

Abstract
X‐linked adrenoleukodystrophy (X‐ALD) and metachromatic leukodystrophy (MLD) are two relatively common examples of hereditary demyelinating diseases caused by a dysfunction of peroxisomal or lysosomal lipid degradation. In both conditions, accumulation of nondegraded lipids leads to the destruction of cerebral white matter. Because of their high lipid content, oligodendrocytes are considered key to the pathophysiology of these leukodystrophies. However, the response to allogeneic stem cell transplantation points to the relevance of cells related to the hematopoietic lineage. In the present study, we aimed to better characterize the pathogenetic role of microglia in the above‐mentioned diseases. Applying recently established microglia markers to human autopsy cases of X‐ALD and MLD we were able to delineate distinct lesion stages in evolving demyelinating lesions. The immune‐phenotype of microglia was altered already early in lesion evolution, and microglia loss preceded full‐blown myelin degeneration both in X‐ALD and MLD. DNA fragmentation indicating phagocyte death was observed in areas showing microglia loss. The morphology and dynamics of phagocyte decay differed between the diseases and between lesion stages, hinting at distinct pathways of programmed cell death. In summary, the present study shows an early and severe damage to microglia in the pathogenesis of X‐ALD and MLD. This hints at a central pathophysiologic role of these cells in the diseases and provides evidence for an ongoing transfer of toxic substrates primarily enriched in myelinating cells to microglia.
Keywords: cell death, demyelination, metachromatic leukodystrophy, microglia, X‐linked adrenoleukodystrophy
1. INTRODUCTION
X‐linked adrenoleukodystrophy (X‐ALD), the most common peroxisomal storage disease (Berger, Pujol, Aubourg, & Forss‐Petter, 2010), is caused by a dysfunction of ABCD1 transporters, which normally shuttle activated very long chain fatty acids (VLCFA) into the peroxisome for further degradation. At least 60% of affected male individuals develop cerebral adrenoleukodystrophy (Beer, Engelen, & van Geel, 2014; Wiesinger, Eichler, & Berger, 2015). Left untreated, the disease has an infaust prognosis, resulting in severe destruction of cerebral white matter (Mahmood, Raymond, Dubey, Peters, & Moser, 2007). Metachromatic leukodystrophy (MLD) is an autosomal recessive lysosomal storage disorder with a mutation in the gene encoding the enzyme arylsulfatase A (ARSA) (Gieselmann et al., 1991). This leads to an accumulation of sulfatides, with destruction of cerebral white matter as a salient end‐stage pathology.
Mature oligodendrocytes sustain myelin sheaths with a high lipid content, representing 70–75% of their dry weight (Nave & Werner, 2014). In addition, most sulfatides in the CNS are located in the myelin sheaths and oligodendrocytes (Eckhardt, 2008). Accordingly, this cell type is considered central to the pathogenesis of leukodystrophies (Gieselmann, 2008). However, the fact that these diseases partly respond to allogeneic or genetically modified autologous stem cell transplantation (Eichler et al., 2017; Kidd et al., 1998; Peters et al., 2004) underscores the fundamental importance of cells derived from—or replaceable by—descendants of the hematopoietic lineage. In addition, some more recent studies in animal models and in human tissue highlight early changes of microglia in lesion development (Eichler et al., 2008; Gong et al., 2017; Wittke, Hartmann, Gieselmann, & Lüllmann‐Rauch, 2004).
The transmembrane protein 119 (Tmem119) and the purinergic receptor P2ry12 are two recently discovered cell surface proteins highly expressed in brain resident microglia under homeostatic conditions as opposed to peripheral macrophages (Bennett et al., 2016; Butovsky et al., 2014; Sasaki et al., 2003). In the present study, we sought to characterize and compare human cases of X‐ALD and MLD by using these new in addition to established phagocyte markers.
We observed striking early changes in the immune phenotype of microglia in X‐ALD and MLD that preceded signs of overt myelin damage. These changes in microglia expression patterns allowed a more precise assessment of lesion evolution. In X‐ALD, in addition, a severe reduction in microglia density preceded full‐blown oligodendrocyte and myelin degeneration. In MLD, phagocyte death was accompanied by lysosomal breakdown and cell membrane lysis and was observed before oligodendrocyte damage and reduction in myelin density. Morphological and histochemical features of cell death varied between the diseases and between lesion stages in a particular disease.
The finding of primary microglia damage in X‐ALD and MLD, where toxic substrates are known to be mainly synthesized in oligodendrocytes and myelin, indicates a shed of undegradable and toxic lipids from myelinating cells and supports the hypothesis that turnover of myelin is outsourced to microglia cells.
2. MATERIALS AND METHODS
2.1. Human tissue
Demyelinating lesion evolution was studied in formalin‐fixed, paraffin‐embedded (FFPE) brain tissue from seven autopsy cases: four cases of X‐ALD and three cases of MLD obtained from the archives of the Institute of Neuropathology at the University Medical Center Göttingen and of the Department of Neuropathology at the Charité Berlin. The patient data are summarized in Table 1. For X‐ALD, patients with infantile, adolescent and adult variants were analyzed, for MLD we examined two patients with the adult and one with the late infantile variant. We defined the following lesion areas for X‐ALD: normal appearing white matter, prelesional areas with changes in microglia number and immune phenotype but largely unaltered myelin and oligodendrocytes, actively demyelinating areas where major myelin breakdown occurred, and early and advanced gliotic areas characterized by progressive astrocytic scarring. In MLD prelesional areas as described above, transition areas, defined by a characteristic enlarged phagocyte morphology and early and advanced gliotic areas were distinguished. In cases of very advanced disease, the entire white matter was demyelinated and dominated by fibrous astrogliosis. These cases were classified as containing predominantly late lesion areas (early and advanced gliotic areas). Cases in early disease stages, in contrast, showed subcortical white matter without overt myelin alterations, and revealed different areas of lesion development with a gradual increase in tissue pathology. These cases with evolving lesion areas (normal appearing white matter, prelesional, actively demyelinating and transition areas) as well as scar tissue areas (early and advanced gliotic areas) were classified as containing early and late stage lesion areas (Table 1). Tissue of 14 cases without confounding CNS pathology, aged between 8 and 65 years, served as controls (Table 1). The study was approved by the ethical review committee of the University Medical Center Göttingen (#24/4/15).
Table 1.
Clinical patient data
| Case | Diagnosis | Sex | Age (ys), disease variant | Predominant lesion area |
|---|---|---|---|---|
| LD1 | X‐ALD | M | 9, juvenile | Early and late lesion areas |
| LD2 | X‐ALD | M | 16, adolescent | Late lesion areas |
| LD3 | X‐ALD | M | 28, adult | Late lesion areas |
| LD4 | X‐ALD | M | 55, adult | Early and late lesion areas |
| LD5 | MLD | F | 2, late infantile | Early and late lesion areas |
| LD6 | MLD | n. k. | n. k., adult | Late lesion areas |
| LD7 | MLD | n. k. | n. k., adult | Early and late lesion areas |
| C1 | Control | M | 8 | NWM |
| C2 | Control | F | 11 | NWM |
| C3 | Control | M | 13 | NWM |
| C4 | Control | M | 26 | NWM |
| C5 | Control | F | 38 | NWM |
| C6 | Control | F | 38 | NWM |
| C7 | Control | F | 39 | NWM |
| C8 | Control | F | 40 | NWM |
| C9 | Control | M | 46 | NWM |
| C10 | Control | F | 57 | NWM |
| C11 | Control | M | 58 | NWM |
| C12 | Control | M | 60 | NWM |
| C13 | Control | M | 62 | NWM |
| C14 | Control | M | 65 | NWM |
LD: leukodystrophy case; C: control case; X‐ALD: X‐linked adrenoleukodystrophy; MLD: metachromatic leukodystrophy; M: Male; F: Female; n. k.: not known; ys: years; NWM: normal white matter.
2.2. Histology and immunohistochemistry
All sections were stained with haematoxylin and eosin (HE) and Luxol fast blue/periodic‐acid Schiff (LFB/PAS) and Bielschowsky silver‐impregnated according to standard procedures to assess cellularity, myelin, and axonal density, respectively. Immunohistochemistry (IHC) was performed with primary antibodies against proteolipid protein (PLP, kind gift of Dr. Klaus‐Armin Nave, Göttingen, Germany, 1:500), myelin basic protein (MBP, Abcam, Cambridge, UK, ab7349, 1:200) myelin associated glycoprotein (MAG, Abcam, ab89780, 1:500), myelin oligodendrocyte protein (MOG, purified in our laboratory), and CNPase (CNP, Biolegend, San Diego, CA, USA, SMI‐91, 1:200) for visualization of myelin. Tubulin polymerization promoting protein (TPPP/p25, Abcam, ab92305, 1:500) and breast carcinoma amplified sequence 1 (BCAS1, Santa Cruz, Dallas, TX, USA, sc‐136342, 1:500) IHC was performed for visualization of oligodendrocytes. Subsets of phagocytes were detected by primary antibodies against ionized calcium‐binding adaptor molecule 1 (Iba1, Wako Pure Chemical Industries, Tokyo, Japan, 019‐19741, 1:500), transmembrane protein 119 (Tmem119, Merck, Darmstadt, Germany, HPA051870, 1:250), P2y12 receptor (P2ry12, Novusbio, Centennial, CO, USA, NBP2‐33870, 1:200) and CD68 equivalent clone Ki‐M1P (1:5000, Radzun et al., 1991). Antibodies against chemokine receptor 2 (CCR2, Abcam, ab32144, 1:4000) and migration inhibitor factor protein 14 (MRP 14, OriGene Technologies, Rockville, MD, USA, BM4026B, 1:500) were used to identify inflammatory monocytes and recently invaded peripheral phagocytes. Cell proliferation was investigated using primary antibodies against Ki67 (Agilent, Santa Clara, CA, USA, M7240, 1:200). Caspase 1 (Abcam, ab54932, 1:100) and activated caspase 3 (BD Biosciences, San Jose, CA, USA, 559565, 1:150) IHC was performed to identify cells undergoing pyroptosis or apoptosis. IHC for amyloid precursor protein (APP, Merck, MAB 348, 1:1000) served as an indicator of acute axonal damage. Glial fibrillary acidic protein (GFAP, Agilent, Z0334, 1:1000) IHC helped to determine the extent of cellular and fibrous astrogliosis. Biotinylated secondary antibodies (Merck, Jackson Immuno Research, Ely, UK, and DCS Innovative diagnostic systems, Hamburg, Germany), peroxidase‐conjugated avidin, and diaminobenzidine (DAB, Merck) were used for immunohistochemistry. Fluorescence‐labeled secondary antibodies (Alexa555‐conjugated goat anti‐mouse IgG, 1:200, and Alexa488‐conjugated goat anti‐rabbit IgG, 1:200, Molecular Probes, Eugene, OR, USA, Life technologies, Carlsbad, CA, USA) were used for fluorescence immunohistochemistry. Terminal deoxynucleotidyl transferase (TdT) mediated deoxyuridine triphosphate (dUTP) nick end labeling (TUNEL) was used to detect DNA fragmentation as described previously (Stadelmann, Bruck, Bancher, Jellinger, & Lassmann, 1998).
2.3. Morphometric analysis
Lesion areas were selected on the basis of the characteristics described below using LFB/PAS staining and immunohistochemistry for Ki‐M1P and myelin proteins (MBP, MAG, MOG, CNPase, and PLP). Borders of specific lesion areas were outlined on sections where IHC for microglia/macrophage markers had been performed, and cell densities were determined using a 10 × 10 ocular morphometric grid at a 400× magnification (Olympus, Japan). Comparable regions of normal white matter were analyzed in age‐matched controls. Results are expressed as cells/mm2. To determine ongoing phagocytosis, Iba1 or Ki‐M1P and MBP‐double‐labeled sections were scanned for intracytoplasmic myelin fragments. Co‐expression of marker proteins on phagocytes was investigated on sections double‐labeled for Tmem119 or P2ry12 and Iba1. Fluorescently‐labeled sections were scanned using the virtual slide microscope VS 120 and visualized using the VS‐ASW software (at 200× magnification) (Olympus, Tokyo, Germany). Immunopositive cells were counted manually using Image J (v. 1.51w NIH, USA) to determine the percentage of Tmem119/Iba1 and P2ry12/Iba1 double‐positive cells.
2.4. Statistical analysis
All statistics were calculated using the GraphPad Prism 6 software (GraphPad Software, La Jolla, CA). For lesion areas identified only in one patient (normal appearing white matter and actively demyelinating area in X‐ALD and prelesional, transition and advanced gliotic areas in MLD) data are represented as mean ± standard error of the mean (SEM) computed from quantifications of randomly selected parts of the lesion areas within the indicated patient. For lesion areas found in more than one patient (prelesional area, early and advanced gliotic areas in X‐ALD and early gliotic areas in MLD) and in controls, data are represented as mean ± SEM computed from average quantifications of the different patients. Here, the number of analyzed patients is indicated. In the graphical representations, average counts from different lesion areas within the same patient are represented by partly filled symbols and without standard errors of the mean. Average counts of the entire dataset of a patient are represented by filled symbols, and SEM is given for multiple analyzed patients. In general, 10 and at least seven randomly sampled parts of a lesion area were quantified for the computation of average counts. To compare differences between cell counts in different lesion areas of the same patient, a paired two‐tailed t‐test was performed. Distributions were tested for normality by D'Agostino–Pearson's test. A p < .05 was considered statistically significant.
3. RESULTS
In the present study, we took advantage of a cohort of patients with X‐ALD and MLD and applied new and established microglia markers in combination with oligodendrocyte, myelin, axon, and astrocyte histo‐ and immunohistochemistry for a better understanding of lesion development with a particular focus on phagocyte immune phenotypes. In our X‐ALD and MLD cases we observed extensive, confluent white matter pathology extending from periventricular brain regions to the cortex, with central brain regions showing the most advanced tissue destruction. Lesion characteristics and lesion evolution are described and compared separately for the diseases.
3.1. Microglia are reduced in prelesional areas of X‐ALD
In two of our 4 X‐ALD patients only areas of chronic tissue destruction were available (leukodystrophy cases 3 and 4 [LD3 and LD4]). Here the completely demyelinated and astrogliotic scar tissue abutted the cortex and white matter was reduced to a small subcortical rim. In contrast, the most acute case available for analysis (LD1) showed large areas of normal‐appearing white matter (area NA in Figures 1a, b and 2a‐d) adjacent to the cortex. Here, the shape and distribution of Iba1+ cells were comparable to age‐matched controls. However, the density of Iba1+ cells was elevated compared with age‐matched controls (180.2 ± 14.0 cells/mm2 for X‐ALD, patient LD1 vs. 49.1 +/−10.1 cells/mm2 for age‐matched controls [n = 3], Figure 1d), and immunoreactivity to Ki‐M1P (CD68 equivalent), highlighting the lysosomal compartments of monocytes, macrophages, and activated microglia, was clearly increased in X‐ALD compared with controls. Nevertheless, we found co‐expression of Tmem119 and P2ry12 on all Iba1+ cells in the normal appearing white matter (Figure 3a–c, P2ry12). Mature oligodendrocytes (TPPP/p25 IHC), myelin (LFB and myelin protein IHC) and axons (Bielschowsky silver impregnation) were not apparently altered in this region. Microglia located directly at the border to the next adjacent region towards the lesion center showed a slightly activated morphology including enlarged cell bodies and fewer and thickened processes (Figure 1a, b).
Figure 1.
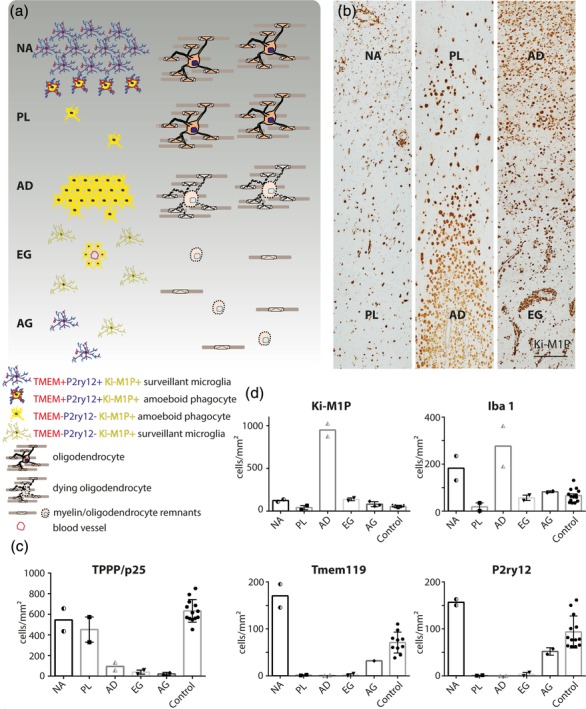
Lesion evolution in X‐ALD. (a) Schematic representation of phagocyte immune phenotypes and density in relation to myelin and oligodendrocyte pathology. NA = normal appearing white matter; PL = prelesional area; AD = actively demyelinating area; EG = early gliotic scar; AG = advanced gliotic scar. Left: Morphology and immune phenotype of Ki‐M1P+ (=CD68 equivalent) phagocytes. P2ry12 and Tmem119 are largely absent in areas PL, AD and EG but are re‐expressed in AG. Right: Oligodendrocyte and myelin alterations start in PL with condensed nuclei observed in some cells. However, cell death and reduction of cell density and myelin are not observed until AD. (b) Patient tissue (LD1) stained with Ki‐M1P. The respective lesion areas are highlighted. Scale bar: 250 μm. Quantification of (c) TPPP/p25+ mature oligodendrocytes and (d) phagocytes expressing Ki‐M1P, Iba1, Tmem119, and P2ry12 in the different lesion areas. Half‐filled symbols represent average cell counts from different lesion areas within one patient (areas NA, AD [LD1]). Filled symbols represent average cell counts computed from all quantifications of the respective marker in a patient (area PL, EG, AG; controls). The values are cells/mm2
Figure 2.
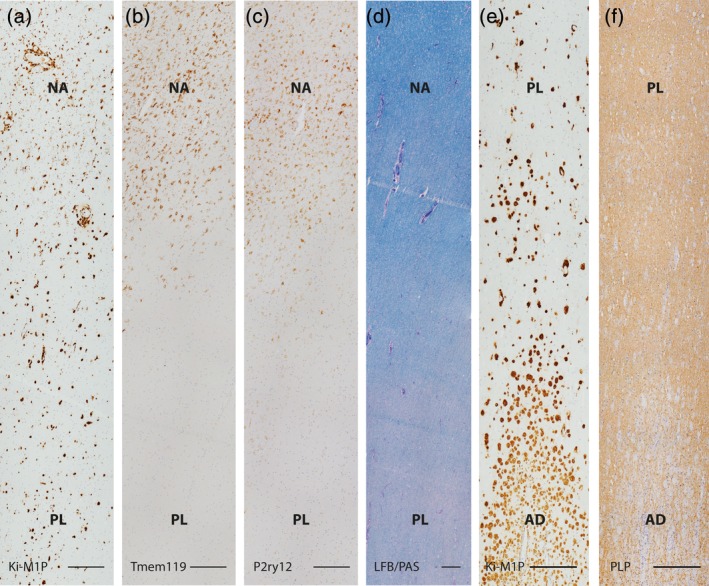
Comparison of marker expression in early X‐ALD lesion areas. (a) Ki‐M1P positive phagocytes in normal appearing white matter and prelesional area as shown in the first panel of Figure 1b. Serial sections of the same area stained for (b) Tmem119, (c) P2ry12 and, in lower magnification, for (d) myelin lipids (LFB/PAS). (e) Lesion area shown in the second panel of Figure 1b depicting invading Ki‐M1P+ phagocytes in prelesional and actively demyelinating areas in comparison to myelin alterations on serial section of the same region (PLP, f). Note the complete loss of Tmem119 and P2ry12 expression in the prelesional area (b, c) and a progressive decrease in LFB staining intensity from the normal appearing white matter to the prelesional area (d). In contrast, the intensity of PLP IHC in the prelesional area is not reduced (f). Scale bar: 250 μm
Figure 3.
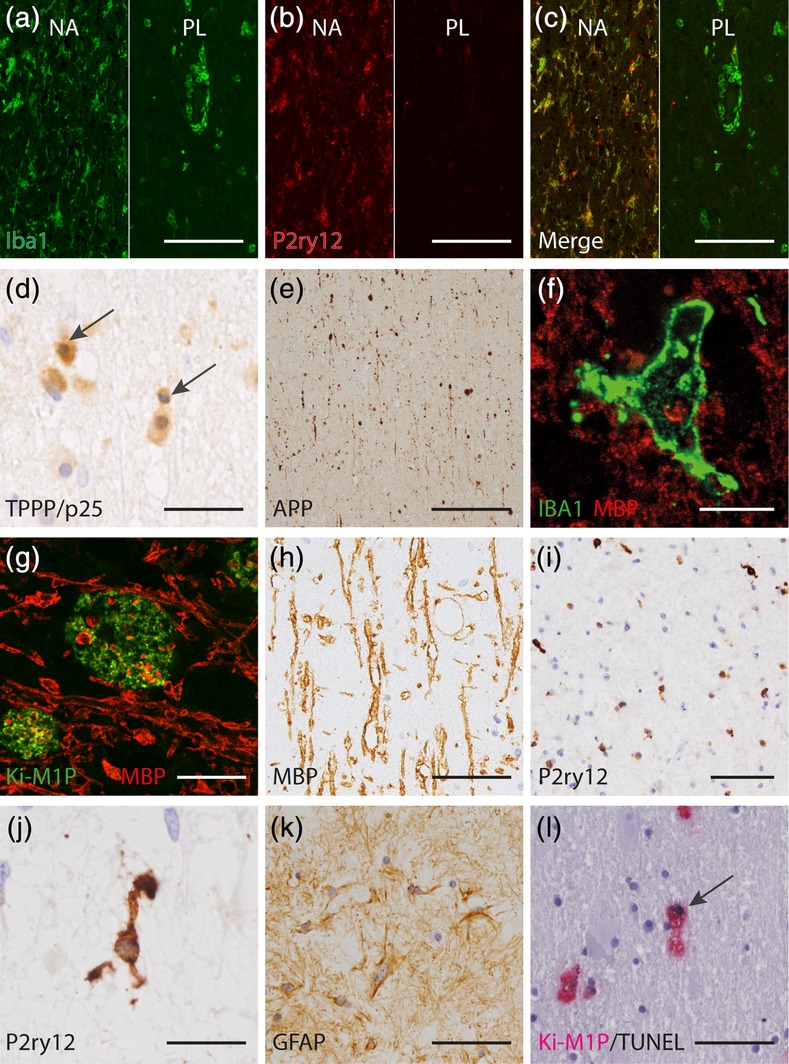
Microglia and oligodendrocyte pathology in X‐ALD. (a–c) Downregulation of homeostatic microglia markers and loss of microglia in the prelesional area compared with the normal appearing white matter (Iba1 [green], P2ry12 [red]). Left: normal appearing white matter (NA) with ramified and evenly distributed surveillant microglia expressing P2ry12. Right: prelesional area (PL) with some P2ry12‐negative amoeboid phagocytes enriched in the perivascular space and severely reduced microglia density within the tissue. (d) Condensed oligodendrocyte nuclei (TPPP/p25 IHC, arrow) and APP+ axonal profiles (APP, e) in the prelesional area of X‐ALD. (f) Recent myelin phagocytosis as evidenced by MBP+ intracellular inclusions in a rare Iba1+ cell in the prelesional area (MBP [red], Iba1 [green]). (g) Ki‐M1P+ phagocytes with intracellular MBP+ inclusions in the actively demyelinating area (MBP [red], Ki‐M1P [green]). (h) Distended, coarse and in part vacuolated myelin remnants in an early gliotic scar (MBP). (i, j) Advanced gliotic scar with microglia expressing the homeostatic marker P2ry12. Cells were evenly distributed, but with fewer and shorter processes compared with the normal appearing white matter and control tissue. (k) Mesh of reactive astrocytes (GFAP) in an advanced gliotic scar. (l) TUNEL+ phagocyte in the prelesional area in X‐ALD (arrow, TUNEL [black], Ki‐M1P [red]). Scale bar: a–c, i: 100 μm; d, h, k, l: 50 μm; e: 200 μm; f: 12,5 μm; g, j: 20 μm
Bordering the normal appearing white matter, we observed a prelesional region with a striking reduction in the number of phagocytes (area PL in Figures 1a, b and 2a‐c). Ki‐M1P+ and Iba1+ cells were mainly observed in the perivascular space and only rarely found within the tissue (18.4 ± 16.8 Iba1+ cells/mm2, [n = 2] Figure 1b, d). Tmem119 and P2ry12 expression was completely absent from the few remaining cells, and all were of amoeboid phenotype (Figure 2b [Tmem119], Figure 2c [P2ry12], Figure 3a–c, [P2ry12 [red] and Iba1 [green]]). In contrast, myelin and oligodendrocytes showed only minor phenotypic alterations, though increasing in severity towards the demyelinated lesion center but not yet leading to a significant reduction in oligodendrocyte cell number (536.8 ± 37.9 TPPP/p25+ cells/mm2 in normal appearing white matter areas versus 574.1 ± 22.95 cells/mm2 in prelesional areas, patient LD1). These alterations included pale myelin lipid‐staining (LFB, Figure 2d), progressive myelin vacuolization and condensed oligodendrocyte nuclei (Figures 2f and 3d). In contrast to the preceding normal‐appearing area, many axons here showed an accumulation of APP as an early indication of impaired axonal transport (Figure 3e). Double labeling of phagocytes (Iba1) and myelin (MBP) depicted only very few Iba1+ cells, and only a minority thereof harbored MBP+ inclusions (Figure 3f).
In contrast, the adjacent actively demyelinating lesion area (area AD in Figures 1a, b and 2e,f) was characterized by an extensive but not sharply demarcated breakdown of myelin and loss of oligodendrocytes (96.8 ± 11.7 TPPP/p25+ cells/mm2 in actively demyelinating areas, patient LD1, Figure 1c). This was observed in myelin protein immunohistochemistry (MAG, MOG, CNPase, MBP, PLP; Figure 2f [PLP]) and myelin lipid (LFB) histochemistry. The myelin loss corresponded to a drastic increase in phagocyte numbers, resulting in a sea of Ki‐M1P+ cells that appeared to protrude into the tissue (949.6 ± 58.7 Ki‐M1P+ cells/mm2, patient LD1, area AD in Figure 1a,b). Most of the cells showed a foamy morphology and all were negative for Tmem119 and P2ry12. About one third of the Ki‐M1P+ cells expressed Iba1. These cells were smaller and less foamy in appearance. Here, MBP+ as well as MAG+ or MOG+ myelin debris was readily found within phagocytes (Figure 3g), indicating recent myelin phagocytosis. Axons in this area were substantially reduced in number, and APP+ axonal profiles were less frequently seen than in the prelesional area, most likely also reflecting the reduction in axon number.
3.2. Phagocytes in the demyelinated lesion center re‐adopt a surveillant microglia shape and marker profile in X‐ALD
Adjacent to this frequently large zone of active demyelination, we observed areas of intense astrocytic scarring. Early gliotic scar regions (area EG in Figure 1a,b) showed Ki‐M1P+ phagocytes that were mostly amoeboid in shape and often enriched within the perivascular spaces. In this region and even more in fully demyelinated, advanced gliotic scar areas (AG in Figure 1a) of patients in advanced disease stages, phagocyte numbers progressively returned to levels of normal‐appearing white matter and age‐matched controls (81.3 ± 17.4 Ki‐M1P+ cells/mm2 in advanced gliotic areas [n = 3] vs. 59.2 +/− 6.3 cells/mm2 in age‐matched controls [n = 6]). Remnants of myelin in early gliotic scar areas were distended and vacuolized, and we did not find any evidence of remyelination in this region (Figure 3h [MBP]).
In the advanced gliotic scar there were less amoeboid phagocytes in the perivascular space compared with early gliotic regions, and cells within the tissue adopted a slender morphology and were much more evenly distributed (Figure 3i [P2ry12] and Figure 1a, AG). However, these cells appeared morphologically altered compared with normal appearing and control white matter microglia, with fewer and shorter branches (Figure 3j [P2ry12]) and a persistently strong Ki‐M1P immunolabeling. Interestingly, these cells expressed the homeostatic microglia markers Tmem119 and P2ry12 that were absent from other lesional and prelesional areas. The fully demyelinated and most advanced lesion areas were dominated by a mesh of reactive astrocytes (Figure 3k [GFAP]). This relatively loose scar tissue contained very few scattered axons, myelin sheaths and oligodendrocyte cell bodies that were severely reduced in number (22.4 ± 9.6 TPPP/p25+ cells/mm2, [n = 2], Figure 1c).
3.3. Phagocytes adopt an amoeboid morphology very early in MLD lesion development
Myelin histo‐ and immunohistochemistry in the acutest case of MLD (LD7) revealed a large area in the subcortex and adjacent white matter that appeared morphologically unaltered (LFB and myelin protein IHC, Figure 5m [MBP]). However, when we examined microglia within this region (Ki‐M1P IHC), we found groups of activated, amoeboid cells between surveillant microglia even in the seemingly least affected subcortical white matter parts. The total number of Ki‐M1P+ phagocytes was increased in this area as compared with white matter of age‐matched controls (247.2 ± 21.2 Ki‐M1P+ cells/mm2, patient LD7 vs. 52.92 ± 4.32 cells/mm2 in controls [n = 11], Figure 4d). Therefore, we classified this area as prelesional (and not normal appearing white matter, area PL in Figure 4a,b). Surveillant microglia that were nevertheless numerous in the white matter adjacent to the cortex expressed Tmem119 and P2ry12 (Figure 5a [P2ry12]). Within the prelesional region the percentage of amoeboid Ki‐M1P+ cells increased continuously towards the center of the lesion until ramified microglia had completely disappeared. While progressively adopting an amoeboid shape, Ki‐M1P+ cells downregulated and finally lost microglia markers, including Iba1. The frequency of co‐expression on phagocytic cells in the prelesional area was comparable for Iba1 and Tmem119 (92.1 ± 5.1% of Iba1+ cells expressed Tmem119, patient LD7), but lower for P2ry12 (37.4 ± 8.5% of Iba1+ cells expressed P2ry12, patient LD7). It is noteworthy that we found P2ry12 expressed in a small proportion of amoeboid cells (Figure 5b).
Figure 5.
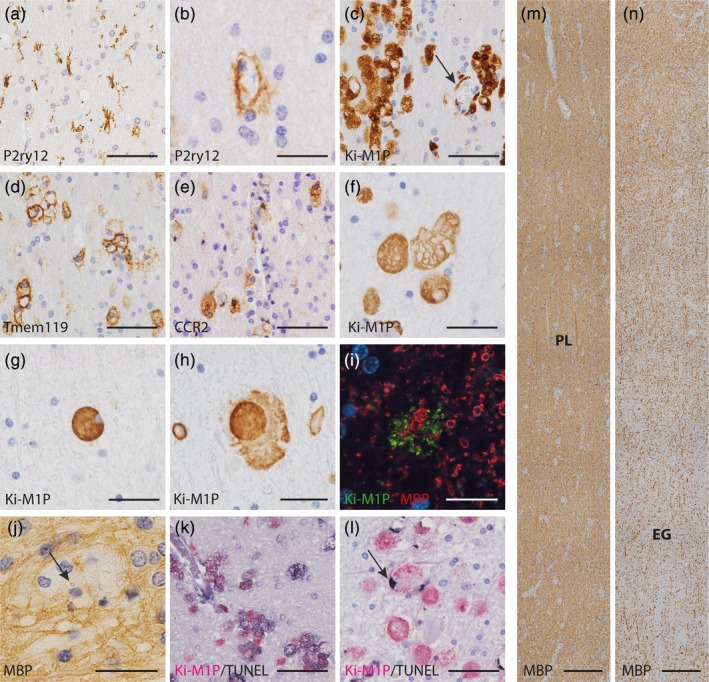
Microglia and oligodendrocyte pathology in MLD. (a) Exemplary P2ry12+ surveillant microglia and P2ry12+ amoeboid phagocytes (b) in the prelesional area. (c) Clusters of Ki‐M1P+ amoeboid phagocytes in the prelesional area; note that some but not all clusters were associated with vessels (arrow). Cells in clusters partly expressed Tmem119 (d) and partly CCR2 (e). (f–h) Morphological changes in Ki‐M1P immunostained phagocytes in prelesional, transition and early gliotic areas. (f) Enlarged intracellular vesicles, putative lysosomes, lined by Ki‐M1P‐IHC in prelesional areas. In contrast, in the transition area, lysosomal compartments lost their demarcation (g), and phagocytes appeared leaky and disintegrated in early gliotic scar areas (h, see also inset Figure 4b). (i) Indication of recent myelin phagocytosis before phagocyte lysis in early gliotic area. Ki‐M1P+ cell remnants (green) were clustered around MBP+ myelin fragments (red). (j) MBP+ inclusions in phagocytes in prelesional areas (MBP, arrow). (k) TUNEL+ cell death of phagocytes in prelesional area (TUNEL [black], Ki‐M1P [red]) with diffuse cytoplasmic stain. In contrast, in the early gliotic scar area TUNEL reactivity was mostly restricted to the nucleus (l, arrow). (m, n) Myelin diminution in MLD occurred without clear demarcation between prelesional and early gliotic areas (MBP). Lesion areas are indicated on the two consecutive tile scans that show serial sections of tissue depicted in the first two panels in Figure 4b. Scale bar: a, c, d e, k l: 50 μm; b, f–h, j: 25 μm; i: 12.5 μm; m, n: 250 μm
Figure 4.
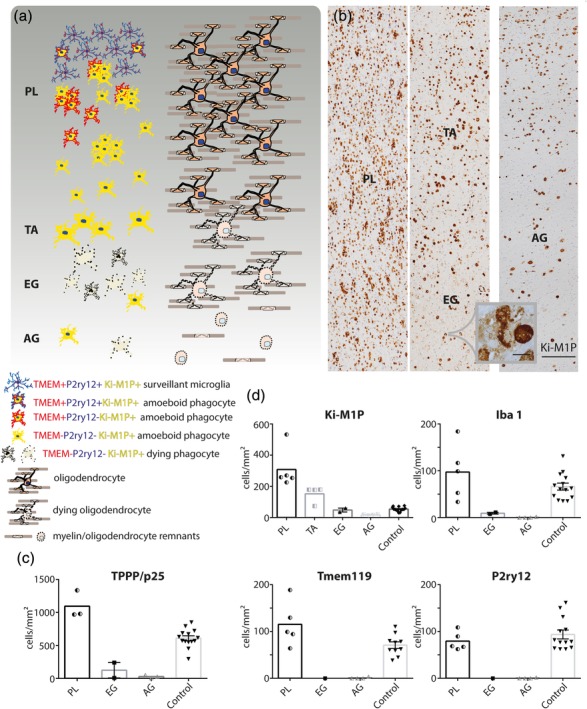
Lesion evolution in MLD. (a) Schematic representation of phagocyte immune phenotypes and density in relation to myelin and oligodendrocyte pathology. Extended areas of normal appearing white matter, with completely unaltered microglia, were not found in the cases studied. PL = prelesional area; TA = transition area; EG = early gliotic scar; AG = advanced gliotic scar. Left: Overall morphology, size, immune phenotype, and density of phagocytes with indication of membrane destruction and cell death. Right: Corresponding oligodendrocyte and myelin pathology. Oligodendrocyte density was elevated in PL, and, similar to myelin, progressively decreased towards later lesion stages. (b) Patient tissue (LD7) stained with Ki‐M1P. The rightmost panel shows a later stage lesion from a second patient (LD6). Lesion areas are indicated, scale bar: 250 μm. Inset: Phagocyte breakdown in area EG, scale bar: 20 μm. Quantification of (c) TPPP/p25+ mature oligodendrocytes and (d) phagocytes expressing Ki‐M1P, Iba1, Tmem119, and P2ry12 in the different lesion areas. TA was very small and only well discernible in Ki‐M1P IHC; therefore, no quantitation of Iba1, Tmem119, and P2ry12 was performed. Half‐filled symbols represent average cell counts from different lesion areas within one patient (areas PL, TA [LD7]; area AG [LD6]). Filled symbols represent average cell counts from different patients (area EG; controls). The values are cells/mm2
Amoeboid cells within the prelesional area were often grouped in clusters (Figure 5c [Ki‐M1P]). The cellular origin of these clusters was difficult to determine. Although many clusters were in the vicinity of vessels (Figure 5c, arrow), this was clearly not the case for all of them, and some of the phagocyte clusters were immunoreactive for Tmem119 (Figure 5d). However, a proportion of phagocytes, especially around vessels, expressed CCR2 (Figure 5e) a marker of inflammatory monocytes, while no expression of MRP14, indicating very recent monocyte invasion, was found. All cells were negative for Ki67 IHC, rendering recent cell division unlikely. Amoeboid phagocytes in this region showed large intracellular vesicles that were lined by Ki‐M1P immunohistochemistry (Figure 5f). The size of these lysosomal vesicles increased when approaching the border to the next, more severely affected region, and cells were often irregularly bulged by them.
3.4. Phagocyte lysis and loss occurs in more advanced lesion areas of MLD
Moving further towards the lesion center, we observed a small transition area (area TA in Figure 4a,b) where Ki‐M1P+ cells showed characteristic alterations in morphology with increased cell volume and often strong PAS staining. The aforementioned intracytoplasmic vesicular structures were not observed and instead, Ki‐M1P IHC showed a diffuse and rather homogeneous cytoplasmic labeling (Figure 5g). The density of phagocytes in this transition area was reduced compared with lesion areas further away from the lesion center (149.3 ± 11,0 Ki‐M1P+ cells/mm2, patient LD7, Figure 4d).
Even closer to the lesion center in the early gliotic scar regions (EG in Figure 4a,b), lesions were characterized by an extensive decay of Ki‐M1P+ phagocytes. We observed only few intact cells compared with preceding, more peripherally located lesion areas (48.3 ± 17.8 Ki‐M1P+ cells/mm2 [n = 2], Figure 4d) between remnants of phagocytes with condensed nuclei and cytoplasmic leakage apparent in PAS histo‐ and Ki‐M1P immunohistochemistry, or even dissolving into diffuse nucleus‐free material in the tissue (Figures 4b and 5h [Ki‐M1P]). Interestingly, we found evidence here of recent myelin phagocytosis before cell dissolution, since phagocyte remnants were often centered around myelin debris (Figure 5i). In the case with the most progressed tissue damage and advanced gliotic scarring (AG in Figure 4a,b, right panel), the phagocyte remnants were mostly absorbed, and only few intact amoeboid cells remained scattered throughout the tissue. The number of phagocytes progressively declined during lesion evolution in MLD to levels far below those of age‐matched controls in the advanced gliotic scar (17.2 ± 3.1 Ki‐M1P+ cells/mm2 in advanced gliotic areas, patient LD6 vs. 53 +/− 4.3 cells/mm2 in age‐matched controls [n = 11], Figure 4d). Remaining phagocytes in scar regions (early and advanced gliotic areas) showed exclusively an amoeboid morphology. In contrast to X‐ALD, we were not able to identify an area where phagocytes returned to a homeostatic marker expression, even in very advanced demyelinated and gliotic regions.
The sequence of lesion evolution in MLD appeared less orderly than in X‐ALD, with patches of earlier lesion stages interspersed between more advanced lesion areas. Moreover, in the vicinity of vessels in early gliotic regions, a higher density of phagocytes with a more ramified shape and rare expression of homeostatic markers was observed.
3.5. Myelin and oligodendrocyte pathology in MLD
In contrast to the clearly discernible border of myelin degeneration in X‐ALD, in MLD, demyelination appeared rather as a continuous diminution of myelin density, starting in late prelesional regions and continuing into early gliotic scar areas. The above‐mentioned transitions in phagocyte morphology between prelesional and early gliotic areas could only be roughly traced in LFB staining, and even less so with myelin IHC (including MAG and CNP, Figure 5m,n [MBP]). Already in prelesional areas we found evidence of ongoing myelin phagocytosis, with intracellular MBP+ inclusions in phagocytes (Figure 5j). However, myelin density was not relevantly altered here. Interestingly, the density of mature oligodendrocytes (TPPP/p25) was slightly increased in the prelesional area compared with age‐matched controls (1095 ± 45.3 TPPP/p25+ cells/mm2 in MLD, patient LD7 vs. 620.4 ± 34.5 cells/mm2 in controls [n = 11], Figure 4c). However, on a centripetal lesion axis, oligodendrocyte numbers progressively declined, eventually reaching levels comparable to advanced gliotic lesion areas in X‐ALD (28.6 ± 5.2 TPPP/p25+ cells/mm2 in MLD advanced gliotic areas, patient LD6). In the early and advanced gliotic scar areas, both myelin and axons were severely reduced. There were no apparent signs of remyelination in early or advanced gliotic areas.
3.6. Characterization of cell death in different lesion areas
In both leukodystrophies we found a clear reduction in phagocyte density in specific lesion areas. We were interested to detect whether this was due to phagocyte death. TUNEL staining, initially viewed as an indicator of apoptosis, appears to be sensitive to different forms of newly discovered programmed cell death pathways (Tonnus & Linkermann, 2017). We therefore applied the TUNEL method as a screening tool of phagocyte cell death in combination with Ki‐M1P‐immunohistochemistry in different lesion areas. In addition, to dissect which cell death pathways were activated in the respective diseases we applied antibodies against caspase 1 for caspase 1‐dependent pyroptosis and active caspase 3 for classical apoptosis.
In X‐ALD we did not find relevant signs of TUNEL‐sensitive cell death in the normal appearing white matter. In contrast, many phagocytes harbored DNA fragmentation in the prelesional area (52.4 ± 11.8% of Ki‐M1P+ cells in prelesional area, patient LD1), but only very few oligodendrocytes or astrocytes, situated mostly at the border to the next following area, were TUNEL+. Staining was often rather diffuse, including parts of the cytoplasm. Cells were, however, negative for caspase 1 and active caspase 3 IHC, and the nucleus was typically not condensed (Figure 3l (Ki‐M1P [red]/TUNEL [black])). These observations suggest that the reduction in microglia density in the prelesional area in X‐ALD is largely due to microglia death that precedes the decay of oligodendrocytes.
In MLD, in the prelesional areas, moving towards the center of the lesion, progressively more cells labelled with TUNEL (47 ± 7.5% of Ki‐M1P+ cells in late prelesional area, patient LD7). Here, the staining pattern in Ki‐M1P+ phagocytes was diffuse and cytoplasmic and there was hardly any labeling of oligodendrocytes or astrocytes (Figure 5k). In the transition area we found much fewer TUNEL+ cells.
In the early gliotic scar areas, however, we again observed many TUNEL+ phagocytes, but with a different phenotype. Cells showed TUNEL reactivity restricted to the nucleus with clear, TUNEL‐negative cytoplasm (Figure 5l, nuclear TUNEL staining in 36.4 ± 7,12% of Ki‐M1P+ cells, cytoplasmic TUNEL staining in 5.6 ± 3.5% of Ki‐M1P+ cells in early gliotic areas, patient LD7). Caspase 1 and active caspase 3 immunoreactivity were not detected in these cells. Here in early gliotic areas, in addition many oligodendrocytes and astrocytes were TUNEL+.
4. DISCUSSION
A central issue in research on microglia and monocytes in the brain is the enormous plasticity of these cells. It is thus a considerable challenge to define the specific molecular signatures with which brain phagocytes can be allocated to cellular subcategories, such as activation status (Hanisch & Kettenmann, 2007; Martinez & Gordon, 2014), stage of maturation or aging (Butovsky et al., 2014; Matcovitch‐Natan et al., 2016; Streit, Xue, Tischer, & Bechmann, 2014), and location and origin (Brendecke & Prinz, 2015; Gomez Perdiguero et al., 2015; Prinz & Priller, 2014; Silvin & Ginhoux, 2018; Tay, Hagemeyer, & Prinz, 2016). Classic microglia markers such as Iba1 (Ito et al., 1998) are expressed in a proportion of invading monocytes as well and are therefore unspecific as to origin. In addition, we observed an early diminution of Iba1 in phagocytes in MLD. This could indicate that under certain conditions, Iba1 is downregulated in activated, phagocytosing or damaged microglia in the human brain and is therefore not fully sensitive to detect all microglia cells. New microglia markers such as the purinergic receptor P2ry12 and the transmembrane protein 119 (Tmem119) have recently emerged as important tools for the identification of brain‐resident microglia in mice and humans (Bennett et al., 2016; Butovsky et al., 2014; Mildner, Huang, Radke, Stenzel, & Priller, 2017; Zrzavy et al., 2017; Zrzavy et al., 2017). Experimental manipulations, however, have demonstrated that their expression is not only dependent on origin, but also on activation status, age and environment (Bennett et al., 2016; Bohlen et al., 2017). Moreover, it was demonstrated that phagocytes transplanted from other tissues can express these markers after engraftment of the brain (Bennett et al., 2018), leading to ambiguity in their interpretation in the disease context.
Importantly, in our study these new markers nevertheless enabled the detection and demarcation of early lesion areas in X‐ALD and MLD that were not discernible when applying established phagocyte markers, such as Ki‐M1P or Iba1, or by relying on myelin and oligodendrocyte phenotype alone. In early X‐ALD surveillant microglia in the comparatively large areas of normal appearing white matter did not significantly downregulate the homeostatic marker proteins Tmem119 and P2ry12. In adjacent prelesional areas we observed, apart from substantial microglia reduction, phagocytes that completely lost Tmem119 and P2ry12 expression. The prelesional area was sharply separated from normal appearing white matter by these alterations in phagocyte immune‐phenotype. In MLD, in contrast, there was hardly any normal appearing white matter without interspersed groups of amoeboid microglia that downregulated homeostatic markers and Iba1 and showed some evidence of myelin phagocytosis. This indicates a widespread and diffuse microglia affection in MLD in comparison to X‐ALD.
The most striking result of our study, however, was the drastic reduction in and death of microglia in certain evolving lesion areas in X‐ALD and MLD that exceeded and preceded oligodendrocyte decay and overt myelin degeneration. Interestingly, the phenotypes of cell death were intrinsically different among the diseases and between lesion stages.
In regard to the early reduction of microglia in prelesional areas in X‐ALD, we found that dying cells were TUNEL+, favoring a programmed cell death pathway. Evidence is provided by previous pathological observations (Eichler et al., 2008) that microglia loss occurs early in the disease course in X‐ALD. Furthermore, Eichler et al. showed toxic effects of locally applied VLCFAs known to accumulate in X‐ALD brain tissue, on microglia. In their study signs of apoptosis with detection of active caspase 3 in the mouse model as well as in human tissue of cerebral X‐ALD were found. Another report recently demonstrated that intraparenchymal injections of 25‐hydroxycholesterol (25OH) into the mouse brain lead to an activation of inflammasomes and caspase 1 via NLRP3, followed by an increase in IL‐1b and recruitment of phagocytes, indicating ongoing pyroptosis. 25OH was shown to be elevated in induced pluripotent stem cells derived from patients suffering from the cerebral form of X‐ALD (Jang et al., 2016). In our study, we did not observe condensed and fragmented phagocyte nuclei typical of apoptosis in the prelesional area. TUNEL+ phagocytic cells were not labelled by either active caspase 3 or caspase 1 IHC. While this clearly may be due to technical limitations in formalin‐fixed paraffin‐embedded autopsy material or capase‐1 independent pyroptosis‐pathways, we doubt that pyroptosis, a proinflammatory type of cell death, is likely a major pathway occurring in the earliest phase of X‐ALD, where no immune cell infiltration is yet apparent. Using magnetic resonance perfusion imaging in cerebral X‐ALD, it was possible to identify areas of reduced perfusion, probably corresponding to prelesional areas, in front of the contrast enhancing lesion edge (actively demyelinating area, Musolino, Rapalino, Caruso, Caviness, & Eichler, 2012). Importantly, the spread of contrast enhancement into this prelesional area on magnetic resonance imaging occurs on a time scale of months rather than days, as does clinical deterioration (Loes et al., 2003; Melhem, Loes, Georgiades, Raymond & Moser, 2000; Musolino et al., 2012). This may indicate that the pathology leading to microglia cell death in the prelesional area and the concomitant peripheral phagocyte and lymphocyte invasion in the next adjacent actively demyelinating area develop slowly. The literature (Eichler et al., 2008; Loes et al., 2003; Melhem et al., 2000; Musolino et al., 2012) and the present findings revealing large prelesional areas of reduced phagocyte densities thus suggest that microglia cell death in the early evolving lesions in X‐ALD does not result in a significant immediate recruitment of phagocytes, as would be expected in pyroptosis. Instead, immunologically silent or inefficient pathways seem to take place, and substantial phagocyte invasion as well as major tissue destruction only occur in later stages of lesion formation (actively demyelinating area). Potentially, the findings of Jang et al. may apply to these later lesion stages or concur with a tissue microenvironment, where inflammatory cascades are transiently blocked and peripheral cell recruitment is inhibited. In this context it should be noted that the authors report that in vitro 25OH treatment of microglia in the absence of lipopolysaccharides failed to induce IL‐1b secretion, thus requiring a strictly pro‐inflammatory priming signal. Further research aiming to identify the specific cascades leading to phagocyte death in early X‐ALD is crucially needed as this may help understand yet unknown triggers of lesion development, and unravel the mystery of the lacking correlation of genotype and cerebral phenotype in the disease (Berger et al., 2010; Berger, Dorninger, Forss‐Petter, & Kunze, 2016; Schluter et al., 2018).
In MLD we found the alterations in phagocyte density, morphology and immune phenotype in the different stages of lesion development were accompanied by different types of TUNEL staining. Phagocytes in prelesional areas contained many enlarged vesicles lined with Ki‐M1P (CD68), a marker typically found on lysosomal membranes. These cells showed diffuse cytoplasmic TUNEL staining, indicating a leakage of DNA fragments into the cell soma. In contrast, phagocytes in early gliotic areas showed a homogenous Ki‐M1P expression pattern in the cell soma but TUNEL staining was restricted to the nucleus. Cells often appeared leaky or even breaking up, and cellular Ki‐M1P+ remnants were spread throughout the tissue. There was a continuous and severe diminution in phagocytes across the lesion stages and towards the center of the lesion. Even though we were not able to molecularly identify the specific pathways of programmed cell death, the differences in phenotype and TUNEL pattern described here support the notion that different cascades of phagocyte decay occur in prelesional and early gliotic areas. Interestingly, a diffuse cytoplasmic TUNEL staining in ramified microglia has previously been described in the resolution phase of brain injury when cell numbers return to prior levels (Streit & Xue, 2009). In addition, on the basis of the described morphological features alone, it seems likely that in MLD, a massive enlargement of lysosomes in prelesional areas as known to occur in different lysosomal storage diseases (Lüllmann‐Rauch, 2005; Xu & Ren, 2015) is followed by a loss of lysosomal compartmentalization accompanied by disintegration of the plasma membrane and breakup of the cell. In early gliotic areas we found a clearly deficient and delayed efferocytosis of cellular remnants. While defective removal of dead phagocytes is assumed to be detrimental in other conditions such as atherosclerosis (Moore & Tabas, 2011), its reasons and significance in this disease await further clarification. In a mutant zebrafish model leading to lysosomal dysfunction, an increased volume of vacuolated lysosomes as found in MLD phagocytes had a negative impact on cell motility and clearance function (Berg et al., 2016; Shen, Sidik, & Talbot, 2016). Furthermore, it has been demonstrated that defects of lysosomes in microglia lead to the accumulation of lipofuscin and impaired phagocytic function (Safaiyan et al., 2016) and that defective lysosomal lipid degradation and removal can hinder efficient tissue regeneration (Cantuti‐Castelvetri et al., 2018). These findings support the notion that the observed lysosome dysfunction in phagocytes could be a major driver of the observed tissue pathology in MLD.
Recently it has been shown in X‐ALD that monocytes/macrophages are the metabolically most affected peripheral immune cell type (Weber et al., 2014) and that switches to an anti‐inflammatory M2 phenotype after activation are impaired (Weinhofer et al., 2018). It is assumed that this functional defect contributes to the prolonged inflammation in cerebral lesions. In line with this we found large actively demyelinating areas in our most acute case of X‐ALD (LD1). However, in advanced gliotic areas, largely devoid of myelin, cells expressing homeostatic microglia markers eventually reappeared that were comparable in number to normal‐appearing white matter and controls. The origin of these cells, which appeared slightly dysmorphic compared with normal white matter microglia, is difficult to determine. New microglia may have been generated from brain‐resident progenitors or clonally expanding multi‐potent microglia cells (Bruttger et al., 2015; Elmore et al., 2014; Huang et al., 2018; Tay et al., 2017). Another possibility is that invaded monocytes express Tmem119 and P2ry12 after differentiation into CNS‐resident macrophages as demonstrated recently after irradiation‐ and chemotherapy‐free monocyte transplantation into the brain (Bennett et al., 2018).
In MLD in contrast, phagocyte alterations, including downregulation of homeostatic markers, reduction in number and amoeboid shape persisted up to advanced gliotic lesion stages. It is not clear why the reappearance of homeostatic microglia did not happen in MLD, even in end stages when myelin loss and astrogliosis reached a comparable level in MLD and X‐ALD. We cannot rule out that the lesions examined in our study were still not advanced enough. However, the overall impression of the tissue does not make this likely. In our view, in chronic MLD lesions severe metabolic alterations of microglia or a tissue environment remaining hostile to the proliferation and differentiation but also deactivation of cells into homeostatic microglia, may lead to a long‐lasting impairment in phagocyte function. This prolonged phagocyte impediment most likely plays a central part in the pathogenic cascade leading to tissue destruction and might also contribute to a seemingly lesser effectiveness of hematopoietic cell replacement therapies. Understanding the factors involved here could be crucial for elucidating the requirements for microglia repopulation and the cellular disease pathogenesis, possibly not only of MLD.
In X‐ALD and MLD, the changes in microglia immune‐phenotype and phagocyte death in prelesional areas preceded a major destruction of oligodendrocytes and myelin. However, in X‐ALD it was shown recently that cellular stress, indicated by marked upregulation of heat shock proteins, occurs in astrocytes in prelesional regions and before active demyelination (Gortz et al., 2018). Also, it has to be noted that within prelesional areas of X‐ALD (but not MLD), we found tissue alterations such as myelin pallor and vacuolization, condensed though TUNEL‐negative oligodendrocyte nuclei, and APP‐positive axons. The methods applied here are in addition not suited to detect all subtle metabolic insults to oligodendrocytes and myelin, for example, changes in membrane lipid composition, changes in lipid raft structure or early de‐compaction of myelin, which potentially could influence microglia phenotype and disease progression (Peferoen, Kipp, van der Valk, van Noort, & Amor, 2014). Undegradable substrates in X‐ALD and MLD are mainly synthesized in oligodendrocytes and the minor myelin alterations observed in the prelesional area might already reflect a pathology leading to increased myelin turnover. Evidence accumulates that defect and aged myelin is shed from oligodendrocytes and taken up by microglia (Hill, Li, & Grutzendler, 2018; Safaiyan et al., 2016). Our findings of severe damage and cell death occurring first in microglia supports the hypothesized outsourcing of myelin degradation by oligodendrocytes in the CNS. Furthermore, we find evidence in X‐ALD and MLD that, while such a strategy might in early stages lead to the sequestration of toxic material and thereby protection of the surrounding tissue, the ensuing destruction of microglia severely drives tissue pathology.
A general drawback of postmortem autopsy studies is the need to draw conclusions about the temporal sequence of lesion evolution from spatial information of lesion areas, since only one timepoint is available. No MRI scans of any of the patients in this study were available, so the assumptions about the development of the demyelinating lesions remain in part speculative. However, in X‐ALD we were able to examine all lesion stages within one patient, which allowed a good correlation with data from prior autopsy and longitudinal MRI studies with regard to the typical evolution and spread of lesions (Eichler et al., 2009; Loes et al., 1994; Loes et al., 2003; Musolino et al., 2012; Schaumburg, Powers, Raine, Suzuki, & Richardson, 1975; van der Knaap & Valk, 2005). Moreover, in X‐ALD and MLD the observation of cellular features comparable to defined stages of CNS tissue damage in other diseases as for example, the acuteness of monocyte infiltration in necrotic brain areas and in autoimmune demyelinating diseases (Brück et al., 1995), and stage‐dependent astrocyte morphology in scar regions (Sofroniew & Vinters, 2010) provide further evidence that our spatial sequence of lesion areas indeed corresponds to a temporal order of lesion evolution.
In the last century, seminal work on MLD and X‐ALD established a first subdivision of lesions into different areas according to phagocyte and myelin morphology (Peiffer, 1959; Schaumburg et al., 1975). New microglia markers help us refine these descriptions, with surprising results. It is striking that in MLD, the loss of phagocytes is more dramatic in many lesion stages than that of mature oligodendrocytes and that in MLD and X‐ALD phagocyte death precedes the decay of oligodendrocytes. We believe this indicates a pivotal—and underestimated—role of microglia in these diseases.
In conclusion, our data are not only relevant for the histopathological staging of lesion evolution in these diseases but may also serve as a basis for further research into the specific pathology of microglia in hereditary leukodystrophies, with a focus on the nature of the initial microglia alterations and the molecular pathways leading to their demise.
ACKNOWLEDGMENTS
We appreciate the excellent technical support by Katja Schulz, Uta Scheidt, Brigitte Maruschak, Jasmin Reichl, Angela Dettmar, Olga Kowatsch, and Heidi Brodmerkel. We thank Cynthia Bunker for language editing. CGB is supported by a Ministry for Science and Culture of Lower Saxony clinician scientist program. CS is supported by the DFG, the Gemeinnützige Hertie‐Stiftung, the Deutsche MS‐Gesellschaft (DMSG) and the National MS Society USA (NMSS).
Bergner CG, van der Meer F, Winkler A, et al. Microglia damage precedes major myelin breakdown in X‐linked adrenoleukodystrophy and metachromatic leukodystrophy. Glia. 2019;67:1196–1209. 10.1002/glia.23598
REFERENCES
- Beer, M. d. , Engelen, M. , & van Geel, B. M. (2014). Frequent occurrence of cerebral demyelination in adrenomyeloneuropathy. Neurology, 83, 2227–2231. 10.1212/WNL.0000000000001074 [DOI] [PubMed] [Google Scholar]
- Bennett, F. C. , Bennett, M. L. , Yaqoob, F. , Mulinyawe, S. B. , Grant, G. A. , Hayden Gephart, M. , … Barres, B. A. (2018). A combination of ontogeny and CNS environment establishes microglial identity. Neuron, 98, 1170–1183.e8. 10.1016/j.neuron.2018.05.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bennett, M. L. , Bennett, F. C. , Liddelow, S. A. , Ajami, B. , Zamanian, J. L. , Fernhoff, N. B. , … Barres, B. A. (2016). New tools for studying microglia in the mouse and human CNS. Proceedings of the National Academy of Sciences of the United States of America, 113, E1738–E1746. 10.1073/pnas.1525528113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berg, R. D. , Levitte, S. , O'Sullivan, M. P. , O'Leary, S. M. , Cambier, C. J. , Cameron, J. , … Ramakrishnan, L. (2016). Lysosomal disorders drive susceptibility to tuberculosis by compromising macrophage migration. Cell, 165, 139–152. 10.1016/j.cell.2016.02.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, J. , Dorninger, F. , Forss‐Petter, S. , & Kunze, M. (2016). Peroxisomes in brain development and function. Biochimica et Biophysica Acta, 1863, 934–955. 10.1016/j.bbamcr.2015.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger, J. , Pujol, A. , Aubourg, P. , & Forss‐Petter, S. (2010). Current and future pharmacological treatment strategies in X‐linked adrenoleukodystrophy. Brain Pathology (Zurich, Switzerland), 20, 845–856. 10.1111/j.1750-3639.2010.00393.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bohlen, C. J. , Bennett, F. C. , Tucker, A. F. , Collins, H. Y. , Mulinyawe, S. B. , & Barres, B. A. (2017). Diverse requirements for microglial survival, specification, and function revealed by defined‐medium cultures. Neuron, 94, 759–773.e8. 10.1016/j.neuron.2017.04.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brendecke, S. M. , & Prinz, M. (2015). Do not judge a cell by its cover – Diversity of CNS resident, adjoining and infiltrating myeloid cells in inflammation. Seminars in Immunopathology, 37, 591–605. 10.1007/s00281-015-0520-6 [DOI] [PubMed] [Google Scholar]
- Brück, W. , Porada, P. , Poser, S. , Rieckmann, P. , Hanefeld, F. , Kretzschmar, H. A. , … Lassmann, H. (1995). Monocyte/macrophage differentiation in early multiple sclerosis lesions. Annals of Neurology, 38, 788–796. 10.1002/ana.410380514 [DOI] [PubMed] [Google Scholar]
- Bruttger, J. , Karram, K. , Wortge, S. , Regen, T. , Marini, F. , Hoppmann, N. , … Waisman, A. (2015). Genetic cell ablation reveals clusters of local self‐renewing microglia in the mammalian central nervous system. Immunity, 43, 92–106. 10.1016/j.immuni.2015.06.012 [DOI] [PubMed] [Google Scholar]
- Butovsky, O. , Jedrychowski, M. P. , Moore, C. S. , Cialic, R. , Lanser, A. J. , Gabriely, G. , … Weiner, H. L. (2014). Identification of a unique TGF‐beta‐dependent molecular and functional signature in microglia. Nature Neuroscience, 17, 131–143. 10.1038/nn.3599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantuti‐Castelvetri, L. , Fitzner, D. , Bosch‐Queralt, M. , Weil, M.‐T. , Su, M. , Sen, P. , … Simons, M. (2018). Defective cholesterol clearance limits remyelination in the aged central nervous system. Science (New York, N.Y.), 359, 684–688. 10.1126/science.aan4183. [DOI] [PubMed] [Google Scholar]
- Eckhardt, M. (2008). The role and metabolism of sulfatide in the nervous system. Molecular Neurobiology, 37, 93–103. 10.1007/s12035-008-8022-3. [DOI] [PubMed] [Google Scholar]
- Eichler, F. , Duncan, C. , Musolino, P. L. , Orchard, P. J. , de, O. S. , Thrasher, A. J. , … Williams, D. A. (2017). Hematopoietic stem‐cell gene therapy for cerebral adrenoleukodystrophy. New England Journal of Medicine, 377, 1630–1638. 10.1056/NEJMoa1700554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler, F. , Grodd, W. , Grant, E. , Sessa, M. , Biffi, A. , Bley, A. , … Kraegeloh‐Mann, I. (2009). Metachromatic leukodystrophy: A scoring system for brain MR imaging observations. American Journal of Neuroradiology, 30, 1893–1897. 10.3174/ajnr.A1739 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eichler, F. S. , Ren, J.‐Q. , Cossoy, M. , Rietsch, A. M. , Nagpal, S. , Moser, A. B. , … Ransohoff, R. M. (2008). Is microglial apoptosis an early pathogenic change in cerebral X‐linked adrenoleukodystrophy? Annals of Neurology, 63, 729–742. 10.1002/ana.21391 [DOI] [PubMed] [Google Scholar]
- Elmore, M. R. P. , Najafi, A. R. , Koike, M. A. , Dagher, N. N. , Spangenberg, E. E. , Rice, R. A. , … Green, K. N. (2014). Colony‐stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron, 82, 380–397. 10.1016/j.neuron.2014.02.040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gieselmann, V. (2008). Metachromatic leukodystrophy: Genetics, pathogenesis and therapeutic options. Acta Paediatrica (Oslo, Norway : 1992), 97, 15–21. 10.1111/j.1651-2227.2008.00648.x. [DOI] [PubMed] [Google Scholar]
- Gieselmann, V. , Polten, A. , Kreysing, J. , Kappler, J. , Fluharty, A. , & Figura, K. v. (1991). Molecular genetics of metachromatic leukodystrophy. Developmental Neuroscience, 13, 222–227. 10.1159/000112164 [DOI] [PubMed] [Google Scholar]
- Gomez Perdiguero, E. , Klapproth, K. , Schulz, C. , Busch, K. , Azzoni, E. , Crozet, L. , … Rodewald, H.‐R. (2015). Tissue‐resident macrophages originate from yolk‐sac‐derived erythro‐myeloid progenitors. Nature, 518, 547–551. 10.1038/nature13989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong, Y. , Sasidharan, N. , Laheji, F. , Frosch, M. , Musolino, P. , Tanzi, R. , … Eichler, F. (2017). Microglial dysfunction as a key pathological change in adrenomyeloneuropathy. Annals of Neurology, 82, 813–827. 10.1002/ana.25085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gortz, A. L. , Peferoen, L. A. N. , Gerritsen, W. H. , van Noort, J. M. , Bugiani, M. , & Amor, S. (2018). Heat shock protein expression in cerebral X‐linked adrenoleukodystrophy reveals astrocyte stress prior to myelin loss. Neuropathology and Applied Neurobiology, 44, 363–376. 10.1111/nan.12399 [DOI] [PubMed] [Google Scholar]
- Hanisch, U.‐K. , & Kettenmann, H. (2007). Microglia: Active sensor and versatile effector cells in the normal and pathologic brain. Nature Neuroscience, 10, 1387–1394. 10.1038/nn1997 [DOI] [PubMed] [Google Scholar]
- Hill, R. A. , Li, A. M. , & Grutzendler, J. (2018). Lifelong cortical myelin plasticity and age‐related degeneration in the live mammalian brain. Nature Neuroscience, 21, 683–695. 10.1038/s41593-018-0120-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang, Y. , Xu, Z. , Xiong, S. , Sun, F. , Qin, G. , Hu, G. , … Peng, B. (2018). Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nature Neuroscience, 21, 530–540. 10.1038/s41593-018-0090-8 [DOI] [PubMed] [Google Scholar]
- Ito, D. , Imai, Y. , Ohsawa, K. , Nakajima, K. , Fukuuchi, Y. , & Kohsaka, S. (1998). Microglia‐specific localisation of a novel calcium binding protein, Iba1. Brain Research. Molecular Brain Research, 57, 1–9. [DOI] [PubMed] [Google Scholar]
- Jang, J. , Park, S. , Hur, H. J. , Cho, H.‐J. , Hwang, I. , Kang, Y. P. , … Kim, D.‐W. (2016). 25‐hydroxycholesterol contributes to cerebral inflammation of X‐linked adrenoleukodystrophy through activation of the NLRP3 inflammasome. Nature Communications, 7, 13129 10.1038/ncomms13129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kidd, D. , Nelson, J. , Jones, F. , Dusoir, H. , Wallace, I. , McKinstry, S. , … Patterson, V. (1998). Long‐term stabilization after bone marrow transplantation in juvenile metachromatic leukodystrophy. Archives of Neurology, 55, 98–99. [DOI] [PubMed] [Google Scholar]
- Loes, D. J. , Fatemi, A. , Melhem, E. R. , Gupte, N. , Bezman, L. , Moser, H. W. , … Raymond, G. V. (2003). Analysis of MRI patterns aids prediction of progression in X‐linked adrenoleukodystrophy. Neurology, 61, 369–374. [DOI] [PubMed] [Google Scholar]
- Loes, D. J. , Hite, S. , Moser, H. , Stillman, A. E. , Shapiro, E. , Lockman, L. , … Krivit, W. (1994). Adrenoleukodystrophy: A scoring method for brain MR observations. American Journal of Neuroradiology, 15, 1761–1766. [PMC free article] [PubMed] [Google Scholar]
- Lüllmann‐Rauch, R. (2005). History and morphology of the lysosome In Saftig P. (Ed.), Lysosomes (pp. 1–16). Boston, MA: Eurekah.com and Springer Science+Business Media Inc. [Google Scholar]
- Mahmood, A. , Raymond, G. V. , Dubey, P. , Peters, C. , & Moser, H. W. (2007). Survival analysis of haematopoietic cell transplantation for childhood cerebral X‐linked adrenoleukodystrophy: A comparison study. Lancet Neurology, 6, 687–692. 10.1016/S1474-4422(07)70177-1 [DOI] [PubMed] [Google Scholar]
- Martinez, F. O. , & Gordon, S. (2014). The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Reports, 6, 13 10.12703/P6-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matcovitch‐Natan, O. , Winter, D. R. , Giladi, A. , Vargas, A. S. , Spinrad, A. , Sarrazin, S. , … Amit, I. (2016). Microglia development follows a stepwise program to regulate brain homeostasis. Science (New York, N.Y.), 353, aad8670 10.1126/science.aad8670 [DOI] [PubMed] [Google Scholar]
- Melhem, E. R. , Loes, D. J. , Georgiades, C. S. , Raymond, G. V. , & Moser, H. W. (2000). X‐linked adrenoleukodystrophy: The role of contrast‐enhanced MR imaging in predicting disease progression. AJNR. American Journal of Neuroradiology, 21, 839–844. [PMC free article] [PubMed] [Google Scholar]
- Mildner, A. , Huang, H. , Radke, J. , Stenzel, W. , & Priller, J. (2017). P2Y12 receptor is expressed on human microglia under physiological conditions throughout development and is sensitive to neuroinflammatory diseases. Glia, 65, 375–387. 10.1002/glia.23097 [DOI] [PubMed] [Google Scholar]
- Moore, K. J. , & Tabas, I. (2011). Macrophages in the pathogenesis of atherosclerosis. Cell, 145, 341–355. 10.1016/j.cell.2011.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Musolino, P. L. , Rapalino, O. , Caruso, P. , Caviness, V. S. , & Eichler, F. S. (2012). Hypoperfusion predicts lesion progression in cerebral X‐linked adrenoleukodystrophy. Brain: A Journal of Neurology, 135, 2676–2683. 10.1093/brain/aws206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nave, K.‐A. , & Werner, H. B. (2014). Myelination of the nervous system: Mechanisms and functions. Annual Review of Cell and Developmental Biology, 30, 503–533. 10.1146/annurev-cellbio-100913-013101 [DOI] [PubMed] [Google Scholar]
- Peferoen, L. , Kipp, M. , van der Valk, P. , van Noort, J. M. , & Amor, S. (2014). Oligodendrocyte‐microglia cross‐talk in the central nervous system. Immunology, 141, 302–313. 10.1111/imm.12163 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peiffer, J. (1959). On metachromatic leukodystrophy (Scholz type). Archiv Fur Psychiatrie Und Nervenkrankheiten, Vereinigt Mit Zeitschrift Fur Die Gesamte Neurologie Und Psychiatrie, 199, 386–416. [DOI] [PubMed] [Google Scholar]
- Peters, C. , Charnas, L. R. , Tan, Y. , Ziegler, R. S. , Shapiro, E. G. , DeFor, T. , … Krivit, W. (2004). Cerebral X‐linked adrenoleukodystrophy: The international hematopoietic cell transplantation experience from 1982 to 1999. Blood, 104, 881–888. 10.1182/blood-2003-10-3402. [DOI] [PubMed] [Google Scholar]
- Prinz, M. , & Priller, J. (2014). Microglia and brain macrophages in the molecular age: From origin to neuropsychiatric disease. Nature Reviews. Neuroscience, 15, 300–312. 10.1038/nrn3722 [DOI] [PubMed] [Google Scholar]
- Radzun, H. J. , Hansmann, M. L. , Heidebrecht, H. J. , Bodewadt‐Radzun, S. , Wacker, H. H. , Kreipe, H. , … Parwaresch, M. R. (1991). Detection of a monocyte/macrophage differentiation antigen in routinely processed paraffin‐embedded tissues by monoclonal antibody Ki‐M1P. Laboratory Investigation; A Journal of Technical Methods and Pathology, 65, 306–315. [PubMed] [Google Scholar]
- Safaiyan, S. , Kannaiyan, N. , Snaidero, N. , Brioschi, S. , Biber, K. , Yona, S. , … Simons, M. (2016). Age‐related myelin degradation burdens the clearance function of microglia during aging. Nature Neuroscience, 19, 995–998. 10.1038/nn.4325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki, Y. , Hoshi, M. , Akazawa, C. , Nakamura, Y. , Tsuzuki, H. , Inoue, K. , … Kohsaka, S. (2003). Selective expression of Gi/o‐coupled ATP receptor P2Y12 in microglia in rat brain. Glia, 44, 242–250. 10.1002/glia.10293 [DOI] [PubMed] [Google Scholar]
- Schaumburg, H. H. , Powers, J. M. , Raine, C. S. , Suzuki, K. , & Richardson, E. P. (1975). Adrenoleukodystrophy. A clinical and pathological study of 17 cases. Archives of Neurology, 32, 577–591. [DOI] [PubMed] [Google Scholar]
- Schluter, A. , Sandoval, J. , Fourcade, S. , Diaz‐Lagares, A. , Ruiz, M. , Casaccia, P. , … Pujol, A. (2018). Epigenomic signature of adrenoleukodystrophy predicts compromised oligodendrocyte differentiation. Brain Pathology, 28, 902–919. 10.1111/bpa.12595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen, K. , Sidik, H. , & Talbot, W. S. (2016). The rag‐Ragulator complex regulates lysosome function and phagocytic flux in microglia. Cell Reports, 14, 547–559. 10.1016/j.celrep.2015.12.055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silvin, A. , & Ginhoux, F. (2018). Microglia heterogeneity along a spatio‐temporal axis: More questions than answers. Glia, 66, 2045–2057. 10.1002/glia.23458 [DOI] [PubMed] [Google Scholar]
- Sofroniew, M. V. , & Vinters, H. V. (2010). Astrocytes: Biology and pathology. Acta Neuropathologica, 119, 7–35. 10.1007/s00401-009-0619-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadelmann, C. , Bruck, W. , Bancher, C. , Jellinger, K. , & Lassmann, H. (1998). Alzheimer disease: DNA fragmentation indicates increased neuronal vulnerability, but not apoptosis. Journal of Neuropathology and Experimental Neurology, 57, 456–464. [DOI] [PubMed] [Google Scholar]
- Streit, W. J. , & Xue, Q.‐S. (2009). Life and death of microglia. Journal of Neuroimmune Pharmacology, 4, 371–379. 10.1007/s11481-009-9163-5. [DOI] [PubMed] [Google Scholar]
- Streit, W. J. , Xue, Q.‐S. , Tischer, J. , & Bechmann, I. (2014). Microglial pathology. Acta Neuropathologica Communications, 2, 142 10.1186/s40478-014-0142-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tay, T. L. , Hagemeyer, N. , & Prinz, M. (2016). The force awakens: Insights into the origin and formation of microglia. Current Opinion in Neurobiology, 39, 30–37. 10.1016/j.conb.2016.04.003 [DOI] [PubMed] [Google Scholar]
- Tay, T. L. , Mai, D. , Dautzenberg, J. , Fernández‐Klett, F. , Lin, G. , Sagar, D. M. , … Prinz, M. (2017). A new fate mapping system reveals context‐dependent random or clonal expansion of microglia. Nature Neuroscience, 20, 793–803. 10.1038/nn.4547 [DOI] [PubMed] [Google Scholar]
- Tonnus, W. , & Linkermann, A. (2017). The in vivo evidence for regulated necrosis. Immunological Reviews, 277, 128–149. 10.1111/imr.12551 [DOI] [PubMed] [Google Scholar]
- van der Knaap, M. S. , & Valk, J. (2005). Magnetic resonance of myelination and myelin disorders. Berlin, Heidelberg: Springer Berlin Heidelberg. [Google Scholar]
- Weber, F. D. , Wiesinger, C. , Forss‐Petter, S. , Regelsberger, G. , Einwich, A. , Weber, W. H. A. , … Berger, J. (2014). X‐linked adrenoleukodystrophy: Very long‐chain fatty acid metabolism is severely impaired in monocytes but not in lymphocytes. Human Molecular Genetics, 23, 2542–2550. 10.1093/hmg/ddt645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinhofer, I. , Zierfuss, B. , Hametner, S. , Wagner, M. , Popitsch, N. , Machacek, C. , … Berger, J. (2018). Impaired plasticity of macrophages in X‐linked adrenoleukodystrophy. Brain: A Journal of Neurology., 141, 2329–2342. 10.1093/brain/awy127 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiesinger, C. , Eichler, F. S. , & Berger, J. (2015). The genetic landscape of X‐linked adrenoleukodystrophy: Inheritance, mutations, modifier genes, and diagnosis. Application of Clinical Genetics, 8, 109–121. 10.2147/TACG.S49590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wittke, D. , Hartmann, D. , Gieselmann, V. , & Lüllmann‐Rauch, R. (2004). Lysosomal sulfatide storage in the brain of arylsulfatase A‐deficient mice: Cellular alterations and topographic distribution. Acta Neuropathologica, 108, 261–271. 10.1007/s00401-004-0883-6 [DOI] [PubMed] [Google Scholar]
- Xu, H. , & Ren, D. (2015). Lysosomal physiology. Annual Review of Physiology, 77, 57–80. 10.1146/annurev-physiol-021014-071649 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zrzavy, T. , Hametner, S. , Wimmer, I. , Butovsky, O. , Weiner, H. L. , & Lassmann, H. (2017). Loss of 'homeostatic' microglia and patterns of their activation in active multiple sclerosis. Brain: A Journal of Neurology, 140, 1900–1913. 10.1093/brain/awx113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zrzavy, T. , Machado‐Santos, J. , Christine, S. , Baumgartner, C. , Weiner, H. L. , Butovsky, O. , … Lassmann, H. (2017). Dominant role of microglial and macrophage innate immune responses in human ischemic infarcts. Brain Pathology, 28, 791–805. 10.1111/bpa.12583 [DOI] [PMC free article] [PubMed] [Google Scholar]