1B and in other genes, such as those associated with ADPLD that result in an ADPKD-like phenotype173,174. Parental renal ultrasonography should be carried out for family members of children who have cystic kidney disease of unknown origin. However, the family history is often unremarkable because of de novo mutations and combinations of incompletely penetrant (hypomorphic) alleles.
Asymptomatic at-risk individuals <18 years of age are usually not screened but decide whether and when to be tested as an adult31,175. The rationale for this recommendation is based on the current lack of disease-specific treatments for those individuals <18 years of age and, in the USA at least, problems can occur with obtaining medical insurance and with workplace issues, despite antidiscrimination legislation. When necessary, monitoring and treatment of arterial hypertension and UTIs in at-risk children are recommended. However, the recommended age of testing may change if therapies become available for those <18 years of age after the completion of clinical trials in children and teenagers. Treatment of young individuals (even children) before irreparable kidney damage has occurred might be particularly effective. Increasing evidence exists that important predictors of future cardiovascular events and mortality (such as left ventricular hypertrophy, biventricular diastolic dysfunction, endothelial dysfunction, increased carotid intima–media thickness and impaired coronary flow velocity reserve) are evident in children and young individuals with normotensive blood pressure and normal kidney function very early in the disease course and that patients might benefit from an early diagnosis75,176,177.
Prognosis.
Identifying patients with rapidly progressing disease would facilitate selection for clinical trials and treatment. Separating patients into classes on the basis of age-related htTKV (the Mayo Classification), and exclusion of atypical patients, have been proposed as an efficient means to identify patients with the most rapidly progressing disease178. Combining genic and allelic information with clinical indications (onset of hypertension at <35 years of age and/or first urological event at <35 years of age) has also enabled identification of a population with rapidly progressing disease (the PROPKD score, a prognostic algorithm to predict renal outcomes in patients with ADPKD on the basis of genetic and clinical data)67,179. European recommendations have highlighted a demonstrated rapid decline in GFR in younger patients and/or a recorded rapid increase in htTKV as a means to identify patients with rapidly progressing disease who are suitable for treatment179.
Prenatal screening.
As ADPKD is an adult-onset disease, prenatal screening is not commonly carried out, but there is increasing interest in this procedure, especially from families with experience of early-onset renal disease or early ICAs. Of note, affected families with children with early-onset ADPKD have a high risk of subsequent children also having early and severe ADPKD, and this information should be shared with affected families1. Preimplantation genetic diagnosis is available for ADPKD, including analysis of the duplicated PKD1 locus, and may be more commonly sought in the future44.
ARPKD
Diagnosis.
ARPKD is phenotypically highly variable: at its most severe, ARPKD presents in utero, at birth or in infancy and is characterized by bilaterally enlarged, echogenic kidneys with poor corticomedullary differentiation, retained reniform contour and multiple tiny cysts that are confined to the distal tubules and collecting ducts (FIG. 8); presentation in older children, teenagers or young adults includes manifestations of portal hypertension or cholangitis; and moderate presentation is seen rarely in older adults with complications of liver disease or with renal manifestations, such as proteinuria, nephrolithiasis and renal insufficiency180–183. Those patients who survive the perinatal period then transition from paediatric to adult care with internal medicine specialists184. With the advent of NGS-based molecular diagnostic testing, it has become evident that mutations in various other genes, such as HNF1B and those associated with ADPKD and many ciliopathies, can produce a phenotype that mimics the symptoms in ARPKD141.
Fig. 8 |. Diagnosis of autosomal recessive polycystic kidney disease using MRI.
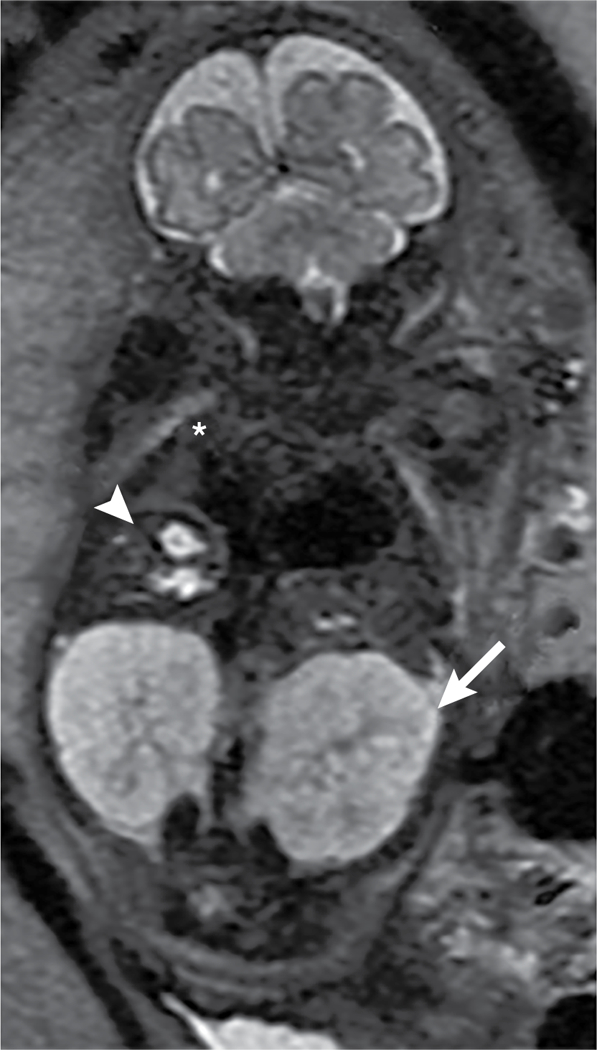
MRI can be used to detect renal and extrarenal manifestations of autosomal recessive polycystic kidney disease (ARPKD). In this coronal, T2-weighted MRI scan of a 32-week-old fetus, the kidneys (arrow) are massively enlarged and are abnormally bright with innumerable tiny cysts, most of which cannot be individually resolved. Cystic biliary disease (arrowhead) and extremely low lung volumes (asterisk) in patients with ARPKD are due to oligohydramnios (deficiency of amniotic fluid).
Potter sequence consists of pulmonary hypoplasia, characteristic facies and spine and limb abnormalities and can be present in severe neonatal cases, with death from respiratory distress occurring in at least 20% of cases26,30,32. Surviving infants can experience childhood ESRD as well as liver complications owing to CHF that worsens with age. Patients who present with ARPKD later in childhood or as adults are more likely to have complications of portal hypertension, whereas renal enlargement is less evident than in patients who present earlier32,181. With advancing clinical course, the kidney structure might increasingly resemble the symptoms observed in ADPKD, including renal cysts that vary considerably in size and appearance, often accompanied by some interstitial fibrosis185. However, in contrast to the persistent renal enlargement that occurs in ADPKD, in ARPKD the kidneys decrease in size as the amount of fibrosis increases.
A diagnosis of ARPKD is likely to be based on clinical imaging in severe neonatal or infantile cases on the basis of enlarged echogenic kidneys, a negative family history of PKD and a lack of additional features (such as central nervous system abnormalities, eye defects and/or polydactyly) that may suggest a syndromic ciliopathy1. However, molecular diagnostic analysis is the gold standard for diagnosing this disorder, and a number of pheno-copying disorders or syndromes must be considered1. Genetic testing can be particularly important in the diagnostic evaluation of patients with early-onset bilateral renal cystic disease. In older patients, CHF with or even without renal cysts may indicate ARPKD and molecular diagnostic analysis may help confirm the diagnosis47.
The large size of the PKHD1 gene (12,222 bp coding sequence, 67 exons and spans ~470 kb)142,144 and a high level of allelic heterogeneity (~60% of mutations are inframe and 40% of mutations are truncating150,186) complicate the molecular diagnosis of ARPKD. Some mutations in PKHD1 are more common than others and some are enriched in specific populations; for example, T36M is the most common mutation and accounts for ~10–15% of mutant alleles in European populations150,186,187. Analysis of DZIP1L is much easier than for PKHD1, as it has a 2,301 bp coding sequence, 16 exons and spans only ~53 kb. Mutations in DZIP1L are a considerably rarer cause of ARPKD than PKHD1 mutations132, perhaps in part owing to the larger size of PKHD1 compared with DZIP1L. Furthermore, limited data suggest that the genomic region encoding the N terminus of DZIP1L may be more susceptible to mutations, which is supported by in silico data that predict that pathogenicity scores decay towards the C terminus132.
In practice, and reflecting the phenotypic overlap of ARPKD with syndromic ciliopathies as well as other forms of PKD, including ADPKD, screening a panel of genes is now often considered to be the gold standard for diagnostics147,167. Advances in NGS now enable simultaneous analysis of a large group of genes in a single test at fairly low cost. We expect that single-gene testing will soon be the exception rather than the rule, especially for genetically heterogeneous disorders with a broad phenotypic spectrum such as PKD. However, at present, targeted NGS panel testing might be the most efficient diagnostic approach. Whichever primary strategy is chosen for PKD diagnosis, it should be able to detect copy number variations such as heterozygous deletions (for example, in HNF1B) and to cover complex genomic regions, such as PKD1.
Prognosis.
In accordance with autosomal recessive inheritance, the recurrence risk for subsequent pregnancies of parents of an affected child with ARPKD is 25%. Males and females are equally affected30. By definition, heterozygous carriers should not show any clinical disease manifestation, but data suggest that individuals who are heterozygous for PKHD1 mutations have an increased risk of PLD and mild PKD35,41. Other relatives (such as healthy siblings) or the affected patients themselves may seek genetic counselling and can usually be reassured of a very low risk (<1%) for ARPKD for their own offspring if their partner is not affected by ARPKD and has no family history of PKD.
Prenatal screening.
Many parents of children with ARPKD seek early and reliable prenatal diagnosis of subsequent children to guide future family planning. Frequently, patients with ARPKD are diagnosed by ultrasonography only late in pregnancy or at birth. However, fetal sonography at the time when pregnancies are usually terminated may fail to detect enlargement and increased echogenicity of the kidneys or oligohydramnios (deficiency of amniotic fluid) secondary to poor fetal urine output. Thus, an early and reliable prenatal diagnosis of ARPKD in at-risk families is feasible only by molecular genetic analysis. Prenatal diagnosis should be offered only to families for which there is an unequivocal diagnosis in an affected family member. However, prenatal molecular genetic analysis is usually done using chorionic villus sampling, which is an invasive procedure that is carried out quite late in pregnancy (no earlier than week 10–12 of gestation). Interested families should also be informed about the possibility of preimplantation genetic diagnosis with in vitro fertilization at some diagnostic centres. Preimplantation genetic diagnosis always requires a high degree of coordination between different medical specialists and a health assessment in advance. In the future, it might be possible to conduct non-invasive prenatal testing of the fetal genotype by screening a maternal blood sample188.
When considering prenatal testing, medical and ethical implications require a careful review by attending physicians and the parents. These tests should be embedded in an ethical framework based on the principles of beneficence and respect for autonomy. Beneficence-based clinical judgements should be evidence-based (that is, they should be based on the best available evidence), and clinical strategies should be identified that are reliably expected to result in the greatest balance of benefits (that is, the protection and promotion of health-related interests) over clinical harms (that is, impairments of those interests)189. Respect for autonomy and the patient’s perspective is another key issue in the context of perinatal genetic testing. The physician is obliged to respect the parents’ values and beliefs, to respect their perspective on their interests and to implement only those clinical strategies that are authorized as a result of informed consent30,189.
Management
ADPKD
ADPKD is a systemic disorder with renal manifestations and an increasing number of extrarenal manifestations (TABLE 1). Management of ADPKD was the subject of a report of a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference44, Spanish and Kidney Health Australia-Caring for Australasians with Kidney Impairment (KHA-CARI) management guidelines and a Core Curriculum190. Additionally, a classification of ADPKD disease severity into different classes (1A–1E) was developed on the basis of htTKV (which provides an accurate estimate of kidney cyst burden in ADPKD) and age178. The classification stratifies patients on the basis of the rate of GFR decline and is useful in clinical trials to identify patients with rapid disease progression who might benefit most from therapy, as opposed to patients whose disease progresses slowly and who might not need therapy191.
Lifestyle interventions to enhance hydration, limit dietary sodium and protein intake and maintain a healthy weight, as well as early detection and rigorous treatment of hypertension and possibly dyslipidaemia, might have a beneficial effect on the course of the disease. Patients with ADPKD often have a mild urinary concentration defect that usually goes undetected, which is consistent with the inappropriate antidiuresis that is observed in Pkd1 heterozygous mice192. Plasma vasopressin and its precursor copeptin, which serves as a marker of vasopressin, may be increased in patients with ADPKD, and plasma copeptin levels correlate with the severity of ADPKD in cross-sectional studies and with the rate of disease progression in prospective studies193. Increasing evidence has shown that the action of vasopressin on the kidneys may contribute not only to the progression of ADPKD but also to the progression of CKD in general194. Additionally, two large studies on cyst and kidney growth (the CRISP study) and hypertension (the HALT study) in ADPKD have shown an association between dietary salt intake and the rate of progression of ADPKD195. The CRISP and the Modification of Diet in Renal Disease (MDRD) studies suggested an association between low levels of high-density lipoprotein (HDL) cholesterol and faster rates of kidney growth or GFR decline, respectively, in patients with ADPKD195. Experimental studies have shown that moderate food or caloric restriction substantially inhibits disease progression in orthologous mouse models of ADPKD, and high body mass index in patients with ADPKD is associated with a poor outcome196.
Treatments that directly target cystogenic mechanisms (such as decreasing cAMP levels in cystic tissues) are now available. For example, the Tolvaptan Efficacy and Safety in Management of Autosomal Dominant Polycystic Kidney Disease and its Outcomes (TEMPO) 3:4 clinical trial showed that the vasopressin receptor 2 antagonist tolvaptan slowed the rate of kidney growth and estimated GFR (eGFR) decline in patients with early ADPKD (age 18–50 years, estimated creatinine clearance ≥60 ml min–1)197; tolvaptan has received regulatory approval in Japan, Canada, the European Union, Switzerland and South Korea for the treatment of ADPKD in patients with rapidly progressive disease. In the USA, approval from the US FDA was deferred until further data are available on the potential benefits and risks of tolvaptan treatment. The Replicating Evidence of Preserved Renal Function: an Investigation of Tolvaptan Safety and Efficacy in ADPKD (REPRISE) clinical trial showed that tolvaptan also slows the eGFR decline in later-stage ADPKD (age 18–65 years, eGFR 25–65 ml/min/1.73 m2)198, and the FDA has now approved the therapy in the USA. In both the TEMPO 3:4 and REPRISE trials, clinically significant elevation of serum transaminases was detected in ~5% of patients who were treated with tolvaptan; therefore, strict instructions to regularly monitor liver enzymes are incorporated into the treatment regimen. Potential benefits and adverse effects of tolvaptan treatment should be considered before prescribing tolvaptan, and the treatment is also recommended only for patients with rapidly progressive disease199.
In the ALADIN study on kidney and cyst growth, patients with ADPKD were randomly assigned to octreotide (a long-acting somatostatin analogue that also decreases cAMP levels) or placebo treatment groups, and a favourable trend of reduced TKV increase and eGFR decline was observed, although statistical significance was not reached and thus larger trials are required200. DIPAK1 and LIPS are ongoing clinical trials to evaluate the effect of the somatostatin analogue lanreotide on disease progression in patients with ADPKD and stage 3 CKD or stage 2–3 CKD, respectively. Clinical trials of the mTOR inhibitors sirolimus and everolimus failed to show benefit despite encouraging results in animal models201–203, which is probably owing to an inability to administer sufficient levels of these drugs to inhibit mTOR in the kidneys without causing systemic toxic effects. Pravastatin, an HMG-CoA reductase inhibitor, has shown promising results in slowing cyst growth in a small randomized clinical trial in children with ADPKD204.
Cyst growth and disease monitoring.
Cyst development and growth varies widely between patients. Furthermore, even in patients with severe disease, GFR remains within the normal range for decades, limiting its value to estimate the prognosis or monitor the progression of the disease in its early stages. TKV correlates with disease manifestations such as hypertension, pain, haematuria and proteinuria159; TKV and cyst volume increase exponentially but at variable rates, averaging 5–6% per year. Accordingly, measurement of TKV is beneficial if the data could influence a medical decision, such as enrolment in a clinical trial or initiation of treatment in countries in which medications are approved for the treatment of ADPKD. MRI is the preferred method to measure TKV, as it is more accurate than ultrasonography and safer than CT, but its cost may be a limitation in some countries.
Hypertension.
A recent meta-analysis concluded that at least 20% of children with ADPKD have hypertension and that many of those children are undiagnosed and do not attend regular follow-ups177. A recent retrospective multicentre study of ambulatory blood pressure monitoring even found a higher prevalence of hypertension in children with ADPKD176. In this study, 31%, 42% and 35% of a cohort of 310 children with ADPKD (mean age 11.5 ± 4.1 years) were hypertensive during the daytime, the night-time or the entire 24-hour cycle, respectively. More than half of the patients (52%) lacked physiological nocturnal blood pressure dipping. Thus, children with a family history of ADPKD should be screened regularly for hypertension from the age of 5 years and at 3-year intervals when no hypertension is found. The HALT PKD placebo-controlled clinical trial examined the role of blockade of the RAAS in patients with early and advanced ADPKD. In patients with early ADPKD (15–49 years of age, eGFR >60 ml/min/1.73 m2), rigorous blood pressure control (blood pressure target range 95/60 to 110/75 mmHg) was associated with slower rates of increase in TKV and renal vascular resistance and with a greater decline in left ventricular mass index (faster during the first 4 months and slower decline there-after), without an overall effect on eGFR, than standard blood pressure control (120/70 to 130/80 mmHg)205,206. Rigorous blood pressure control was not studied in patients with more advanced ADPKD (18–64 years of age, eGFR 25–60 ml/min/1.73 m2) because it was considered to be detrimental in these patients207. At both early and late stages of the disease, monotherapy with an angiotensin-converting-enzyme (ACE) inhibitor achieved good blood pressure control (<130/85 mmHg) in most patients. Therefore, ACE inhibitors (or angiotensin receptor blockers (ARBs)) are considered to be the first-line therapy for hypertension in ADPKD.
Pain.
At least 60% of patients with ADPKD eventually experience acute or chronic abdominal or flank pain of renal origin. Acute pain is often associated with kidney cyst haemorrhage, infection or nephrolithiasis (kidney stones). Chronic pain is usually attributable to cyst enlargement that causes kidney capsule stretching or to marked enlargement of the kidneys and/or liver that causes musculoskeletal back pain. Managing chronic pain can be challenging and requires a multidisciplinary stepwise approach with multiple strategies, including physical therapy interventions, pain medications, cyst sclerosis and laparoscopic cyst fenestrations and renal denervation procedures208–210.
Haematuria.
Gross haematuria may be associated with cyst haemorrhage or infection, passage of kidney stones or kidney or urinary tract neoplasms. Cyst haemorrhage can be associated with fever, which might be difficult to differentiate from cyst infection. With conservative management (rest, holding anticoagulants or antiplatelet drugs, and analgesics if needed), most haemorrhages resolve within 2–7 days. If symptoms persist for >1 week or if the initial episode occurs after the age of 50 years, investigation to exclude neoplasm should be undertaken. The management of gross haematuria is mostly conservative — rest, hydration and avoidance or temporary holding of medications that facilitate bleeding. The antifibrinolytic agent tranexamic acid can be considered when conservative therapy fails211. Selective arterial embolization or nephrectomy may be necessary in cases of life-threatening haemorrhage.
Nephrolithiasis.
Increased frequency of nephrolithiasis in patients with ADPKD is attributed to increased urinary stasis and metabolic factors, which include low urinary pH, low citrate concentration and decreased ammonia excretion. These factors predispose to the development of uric acid and calcium oxalate stones. The medical treatment of nephrolithiasis in patients with ADPKD is similar to that in the general population; for example, extracorporeal shock wave lithotripsy and percutaneous nephrolithotomy in patients with early disease and normal kidney function have been used successfully without increased risk of complications212. Flexible ureterorenoscopy with laser fragmentation has also been used successfully213.
Cyst infection.
The triad of abdominal or flank pain and/or tenderness, fever and elevated erythrocyte sedimentation rate or C-reactive protein (CRP) should raise suspicion of cyst infection. Initial evaluation should include urinalysis, urine culture with antimicrobial susceptibility testing, blood culture and imaging with CT scan or MRI214,215. When available, an 18F-fluorodeoxyglucose (FDG)–PET scan is more sensitive than CT or MRI for detecting cyst infections216. If a complex cyst (that is, a cyst that does not meet the diagnostic criteria of a benign simple cyst) is detected, aspiration of cyst fluid should be considered if clinically feasible and the fluid should be sent for microbiology studies. Treatment of cyst infections should include intravenous broad-spectrum antibiotics if the patient has systemic symptoms, followed by a transition to oral antibiotics that have good cyst penetration, such as ciprofloxacin or other quinolones (total duration of antibiotic treatment should be at least 4 weeks). If the fever persists unabated for >72 hours, repeat imaging should be obtained to exclude complications that may require intervention (for example, perinephric abscess or urinary tract obstruction). Infected cysts >5 cm may be resistant to antibiotic therapy and require percutaneous or surgical drainage217. Perinephric abscess requires prolonged intravenous antibiotic therapy and drainage should be considered. Nephrectomy is rarely needed but may be considered in cases of perinephric abscess or emphysematous pyelonephritis that are resistant to drainage and/or antibiotic therapy218.
Renal cell carcinoma.
A high index of suspicion is required to make the diagnosis of renal cell carcinoma (RCC) in the rare cases when a tumour develops in a polycystic kidney, as RCC does not occur more frequently in ADPKD than in other renal diseases219. Rapid growth of a complex cyst, a solid mass on ultrasonography, speckled calcifications on CT imaging, contrast enhancement, tumour thrombus and regional lymphadenopathies on CT or MRI scans should raise suspicion of RCC. Fever and systemic symptoms (anorexia, fatigue and weight loss) are frequent presenting symptoms220. Nephronsparing interventions should be used whenever feasible and safe221.
ESRD.
Kidney transplantation is the optimal kidney replacement treatment and the results in ADPKD patients are better than in the general ESRD population222. Given the natural course of the disease, preemptive transplantation can be planned and is associated with better outcomes. Radical nephrectomy for space considerations is carried out in 20–25% of patients with ADPKD and may be done before, at the time of or several months after the transplantation. During the first year following a successful kidney transplantation, the volume of the native polycystic kidneys decreases by 30–40%223 for reasons that are unclear but might include reduced blood flow and resolution of uraemic factors that stimulate renal growth. Indications for nephrectomy also include recurrent and/or severe infection, bleeding, intractable pain, symptomatic nephrolithiasis and suspicion of kidney malignancy. Hand-assisted laparoscopic nephrectomy is less invasive than an open approach and the preferred timing varies between centres224.
Although ADPKD is not a contraindication for peritoneal dialysis225, it is less commonly used than haemo-dialysis given the challenge of accommodating a large volume of peritoneal dialysate and an increased risk of abdominal wall hernias. In haemodialysis treatment, patients with ADPKD may have an increased risk of aneurysmal dilatation of arteriovenous fistulas225.
Hepatic cysts.
In the majority of patients with ADPKD, liver cysts remain asymptomatic. Because of the increased survival of patients with ADPKD on RRT, the prevalence of clinically significant PLD is increasing226. Manifestations of PLD may be due to mass effects from organomegaly or cyst complications such as haemorrhage, infection or rupture. In addition to female sex, the main risk factors for severe PLD include multiple pregnancies and prolonged use of estrogens and, possibly, progestogens227.
Surgical interventions for severe PLD should target symptom alleviation and improvement of QOL and should be tailored to the characteristics of individual patients and to the natural history of PLD68. Surgical options include percutaneous aspiration and sclerosis with ethanol or other agents, deroofing of cysts laparoscopically or by open surgery and combined liver resection and cyst fenestration228. The latter procedure should be reserved for patients with severe symptomatic PLD and a liver anatomy that allows the preservation of at least two contiguous liver segments with adequate hepatic venous drainage229. Transcatheter arterial embolization results in only a modest volume reduction and is associated with substantial morbidity and mortality230,231. Liver transplantation is considered for patients when partial liver resection is not feasible. Somatostatin analogues, such as octreotide and lanreotide, can reduce liver cyst volume232–236.
Liver cyst infections are a common complication of PLD and are diagnosed, evaluated and managed similarly to infected kidney cysts. Diverticular disease and ascending cholangitis should be considered as a possible source of liver cyst infections. The presentation of ascending cholangitis is characterized by cholestasis with elevation of conjugated bilirubin. Some patients may experience recurrent episodes of cholangitis that may be difficult to treat, sometimes requiring monthly rotating antibiotics to prevent recurrences. Endoscopic sphincterotomy may increase the risk of recurrent cholangitis and should be avoided if possible. It is important to recognize that mild dilatation of the extrahepatic common bile duct can be observed in up to 40% of patients with ADPKD and should not lead to an unnecessary and dangerous sphincterotomy237.
Intracranial aneurysms.
ICAs are detected in ~8% of patients with ADPKD, which is approximately fivefold higher than in the general population but is further increased (approximately threefold higher) in patients with a relative who has had an ICA238.
The rupture of ICAs is associated with a mortality of up to 50% and a mean age at rupture of 40 years, which is lower than in the general population238. Rupture usually presents as sudden onset of a severe headache (described as a thunder clap headache). Increased size, posterior location, family history of ICAs and a personal history of subarachnoid haemorrhage are potential risks for rupture. The management of unruptured ICAs is the same as that in the general population69,239,240.
Given the frequency of ICAs in patients with ADPKD and that they increase morbidity and mortality risk, whether patients with ADPKD should undergo presymptomatic screening for ICAs has been intensely debated and universal consensus has not been reached239,241. The majority of ICAs that were detected by presymptomatic screening in patients with ADPKD (which is generally carried out using magnetic resonance angiography (MRA)) are <7 mm in diameter and are in the anterior circulation (80–90% of cases), which in the general population have a very small risk of rupture239. Few longitudinal studies of asymptomatic, unruptured ICAs in patients with ADPKD have been carried out, but the few that have suggest that their natural history is not different from that of ICAs in the general population69,242. Accordingly, although we do not recommend presymptomatic screening for all patients with ADPKD, screening might be appropriate in patients with a family history of aneurysmal rupture owing to the higher prevalence of unruptured ICAs and possible increased risk of rupture243,244. Screening may also be appropriate in patients with ADPKD who have a family history of ICAs and a personal history of intracranial haemorrhage, as well as in individuals in high-risk professions such as pilots and before major elective surgeries. Given the substantial risk of intervention, it is our opinion that treatment is not usually recommended for ICAs <7 mm in size; the frequency of follow-up MRAs for smaller ICAs depends on information about stability. If screening is negative, rescreening of patients with good life expectancy at 5-year intervals seems reasonable in our opinion. MRA for the detection of ICAs does not require administration of gadolinium and is preferable to CT angiography (CTA), especially in patients with reduced eGFR to avoid iodinated contrast245.
ARPKD
In the most severe cases of ARPKD, poor urine output in utero results in oligohydramnios (deficiency of amniotic fluid) and Potter sequence. The ARPKD presentation at birth may be dominated by respiratory difficulties from pulmonary hypoplasia and/or from restrictive disease caused by massive kidney enlargement1. In patients with respiratory insufficiency, the cause should be identified and aggressive resuscitative measures, including artificial ventilation if needed, should be implemented. In some severe cases, unilateral or bilateral nephrectomies may be considered, although the evidence to support this approach has been questioned183,246. In patients who survive infancy, renal function may improve or remain stable for many years and slowly progress to renal failure only later in life as the kidneys develop macroscopic cysts and interstitial fibrosis, and the size ofthe kidneys relative to body mass often decreases182,183.
Hypertension.
Arterial hypertension often develops during the first months of life and affects up to 80% of children with ARPKD32. Although arterial hypertension is sometimes difficult to control, even with multidrug treatment, it generally responds well to salt restriction and antihypertensive drugs (preferably an ACE inhibitor or ARB). After the first year of life, hypertension may become easier to control for reasons that are still poorly understood.
Electrolyte, nutritional and growth abnormalities.
Electrolyte abnormalities due to the inability to concentrate and dilute urine are very common in the early, severe presentations of ARPKD and should be anticipated and treated183. An assertive nutritional programme and correction of acidosis are important to optimize linear renal growth. If renal growth impairment occurs, treatment with growth hormone can be considered247.
ESRD.
For infants with ESRD, peritoneal dialysis or haemodialysis can be used. Renal transplantation is limited by body size, but in centres that are well experienced in paediatric surgery, it can be carried out even in small children with a minimum weight of 7 kg248,249. Rejection rates and survival beyond 3 years of age are not different from patients with most other renal diseases who undergo transplant surgery. Biliary sepsis can cause mortality in patients with ARPKD after renal transplantation249.
Congenital hepatic fibrosis.
Although CHF invariably occurs in children with ARPKD and can be associated with non-obstructive intrahepatic bile duct dilatation (Caroli disease), clinical manifestations of portal hypertension typically manifest as the children age. Some adolescents and adults without a previous diagnosis of ARPKD may present with hepatic manifestations, most often with complications of portal hypertension (such as variceal oesophageal bleeding, splenomegaly and hypersplenism with leukopenia, thrombocytopenia or anaemia) or episodes of cholangitis or sepsis and complications of biliary sludge or lithiasis. Liver enzyme values can occasionally be mildly elevated, but hepatocellular function is usually normal. The kidneys in these patients may be normal or exhibit varying degrees of medullary collecting duct ectasia or macrocystic disease without marked renal enlargement. Patients with ARPKD who survive infancy and those who present during adolescence may require a portosystemic shunt to prevent life-threatening haemorrhages from oesophageal varices. Patients with associated Caroli disease might have recurrent episodes of cholangitis. Although routine antibiotic prophylaxis for cholangitis is not recommended, antibiotic prophylaxis for 6–12 weeks after a cholangitis episode, immediately after transplantation or in the context of increased immunosuppression may be considered249. Adult patients with ARPKD >40 years of age may have a slightly increased risk of developing hepatic tumours, especially cholangiocarcinomas250.
Combined kidney and liver transplantation has been advocated for selected patients with ESRD who have substantial bile duct dilatation and episodes of cholangitis251–253. In addition to primary hyperoxaluria, ARPKD can certainly be considered an indication for combined liver and kidney transplantation during childhood. The best timing and strategy for combined transplantation is still a matter of debate and usually requires decision-making based on individual circumstances.
Quality of life
In the personalized medicine approach, it is no longer sufficient to treat the disease — instead, clinicians must treat their patients. Consequently, disease management paradigms should include biological, physical and psychological assessments, which seems to be particularly important for patients with multifaceted diseases such as ADPKD and further underscores the importance of including patient-reported outcomes in routine clinical care. In parallel, sensitive and appropriate kidney disease measures should be developed alongside other established tools to assess the burden of ADPKD in an individualized, personalized manner. Health-related QOL (HRQOL), which encompasses physical and mental health, social issues as well as pain and discomfort254, facilitates patient-centred care and encourages shared decision-making among medical professionals and patients. The specific issues of QOL have not been systemically studied in patients with ARPKD or their families; accordingly, we focus on only ADPKD.
ADPKD affects the biological, physical and psychological aspects of patient QOL. For example, physicians underestimate the effect of renal pain in patients with ADPKD, despite its high prevalence in patients with chronic kidney diseases255. Pain and abdominal fullness, which are both common symptoms of ADPKD, are often associated with lower eGFR256. Furthermore, pain is independently associated with clinical depression257, and although evidence exists that depression is common among patients with ADPKD257, studies that explicitly address the treatment of depression in patients with ADPKD are lacking. Accordingly, HRQOL assessment might help guide treatment decisions. Indeed, HRQOL scores have been shown to predict risk of increased hospitalization and mortality in patients with chronic kidney diseases258. However, only a limited number of HRQOL studies of patients with ADPKD have analysed possible correlations between disease progression, psychosocial burden and their effect on daily living.
Generic QOL measures (such as the 36-item Short Form Health Survey (SF-36)) are fairly insensitive, especially in patients with early-stage ADPKD255,256,259. In a recent study of HRQOL in patients with ADPKD who encompassed all stages of the disease (from patients with early CKD to those on dialysis or undergoing renal transplantation), overall HRQOL was generally highest in patients with CKD stage 1–3, followed by renal transplant recipients, patients with stage 4–5 CKD and patients on dialysis255. Progressive disease predominantly had an effect on physical health, whereas mental health varied less between patients with different stages of the disease255.
A new self-administered patient-reported outcome questionnaire, the ADPKD Impact Scale (ADPKD-IS), which consists of 14 items representing 3 conceptual domains (physical, emotional and fatigue) and 4 additional questions, was developed on the basis of the information (see below) of 1,674 patients worldwide260. The ADPKD-specific conceptual framework (a model representing concepts and themes to be measured and their relationships) in this questionnaire was created on the basis of information from patients and clinical experts about how ADPKD affected the general health, daily activities, physical or social activities, pain experience, urinary issues (such as urgency and frequency of urination as well as nocturia) and emotional well-being of patients. Scores on the physical, emotional and fatigue domains of the ADPKD-IS differed substantially between patients with stage 3b CKD versus those with stage 1 CKD at baseline260.
Of note, effective treatment of ADPKD can improve HRQOL — a 1-year trial with octreotide resulted in modestly reduced liver volumes (4.95% ± 6.77%) and was associated with improvements in pain and emotional scales on the SF-36 (REF.234). Perceived social support and the psychosocial impact of coping with a diagnosis of ADPKD were analysed in another cross-sectional study257, which found that lower eGFR was associated with less perceived social support. Depression, increased kidney size and loss of a first-degree relative to ADPKD were identified as independent risk factors for increased psychosocial risk. Of note, women were more likely to be predisposed to poorer overall psychosocial well-being than men257.
Given the genetic basis of ADPKD, patients are often faced with major psychological challenges related to the hereditary nature of the disorder and its potential effect on their family and offspring. ‘Genetic guilt’ may be the reason many patients hide their diagnosis from their spouse and family, even when these people are usually central to their emotional support network. Hidden genetic guilt substantially affects the psychosocial well-being of patients261.
Outlook
Although the prospects for better outcome predictions and treatments have dramatically improved over the past decade, an incomplete understanding of the function of the polycystin proteins remains a substantial impediment to understanding the pathogenesis of PKD and developing rational therapies to treat these diseases. For ARPKD in particular, the limitations of experimental models and the paucity of biomarkers and imaging modalities to enable prediction of the disease course limit current efforts to design clinical trials to test the efficacy of potential new therapies.
Pathogenesis
The greatest challenge in research on PKD, and that which is likely to be of greatest benefit if overcome, is understanding the pathogenesis of PKD. Although the aetiologies of ADPKD and ARPKD are mostly understood, many questions about the pathogenesis of these diseases are still intensely disputed. For example, although PC2 is a TRP-like calcium channel, a definite role for Ca2+ in the pathogenesis of ADPKD has not been established, and the role of PC2 as a chaperone for PC1 may be important262,263. The role of somatic changes at the disease-causing locus in cystic epithelia, which are required for cyst initiation and, more importantly, for cyst expansion and/or persistence, is another highly debated area of research that would benefit from additional studies56,62,63,264.
The ciliary basis of PKD pathogenesis has been the predominant hypothesis for >15 years, but studies have cast doubt on this hypothesis. The proposed ciliary localization (among others) of PC1, PC2, fibrocystin and DZIP1L132,145 and the observations that a loss of ciliary function265 or the abnormal ciliary composition in some syndromic ciliopathies266,267 cause PKD are the strongest evidence supporting the cilia hypothesis. However, the role of the polycystin complex in cilia is not clear; for example, the hypothesis that the polycystin complex functions as a mechanosensor in cilia is not supported by recent data86,87. Indeed, despite a study suggesting that the polycystin complex is a WNT receptor, the function of the polycystin complex in healthy tissue seems far from resolved and this uncertainty remains a major impediment to understanding the pathogenesis of ADPKD268. As discussed earlier, cilia certainly have a role in PKD, as concomitant ablation of Pkd1 and loss of cilia in mice results in less severe cystic disease than ablation of Pkd1 alone103. Alternatively, separate cystic (polycystin complex-related) and proliferation pathways required for cystic growth are ciliary-based. In fact, the ciliary localization of PC1 has been questioned (a specific stimulus might be required for PC1 to get to the cilium), and other polycystin-like proteins (such as PKD1-like protein 1 (PKD1L1) and PKD2L1), rather than PC1 and PC2, might regulate ciliary Ca2+ levels87,269,270. In addition, conflicting data exist about whether calcium influx into the cilium is sufficient to activate a cytoplasmic calcium response87,271.
Although a ciliary function has been proposed for both fibrocystin and DZIP1L, and genetic studies suggest that they are associated with the polycystin complex121,132, the precise role of these proteins remains unresolved. That fibrocystin seems much more important for kidney tubule integrity in humans than in mice further complicates experimental resolution272,273. The functions of PC1, PC2, fibrocystin and DZIP1L, or those of the cleavage products of these proteins, when secreted into urine in soluble form148 or in urinary extracellular vesicles exocytosed by the multivesicular body pathway274, or even possibly from cilia275, require further study. Other unresolved questions include whether the C terminus of PC1 (and fibrocystin) is cleaved and translocates to the nucleus, how much it contributes to the signalling by the polycystin complex and whether it is relevant for preventing PKD276. Additionally, ciliary-based signalling, such as that mediated by sonic hedgehog, has also been suggested to be important in PKD, although its precise role in ADPKD and ARPKD is not clear277.
Diagnosis and prognosis
Further research is necessary to improve the identification of patients who require treatment and especially the monitoring of treatment efficacy. It is widely accepted that current methods to monitor kidney function are not very useful for diagnosis in the early stages of disease when treatment is likely to be the most efficacious. Determination of htTKV by imaging is currently the best method to predict disease outcomes and to monitor treatment efficacy, and further quantitative image analysis of kidney composition is likely to improve its predictive and monitoring value278. Research to identify potential biochemical biomarkers is still in the early stages, but with an easily accessible supply of urine from patients to monitor kidney function, it seems likely that this area of research is ripe for development279,280. Together with the present genic and allelic predictive value of the germline mutation, better in silico analyses and cellular assays should enable the identity and penetrance of in-frame mutations to be more accurately predicted262. Other genetic factors that are associated with disease progression (identified using genome-wide association studies, WES and WGS) are also likely to be of prognostic value and to improve our understanding of the pathogenesis of PKD79.
Therapies
This is an exciting time for PKD studies, as the first drug specifically for the treatment ADPKD, tolvaptan, has received regulatory approval in many parts of the world179,197. It seems likely that other treatments targeting signalling pathways downstream of PKD-causing genetic lesions, including inhibitors of cAMP generation (for example, somatostatin analogues such as octreotide200), more effective inhibitors of mTOR activity in the kidneys (for example, rapamycin conjugates281), remedying metabolic abnormalities (for example, by food restriction282) and other therapies that were effective in rodent studies (for example, reducing inflammation or calcium signalling; reviewed elsewhere33), are likely to be approved for human treatment in the next few years. There are prospects for combination treatments283, and some of these therapies may also be effective in treating severe PLD284. Furthermore, although the development of treatments that more directly target the basic defect (deficiency of PCI or PC2) in PKD has not been undertaken, evidence exists for other genetic diseases that this approach could be productive285–287. These treatments could include chaperone treatment for some missense mutations288, targeting nonsense mutations using translational readthrough-inducing drugs289, boosting the levels of functional PC1 and somatic gene editing using CRISPR–Cas9 (REF.290) to repair the underlying defect in the kidneys. To date, little progress has been made in developing treatments for ARPKD, but given the related disease mechanisms in ADPKD and ARPKD, there is the hope that at least some therapies for ADPKD might also be effective in treating ARPKD.
Acknowledgements
C.B. receives research support for his laboratory from the Deutsche Forschungsgemeinschaft (DFG) Collaborative Research Centre (SFB) KIDGEM 1140 and the Federal Ministry of Education and Research (BMBF, 01GM1515C). L.M.G.-W. is supported by US National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) funding of the University of Alabama–Birmingham Hepato-Renal Fibrocystic Disease Core Center (DK074038). D.J.M.P. receives financial support from the Dutch Kidney Foundation and the Netherlands Organisation for Scientific Research. S.H. is supported by the Japan Society for the Promotion of Science KAKENHI grant number JP15K10632. P.C.H. and V.E.T. are supported by NIDDK funding of the Mayo Translational Polycystic Kidney Disease Center (DK090728). The authors thank J. Smith, T. Kline and M. Edwards (all at the Mayo Clinic, MN, USA) for their assistance with figures 1 and 7.
Footnotes
Competing interests
C.B. is an employee of Bioscientia/Sonic Healthcare and holds a part-time faculty appointment at the University of Freiburg, Germany.
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Reviewer information
Nature Reviews Disease Primers thanks J. Calvet and the other anonymous reviewer(s) for their contribution to the peer review of this work.
RELATED LIKS
ADPKD Mutation Database (PKDB): http://pkdb.mayo.edu/ Genome aggregation database (gnomAD): http://gnomad.broadinstitute.org/
References
- 1.Bergmann C ARPKD and early manifestations of ADPKD: the original polycystic kidney disease and phenocopies. Pediatr. Nephrol. 30, 15–30 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Torres VE, Harris PC & Pirson Y Autosomal dominant polycystic kidney disease. Lancet 369, 1287–1301 (2007). [DOI] [PubMed] [Google Scholar]
- 3.Bergmann C & Weiskirchen R It’s not all in the cilium, but on the road to it: genetic interaction network in polycystic kidney and liver diseases and how trafficking and quality control matter. J. Hepatol.56, 1201–1203 (2012). [DOI] [PubMed] [Google Scholar]
- 4.Gerdes JM, Davis EE & Katsanis N The vertebrate primary cilium in development, homeostasis, and disease. Cell 137, 32–45 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hildebrandt F, Benzing T & Katsanis N Ciliopathies. N. Engl. J. Med. 364, 1533–1543 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dalgaard OZ Bilateral polycystic disease of the kidneys; a follow-up of two hundred and eighty-four patients and their families. Acta Med. Scand. Suppl. 328, 1–255 (1957). [PubMed] [Google Scholar]
- 7.Iglesias CG et al. Epidemiology of adult polycystic kidney disease, Olmsted County, Minnesota: 1935–1980. Am. J. Kidney Dis. 2, 630–639 (1983). [DOI] [PubMed] [Google Scholar]
- 8.Levy M & Feingold J Estimating prevalence in single-gene kidney diseases progressing to renal failure. Kidney Int. 58, 925–943 (2000). [DOI] [PubMed] [Google Scholar]
- 9.Davies F et al. Polycystic kidney disease re-evaluated: a population-based study. Q. J. Med. 79, 477–485 (1991). [PubMed] [Google Scholar]
- 10.Simon P et al. [Epidemiologic data, clinical and prognostic features of autosomal dominant polycystic kidney disease in a French region]. Nephrologie 17, 123–130 (1996). [PubMed] [Google Scholar]
- 11.Yersin C et al. Frequency and impact of autosomal dominant polycystic kidney disease in the Seychelles (Indian Ocean). Nephrol. Dial. Transplant. 12, 2069–2074 (1997). [DOI] [PubMed] [Google Scholar]
- 12.Higashihara E et al. Prevalence and renal prognosis of diagnosed autosomal dominant polycystic kidney disease in Japan. Nephron 80, 421–427 (1998). [DOI] [PubMed] [Google Scholar]
- 13.de Almeida E et al. Prevalence of autosomaldominant polycystic kidney disease in Alentejo. Portugal. Kidney Int. 59, 2374 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Neumann HP et al. Epidemiology of autosomal-dominant polycystic kidney disease: an in-depth clinical study for south-western Germany. Nephrol. Dial. Transplant. 28, 1472–1487 (2013). [DOI] [PubMed] [Google Scholar]
- 15.McGovern AP et al. Identification of people with autosomal dominant polycystic kidney disease using routine data: a cross sectional study. BMC Nephrol.15, 182 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Willey CJ et al. Prevalence of autosomal dominant polycystic kidney disease in the European Union. Nephrol. Dial. Transplant. 32, 1356–1363 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chan KW Adult polycystic kidney disease in Hong Kong Chinese: an autopsy study. Pathology 25, 229–232 (1993). [DOI] [PubMed] [Google Scholar]
- 18.Cornec-Le Gall E, Torres VE & Harris PC Genetic complexity of autosomal dominant polycystic kidney and liver diseases. J. Am. Soc. Nephrol. 29, 13–23 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wakai K et al. Trends in incidence of end-stage renal disease in Japan, 1983–2000: age-adjusted and age-specific rates by gender and cause. Nephrol. Dial. Transplant. 19, 2044–2052 (2004). [DOI] [PubMed] [Google Scholar]
- 20.The United States Renal Data System (USRDS).USRDS 1999 Annual Data Report (National Institute of Diabetes and Digestive and Kidney Diseases,1999). [Google Scholar]
- 21.Stengel B et al. Trends in the incidence of renal replacement therapy for end-stage renal disease in Europe, 1990–1999. Nephrol. Dial. Transplant. 18, 1824–1833 (2003). [DOI] [PubMed] [Google Scholar]
- 22.Parfrey PS et al. The diagnosis and prognosis of autosomal dominant polycystic kidney disease.N. Engl. J. Med. 323, 1085–1090 (1990). [DOI] [PubMed] [Google Scholar]
- 23.Simon P Prognosis of autosomal dominant polycystic kidney disease. Nephron 71, 247–248 (1995). [DOI] [PubMed] [Google Scholar]
- 24.Spithoven EM et al. Analysis of data from the ERA-EDTA registry indicates that conventional treatments for chronic kidney disease do not reduce the need for renal replacement therapy in autosomal dominant polycystic kidney disease. Kidney Int. 86, 1244–1252 (2014). [DOI] [PubMed] [Google Scholar]
- 25.Shaw C, Simms RJ, Pitcher D & Sandford R Epidemiology of patients in England and Wales with autosomal dominant polycystic kidney disease and end-stage renal failure. Nephrol. Dial. Transplant. 29, 1910–1918 (2014). [DOI] [PubMed] [Google Scholar]
- 26.Alzarka B, Morizono H, Bollman JW, Kim D & Guay-Woodford LM Design and Implementation of the Hepatorenal Fibrocystic Disease Core Center Clinical Database: a centralized resource for characterizing autosomal recessive polycystic kidney disease and other hepatorenal fibrocystic diseases. Front. Pediatr. 5, 80 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kääriäinen H Polycystic kidney disease in children: a genetic and epidemiological study of 82 Finnish patients. J. Med. Genet. 24, 474–481 (1987). [PMC free article] [PubMed] [Google Scholar]
- 28.Bergmann C & Zerres K Early manifestations of polycystic kidney disease. Lancet 369, 2157 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Guay-Woodford LM & Desmond RA Autosomal recessive polycystic kidney disease: the clinical experience in North America. Pediatrics 111, 1072–1080 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Bergmann C et al. PKHD1 mutations in families requesting prenatal diagnosis for autosomal recessive polycystic kidney disease (ARPKD). Hum. Mutat. 23, 487–495 (2004). [DOI] [PubMed] [Google Scholar]
- 31.Gimpel C et al. Perinatal diagnosis, management, and follow-up of cystic renal diseases: a clinical practice recommendation with systematic literature reviews. JAMA Pediatr. 172, 74–86 (2018). [DOI] [PubMed] [Google Scholar]
- 32.Bergmann C et al. Clinical consequences of PKHD1 mutations in 164 patients with autosomal-recessive polycystic kidney disease (ARPKD). Kidney Int. 67, 829–848 (2005). [DOI] [PubMed] [Google Scholar]
- 33.Harris PC & Torres VE Genetic mechanisms and signaling pathways in autosomal dominant polycystic kidney disease (ADPKD). J. Clin. Invest. 124, 2315–2324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Porath B et al. Mutations in GANAB, encoding the glucosidase Ila subunit, cause autosomal-dominant polycystic kidney and liver disease. Am. J. Hum. Genet. 98, 1193–1207 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Besse W et al. Isolated polycystic liver disease genes define effectors of polycystin-1 function. J. Clin. Invest. 127, 3558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cornec-Le Gall E et al. Monoallelic mutations to DNAJB11 cause atypical autosomal-dominant polycystic kidney disease. Am. J. Hum. Genet. 102, 832–844 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Drenth JP, Te Morsche RH, Smink R, Bonifacino JS & Jansen JB Germline mutations in PRKCSH are associated with autosomal dominant polycystic liver disease. Nat. Genet. 33, 345–347 (2003). [DOI] [PubMed] [Google Scholar]
- 38.Davila S et al. Mutations in SEC63 cause autosomal dominant polycystic liver disease. Nat. Genet. 36, 575–577 (2004). [DOI] [PubMed] [Google Scholar]
- 39.Li A et al. Mutations in PRKCSH cause isolated autosomal dominant polycystic liver disease. Am. J. Hum. Genet. 72, 691–703 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cnossen WR et al. Whole-exome sequencing reveals LRP5 mutations and canonical Wnt signaling associated with hepatic cystogenesis. Proc. Natl Acad. Sci. USA 111, 5343–5348 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gunay-Aygun M et al. Hepatorenal findings in obligate heterozygotes for autosomal recessive polycystic kidney disease. Mol. Genet. Metab. 104, 677–681 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Hart TC et al. Mutations of the UMOD gene are responsible for medullary cystic kidney disease 2 and familial juvenile hyperuricaemic nephropathy. J. Med. Genet. 39, 882–892 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kirby A et al. Mutations causing medullary cystic kidney disease type 1 lie in a large VNTR in MUC1 missed by massively parallel sequencing. Nat. Genet. 45, 299–303 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chapman AB et al. Autosomal dominant polycystic kidney disease (ADPKD): executive summary from a Kidney Disease: Improving Global Outcomes (KDIGO) Controversies Conference. Kidney Int. 88, 17–27 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Bolar NA et al. Heterozygous loss-of-function SEC61A1 mutations cause autosomal-dominant tubulo-interstitial and glomerulocystic kidney disease with anemia. Am. J. Hum. Genet. 99, 174–187 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Plaisier E et al. COL4A1 mutations and hereditary angiopathy, nephropathy, aneurysms, and muscle cramps. N. Engl. J. Med. 357, 2687–2695 (2007). [DOI] [PubMed] [Google Scholar]
- 47.Bergmann C Recent advances in the molecular diagnosis of polycystic kidney disease. Expert Rev.Mol. Diagn. 17, 1037–1054 (2017). [DOI] [PubMed] [Google Scholar]
- 48.Hughes J et al. The polycystic kidney disease 1 (PKD1) gene encodes a novel protein with multiple cell recognition domains. Nat. Genet. 10, 151–160 (1995). [DOI] [PubMed] [Google Scholar]
- 49.Qian F et al. Cleavage of polycystin-1 requires the receptor for egg jelly domain and is disrupted by human autosomal-dominant polycystic kidney disease 1-associated mutations. Proc. Natl Acad. Sci. USA 99, 16981–16986 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kurbegovic A et al. Novel functional complexity of polycystin-1 by GPS cleavage in vivo: role in polycystic kidney disease. Mol. Cell. Biol. 34, 3341–3353 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Low SH et al. Polycystin-1, STAT6, and P100 function in a pathway that transduces ciliary mechanosensation and is activated in polycystic kidney disease. Dev. Cell 10, 57–69 (2006). [DOI] [PubMed] [Google Scholar]
- 52.Xu Y et al. The polycystin-1, lipoxygenase, and a-toxin domain regulates polycystin-1 trafficking. J. Am. Soc. Nephrol. 27, 1159–1173 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Tsiokas L, Kim E, Arnould T, Sukhatme VP & Walz, G. Homo- and heterodimeric interactions between the gene products of PKD1 and PKD2.Proc. Natl Acad. Sci. USA 94, 6965–6970 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shen PS et al. The structure of the polycystic kidney disease channel PKD2 in lipid nanodiscs. Cell 167, 763–773 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Grieben M et al. Structure of the polycystic kidney disease TRP channel Polycystin-2 (PC2). Nat. Struct. Mol. Biol. 24, 114–122 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Qian F, Watnick TJ, Onuchic LF & Germino GG The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell 87, 979–987 (1996). [DOI] [PubMed] [Google Scholar]
- 57.Watnick T et al. Mutations of PKD1 in ADPKD2 cysts suggest a pathogenic effect of trans-h eterozygous mutations. Nat. Genet. 25, 143–144 (2000). [DOI] [PubMed] [Google Scholar]
- 58.Pei Y et al. Somatic PKD2 mutations in individual kidney and liver cysts support a ‘two-hit’ model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 10, 1524–1529 (1999). [DOI] [PubMed] [Google Scholar]
- 59.Bergmann C et al. Mutations in multiple PKD genes may explain early and severe polycystic kidney disease. J. Am. Soc. Nephrol. 22, 2047–2056 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Happe H et al. Toxic tubular injury in kidneys from Pkd1-deletion mice accelerates cystogenesis accompanied by dysregulated planar cell polarity and canonical Wnt signaling pathways. Hum. Mol. Genet. 18, 2532–2542 (2009). [DOI] [PubMed] [Google Scholar]
- 61.Patel V et al. Acute kidney injury and aberrant plana cell polarity induce cyst formation in mice lacking rena cilia. Hum. Mol. Genet. 17, 1578–1590 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lantinga-van Leeuwen IS et al. Lowering of Pkd1 expression is sufficient to cause polycystic kidney disease. Hum. Mol. Genet. 13, 3069–3077 (2004). [DOI] [PubMed] [Google Scholar]
- 63.Hopp K et al. Functional polycystin-1 dosage govern: autosomal dominant polycystic kidney disease severity. J. Clin. Invest. 122, 4257–4273 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cornec-Le Gall E et al. Type of PKD1 mutation influences renal outcome in ADPKD. J. Am. Soc. Nephrol. 24, 1006–1013 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Leonhard WN et al. Scattered deletion of PKD1 in kidneys causes a cystic snowball effect and recapitulates polycystic kidney disease. J. Am. Soc. Nephrol. 26, 1322–1333 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Heyer CM et al. Predicted mutation strength of nontruncating PKD1 mutations aids genotype-phenotype correlations in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 27, 2872–2884 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Cornec-Le Gall E et al. The PROPKD score: a new algorithm to predict renal survival in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 27, 942–951 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chebib FT et al. Effect of genotype on the severity and volume progression of polycystic liver disease in autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 31, 952–960 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Irazabal MV et al. Extended follow-up of unrupture intracranial aneurysms detected by presymptomatic screening in patients with autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 6 1274–1285 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Vujic M et al. Incompletely penetrant PKD1 alleles mimic the renal manifestations of ARPKD. J. Am. Soc Nephrol. 21, 1097–1102 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Audrezet M-P et al. Comprehensive PKD1 and PKD mutation analysis in prenatal autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 27, 722–729 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Losekoot M et al. Neonatal onset autosomal dominant polycystic kidney disease (ADPKD) in a patient homozygous for a PKD2 missense mutation due to uniparental disomy. J. Med. Genet. 49, 37–40 (2012). [DOI] [PubMed] [Google Scholar]
- 73.Pei Y et al. Bilineal disease and trans-heterozygotes in autosomal dominant polycystic kidney disease. Am J. Hum. Genet. 68, 355–363 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Rossetti S et al. Mutation analysis of the entire PKD1 gene: genetic and diagnostic implications. Am. J. Hum Genet. 68, 46–63 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Iliuta IA et al. Polycystic kidney disease without an apparent family history. J. Am. Soc. Nephrol. 28, 2768–2776 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Persu A et al. Comparison between siblings and twins supports a role for modifier genes in ADPKD. Kidney Int. 66, 2132–2136 (2004). [DOI] [PubMed] [Google Scholar]
- 77.Paterson AD et al. Progressive loss of renal function is an age-dependent heritable trait in type 1 autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 16, 755–762 (2005). [DOI] [PubMed] [Google Scholar]
- 78.Fain PR et al. Modifier genes play a significant role in the phenotypic expression of PKD1. Kidney Int. 67, 1256–1267 (2005). [DOI] [PubMed] [Google Scholar]
- 79.Liu X-G et al. Genetic variation of DKK3 may modify renal disease severity in PKD1. J. Am. Soc. Nephrol. 21, 1510–1520 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hogan MC et al. Characterization of PKD protein¬positive exosome-like vesicles. J. Am. Soc. Nephrol. 20, 278–288 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Drummond IA Polycystins, focal adhesions and extracellular matrix interactions. Biochim. Biophys. Acta 1812, 1322–1326 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lee K, Battini L & Gusella GL Cilium, centrosome and cell cycle regulation in polycystic kidney disease. Biochim. Biophys. Acta 1812, 1263–1271 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Rowe I et al. Defective glucose metabolism in polycystic kidney disease identifies a new therapeutic strategy. Nat. Med. 19, 488–493 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Yamaguchi T et al. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 63, 1983–1994 (2003). [DOI] [PubMed] [Google Scholar]
- 85.Fischer E et al. Defective planar cell polarity in polycystic kidney disease. Nat. Genet. 38, 21–23 (2006). [DOI] [PubMed] [Google Scholar]
- 86.Nauli SM et al. Polycystins 1 and 2 mediate mechanosensation in the primary cilium of kidney cells. Nat. Genet. 33, 129–137 (2003). [DOI] [PubMed] [Google Scholar]
- 87.Delling M, DeCaen PG, Doerner JF, Febvay S & Clapham DE Primary cilia are specialized calcium signalling organelles. Nature 504, 311–314 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Du J & Wilson PD Abnormal polarization of EGF receptors and autocrine stimulation of cyst epithelial growth in human ADPKD. Am. J. Physiol. 269, C487–C495 (1995). [DOI] [PubMed] [Google Scholar]
- 89.MacRae Dell K, Nemo R, Sweeney WE Jr. & Avner ED EGF-related growth factors in the pathogenesis of murine ARPKD. Kidney Int. 65, 2018–2029 (2004). [DOI] [PubMed] [Google Scholar]
- 90.Arnould T et al. The polycystic kidney disease 1 gene product mediates protein kinase C α-dependent and c-Jun N-terminal kinase-dependent activation of the transcription factor AP-1. J. Biol. Chem. 273, 6013–6018 (1998). [DOI] [PubMed] [Google Scholar]
- 91.Parnell SC et al. The polycystic kidney disease-1 protein, polycystin-1, binds and activates heterotrimeric G-proteins in vitro. Biochem. Biophys. Res. Commun. 251, 625–631 (1998). [DOI] [PubMed] [Google Scholar]
- 92.Bhunia AK et al. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell 109, 157–168 (2002). [DOI] [PubMed] [Google Scholar]
- 93.Boca M et al. Polycystin-1 induces resistance to apoptosis through the phosphatidylinositol 3-kinase/Akt signaling pathway. J. Am. Soc. Nephrol. 17, 637–647 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Takiar V et al. Activating AMP-activated protein kinase (AMPK) slows renal cystogenesis. Proc. Natl Acad. Sci. USA 108, 2462–2467 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Shillingford JM et al. The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease.Proc. Natl Acad. Sci. USA 103, 5466–5471 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Renken C, Fischer D-C, Kundt G, Gretz N & Haffner D Inhibition of mTOR with sirolimus does not attenuate progression of liver and kidney disease in PCK rats. Nephrol. Dial. Transplant. 26, 92–100 (2011). [DOI] [PubMed] [Google Scholar]
- 97.Leonhard WN et al. Curcumin inhibits cystogenesis by simultaneous interference of multiple signaling pathways: in vivo evidence from a Pkd1-deletion model. Am. J. Physiol. Renal Physiol. 300, F1193–F1202 (2011). [DOI] [PubMed] [Google Scholar]
- 98.Menezes LF et al. Network analysis of a Pkd1-mouse model of autosomal dominant polycystic kidney disease identifies HNF4alpha as a disease modifier. PLOS Genet. 8, e1003053 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Chiaravalli M et al. 2-deoxy-d-glucose ameliorates PKD progression. J. Am. Soc. Nephrol. 27, 1958–1969 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Riwanto M et al. Inhibition of aerobic glycolysis attenuates disease progression in polycystic kidney disease. PLOS ONE 11, e0146654 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rodriguez D et al. Inhibition of sodium-glucose cotransporter 2 with dapagliflozin in han: SPRD rats with polycystic kidney disease. Kidney Blood Press Res. 40, 638–647 (2015). [DOI] [PubMed] [Google Scholar]
- 102.Menezes LF, Lin CC, Zhou F & Germino GG Fatty acid oxidation is impaired in an orthologous mouse model of autosomal dominant polycystic kidney disease. EBioMedicine 5, 183–192 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ma M, Tian X, Igarashi P, Pazour GJ & Somlo S Loss of cilia suppresses cyst growth in genetic models of autosomal dominant polycystic kidney disease. Nat. Genet. 45, 1004–1012 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Lee K, Boctor S, Barisoni LMC & Gusella GL Inactivation of integrin-beta1 prevents the development of polycystic kidney disease after the loss of polycystin-1. J. Am. Soc. Nephrol. 26, 888–895 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Wilson PD, Geng L, Li X & Burrow CR The PKD1 gene product, ‘polycystin-1,’ is a tyrosine-phosphorylated protein that colocalizes with alpha2beta1-integrin in focal clusters in adherent renal epithelia. Lab Invest. 79, 1311–1323 (1999). [PubMed] [Google Scholar]
- 106.Silberberg M, Charron AJ, Bacallao R & Wandinger-Ness A Mispolarization of desmosomal proteins and altered intercellular adhesion in autosomal dominant polycystic kidney disease.Am. J. Physiol. Ren. Physiol. 288, F1153–F1163 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Castelli M et al. Regulation of the microtubular cytoskeleton by Polycystin-1 favors focal adhesions turnover to modulate cell adhesion and migration. BMC Cell Biol. 16, 15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Luyten A et al. Aberrant regulation of planar cell polarity in polycystic kidney disease. J. Am. Soc. Nephrol. 21, 1521–1532 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Kunimoto K et al. Disruption of core planar cell polarity signaling regulates renal tubule morphogenesis but is not cystogenic. Curr. Biol. 27, 3120–3131 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Griffin MD, Torres VE, Grande JP & Kumar R Vascular expression of polycystin. J. Am. Soc. Nephrol. 8, 616–626 (1997). [DOI] [PubMed] [Google Scholar]
- 111.Huang JL et al. Vascular endothelial growth factor C for polycystic kidney diseases. J. Am. Soc. Nephrol. 27, 69–77 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Outeda P et al. Polycystin signaling is required for directed endothelial cell migration and lymphatic development. Cell Rep. 7, 634–644 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Grantham JJ, Geiser JL & Evan AP Cyst formation and growth in autosomal dominant polycystic kidney disease. Kidney Int. 31, 1145–1152 (1987). [DOI] [PubMed] [Google Scholar]
- 114.Liu D et al. A Pkd1-Fbn1 genetic interaction implicates TGF-beta signaling in the pathogenesis of vascular complications in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 25, 81–91 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Grantham JJ Clinical practice. Autosomal dominant polycystic kidney disease. N. Engl. J. Med. 359, 1477–1485 (2008). [DOI] [PubMed] [Google Scholar]
- 116.Swenson-Fields KI et al. Macrophages promote polycystic kidney disease progression. Kidney Int. 83, 855–864 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Ta MH, Harris DC & Rangan GK Role of interstitial inflammation in the pathogenesis of polycystic kidney disease. Nephrology (Carlton). 18, 317–330 (2013). [DOI] [PubMed] [Google Scholar]
- 118.Karihaloo A et al. Macrophages promote cyst growth in polycystic kidney disease. J. Am. Soc. Nephrol. 22, 1809–1814 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Chen L et al. Macrophage migration inhibitory factor promotes cyst growth in polycystic kidney disease.J. Clin. Invest. 125, 2399–2412 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Watnick TJ et al. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol. Cell 2, 247–251 (1998). [DOI] [PubMed] [Google Scholar]
- 121.Fedeles SV et al. A genetic interaction network of five genes for human polycystic kidney and liver diseases defines polycystin-1 as the central determinant of cyst formation. Nat. Genet. 43, 639–647 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Ecder T & Schrier RW Cardiovascular abnormalities in autosomal-dominant polycystic kidney disease. Nat. Rev. Nephrol. 5, 221–228 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Belz MM et al. Familial clustering of ruptured intracranial aneurysms in autosomal dominant polycystic kidney disease. Am. J. Kidney Dis. 38, 770–776 (2001). [DOI] [PubMed] [Google Scholar]
- 124.Lorthioir A et al. Polycystin deficiency induces dopamine-reversible alterations in flow-mediated dilatation and vascular nitric oxide release in humans. Kidney Int. 87, 465–472 (2015). [DOI] [PubMed] [Google Scholar]
- 125.Sharif-Naeini R et al. Polycystin-1 and −2 dosage regulates pressure sensing. Cell 139, 587–596 (2009). [DOI] [PubMed] [Google Scholar]
- 126.Morel N et al. PKD1 haploinsufficiency is associated with altered vascular reactivity and abnormal calcium signaling in the mouse aorta. Pflugers Arch. 457, 845–856 (2009). [DOI] [PubMed] [Google Scholar]
- 127.Qian Q et al. Pkd2 haploinsufficiency alters intracellular calcium regulation in vascular smooth muscle cells. Hum. Mol. Genet. 12, 1875–1880 (2003). [DOI] [PubMed] [Google Scholar]
- 128.Hassane S et al. Pathogenic sequence for dissecting aneurysm formation in a hypomorphic polycystic kidney disease 1 mouse model. Arter. Thromb. Vasc. Biol. 27, 2177–2183 (2007). [DOI] [PubMed] [Google Scholar]
- 129.Kim K, Drummond I, Ibraghimov-Beskrovnaya O, Klinger K & Arnaout MA Polycystin 1 is required for the structural integrity of blood vessels. Proc. Natl Acad. Sci. USA 97, 1731–1736 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Boulter C et al. Cardiovascular, skeletal, and renal defects in mice with a targeted disruption of the Pkd1 gene. Proc. Natl Acad. Sci. USA 98, 12174–12179 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pennekamp P et al. The ion channel polycystin-2 is required for left-right axis determination in mice.Curr. Biol. 12, 938–943 (2002). [DOI] [PubMed] [Google Scholar]
- 132.Lu H et al. Mutations in DZIP1L, which encodes a ciliary-transition-zone protein, cause autosomal recessive polycystic kidney disease. Nat. Genet. 49, 1025–1034 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Ahrabi AK et al. Glomerular and proximal tubule cysts as early manifestations of Pkd1 deletion. Nephrol. Dial. Transplant. 25, 1067–1078 (2010). [DOI] [PubMed] [Google Scholar]
- 134.Garcia-Gonzalez MA et al. Pkd1 and Pkd2 are required for normal placental development. PLOS ONE 5, e12821 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lu W et al. Perinatal lethality with kidney and pancreas defects in mice with a targeted Pkdl mutation. Nat. Genet. 17, 179–181 (1997). [DOI] [PubMed] [Google Scholar]
- 136.Lu W et al. Late onset of renal and hepatic cysts in Pkd1-targeted heterozygotes. Nat. Genet. 21, 160–161 (1999). [DOI] [PubMed] [Google Scholar]
- 137.Piontek KB et al. A functional floxed allele of Pkd1 that can be conditionally inactivated in vivo. J. Am. Soc. Nephrol. 15, 3035–3043 (2004). [DOI] [PubMed] [Google Scholar]
- 138.Lantinga-van Leeuwen IS et al. Kidney-specific inactivation of the Pkd1 gene induces rapid cyst formation in developing kidneys and a slow onset of disease in adult mice. Hum. Mol. Genet. 16, 3188–3196 (2007). [DOI] [PubMed] [Google Scholar]
- 139.Wu G et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell 93, 177–188 (1998). [DOI] [PubMed] [Google Scholar]
- 140.Bastos AP et al. Pkd1 haploinsufficiency increases renal damage and induces microcyst formation following ischemia/reperfusion. J. Am. Soc. Nephrol.20, 2389–2402 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bergmann C Genetics of autosomal recessive polycystic kidney disease and its differential diagnoses. Front. Pediatr. 10.3389/fped.2017.00221 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Onuchic LF et al. PKHD1, the polycystic kidney and hepatic disease 1 gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel β-helix 1 repeats. Am. J. Hum. Genet. 70, 1305–1317 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Boddu R et al. Intragenic motifs regulate the transcriptional complexity of Pkhd1/PKHD1. J. Mol. Med. 92, 1045–1056 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ward CJ et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large, receptor-like protein. Nat. Genet. 30, 259–269 (2002). [DOI] [PubMed] [Google Scholar]
- 145.Ward CJ et al. Cellular and subcellular localization of the ARPKD protein; fibrocystin is expressed on primary cilia. Hum. Mol. Genet. 12, 2703–2710 (2003). [DOI] [PubMed] [Google Scholar]
- 146.Follit JA, Li L, Vucica Y & Pazour GJ The cytoplasmic tail of fibrocystin contains a ciliary targeting sequence. J. Cell Biol. 188, 21–28 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Hiesberger T et al. Proteolytic cleavage and nuclear translocation of fibrocystin is regulated by intracellular Ca2+ and activation of protein kinase C. J. Biol. Chem. 281, 34357–34364 (2006). [DOI] [PubMed] [Google Scholar]
- 148.Kaimori JY et al. Polyductin undergoes notch-like processing and regulated release from primary cilia. Hum. Mol. Genet. 16, 942–956 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Cabezas OR et al. Polycystic kidney disease with hyperinsulinemic hypoglycemia caused by a promoter mutation in phosphomannomutase 2. J. Am. Soc. Nephrol. 28, 2529–2539 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bergmann C et al. Spectrum of mutations in the gene for autosomal recessive polycystic kidney disease (ARPKD/PKHD1). J. Am. Soc. Nephrol. 14, 76–89 (2003). [DOI] [PubMed] [Google Scholar]
- 151.Menezes LFC et al. Polyductin, the PKHD1 gene product, comprises isoforms expressed in plasma membrane, primary cilium, and cytoplasm. Kidney Int. 66, 1345–1355 (2004). [DOI] [PubMed] [Google Scholar]
- 152.Zhang MZ et al. PKHD1 protein encoded by the gene for autosomal recessive polycystic kidney disease associates with basal bodies and primary cilia in renal epithelial cells. Proc. Natl Acad. Sci. USA 101, 2311–2316 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wang S et al. Fibrocystin/polyductin, found in the same protein complex with polycystin-2, regulates calcium responses in kidney epithelia. Mol. Cell. Biol. 27, 3241–3252 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Fischer DC et al. Activation of the AKT/mTOR pathway in autosomal recessive polycystic kidney disease (ARPKD). Nephrol. Dial. Transplant. 24, 1819–1827 (2009). [DOI] [PubMed] [Google Scholar]
- 155.Garcia-Gonzalez MA et al. Genetic interaction studies link autosomal dominant and recessive polycystic kidney disease in a common pathway. Hum. Mol. Genet. 16, 1940–1950 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lager DJ, Qian Q, Bengal RJ, Ishibashi M & Torres VE The pck rat: a new model that resembles human autosomal dominant polycystic kidney and liver disease. Kidney Int. 59, 126–136 (2001). [DOI] [PubMed] [Google Scholar]
- 157.O’Meara CC et al. Role of genetic modifiers in an orthologous rat model of ARPKD. Physiol. Genom. 44, 741–753 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Reeders ST et al. Prenatal diagnosis of autosomal dominant polycystic kidney disease with a DNA probe. Lancet 328, 6–8 (1986). [DOI] [PubMed] [Google Scholar]
- 159.Grantham JJ et al. Volume progression in polycystic kidney disease. N. Engl. J. Med. 354, 2122–2130 (2006). [DOI] [PubMed] [Google Scholar]
- 160.Rule AD et al. Characteristics of renal cystic and solid lesions based on contrast-enhanced computed tomography of potential kidney donors. Am. J. Kidney Dis. 59, 611–618 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Pei Y et al. Unified criteria for ultrasonographic diagnosis of ADPKD. J. Am. Soc. Nephrol. 20, 205–212 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Belibi FA & Edelstein CL Unified ultrasonographic diagnostic criteria for polycystic kidney disease. J. Am. Soc. Nephrol. 20, 6–8 (2009). [DOI] [PubMed] [Google Scholar]
- 163.Rossetti S et al. Comprehensive molecular diagnostics in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 18, 2143–2160 (2007). [DOI] [PubMed] [Google Scholar]
- 164.Audrezet MP et al. Autosomal dominant polycystic kidney disease: comprehensive mutation analysis of PKD1 and PKD2 in 700 unrelated patients. Hum. Mutat. 33, 1239–1250 (2012). [DOI] [PubMed] [Google Scholar]
- 165.Rossetti S et al. Identification of gene mutations in autosomal dominant polycystic kidney disease through targeted resequencing. J. Am. Soc. Nephrol. 23, 915–933 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Trujillano D et al. Diagnosis of autosomal dominant polycystic kidney disease using efficient PKD1 and PKD2 targeted next-generation sequencing. Mol. Genet. Genom. Med. 2, 412–421 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Eisenberger T et al. An efficient and comprehensive strategy for genetic diagnostics of polycystic kidney disease. PLOS ONE 10, e0116680 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mallawaarachchi AC et al. Whole-genome sequencing overcomes pseudogene homology to diagnose autosomal dominant polycystic kidney disease. Eur. J. Hum. Genet. 24, 1584–1590 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lanktree MB et al. Prevalence estimates of polycystic kidney and liver disease by population sequencing. J. Am. Soc. Nephrol. 29, 2593–2600 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Rossetti S et al. Incompletely penetrant PKD1 alleles associated with mild, homozygous or in utero onset PKD. J. Am. Soc. Nephrol. 18, 848–855 (2009). [Google Scholar]
- 171.Consugar MB et al. Characterization of large rearrangements in autosomal dominant polycystic kidney disease and the PKD1/TSC2 contiguous gene syndrome. Kidney Int. 74, 1468–1479 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Tan AY et al. Autosomal dominant polycystic kidney disease caused by somatic and germline mosaicism. Clin. Genet. 87, 373–377 (2015). [DOI] [PubMed] [Google Scholar]
- 173.Cnossen WR & Drenth JP Polycystic liver disease: an overview of pathogenesis, clinical manifestations and management. Orphanet J. Rare Dis. 9, 69 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Drenth JPH, Chrispijn M & Bergmann C Congenital fibrocystic liver diseases. Best Pract. Res. Clin. Gastroenterol. 24, 573–584 (2010). [DOI] [PubMed] [Google Scholar]
- 175.De Rechter S et al. Clinicians’ attitude towards family planning and timing of diagnosis in autosomal dominant polycystic kidney disease. PLOS ONE 12, e0185779 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Massella L et al. Prevalence of hypertension in children with early-stage ADPKD. Clin. J. Am. Soc. Nephrol. 13, 874–883 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Marlais M et al. Hypertension in autosomal dominant polycystic kidney disease: a meta-analysis. Arch. Dis. Child 101, 1142–1147 (2016). [DOI] [PubMed] [Google Scholar]
- 178.Irazabal MV et al. Imaging classification of autosomal dominant polycystic kidney disease: a simple model for selecting patients for clinical trials. J. Am. Soc. Nephrol. 26, 160–172 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gansevoort RT et al. Recommendations for the use of tolvaptan in autosomal dominant polycystic kidney disease: a position statement on behalf of the ERA-EDTA Working Groups on Inherited Kidney Disorders and European Renal Best Practice. Nephrol. Dial Transpl. 31, 337–348 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Gunay-Aygun M et al. Autosomal recessive polycystic kidney disease and congenital hepatic fibrosis: summary statement of a first National Institutes of Health/Office of Rare Diseases conference. J. Pediatr. 149, 159–164 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Adeva M et al. Clinical and molecular characterization defines a broadened spectrum of autosomal recessive polycystic kidney disease (ARPKD). Medicine (Baltimore). 85, 1–21 (2006). [DOI] [PubMed] [Google Scholar]
- 182.Dell KM et al. Kidney disease progression in autosomal recessive polycystic kidney disease.J. Pediatr. 171, 196–201 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Guay-Woodford LM et al. Consensus expert recommendations for the diagnosis and management of autosomal recessive polycystic kidney disease: report of an international conference. J. Pediatr. 165, 611–617 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Fonck C et al. Autosomal recessive polycystic kidney disease in adulthood. Nephrol. Dial. Transplant. 16, 1648–1652 (2001). [DOI] [PubMed] [Google Scholar]
- 185.Avni FE et al. Hereditary polycystic kidney diseases in children: changing sonographic patterns through childhood. Pediatr. Radiol. 32, 169–174 (2002). [DOI] [PubMed] [Google Scholar]
- 186.Bergmann C et al. PKHD1 mutations in autosomal recessive polycystic kidney disease (ARPKD).Hum. Mutat. 23, 453–463 (2004). [DOI] [PubMed] [Google Scholar]
- 187.Consugar MB et al. Haplotype analysis improves molecular diagnostics of autosomal recessive polycystic kidney disease. Am. J. Kidney Dis. 45, 77–87 (2005). [DOI] [PubMed] [Google Scholar]
- 188.Liu L, Li K, Fu X, Chung C & Zhang KA Forward look at noninvasive prenatal testing. Trends Mol. Med. 22, 958–968 (2016). [DOI] [PubMed] [Google Scholar]
- 189.Chervenak FA & McCullough LB Ethical issues in perinatal genetics. Semin. Fetal Neonatal Med. 16, 70–73 (2011). [DOI] [PubMed] [Google Scholar]
- 190.Chebib FT & Torres VE Autosomal dominant polycystic kidney disease: core curriculum 2016. Am. J. Kidney Dis. 67, 792–810 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Dhariwal M, Rasmussen M & Holstein BE Body mass index and smoking: cross-sectional study of a representative sample of adolescents in Denmark. Int. J. Publ. Heal. 55, 307–314 (2010). [DOI] [PubMed] [Google Scholar]
- 192.Ahrabi AK et al. PKD1 haploinsufficiency causes a syndrome of inappropriate antidiuresis in mice. J. Am. Soc. Nephrol. 18, 1740–1753 (2007). [DOI] [PubMed] [Google Scholar]
- 193.Boertien WE et al. Relationship of copeptin, a surrogate marker for arginine vasopressin, with change in total kidney volume and GFR decline in autosomal dominant polycystic kidney disease: results from the CRISP cohort. Am. J. Kidney Dis. 61, 420–429 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Bankir L, Bouby N & Ritz E Vasopressin: a novel target for the prevention and retardation of kidney disease? Nat. Rev. Nephrol. 9, 223–239 (2013). [DOI] [PubMed] [Google Scholar]
- 195.Torres VE et al. Potentially modifiable factors affecting the progression of autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 6, 640–647 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Nowak KL et al. Overweight and obesity are predictors of progression in early autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 29, 571–578 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Torres VE et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 367, 2407–2418 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Torres VE et al. Tolvaptan in later-stage autosomal dominant polycystic kidney disease. N. Engl. J. Med. 377, 1930–1942 (2017). [DOI] [PubMed] [Google Scholar]
- 199.Chebib FT et al. A practical guide for treatment of rapidly progressive ADPKD with tolvaptan. J. Am. Soc. Nephrol. 29, 2458–2470 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Caroli A et al. Effect of longacting somatostatin analogue on kidney and cyst growth in autosomal dominant polycystic kidney disease (ALADIN): a randomised, placebo-controlled, multicentre trial. Lancet 382, 1485–1495 (2013). [DOI] [PubMed] [Google Scholar]
- 201.Serra AL et al. Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N. Engl. J. Med. 363, 820–829 (2010). [DOI] [PubMed] [Google Scholar]
- 202.Perico N et al. Sirolimus therapy to halt the progression of ADPKD. J. Am. Soc. Nephrol. 21, 1031–1040 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Walz G et al. Everolimus in patients with autosomal dominant polycystic kidney disease. N. Engl. J. Med. 363, 830–840 (2010). [DOI] [PubMed] [Google Scholar]
- 204.Cadnapaphornchai MA et al. Effect of pravastatin on total kidney volume, left ventricular mass index, and microalbuminuria in pediatric autosomal dominant polycystic kidney disease. Clin. J. Am. Soc. Nephrol. 9, 889–896 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Schrier RS et al. Blood pressure in early autosomal dominant polycystic kidney disease. N. Engl. J. Med. 371, 2255–2266 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Irazabal MV et al. Prognostic enrichment design in clinical trials for ADPKD: the HALT PKD clinical trial. Nephrol. Dial. Transplant. 32, 1857–1865 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Torres VE et al. Angiotensin blockade in late autosomal dominant polycystic kidney disease. N. Engl. J. Med. 371, 2267–2276 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Tellman MW, Bahler CD, Shumate AM, Bacallao RL & Sundaram CP Management of pain in autosomal dominant polycystic kidney disease and anatomy of renal innervation. J. Urol. 193, 1470–1478 (2015). [DOI] [PubMed] [Google Scholar]
- 209.Casteleijn NF et al. A stepwise approach for effective management of chronic pain in autosomal-dominant polycystic kidney disease. Nephrol. Dial. Transplant. 29(Suppl. 4), iv142–iv153 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.de Jager RL et al. Catheter-based renal denervation as therapy for chronic severe kidney-related pain. Nephrol. Dial. Transplant. 33, 614–619 (2017). [DOI] [PubMed] [Google Scholar]
- 211.Hulme P & Wylie K Towards evidence based emergency medicine: best BETs from the Manchester Royal Infirmary. BET 1: tranexamic acid in life-threatening haematuria. Emerg. Med. J. 32, 168–169 (2015). [DOI] [PubMed] [Google Scholar]
- 212.Mallett A, Patel M, Tunnicliffe DJ & Rangan GK KHA-CARI autosomal dominant polycystic kidney disease guideline: management of renal stone disease. Semin. Nephrol. 35, 603–606 (2015). [DOI] [PubMed] [Google Scholar]
- 213.Yili L et al. Flexible ureteroscopy and holmium laser lithotripsy for treatment of upper urinary tract calculi in patients with autosomal dominant polycystic kidney disease. Urol. Res. 40, 87–91 (2012). [DOI] [PubMed] [Google Scholar]
- 214.Jouret F et al. Diagnosis of cyst infection in patients with autosomal dominant polycystic kidney disease: attributes and limitations of the current modalities. Nephrol. Dial. Transplant. 27, 3746–3751 (2012). [DOI] [PubMed] [Google Scholar]
- 215.Lantinga MA, Drenth JP & Gevers TJ Diagnostic criteria in renal and hepatic cyst infection. Nephrol. Dial. Transplant. 30, 744–751 (2014). [DOI] [PubMed] [Google Scholar]
- 216.Neuville M, Hustinx R, Jacques J, Krzesinski JM & Jouret F Diagnostic algorithm in the management of acute febrile abdomen in patients with autosomal dominant polycystic kidney disease. PLOS ONE 11, e0161277 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Lantinga MA et al. Management of renal cyst infection in patients with autosomal dominant polycystic kidney disease: a systematic review. Nephrol. Dial. Transplant. 32, 144–150 (2017). [DOI] [PubMed] [Google Scholar]
- 218.Watanabe K et al. A case of autosomal dominant polycystic kidney disease with emphysematous polycystic renal infection that required surgical treatment. Intern. Med. 10.2169/internalmedicine.1257-18 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Karami S et al. Risk of renal cell carcinoma among kidney transplant recipients in the United States. Am. J. Transplant. 16, 3479–3489 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Keith DS, Torres VE, King BF, Zincki H & Farrow GM Renal cell carcinoma in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 4, 1661–1669 (1994). [DOI] [PubMed] [Google Scholar]
- 221.Xu L et al. Percutaneous radiofrequency ablation with contrast-enhanced ultrasonography for solitary and sporadic renal cell carcinoma in patients with autosomal dominant polycystic kidney disease. World J. Surg. Oncol. 14, 193 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Spithoven EM et al. Renal replacement therapy for autosomal dominant polycystic kidney disease (ADPKD) in Europe: prevalence and survival-an analysis of data from the ERA-EDTA Registry. Nephrol. Dial. Transplant. 29, 15–25 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Jung Y et al. Volume regression of native polycystic kidneys after renal transplantation. Nephrol. Dial. Transplant. 31, 73–79 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Neeff HP et al. One hundred consecutive kidney transplantations with simultaneous ipsilateral nephrectomy in patients with autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 28, 466–471 (2013). [DOI] [PubMed] [Google Scholar]
- 225.Courivaud C et al. Polycystic kidney size and outcomes on peritoneal dialysis: comparison with haemodialysis. Clin. Kidney J. 7, 138–143 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Perrone RD, Ruthazer R & Terrin NC Survival after end-stage renal disease in autosomal dominant polycystic kidney disease: contribution of extrarenal complications to mortality. Am. J. Kidney Dis. 38, 777–784 (2001). [DOI] [PubMed] [Google Scholar]
- 227.Hogan MC et al. Liver involvement in early autosomal-dominant polycystic kidney disease.Clin. Gastroenterol. Hepatol. 13, 155–164 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Drenth JP, Chrispijn M, Nagorney DM, Kamath PS & Torres VE Medical and surgical treatment options for polycystic liver disease. Hepatology 52, 2223–2230 (2010). [DOI] [PubMed] [Google Scholar]
- 229.Chebib FT et al. Outcomes and durability of hepatic reduction after combined partial hepatectomy and cyst fenestration for massive polycystic liver disease. J. Am. Coll. Surg. 223, 118–126 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hoshino J et al. Intravascular embolization therapy in patients with enlarged polycystic liver. Am. J. Kidney Dis. 63, 937–944 (2014). [DOI] [PubMed] [Google Scholar]
- 231.Yang J et al. Comparison of volume-reductive therapies for massive polycystic liver disease in autosomal dominant polycystic kidney disease. Hepatol. Res. 46, 183–191 (2016). [DOI] [PubMed] [Google Scholar]
- 232.van Keimpema L, de Man RA & Drenth JP Somatostatin analogues reduce liver volume in polycystic liver disease. Gut 57, 1338–1339 (2008). [DOI] [PubMed] [Google Scholar]
- 233.Caroli A et al. Reducing polycystic liver volume in ADPKD: effects of somatostatin analogue octreotide. Clin. J. Am. Soc. Nephrol. 5, 783–789 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Hogan MC et al. Randomized clinical trial of long-acting somatostatin for autosomal dominant polycystic kidney and liver disease. J. Am. Soc. Nephrol. 21, 1052–1061 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hogan MC et al. Efficacy of 4 years of octreotide long-acting release therapy in patients with severe polycystic liver disease. Mayo Clin. Proc. 90, 1030–1037 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Gevers TJ et al. Young women with polycystic liver disease respond best to somatostatin analogues: a pooled analysis of individual patient data. Gastroenterology 145, 352–357 (2013). [DOI] [PubMed] [Google Scholar]
- 237.Ishikawa I et al. High incidence of common bile duct dilatation in autosomal dominant polycystic kidney disease patients. Am. J. Kidney Dis. 27, 321–326 (1996). [DOI] [PubMed] [Google Scholar]
- 238.Pirson Y, Chauveau D & Torres V Management of cerebral aneurysms in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 13, 269–276 (2002). [DOI] [PubMed] [Google Scholar]
- 239.Brown RD & Torner J Unruptured intracranial aneurysms: some questions answered, many questions remain. Re: Pelz D. CURES and the dilemma of unruptured intracranial aneurysms. Can J Neuro Sci. 2011 Mar;38(2):191–2. Can. J. Neurol. Sci. 38, 785–787 (2011). [PubMed] [Google Scholar]
- 240.Rozenfeld MN et al. Autosomal dominant polycystic kidney disease and intracranial aneurysms: is there an increased risk of treatment? AJNR Am. J. Neuroradiol 37, 290–293 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Flahault A et al. Screening for intracranial aneurysms in autosomal dominant polycystic kidney disease is cost-effective. Kidney Int. 93, 716–726 (2018). [DOI] [PubMed] [Google Scholar]
- 242.Jiang T et al. A follow-up study of autosomal dominant polycystic kidney disease with intracranial aneurysms using 3.0 T three-dimensional time-of-flight magnetic resonance angiography. Eur. J. Radiol 82, 1840–1845 (2013). [DOI] [PubMed] [Google Scholar]
- 243.Ring T & Spiegelhalter D Risk of intracranial aneurysm bleeding in autosomal-dominant polycystic kidney disease. Kidney Int. 72, 1400–1402 (2007). [DOI] [PubMed] [Google Scholar]
- 244.Xu HW, Yu SQ, Mei CL & Li MH Screening for intracranial aneurysm in 355 patients with autosomal-dominant polycystic kidney disease. Stroke 42, 204–206 (2011). [DOI] [PubMed] [Google Scholar]
- 245.Flahault A et al. Screening for unruptured intracranial aneurysms in autosomal dominant polycystic kidney disease: a survey of 420 nephrologists. PLOS ONE 11, e0153176 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Hartung EA & Guay-Woodford LM Autosomal recessive polycystic kidney disease: a hepatorenal fibrocystic disorder with pleiotropic effects. Pediatrics 134, e833–e845 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Lilova M, Kaplan BS & Meyers KEC Recombinant human growth hormone therapy in autosomal recessive polycystic kidney disease. Pediatr. Nephrol. 18, 57–61 (2003). [DOI] [PubMed] [Google Scholar]
- 248.Becker T et al. Paediatric kidney transplantation in small children— a single centre experience. Transpl. Int. 19, 197–202 (2006). [DOI] [PubMed] [Google Scholar]
- 249.Davis ID, Ho M, Hupertz V & Avner ED Survival of childhood polycystic kidney disease following renal transplantation: the impact of advanced hepatobiliary disease. Pediatr. Transplant. 7, 364–369 (2003). [DOI] [PubMed] [Google Scholar]
- 250.Srinath A & Shneider BL Congenital hepatic fibrosis and autosomal recessive polycystic kidney disease. J. Pediatr. Gastroenterol. Nutr. 54, 580–587 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Chapal M et al. Kidney and liver transplantation in patients with autosomal recessive polycystic kidney disease: a multicentric study. Nephrol. Dial. Transplant. 27, 2083–2088 (2012). [DOI] [PubMed] [Google Scholar]
- 252.Telega G, Cronin D & Avner ED New approaches to the autosomal recessive polycystic kidney disease patient with dual kidney-liver complications. Pediatr. Transplant. 17, 328–335 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Brinkert F et al. Combined liver-kidney transplantation for children with autosomal recessive polycystic kidney disease (ARPKD): indication and outcome. Transpl. Int. 26, 640–650 (2013). [DOI] [PubMed] [Google Scholar]
- 254.Patrick DL & Erickson P Health Status and Health Policy: Quality of Life in Health Care Evaluation and Resource Allocation (Oxford Univ. Press Inc., 1993). [Google Scholar]
- 255.Eriksson D et al. Health-related quality of life across all stages of autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 32, 2106–2111 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Miskulin DC et al. Health-related quality of life in patients with autosomal dominant polycystic kidney disease and CKD stages 1–4: a cross-sectional study. Am. J. Kidney Dis. 63, 214–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Simms RJ, Thong KM, Dworschak GC & Ong AC Increased psychosocial risk, depression and reduced quality of life living with autosomal dominant polycystic kidney disease. Nephrol. Dial. Transplant. 31, 1130–1140 (2016). [DOI] [PubMed] [Google Scholar]
- 258.Mujais SK et al. Health-related quality of life in CKD patients: correlates and evolution over time. Clin. J. Am. Soc. Nephrol. 4, 1293–1301 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Rizk D et al. Quality of life in autosomal dominant polycystic kidney disease patients not yet on dialysis. Clin. J. Am. Soc. Nephrol. 4, 560–566 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Oberdhan D et al. Development of the autosomal dominant polycystic kidney disease impact scale: a new health-related quality-of-life instrument. Am. J. Kidney Dis. 71, 225–235 (2018). [DOI] [PubMed] [Google Scholar]
- 261.Tong A et al. A painful inheritance-patient perspectives on living with polycystic kidney disease: thematic synthesis of qualitative research. Nephrol. Dial. Transplant. 30, 790–800 (2015). [DOI] [PubMed] [Google Scholar]
- 262.Gainullin VG, Hopp K, Ward CJ, Hommerding CJ & Harris PC Polycystin-1 maturation requires polycystin-2 in a dose-dependent manner. J. Clin. Invest. 125, 607–620 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Kim H et al. Ciliary membrane proteins traffic through the Golgi via a Rabep1/GGA1/Arl3-dependent mechanism. Nat. Commun. 5, 5482 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Tan AY et al. Somatic mutations in renal cyst epithelium in autosomal dominant polycystic kidney disease. J. Am. Soc. Nephrol. 29, 2139–2156 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Lin F et al. Kidney-specific inactivation of the KIF3A subunit of kinesin-II inhibits renal ciliogenesis and produces polycystic kidney disease. Proc. Natl Acad. Sci. USA 100, 5286–5291 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Garcia-Gonzalo FR et al. A transition zone complex regulates mammalian ciliogenesis and ciliary membrane composition. Nat. Gen. 43, 776–784 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Chih B et al. A ciliopathy complex at the transition zone protects the cilia as a privileged membrane domain. Nat. Cell Biol. 14, 61–72 (2012). [DOI] [PubMed] [Google Scholar]
- 268.Kim S et al. The polycystin complex mediates Wnt/Ca2+ signalling. Nat. Cell Biol. 18, 752–764 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.DeCaen PG, Delling M, Vien TN & Clapham DE Direct recording and molecular identification of the calcium channel of primary cilia. Nature 504, 315–318 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Mick DU et al. Proteomics of primary cilia by proximity labeling. Dev. Cell 35, 497–512 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Yuan S, Zhao L, Brueckner M & Sun Z Intraciliary calcium oscillations initiate vertebrate left-right asymmetry. Curr. Biol. 25, 556–567 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Moser M et al. A mouse model for cystic biliary dysgenesis in autosomal recessive polycystic kidney disease (ARPKD). Hepatology 41, 1113–1121 (2005). [DOI] [PubMed] [Google Scholar]
- 273.Bakeberg JL et al. Epitope-tagged Pkhd1 tracks the processing, secretion, and localization of fibrocystin.J. Am. Soc. Nephrol. 22, 2266–2277 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Galarreta CI et al. Tubular obstruction leads to progressive proximal tubular injury and atubular glomeruli in polycystic kidney disease. Am. J. Pathol. 184, 1957–1966 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Wood CR & Rosenbaum JL Ciliary ectosomes: transmissions from the cell’s antenna. Trends Cell Biol. 25, 276–285 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Chauvet V et al. Mechanical stimuli induce cleavage and nuclear translocation of the polycystin-1 C terminus. J. Clin. Invest. 114, 1433–1443 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Tran PV et al. Downregulating hedgehog signaling reduces renal cystogenic potential of mouse models. J. Am. Soc. Nephrol. 25, 2201–2212 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Kline TL et al. Image texture features predict renal function decline in patients with autosomal dominant polycystic kidney disease. Kidney Int. 92, 1206–1216 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Hogan MC et al. Identification of biomarkers for PKD1 using urinary exosomes. J. Am. Soc. Nephrol. 26, 1661–1670 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Pejchinovski M et al. Urine peptidome analysis predicts risk of end-stage renal disease and reveals proteolytic pathways involved in autosomal dominant polycystic kidney disease progression. Nephrol. Dial. Transplant. 32, 487–497 (2016). [DOI] [PubMed] [Google Scholar]
- 281.Shillingford JM, Leamon CP, Vlahov IR & Weimbs T Folate-conjugated rapamycin slows progression of polycystic kidney disease. J. Am. Soc. Nephrol. 23, 1674–1681 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Warner G et al. Food restriction ameliorates the development of polycystic kidney disease. J. Am. Soc. Nephrol. 27, 1437–1447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Hopp K et al. Tolvaptan plus pasireotide shows enhanced efficacy in a PKD1 model. J. Am. Soc. Nephrol. 26, 39–47 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Masyuk TV, Masyuk AI & La Russo NF Therapeutic targets in polycystic liver disease. Curr. Drug Targets 18, 950–957 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Wainwright CE et al. Lumacaftor-ivacaftor in patients with cystic fibrosis homozygous for Phe508del CFTR. N. Engl. J. Med. 373, 220–231 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Nelson CE et al. In vivo genome editing improves muscle function in a mouse model of Duchenne muscular dystrophy. Science 351, 403–407 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Tabebordbar M et al. In vivo gene editing in dystrophic mouse muscle and muscle stem cells. Science 351, 407–411 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 288.Fedeles SV et al. Sec63 and Xbp1 regulate IRE1a activity and polycystic disease severity. J. Clin. Invest. 125, 1955–1967 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Nagel-Wolfrum K, Moller F, Penner I, Baasov T& Wolfrum U Targeting nonsense mutations in diseases with translational read-through-inducing drugs (TRIDs). BioDrugs 30, 49–74 (2016). [DOI] [PubMed] [Google Scholar]
- 290.Wojtal D et al. Spell checking nature: versatility of CRISPR/Cas9 for developing treatments for inherited disorders. Am. J. Hum. Genet. 98, 90–101 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Huang E et al. DNA testing for live kidney donors at risk for autosomal dominant polycystic kidney disease. Transplantation 87, 133–137 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Harris PC & Rossetti S Molecular diagnostics for autosomal dominant polycystic kidney disease. Nat. Rev. Nephrol. 6, 197–206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Chebib FT, Sussman CR, Wang X, Harris PC & Torres VE Vasopressin and disruption of calcium signalling in polycystic kidney disease. Nat. Rev. Nephrol. 11, 451–464 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Ong ACM & Harris PC A polycystin-centric view of cyst formation and disease: the polycystins revisited. Kidney Int. 88, 699–710 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Streets AJ, Wessely O, Peters DJ & Ong AC Hyperphosphorylation of polycystin-2 at a critical residue in disease reveals an essential role for polycystin-1-regulated dephosphorylation. Hum. Mol. Genet. 22, 1924–1939 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Kim I et al. Fibrocystin/polyductin modulates renal tubular formation by regulating polycystin-2 expression and function. J. Am. Soc. Nephrol. 19, 455–468 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 297.Koulen P et al. Polycystin-2 is an intracellular calcium release channel. Nat. Cell Biol. 4, 191–197 (2002). [DOI] [PubMed] [Google Scholar]
- 298.Hartung EA & Guay-Woodford LM Polycystic kidney disease: DZIP1L defines a new functional zip code for autosomal recessive PKD. Nat. Rev. Nephrol. 13, 519–520 (2017). [DOI] [PubMed] [Google Scholar]
- 299.Song CJ, Zimmerman KA, Henke SJ & Yoder BK Inflammation and fibrosis in polycystic kidney disease. Results Probl. Cell Differ. 60, 323–344 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Qian Q, Harris PC & Torres VE Treatment prospects for autosomal-dominant polycystic kidney disease. Kidney Int. 59, 2005–2022 (2001). [DOI] [PubMed] [Google Scholar]
- 301.Brancatelli G et al. Fibropolycystic liver disease: CT and MR imaging findings. RadioGraphics 25, 659–670 (2005). [DOI] [PubMed] [Google Scholar]
- 302.Ho TA et al. Autosomal dominant polycystic kidney disease is associated with central and nephrogenic defects in osmoregulation. Kidney Int. 82, 1121–1129 (2012). [DOI] [PubMed] [Google Scholar]
- 303.O’Brien K et al. Congenital hepatic fibrosis and portal hypertension in autosomal dominant polycystic kidney disease. J. Pediatr. Gastroenterol. Nutr. 54, 83–89 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 304.Luciano RL & Dahl NK Extra-renal manifestations of autosomal dominant polycystic kidney disease (ADPKD): considerations for routine screening and management. Nephrol. Dial. Transplant. 29, 247–254 (2014). [DOI] [PubMed] [Google Scholar]