

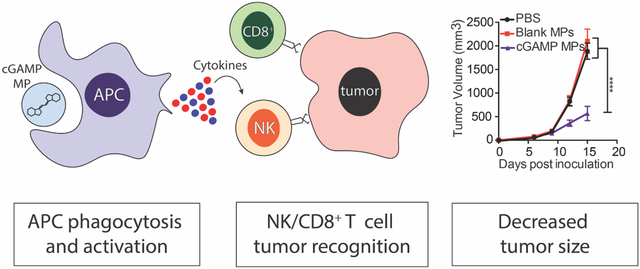
Abstract
Immunotherapies have significantly improved cancer patient survival, but response rates are still limited. Thus, novel formulations are needed to expand the breadth of immunotherapies. Pathogen associated molecular patterns (PAMPs) can be used to stimulate an immune response, but several pathogen recognition receptors are located within the cell, making delivery challenging. We have employed the biodegradable polymer acetalated dextran (Ace-DEX) to formulate PAMP microparticles (MPs) in order to enhance intracellular delivery. While treatment with four different PAMP MPs resulted in tumor growth inhibition, cyclic GMP-AMP (cGAMP) MPs were most effective. cGAMP MPs showed anti-tumor efficacy at doses 100–1,000 fold lower than published doses of soluble cGAMP in two murine tumor models. Treatment with cGAMP MPs resulted in increased natural killer cell numbers in the tumor environment. Immune cell depletion studies confirmed that NK cells were responsible for the anti-tumor efficacy in an aggressive mouse melanoma model. NK cell and CD8+ T cells were both required for early anti-tumor function in a triple negative breast cancer model. In summary, cGAMP MP treatment results in NK and T cell-dependent anti-tumor immune response.
Keywords: Acetalated Dextran, Immunotherapy, STING, Microparticle, Natural Killer cells, CD8+ T Cells
Graphical Abstract
Introduction
Cancer growth is enhanced through uncontrolled proliferation and an immunosuppressive environment1. Cancer cells often produce immune suppressive factors, such as interleukin (IL)-10 and TGF-β, as well as upregulate immunosuppressive receptors, such as programmed deathligand 1 (PD-L1). This can allow the cancer cells to escape immune surveillance and proliferate uncontrollably. An anti-tumor immune response can be activated through delivering immune stimulating molecules to overcome these immunosuppressive signals2. Recently, recombinant cytokines, such as, granulocyte-macrophage colony-stimulating factor (GM-CSF), multiple interleukins, type I interferons (IFN-I), and tumor necrosis factor (TNF) have all shown promise as cancer immunotherapies3–5. Indeed, recombinant IFNa and IL-2 have both received FDA approval as cancer immunotherapies, with IFNa being the only therapy approved for patients with high risk stage II and III melanoma3. However, there are many challenges associated with clinical application of recombinant cytokines, including short half-lives and high patient noncompliance due to negative side effects after systemic delivery6,7. To overcome some of the shortcomings of recombinant cytokines, pathogen associated molecular patterns (PAMPs) have been used to generate robust immune cell activation and endogenous cytokine production through binding and activating pathogen recognition receptors (PRRs)8. PAMPs have been shown to induce strong immune responses with vaccines9,10, as well as, generate robust immune responses against tumors11.
In order for production of downstream cytokines and activation of immune cells to occur, PAMPs must be delivered to their respective PRRs. Poly (I:C) and imiquimod bind and activate endosomal toll-like receptors (TLRs) 3 and 7, respectively12,13. Murabutide, poly (I:C), and the STING agonist 3’3’-cyclic GMP-AMP (cGAMP) bind and activate cytosolic nucleotide-binding oligomerization domain-containing protein 2 (NOD2), retinoic acid-inducible gene I (RIG-I), and STING, respectively7,14. These PAMPs have been used to activate sentinel cells leading to the production of pro-inflammatory cytokines and have been investigated in clinical trials12–15. However, due to the localization of these PRRs within the cell, large doses of soluble drug are often required. However, large quantities of STING agonists have been associated with toxicity, such as T cell apoptosis16–18. We, and many others, have shown encapsulation of PAMPs into a particle delivery system leads to improved activity19,20, however, not all encapsulation systems are equal. Liposomes are a common particle delivery system and a number of liposome formulations are FDA approved. However, liposomes can have large batch-to-batch variation and poor stability during storage21. Furthermore, common liposomal manufacturing methods are difficult to scale up to produce amounts needed clinically22. Alternatively, poly (lactic-coglycolic acid) (PLGA) polymeric microparticles (MPs), which are used in many FDA-approved products, can be made by various methods. However, these often result in low encapsulation efficiencies (EEs) for hydrophilic drugs23 and have potentially detrimental acidic byproducts that can damage both the cargo and the host cell19.
To overcome the shortcomings of these common formulations, we, and other groups have investigated other PAMP delivery systems9,24–26. We have utilized a previously published on, acetalated dextran (Ace-DEX) polymeric MP platform. Ace-DEX MPs are acid-sensitive, biodegradable, and have tunable degradation kinetics as well as pH-neutral, hydrolytic byproducts27–29. We have previously shown that various PAMPs can be encapsulated within Ace-DEX MPs by several formulation methods30–37. In particular, we have used an electrospray (ES) formulation process to formulate PAMPs into Ace-DEX MPs to improve their efficacy as an anti-infective therapy38 or as vaccine adjuvants30–32. However, the Ace-DEX cGAMP MPs have not been investigated as a cancer immunotherapy. Utilizing Ace-DEX MPs, we are able to achieve the following benefits: (a) passive targeting to antigen presenting cells (APCs) due to the micron size (~0.5–2 μm) of Ace-DEX MPs30; (b) rapid release of cargo in acidic endosomes (pH=5.0) and slower release at a neutral pH30,39; (c) cargo stability for long-term storage outside of cold chain40; (d) terminally sterilizable formulation30. In addition, the ES technique results in higher encapsulation efficiencies than other formulation methods,30 is scalable32,38,41, and results in more monodispersed particles30,32,38,41,42. We have shown previously that encapsulating PAMPs in ES Ace-DEX MPs results in higher pro-inflammatory cytokine production and immune system activation30,31.
Robust pro-inflammatory cytokine production can activate both the innate and adaptive immune responses against a tumor. With this manuscript, we will investigate the, previously unknown, anti-tumor mechanism of the Ace-DEX cGAMP MPs. CD8+ T cells and natural killer (NK) cells are the primary immune cells responsible for direct tumor killing. CD8+ T cells are a member of the adaptive immune system and recognize tumor neoantigen presentation on the major histocompatibility complex (MHC) I, leading to tumor death. NK cells are the innate counterpart to CD8+ T cells. NK cells recognize and kill cells that downregulate MHC I to evade a CD8+ response, a common phenomenon in tumors43. Both CD8+ T cells and NK cells kill target cells through the release of perforin and granzymes. Perforin creates pro-apoptotic pores in the cell membrane and granzymes triggers caspase-dependent apoptosis44. Both molecules are upregulated in the presence of pro-inflammatory cytokines45,46. Thus, we hypothesize that delivering PAMPs that promote immune activation will increase the killing potential of CD8+ T cells and/or NK cells, resulting in significantly decreased tumor size.
Here we investigated Ace-DEX MPs encapsulated PAMPs as a cancer immunotherapy. We compared the PAMP MPs to determine the optimal PAMP Ace-DEX combination. Route of administration and dose studies were also performed. Finally, we determined the cellular mechanism of the optimal PAMP MPs.
Materials and Methods
Microparticle Formulation Materials
The materials used for polymer synthesis and microparticle production were purchased from Sigma Aldrich (St. Louis, MO) unless otherwise stated. 2-ethoxy propene was purchased from Matrix Scientific (Elgin, SC). Imiquimod, 3’3’-cGAMP, murabutide, and poly(I:C) were purchased from Invivogen (San Diego, CA).
Fabrication and Quantification of Electrosprayed PAMP Microparticles
Ace-DEX was synthesized and characterized according to Kauffman et al. using dextran with a 70 kDa average molecular weight39. After synthesis, Ace-DEX was rapidly hydrolyzed in deuterium oxide with the addition of 10% v/v deuterium chloride and analyzed by 1H NMR spectroscopy (Inova 400 MHz spectrometer; Varian Medical Systems, Palo Alto, CA). The relative cyclic acetal coverage was determined to be 40 ± 3%. Ace-DEX MPs loaded with cGAMP, imiquimod, murabutide, or poly(I:C) were fabricated by using an electrohydrodynamic spraying (ES) method. Ace-DEX was dissolved in ethanol at either 20 or 30 mg/mL. cGAMP, imiquimod, murabutide, or poly(I:C) were dissolved in molecular grade water. Ace-DEX (20 mg/mL) and cGAMP or poly(I:C) (1% w/w) mixtures were dissolved within a 90:10 ethanol:water (% v/v) solvent. Ace-DEX (30 mg/mL) and imiquimod or murabutide (1% w/w) mixtures were dissolved within a 95:5 ethanol:water (% v/v) solvent. Blank MPs were fabricated by the same process using just pure molecular grade water (20 mg/mL Ace-DEX, 90:10 ethanol:water).
cGAMP loading was quantified by high performance liquid chromatography (HPLC, Agilent 1100 series, Santan Clara, CA) using a water/methanol gradient method through an Aquasil C18 column (150 mm length, 4.6 mm inner diameter, 5μm pore size, Thermo Fisher, Waltham, MA).
Imiquimod, murabutide, and poly (I:C) were quantified by dissolving MPs in dimethyl sulfoxide (DMSO). Imiquimod loading was quantified using its autofluorescence (excitation: 325 nm, emission: 365 nm)36. Murabutide loading was quantified using a fluorescamine assay (excitation: 390 nm, emission: 460 nm)32. Poly(I:C) loading was quantified using a Quant-iT OliGreen kit (Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s specifications. All three assays used a standard curve treated under the same conditions and analyzed using a microplate reader (SpectraMax M2, Molecular Devices, San Jose, CA).
Loading capacity and encapsulation efficiency of PAMPs in MPs were calculated as shown:
Physical Characterization of Electrosprayed PAMP Microparticles
The hydrodynamic diameter, polydispersity index (PDI), and zeta potential of all MPs (n=3) were measured using a Brookhaven NanoBrook 90Plus Zeta Particle Size Analyzer (Holtsville, NY).
For scanning electron microscopy (SEM) imaging, Ace-DEX MPs were resuspended in water and pipetted onto the surface of an aluminum pin stub (Ted Pella, Inc., Redding, CA). This was incubated at 100°C for 20 minutes before sputter-coating with 6 nm of AuPd. SEM images were acquired using an S-4700 scanning electron microscope. SEM images were taken of all MPs to determine morphology using an S-4700 scanning electron microscope (Hitachi High Technologies America, Schaumburg, IL).
In Vitro Studies
Bone marrow-derived dendric cells (BMDCs) were prepared as previously described30. Cells were then treated with 1 μg/mL of the respective PAMP, either soluble of encapsulated. After 24 hours, supernatants were taken and run on either TNF (BD Bioscience cat. 558534), IL-6 (BD Bioscience cat. 555240), or IFN-b (described previously30) ELISAs.
Murine Tumor Experiments
All studies were conducted in accordance with National Institutes of Health’s guidelines for the care and use of laboratory animals and approved by the Institutional Animal Care and Use Committee (IACUC) at the University of North Carolina-Chapel Hill. C57BL/6J and B6(Cg)-Tmem173tm1.2Camb/J (Tmem173−/−) mice were purchased from Jackson Laboratories (Sacramento, California). Mice were inoculated with tumors between 8–12 weeks of age. The B16F10 cell line was obtained from ATCC (Manassas, VA cat. CRL-6475) and cultured in DMEM medium with 10% fetal bovine serum, 100 U/mL penicillin, and 100 μg/mL streptomycin. Male and female mice were inoculated subcutaneously with 200,000 B16F10 cells in 50% matrigel (Corning, Corning, NY, cat. 354234) on the flank. The E0771 cell line was obtained from CH3 Biosystems (Buffalo, NY cat. 940001) and cultured in RPMI 1640 medium with 10% fetal bovine serum, 10 mM HEPES, 100 U/mL penicillin, and 100 μg/mL streptomycin. Female mice were inoculated with 750,000 E0771 cells in 50% matrigel into the mammary fat pad. Tumors were allowed to grow for six days before the first treatment, resulting in tumor volumes around 100 mm3. All MPs were injected using 1 mg/mL egg phosphatidylcholine (Avanti Polar Lipids, Alabaster, AL) in phosphate-buffered saline (PBS) to aid in MPs suspension. Mice were injected every three days, for a total of three (B16F10) or seven (E0771) injections. Treatments were delivered by either intraperitoneal (i.p), intramuscular (i.m.), intravenous (i.v.), or intratumoral (i.t) route of administration. Tumor volume was measured every three days and was calculated using the formula V=½W2*L47. Survival was reported when the tumor reached the humane endpoint, 20 mm in diameter, in accordance with the IACUC protocol.
Biodistribution
Dextran was mixed at a 1:3 ratio of 70kDa Texas-red labeled dextran to 70 kDa dextran (ThermoFisher cat. D1830). The mixture of dextran was reacted with 2-ethoxypropene overnight. Mice were inoculated with 200,000 B16F10 tumor cells on day 0. On day 10, mice were treated with 10 μg of cGAMP encapsulated in Texas-red labeled Ace-DEX polymer. Texas-red Axe-DEX was made as described above with Texas-red labeled dextran obtained from Thermo Fisher (Waltham, MA cat. D1830). Twenty-four hours after treatment, mice were sacrificed the following organs were harvested: brain, lung, liver, kidney, spleen, lymph nodes, and tumor. The organs were then imaged using the IVIS Kinetic (PerkinElmer Waltham, MA). Samples were excited for 1 second at an excitation wavelength of 570 nm and detected using the Cy5.5 emission filter. The spleen was then taken and made into a single cell suspension. The splenocytes were stained (CD45 (BV-421), CD3 (PE-Cy7), CD4 (PerCP-cy5.5), CD8 (AF700), CD11b (APC), and CD11c (Pac-Blue); Biolegend, San Diego, CA), analyzed on a LSR II (BD Biosciences, San Jose, CA) and analyzed by FlowJo software (Tree Star, Ashland, OR).
Tumor Cytokines
Mice were inoculated with 200,000 B16F10 tumor cells on day 0. On day 10, mice were treated with 10 μg of cGAMP encapsulated in Ace-DEX polymer. Three hours later, mice were sacrificed and tumors were harvested. The cOmplete EDTA-free protease inhibitor cocktail (Sigma cat. COEDTAF-RO) in PBS was added to tumor (at 4 μL/mg of tissue) to normalize volume. Tissues were homogenized and supernatants were used to run IFN-b (described previously30) and IL-6 ELISAs (BD Bioscience cat. 558534).
Flow Cytometry
Tumors were harvested from treated mice and prepared into single cell suspensions. Intracellular cytokines were stained using the intracellular staining kit (Biolegend cat. 420201) and re-stimulated using 1x PMA/ionomycin with brefeldin A (Biolegend cat. 423303). Immune cell populations were identified by flow cytometry using either an LSRII or LSRFortessa (BD Biosciences) and analyzed by FlowJo software.
T cell and NK cell Depletion
Cellular subsets were depleted as described previously48. One day before tumor inoculation, the following cell types were depleted with 400 μg of the following antibodies: NK cells with anti-NK1.1 antibody (BioXcell, West Lebanon, NH, clone PK136, cat. BE0036), CD8+ T cells with anti-CD8alpha antibody (BioXcell, clone 2.43 cat. BE0061), and CD4+ T cells with anti-CD4 antibody (BioXcell, clone GK1.5 cat. BE0003). Mice were then inoculated with either 200,000 B16F10 cells or 750,000 E0771 cells and received three or five cGAMP MP treatments, respectively. To confirm depletion, blood was collected on day 15 and prepared for flow cytometry using a red blood cell lysis/fixation buffer kit (Biolegend, cat. 422401).
Statistics
All tumor volume experiments were analyzed with a two-way ANOVA with post-hoc Tukey’s multiple comparisons. Survival studies were analyzed using survival statistics, and p-values adjusted for multiple comparisons. Comparisons between flow cytometry data were analyzed with one-way ANOVA with Post-doc Tukey’s multiple comparison test. Outliers were identified with ROUT outlier test.
For additional methods see supplementary information.
Results
Electrohydrodynamic spraying of Ace-DEX MPs results in high drug encapsulation for four different PAMPs.
Immune PAMPs cGAMP, imiquimod, murabutide, or poly(I:C) were encapsulated at ~1% w/w in the previously published Ace-DEX MPs by electrohydrodynamic spraying (ES)30,37. Scanning electron microscopy images demonstrate the formation of MPs (Fig. 1A–E). All of the encapsulated PAMP MPs were semi-spherical in shape and had a hydrodynamic diameter ranging between 0.687 and 1.12 μm (Fig. 1F). All PAMP MPs had a negative surface charge and had similar final loading (Fig. 1F). Batch-to-batch variability was assessed (Fig. 1G), as well as, storage stability (Fig. S1). Polydispersity index was measured over a 60-minute period after particles were resuspended in injection solution to assess particle stability before injections (Fig. 1G). There was very little batch-to-batch variability between the four batches both in size and encapsulation efficiency.
Figure 1. Ace-DEX MPs characteristics.

(A-E) Scanning electron microscopic images of PAMP microparticles (MPs). (F) Table of MPs characteristics. (G) Batch-to-batch size, loading, and encapsulation efficiency data for cGAMP loaded MPs. Data are displayed as mean ± S.D.
cGAMP improves anti-tumor efficacy over other PAMPs in Ace-DEX MPs
To assess the anti-tumor activity of PAMP MPs, mice were inoculated with B16F10 melanoma cells. At day six, when tumors were palpable and around 100 mm3, mice were treated intratumorally (i.t.) with PAMPs loaded into Ace-DEX MPs (10 μg PAMP/dose). Mice were then treated every three days for a total of three injections. Once the last injection was complete, mice were continually monitored until they reached the study endpoint. Treatment with one of each of the four PAMP MPs showed significant tumor inhibition compared to PBS and Blank MP-treated mice (Fig. 2A). Mean survival was also increased in mice treated with cGAMP MPs, imiquimod MPs, and poly IC MPs (Fig. 2B). Although all compounds resulted in a decrease in tumor size, mice treated with the cGAMP MPs resulted in significantly smaller tumors compared to other PAMP MPs (p<0.05) (Fig. 2A). Based on these data, cGAMP MPs were selected as the optimal anti-tumor formulation and were used for all subsequent studies.
Figure 2. cGAMP MPs enhance anti-tumor immune response over other PAMPs and increase biological activity in vitro.

(A-B) Mice were inoculated with B16F10 cells on day 0. Tumors were treated with either PBS, Blank MPs, or 10 μg of encapsulated PAMPs by i.t. administration on day 6, 9, and 12. Tumors were measured until diameter >20mm. (C-E) BMDCs were treated with either 0.1 or 1 μg/mL of cGAMP (either as free drug or encapsulated in Ace-DEX MPs) for 24 hours. Supernatants were then collected and run on ELISA. (A-B. n=10 over 2 experiments, (C-E) n=6 over 3 experiments ± SEM). A. Two-way ANOVA, B. Mantel Cox test, C-E. One-way ANOVA. * p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001
Anti-tumor activity of cGAMP is enhanced by Ace-DEX MP delivery in vitro
STING agonists, such as cyclic dinucleotides (CDNs) have recently gathered a great deal of interest as a cancer immunotherapy and are currently in clinical trials for treatment of solid tumors7,49–51[ NCT02675439, NCT03010176]. However, due to the hydrophilic, charged nature of STING agonists and STING’s localization in the cytosol, large quantities are needed to achieve biological effects7,52. Large quantities of STING agonists have caused T cell apoptosis16–18. Thus, there is interest in improving the delivery of STING agonists. In this study, we tested three doses of both encapsulated and soluble cGAMP in vitro. In agreement with previous literature, cGAMP Ace-DEX MPs enhance the pro-inflammatory cytokine secretion over soluble cGAMP at two doses in BMDCs (Fig. 2C–E).
cGAMP MP treatment inhibits tumor burden using four different routes of administration.
Cyclic dinucleotides have been delivered as anti-tumor therapeutics via several routes of administration7,49–51. However, the optimal route of administration for an immunotherapy has not been extensively investigated. To determine the optimal route of cGAMP MPs delivery, biodistribution experiments were performed on mice bearing B16F10 melanoma tumors. Texas-red labeled Ace-DEX cGAMP MPs were administered by i.p., i.m., i.v., or i.t. route. Mice were harvested 24 hours later and organs were imaged. Averaged radiance was measured and compared to other routes of administration (Fig. 3A, S2). As expected, i.m. resulted in the highest concentration of particles in the injection leg and i.t. resulted in the highest concentration of particles in the tumor (Fig. 3A, S2). The two systemic routes (i.p. and i.v.) resulted in significantly higher particles in the spleen. Only the i.v. route resulted in particles in the lung and liver (Fig. 3A, S2). After all four routes, particles were taken up preferentially by CD3−CD11b+ and CD3−CD11c+ cells, which are APCs (Fig. S3). To assess the tumor immune environment after the four routes of administration, mice were treated with 10 μg of MPs-encapsulated cGAMP administered by an i.p., i.m., i.v., or i.t. route. Three hours later, tumors and serum were harvested and an IFN-β ELISA was run on either tumor homogenate or serum. IFN-β levels were only increased in the tumor after i.t. administration and only increased in the serum after i.v. administration (Fig. 3B). Next, biological efficacy was determined by treating tumor bearing mice with 10 μg of encapsulated cGAMP administered by the four routes a total of three times. All routes of administration resulted in significant tumor inhibition compared to untreated mice (Fig. 3C). Moreover, the i.t. route of cGAMP MP administration resulted in significantly smaller tumor volume compared to both the i.p. or i.m. routes (Fig. 3C). However, tumor growth inhibition between the i.v. and i.t. treated mice was not significantly different (Fig. 3C). As systemic administration of STING agonists may be associated with deleterious effects16–18, further studies were conducted using an i.t. route of administration.
Figure 3. Intratumoral (i.t.) delivery is the optimal route for cGAMP MPs.

Mice were inoculated with B16F10 cells on day 0. (A) On day 10, mice were injected by four routes of administration with 1 mg of Texas-red labeled cGAMP Ace-DEX MPs. Twenty-four hours later, mice were sacrificed and brain, liver, kidneys, spleen, inguinal lymph nodes, lung, and tumors were removed. Average radiance was detected by the IVIS Kinetic at an excitation wavelength of 570nm. (B) On day 10, mice were treated with 10 μg of encapsulated cGAMP. Three hours later, mice were sacrificed and tumor was removed. And homogenized. Supernatants were taken and measured for IFN-β or IL-6 ELISA. (C) On day 6, 9, and 12, mice were injected i.p., i.m., i.v., or i.t. with 10 μg of encapsulated cGAMP. Tumor were monitored every 3 days. (A. n=7–8 mice over 2 experiments, B. n=8 mice over 2 experiments, C. n=10–12 mice over 3 experiments ± SEM). A, B. One-way ANOVA, C. Two-way ANOVA. **p < 0.01, ***p < 0.001, ****p < 0.0001
Anti-tumor activity of cGAMP is enhanced by Ace-DEX MP delivery in vivo
We next compared the biological activity of encapsulated and soluble cGAMP in the B16F10 tumor model. In vivo dose studies illustrate that encapsulated cGAMP significantly inhibited tumor growth at the 0.1 μg cGAMP dose compared to soluble drug (Fig. 4). There was not a significant difference in tumor size between the soluble and encapsulated cGAMP at the 1 and 10 μg doses (Fig. 4) and there was not a significant difference between the tumor growth in three cGAMP MP treated groups.
Figure 4. Ace-DEX MPs enhance cGAMPs biological activity at lower doses.

Mice were inoculated with B16F10 cells on day 0. On day 6, 9, and 12 mice were treated with either PBS, blank Ace-DEX MPs, and 0.1 μg, 1 μg, or 10 μg of either soluble (Sol) or encapsulated cGAMP. Tumor volume was monitored every 3 days (n = 10–12 mice, mean ± SEM over 3 experiments). Two-way ANOVA ***p < 0.001, ****p < 0.0001
The anti-tumor immune response produced by cGAMP MPs is dependent on host cell STING expression
To determine whether the effect of cGAMP MPs are on the host immune cells or directly on the tumor cells, treatment was performed in Tmem173−/− mice, which lack functional STING. Both wild-type (WT) and Tmem173−/− mice were inoculated with B16F10 melanoma cells and then treated with either PBS or cGAMP MPs. Wild-type mice treated with cGAMP MPs had significant tumor growth inhibition compared to mice receiving PBS treatment, irrespective of genotype (Fig. 5). Importantly, cGAMP MPs treated Tmem173−/− mice did not demonstrate impairment of tumor growth (Fig. 5)50. These data demonstrate that the cGAMP MPs anti-tumor efficacy was dependent on STING expression.
Figure 5. cGAMP MPs anti-tumor efficacy is STING dependent.

(A-B) C57BL6/J mice or B6(Cg)-Tmem173tm1.2Camb/J (Tmem173−/−) were inoculated with B16F10 tumors on day 0. Starting on day 6, 9, and 12 mice were treated with either PBS or 10 μg cGAMP encapsulated in Ace-DEX MPs by i.t. administration. Tumor volume was monitored every 3 days and mice were sacrificed when tumors reached 20 mm in diameter (n = 8–10 mice ± mean SEM over 2 experiments). A. Two-way ANOVA, B. Mantel Cox test (B) ***p < 0.001, ****p < 0.0001
Natural killer cells are responsible for the anti-melanoma immune response generated by cGAMP MPs in the B16F10 melanoma model
In agreement with previous literature, we have shown that cGAMP MPs are primarily taken up by the passive targeting of phagocytes53,54 (Fig. S3). We next investigated the changes in tumor infiltrating leukocyte populations after cGAMP MP treatment. Mice were inoculated with the B16F10 tumor cells and treated every three days for three total injections. Mice were then sacrificed on day 15 and tumors were harvested. Tumor infiltrating leukocytes were analyzed by flow cytometry. T cells were defined as CD3+, NK cells were defined as CD3-NK1.1+, and APCs were defined as CD3−CD11b+/CD11c+. Mice treated with cGAMP MPs had a significant increase in tumor infiltrating leukocytes (Fig. 6, Fig. S4 A–B). As expected, tumor infiltrating APCs marked by CD11b and CD11c were increased while increases in CD4+ and CD8+ T cells did not reach statistical significance (Fig. S4 A–D). Additionally, the percentage of tumor infiltrating NK cells was significantly increased after cGAMP MP treatment (Fig. 6A). Interestingly, the percentage of granzyme B+ NK cells was also increased (Fig. 6B), suggesting that the cGAMP MP treatment resulted in an increased recruitment of cytotoxic NK cells to the tumor environment. When treated with 0.1 μg of cGAMP encapsulated in Ace-DEX MPs, the number of NK cells was not significantly increased, however, the percentage of activated NK cells was significantly higher compared to PBS, blank MP, and an equivalent amount of soluble cGAMP (Fig. 6C, D). There was a negative correlation between NK cell number and tumor volume (Fig. S5) that was only significant for the mice treated with cGAMP MPs (Fig. S5).
Figure 6. cGAMP MP anti-tumor efficacy is NK cell dependent in melanoma model.

(A-J) Mice were inoculated with B16F10 cells on day 0. (A-D) On day 6, 9, and 12 mice were treated with cGAMP (A-B 10 μg, C-D 0.1 μg) encapsulated in Ace-DEX microparticles (MPs) by i.t. administration. On day 15, mice were sacrificed and tumors were processed and stained for the presence of tumor infiltrating leukocytes by flow cytometry (n=9–10 mice, mean ± SEM over 2 experiments). (E-J) On day 5, mice received their first i.p. injection of 400 μg of either an isotype control, or an αCD4, αCD8, or αNK1.1 depletion antibody. Mice continued to receive this dose twice a week for the reminder of the study. On day 6, 9, and 12 mice were treated with either PBS or 10 μg cGAMP encapsulated in Ace-DEX MPs. Tumor volume was monitored every 3 days for 30 days. The isotype control groups are repeated in panels E-G. (n=10 mice, mean ± SEM over 2 experiments). A-D. One-way ANOVA, E-G. Two-way ANOVA, *p < 0.05, **p < 0.01, ****p < 0.0001, #p< 0.05, ##p < 0.01, ###p<0.001, †p< 0.05
To determine if NK cells, CD4+ T cells, or CD8+ T cells were functionally necessary for the anti-tumor response after cGAMP MP treatment, cell specific depletion antibodies were used. Mice were treated with either an isotype control IgG antibody, anti-NK1.1, anti-CD4, or anti-CD8 depletion antibodies. The efficiency of depletion was empirically confirmed by flow cytometry (Fig. S6). Consistent with the increased percentages of total NK cells (Fig. 6A) and cytotoxic NK cells (Fig. 6B) in the tumor microenvironment, only the NK cell depletion with anti-NK1.1 antibody reduced cGAMP MP-mediated tumor growth inhibition (Fig. 6E–G). The 30-day survival curves support the tumor volume data where anti-NK1.1 negated the benefit of cGAMP MPs and animals in this group showed significantly lower mean survival (Fig. 6H–J). Mice treated with cGAMP MPs after CD4+ or CD8+ T cell depletion did not have significantly altered mean survival times compared to the mice treated with cGAMP MPs with the isotype control (Fig. 6H–J). These data demonstrate that NK cells are responsible for direct tumor killing after cGAMP MP treatment in the B16F10 melanoma model.
A mixture of natural killer cells and T cells are responsible for the anti-tumor immune response generated by cGAMP MPs in a triple negative breast cancer (TNBC) model
The above experiments demonstrate that tumor growth inhibition in B16F10 inoculated mice treated with cGAMP MPs was mediated by NK cells. However, the B16F10 melanoma model is an aggressive and fast-growing cancer model. When no treatment was administered, only 50% survival remained at around day 15. The rapid kinetics of the B16F10 model may not be optimal for the assessment of a T cell role after cGAMP MP treatment. To determine if T cells played a role in the anti-tumor efficacy of cGAMP MPs, we treated mice with 0.1 μg of encapsulated cGAMP in a second murine tumor model with slower growth kinetics. We selected the E0771 TNBC model, where 50% survival occurs around day 28. This allowed for seven injections of the cGAMP MPs compared to three in the B16F10 model. It gave more time for a T cell response to develop, as an antigen specific T cell response takes days to weeks55,56. We also selected a TNBC model because this subtype is the most aggressive form of breast cancer and no effective targeted therapies have been developed57. Thus, TNBC is a good candidate for intervention with novel immunotherapies. The anti-tumor effect was recapitulated in the E0771 mouse model (Fig. 7A, B). Tumor growth was inhibited after cGAMP MP treatment comparedto PBS, blank MP, and soluble cGAMP treatments (Fig. 7A), and cGAMP MP treatment resulted in a significantly increased mean survival time (Fig. 7B). These data confirm that the tumor growth inhibition by cGAMP MP was not a model dependent phenomenon.
Figure 7. cGAMP MP anti-tumor efficacy is NK and CD8+ T cell dependent in a TNBC model.

(A-E) Mice were inoculated with E0771 cells on day 0. Starting on day 6, mice were treated with either PBS or 0.1 μg cGAMP encapsulated in Ace-DEX microparticles (MPs) by i.t. administration for a total of 5 (C-E) or 7 injections (A-B). Tumor volume was monitored every 3 days. The isotype control groups are repeated in panels C-E. On day 21 mice were sacrificed and tumor infiltrated leukocytes were analyzed by flow cytometry (F-G) (A-B. n = 9–10 mice, C-E. n=8–10 mice, F-G. n=3–5 mice mean ± SEM over 2 experiments). A,C-E. Two-way ANOVA, B. Mantel Cox test, F-G. One-way ANOVA. *p < 0.05, **p < 0.01, ****p < 0.0001, ####p<0.0001.
We next performed antibody depletion studies in the E0771 TNBC mouse model to define the immune cell types that conferred anti-tumor immunity upon cGAMP MPs treatment. During the first 12 days of tumor growth assessment, mice treated with cGAMP MPs + αNK1.1 depletion antibody resulted in significantly smaller tumors than those treated with PBS + αNK1.1, but significantly larger tumors then mice treated with cGAMP MPs + Isotype antibody (Fig. S7A). A similar outcome was seen with the mice treated with cGAMP MPs + αCD8 depletion antibody during the first 12 days of tumor assessment (Fig. S7B). This indicates that the reduction of tumor size in the presence of cGAMP MPs was mediated by both NK cells and CD8+ T cells. In contrast to NK or CD8 depletion, CD4 depletion resulted in better tumor control, perhaps due to the removal of CD4+ T regulatory cells (Fig. S7C). By day 18, the size of the tumors from the cGAMP MPs + αNK1.1 depletion was similar to those from the cGAMP MPs + isotype group, indicating that NK cells were no longer contributing to anti-tumor immunity at this later timepoint (Fig. 7C). By contrast, the cGAMP MPs + αCD8 depletion group had significantly larger tumor volume than mice treated with cGAMP MPs + Isotype antibody, indicating the importance of CD8 T cells at this point for tumor control (Fig. 7D). Thus, both NK cells and CD8+ T cells were important in the early cGAMP MP anti-tumor efficacy but at later treatment times, CD8+ T cells dominated the antitumor activity. The mice given the cGAMP MPs + αCD4 depletion antibody had significantly smaller tumors compared to mice given PBS + αCD4 depletion antibody at early timepoints, however, as the depletion continued, both groups resulted in decreasing tumor volume (Fig. 7E). When tumor infiltrating leukocytes were analyzed, tumors from the cGAMP MPs + αNK1.1 depletion had significantly higher CD8+ T cell numbers compared to the cGAMP MPs + isotype control (Fig. 7F). A similar compensation was seen after CD8+ T cell depletion with the activated NK cells, however, this phenomenon was not dependent on cGAMP MP treatment (Fig. 7G).
Discussion
While PAMPs represent attractive adjuvants for cancer immunotherapy, many PRRs reside within the cell and present a challenge for efficient loading and delivery. To determine if delivery with Ace-DEX MPs enhances the biological activity of immune compounds, we tested four encapsulated PAMPs. The four PAMPs were selected because each utilizes a different pathway and each has been used in clinical trials as potential cancer treatments12–15[ NCT02276300, NCT02423863, NCT02675439]. In agreement with previously published literature, all PAMPs tested resulted in inhibited tumor growth12–14,31,58. However, cGAMP MP treated mice had significantly smaller tumors than all other treatments.
CDNs have been successfully delivered as immunotherapies using i.t., s.c., i.m., and i.v. routes of administration7,49–51. Route of administration has also been shown to affect cGAMP activity59. When testing cGAMP as a flu vaccine adjuvant, an intradermal injection in combination with a flu antigen resulted in enhanced protection compared to i.m. delivery59. However, the route of administration of cGAMP has not been directly compared within the context of tumor immunotherapy. Here we have shown that route of administration significantly changes the biodistribution and cytokine profile of the cGAMP MPs. We then demonstrated that cGAMP MPs significantly reduce tumor growth via multiple routes of administration, of which i.t. injections resulted in the most significant tumor growth inhibition. CDNs have been given by i.t. injection for the treatment of melanoma previously and it is currently being investigated in clinical trials7,50,51,60,61 [ NCT02675439]. Thus, for accessible tumors, i.t. injections are a translationally relevant route of administration for the cGAMP MPs. For inaccessible tumors, our results indicate that cGAMP MPs delivered by an i.v., i.p., or an i.m route of administration also retain significant anti-tumor efficacy.
Previously reported i.t. delivery of soluble STING agonists has resulted in tumor growth inhibition7,50,51,60,61. Although anti-tumor efficacy has been achieved through this local delivery of soluble cGAMP, high soluble concentrations (100–400 μg) are required for in vivo efficacy7,52. The high concentrations of STING agonists are required due to the cytosolic localization of the PRR. CDNs, such as cGAMP, are hydrophilic/charged molecules that cannot easily transverse the cell membrane to activate STING. The low molecular weight of cGAMP may also results in rapid diffusion from the injection site, decreasing potency22. For example, i.t. injections of STING agonists in clinical trials has begun at 50 μg per human dose [ NCT02675439]. Other groups have previously shown that incorporation of cGAMP into liposomes reduced the cGAMP dose required to inhibit tumor growth to 10 μg cGAMP, a 10-fold dose reduction compared to soluble CDN51,61. However, Ace-DEX MPs have several advantages over other formulations. Electrospraying cGAMP into Ace-DEX MPs results in high (>90%) encapsulation efficiency of the molecule, resulting in less wasted adjuvant (Fig. 1). The particles are stable over months of storage and are consistent across batches. Here, we achieve tumor growth inhibition in two challenging murine tumor models and increase mean survival time in a TNBC murine model at 0.1 μg dose of cGAMP delivered using Ace-DEX MPs, resulting in a 100-fold dose sparing over published cGAMP liposomes51,61 and up to 1,000-fold dose sparing compared to literature reports for soluble cGAMP7,52. The 0.1 μg cGAMP dose is the lowest reported amount that results in significant tumor inhibition62. A reduced amount of drug necessary to obtain a biological effect is desirable for translational application as high doses of STING agonists has been reported to cause T cell apoptosis.16–18. Encapsulation also achieves targeted delivery, mitigating effects on off target cells.
STING agonists have been shown to depend on M1 macrophage polarization, as well as, T cell and NK cell activation for anti-tumor efficacy63–65. The effect of cGAMP on macrophages has been investigated elsewhere63 and was not addressed in this paper. CD8+ T cells and NK cells are the two primary immune cell types responsible for direct tumor killing. We have shown that tumor infiltrating NK cells play a significant role in the anti-tumor immune response generated by cGAMP MPs. Although the cGAMP MPs may be mechanistically similar to soluble cGAMP, our cGAMP MPs have enhanced the biological efficacy of cGAMP and thus, we achieved NK cell activation at low doses cGAMP (Fig. 6D). The MPs alone have no effect on the anti-tumor immunity (Fig. 1), illustrating that the Ace-DEX MPs are inert and it is the encapsulation of cGAMP enhances the biological activity. After treatment with cGAMP MPs, the tumor infiltrating NK cells appeared to have enhanced killing function, as evidenced by an increase in NK cell granzyme B expression. Furthermore, depletion of NK cells significantly reduced the efficacy of cGAMP MPs directly implicating NK cells in the mechanism of action.
The cGAMP MP anti-tumor efficacy was completely NK cell-dependent in the B16F10 melanoma model, but dependent on both NK cells and CD8+ T cells in the E0771 TNBC model, which had a slower tumor growth kinetics. Both NK cells and CD8+ T cells have previously been partially implicated in the anti-tumor immune response after CDN delivery,44,45,64. For example, i.t. injection of soluble cGAMP in a B16F10 melanoma model resulted in tumor growth inhibition. Depletion of CD8+ T cells during cGAMP treatment resulted in increased tumor size. However, tumor growth inhibition was still observed when compared to untreated tumors45. A partial dependence upon CD8+ T cells after cGAMP treatment was also observed in the 4T1 TNBC model and a B16F10 metastasis model as well50,61. We have shown that, depending on the model, STING-dependent anti-tumor function can be dependent on NK cells alone or a combination of NK cells and CD8+ T cells. The discrepancies between the different tumor models can be explained by the CDN formulation and/or timing of the mouse model. In two of the studies, soluble or micellar cGAMP formulations were delivered by i.t. injections. Due to the smaller size of the particles, they have less selectivity then the MPs. We have shown that cGAMP Ace-DEX MPs are preferentially taken up by APCs, allowing for a more targeted delivery to APCs. Targeted delivery can mitigate the effects on off target cells, thus adding an advantage to the cGAMP MPs16–18. The mechanistic differences could also be due to a model dependent phenomenon. The B16F10 melanoma model used in our study was fast growing and highly necrotic, whereas, the E0771 TNBC was a slower growing, and more solid tumor. It can take days to weeks to mount a full, robust adaptive immune response55,56. Therefore, the B16F10 model may suggest the importance for NK cells in early tumor development before CD8+ T cells can begin to respond. This was also observed in the two-phase response to the E0771 TNBC model. At early stages, depletion of NK cells with cGAMP MP treatment results in significantly larger tumors compared to isotype control mice treated with cGAMP MPs (Fig. S6). However, as the model progresses, NK cell depletion plays a less significant role as CD8+ T cells take over (Fig 7C–E). The T cell dependence of the E0771 model was also observed in the CD4 depletion groups. In the later stage tumor development, both PBS and cGAMP MPs treated mice given CD4 depletion resulted in shrinking tumors. We hypothesize that this was due to the importance of regulatory T cells in the E0771 model, as previously reported66. Interestingly, when mice were treated with cGAMP MPs and depleted of NK cells, the number of CD8+ T cells increased within the tumor compared to mice treated with cGAMP MPs and the isotype control. This suggests that when NK cells are absent, the CD8+ T cells compensate for the NK cells absence after cGAMP MP treatment. The same was seen with activated NK cells after CD8+ T cells depletion, however, this occurs regardless of the cGAMP MP treatment. All of these data indicate the importance of NK cells involvement in early stages of tumor immunotherapy after cGAMP MP treatment.
Until recently, NK cells have been overlooked in regard to cancer immunotherapy. The past few years has seen increased interest in NK cells targeted therapies, many of which have entered clinical trials. One such therapy is NK cell adoptive transfer [UMIN000007527, NCT00187096]. However, NK cell adoptive transfer has low efficacy and can result in graft versus host disease if T cells are not properly suppressed67. Recombinant cytokine therapies, such as IL-2 and IL-15, are currently either approved for clinical use or undergoing clinical trials. IL-2 treatment is an FDA approved therapy that effects both T cells and NK cells. IL-15 therapy, which more specifically targets NK cells, is currently in clinical trials [ NCT01385423, NCT01875601]. Both IL-2 and IL-15 therapies have toxicity issues and severe side effects that lead to patient noncompliance67. Checkpoint inhibition has also been used to improve NK cell anti-tumor efficacy. This includes monoclonal antibodies against killer-cell immunoglobulin-like receptor (KLR), a family of receptors highly expressed in NK cells, and NK2GA, a receptor expressed predominantly on NK cells67. However, these checkpoint inhibitors have low efficacy67. In this study, cGAMP MP treatment resulted in a robust NK cell mediated cancer immunotherapy. This NK cell dependent therapy has several advantages over solely NK targeted therapies. We achieved significant tumor growth inhibition and increased mean survival time after cGAMP MP delivery alone. Moreover, the ability to locally deliver cGAMP MPs decreases the likelihood for off target effects often seen with recombinant cytokines (e.g. IL-2, IL-15, and IFN-α).
These data demonstrate that treatment with cGAMP MPs leads to generation of a robust innate and adaptive immune-mediated anti-cancer response. The innate and adaptive immune response are both important when responding to cancer. The majority of cancer immunotherapies have focused on the adaptive immune response because of the ability to generate a memory response. Innate immune cells, such as NK cells, respond faster than the adaptive response. This was illustrated by the significant NK cell reliance in the fast growing B16F10 model and in the early stages of the E0771 TNBC model. This robust innate immune activation in combination with cGAMP’s ability to activate CD8+ T cells creates a balanced anti-tumor immune response, combining a rapid anti-tumor innate immune response, along with the memory and longevity of an adaptive immune response.
Conclusions
In summary, we demonstrated that Ace-DEX MPs encapsulating cGAMP resulted in the most efficacious tumor growth inhibition compared to three clinically relevant PAMP MPs. The cGAMP MPs also enhanced tumor growth inhibition in four routes of administration, but the intratumoral route was the most effective. Ace-DEX MPs enhanced the biological activity of cGAMP both in vitro and in vivo in two, difficult to treat, murine tumor models. We showed that a 0.1 μg dose of cGAMP was needed to obtain this tumor growth inhibition. This was the lowest dose of cyclic dinucleotides reported that results in an anti-tumor immune response. The resulting anti-tumor efficacy was NK cell dependent in the fast growing B16F10 melanoma model while the anti-tumor efficacy of the slower growing TNBC model was dependent on both NK cells and CD8+ T cells. This works shows that NK cells are important in the initial stages of the STING-dependent, anti-tumor immune response. Overall, these data provide new insights into patient treatment options and the mechanism of STING dependent anti-tumor immune response.
Supplementary Material
Acknowledgments
This work was supported by the National Institute of Health [U19AI109784 (JPT and EMB), U54CA198999 (JPT), U19AI067798 (JPT), T32AI007273 (RWS), T32CA196589 (PT), and T32AI007151 (RDJ)], the North Carolina Biotechnology Center Biotechnology Innovation (BIG) Grant, and a University of Cancer Research Fund Lineberger development grant. We would like to thank the UNC Flow Cytometry Core Facility, supported in part by P30 CA016086 Cancer Center Core Support Grant to the UNC Lineberger Comprehensive Cancer Center. We would also like to thank the Chapel Hill Analytical and Nanofabrication Laboratory, CHANL, a member of the North Carolina Research Triangle Nanotechnology Network, RTNN. CHANL is supported by the National Science Foundation, Grant ECCS-1542015, as part of the National Nanotechnology Coordinated Infrastructure, NNCI. We would also like to thank members of Jenny Ting’s lab, especially Drs. Dana Elmore, Sara Gibson, and Jason Tam for their input.
Footnotes
Conflict of interest: Drs. Ainslie, Bachelder, and Ting are founders and serve on the advisory board for IMMvention Therapeutix, Inc. Although a financial conflict of interest was identified for management based on the overall scope of the project and its potential benefit to IMMvention Therapeutix, Inc., the research findings included in the publication may not necessarily relate to the interests of IMMvention Therapeutix, Inc. The terms of this arrangement have been reviewed by the University of North Carolina at Chapel Hill in accordance with its policy on objectivity in research.
References
- 1.Vesely MD, Kershaw MH, Schreiber RD & Smyth MJ Natural Innate and Adaptive Immunity to Cancer. Annu. Rev. Immunol 29, 235–271 (2011). [DOI] [PubMed] [Google Scholar]
- 2.Blattman JN & Greenberg PD Cancer immunotherapy: a treatment for the masses. Science 305, 200–5 (2004). [DOI] [PubMed] [Google Scholar]
- 3.Lee S & Margolin K Cytokines in cancer immunotherapy. Cancers (Basel). 3, 3856–93 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lejeune FJ & Rüegg C Recombinant human tumor necrosis factor: an efficient agent for cancer treatment. Bull. Cancer 93, E90–100 (2006). [PubMed] [Google Scholar]
- 5.Taeger G et al. Effectiveness of regional chemotherapy with TNF-α/Melphalan in advanced soft tissue sarcoma of the extremities. Int. J. Hyperth 24, 193–203 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Trinchieri G Type I interferon: friend or foe? J Exp Med 207, 2053–2063 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Li T et al. Antitumor Activity of cGAMP via Stimulation of cGAS-cGAMP-STING-IRF3 Mediated Innate Immune Response. Sci Rep 6, 19049 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mogensen TH Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev 22, 240–73, Table of Contents (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li J et al. Self-Assembled Multivalent DNA Nanostructures for Noninvasive Intracellular Delivery of Immunostimulatory CpG Oligonucleotides. ACS Nano 5, 8783–8789 (2011). [DOI] [PubMed] [Google Scholar]
- 10.Amorij J-P et al. Towards tailored vaccine delivery: Needs, challenges and perspectives. J. Control. Release 161, 363–376 (2012). [DOI] [PubMed] [Google Scholar]
- 11.Hobohm U, Stanford JL & Grange JM Pathogen-associated molecular pattern in cancer immunotherapy. Crit. Rev. Immunol 28, 95–107 (2008). [DOI] [PubMed] [Google Scholar]
- 12.Ammi R et al. Poly(I:C) as cancer vaccine adjuvant: Knocking on the door of medical breakthroughs. Pharmacol. Ther 146, 120–131 (2015). [DOI] [PubMed] [Google Scholar]
- 13.Schön MP & Schön M TLR7 and TLR8 as targets in cancer therapy. Oncogene 27, 190–199 (2008). [DOI] [PubMed] [Google Scholar]
- 14.BAHR GM, DARCISSAC E, POUILLART PR & CHEDID LA Synergistic Effects Between Recombinant Interleukin-2 and the Synthetic Immunomodulator Murabutide: Selective Enhancement of Cytokine Release and Potentiation of Antitumor Activity. J. Interf. Cytokine Res 16, 169–178 (1996). [DOI] [PubMed] [Google Scholar]
- 15.Corrales L et al. Direct Activation of STING in the Tumor Microenvironment Leads to Potent and Systemic Tumor Regression and Immunity. Cell Rep 11, 1018–1030 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Larkin B et al. Cutting Edge: Activation of STING in T Cells Induces Type I IFN Responses and Cell Death. J. Immunol 199, 397–402 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gulen MF et al. Signalling strength determines proapoptotic functions of STING. Nat. Commun 8, 427 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu Y et al. Activated STING in a Vascular and Pulmonary Syndrome. N. Engl. J. Med 371, 507–518 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bachelder EM, Pino EN & Ainslie KM Acetalated Dextran: A Tunable and Acid-Labile Biopolymer with Facile Synthesis and a Range of Applications. Chem Rev 117, 1915–1926 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Zhu G, Zhang F, Ni Q, Niu G & Chen X Efficient Nanovaccine Delivery in Cancer Immunotherapy. ACS Nano 11, 2387–2392 (2017). [DOI] [PubMed] [Google Scholar]
- 21.Huang X; Zhang T; Song Y; She Z; Li J; Deng Y, Z. L Progress involving new techniques for liposome preparation. Asian J Pharm Sci 9, 176–182 (2014). [Google Scholar]
- 22.Hanson MC et al. Nanoparticulate STING agonists are potent lymph node-targeted vaccine adjuvants. J Clin Invest 125, 2532–2546 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Makadia HK & Siegel SJ Poly Lactic-co-Glycolic Acid (PLGA) as Biodegradable Controlled Drug Delivery Carrier. Polym. 3, 1377–1397 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mammadov R et al. Virus-like nanostructures for tuning immune response. Sci. Rep 5, 16728 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wilson KD, de Jong SD & Tam YK Lipid-based delivery of CpG oligonucleotides enhances immunotherapeutic efficacy. Adv. Drug Deliv. Rev 61, 233–242 (2009). [DOI] [PubMed] [Google Scholar]
- 26.Gunay G et al. Antigenic GM3 Lactone Mimetic Molecule Integrated Mannosylated Glycopeptide Nanofibers for the Activation and Maturation of Dendritic Cells. ACS Appl. Mater. Interfaces 9, 16035–16042 (2017). [DOI] [PubMed] [Google Scholar]
- 27.Bachelder EM, Beaudette TT, Broaders KE, Dashe J & Fréchet JMJ Acetalderivatized dextran: an acid-responsive biodegradable material for therapeutic applications. J. Am. Chem. Soc 130, 10494–5 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Broaders KE, Cohen JA, Beaudette TT, Bachelder EM & Frechet JM Acetalated dextran is a chemically and biologically tunable material for particulate immunotherapy. Proc Natl Acad Sci U S A 106, 5497–5502 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kauffman KJ et al. Synthesis and characterization of acetalated dextran polymer and microparticles with ethanol as a degradation product. ACS Appl Mater Interfaces 4, 4149–4155 (2012). [DOI] [PubMed] [Google Scholar]
- 30.Junkins RD et al. A robust microparticle platform for a STING-targeted adjuvant that enhances both humoral and cellular immunity during vaccination. J Control Release (2017). doi: 10.1016/j.jconrel.2017.11.030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen N et al. Tunable degradation of acetalated dextran microparticles enables controlled vaccine adjuvant and antigen delivery to modulate adaptive immune responses. J. Control. Release 273, 147–159 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gallovic MD et al. Acetalated Dextran Microparticulate Vaccine Formulated via Coaxial Electrospray Preserves Toxin Neutralization and Enhances Murine Survival Following Inhalational Bacillus Anthracis Exposure. Adv. Healthc. Mater 5, 2617–2627 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Schully KL et al. Rapid vaccination using an acetalated dextran microparticulate subunit vaccine confers protection against triplicate challenge by bacillus anthracis. Pharm Res 30, 1349–1361 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Schully KL et al. Evaluation of a biodegradable microparticulate polymer as a carrier for Burkholderia pseudomallei subunit vaccines in a mouse model of melioidosis. Int. J. Pharm 495, 849–61 (2015). [DOI] [PubMed] [Google Scholar]
- 35.Peine KJ et al. Efficient delivery of the toll-like receptor agonists polyinosinic:polycytidylic acid and CpG to macrophages by acetalated dextran microparticles. Mol Pharm 10, 2849–2857 (2013). [DOI] [PubMed] [Google Scholar]
- 36.Bachelder EM et al. In vitro analysis of acetalated dextran microparticles as a potent delivery platform for vaccine adjuvants. Mol. Pharm 7, 826–35 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Collier MA et al. Acetalated Dextran Microparticles for Codelivery of STING and TLR7/8 Agonists. Mol. Pharm 15, 4933–4946 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Duong AD et al. Electrospray Encapsulation of Toll-Like Receptor Agonist Resiquimod in Polymer Microparticles for the Treatment of Visceral Leishmaniasis. Mol. Pharm 10, 1045–1055 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kauffman KJ et al. Synthesis and Characterization of Acetalated Dextran Polymer and Microparticles with Ethanol as a Degradation Product. ACS Appl. Mater. Interfaces 4, 4149–4155 (2012). [DOI] [PubMed] [Google Scholar]
- 40.Kanthamneni N et al. Enhanced stability of horseradish peroxidase encapsulated in acetalated dextran microparticles stored outside cold chain conditions. Int J Pharm 431, 101–110 (2012). [DOI] [PubMed] [Google Scholar]
- 41.Almeria B, Fahmy TM & Gomez A A multiplexed electrospray process for singlestep synthesis of stabilized polymer particles for drug delivery. J Control Release 154, 203–210 (2011). [DOI] [PubMed] [Google Scholar]
- 42.Gallovic MD et al. Chemically modified inulin microparticles serving dual function as a protein antigen delivery vehicle and immunostimulatory adjuvant. Biomater. Sci 4, 483–93 (2016). [DOI] [PubMed] [Google Scholar]
- 43.Bubeník J MHC class I down-regulation: tumour escape from immune surveillance? (review). Int. J. Oncol 25, 487–91 (2004). [PubMed] [Google Scholar]
- 44.Afonina IS, Cullen SP & Martin SJ Cytotoxic and non-cytotoxic roles of the CTL/NK protease granzyme B. Immunol. Rev 235, 105–116 (2010). [DOI] [PubMed] [Google Scholar]
- 45.Kohlmeier JE, Cookenham T, Roberts AD, Miller SC & Woodland DL Type I interferons regulate cytolytic activity of memory CD8(+) T cells in the lung airways during respiratory virus challenge. Immunity 33, 96–105 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Madera S et al. Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J. Exp. Med 213, 225–33 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Faustino-Rocha A et al. Estimation of rat mammary tumor volume using caliper and ultrasonography measurements. Lab Anim. (NY). 42, 217–224 (2013). [DOI] [PubMed] [Google Scholar]
- 48.Moynihan KD et al. Eradication of large established tumors in mice by combination immunotherapy that engages innate and adaptive immune responses. Nat. Med 22, 1402–1410 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Wang H et al. cGAS is essential for the antitumor effect of immune checkpoint blockade. Proc Natl Acad Sci U S A 114, 1637–1642 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohkuri T et al. Intratumoral administration of cGAMP transiently accumulates potent macrophages for anti-tumor immunity at a mouse tumor site. Cancer Immunol Immunother 66, 705–716 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koshy Alexander S.; Gu Lu; Graveline Amanda R.; Mooney David J., S. T. C Liposomal Delivery Enhances Immune Activation by STING Agonists for Cancer Immunotherapy. Adv. Biosys 1, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Tao J, Zhou X & Jiang Z cGAS-cGAMP-STING: The three musketeers of cytosolic DNA sensing and signaling. IUBMB Life 68, 858–870 (2016). [DOI] [PubMed] [Google Scholar]
- 53.Foged C, Brodin B, Frokjaer S & Sundblad A Particle size and surface charge affect particle uptake by human dendritic cells in an in vitro model. Int J Pharm 298, 315–322 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Hirota K et al. Optimum conditions for efficient phagocytosis of rifampicin-loaded PLGA microspheres by alveolar macrophages. J Control Release 119, 69–76 (2007). [DOI] [PubMed] [Google Scholar]
- 55.Gajewski TF, Schreiber H & Fu Y-X Innate and adaptive immune cells in the tumor microenvironment. Nat. Immunol 14, 1014–1022 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Restifo NP, Dudley ME & Rosenberg SA Adoptive immunotherapy for cancer: harnessing the T cell response. Nat. Rev. Immunol 12, 269–281 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Neophytou C, Boutsikos P & Papageorgis P Molecular Mechanisms and Emerging Therapeutic Targets of Triple-Negative Breast Cancer Metastasis. Front. Oncol 8, 31(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Drobits B et al. Imiquimod clears tumors in mice independent of adaptive immunity by converting pDCs into tumor-killing effector cells. J. Clin. Invest 122, 575–85 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang J, Li P & Wu MX Natural STING Agonist as an ‘Ideal’ Adjuvant for Cutaneous Vaccination. J. Invest. Dermatol 136, 2183–2191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Demaria O et al. STING activation of tumor endothelial cells initiates spontaneous and therapeutic antitumor immunity. Proc. Natl. Acad. Sci 112, 15408–15413 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Nakamura T et al. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma. J. Control. Release 216, 149–157 (2015). [DOI] [PubMed] [Google Scholar]
- 62.Wilson DR et al. Biodegradable STING agonist nanoparticles for enhanced cancer immunotherapy. Nanomedicine Nanotechnology, Biol. Med 14, 237–246 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ohkuri T, Kosaka A, Nagato T & Kobayashi H Effects of STING stimulation on macrophages: STING agonists polarize into ‘classically’ or ‘alternatively’ activated macrophages? Hum Vaccin Immunother 0 (2017). doi: 10.1080/21645515.2017.1395995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nakamura T et al. Liposomes loaded with a STING pathway ligand, cyclic di-GMP, enhance cancer immunotherapy against metastatic melanoma ☆. J. Control. Release 216, 149–157 (2015). [DOI] [PubMed] [Google Scholar]
- 65.Takashima K et al. STING in tumor and host cells cooperatively work for NK cell-mediated tumor growth retardation. (2016). doi: 10.1016/j.bbrc.2016.09.021 [DOI] [PubMed]
- 66.Huang Y et al. CD4+and CD8+ T cells have opposing roles in breast cancer progression and outcome. Oncotarget 6, 17462–78 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Guillerey C, Huntington ND & Smyth MJ Targeting natural killer cells in cancer immunotherapy. Nature Immunology (2016). doi: 10.1038/ni.3518 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.