

Abstract
The long‐sought carbene–bismuthinidene, (CAAC)Bi(Ph), has been synthesized. Notably, this represents both the first example of a carbene‐stabilized subvalent bismuth complex and the extension of the carbene‐pnictinidene concept to a non‐toxic metallic element (Bi). The bonding has been investigated by single‐crystal X‐ray diffraction studies and DFT calculations. This report also highlights the hitherto unknown reducing and ligand transfer capability of a beryllium(0) complex.
Keywords: beryllium, bismuth, bismuthinidenes, carbenes, subvalency
Carbene–pnictinidenes represent a class of molecules, which feature a carbene bound to an electron‐rich Group 15 element.1 Interest in these types of molecules has grown substantially in recent years, and applications in chemical synthesis and catalysis continue to be discovered.2 These pnictinidene adducts may be characterized as a hybrid of three resonance structures (Figure 1).3 However, the specific structure is dependent on the type of carbene, the pnictogen (Pn) element, and the substituent (R) on Pn. Among this class of compounds, carbene–phosphinidenes are the most well‐established and the electron‐rich phosphorus center has been used to coordinate to main‐group elements and transition metals.1, 2, 3, 4 The resulting compounds have even been employed in homogenous catalysis.5 It is noteworty that all of the carbene–pnctinidenes that have been reported to date feature non‐metal (P)1, 2, 3, 4 or metalloids (As, Sb) as the Pn element.6, 7 We sought to extend the carbene–pnictinidene concept to a fully metallic element (Bi).
Figure 1.
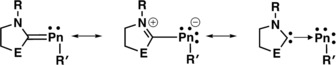
Resonance structures of carbene–pncitinidene complexes, (E=NR or CR2).
Bismuthinidenes are extremely rare, and dicoordinate bismuth compounds are typically highly reactive. As such, chemists have utilized sterically bulky groups to protect the bismuth center.8, 9, 10 A prominent example is the isolation of the first compound containing a Bi=Bi double bond by Tokitoh et al. in 1997.8 Since then, a number of compounds have been reported, which feature Bi=Bi double bonds stabilized by a variety of formally anionic ligands (Figure 2, A).9
Figure 2.
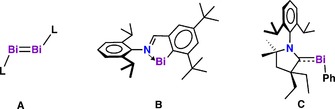
Dicoordinate bismuth complexes, A (previous work, L=monoanionic ligands), B (previous work, bismuth metal in five‐membered ring), and C (this work).
Additionally, a dicoordinate monomeric bismuth complex was synthesized by Dostal ultilizating an N–C chelating ligand, in which the Bi atom is stabilized by an N‐donor, which results in the incoorpation of the Bi atom in a ring (Figure 2, B).10 The resulting C3NBi ring was reported to possess aromatic character. In contrast, the synthesis of a carbene‐bismuthinidene would result in an exocyclic subvalent bismuth center (Figure 2, C). These types of complexes have eluded chemists. Indeed, current knowledge of carbene–bismuth chemistry pales in comparison to the chemistry of carbene‐phosphinidene,4 ‐arsinidene,6 and ‐stibinidene.7 This is in part due to metallic nature of bismuth, which results in significant differences in stability and reactivity compared to light Pn elements. Dutton and co‐workers reported the first examples of N‐heterocyclic carbene–bismuth complexes; however, attempts to reduce the bismuth(III) chloride adducts to subvalent species were unsuccessful.11 Goicoechea and co‐workers synthesized NHC–bismuth(III) tribromides,12 but the reactivity with reducing agents was not reported. A recent comprehensive review of the field highlighted the difficulty in synthesizing NHC adducts of heavier Pn elements, which was partly attributed to the destabilization of the 2pπ–npπ interaction as you descend the group.13 Consequently, we sought to exploit the stronger σ‐donating and π‐accepting properties of cyclic(alkyl)(amino) carbene (CAAC), which was first prepared by Bertrand and co‐workers.14
Recently, we synthesized a series of bismuth(III) complexes [i.e., carbene–Bi(Ph)Cl2)] by combining NHCs and CAACs with phenylbismuth dichloride, PhBiCl2(THF).15 The CAAC BiIII compounds were obtained in higher yield and were more resistant to isomerization or decomposition compared to the analogous NHC complexes. It is also noteworthy that the (CAAC)Bi(Ph)Cl2 complexes represented the first examples of compounds with CAAC coordination to bismuth. In an effort to advance carbene–bismuth chemistry, we sought to synthesize a subvalent bismuth compound stabilized by CAAC. Herein, we report the synthesis of the first carbene‐stabilized bismuthinidene complex (Et2CAAC)Bi–Ph (3), which was formed by hitherto unknown methods, which utilize a beryllium(0) complex as a reducing agent and ligand transfer reagent.
Reduction of (Et2CAAC)Bi(Ph)Cl2 with two equivalents of potassium graphite (KC8) in toluene at −37 °C resulted in a color change from pale yellow to red over one hour (Scheme 1). The solvent was removed, and the residue was extracted with hexane to give a red solution. The UV/Vis spectrum recorded at room temperature of the red solution in hexane showed a new absorption band at λ max 516 nm, which was attributed to the formation of a reduced bismuth species (Figure 3). However, the red solution rapidly decolorizes, and an insoluble black precipitate was observed. This precipitate was attributed to bismuth metal, obtained through decomposition of the target compound. Therefore, we reasoned that long reaction times associated with using KC8, potential reactive intermediates, or the strongly reducing nature of KC8 contributed to the decomposition of the Bi species. Though the solution was initially colored, it is likely that the true quantity of reduced Bi species in solution was actually very small. Therefore, only free CAAC ligand could be isolated. We then turned our attention to soluble reducing agents that would rapidly produce an insoluble by‐product to make isolation facile.
Scheme 1.
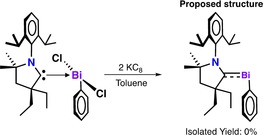
Attempt to reduce BiIII starting material with KC8.
Figure 3.
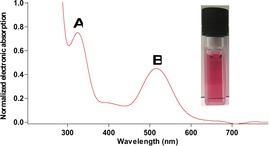
Normalized electronic absorption spectrum. Transition A, λ=324 nm (free CAAC). Transition B, λ=516 nm (new Bi species).
Recently, Braunschweig and co‐workers seminally reported the synthesis of a Be0 compound, Be(Me2CAAC)2.16 Though there were no reports of Be0 being used as a reducing agent, we speculated that a Be0 compound might facilitate the removal of the chloride atoms. Additionally, our previous calculations showed that coordination of the CAAC results in the weakening of the Bi−Cl bonds, indicating that a strong reducing agent may not be necessary.15 The Be0 complex was also selected due to its high solubility in most organic solvents including hexanes.16 Therefore, we prepared compounds 1 and 2 in 85 % and 79 % yield, respectively, according to a modified procedure (Scheme 2). Colorless rod‐shaped crystals of 1 suitable for a single crystal X‐ray diffraction study were grown from toluene at −37 °C (Figure S11 in the Supporting Information). Purple single crystals of compound 2 suitable for X‐ray diffraction analysis were obtained from a hexane solution at −37 °C. The molecular structure of 2 in Figure 4 shows two Et2CAAC coordinated to one Be atom and the bonding is similar to Be(Me2CAAC)2.
Scheme 2.
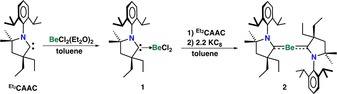
Synthesis of beryllium(0) reducing agent.
Figure 4.
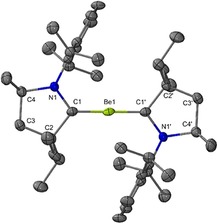
Molecular structure of compound 2.17
A yellow suspension of (Et2CAAC)Bi(Ph)Cl2 was added to a purple solution of Be(Et2CAAC)2 in toluene at −37 °C (Scheme 3). The combined homogeneous reaction mixture was kept at −37 °C for 30 minutes to give a dark red solution containing some bismuth metal. After workup, the first carbene–bismuthinidene complex was isolated as red single crystals in 10 % yield. To the best of our knowledge, this is the first example of a Be0 complex being used as a reducing agent. Surprisingly, hexane‐ and toluene‐soluble 2 can also act as a ligand‐transfer reagent (Scheme 3). Indeed, red crystals of compound 3 were obtained from the direct reaction of PhBiCl2(THF) and 2, which results in a deep red color after 30 minutes at −37 °C. However, there was no significant change in the reaction yield. The low isolated yield is due to the extreme reactivity of compound 3 in solution. The 1H NMR spectrum of 3 in [D8]toluene at 0 °C showed a characteristic doublet at 8.91 ppm for the Bi–Ph(ortho‐H) (Figure S6 in the Supporting Information). However, the [D8]toluene solution shows decomposition within five minutes (Figure S6b in the Supporting Information), indicated by gradual decolorizing of the solution and concomitant formation of bismuth metal and the neutral Et2CAAC ligand. The 13C NMR spectrum of 3 in [D8]toluene at 0 °C showed a characteristic singlet at 190 ppm for the carbene carbon, compared to 317 ppm for the free Et2CAAC (Figure S7 in the Supporting Information). Indeed, this 13C NMR resonance is in accord with (CAAC)P(Ph) complexes 191–208 ppm.4a Compound 3 is a thermally stable crystalline material which decomposes at 100 °C. Solid samples stored at room temperature under a dry argon atmosphere retain their color and crystallinity for approximately two weeks.
Scheme 3.
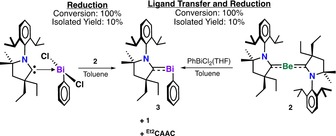
Synthesis of cyclic(alkyl)(amino) carbene‐bismuthinidene, 3. Reduction and ligand transfer mediated by a Be0 complex.
Red air‐ and moisture‐sensitive rod‐shaped crystals of 3 suitable for a single‐crystal X‐ray diffraction study were grown from hexane solution at −37 °C. The structure of 3 shows a dicoordinate bismuth center in a bent arrangement, with two Bi−C bonds (Figure 5). Unlike previously reported bismuthinidene complexes stabilized by monoanionic chelating NCN ligands,10 3 features an exocyclic monomeric bismuth center and is stabilized by the formation of a carbene–metal interaction with partial double bond character. Remarkably, there are no additional interactions with the bismuth atom and it remains open without dimerization (Figure 6). There is no example of a carbene‐bismuthinidene compound that can be used to compare to compound 3. The Bi−Ccarbene bond in 3 [Bi1−C1: 2.199(2) Å] is the shortest bond in all reported Ccarbene−Bi bonds (2.35–2.4566 Å) and significantly shorter than that in the BiIII compound Et2CAAC‐Bi(Ph)Cl2 [2.4566(15) Å].11, 12, 15 Notably, the Bi1−C1 bond [2.199(2) Å] is shorter than the covalent Bi−CPh bond [Bi1‐C23: 2.278(2) Å] in compound 3, which is indicative of partial double bond character of between CAAC and bismuth metal center. There is no significant difference in the Bi−CPh bond in compound 3 [2.278(2) Å] and Et2CAAC−Bi(Ph)Cl2 [2.2732(16) Å].15
Figure 5.
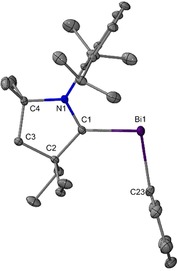
Molecular structure of 3 (thermal ellipsoids at 50 % probability; H atoms omitted for clarity). Selected bond lengths [Å] and angles [°]: Bi1−C1: 2.199(2); Bi1−C23: 2.278(2); N1−C1: 1.334(3); N1−C11: 1.452(2); N1−C4: 1.516(3); C1−C2: 1.519(3); N1‐C1‐Bi1: 119.65(14); C1‐Bi1‐C23: 99.60(8); C1‐N1‐C11:120.45(17); C1‐N1‐C4: 115.43(16); C11‐N1‐C4: 124.08(16); N1‐C1‐C2: 109.53(17).17
Figure 6.
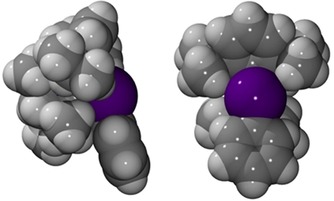
Space filling model: front view (left) and side view (right).
To gain further insight into the bonding of 3, we carried out DFT calculations at the ωB97XD/cc‐pVTZ(‐PP) level of theory (for details see Supporting Information). The gas‐phase optimized structure of 3 is in agreement with the X‐ray structure (e.g., Bi−Ccarbene distances: 2.195 vs. 2.199 Å, respectively). We also performed computations on a simplified system (3M), in which the Dipp, Ph, and Et substituents were replaced by methyl groups (Figure 7). The geometrical parameters of 3M are comparable to compound 3. On the optimized geometries natural bond orbital (NBO) calculations, natural population analysis (NPA), and atoms‐in‐molecules (AIM) analysis have been carried out (see the Supporting Information for details).
Figure 7.
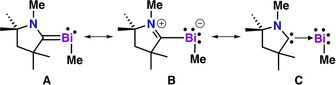
Resonance structures of CAAC–bismuthinidene compound 3M.
The Bi−Ccarbene distance of 2.182 Å in 3M is significantly shorter than the Bi−Cmethyl distance (2.286 Å). The Wiberg bond index (WBI) value of the Bi−Ccarbene bond in 3M (1.28) shows partial double bond character [WBI: Bi−C (0.87), Bi=C bond (1.84)]. The electron density value of 0.107 a.u. at the bond critical point also indicates the strengthening of the Bi−Ccarbene bond [H2C=Bi−Me (0.136 a.u.); Me3Bi (0.099 a.u.)]. Furthermore, the ellipticity of this bond (ϵ=0.282), which accounts for the π character, equals that of a prototype Bi=C bond (ϵ=0.282 in H2C=Bi−Me). The relative weakness of the Bi−Ccarbene bond compared to a true Bi=C bond necessitates the consideration of additional resonance structures.
Based on natural resonance theory (NRT) analysis of the electronic structure of 3M, the weight of the double‐bonded species A (42.4 %) exceeds that of zwitterionic B (20.6 %), with the remaining 37 % comprised of a series of minor contributors (less than 2 % each). Representation C (Figure 7) depicts 3M as a donor–acceptor complex between a carbene and a low‐coordinate Bi−Me moiety. However, the Bi−Ccarbene bond (WBI: 1.28 in 3M) is much stronger than the dative interaction observed for an adduct of CAAC and a Bi(Ph)Cl2 unit (WBI: 0.60).15 This representation does however reflect the fragmentation of 3 into free CAAC. Collectively, the data for 3M supports a hybrid structure with a Bi−Ccarbene interaction that is between a single and a double bond.
Further information may be deduced from the frontier molecular orbitals of 3M and H2C=Bi−Me (Figure 8). The HOMO of H2C=Bi−Me represents a π(Bi−C) interaction, whereas the LUMO is a π*(Bi−C) antibonding orbital. In contrast, the HOMO of 3M can be interpreted as a partial π bond or as a p‐type lone pair of electrons on Bi showing interaction towards Ccarbene, and the LUMO is a π*(C−N) antibonding orbital.
Figure 8.
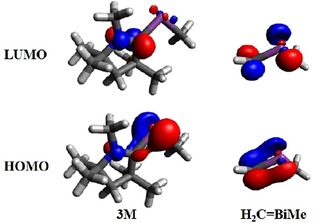
LUMO and HOMO orbitals of 3M and H2C=BiMe at the ωB97XD/cc‐pVDZ(‐PP) level (contour value at 0.07).
In summary, we have synthesized and structurally characterized the first carbene‐bismuthinidene complex, (CAAC)Bi(Ph), which contains the shortest Bi−Ccarbene bond. This represents the first example of a carbene‐stabilized subvalent bismuth complex. We have also reported the first example of a beryllium(0) complex being used as a reducing agent and a ligand‐transfer reagent. This may open the door to new reduction chemistry and allow access to other highly reactive main‐group molecules. Because the chemistry of carbene‐pnictinidene has grown to be a thriving area of active research,4 we expect that the synthesis of the missing metallic analogue will spark new research into heavier Group 15 carbene chemistry. We are currently investigating the reactivity of the title compound and utilization of the electron‐rich Bi center as neutral ligand to form metal–metal bonds.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
We are grateful to the University of Virginia for support of this work. Z.B. acknowledges the Hungarian National Research, Development, and Innovation Office (NKFIH, PD 116329). Varga József Alapítvány is also acknowledged (R.M.).
G. Wang, L. A. Freeman, D. A. Dickie, R. Mokrai, Z. Benkő, R. J. Gilliard, Chem. Eur. J. 2019, 25, 4335.
Contributor Information
Prof. Dr. Zoltán Benkő, Email: zbenko@mail.bme.hu
Prof. Dr. Robert J. Gilliard, Jr., Email: rjg8s@virginia.edu.
References
- 1.
- 1a. Schmidpeter A., Gebler W., Zwaschka F., Sheldrick W. S., Angew. Chem. Int. Ed. Engl. 1980, 19, 722–723; [Google Scholar]; Angew. Chem. 1980, 92, 767–768; [Google Scholar]
- 1b. A. J. Arduengo III , Dias H. V. R., Calabrese J. C., Chem. Lett. 1997, 26, 143–144. [Google Scholar]
- 2.
- 2a. Floch P. L., Coord. Chem. Rev. 2006, 250, 627–681; [Google Scholar]
- 2b. Krachko T., Bispinghoff M., Tondreau A. M., Stein D., Baker M., Ehlers A. W., Slootweg J. C., Grützmacher H., Angew. Chem. Int. Ed. 2017, 56, 7948–7951; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 8056–8059; [Google Scholar]
- 2c. Dostál L., Coord. Chem. Rev. 2017, 353, 142–158; [Google Scholar]
- 2d. Balmer M., Gottschling H., von Hänisch C., Chem. Commun. 2018, 54, 2659–2661; [DOI] [PubMed] [Google Scholar]
- 2e. Kundu S., Sinhababu S., Luebben A. V., Mondal T., Koley D., Dittrich B., Roesky H. W., J. Am. Chem. Soc. 2018, 140, 151–154. [DOI] [PubMed] [Google Scholar]
- 3. A. J. Arduengo III , Calabrese J. C., Cowley A. H., Dias H. V. R., Goerlich J. R., Marshall W. J., Riegel B., Inorg. Chem. 1997, 36, 2151–2158. [DOI] [PubMed] [Google Scholar]
- 4.For a comprehensive review on the chemistry of carbene-phosphinidenes and their applications:
- 4a. Krachko T., Slootweg J. C., Eur. J. Inorg. Chem. 2018, 2734–2754. For reviews on phosphinidenes, see: [Google Scholar]
- 4b. Lammertsma K., Top. Curr. Chem. 2003, 229, 95–119; [Google Scholar]
- 4c. Slootweg J. C., Lammertsma K. in Science of Synthe- sis Vol. 42 (Eds.: B. M. Trost, F. Mathey), Thieme, Stuttgart, 2009, pp. 15–36; [Google Scholar]
- 4d. Mathey F., Tran Huy N. H., Marinetti A., Helv. Chim. Acta 2001, 84, 2938–2957; [Google Scholar]
- 4e. Lammertsma K., Vlaar M. J. M., Eur. J. Org. Chem. 2002, 1127–1138; [Google Scholar]
- 4f. Aktaş H., Slootweg J. C., Lammertsma K., Angew. Chem. Int. Ed. 2010, 49, 2102–2113; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2010, 122, 2148–2159; for acyclic carbene-phosphinidene adducts, see: [Google Scholar]
- 4g. Weber L., Eur. J. Inorg. Chem. 2000, 2425–2441; [Google Scholar]
- 4h. Weber L., Eur. J. Inorg. Chem. 2007, 4095–4117. [Google Scholar]
- 5. Doddi A., Bockfeld D., Nasr A., Bannenberg T., Jones P. G., Tamm M., Chem. Eur. J. 2015, 21, 16178–16189. [DOI] [PubMed] [Google Scholar]
- 6. Doddi A., Weinhart M., Hinz A., Bockfeld D., Goicoechea J. M., Scheer M., Tamm M., Chem. Commun. 2017, 53, 6069–6072. [DOI] [PubMed] [Google Scholar]
- 7.
- 7a. Kretschmer R., Ruiz D. A., Moore C. E., Rheingold A. L., Bertrand G., Angew. Chem. Int. Ed. 2014, 53, 8176–8179; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 8315–8318; [Google Scholar]
- 7b. Dorsey C. L., Mushinski R. M., Hudnall T. W., Chem. Eur. J. 2014, 20, 8914–8917. [DOI] [PubMed] [Google Scholar]
- 8. Tokitoh N., Arai Y., Okazaki R., Nagase S., Science 1997, 277, 78. [Google Scholar]
- 9.
- 9a. Twamley B., Solfield C. D., Olmstead M. M., Power P. P., J. Am. Chem. Soc. 1999, 121, 3357; [Google Scholar]
- 9b. Sasamori T., Mieda E., Nagahora N., Sato K., Shiomi D., Takui T., Hosoi Y., Furukawa Y., Takagi N., Nagase S., Tokitoh N., J. Am. Chem. Soc. 2006, 128, 12582; [DOI] [PubMed] [Google Scholar]
- 9c. Sasamori T., Tokitoh N., Dalton Trans. 2008, 1395; [DOI] [PubMed] [Google Scholar]
- 9d. Prabusankar G., Gemel C., Parameswaran P., Flener C., Frenking G., Fischer R. A., Angew. Chem. Int. Ed. 2009, 48, 5526–5529; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2009, 121, 5634–5637; [Google Scholar]
- 9e. Sakagami M., Sasamori T., Sakai H., Furukawa H., Tokitoh N., Chem. Asian J. 2013, 8, 690; [DOI] [PubMed] [Google Scholar]
- 9f. Sasamori T., Sakagami M., Tokitoh N., Heteroat. Chem. 2014, 25, 306; [Google Scholar]
- 9g. Dange D., Davey A., Abdalla J. A. B., Aldridge S., Jones C., Chem. Commun. 2015, 51, 7128; [DOI] [PubMed] [Google Scholar]
- 9h. Majhi P. K., Ikeda H., Sasamori T., Tsurugi H., Mashima K., Tokitoh N., Organometallics 2017, 36, 1224–1226. [Google Scholar]
- 10. Vránová I., Alonso M., Lo R., Sedlák R., Jambor R., Růžička A., De Proft F., Hobza P., Dostál L., Chem. Eur. J. 2015, 21, 16917–16928. [DOI] [PubMed] [Google Scholar]
- 11. Aprile A., Corbo R., Vin Tan K., Wilson D. J., Dutton J. L., Dalton Trans. 2014, 43, 764–768. [DOI] [PubMed] [Google Scholar]
- 12. Waters J. B., Chen Q., Everitt T. A., Goicoechea J. M., Dalton Trans. 2017, 46, 12053–12066. [DOI] [PubMed] [Google Scholar]
- 13. Nesterov V., Reiter D., Bag P., Frisch P., Holzner R., Porzelt A., Inoue S., Chem. Rev. 2018, 118, 9678–9842. [DOI] [PubMed] [Google Scholar]
- 14.
- 14a. Lavallo V., Canac Y., Präsang C., Donnadieu B., Bertrand G., Angew. Chem. Int. Ed. 2005, 44, 5705; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2005, 117, 5851; [Google Scholar]
- 14b. Melaimi M., Jazzar R., Soleilhavoup M., Bertrand G., Angew. Chem. Int. Ed. 2017, 56, 10046–10068; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10180–10203. [Google Scholar]
- 15. Wang G., Freeman L. A., Dickie D. A., Mokrai R., Benkő Z., Gilliard R. J., Inorg. Chem. 2018, 57, 11687–11695. [DOI] [PubMed] [Google Scholar]
- 16. Arrowsmith M., Braunschweig H., Celik M. A., Dellermann T., Dewhurst R. D., Ewing W. C., Hammond K., Kramer T., Krummenacher I., Mies J., Radacki K., Schuster J. K., Nat. Chem. 2016, 8, 890–894. [DOI] [PubMed] [Google Scholar]
- 17. CCDC 1879088 (1), 1879089 (2), and 1879090 (3) contain the supplementary crystallographic data. These data can be obtained free of charge by The Cambridge Crystallographic Data Centre.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary