

Abstract
Objective
The goal of this narrative review is to provide an overview of migraine pathophysiology, with an emphasis on the role of calcitonin gene‐related peptide (CGRP) within the context of the trigeminovascular system.
Background
Migraine is a prevalent and disabling neurological disease that is characterized in part by intense, throbbing, and unilateral headaches. Despite recent advances in understanding its pathophysiology, migraine still represents an unmet medical need, as it is often underrecognized and undertreated. Although CGRP has been known to play a pivotal role in migraine for the last 2 decades, this has now received more interest spurred by the early clinical successes of drugs that block CGRP signaling in the trigeminovascular system.
Design
This narrative review presents an update on the role of CGRP within the trigeminovascular system. PubMed searches were used to find recent (ie, 2016 to November 2018) published articles presenting new study results. Review articles are also included not as primary references but to bring these to the attention of the reader. Original research is referenced in describing the core of the narrative, and review articles are used to support ancillary points.
Results
The trigeminal ganglion neurons provide the connection between the periphery, stemming from the interface between the primary afferent fibers of the trigeminal ganglion and the meningeal vasculature and the central terminals in the trigeminal nucleus caudalis. The neuropeptide CGRP is abundant in trigeminal ganglion neurons, and is released from the peripheral nerve and central nerve terminals as well as being secreted within the trigeminal ganglion. Release of CGRP from the peripheral terminals initiates a cascade of events that include increased synthesis of nitric oxide and sensitization of the trigeminal nerves. Secreted CGRP in the trigeminal ganglion interacts with adjacent neurons and satellite glial cells to perpetuate peripheral sensitization, and can drive central sensitization of the second‐order neurons. A shift in central sensitization from activity‐dependent to activity‐independent central sensitization may indicate a mechanism driving the progression of episodic migraine to chronic migraine. The pathophysiology of cluster headache is much more obscure than that of migraine, but emerging evidence suggests that it may also involve hypersensitivity of the trigeminovascular system. Ongoing clinical studies with therapies targeted at CGRP will provide additional, valuable insights into the pathophysiology of this disorder.
Conclusions
CGRP plays an essential role in the pathophysiology of migraine. Treatments that interfere with the functioning of CGRP in the peripheral trigeminal system are effective against migraine. Blocking sensitization of the trigeminal nerve by attenuating CGRP activity in the periphery may be sufficient to block a migraine attack. Additionally, the potential exists that this therapeutic strategy may also alleviate cluster headache as well.
Keywords: calcitonin gene‐related peptide, migraine, nitric oxide, sensitization, trigeminal system
Abbreviations
- BOLD
blood oxygenation level‐dependent
- CGRP
calcitonin gene‐related peptide
- CLR
calcitonin receptor‐like receptor
- CNS
central nervous system
- CSD
cortical spreading depression
- DRG
dorsal root ganglion/ganglia
- fMRI
functional magnetic resonance imaging
- HT
high threshold
- NO
nitric oxide
- PAG
periaqueductal gray
- PKA
protein kinase A
- PKC
protein kinase C
- RAMP
receptor activity modifying protein
- ROS
reactive oxygen species
- RVM
rostral ventromedial medulla
- TG
trigeminal ganglion
- TNC
trigeminal nucleus caudalis
- TNF
tumor necrosis factor
- WDR
wide dynamic range
Background
Migraine is probably one of the oldest ailments, known since ancient times, as noted by the Egyptians, and yet even now, it remains an unmet medical need that is often inadequately recognized and undertreated.1, 2, 3, 4, 5 The second largest cause of disability in the world, migraine is a complex neurological disease is characterized by attacks lasting between 4 hours and 3 days, and is characterized primarily by a moderate‐to‐severe paroxysmal, unilateral headache that is aggravated by movement, and may be accompanied by associated symptoms of photophobia, phonophobia, osmophobia, allodynia, pain on movement, and nausea and vomiting.4, 6, 7 First described by Hippocrates over 2400 years ago, aura that commonly manifest as scintillations or expanding circle of scotoma may precede migraine headache in ~20‐30% of cases. However, aura can also manifest as somatosensory, auditory, or olfactory. Aura may last from 5 to 60 minutes, and is usually followed by a headache within 60 minutes.6 Premonitory symptoms that include fatigue, difficulty in concentrating, neck stiffness, sensitivity to light and/or sound, nausea, blurred vision, yawning, and pallor may appear as early as 72 hours before a migraine attack, signaling early‐onset changes in the central nervous system (CNS).8, 9, 10
Migraine was long considered to be primarily a vascular disorder, resulting from meningeal vasodilation. However, advances made in the past 2 decades show that migraine is a complex neurological disease that includes the participation of multiple cortical, subcortical, and brainstem areas that regulate autonomic, affective, cognitive, and sensory functions.9, 10, 11 This view is consistent with the complex and varied symptomology associated with migraine, including the premonitory and aura phases. However, it is still unclear what mechanisms are invoked to initiate the activation of these brain regions, or how the progression of activation of central sites occur to produce migraine.
Technological advances that allow greater precision in brain imaging techniques have allowed the visualization of brain regions that may be involved in the generation and progression of migraine. Early studies using positron emission tomography (PET) found that the brainstem regions including the periaqueductal gray (PAG), dorsal raphe (DR), and locus coeruleus (LC) are activated during a migraine attack, and might represent a brainstem migraine generator.12, 13 Since then, the concept of the brainstem as a migraine generator has fallen into disfavor, in part because this region remains activated even after the headache is resolved with triptans and because this region shows increased activity during, but not immediately before, a migraine attack.14 However, expanding the scope of imaging studies from these earlier investigations revealed that the hypothalamus may play a prominent role in the genesis of migraine attacks. Premonitory symptoms such as food craving, fatigue, nausea, and yawning suggest hypothalamic involvement.15, 16 Nitroglycerin‐induced migraine increased hypothalamic activity during the early premonitory phase in patients undergoing H2 15O PET cerebral blood flow scans.17 In an attempt to capture changes in brain activity prior to a migraine attack, a patient who experienced 2‐3 migraine headaches per month was subjected to fMRI every morning for 30 days.18 This study showed that the hypothalamus increases activity during the 24 hours prior to migraine headache pain, along with increased functional coupling to the trigeminal nucleus caudalis (TNC).18 During the ictal phase, functional coupling of the hypothalamus to the TNC decreases and coupling to the dorsal pons is strengthened. By altering its connectivity with other brain regions, the hypothalamus may be able to alter the activity of regions of migraine pathophysiology and could function as a migraine generator.18 In a more recent study performed with healthy controls and with patients with episodic or chronic migraine, standardized trigeminal nociceptive stimulation was used along with fMRI using a high‐resolution echo‐planar imaging protocol in order to better understand the role of the hypothalamus in migraine.16 In that study, painful trigeminal stimulation produced greater activation of the hypothalamus of patients with chronic migraine than in the healthy control subjects. Moreover, the posterior part of the hypothalamus was more activated in patients with either chronic or episodic migraine when compared to healthy control subjects during the headache‐free phase. It is believed that the anterior part of the hypothalamus is implicated in the initiation of migraine attacks and the posterior part in migraine headache pain.16
Hypothalamic nuclei (eg, arcuate, dorsomedial, suprachiasmatic, median eminence/pars tuberalis) are important regions contributing to internal homeostatic regulation, and contain neurons that participate in expression of clock genes controlling diurnal, circadian, and circannual rhythms, as well as regulation of hormonal cycles.19 Oscillations in hypothalamic activity temporally alter functional connections among the hypothalamus, and brainstem, and dopaminergic networks and lead to changes in subcortical and brainstem regions, altering susceptibility thresholds to sensory stimuli, thus initiating and ending a migraine attack.20 A recent fMRI study showed increased infra‐slow oscillatory activity, increased connectivity strengths, and regional homogeneity in the TNC, dorsal pons, and hypothalamus immediately prior to a migraine attack.21 Likewise, imaging studies have also shown that the hypothalamic nuclei are activated during the pain phase of cluster headache attacks.22 The cyclic nature and circadian predictability of cluster headache is consistent with the pacemaking functions of hypothalamic nuclei.22, 23, 24
As described in a recent review, some investigators hold that cortical spreading depression (CSD) can initiate a migraine attack.25 This view is not inconsistent with studies showing excitation of cortical and subcortical structures preceding or concomitant with migraine attacks, and supports the concept that migraine is due in part to cortical hyperexcitability.26, 27 However, whether CSD has a role in the initiation of a migraine attack has not been demonstrated definitively, and the role of CSD in migraine remains a topic of vigorous debate. The possible role of CSD in migraine is further discussed below.
An emerging view9, 15, 28 suggests that cortical excitability can perturb other central sites (eg, descending pain modulatory systems), such that the migraine attack is initiated centrally, as suggested by the prodromal symptoms that precede migraine headache pain by many hours. This excitability leads to peripheral sensitization, and a CSD could be generated as the hyperexcitability progresses. This enhanced activity, with or without CSD, can, through mechanisms not yet determined, lead to peripheral sensitization and the development of migraine headache pain.9, 15, 28
Objective of the Review
Recent advances in preclinical and clinical investigations have provided convergent evidence to refine our understanding of mechanisms driving the pathophysiology of migraine. The past decade has seen an increased emphasis on the role of the trigeminovascular system in connecting peripheral events with central consequences, and the promising results from recent clinical trials provide a strong indication that calcitonin gene‐related peptide (CGRP) is a critical element in migraine generation and progression. This narrative review was undertaken to describe the role of CGRP in the trigeminovascular system in the context of recent clinical and preclinical investigations, and to update the discussion of mechanisms underlying migraine pathophysiology. PubMed searches were performed to find the most recent (ie, 2016 to end of November, 2018) comprehensive reviews of migraine and to identify the most recently published clinical and preclinical investigations related to migraine pathophysiology. References to the initial studies are used in providing background and context, and reviews are used to support additional, ancillary points. Finally, we bring the reader's attention to recent reviews addressing migraine pathophysiology globally.
Trigeminal Neurovascular System
The meningeal vasculature is densely innervated with primary afferent nociceptive unmyelinated C‐fibers and thinly myelinated Aδ fibers arising chiefly from the ophthalmic (V1) division of the trigeminal nerve, although some innervation through the maxillary (V2) and mandibular divisions (V3) also occur.9, 29 These sensory nerves are pseudo‐unipolar nerves, with cell bodies in the trigeminal ganglion (TG) and bifurcating axons that project to peripheral and central sites. In the periphery, the peripheral fibers from the TG neurons innervate the cerebral arteries, meningeal arteries, and project into parts of the dura that are not highly vascularized. Immunohistochemical studies of rat and human tissue revealed that the unmyelinated C‐fibers innervating meningeal and cerebral arteries express CGRP as well as substance P, VIP, and nNOS.30 All of the fibers that expressed substance P also expressed CGRP, but there were many CGRP fibers that did not express substance P. CGRP expression was limited to the nociceptive C‐fibers. The Aδ fibers coexpressed immunofluorescence for calcitonin receptor‐like receptor (CLR) and (receptor activity‐modifying protein 1) RAMP1, indicating the presence of functional CGRP receptors. Thus, CGRP released from terminals of C‐fibers can sensitize the adjacent Aδ nerve terminals.30 These observations are consistent with recent findings that the humanized monoclonal anti‐CGRP antibody fremanezumab inhibits evoked firing of Aδ, not of C, fibers.31
Mast cells present in the dura of rats express the CGRP receptor whereas those of humans do not.30 This observation is consistent with earlier studies showing CGRP‐evoked histamine release from mast cells in rat, but not human, dural tissue.32 The CGRP receptor components were also detected in the vascular smooth muscle cells, and the blood vessels were all innervated by CGRP‐positive nerve fibers. The dura is sparsely innervated by afferent fibers that express nNOS in close proximity to CGRP‐expressing fibers. No colocalization of nNOS with CGRP was detected.30
Immunohistochemical studies performed with rat and human tissue revealed that approximately one‐half of TG neurons express CGRP and about one‐third express both the CLR and RAMP1 components of the CGRP receptor.33 There was very little colocalization of CGRP with CGRP receptors, which, combined with studies using NF markers showed that CGRP is expressed in unmyelinated C‐fibers and the receptor in the myelinated Aδ fibers.30, 33 Moreover, staining for the CGRP receptor was also found on satellite cells within the TG, indicating that release of CGRP within the ganglion can activate both neurons and glial cells.33 However, a different study using antibodies raised against a fusion protein of the extracellular domains of CLR and RAMP1 failed to find CGRP receptors on satellite cells of human TG tissue.34 Expression of synaptosomal‐associated protein of 25 kDa (SNAP‐25) in vesicular structures, some of which contained CGRP, indicates that neuronally mediated paracrine signaling can occur within the TG.33
The central projections of the TG neurons project to the trigeminal nucleus caudalis (TNC; Sp5C) in the caudal medulla and the cervical spinal cord where they terminate in the outer (I‐II) and inner (V‐VI) laminae. Electrophysiologic studies performed in rats indicated that activation of the trigeminal Aδ afferent nerves activated high‐threshold TNC neurons whereas the C‐fibers activated the wide dynamic range neurons.31, 35 Immunofluorescence for CGRP was detected in C‐fiber terminals principally in the outer laminae of human TNC.30 The CGRP receptor components were detected on Aδ fibers in the outer laminae and spinal trigeminal tract of human TNC.30 No neuronal cell bodies were found to express either CGRP or its receptor in the outer laminae of human TNC tissue, suggesting that CGRP acts on presynaptic receptors on adjacent nerve terminals.30, 36, 37 In contrast, when antibodies to the fusion protein were used, CGRP receptors were detected on dendrites and cell bodies in the TNC of the cynomolgus monkey.34
Nociceptive inputs from the dura are relayed by the second‐order TNC neurons to the ventroposteromedial thalamus and the medial nucleus of the posterior complex. The thalamus has bidirectional connections with several cortical regions, including, but not limited to, the somatosensory cortex, insula, amygdala, and limbic regions, thus integrating nociceptive inputs with cognition, emotion, and autonomic responses as part of a complex “pain matrix.”38, 39 The hypothalamus receives trigeminovascular inputs via the thalamus, and sends bilateral projections to the outer laminae of the TNC. Retrograde labeling studies in rats revealed that the paraventricular nucleus, the lateral hypothalamic area, the perifornical hypothalamic area, the A11 nucleus, and the retrochiasmatic area of the hypothalamus project bilaterally to the outer laminae of the TNC.40 In studies with rodents, inhibition of hypothalamic activity with the GABAA agonist muscimol microinjected into the paraventricular nucleus of the hypothalamus (PVN) attenuated meningeal‐evoked activities of TNC neurons, whereas the GABAA antagonist gabazine enhanced these responses.41 The PVN of the hypothalamus may be able to modulate trigeminal autonomic cephalalgias by integrating nociceptive mechanisms with autonomic, and stress‐processing mechanisms.41 Moreover, the A11 nucleus of the posterior hypothalamus sends dopaminergic projections to the superficial laminae of the TNC, and all dopaminergic neurons of this nucleus co‐express CGRP, although there are also many non‐dopaminergic neurons that express only CGRP.42 Stimulation of the A11 nucleus inhibits firing of TNC neurons evoked by stimulation of the middle meningeal artery, and this effect is blocked by the D2 receptor antagonist eticlopride.42 Through the release of CGRP and dopamine in the TNC, the A11 nucleus can exert positive and negative modulatory influences on TNC activity.
Projections from the thalamus, as well as hypothalamus and cortical sites, to midbrain and medullary sites form the descending pain modulatory system. The periaqueductal gray (PAG) and the locus coeruleus receive these pain modulatory signals from superior sites as well as nociceptive inputs from the TNC and spinal cord, and are in bilateral communication with the rostral ventromedial medulla (RVM), which represents the final common pathway in descending pain modulation. Studies in cats showed that electrical stimulation of the PAG abolished the responses of TNC neurons to noxious stimulation of the dura.43 Descending serotonergic neurons from the RVM can inhibit or facilitate nociceptive inputs, depending on the specific serotonergic receptor subtypes activated, with the 5‐HT3 receptor mediating nociception and the 5‐HT7 receptor producing antinociception. Collectively, studies in animal models suggest that enhanced pain states result from a shift in descending modulation to favor pain facilitation over inhibition, thus enhancing pain. Inhibition of RVM activity abolished enhanced nociceptive responses, including responses to stimuli applied to the dura.44, 45, 46, 47 The role of descending pain modulation in central sensitization and migraine pain is elaborated further below.
Imaging studies performed in patients with migraine, with or without allodynia, showed that atypical resting state functional connectivity between the PAG and higher pain‐processing regions is associated with allodynia, and further demonstrate a role for this region in pain modulation.48 Clinically, the presence of descending pain modulation can be demonstrated with conditioned pain modulation (CPM [formerly called diffuse noxious inhibitory controls, or DNIC]), where “pain inhibits pain.”49, 50 In order to demonstrate CPM, a subject typically is exposed to a persistent painful stimulus (eg, mild ischemia with an inflatable cuff, placing a hand or foot in cold or hot water), and is then challenged with an acute noxious stimulus (eg, pinch or applied heat probe).49, 50, 51 When CPM is present, the introduction of the acute stimulus causes a reduction in the perceived painfulness of the water bath. This is believed to be due to activation of descending pain inhibition.49, 50 Patients with idiopathic pain states usually demonstrate diminished or absent CPM, suggesting a shift away from descending inhibition and toward facilitation. In a recent study of patients with episodic and chronic migraine, pain thresholds were determined to repetitive mechanical and laser‐evoked thermal stimuli over a period of 30 minutes.52 The patients then presented with a conditioning stimulus by immersing the contralateral foot in a warm (30°C) or noxious cold (8°C) water bath between 10 and 20 minutes of the repetitive stimuli. In healthy control subjects, the conditioning stimulus significantly reduced perceived pain intensity of the repetitive nociceptive stimuli, indicating a robust CPM. Patients with episodic migraine showed a reduced CPM, whereas those with chronic migraine showed no CPM, but rather pain facilitation when exposed to the conditioning stimulus.52 These results are consistent with the idea that migraine headache pain may be due in part to a loss of descending pain inhibitory influence on the trigeminovascular system.
The Trigeminovascular System Mediates Dural Nociception
Early studies performed with conscious patients undergoing brain surgeries under local anesthesia showed that noxious stimuli applied to meningeal arteries or to the dura in the vicinity of the arteries produced headache pain that was sometimes accompanied by nausea.53, 54 The pain produced by dural stimulation was localized and referred to the ipsilateral temporal region, the ipsilateral eye, or to the forehead.53, 54 Patients who had previous section of the trigeminal nerve or who received local anesthetic injection into the TG reported no pain upon dural stimulation.53 These experiments provided early clinical evidence that dural innervation via the trigeminal nerve could produce headache and may underlie migraine headache pain. Later studies in animal models showed that the perivascular regions of the dura were richly innervated with peripheral fibers from the TG whereas the dura regions with sparse vasculature were sparsely innervated.29 Several electrophysiologic and immunohistologic studies have shown that noxious stimulation of the dura activates nociceptive neurons of the trigeminocervical complex. Stimulation of the dura or of the periorbital region excites trigeminal neurons that in turn stimulate the second‐order TNC neurons.55, 56 In one study, TNC activity elicited by periorbital nerve stimulation was enhanced by infusion of nitroglycerin in rats.57 The application of an inflammatory “soup” to the dura produced a local neurogenic inflammation, which was associated with enhanced neuronal responses to stimuli.56, 58 Light mechanical stimuli applied to the cutaneous receptive field or to the dura resulted in increased TG activity, which was followed by enhanced responses of second‐order TNC neurons. Mechanical stimuli that were unable to evoke responses were active after inflammation.56, 58 Such studies led to the idea that neurogenic inflammation of the meninges could sensitize the peripheral afferent TG fibers (ie, peripheral sensitization), which manifests as migraine headache.56, 58 However, neurogenic inflammation of the dura has not been observed in humans. Moreover, although CGRP‐mediated mast cell degranulation has been observed in animal models, human mast cells do not express CGRP receptors. On the other hand, in vitro studies with human dural tissue showed that substance P, which is co‐expressed in some CGRP afferents, is able to elicit mast cell degranulation in human dural tissue.59 However, it is doubtful that substance P can evoke sufficient neurogenic inflammation to be a factor in migraine genesis. Based on animal studies showing high efficacy of antagonists for neurokinin‐1 (NK‐1), the substance P receptor, in blocking neurogenic inflammation, several of these antagonists, GR205171, RPR 100893, and lanepitant (LY‐303870) were advanced to clinical trials, but they all failed in double‐blind, randomized clinical trials for episodic migraine.60 In a separate randomized clinical trial, lanepitant was not effective in preventing migraine.61 That peripheral sensitization is an important factor in the progression of a migraine attack is generally well accepted, but what remains unknown is the mechanisms that produce peripheral sensitization in patients with migraine.9
As the TNC receives repetitive, persistent nociceptive inputs, the TNC neurons become sensitized to further inputs, responding to lower threshold stimuli and increasing their receptive fields, indicating activity‐dependent central sensitization. With central sensitization, the perceptual responses to noxious inputs are exaggerated, prolonged, and spread widely. Over extended periods of time, neuroplastic adaptations occur at medullary and cortical pain modulatory sites, resulting in a shift in the pain modulatory systems to favor a net descending pain facilitation. This latter phase of central sensitization is activity‐independent and may underlie the conversion of episodic migraine to chronic migraine.62 The role of descending pain modulation and of central sensitization in the progression of a migraine attack, as well as its contribution to chronic migraine has been discussed in several comprehensive reviews.9, 63, 64, 65, 66, 67, 68, 69, 70
In a series of elegant experiments, Burstein and colleagues paired observations from animal studies with clinical presentation of migraine and suggested that as the migraine attack progresses, the pathophysiology of migraine progresses from peripheral sensitization of the primary afferent TG neurons to central sensitization of TNC second‐order neurons and ultimately to central sensitization of third‐order neurons in the thalamus.58, 68, 70, 71, 72, 73 In one such study, fMRI performed in patients during a migraine attack showed enhanced activation of the posterior thalamus in response to light brush or thermal stimuli applied to the dorsum of the hand in patients with extracephalic cutaneous allodynia.73 In the same study, it was also reported that rats with dural inflammation showed activation of thalamic neurons in response to innocuous stimulation of the paws after onset of central sensitization, whereas no responses were elicited by the same stimuli at baseline.73 Based on these studies, it was hypothesized that early in a migraine attack, patients may have the throbbing migraine headache without cutaneous allodynia, driven by peripheral sensitization of the TG, and triptans are effective at that stage. As the migraine attack progresses, cutaneous allodynia develops in the periorbital area, signaling central sensitization of convergent TNC neurons and an increased resistance to the efficacy of triptans. Widespread cutaneous allodynia extending to extracephalic sites suggests central sensitization of third‐order thalamic neurons, and resistance to treatment with triptans.68, 69, 70, 71, 73, 74
Establishment of activity‐independent central sensitization, driven by descending pain facilitation, was suggested in an animal model of dural inflammation that induced periorbital cutaneous allodynia.75 In that study, the microinjection of bupivacaine into the RVM to block descending modulation did not abolish allodynia when given 30 minutes after the start of inflammation and presumably during the period of peripheral sensitization.75 However, cutaneous allodynia was abolished when bupivacaine was administered at 1.5 hours after the start of inflammation, and during the period central sensitization might be driven in part by net descending facilitation from the RVM.75
Role of Trigeminal CGRP in Migraine – Clinical Evidence
The neuropeptide CGRP is widely expressed in the peripheral and CNS. The α‐isoform of the neuropeptide CGRP is highly expressed in somatosensory peripheral nerves and in the CNS whereas the β‐CGRP isoform is predominantly expressed in motor neurons and in the enteric nervous system.76 A growing body of evidence indicates that CGRP in the trigeminovascular system is a principal mediator of migraine. Immunohistochemical studies determined that approximately 50% of human TG neurons express CGRP, almost exclusively on thin, unmyelinated nociceptive C‐fibers, whereas the myelinated A‐fibers and vascular smooth muscle cells of the dura expressed the CGRP receptor.33 CGRP is not directly algogenic, since intradermal injections of the neuropeptide produces an erythemic flare but without pain.77 The i.v. infusion of CGRP produces a delayed migraine attack in a majority of individuals with a past history of migraine.78 Importantly, CGRP does not produce a delayed migraine‐like headache in healthy individuals, nor does it evoke any somatic pain other than headache. These observations suggest that CGRP induces migraine attacks through activation of downstream signaling cascades. In contrast, the i.v. infusion of PGE2, a well‐known proinflammatory agent with direct nociceptive effects, produces an immediate migraine attack in susceptible individuals.79
Several studies demonstrated that blood levels of CGRP obtained from the jugular vein of people with migraine are elevated during a migraine attack.80, 81, 82, 83 Moreover, the interictal CGRP blood levels of patients with migraine are elevated when compared to individuals without migraine.83 Additionally, the interictal levels of CGRP of people with chronic migraine are significantly elevated when compared to those of patients with episodic migraine, and it was suggested that these elevated interictal CGRP levels in people with migraine could be a biomarker aiding the diagnosis of chronic migraine.83 Furthermore, treatment with onabotulinumtoxin A over 1 month reduced interictal CGRP blood levels in people with chronic migraine who were responsive to the treatment but not in nonresponders.84, 85 Induction of migraine with nitroglycerin infusion increases jugular blood levels of CGRP.86 Sumatriptan inhibits release of CGRP from trigeminal nerve terminals and in that study, produced parallel reductions in migraine headache pain intensity and blood levels of CGRP.86 In another study, migraine attacks were associated with elevated levels of CGRP in saliva, which were blocked by efficacious rizatriptan treatment.87 In that study, patients with higher elevations of CGRP also showed better responsiveness to rizatriptan.87 Collectively, these studies suggest a prominent role of CGRP in the trigeminovascular system regarding migraine pathophysiology.
Indeed, randomized clinical studies with small‐molecule CGRP antagonists or with humanized monoclonal antibodies to CGRP or to the CGRP receptor have shown encouraging results in relieving migraine attacks. Six small‐molecule CGRP antagonists (olcegepant, telcagepant, MK‐3207, BI 44370 TA, rimegepant, and ubrogepant) were efficacious in the acute treatment of migraine.88, 89, 90, 91, 92, 93, 94 More recently, the CGRP monoclonal antibody galcanezumab (LY2951742) reduced the frequency of migraine headache days in patients with episodic migraine with low incidence of adverse events in a phase 2 and two phase 3 randomized clinical trials.95, 96, 97 Galcanezumab was efficacious and well tolerated in patients with chronic migraine as well.98 Eptinezumab (ALD403) and fremanezumab (TEV‐48125) showed efficacy in episodic migraine, and fremanezumab in chronic migraine, with favorable safety profiles.7, 99, 100 Erenumab (AMG 334), the humanized monoclonal antibody to the CGRP receptor, was also effective in episodic and chronic migraine.101, 102 Between mid‐May, 2018, and the time of this writing, the U.S. Food and Drug Administration (FDA) approved galcanezumab, fremanezumab, and erenumab for the prevention of migraine.
Role of Trigeminal CGRP in Migraine – Preclinical Evidence
Nociceptor Sensitization
However, despite strong evidence that CGRP is a key component of migraine pathophysiology, the mechanisms through which CGRP could elicit migraine attacks are largely unknown. Nearly all understanding of potential mechanisms is derived from preclinical studies in animal models. In studies performed in rats or cats, electrical stimulation of the sagittal sinus or of the TG released CGRP into the jugular bloodstream and, acting on presynaptic 5‐HT1A/1D receptors, was able to inhibit CGRP release.56, 80, 82, 103, 104 In cultured TG neurons, CGRP induced delayed and long‐lasting upregulations gene transcriptions via protein kinase A (PKA) and protein kinase C (PKC) to produce delayed and long‐lasting sensitization.105, 106 In a pair of investigations using dural stimulation, Melo‐Carrillo and coworkers made the serendipitous discovery that the peripheral Aδ fibers express the CGRP receptor and activate high‐threshold (HT) TNC second‐order neurons, while the C‐fibers do not express CGRP receptors and activate wide‐dynamic range (WDR) TNC neurons.31, 35 Moreover, the role of CGRP was found to differ between meningeal and non‐meningeal peripheral nerves in the generation of pain.35 Studies showing an absence of somatic pain after CGRP i.v. or intradermal administration to human subjects, differential terminations of meningeal and extra meningeal afferents in the TNC suggest that meningeal and cutaneous inputs are processed differently in the TNC.77, 107
The proposition that the anti‐CGRP antibodies act peripherally, at the trigeminal–meningeal interface, rather than centrally is also supported by a recent clinical PET study that showed very low (<10%) receptor occupancy of central CGRP receptors by the CGRP receptor antagonist telcagepant at doses that blocked migraine attacks, suggesting a predominantly peripheral effect.108 Based on the results of recent electrophysiologic studies in rats where CSD was evoked to sensitize meningeal afferents, Melo‐Carrillo et al31 proposed that CSD evokes the release of CGRP from meningeal C‐fiber terminals, which in turn activates adjacent Aδ afferent fibers that express CGRP receptors. Thus, CGRP antibodies can prevent the activation of the Aδ afferent fibers at a peripheral site to prevent the development of migraine headache.31
CSD May Initiate Trigeminal Sensitization
Since CSD was first described by Leao as a slow‐moving (3‐5 mm/min) wave of depolarization accompanied by reduced electrocorticogram activity, vasodilation, and hyperemia,109, 110 it was regarded as the electrophysiologic correlate of migraine aura.111, 112 Electrophysiologic studies have shown that CSD can sensitize peripheral and central trigeminovascular neurons,113 although others have shown no effect of CSD on peripheral trigeminal or TNC neurons.114 Excitation of the trigeminal terminals of the dura can elicit a release of transmitters, including CGRP, which results in meningeal vasodilation and a neurogenic inflammation. The application of K+ to the cortex of anesthetized rats produced CSD, which resulted in oxidative stress and increased production of reactive oxygen species (ROS) in not only the cortex and meninges but also of the TG.115, 116 It was also determined that ROS can directly acutely excite trigeminal nociceptors and also promote the release of CGRP from trigeminal terminals to promote sensitization of TG neurons.115
In a recent study, the activities of WDR and HT lamina I‐II and lamina IV‐V TNC neurons with convergent inputs from dural and facial regions were examined in response to CSD.35 Induction of CSD by pinprick stimulation of the visual cortex was followed 2 hours later by an increase in spontaneous firing rates of HT, but not WDR, neurons.35 In that study, CSD also elicited increased responses and expansion of receptive fields of the HT neurons in response to mechanical stimulation of the dura, brush or pressure stimulus applied to the periorbital skin and brush of the cornea, indicating central sensitization. Moreover, pretreatment with the humanized monoclonal antibody to CGRP (CGRP‐mAb) fremanezumab prevented or markedly attenuated the development of these signs of sensitization. Additionally, CGRP‐mAb profoundly and significantly reduced the spontaneous activity and evoked responses to dural stimulation of HT, but not WDR, TNC neurons, but did not alter the responses of TNC neurons to periorbital cutaneous or corneal stimuli in the absence of CSD.35 Taken together, these results from animal studies strongly suggested that CSD can lead to a delayed central sensitization of TNC neurons through a mechanism dependent in part on CGRP. It was also shown that CGRP‐mAb can prevent the development of central sensitization and cutaneous allodynia elicited by CSD.
Although numerous imaging studies showing a wave of oligemia across the occipital cortex that correlates with aura support a role for CSD in aura,117, 118, 119 its relationship to migraine attacks is still under debate. For example, the majority of people with migraine do not experience aura.6 Moreover, there is no satisfactory explanation for the genesis of CSD in migraine. CSD in animal studies in induced by damaging stimuli applied to the cortex. Clinically, CSD may be observed with traumatic brain injury or infarct, and is associated with progression of ischemia and of brain injury, and worsening of patient outcomes.25 There is no evidence of injury or trauma to cause CSD in migraine.25, 120 Spontaneous CSD was inferred in one study, where functional magnetic resonance imaging (fMRI) performed in a patient with migraine with aura induced by strenuous exercise and photostimulation showed changes in blood oxygenation level‐dependent (BOLD) signal suggestive of CSD.121Also, in a study of patients with migraine using magnetoencephalographic waveforms, patients with migraine with aura induced by a visual stimulus showed spreading depression‐like neuroelectric event.122 These studies suggest that spontaneous CSD‐like events are possible. Nonetheless, there is insufficient evidence to show that the hyperemia and oligemia observed in imaging studies truly represent an electrophysiologic event. Importantly, a randomized clinical trial showed that tonabersat, a drug that inhibits CSD, did not reduce the number of migraine headache days patients experienced, but significantly reduced the number of days with aura.123
CGRP Actions on Glia
There is a growing body of evidence that shows that CGRP from TG neurons can interact with the TG satellite glial cells to promote sensitization. Capsaicin‐induced activation of TG neurons stimulates the secretion of nitric oxide (NO) and CGRP from the TG soma, which stimulate adjacent satellite glial cells to release interleukin (IL)‐1β and increase cyclooxygenase activity, increasing PGE2 production.124, 125, 126 In this way, TG satellite cells promote sensitization of the TG neurons.124, 125, 126 Chemically induced inflammation of the dura in rats increased the expression of pERK1/2 in TG satellite cells and of interleukin‐1β (IL‐1β) and CGRP in TG neurons, demonstrating a long‐term sensitization of the TG due in part to neuronal–glial interactions.127 Other studies have shown that CGRP released from TG neurons stimulate production of tumor necrosis factor‐α (TNF‐α) in satellite cells, which then acts on TG neurons to increase both the production and the release of CGRP.128 Moreover, TNF‐α can acutely sensitize TG neurons directly, as well as evoke the upregulation of several proinflammatory cytokines in these neurons, likely through the TNF‐α receptors.129 Together, these studies demonstrate that CGRP secreted from TG neurons can increase cytokine production in TG neurons and satellite glial cells and increase its own production and release from neurons, maintaining a sensitized state and enhancing pain.124, 126, 129, 130
Central CGRP in Migraine
As discussed in several recent reviews, it is generally accepted that anti‐CGRP therapeutics most likely act peripherally to relieve migraine attacks, since the small‐molecule CGRP receptor antagonists and the anti‐CGRP antibodies have very little ability to cross the blood–brain barrier.9, 108, 131, 132, 133, 134, 135 Nonetheless, a central role of CGRP in migraine is still possible, since several studies reveal a widespread distribution of the neuropeptide and the receptor components CLR and RAMP1 throughout the CNS. A recent study performed in rat brain using immunoflurescent antibodies raised against CLR, RAMP1, and CGRP revealed extensive labeling for CGRP in many neurons throughout the cerebral cortex, and labeling for the CGRP receptor components in surrounding nerve fibers.136 Immunoflurescence for CGRP or for its receptor components were found in the thalamus and hypothalamus, and generally agreed with earlier immunohistochemical and in situ hybridization studies that found evidence for either CGRP or CGRP receptor components at numerous brain sites in addition to the TNC, including the cerebellum, hippocampus, hypothalamus, amygdala, basal ganglia, parabrachial nucleus, Kölliker‐Fuse nucleus, striatum, colliculi, medullary cranial motor nuclei, nucleus ambiguous, peripeduncular, posterior, centromedial thalamic nuclei, central gray, and the inferior colliculus.65, 136, 137, 138, 139, 140, 141, 142, 143, 144 Studies performed in rat, primate, and human cerebellar tissue revealed the existence of CGRP in Purkinje cell bodies and of CGRP receptor elements, suggesting functional receptors, in Purkinje cell bodies and processes.138, 145 Moreover, glia cells that tightly surround the Purkinje cells do not express CLR or RAMP1. These regions, including the cerebellum, are believed to be involved in some aspects of pain processing or pain modulation.
However, the function of CGRP at these sites remains to be fully understood regarding pain and migraine headache, since both antinociceptive and pronociceptive functions of CGRP at central sites have been demonstrated in animal models.144 The microinjection of CGRP into the PAG was antinociceptive in normal rats as well as rats with mononeuropathy.146, 147 In those studies, antinociception was abolished by the CGRP receptor antagonist CGRP8‐37. However, in another study, CGRP applied in the ventrolateral PAG enhanced responses of TNC neurons to stimulation of the dura or of the convergent cutaneous receptive fields in rats.148 Other studies showed that descending spinopetal projections from the NRM express CGRP and can inhibit nociceptive inputs in the spinal cord. A study employing behavioral and immunohistochemical techniques found that met‐enkephalinergic neurons projecting from the central nucleus of amygdala (CeA) to the PAG are activated by CGRP in the CeA and thus produce analgesia via the PAG.149 Microinjection of CGRP into the serotonergic nucleus raphe magnus produced antinociception that was blocked by intra‐NRM CGRP8‐37 or naloxone, indicating an enkephalinergic link.150 The wide distribution of CGRP and the CGRP receptor throughout the CNS, and the potential for myriad interactions with numerous transmitter systems presents a very complex picture. A considerable amount of future studies are needed in order to figure out the possible role of CGRP at central sites with regard to migraine.65, 144
Contribution of Neuronal–Glial Interactions in Migraine
Glial activation associated with the central terminals of TG neurons in the TNC has also been observed. Immunofluorescence studies indicated that primary afferent terminals expressing CGRP were in contact with astrocytes in laminae I and II of the TNC.151 Injecting CGRP into the temporomandibular joint of rats activated astrocytes and microglia in the TNC, which can help maintain central sensitization.64 In another study, the intracisternal injection of CGRP enhanced nocifensive responses in rats and increased expression of PKA and glial fibrillary acidic protein (GFAP) in the medullary dorsal horn, indicating central sensitization of second‐order neurons and activation of astrocytes, but not of microglia, since there was no increase in ionized calcium‐binding adapter molecule 1 (Iba1).152 Neuronal–glial interactions contribute to both peripheral and central sensitization, through which nociception can be amplified. However, the clinical importance of these interactions regarding migraine remains to be determined, since there are virtually no clinical studies that have been designed to explore this aspect. Very recently, a double‐blind, randomized, placebo‐controlled trial was conducted in patients with chronic migraine who received ibudilast, a nonspecific inhibitor of glial cells, for 8 weeks.153 There were no changes in frequency or intensity of migraine attacks, and no changes in the secondary outcome measures of migraine headache index, medication use, allodynia, or quality of life.153 Because this study was performed in patients with chronic migraine, it is unknown if glial cell inhibitors would prove clinically useful in episodic migraine, or if they could prevent the progression of episodic migraine to chronic migraine.
Interaction of CGRP with Nitric Oxide in the Trigeminovascular System
As recently reviewed, an interesting, although perhaps still somewhat speculative, interaction that deserves further investigation is the possibility that CGRP and NO can amplify each other's activity in a reciprocal fashion throughout the trigeminovascular system, acting at the peripheral neurovascular interface, within the TG, and in the TNC.154 Infusions of both CGRP or nitroglycerin to patients with migraine induce migraine attacks and elevate jugular blood CGRP levels.155 Activation of CGRP receptors on vascular smooth muscle cells can lead to enhanced NO production via cyclic adenosine monophosphate (cAMP) and endothelial nitric oxide synthase (eNOS), which is possibly able to sensitive adjacent trigeminal nociceptive nerve endings, thus maintaining a state of peripheral sensitization (Fig. 1).156, 157, 158, 159, 160
Figure 1.
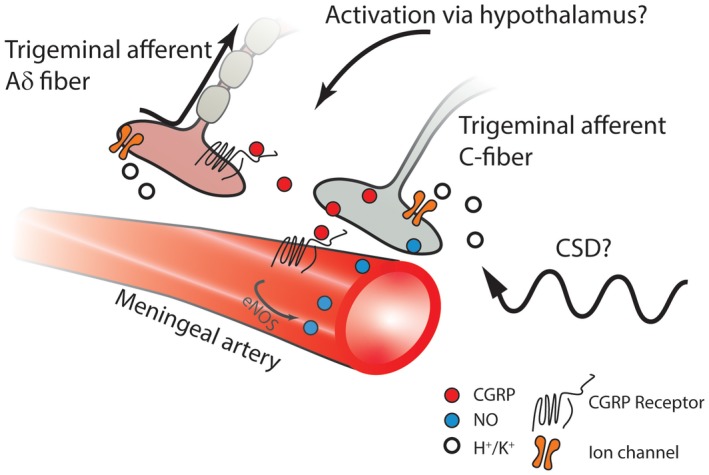
Some event occurring centrally, such as oscillations in hypothalamic activity and/or increased cortical excitability, can activate the trigeminovascular system to cause release of CGRP from CGRP‐ergic trigeminal afferent C‐fibers. In addition, CSD, regardless of its possible role in migraine pathogenesis, can cause liberation of extracellular potassium (K+) and hydrogen (H+) ions as well as proinflammatory substances from meningeal glia cells and nerve terminals. These substances can activate trigeminal nerve endings, resulting in the release of CGRP. Thus, whether the initiating event is central or peripheral, CGRP can act on its receptors on neighboring terminals of trigeminal afferent myelinated Aδ fibers, leading to sensitization of these sensory neurons. Moreover, acting on endothelial CGRP receptors, released CGRP can produce direct vasodilation and activate signaling cascades resulting in activation of eNOS and NO production. NO causes vasodilation and diffuses to the trigeminal nerve terminal, where it can enhance the further release of CGRP. Calcitonin gene‐related peptide (CGRP); endothelial nitric oxide synthase (eNOS); nitric oxide (NO).
In the TG, CGRP secreted from neurons can activate signaling cascades by interacting with the CGRP receptors present on satellite glia and on adjacent non‐CGRPergic TG neurons, inducing NO production in these glia and neurons to cause release of additional CGRP and the activation of signaling cascades to increase production of proinflammatory mediators, thus sensitizing TG neurons.161, 162, 163 Studies performed with primary TG cultures suggested the presence of a feed‐forward loop that can increase both CGRP and NO production (Fig. 2).161, 164
Figure 2.
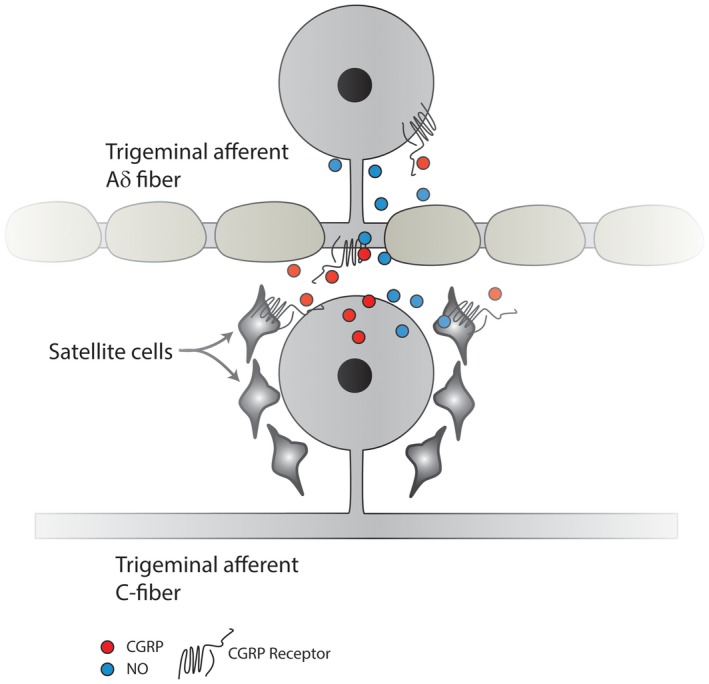
Trigeminal CGRP‐ergic C‐fiber nociceptive neurons can secrete CGRP in the trigeminal ganglion, which can diffuse to satellite cells, evoking the release of NO. NO can diffuse back to the TG neuron, as well as to adjacent non‐CGRP neurons, thereby enhancing their activity. Moreover, CGRP can act directly on adjacent non‐CGRP Aδ sensory neuronal cell bodies that express the CGRP receptor, resulting in their sensitization. Calcitonin gene‐related peptide (CGRP); CGRP receptor (CGRP‐R); nitric oxide (NO); neuronal nitric oxide synthase (nNOS); trigeminal ganglion (TG).
The increased activity of central terminals of sensitized TG afferents can promote the upregulation of neuronal NOS (nNOS) in the postsynaptic neurons, which can both sensitize the second‐order neurons as well as the primary afferent terminals by retrograde diffusion of NO.57, 163, 165 Moreover, both NO and CGRP activate astrocytes and microglia.57, 163, 165, 166 Taken together, these studies indicate that both CGRP and NO contribute to central sensitization, and they do so in a way that can potentiate each other's activity (Fig. 3).
Figure 3.
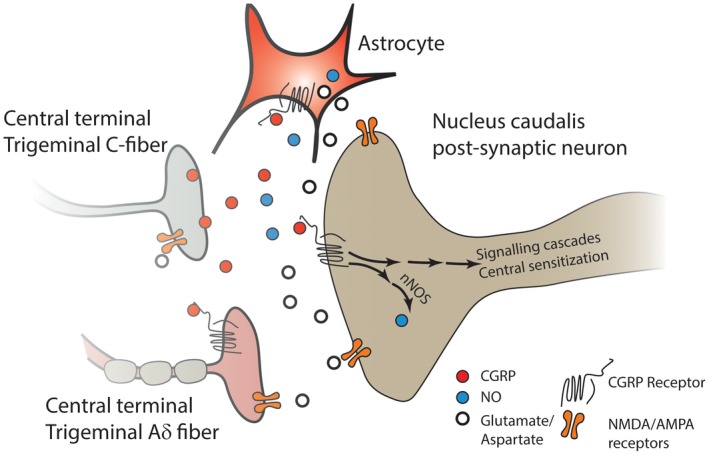
CGRP released from the central terminals of unmyelinated nociceptive C‐fiber TG neurons can activate the CGRP receptors of the second‐order neurons, and elicit production of NO via nNOS. NO acts as a retrograde neuromodulator and enhances the activity of both the CGRP and non‐CGRP nerve terminals synapsing with the second‐order neuron, resulting in enhanced transmitter release. CGRP released from the TG neuron can also activate astrocytes, eliciting the release of NO and other inflammatory mediators, and act on receptors on the terminals of neighboring Aδ neurons that express the CGRP receptor, leading to their sensitization. Moreover, the release of excitatory transmitters such as glutamate from second‐order neurons and astrocytes results in activation of NMDA and AMPA receptors on second‐order neurons, on primary afferent nerve terminals, and on astrocytes to further promote release of excitatory substances, thus further enhancing the activity of the second‐order neuron and leading to a state of central sensitization. α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid receptor (AMPA‐R); calcitonin gene‐related peptide (CGRP); N‐methyl‐D‐aspartate (NMDA); nitric oxide (NO); neuronal nitric oxide synthase (nNOS); trigeminal ganglion (TG).
In an animal model designed to mimic triptan‐induced medication overuse headache, which may be considered to be a type of chronic migraine, persistent triptan exposure produced cutaneous allodynia and upregulated CGRP and nNOS, but not substance P, in rat TG. The increased levels remained even after allodynia resolved. Subsequent exposure to known precipitors of migraine, such as NO donor infusion or bright light stress, provoked cutaneous allodynia and enhanced release of CGRP, and these effects were blocked by a nNOS inhibitor NXN‐232.167, 168, 169 Early clinical trials showed that the nonselective NOS inhibitor L‐NG‐methylarginine was superior to placebo in relieving migraine attacks or tension‐type headache,170, 171 although it did not block histamine‐induced vasodilation or migraine attacks.172 However, nonselective NOS inhibitors are also associated with cardiovascular effects, especially blood pressure increases, and are therefore considered to be unsuitable for development as migraine treatments. 154 There currently are no selective iNOS, eNOS, or nNOS inhibitors in clinical development.154
The Role of CGRP in Non‐Headache Migraine Symptoms
Intravenous infusion of CGRP produces a delayed migraine‐like headache in patients with migraine but does not induce premonitory symptoms.173 The authors suggested that CGRP infusion produced a migraine attack, but did not produce the centrally driven premonitory symptoms, suggesting that peripheral mechanisms of CGRP induced the migraine attacks.173
Photophobia is a distinctive feature of migraine that led to the development of a preclinical model in mice.174 Intracerebroventricular injection of CGRP induces aversion to bright or dim light in transgenic mice that overexpress the human RAMP1 subunit of the CGRP receptor, but not to wild‐type mice, and aversion to bright light in wild‐type mice.174, 175, 176 Co‐administration of olcegepant blocks light aversion.174, 175, 176 Moreover, peripherally administered CGRP also produced aversion to bright light that was blocked by a CGRP antibody, in the wild‐type and transgenic mice, but failed to induce aversion to low light in the transgenic mice.174 It was concluded that CGRP can elicit photophobia through distinct, and perhaps overlapping, peripheral and central mechanisms.174
Role of CGRP in Cluster Headache
Unlike migraine, cluster headache is not common, occurring in <1% of the population, and its pathophysiology remains largely obscure.6, 177, 178 Cluster headache is a trigeminal autonomic cephalalgia characterized by severe, strictly unilateral painful attacks of discrete episodes of 15‐180 minutes each, usually occurring in clusters with long periods of remission between clusters. The trigeminal distribution of pain, the circadian and circannual periodicity of the cluster episodes, and the autonomic symptoms ipsilateral to the pain represent the 3 major features of cluster headache. The current consensus is that cluster headache is a complex disorder that includes activation of the trigemino‐parasympathetic reflex and hypothalamic involvement. Functional and structural imaging studies, along with noted regularity of cluster headache attacks and the neuroendocrine abnormalities associated with them led to the suggestion that the posterior hypothalamus may act as a generator of cluster headache.179, 180, 181 Consequently, deep brain stimulation (DBS) of the hypothalamus was used to treat intractable cluster headache in several uncontrolled studies, and it was reported that overall 66% of patients achieved ≥50% reduction in headache frequency.179, 180 Only 1 randomized clinical study examined hypothalamic DBS in patients with intractable cluster headache.182 In that study, the 1‐month blinded phase of the study was too short to permit a valid evaluation, because prolonged stimulation over weeks to months is required to achieve an effect.179, 180 After the 1‐year open phase of the study, 6 of the 11 patients had >50% reductions in cluster headache frequency, and 3 patients were pain‐free.182 However, the fact that many patients do not benefit from hypothalamic DBS and the long latencies required before an observable benefit is obtained suggest that the hypothalamus is only 1 actor in a complex neural network in generating cluster headache.183 Others suggest that the posterior hypothalamus regulates the duration, rather than the generation, of cluster headache attacks.179 The pathophysiology of cluster headache has been the subject of several recent reviews.178, 181, 183, 184
A role of CGRP in cluster headache was first suggested when elevated levels of CGRP were found in the jugular blood of patients with spontaneous cluster headache attacks that normalized after treatment of cluster headache symptoms with oxygen or sumatriptan, or spontaneous resolution.185 A later study found that baseline jugular CGRP levels were higher in cluster headache patients who were in the active phase when compared to those in remission.186 Cluster headache was induced with sublingual nitroglycerin only in patients in the active phase.186 Moreover, nitroglycerin elevated blood CGRP levels only in the patients in the active phase and normalized after the headaches, spontaneously remitted or were treated with sumatriptan.186 Further studies showed that CGRP plasma levels were not increased during the 27‐minute latent period between nitroglycerin administration and the headache onset.187 It was suggested that the trigeminal system of cluster headache patients was sensitized in the active phase, demonstrated by the sensitivity of these patients to the triggering action of nitroglycerin.186, 187 It was pointed out that none of the 3 studies were placebo‐controlled, that there were inconsistencies in the CGRP blood levels reported among the studies, and that patients in one study185 were not drug‐free; consequently, more rigorous studies are needed to understand the role of CGRP in cluster headache.177 Most recently, a placebo‐controlled, randomized clinical trial showed that intravenous infusions of CGRP provoked cluster headache attacks in 89% of patients with active‐phase episodic cluster headache and in none of the patients with remission‐phase episodic cluster headache.23 Moreover, CGRP infusion provoked a cluster headache attack in 50% of patients with chronic cluster headache. A novel finding in that study was that the patients with chronic cluster headache who reported an attack after CGRP infusion had a median attack frequency of 33, whereas those who reported no cluster headache attack after CGRP had a median attack frequency of 7.5, over the preceding 30 days.23
There are currently 6 clinical trials involving 2 therapies targeting CGRP, galcanezumab (NCT02797951, NCT02397473, and NCT02438826), and fremanezumab (NCT03107052, NCT02945046, and NCT02964338), for episodic and chronic cluster headache.177, 188 Recently reported results from a phase 3 randomized clinical trial (NCT02397473) showed that galcanezumab was significantly superior to placebo in reducing weekly cluster headache attack frequency in patients with episodic cluster headache.189, 190 However, galcanezumab did not meet the primary endpoint in patients with chronic cluster headache.191, 192 The difference in susceptibility to provocation of cluster headache by CGRP infusion, and the differential results between patients with episodic and chronic cluster headache who received galcanezumab, suggests that there may be a stronger link between CGRP and episodic, relative to chronic, cluster headache. Additional results from future studies will provide valuable insights into the pathophysiology of cluster headache.
Conclusion
The past decade has seen considerable progress in understanding the pathophysiology of migraine. Among those, the important role of the peptide, CGRP, has been identified as being released in the trigeminovascular complex, leading to neurogenic inflammation and vasodilation. These advances have led to several promising novel candidates for the treatment and prevention of episodic and chronic migraine. However, the site of action of CGRP to initiate and maintain migraine prevention remains open to debate. A growing body of evidence supports a peripheral site of action, especially since most of the clinically effective treatments (ie, triptans, onabotulinumtoxin type A, and the CGRP antibodies currently in development) do not readily cross the blood–brain barrier. However, the trigeminal nerve spans the interface between peripheral and central components, and provides a mechanism through which drugs acting peripherally can still exert a downstream central effect. By sensitizing the afferent fibers through interactions with NO, and other inflammatory mediators, at the neurovascular interface and within the TG, CGRP can promote enhanced neuronal activity that ultimately drives central sensitization, even at tertiary sites such as the thalamus. These changes can lead to development of activity‐independent central sensitization, which may drive the progression of episodic migraine to the chronic form. The pathophysiology of cluster headache is much more obscure than that of migraine, but emerging evidence suggests a potential role of CGRP in the trigeminovascular system of patients with cluster headache as well. The results of ongoing clinical trials with CGRP antibodies will provide valuable insights into the mechanisms of this still enigmatic disorder.
As our understanding of underlying pathophysiology of migraine and cluster headache develops, a potential series of events emerge to explain the progression from prodrome to postdrome. The intrinsic oscillations of hypothalamic activity and connectivity to pontine and brainstem sites can could be perturbed in susceptible individuals to produce a hyperexcited cortex, a phase that may manifest clinically as the prodrome. This phase might be associated with activation of the TNC, perhaps via the hypothalamus, which has direct CGRP‐ergic projections to this region. Although CGRP does not directly excite TNC neurons, it may lead to peripheral sensitization of the afferent Aδ fibers entering the outer laminae of the TNC. Via cross‐excitation within the TG, the peripheral C‐fibers may also become sensitized, which may occur with or without generation of a CSD. The lag time between the inputs to the trigeminal system and sensitization of the peripheral nerves could explain the long lag time between prodrome and the initiation of a migraine attack, with or without aura. Termination of the migraine headache would occur as the spontaneous oscillations change, resulting in reduced drive that maintains sensitization, and resulting in the postdrome. Thus, whether the excitation of the trigeminovascular system arises from central or peripheral events, disruption of this cycle by blocking even the peripheral effects of CGRP may be sufficient to disrupt this migraine headache cycle. The cyclic nature of cluster headaches suggest that this model may be applicable to understanding its pathophysiology as well.
Statement of Authorship
Category 1
(a) Conception and Design
Smriti Iyengar, Kirk W. Johnson, Sheena K. Aurora
(b) Acquisition of Data
Smriti Iyengar, Kirk W. Johnson, Sheena K. Aurora, Michael H. Ossipov
(c) Analysis and Interpretation of Data
Smriti Iyengar, Kirk W. Johnson, Sheena K. Aurora, Michael H. Ossipov
Category 2
(a) Drafting the Manuscript
Smriti Iyengar, Kirk W. Johnson, Sheena K. Aurora, Michael H. Ossipov
(b) Revising It for Intellectual Content
Smriti Iyengar, Kirk W. Johnson, Sheena K. Aurora, Michael H. Ossipov
Category 3
(a) Final Approval of the Completed Manuscript
Smriti Iyengar, Kirk W. Johnson, Sheena K. Aurora, Michael H. Ossipov
Acknowledgments
Rod Everhart of Syneos Health kindly provided editorial assistance. Eli Lilly and Company contracted Syneos Health, for editorial services.
Conflicts of Interest: SKA and KWJ are employed by and are stockholders of Eli Lilly and Company. MHO is employed by Syneos Health. SI is a past employee of Eli Lilly and Company and a stockholder.
Funding: This review was sponsored by Eli Lilly and Company.
Ethics Approval and Consent to Participate: Not applicable.
References
- 1. Popko L. Some notes on papyrus ebers, ancient egyptian treatments of migraine, and a crocodile on the patient's head. Bull Hist Med. 2018;92:352‐366. [DOI] [PubMed] [Google Scholar]
- 2. Headache Classification Committee of the International Headache Society (IHS) . The International Classification of Headache Disorders. Cephalalgia. 2018;38:1‐211. [DOI] [PubMed] [Google Scholar]
- 3. Lipton RB, Silberstein SD. Episodic and chronic migraine headache: Breaking down barriers to optimal treatment and prevention. Headache. 2015;55(Suppl. 2):103‐122. [DOI] [PubMed] [Google Scholar]
- 4. Peck KR, Johnson YL, Smitherman TA. Migraine. Handb Clin Neurol. 2016;138:283‐293. [DOI] [PubMed] [Google Scholar]
- 5. Tfelt‐Hansen P, Olesen J. Taking the negative view of current migraine treatments: The unmet needs. CNS Drugs. 2012;26:375‐382. [DOI] [PubMed] [Google Scholar]
- 6. Headache Classification Committee of the International Headache Society (IHS) . The International Classification of Headache Disorders, 3rd edition (beta version). Cephalalgia. 2013;33:629‐808. [DOI] [PubMed] [Google Scholar]
- 7. Bigal ME, Edvinsson L, Rapoport AM, et al. Safety, tolerability, and efficacy of TEV‐48125 for preventive treatment of chronic migraine: A multicentre, randomised, double‐blind, placebo‐controlled, phase 2b study. Lancet Neurol. 2015;14:1091‐1100. [DOI] [PubMed] [Google Scholar]
- 8. Giffin NJ, Ruggiero L, Lipton RB, et al. Premonitory symptoms in migraine: An electronic diary study. Neurology. 2003;60:935‐940. [DOI] [PubMed] [Google Scholar]
- 9. Goadsby PJ, Holland PR, Martins‐Oliveira M, et al. Pathophysiology of migraine: A disorder of sensory processing. Physiol Rev. 2017;97:553‐622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Pietrobon D, Moskowitz MA. Pathophysiology of migraine. Annu Rev Physiol. 2013;75:365‐391. [DOI] [PubMed] [Google Scholar]
- 11. Puledda F, Messina R, Goadsby PJ. An update on migraine: Current understanding and future directions. J Neurol. 2017;264:2031‐2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Diener HC, May A. New aspects of migraine pathophysiology: Lessons learned from positron emission tomography. Curr Opin Neurol. 1996;9:199‐201. [DOI] [PubMed] [Google Scholar]
- 13. Weiller C, May A, Limmroth V, et al. Brain stem activation in spontaneous human migraine attacks. Nat Med. 1995;1:658‐660. [DOI] [PubMed] [Google Scholar]
- 14. May A.The initiation of migraine attacks‐revisiting the brainstem. Cephalalgia Conference: 5th European Headache and Migraine Trust International Congress, EHMTIC 2016 United Kingdom. 2016; 36(September). [Google Scholar]
- 15. May A. Understanding migraine as a cycling brain syndrome: Reviewing the evidence from functional imaging. Neurol Sci. 2017;38:125‐130. [DOI] [PubMed] [Google Scholar]
- 16. Schulte LH, Allers A, May A. Hypothalamus as a mediator of chronic migraine: Evidence from high‐resolution fMRI. Neurology. 2017;88: 2011‐2016. [DOI] [PubMed] [Google Scholar]
- 17. Maniyar FH, Sprenger T, Monteith T, et al. Brain activations in the premonitory phase of nitroglycerin‐triggered migraine attacks. Brain. 2014;137:232‐241. [DOI] [PubMed] [Google Scholar]
- 18. Schulte LH, May A. The migraine generator revisited: Continuous scanning of the migraine cycle over 30 days and three spontaneous attacks. Brain. 2016;139:1987‐1993. [DOI] [PubMed] [Google Scholar]
- 19. Guilding C, Hughes AT, Brown TM, et al. A riot of rhythms: Neuronal and glial circadian oscillators in the mediobasal hypothalamus. Mol Brain. 2009;2:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. May A. Understanding migraine as a cycling brain syndrome: Reviewing the evidence from functional imaging. Neurol Sci. 2017;38:125‐130. [DOI] [PubMed] [Google Scholar]
- 21. Meylakh N, Marciszewski KK, Di PF, et al. Deep in the brain: Changes in subcortical function immediately preceding a migraine attack. Hum Brain Mapp. 2018;39:2651‐2663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. May A, Bahra A, Buchel C, et al. Hypothalamic activation in cluster headache attacks. Lancet. 1998;352:275‐278. [DOI] [PubMed] [Google Scholar]
- 23. Vollesen ALH, Snoer A, Beske RP, et al. Effect of infusion of calcitonin gene‐related peptide on cluster headache attacks: A randomized clinical trial. JAMA Neurol. 2018;75:1187‐1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Vollesen AL, Benemei S, Cortese F, et al. Migraine and cluster headache – The common link. J Headache Pain. 2018;19:89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Close LN, Eftekhari S, Wang M, et al. Cortical spreading depression as a site of origin for migraine: Role of CGRP. Cephalalgia. 2019;39:428‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Aurora SK, Welch KM. Brain excitability in migraine: Evidence from transcranial magnetic stimulation studies. Curr Opin Neurol. 1998;11:205‐209. [DOI] [PubMed] [Google Scholar]
- 27. Aurora SK, Cao Y, Bowyer SM, et al. The occipital cortex is hyperexcitable in migraine: Experimental evidence. Headache. 1999;39:469‐476. [DOI] [PubMed] [Google Scholar]
- 28. Schulte LH, May A. Of generators, networks and migraine attacks. Curr Opin Neurol. 2017;30:241‐245. [DOI] [PubMed] [Google Scholar]
- 29. Mayberg M, Langer RS, Zervas NT, et al. Perivascular meningeal projections from cat trigeminal ganglia: Possible pathway for vascular headaches in man. Science. 1981;213:228‐230. [DOI] [PubMed] [Google Scholar]
- 30. Eftekhari S, Warfvinge K, Blixt FW, et al. Differentiation of nerve fibers storing CGRP and CGRP receptors in the peripheral trigeminovascular system. J Pain. 2013;14:1289‐1303. [DOI] [PubMed] [Google Scholar]
- 31. Melo‐Carrillo A, Strassman AM, Nir RR, et al. Fremanezumab‐A humanized monoclonal anti‐CGRP antibody‐inhibits thinly myelinated (Adelta) but not unmyelinated (C) meningeal nociceptors. J Neurosci. 2017;37:10587‐10596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ottosson A, Edvinsson L. Release of histamine from dural mast cells by substance P and calcitonin gene‐related peptide. Cephalalgia. 1997;17:166‐174. [DOI] [PubMed] [Google Scholar]
- 33. Eftekhari S, Salvatore CA, Calamari A, et al. Differential distribution of calcitonin gene‐related peptide and its receptor components in the human trigeminal ganglion. Neuroscience. 2010;169:683‐696. [DOI] [PubMed] [Google Scholar]
- 34. Miller S, Liu H, Warfvinge K, et al. Immunohistochemical localization of the calcitonin gene‐related peptide binding site in the primate trigeminovascular system using functional antagonist antibodies. Neuroscience. 2016;328:165‐183. [DOI] [PubMed] [Google Scholar]
- 35. Melo‐Carrillo A, Noseda R, Nir R, et al. Selective inhibition of trigeminovascular neurons by fremanezumab: A humanized monoclonal anti‐CGRP antibody. J Neurosci. 2017;37:7149‐7163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Eftekhari S, Edvinsson L. Possible sites of action of the new calcitonin gene‐related peptide receptor antagonists. Ther Adv Neurol Disord. 2010;3:369‐378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Eftekhari S, Edvinsson L. Calcitonin gene‐related peptide (CGRP) and its receptor components in human and rat spinal trigeminal nucleus and spinal cord at C1‐level. BMC Neurosci. 2011;12:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Noseda R, Burstein R. Migraine pathophysiology: Anatomy of the trigeminovascular pathway and associated neurological symptoms, cortical spreading depression, sensitization, and modulation of pain. Pain. 2013;154(Suppl. 1):S44‐S53. [DOI] [PubMed] [Google Scholar]
- 39. Noseda R, Jakubowski M, Kainz V, et al. Cortical projections of functionally identified thalamic trigeminovascular neurons: Implications for migraine headache and its associated symptoms. J Neurosci. 2011;31:14204‐14217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Abdallah K, Artola A, Monconduit L, et al. Bilateral descending hypothalamic projections to the spinal trigeminal nucleus caudalis in rats. PLoS ONE. 2013;8:e73022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Robert C, Bourgeais L, Arreto CD, et al. Paraventricular hypothalamic regulation of trigeminovascular mechanisms involved in headaches. J Neurosci. 2013;33:8827‐8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Charbit AR, Akerman S, Holland PR, et al. Neurons of the dopaminergic/calcitonin gene‐related peptide A11 cell group modulate neuronal firing in the trigeminocervical complex: An electrophysiological and immunohistochemical study. J Neurosci. 2009;29:12532‐12541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Strassman A, Mason P, Moskowitz M, et al. Response of brainstem trigeminal neurons to electrical stimulation of the dura. Brain Res. 1986;379:242‐250. [DOI] [PubMed] [Google Scholar]
- 44. De Felice M, Ossipov MH. Cortical and subcortical modulation of pain. Pain Manag. 2016;6:111‐120. [DOI] [PubMed] [Google Scholar]
- 45. Ossipov MH. The perception and endogenous modulation of pain. Scientifica. 2012;2012:1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Meng ID, Dodick D, Ossipov MH, et al. Pathophysiology of medication overuse headache: Insights and hypotheses from preclinical studies. Cephalalgia. 2011;31:851‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Fenton BW, Shih E, Zolton J. The neurobiology of pain perception in normal and persistent pain. Pain Manag. 2015;5:297‐317. [DOI] [PubMed] [Google Scholar]
- 48. Schwedt TJ, Larson‐Prior L, Coalson RS, et al. Allodynia and descending pain modulation in migraine: A resting state functional connectivity analysis. Pain Med. 2014;15:154‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Yarnitsky D. Conditioned pain modulation (the diffuse noxious inhibitory control‐like effect): Its relevance for acute and chronic pain states. Curr Opin Anaesthesiol. 2010;23:611‐615. [DOI] [PubMed] [Google Scholar]
- 50. Villanueva L. Diffuse noxious inhibitory control (DNIC) as a tool for exploring dysfunction of endogenous pain modulatory systems. Pain. 2009;143:161‐162. [DOI] [PubMed] [Google Scholar]
- 51. Lewis GN, Heales L, Rice DA, et al. Reliability of the conditioned pain modulation paradigm to assess endogenous inhibitory pain pathways. Pain Res Manag. 2012;17:98‐102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Guy N, Voisin D, Mulliez A, et al. Medication overuse reinstates conditioned pain modulation in women with migraine. Cephalalgia. 2018;38:1148‐1158. [DOI] [PubMed] [Google Scholar]
- 53. Penfield W, McNaughton F. Dural headache and innervation of the dura mater. Arch Neurol Psychiatry. 1940;44:43‐75. [Google Scholar]
- 54. Ray BS, Wolff HG. Experimental studies on headache. Pain‐sensitive structures of the head and their significance in headache. Arch Surg. 1942;41:813‐856. [Google Scholar]
- 55. Davis KD, Dostrovsky JO. Activation of trigeminal brain‐stem nociceptive neurons by dural artery stimulation. Pain. 1986;25:395‐401. [DOI] [PubMed] [Google Scholar]
- 56. Strassman AM, Raymond SA, Burstein R. Sensitization of meningeal sensory neurons and the origin of headaches. Nature. 1996;384:560‐564. [DOI] [PubMed] [Google Scholar]
- 57. Jones MG, Lever I, Bingham S, et al. Nitric oxide potentiates response of trigeminal neurones to dural or facial stimulation in the rat. Cephalalgia. 2001;21:643‐655. [DOI] [PubMed] [Google Scholar]
- 58. Burstein R, Yamamura H, Malick A, et al. Chemical stimulation of the intracranial dura induces enhanced responses to facial stimulation in brain stem trigeminal neurons. J Neurophysiol. 1998;79:964‐982. [DOI] [PubMed] [Google Scholar]
- 59. Kulka M, Sheen CH, Tancowny BP, et al. Neuropeptides activate human mast cell degranulation and chemokine production. Immunology. 2008;123:398‐410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. May A, Goadsby PJ. Substance P receptor antagonists in the therapy of migraine. Expert Opin Investig Drugs. 2001;10:673‐678. [DOI] [PubMed] [Google Scholar]
- 61. Goldstein DJ, Offen WW, Klein EG, et al. Lanepitant, an NK‐1 antagonist, in migraine prevention. Cephalalgia. 2001;21:102‐106. [DOI] [PubMed] [Google Scholar]
- 62. Burstein R. Deconstructing migraine headache into peripheral and central sensitization. Pain. 2001;89:107‐110. [DOI] [PubMed] [Google Scholar]
- 63. Aurora SK, Brin MF. Chronic migraine: An update on physiology, imaging, and the mechanism of action of two available pharmacologic therapies. Headache. 2017;57:109‐125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Cady RJ, Glenn JR, Smith KM, et al. Calcitonin gene‐related peptide promotes cellular changes in trigeminal neurons and glia implicated in peripheral and central sensitization. Mol Pain. 2011;7:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Iyengar S, Ossipov MH, Johnson KW. The role of calcitonin gene‐related peptide in peripheral and central pain mechanisms including migraine. Pain. 2017;158:543‐559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Moskowitz MA. Defining a pathway to discovery from bench to bedside: The trigeminovascular system and sensitization. Headache. 2008;48:688‐690. [DOI] [PubMed] [Google Scholar]
- 67. Noseda R, Burstein R. Migraine pathophysiology: Anatomy of the trigeminovascular pathway and associated neurological symptoms, CSD, sensitization and modulation of pain. Pain. 2013;154(Suppl. 1):S44‐S53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Bernstein C, Burstein R. Sensitization of the trigeminovascular pathway: Perspective and implications to migraine pathophysiology. J Clin Neurol. 2012;8:89‐99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Malick A, Burstein R. Peripheral and central sensitization during migraine. Funct Neurol. 2000;15(Suppl. 3):28‐35. [PubMed] [Google Scholar]
- 70. Burstein R, Cutrer MF, Yarnitsky D. The development of cutaneous allodynia during a migraine attack clinical evidence for the sequential recruitment of spinal and supraspinal nociceptive neurons in migraine. Brain. 2000;123(Pt 8):1703‐1709. [DOI] [PubMed] [Google Scholar]
- 71. Burstein R, Jakubowski M. Analgesic triptan action in an animal model of intracranial pain: A race against the development of central sensitization. Ann Neurol. 2004;55:27‐36. [DOI] [PubMed] [Google Scholar]
- 72. Burstein R, Levy D, Jakubowski M. Effects of sensitization of trigeminovascular neurons to triptan therapy during migraine. Rev Neurol. 2005;161:658‐660. [DOI] [PubMed] [Google Scholar]
- 73. Burstein R, Jakubowski M, Garcia‐Nicas E, et al. Thalamic sensitization transforms localized pain into widespread allodynia. Ann Neurol. 2010;68:81‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Burstein R, Noseda R, Borsook D. Migraine: Multiple processes, complex pathophysiology. J Neurosci. 2015;35:6619‐6629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Edelmayer RM, Vanderah TW, Majuta L, et al. Medullary pain facilitating neurons mediate allodynia in headache‐related pain. Ann Neurol. 2009;65:184‐193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Muddhrry PK, Ghatki MA, Spokks RA, et al. Differential expression of a‐CGRP and b‐CGRP by primary sensory neurons and enteric autonomic neurons of the rat. Neuroscience. 1988;25:195‐205. [DOI] [PubMed] [Google Scholar]
- 77. Weidner C, Klede M, Rukwied R, et al. Acute effects of substance P and calcitonin gene‐related peptide in human skin – A microdialysis study. J Invest Dermatol. 2000;115:1015‐1020. [DOI] [PubMed] [Google Scholar]
- 78. Thomsen LL, Kruuse C, Iversen H, et al. A nitric oxide donor (nitroglycerine) triggers genuine migraine attacks. Eur J Neurol. 1994;1:73‐80. [DOI] [PubMed] [Google Scholar]
- 79. Antonova M, Wienecke T, Olesen J, et al. Prostaglandin E(2) induces immediate migraine‐like attack in migraine patients without aura. Cephalalgia. 2012;32:822‐833. [DOI] [PubMed] [Google Scholar]
- 80. Goadsby PJ, Edvinsson L, Ekman R. Release of vasoactive peptides in the extracerebral circulation of humans and the cat during activation of the trigeminovascular system. Ann Neurol. 1988;23:193‐196. [DOI] [PubMed] [Google Scholar]
- 81. Goadsby PJ, Edvinsson L, Ekman R. Vasoactive peptide release in the extracerebral circulation of humans during migraine headache. Ann Neurol. 1990;28:183‐187. [DOI] [PubMed] [Google Scholar]
- 82. Goadsby PJ, Edvinsson L. The trigeminovascular system and migraine: Studies characterizing cerebrovascular and neuropeptide changes seen in humans and cats. Ann Neurol. 1993;33:48‐56. [DOI] [PubMed] [Google Scholar]
- 83. Cernuda‐Morollon E, Larrosa D, Ramon C, et al. Interictal increase of CGRP levels in peripheral blood as a biomarker for chronic migraine. Neurology. 2013;81:1191‐1196. [DOI] [PubMed] [Google Scholar]
- 84. Cernuda‐Morollon E, Martinez‐Camblor P, Ramon C, et al. CGRP and VIP levels as predictors of efficacy of Onabotulinumtoxin type A in chronic migraine. Headache. 2014;54:987‐995. [DOI] [PubMed] [Google Scholar]
- 85. Cernuda‐Morollon E, Ramon C, Martinez‐Camblor P, et al. OnabotulinumtoxinA decreases interictal CGRP plasma levels in patients with chronic migraine. Pain. 2015;156:820‐824. [DOI] [PubMed] [Google Scholar]
- 86. Juhasz G, Zsombok T, Jakab B, et al. Sumatriptan causes parallel decrease in plasma calcitonin gene‐related peptide (CGRP) concentration and migraine headache during nitroglycerin induced migraine attack. Cephalalgia. 2005;25:179‐183. [DOI] [PubMed] [Google Scholar]
- 87. Cady RK, Vause CV, Ho TW, et al. Elevated saliva calcitonin gene‐related peptide levels during acute migraine predict therapeutic response to rizatriptan. Headache. 2009;49:1258‐1266. [DOI] [PubMed] [Google Scholar]
- 88. Ho TW, Olesen J, Dodick DW, et al. Antimigraine efficacy of telcagepant based on patient's historical triptan response. Headache. 2011;51:64‐72. [DOI] [PubMed] [Google Scholar]
- 89. Hoffmann J, Goadsby PJ. New agents for acute treatment of migraine: CGRP receptor antagonists, iNOS inhibitors. Curr Treat Options Neurol. 2012;14:50‐59. [DOI] [PubMed] [Google Scholar]
- 90. Olesen J, Diener HC, Husstedt IW, et al. Calcitonin gene‐related peptide receptor antagonist BIBN 4096 BS for the acute treatment of migraine. N Engl J Med. 2004;350:1104‐1110. [DOI] [PubMed] [Google Scholar]
- 91. Silberstein SD. Emerging target‐based paradigms to prevent and treat migraine. Clin Pharmacol Ther. 2013;93:78‐85. [DOI] [PubMed] [Google Scholar]
- 92. Voss T, Lipton RB, Dodick DW, et al. A phase IIb randomized, double‐blind, placebo‐controlled trial of ubrogepant for the acute treatment of migraine. Cephalalgia. 2016;36:887‐898. [DOI] [PubMed] [Google Scholar]
- 93. Diener HC, Barbanti P, Dahlof C, et al. BI 44370 TA, an oral CGRP antagonist for the treatment of acute migraine attacks: Results from a phase II study. Cephalalgia. 2011;31:573‐584. [DOI] [PubMed] [Google Scholar]
- 94. Marcus R, Goadsby PJ, Dodick D, et al. BMS‐927711 for the acute treatment of migraine: A double‐blind, randomized, placebo controlled, dose‐ranging trial. Cephalalgia. 2014;34:114‐125. [DOI] [PubMed] [Google Scholar]
- 95. Stauffer VL, Dodick DW, Zhang Q, et al. Evaluation of galcanezumab for the prevention of episodic migraine: The EVOLVE‐1 randomized clinical trial. JAMA Neurol. 2018;75:1080‐1088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Skljarevski V, Matharu M, Millen BA, et al. Efficacy and safety of galcanezumab for the prevention of episodic migraine: Results of the EVOLVE‐2 Phase 3 randomized controlled clinical trial. Cephalalgia. 2018;38:1442‐1454. [DOI] [PubMed] [Google Scholar]
- 97. Dodick DW, Goadsby PJ, Spierings EL, et al. Safety and efficacy of LY2951742, a monoclonal antibody to calcitonin gene‐related peptide, for the prevention of migraine: A phase 2, randomised, double‐blind, placebo‐controlled study. Lancet Neurol. 2014;13:885‐892. [DOI] [PubMed] [Google Scholar]
- 98. Detke HC, Goadsby PJ, Wang S, et al. Galcanezumab in chronic migraine: The randomized, double‐blind, placebo‐controlled REGAIN study. Neurology. 2018;91:e2211‐e2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Bigal ME, Dodick DW, Rapoport AM, et al. Safety, tolerability, and efficacy of TEV‐48125 for preventive treatment of high‐frequency episodic migraine: A multicentre, randomised, double‐blind, placebo‐controlled, phase 2b study. Lancet Neurol. 2015;14:1081‐1090. [DOI] [PubMed] [Google Scholar]
- 100. Dodick DW, Goadsby PJ, Silberstein SD, et al. Safety and efficacy of ALD403, an antibody to calcitonin gene‐related peptide, for the prevention of frequent episodic migraine: A randomised, double‐blind, placebo‐controlled, exploratory phase 2 trial. Lancet Neurol. 2014;13:1100‐1107. [DOI] [PubMed] [Google Scholar]
- 101. Sun H, Dodick DW, Silberstein S, et al. Safety and efficacy of AMG 334 for prevention of episodic migraine: A randomised, double‐blind, placebo‐controlled, phase 2 trial. Lancet Neurol. 2016;15:382‐390. [DOI] [PubMed] [Google Scholar]
- 102. Tepper S, Ashina M, Reuter U, et al. Safety and efficacy of erenumab for preventive treatment of chronic migraine: A randomised, double‐blind, placebo‐controlled phase 2 trial. Lancet Neurol. 2017;16:425‐434. [DOI] [PubMed] [Google Scholar]
- 103. Buzzi MG, Carter WB, Shimizu T, et al. Dihydroergotamine and sumatriptan attenuate levels of CGRP in plasma in rat superior sagittal sinus during electrical stimulation of the trigeminal ganglion. Neuropharmacology. 1991;30:1193‐1200. [DOI] [PubMed] [Google Scholar]
- 104. Moskowitz MA. Neurogenic inflammation in the pathophysiology and treatment of migraine. Neurology. 1993;43:S16‐S20. [PubMed] [Google Scholar]
- 105. Giniatullin R, Nistri A, Fabbretti E. Molecular mechanisms of sensitization of pain‐transducing P2X3 receptors by the migraine mediators CGRP and NGF. Mol Neurobiol. 2008;37:83‐90. [DOI] [PubMed] [Google Scholar]
- 106. Fabbretti E, D'Arco M, Fabbro A, et al. Delayed upregulation of ATP P2X3 receptors of trigeminal sensory neurons by calcitonin gene‐related peptide. J Neurosci. 2006;26:6163‐6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Melin C, Jacquot F, Vitello N, et al. Different processing of meningeal and cutaneous pain information in the spinal trigeminal nucleus caudalis. Cephalalgia. 2017;37:1189‐1201. [DOI] [PubMed] [Google Scholar]
- 108. Hostetler ED, Joshi AD, Sanabria‐Bohorquez S, et al. In vivo quantification of calcitonin gene‐related peptide receptor occupancy by telcagepant in rhesus monkey and human brain using the positron emission tomography tracer [11C]MK‐4232. J Pharmacol Exp Ther. 2013;347:478‐486. [DOI] [PubMed] [Google Scholar]
- 109. Leao AAP. Spreading depression of activity in cerebral cortex. J Neurophysiol. 1944;7:359‐390. [DOI] [PubMed] [Google Scholar]
- 110. Leao AAP. Further observations on the spreading depression of activity in the cerebral cortex. J Neurophysiol. 1947;10:409‐414. [DOI] [PubMed] [Google Scholar]
- 111. Lauritzen M. Pathophysiology of the migraine aura. The spreading depression theory. Brain. 1994;117(Pt 1):199‐210. [DOI] [PubMed] [Google Scholar]
- 112. Milner PM. Note on a possible correspondence between the scotomas of migraine and spreading depression of Leao. Electroencephalogr Clin Neurophysiol. 1958;10:705. [DOI] [PubMed] [Google Scholar]
- 113. Zhang X, Levy D, Kainz V, et al. Activation of central trigeminovascular neurons by cortical spreading depression. Ann Neurol. 2011;69:855‐865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ebersberger A, Schaible HG, Averbeck B, et al. Is there a correlation between spreading depression, neurogenic inflammation, and nociception that might cause migraine headache? Ann Neurol. 2001;49:7‐13. [PubMed] [Google Scholar]
- 115. Shatillo A, Koroleva K, Giniatullina R, et al. Cortical spreading depression induces oxidative stress in the trigeminal nociceptive system. Neuroscience. 2013;253:341‐349. [DOI] [PubMed] [Google Scholar]
- 116. Russo AF. Calcitonin gene‐related peptide (CGRP): A new target for migraine. Annu Rev Pharmacol Toxicol. 2015;55:533‐552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Olesen J, Friberg L, Olsen TS, et al. Timing and topography of cerebral blood flow, aura, and headache during migraine attacks. Ann Neurol. 1990;28:791‐798. [DOI] [PubMed] [Google Scholar]
- 118. Cao Y, Welch KM, Aurora S, et al. Functional MRI‐BOLD of visually triggered headache in patients with migraine. Arch Neurol. 1999;56:548‐554. [DOI] [PubMed] [Google Scholar]
- 119. Hadjikhani N, Del Sanchez RM, Wu O, et al. Mechanisms of migraine aura revealed by functional MRI in human visual cortex. Proc Natl Acad Sci U S A. 2001;98:4687‐4692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Charles A, Hansen JM. Migraine aura: New ideas about cause, classification, and clinical significance. Curr Opin Neurol. 2015;28:255‐260. [DOI] [PubMed] [Google Scholar]
- 121. Arngrim N, Hougaard A, Ahmadi K, et al. Heterogenous migraine aura symptoms correlate with visual cortex functional magnetic resonance imaging responses. Ann Neurol. 2017;82:925‐939. [DOI] [PubMed] [Google Scholar]
- 122. Bowyer SM, Aurora KS, Moran JE, et al. Magnetoencephalographic fields from patients with spontaneous and induced migraine aura. Ann Neurol. 2001;50:582‐587. [DOI] [PubMed] [Google Scholar]
- 123. Hauge AW, Asghar MS, Schytz HW, et al. Effects of tonabersat on migraine with aura: A randomised, double‐blind, placebo‐controlled crossover study. Lancet Neurol. 2009;8:718‐723. [DOI] [PubMed] [Google Scholar]
- 124. Capuano A, De CA, Lisi L, et al. Proinflammatory‐activated trigeminal satellite cells promote neuronal sensitization: Relevance for migraine pathology. Mol Pain. 2009;5:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. De CA, Lisi L, Capuano A, et al. Trigeminal satellite cells express functional calcitonin gene‐related peptide receptors, whose activation enhances interleukin‐1beta pro‐inflammatory effects. J Neuroimmunol. 2011;237:39‐46. [DOI] [PubMed] [Google Scholar]
- 126. Thalakoti S, Patil VV, Damodaram S, et al. Neuron‐glia signaling in trigeminal ganglion: Implications for migraine pathology. Headache. 2007;47:1008‐1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Lukacs M, Haanes KA, Majlath Z, et al. Dural administration of inflammatory soup or complete freund's adjuvant induces activation and inflammatory response in the rat trigeminal ganglion. J Headache Pain. 2015;16:564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Bowen EJ, Schmidt TW, Firm CS, et al. Tumor necrosis factor‐alpha stimulation of calcitonin gene‐related peptide expression and secretion from rat trigeminal ganglion neurons. J Neurochem. 2006;96:65‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Durham ZL, Hawkins JL, Durham PL. Tumor necrosis factor‐alpha stimulates cytokine expression and transient sensitization of trigeminal nociceptive neurons. Arch Oral Biol. 2017;75:100‐106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Durham PL. Diverse physiological roles of calcitonin gene‐related peptide in migraine pathology: Modulation of neuronal‐glial‐immune cells to promote peripheral and central sensitization. Curr Pain Headache Rep. 2016;20:48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Edvinsson L. The CGRP pathway in migraine as a viable target for therapies. Headache. 2018;58(Suppl. 1):33‐47. [DOI] [PubMed] [Google Scholar]
- 132. Edvinsson L, Haanes KA, Warfvinge K, et al. CGRP as the target of new migraine therapies – Successful translation from bench to clinic. Nat Rev Neurol. 2018;14:338‐350. [DOI] [PubMed] [Google Scholar]
- 133. Barbanti P, Aurilia C, Fofi L, et al. The role of anti‐CGRP antibodies in the pathophysiology of primary headaches. Neurol Sci. 2017;38(Suppl. 1):31–35. [DOI] [PubMed] [Google Scholar]
- 134. Edvinsson L. The trigeminovascular pathway: Role of CGRP and CGRP receptors in migraine. Headache. 2017;57(Suppl. 2):47‐55. [DOI] [PubMed] [Google Scholar]
- 135. Edvinsson L, Warfvinge K. Recognizing the role of CGRP and CGRP receptors in migraine and its treatment. Cephalalgia. 2019;39:366‐373. [DOI] [PubMed] [Google Scholar]
- 136. Warfvinge K, Edvinsson L. Distribution of CGRP and CGRP receptor components in the rat brain. Cephalalgia. 2019;39:342‐353. [DOI] [PubMed] [Google Scholar]
- 137. Wimalawansa SJ. Calcitonin gene‐related peptide and its receptors: Molecular genetics, physiology, pathophysiology, and therapeutic potentials. Endocr Rev. 1996;17:533‐585. [DOI] [PubMed] [Google Scholar]
- 138. Eftekhari S, Salvatore CA, Gaspar RC, et al. Localization of CGRP receptor components, CGRP, and receptor binding sites in human and rhesus cerebellar cortex. Cerebellum. 2013;12:937‐949. [DOI] [PubMed] [Google Scholar]
- 139. Kresse A, Jacobowitz DM, Skofitsch G. Distribution of calcitonin gene‐related peptide in the central nervous system of the rat by immunocytochemistry and in situ hybridization histochemistry. Ann N Y Acad Sci. 1992;657:455‐457. [DOI] [PubMed] [Google Scholar]
- 140. Kruger L, Sternini C, Brecha NC, et al. Distribution of calcitonin gene‐related peptide immunoreactivity in relation to the rat central somatosensory projection. J Comp Neurol. 1988;273:149‐162. [DOI] [PubMed] [Google Scholar]
- 141. Russell FA, King R, Smillie SJ, et al. Calcitonin gene‐related peptide: Physiology and pathophysiology. Physiol Rev. 2014;94:1099‐1142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Russo AF, Dickerson IM. CGRP: A multifunctional neuropeptide In: Lajtha A, Lim R, eds. Handbook of Neurochemistry and Molecular Neurobiology: Neuroactive Proteins and Peptides. Boston, MA: Springer, US; 2006:391‐426. [Google Scholar]
- 143. Tschopp FA, Henke H, Petermann JB, et al. Calcitonin gene‐related peptide and its binding sites in the human central nervous system and pituitary. Proc Natl Acad Sci U S A. 1985;82:248‐252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144. Yu LC, Hou JF, Fu FH, et al. Roles of calcitonin gene‐related peptide and its receptors in pain‐related behavioral responses in the central nervous system. Neurosci Biobehav Rev. 2009;33:1185‐1191. [DOI] [PubMed] [Google Scholar]
- 145. Edvinsson L, Eftekhari S, Salvatore CA, et al. Cerebellar distribution of calcitonin gene‐related peptide (CGRP) and its receptor components calcitonin receptor‐like receptor (CLR) and receptor activity modifying protein 1 (RAMP1) in rat. Mol Cell Neurosci. 2011;46:333‐339. [DOI] [PubMed] [Google Scholar]
- 146. Yu LC, Weng XH, Wang JW, et al. Involvement of calcitonin gene‐related peptide and its receptor in anti‐nociception in the periaqueductal grey of rats. Neurosci Lett. 2003;349:1‐4. [DOI] [PubMed] [Google Scholar]
- 147. Xu S, Lundeberg T, Yu L. Antinociceptive effects of calcitonin gene‐related peptide injected into periaqueductal grey of rats with mononeuropathy. Brain Res. 2000;859:358‐360. [DOI] [PubMed] [Google Scholar]
- 148. Pozo‐Rosich P, Storer RJ, Charbit AR, et al. Periaqueductal gray calcitonin gene‐related peptide modulates trigeminovascular neurons. Cephalalgia. 2015;35:1298‐1307. [DOI] [PubMed] [Google Scholar]
- 149. Xu W, Lundeberg T, Wang YT, et al. Antinociceptive effect of calcitonin gene‐related peptide in the central nucleus of amygdala: Activating opioid receptors through amygdala‐periaqueductal gray pathway. Neuroscience. 2003;118:1015‐1022. [DOI] [PubMed] [Google Scholar]
- 150. Huang Y, Brodda‐Jansen G, Lundeberg T, et al. Anti‐nociceptive effects of calcitonin gene‐related peptide in nucleus raphe magnus of rats: An effect attenuated by naloxone. Brain Res. 2000;873:54‐59. [DOI] [PubMed] [Google Scholar]
- 151. Yihong Z, Tamada Y, Akai K, et al. Morphological interrelationship between astrocytes and nerve endings in the rat spinal trigeminal nucleus caudalis. Okajimas Folia Anat Jpn. 2006;83:91‐96. [DOI] [PubMed] [Google Scholar]
- 152. Cornelison LE, Hawkins JL, Durham PL. Elevated levels of calcitonin gene‐related peptide in upper spinal cord promotes sensitization of primary trigeminal nociceptive neurons. Neuroscience. 2016;339:491‐501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Kwok YH, Swift JE, Gazerani P, et al. A double‐blind, randomized, placebo‐controlled pilot trial to determine the efficacy and safety of ibudilast, a potential glial attenuator, in chronic migraine. J Pain Res. 2016;9:899‐907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Pradhan AA, Bertels Z, Akerman S. Targeted nitric oxide synthase inhibitors for migraine. Neurotherapeutics. 2018;15:391‐401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155. Iversen HK, Olesen J. Headache induced by a nitric oxide donor (nitroglycerin) responds to sumatriptan. A human model for development of migraine drugs. Cephalalgia. 1996;16:412‐418. [DOI] [PubMed] [Google Scholar]
- 156. Koulchitsky S, Fischer MJ, Messlinger K. Calcitonin gene‐related peptide receptor inhibition reduces neuronal activity induced by prolonged increase in nitric oxide in the rat spinal trigeminal nucleus. Cephalalgia. 2009;29:408‐417. [DOI] [PubMed] [Google Scholar]
- 157. Fischer MJ, Koulchitsky S, Messlinger K. The nonpeptide calcitonin gene‐related peptide receptor antagonist BIBN4096BS lowers the activity of neurons with meningeal input in the rat spinal trigeminal nucleus. J Neurosci. 2005;25:5877‐5883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158. Koulchitsky S, Fischer MJ, De CR, et al. Biphasic response to nitric oxide of spinal trigeminal neurons with meningeal input in rat – possible implications for the pathophysiology of headaches. J Neurophysiol. 2004;92:1320‐1328. [DOI] [PubMed] [Google Scholar]
- 159. Ramachandran R, Bhatt DK, Ploug KB, et al. Nitric oxide synthase, calcitonin gene‐related peptide and NK‐1 receptor mechanisms are involved in GTN‐induced neuronal activation. Cephalalgia. 2014;34:136‐147. [DOI] [PubMed] [Google Scholar]
- 160. Akerman S, Williamson DJ, Kaube H, et al. Nitric oxide synthase inhibitors can antagonize neurogenic and calcitonin gene‐related peptide induced dilation of dural meningeal vessels. Br J Pharmacol. 2002;137:62‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161. Messlinger K, Lennerz JK, Eberhardt M, et al. CGRP and NO in the trigeminal system: Mechanisms and role in headache generation. Headache. 2012;52:1411‐1427. [DOI] [PubMed] [Google Scholar]
- 162. Messlinger K, Fischer MJ, Lennerz JK. Neuropeptide effects in the trigeminal system: Pathophysiology and clinical relevance in migraine. Keio J Med. 2011;60:82‐89. [DOI] [PubMed] [Google Scholar]
- 163. Barbanti P, Egeo G, Aurilia C, et al. Drugs targeting nitric oxide synthase for migraine treatment. Expert Opin Investig Drugs. 2014;23:1141‐1148. [DOI] [PubMed] [Google Scholar]
- 164. Bellamy J, Bowen EJ, Russo AF, et al. Nitric oxide regulation of calcitonin gene‐related peptide gene expression in rat trigeminal ganglia neurons. Eur J Neurosci. 2006;23:2057‐2066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165. Lambert GA, Hoskin KL, Zagami AS. Nitrergic and glutamatergic neuronal mechanisms at the trigeminovascular first‐order synapse. Neuropharmacology. 2004;47:92‐105. [DOI] [PubMed] [Google Scholar]
- 166. Reddington M, Priller J, Treichel J, et al. Astrocytes and microglia as potential targets for calcitonin gene related peptide in the central nervous system. Can J Physiol Pharmacol. 1995;73:1047‐1049. [DOI] [PubMed] [Google Scholar]
- 167. De Felice M, Ossipov MH, Wang R, et al. Triptan‐induced latent sensitization: A possible basis for medication overuse headache. Ann Neurol. 2010;67:325‐337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. De Felice M, Ossipov MH, Wang R, et al. Triptan‐induced enhancement of neuronal nitric oxide synthase in trigeminal ganglion dural afferents underlies increased responsiveness to potential migraine triggers. Brain. 2010;133:2475‐2488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169. De Felice M, Ossipov MH, Porreca F. Persistent medication‐induced neural adaptations, descending facilitation, and medication overuse headache. Curr Opin Neurol. 2011;24:193‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Ashina M, Lassen LH, Bendtsen L, et al. Effect of inhibition of nitric oxide synthase on chronic tension‐type headache: A randomised crossover trial. Lancet. 1999;353:287‐289. [DOI] [PubMed] [Google Scholar]
- 171. Lassen LH, Ashina M, Christiansen I, et al. Nitric oxide synthase inhibition: A new principle in the treatment of migraine attacks. Cephalalgia. 1998;18:27‐32. [DOI] [PubMed] [Google Scholar]
- 172. Lassen LH, Christiansen I, Iversen HK, et al. The effect of nitric oxide synthase inhibition on histamine induced headache and arterial dilatation in migraineurs. Cephalalgia. 2003;23:877‐886. [DOI] [PubMed] [Google Scholar]
- 173. Guo S, Vollesen AL, Olesen J, et al. Premonitory and nonheadache symptoms induced by CGRP and PACAP38 in patients with migraine. Pain. 2016;157:2773‐2781. [DOI] [PubMed] [Google Scholar]
- 174. Mason BN, Kaiser EA, Kuburas A, et al. Induction of migraine‐like photophobic behavior in mice by both peripheral and central CGRP mechanisms. J Neurosci. 2017;37:204‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175. Kaiser EA, Kuburas A, Recober A, et al. Modulation of CGRP‐induced light aversion in wild‐type mice by a 5‐HT(1B/D) agonist. J Neurosci. 2012;32:15439‐15449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Recober A, Kuburas A, Zhang Z, et al. Role of calcitonin gene‐related peptide in light‐aversive behavior: Implications for migraine. J Neurosci. 2009;29:8798‐8804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177. Ashina H, Newman L, Ashina S. Calcitonin gene‐related peptide antagonism and cluster headache: An emerging new treatment. Neurol Sci. 2017;38:2089‐2093. [DOI] [PubMed] [Google Scholar]
- 178. Eller M, Goadsby PJ. Trigeminal autonomic cephalalgias. Oral Dis. 2016;22:1‐8. [DOI] [PubMed] [Google Scholar]
- 179. Leone M, Proietti CA. Deep brain stimulation in headache. Cephalalgia. 2016;36:1143‐1148. [DOI] [PubMed] [Google Scholar]
- 180. Leone M, Giustiniani A, Cecchini AP. Cluster headache: Present and future therapy. Neurol Sci. 2017;38:45‐50. [DOI] [PubMed] [Google Scholar]
- 181. May A. Cluster headache: Pathogenesis, diagnosis, and management. Lancet. 2005;366:843‐855. [DOI] [PubMed] [Google Scholar]
- 182. Fontaine D, Lazorthes Y, Mertens P, et al. Safety and efficacy of deep brain stimulation in refractory cluster headache: A randomized placebo‐controlled double‐blind trial followed by a 1‐year open extension. J Headache Pain. 2010;11:23‐31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183. Obermann M, Matharu M. Pathophysiology of cluster headache: Current status and future directions In: Ashina M, Geppetti P, eds. Pathophysiology of Headaches—From Molecule to Man. Cham, Switzerland: Springer International Publishing; 2015:247‐258. [Google Scholar]
- 184. Leone M, Proietti CA. Advances in the understanding of cluster headache. Expert Rev Neurother. 2017;17:165‐172. [DOI] [PubMed] [Google Scholar]
- 185. Goadsby PJ, Edvinsson L. Human in vivo evidence for trigeminovascular activation in cluster headache. Neuropeptide changes and effects of acute attacks therapies. Brain. 1994;117(Pt 3):427‐434. [DOI] [PubMed] [Google Scholar]
- 186. Fanciullacci M, Alessandri M, Figini M, et al. Increase in plasma calcitonin gene‐related peptide from the extracerebral circulation during nitroglycerin‐induced cluster headache attack. Pain. 1995;60:119‐123. [DOI] [PubMed] [Google Scholar]
- 187. Fanciullacci M, Alessandri M, Sicuteri R, et al. Responsiveness of the trigeminovascular system to nitroglycerine in cluster headache patients. Brain. 1997;120(Pt 2):283‐288. [DOI] [PubMed] [Google Scholar]
- 188. Schuster NM, Rapoport AM. Calcitonin gene‐related peptide‐targeted therapies for migraine and cluster headache: A review. Clin Neuropharmacol. 2017;40:169‐174. [DOI] [PubMed] [Google Scholar]
- 189. Martinez JM, Goadsby PJ, Dodick DW, et al. Study CGAL: A phase 3 placebo‐ controlled study of galcanezumab in patients with episodic cluster headache: Results from the 8‐week double‐blind treatment phase. Headache. 2018;58:1289‐1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190. Martinez JM, Goadsby PJ, Dodick DW, et al. Study CGAL: A placebo‐controlled study of galcanezumab in patients with episodic cluster headache: Results from the 8‐week double‐blind treatment phase. Postgrad Med. 2018;130:24‐25.29110567 [Google Scholar]
- 191. Gelfand AA, Goadsby PJ. Cluster headache and calcitonin gene‐related peptide‐more on quantum therapeutics in headache medicine. JAMA Neurol. 2018;75:1179‐1180. [DOI] [PubMed] [Google Scholar]
- 192. Eli Lilly and Company . Lilly's Galcanezumab Meets Primary Endpoint in Phase 3 Study Evaluating Galcanezumab for the Prevention of Episodic Cluster Headache. Eli Lilly and Company; 2018. [cited 2018 Oct 27]; Available at: https://investor.lilly.com/news-releases/news-release-details/lillys-galcanezumab-meets-primary-endpoint-phase-3-study. [Google Scholar]