

Abstract
Using our previously designed biphenyl-2-ylphosphine L2 featuring a remote tertiary amino group as an enabling ligand, we have developed the first gold-catalyzed intermolecular hydroalkenylation of alkynes. Synthetically valuable conjugated dienyl alcohols are formed in moderate to good yields. A range of alkenyltrifluoroborates are allowed as the alkenyl donor, and no erosion of alkene geometry and/or the propargylic configuration are detected. DFT calculations confirm the critical role of the remote basic group as a general base catalyst in promoting this novel gold catalysis in good efficiency.
Keywords: gold, ligand, catalysis, dienol, calculations
Graphical Abstract
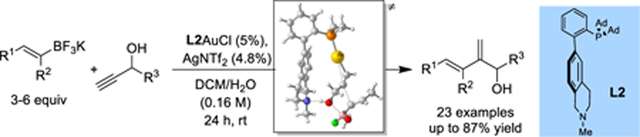
Efficient gold-catalyzed intermolecular hydroalkenylation of propargylic alcohol is enabled by a previously designed biphenyl-2-ylphosphine featuring a remote tertiary amino group. Conjugated dinenol are formed with high regioselectivity at the propargylic alcohols and stereospecificity with regard to alkenyltrifluoroborates and without erosion at the propargylic configuration.
For the past several years we have developed several new phosphine ligands[1] for homogeneous gold catalysis.[2] These ligands, as shown in Figure 1, share the privileged biphenyl-2-ylphosphine framework[3] but are unique and hence novel in their featured basic functional groups at the lower half of the pendant benzene ring. They are designed to achieve beneficial interactions between such a basic group and the substrate or incoming nucleophiles by harnessing the rather unique and robust linear structure of ligand-gold(I)-substrate complexes (Figure 1A). With the amine-based ligands including L1-L4, several gold catalyses are enabled by engaging interactions between the basic nitrogen with alkyne/allene substrates.[1a, 1c, 1e] With WangPhos featuring a remote amide group as ligand, nucleophilic additions to alkynes are accelerated owing to H-bonding between its remote amide group and incoming nucleophiles in the form of general base catalysis.[1b, 1d, 1f] This phenomenon has recently been harnessed to achieve highly enantioselective allenol cyclizations.[1g] However, the nucleophiles are so far limited to heteronucleophiles due to the necessity of H-bonding.
Figure 1.
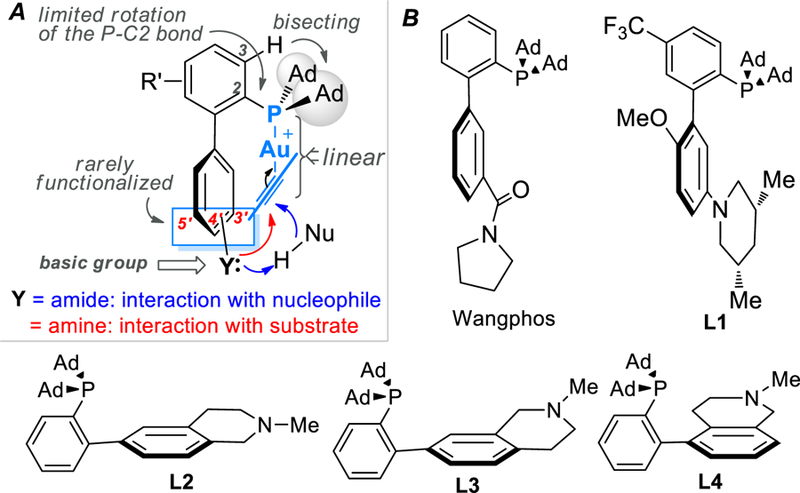
Development of gold-specific biphenyl-2-ylphosphines featuring remote basic groups: A) design; B) selected examples.
It is envisioned, however, that a ligand-enabled relay strategy could enable the use of alkenyl-based nucleophiles. As shown in Scheme 1A, with a propargylic alcohol as the substrate, the H-bonding between the hydroxy group and the remote basic site should form the structure A and enhance the basicity/nucleophilicity of the oxygen atom, which would in turn exhibit increased tendency to recruit an alkenylboron reactant via the formation of a Lewis pair in B. Such an interaction would in a ‘relay’ fashion enhance the nucleophilicity of the alkenyl moiety and achieve intramolecularity of the alkene attack at the C-C triple bond. As such, the reaction would constitute a novel bimolecular synthesis of dienols and conceptually achieve a gold-catalyzed intermolecular hydroalkenylation of C-C triple bond. Notably, intermolecular[4] hydroalkenylation[5],[6] has not been reported in gold catalysis.
Scheme 1.
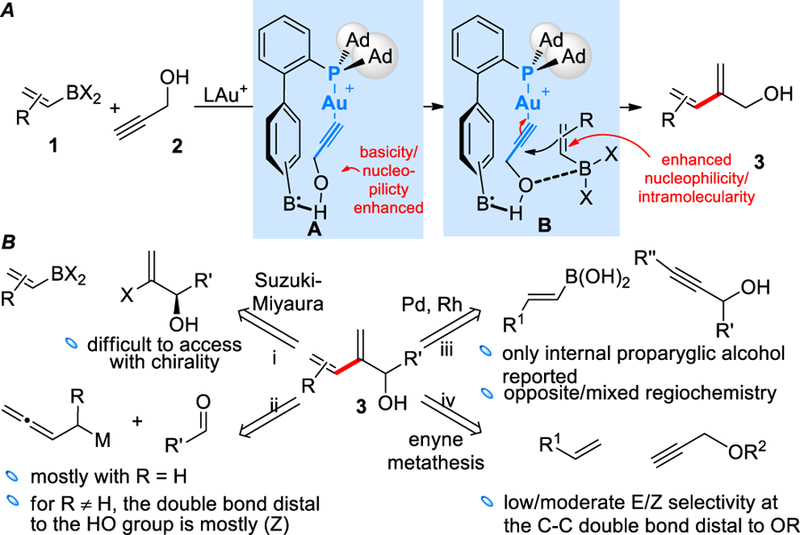
Synthesis of dienols: a) our design, a relay strategy based on ligand-enabled gold catalysis. b) prior arts and their limitations.
The dienol motif of 3 can be found as key structural components of natural products[7] or in key intermediates in natural product synthesis[8] and, due to its densely packed functionalities, are rich with reactivities.[8b, 9] As summarized in Scheme 2B, it can be mainly assembled in the following manners but with limitations: i) the Suzuki-Miyaura coupling,[10] which is straightforward but requires often difficult-to-access chiral α-haloallylic alcohols in order to prepare chiral 3; ii) the additions of buta-2,3-dien-1-ylmetal species to aldehydes, which are mostly explored without substitution (i.e., R = H) [9e, 11] or lead selectively to the formation of products possessing a (Z)-double bond distal to the hydroxy group when R ≠ H[12] and hence display limited product scope; iii) transition metal-catalyzed hydroalkenylations[6b–d, 6f, 13] of propargylic alcohols using boronic acids, which has examples using only internal propargyl alcohol substrates[13c–e] and exhibiting opposite[13c] or poor[13d] regiochemistry; iv) intermolecular enyne metathesis, which displays low/moderate E/Z selectivities;[9d, 14] v) one related case of oxidative Heck (57% yield, not shown).[15]
Scheme 2.
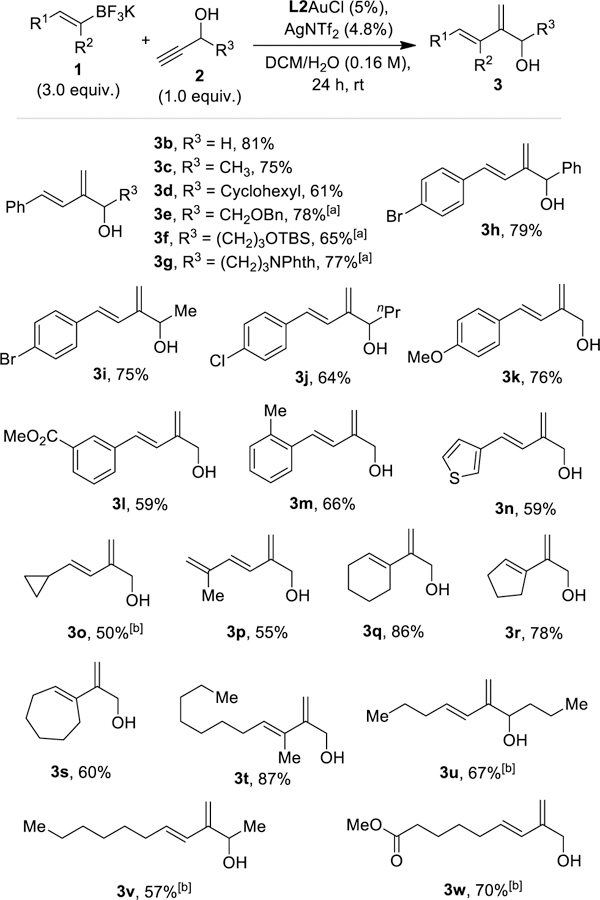
The reaction scope. Isolated yields reported. [a] 8% cat. and 6.0 equiv. of borates were used in 3e-g. [b] 6.0 equiv. of borates were used.
Herein, we report that the implementation of our design (Scheme 1A), which permits for the first time gold-catalyzed intermolecular additions of alkenyltrifluoroborates to C-C triple bonds and, moreover, provides a broadly applicable and expedient access to 2-methylene-3-buten-1-ol derivatives with well-defined regio- and stereochemistry.
At the outset, we chose the terminal secondary propargylic alcohol 1-hexyn-3-ol (2) as the substrate and trans-styryl-based trifluoroborate (E)-1a as the nucleophilic alkenyl species for reaction discovery and optimization (Table 1). Extensive exploration of ligands and reaction conditions revealed that the optimal conditions are: (E)-1a : 2 = 3:1, L2AuCl (5 mol%), AgNTf2 (4.8 mol %), DCM/H2O = 4:1, 24 h, ambient temperature. As shown in entry 1, under these conditions, the desired dienol product 3a was formed in 77% yield. The organic phase could be replaced by DCE (entry 2) or PhCF3 with only minor impact on yield but not by toluene (entry 4) due to its poor solubility of (E)-1a. Besides AgNTf2, other typically employed chloride abstractors including NaBARF (entry 5), AgOTf (entry 6) and AgBF4 (entry 7) were effective but led to slightly lower yields. Decreasing the amount of (E)-1a to 2 equiv resulted a notable drop of yield (56% in entry 8 vs. 69% in entry 2). With 2 equiv of (E)-1a and DCE/H2O (4:1) as reaction media, the performance of various other ligands are shown in entries 9–12. Not surprisingly, commercially available and typically employed ligands such as Ph3P, IPr, JohnPhos and MorDalPhos were largely ineffective (entry 9); in the presence of exogeneous Et3N (1 eq to Au), the yield was notably improved but still poor (entry 10). These results highlight the enabling role of the basic tertiary amino group in L2. Notably, the other tertiary amine-functionalized ligands L3 (entry 11) and L4 (entry 12) we previously developed[1c] along with L2 are inferior to L2, and L4 is only marginally better than the JohnPhos/Et3N combination (cf. entry 10). These results reveal the importance of properly positioning the basic group in this catalysis and affirm the advantage of our designed bifunctional ligands in achieving it. Without adding H2O, no desired product was observed (entry 13), which suggests that that the trifluoroborate salt need to undergo (partial) hydrolysis during the reaction.[16] When the corresponding boronic acid was used in place of (E)-1a, the reaction did occur, but was contaminated by competing hydration and afforded a lower yield (entry 14). In entries 9–12 and 14, protodeboronation leading to styrene is a significant side reaction and responsible for the low yields. When (Z)-1a was used, no dienol 3a or its double isomer was detected, and its protodeboronation appeared to be more facile than that of (E)-1a and is the major process for its decomposition (entry 15).
Table 1.
Reaction conditions optimization.[a]
 | ||
|---|---|---|
| Entry | Deviation from the optimized conditions | Yield(%)[b] |
| 1 | - | 77 |
| 2 | DCE/H2O (4:1) as reaction media | 69 |
| 3 | PhCF3/H2O (4:1) as reaction media | 67 |
| 4 | PhCH3/H2O (4:1) as reaction media | 0 |
| 5 | NaBARF instead of AgNTf2 as chloride abstractor | 70 |
| 6 | AgOTf instead of AgNTf2 as chloride abstractor | 70 |
| 7 | AgBF4 instead of AgNTf2 as chloride abstractor | 61 |
| 8 | 2 equiv of 1a; DCE/H2O (4:1) as reaction media | 56 |
| 9 | Ph3P, IPr, JohnPhos or MorDalPhos as Au ligand; 2 equiv of 1a; DCE/H2O (4:1) as reaction media | ≤6 |
| 10 | JohnPhos as Au ligand, 2 equiv of 1a; DCE/H2O (4:1) as reaction media; 5% Et3N | 16 |
| 11 | L3 as Au ligand; 2 equiv of 1a; DCE/H2O (4:1) as reaction media | 37 |
| 12 | L4 as Au ligand; 2 equiv of 1a; DCE/H2O (4:1) as reaction media | 18 |
| 13 | dry DCM used as solvent | 0 |
| 14 | (E)-styrylboronic acid instead of 1a | 35[c] |
| 15 | (Z)-1a instead | 0[d] |
0.1 mmol of 1-hexyn-3-ol (2) were used.
NMR yields.
27% 2 remained along with 22% of hydration product.
Mostly protodeboronation.
The reaction scope is shown in Scheme 2. Firstly, a range of the terminal secondary propargylic alcohols 2 was examined. Various R3 groups of 2 are allowed, including H, sterically demanding cyclohexyl (in the case of 3d), O-/N-functionalized alkyl groups (in the cases of 3e-3f) and a phenyl (in the case of 3h). Fairly good to excellent yields of 3b-3g were realized. In the series of 3b-3d, it is clear that the reaction efficiency correlates inversely with the steric size of the R3 group. On the other hand, terminal tertiary or internal propargylic alcohols do not lead to observable reaction. The scope of the (E)-styryltrifluoroborates was then examined. As shown in the cases of 3h-3m, substitutions on the benzene ring such as halides, electron-donating MeO, electron-withdrawing ester and a sterically congesting 2-methyl group were all tolerated. Of note are the cases of 3h and 3i, the C-Br bonds of which should be a liability in relevant Ph/Rh catalysis. The scope of the alkenylborate 1 can be substantially broadened beyond styryl ones to include those substituted by a heteroarene, a cyclohexyl, and another alkenyl to afford highly functionalized 3n-3p in serviceably yields. Furthermore, the conjugative substituents on the alkenylborate 1 featured in the cases discussed so far are not prerequisites for this gold catalysis. As shown with the cases of 3q-3w, monoalkyl substitutions (3u-3w) and bisalkyl substitutions in cyclic forms (3q-3s) and an acyclic one (3t) were all smoothly accommodated, and in some cases (e.g., 3q and 3t) the yields were excellent. Of note is that the alkyl substitutions ipso to boron in the cases of 3q-3t could hinder the reaction but quite to contrary appear to be somewhat beneficiary. For the cases of 3o and 3u-3w, 6.0 equiv of the corresponding borates were needed for full conversion, which is attributed to their susceptibility to protodeboration. In all these cases, the double bond geometries of the borates 1 were completely maintained in the dienol products.
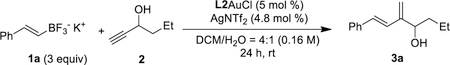
To understand the reaction mechanism and substantiate the essential role of the remote tertiary amino group of L2 in facilitating the reaction, we performed DFT-B3LYP[17] calculations on the energetics of the reaction up to the step resulting in the formation of the dienylgold intermediate. To simplify the calculations, the 1-adamantyl groups of L2 were replaced by methyl groups. As alluded above, the trifluoroborates should undergo hydrolysis[16] at the beginning. We have examined both of the hydrolyzed boron species, RB(OH)F and RB(OH)2. In the case of RB(OH)F, the two diastereomers of the initially formed ternary complexes (e.g., B-F-OH in Scheme 3) differing by the orientation of the boron F and HO substituents are separately calculated. Among the three reaction paths, the one shown in Scheme 3 is energetically most favorable and has the lowest free energy barrier in the critical first step [10.0 kcal/mol vs. 15.3 kcal/mol for RB(OH)2 and 15.2 kcal/mol for the other RB(OH)F path, DCM, for details, see SI]. This finding is consistent with RB(OH)2 leading to a lower yield (see Table 1, entry 14). The initial ternary complex B-F-OH in Scheme 3 exhibits an expected H-bond between L2 amino nitrogen and propargylic hydroxy proton and some electrostatic attraction between propargylic oxygen and the boron center. The early transition state, B-F-OH-TS1, is only 10.0 kcal/mol higher in free energy and reveals the migration of the hydroxy proton from O to N in an early stage and the significant shortening of the nascent B-O bond. The subsequently formed cyclopropyl gold carbene intermediate B-F-OH-Int1 is 21.4 kcal/mol (DCM) more stable than B-F-OH and has the original propargyl hydroxy proton completely migrated to the ligand amino nitrogen while maintaining a H-bond with its initially bonded oxygen. The next step, i.e., the fragmentation of the C-B bond, has an even lower barrier of ∆G = 5.4 kcal/mol, and throughout the H-bond between the protonated ammonium and the original propargylic oxygen is maintained. Subsequent protodeauration of the resulting intermediate B-F-OH-Int2 by the ammonium proton (not calculated) should afford the final product. These theoretical studies largely support our original design and confirm that the properly positioned remote tertiary amino group plays an intimate role of a general base catalyst in proton shuttling. The reaction energetics corresponding to Scheme 3 but with L3 or L4 as ligand (the Ad groups are again replaced by methyl groups, see SI) was also calculated. The first step barrier in DCM is 17.4 kcal/mol and 16.3 kcal/mol for L3 and L4, respectively, which is in line with the observed lower efficiencies in Table 1, entries 11 and 12.
Scheme 3.
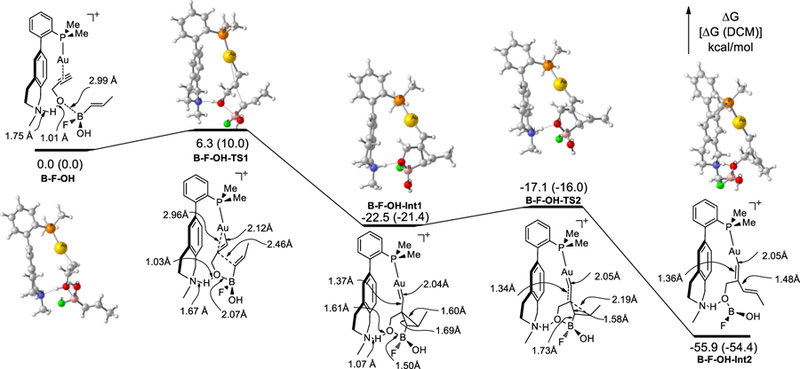
B3LYP/6–31+G(d,p)/LANL2DZ(Au) energetics of the reaction with alkenyl(fluoro)(hydroxy)borane as nucleophilic reagent in gas phase and in DCM (number in parenthesis, calculated with PCM as single point and with def2-TZVP for Au).
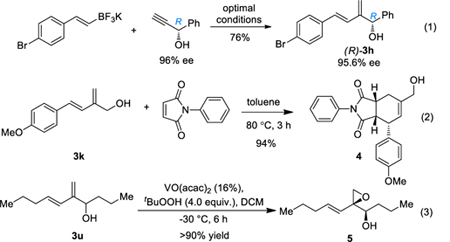
To establish the synthetic utility of this chemistry, we first examined whether the reaction would erode the e.e. of a chiral propargylic center. As shown in Eq. 1, essentially no racemization is detected in the formation of (R)-3h. Combined with readily available chiral propargylic alcohols, this chemistry offers valuable access to chiral dienols. A Diels-Alder cycloaddition was performed by using compound 3k and N-phenylmaleimide as the substrates, and only the endo-cycloadduct was obtained with an excellent yield (Eq. 2). Finally, a selective epoxidation of the allylic C-C double bond was achieved. The epoxy alcohol product 5 is not stable on silica gel column and hence isolated crude yet mostly pure due to the high reaction efficiency (Eq. 3). Only one diastereoisomer of 5 was detected, and the relative stereochemistry was assigned based on literature precedent. [17]
In summary, we have developed the first gold-catalyzed intermolecular hydroalkenylation of propargylic alcohols. A designed biphenyl-2-ylphosphine ligand featuring a remote basic tertiary amine is experimentally proven as critical in enabling this novel gold catalysis with high efficiency. DFT calculations reveal the role of the amino group as a general base catalyst. Synthetically valuable conjugated dienyl alcohols are formed in moderate to good yields. A range of alkenyltrifluoroborates are allowed as the alkenyl donor, and the reaction is highly regioselective with regard to the propargylic alcohol, stereospecific with regard to the alkene geometry, and does not erode the propargylic configuration.
Supplementary Material
Acknowledgements
S Liao thanks China Scholarship Council for scholarship. LZ thank NIGMS R01GM123342 for financial support and NIH shared instrument grant S10OD012077 for the acquisition of a 400 MHz NMR spectrometer. GZ thanks Regione Lombardia – Cariplo Foundation Grant (Sottomisura B/2016), POR FESR 2014-2020/Innovazione e competitività, progetto VIPCAT for financial support.
Footnotes
Supporting information for this article is given via a link at the end of the document
References
- [1].(a) Wang Z, Wang Y, Zhang L, J. Am. Chem. Soc 2014, 136, 8887–8890; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang Y, Wang Z, Li Y, Wu G, Cao Z, Zhang L, Nature Commun 2014, doi: 10.1038/ncomms4470; [DOI]; (c) Wang Z, Ying A, Fan Z, Hervieu C, Zhang L, ACS Catal 2017, 7, 3676–3680; [Google Scholar]; (d) Xu Z, Chen H, Wang Z, Ying A, Zhang L, J. Am. Chem. Soc 2016, 138, 5515–5518; [DOI] [PubMed] [Google Scholar]; (e) Li X, Wang Z, Ma X, Liu P.-n., Zhang L, Org. Lett 2017, 19, 5744–5747; [DOI] [PubMed] [Google Scholar]; (f) Li X, Liao S, Wang Z, Zhang L, Org. Lett 2017, 19, 3687–3690; [DOI] [PubMed] [Google Scholar]; (g) Wang Z, Nicolini C, Hervieu C, Wong Y-F, Zanoni G, Zhang L, J. Am. Chem. Soc 2017, 139, 16064–16067. [DOI] [PubMed] [Google Scholar]
- [2].For reviews on gold-catalyzed enyne cycloisomerizaitons, see: (a) Sun J, Conley MP, Zhang L, Kozmin SA, J. Am. Chem. Soc 2006, 128, 9705–9710; [DOI] [PubMed] [Google Scholar]; (b) Jiménez-Núñez E, Echavarren AM, Chem. Rev 2008, 108, 3326–3350; [DOI] [PubMed] [Google Scholar]; (c) Fürstner A, Davies PW, Angew. Chem., Int. Ed 2007, 46, 3410–3449; [DOI] [PubMed] [Google Scholar]; (d) Hashmi ASK, Chem. Rev 2007, 107, 3180–3211; [DOI] [PubMed] [Google Scholar]; (e) Abu Sohel SM, Liu R-S, Chem. Soc. Rev 2009, 38, 2269–2281; [DOI] [PubMed] [Google Scholar]; (f) Sengupta S, Shi X, Chemcatchem 2010, 2, 609–619; [Google Scholar]; (g) Zhang D-H, Tang X-Y, Shi M, Acc. Chem. Res 2014, 47, 913–924. [DOI] [PubMed] [Google Scholar]
- [3].Surry DS, Buchwald SL, Angew. Chem., Int. Ed 2008, 47, 6338–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].For selected studies, see: (a) López-Carrillo V. n., Echavarren AM, J. Am. Chem. Soc 2010, 132, 9292–9294; [DOI] [PubMed] [Google Scholar]; (b) Yeom H-S, Koo J, Park H-S, Wang Y, Liang Y, Yu Z-X, Shin S, J. Am. Chem. Soc 2012, 134, 208–211. [DOI] [PubMed] [Google Scholar]
- [5].For a gold-catalyzed intramolecular case, see: Cheong PH-Y, Morganelli P, Luzung MR, Houk KN, Toste FD, J. Am. Chem. Soc 2008, 130, 4517–4526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].For selected studies of hydroalkenylation of alkyne catalyzed by other metals, see: (a) Arcadi A, Cacchi S, Fabrizi G, Marinelli F, Pace P, Eur. J. Org. Chem 1999, 3305–3313;; (b) Hayashi T, Inoue K, Taniguchi N, Ogasawara M, J. Am. Chem. Soc 2001, 123, 9918–9919; [DOI] [PubMed] [Google Scholar]; (c) Lautens M, Yoshida M, Org. Lett 2002, 4, 123–125; [DOI] [PubMed] [Google Scholar]; (d) Oh CH, Jung HH, Kim KS, Kim N, Angew. Chem., Int. Ed 2003, 42, 805–808; [DOI] [PubMed] [Google Scholar]; (e) Tsukada N, Setoguchi H, Mitsuboshi T, Inoue Y, Chem. Lett 2006, 35, 1164–1165; [Google Scholar]; (f) Lin PS, Jeganmohan M, Cheng CH, Chem. - Eur. J 2008, 14, 11296–11299; [DOI] [PubMed] [Google Scholar]; (g) Neisius NM, Plietker B, Angew. Chem., Int. Ed 2009, 48, 5752–5755, S5752/5751-S5752/5742. [DOI] [PubMed] [Google Scholar]
- [7].Naodovic M, Xia G, Yamamoto H, Org. Lett 2008, 10, 4053–4055. [DOI] [PubMed] [Google Scholar]
- [8].(a) Molander GA, Cadoret F, Tetrahedron Lett 2011, 52, 2199–2202; [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Sarkar SM, Wanzala EN, Shibahara S, Takahashi K, Ishihara J, Hatakeyama S, Chem. Commun 2009, 5907–5909; [DOI] [PubMed]; (c) Davidson BS, Plavcan KA, Meinwald J, J. Org. Chem 1990, 55, 3912–3917; [Google Scholar]; (d) Ghosh AK, Nyalapatla PR, Org. Lett 2016, 18, 2296–2299; [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Huang JM, Xu KC, Loh TP, Synthesis 2003, 755–764;; (f) White JD, Choi YG, Org. Lett 2000, 2, 2373–2376. [DOI] [PubMed] [Google Scholar]
- [9].(a) Cayzer TN, Paddon-Row MN, Sherburn MS, Eur. J. Org. Chem 2003, 4059–4068;; (b) Tan SM, Willis AC, Paddon-Row MN, Sherburn MS, Angew. Chem., Int. Ed 2016, 55, 3081–3085; [DOI] [PubMed] [Google Scholar]; (c) Nicolaou KC, Brenzovich WE, Bulger PG, Francis TM, Org. Biomol. Chem 2006, 4, 2119–2157; [DOI] [PubMed] [Google Scholar]; (d) Clark DA, Clark JR, Diver ST, Org. Lett 2008, 10, 2055–2058; [DOI] [PubMed] [Google Scholar]; (e) Huang YY, Yang X, Lv ZC, Cai C, Kai C, Pei Y, Feng Y, Angew. Chem., Int. Ed 2015, 54, 7299–7302; [DOI] [PubMed] [Google Scholar]; (f) Hodgson DM, Arif T, Org. Lett 2010, 12, 4204–4207; [DOI] [PubMed] [Google Scholar]; (g) Li YG, Han YL, Xiong HG, Zhu NB, Qian B, Ye CQ, Kantchev EAB, Bao HL, Org. Lett 2016, 18, 392–395; [DOI] [PubMed] [Google Scholar]; (h) Clark JR, Griffiths JR, Diver ST, J. Am. Chem. Soc 2013, 135, 3327–3330. [DOI] [PubMed] [Google Scholar]
- [10].Guo B, Fu C, Ma S, Eur. J. Org. Chem 2012, 2012, 4034–4041. [Google Scholar]
- [11].(a) Duran-Galvan M, Connell BT, Eur. J. Org. Chem 2010, 2445–2448;; (b) Yu CM, Lee SJ, Jeon M, J. Chem. Soc. Perk. T 1 1999, 3557–3558. [Google Scholar]
- [12].Lachance H, Hall DG, Org. React 2008, 73, 1–573. [Google Scholar]
- [13].(a) Genin E, Michelet V, Genêt J-P, J. Organomet. Chem 2004, 689, 3820–3830; [Google Scholar]; (b) Babu MH, Kumar GR, Kant R, Reddy MS, Chem. Commun 2017, 53, 3894–3897; [DOI] [PubMed] [Google Scholar]; (c) Kim N, Kim KS, Gupta AK, Oh CH, Chem. Commun 2004, 618–619; [DOI] [PubMed]; (d) Panteleev J, Huang RY, Lui EK, Lautens M, Org. Lett 2011, 13, 5314–5317; [DOI] [PubMed] [Google Scholar]; (e) Oh CH, Park SJ, Ryu JH, Gupta AK, Tetrahedron Lett 2004, 45, 7039–7042. [Google Scholar]
- [14].Clark JR, French JM, Jecs E, Diver ST, Org. Lett 2012, 14, 4178–4181. [DOI] [PubMed] [Google Scholar]
- [15].Zheng CW, Wang D, Stahl SS, J. Am. Chem. Soc 2012, 134, 16496–16499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].(a) Molander GA, Biolatto B, J. Org. Chem 2003, 68, 4302–4314; [DOI] [PubMed] [Google Scholar]; (b) Butters M, Harvey JN, Jover J, Lennox AJJ, Lloyd‐Jones GC, Murray PM, Angew. Chem., Int. Ed 2010, 49, 5156–5160. [DOI] [PubMed] [Google Scholar]
- [17].Adam W; Corma A; Reddy TI; Renz M J. Org. Chem 1997, 62, 3631 [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.